
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of PEG-lipid anchor length on lipid nanoparticle pharmacokinetics and activity in a mouse model of traumatic brain injury†
Lauren E.
Waggoner‡
 a,
Katelyn F.
Miyasaki‡
b and
Ester J.
Kwon
*b
a,
Katelyn F.
Miyasaki‡
b and
Ester J.
Kwon
*b
aDepartment of Nanoengineering, University of California, San Diego, La Jolla, CA 92093, USA
bDepartment of Bioengineering, University of California, San Diego, La Jolla, CA 92093, USA. E-mail: ejkwon@ucsd.edu
First published on 29th March 2023
Abstract
Traumatic brain injury (TBI) affects millions of people worldwide, yet there are currently no therapeutics that address the long-term impairments that develop in a large portion of survivors. Lipid nanoparticles (LNPs) are a promising therapeutic strategy that may address the molecular basis of TBI pathophysiology. LNPs are the only non-viral gene delivery platform to achieve clinical success, but systemically administered formulations have only been established for targets in the liver. In this work, we evaluated the pharmacokinetics and activity of LNPs formulated with polyethylene glycol (PEG)-lipids of different anchor lengths when systemically administered to a mouse model of TBI. We observed an increase in LNP accumulation and activity in the injured brain hemisphere compared to the uninjured contralateral brain hemisphere. Interestingly, transgene expression mediated by LNPs was more durable in injured brain tissue compared to off-target organs when compared between 4 and 24 hours. The PEG-lipid is an important component of LNP formulation necessary for the stable formation and storage of LNPs, but the PEG-lipid structure and content also has an impact on LNP function. LNP formulations containing various ratios of PEG-lipid with C18 (DSPE-PEG) and C14 (DMG-PEG) anchors displayed similar physicochemical properties, independent of the PEG-lipid compositions. As the proportion of DSPE-PEG was increased in formulations, blood circulation times of LNPs increased and the duration of expression increased. We also evaluated diffusion of LNPs after convection enhanced delivery (CED) in healthy brains and found LNPs distributed >1 mm away from the injection site. Understanding LNP pharmacokinetics and activity in TBI models and the impact of PEG-lipid anchor length informs the design of LNP-based therapies for TBI after systemic administration.
Introduction
Traumatic brain injury (TBI) impacts millions per year globally and there are currently no clinically approved therapeutics that address long-term patient brain health,1–3 with 43% of hospitalized TBI survivors developing long-term disabilities,4 including cognitive, motor, and psychosocial impairments.5 The primary injury causes immediate cellular and structural damage due to the physical impact, and initiates a secondary injury that lasts days to weeks, during which a number of biochemical and physiological responses lead to progressive neurodegeneration and eventual functional impairments.6 Gene therapy mediated by lipid nanoparticles (LNPs) is one potential therapy for TBI that could address disease pathways during the secondary injury. While viral vectors are highly efficient, LNPs are a potential alternative that have lower immunogenicity7 and are less limited by cargo size.8,9 In addition, the activity of LNPs is more transient compared to viral vectors,10–13 making them well-matched to treat the acute phase of TBI within days to weeks of the injury.14–16 LNPs are currently the most clinically advanced non-viral gene delivery vehicle, and multiple formulations have been approved by the FDA.17–19 Patisiran, an LNP treatment for hereditary transthyretin-mediated amyloidosis, was approved in 2018 and was the first LNP and siRNA therapeutic approved for clinical use.17,20,21 In 2020, mRNA LNPs were developed as vaccines against SARS-CoV-2 and received emergency use authorization for the COVID-19 pandemic22–24 followed by full FDA approval shortly after in the subsequent 1–2 years.18,19 There are multiple siRNA and mRNA LNPs in the clinical pipeline as therapeutics for cancer, infections, and genetic disorders.25–27 In this work, we evaluate the potential of LNPs for non-viral gene delivery to the injured brain in a mouse model of TBI.TBI causes dysregulation of the blood–brain barrier (BBB), hallmarks of which include cell death and loss of tight junction proteins,28 both of which cause an increase in BBB permeability; this increased permeability creates an opportunity for access of nanoscale materials into injured brain tissue after systemic delivery. Our group and others have demonstrated increased transport of a variety of nanoparticle types into the injured brain exploiting this transient dysregulation.28–31 Appreciable passive nanoparticle accumulation in the injured tissue has been reported for diameters ∼100 nm in size after intravenous administration up to 24 hours after TBI.32–35 The principle of nanoparticle accumulation in the injured brain is analogous to the enhanced permeation and retention (EPR) effect described in tumors.36,37 Passive accumulation of nanoparticles in tumors and the injured brain is known to be impacted by the physicochemical properties of nanoparticles, including size,33,38 charge,39–41 and surface chemistry.36,42 LNP pharmacokinetics are also impacted by their physicochemical properties. For example, increased lung, liver, and spleen LNP accumulation have been achieved by tuning LNP charge through formulation with specific organ targeting (SORT) cationic, zwitterionic, and anionic lipids, respectively.43 Size-based differences in organ accumulation have also been observed, with 80 nm LNPs preferentially accumulating more in the spleen compared to 45 nm and 30 nm LNPs.44
One aspect of nanoparticle physicochemical property is hydrophilicity, typically tuned in LNPs by the inclusion of polyethylene glycol (PEG)-lipids in the formulation. PEG has been incorporated in nanomedicine for decades in order to increase blood half-life through reducing uptake by the reticuloendothelial system (RES).45 Generally, increased passive accumulation is observed by nanoparticles with longer blood half-lives.39,41,45–47 For example, in a breast cancer model, the addition of PEG to paclitaxel liposomes increased blood area under the curve values ∼4.5-fold, and thus increased paclitaxel tumor accumulation ∼1.5- and 4-fold at 6 and 24 hours after systemic administration.48 Similarly, PEG-lipids are included in LNP formulations to extend their blood circulation, in addition to improving their stability during synthesis and storage.49,50 However, PEG is also a steric barrier that hinders cellular interaction and endosomal escape, crucial steps in payload delivery.51–54 Recent research has demonstrated that PEG mole percent (mol%),55–57 PEG molecular weight,58 and PEG-lipid anchor length55,59,60 affect the activity and pharmacokinetic profiles of LNPs. Reduction in the amount of PEG-lipid in LNP formulations mediated more transfection in hepatocytes in primary cell culture and after systemic administration in vivo due to their increased cellular interactions.56 In studies of LNPs formulated with different PEG-lipid anchor lengths, Mui et al. demonstrated that short C14 anchor PEG-lipids desorb quickly from LNP formulations in the blood, leading to accumulation in the liver, while LNP formulations with long C18 anchor PEG-lipids had longer blood half-lives and subsequently accumulated less in RES organs.55 Previous research has also demonstrated that PEG-lipid anchor length affects passive accumulation into tumors after systemic administration; LNPs with longer anchor PEG-lipids demonstrate greater tumor accumulation due to their longer blood half-lives.59 However, PEG-lipid anchor lengths have yet to be investigated as a design parameter for systemic LNP delivery to the brain in the context of TBI.
In this work, our goal was to investigate PEG-lipid anchor lengths as a design parameter for passive accumulation of LNPs into the injured brain after systemic administration in a mouse model of TBI. We formulated siRNA and mRNA LNPs with various ratios of DMG-PEG, a PEG-lipid with a “short” C14 anchor capable of quickly desorbing from the LNP once in circulation,55,60 and DSPE-PEG, a PEG-lipid with a “long” C18 anchor with slower desorption kinetics.55,59,60 When administered systemically in a mouse model of TBI, we observed greater accumulation and activity of all LNP formulations in the injured brain hemisphere compared to the uninjured contralateral brain hemisphere. In comparing how PEG-lipid composition impacts pharmacokinetics and activity, we found LNP formulations with more DSPE-PEG had longer blood half-lives and reduced activity in off-target organs 4 hours after administration. Interestingly, LNP activity in injured brain tissue remained constant or increased from 4 to 24 hours, while off-target organ LNP activity decreased ∼10-fold and this phenomenon was more pronounced as DSPE-PEG content increased. We next sought to determine if LNPs distributed through the brain microenvironment after convection enhanced delivery (CED) and found that they distributed at least 1 mm away from the injection site and interacted with neurons and microglia. Our results suggest that PEG-lipid anchor length is an important parameter in tuning the pharmacokinetics and activity of LNPs for application in TBI. Understanding LNP pharmacokinetics and activity in TBI models and the impact of PEG-lipid anchor length informs the design of LNP-based therapies for TBI after systemic administration.
Results and discussion
Characterization of LNPs formulated with various PEG-lipid compositions
Clinical formulations have so far been applied to the liver after systemic delivery or vaccinations after intramuscular delivery and therefore LNPs were designed to desorb PEG-lipid quickly in the biological milieu to interact with target cells and tissues. The PEG-lipids used in FDA-approved LNP formulations have two C14 alkyl chains, allowing fast desorption kinetics,55,59,60 and include PEG2000-C-DMG (patisiran), PEG2000-DMG (elasomeran), and ALC-0159 (tozinameran). The only approved systemically administered LNP, patisiran, targets the liver through the desorption of PEG2000-C-DMG,61 allowing for the adsorption of apolipoprotein E (ApoE) that interacts with hepatocytes through the low-density lipoprotein receptor.62 In order to develop LNPs for non-RES targets after systemic delivery, previous studies have evaluated PEG-lipids with longer anchors to tune LNP pharmacokinetics such as extending blood half-lives and therefore increasing their accumulation in non-RES organs or tumor tissue.59,63–65 In the present study, our objective was to analyze the effects of PEG-lipid anchor length on passive accumulation of LNPs into the injured brain after systemic administration in a model of TBI. Our LNP formulation is based on patisiran and its composition is DLin-MC3-DMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DSPC:cholesterol:PEG-lipid at mole ratios of 50:10:38.5:1.5 (Fig. S1†). DLin-MC3-DMA (MC3) is the ionizable lipid essential for endosomal escape, DSPC is the helper lipid, and cholesterol and the PEG-lipid enhance the stability of the LNP.61 With the goal to study whether the PEG-lipid anchor length impacts brain accumulation and LNP pharmacokinetics, we compared LNPs formulated with a fixed total mole percent of 1.5% PEG-lipid but with three ratios of DSPE-PEG:DMG-PEG (0.1:1.4, 0.5:1, 1:0.5) (Fig. 1a Table). Each formulation included 0.1% of DSPE-PEG-Cy7 in order to quantify between formulations. Small and uniform LNPs were formulated through nanoprecipitation in a microfluidic mixer with staggered herringbone features, as described previously (Fig. 1a).66,67 For each of the LNP formulations, lipids were combined in ethanol before formulation in the microfluidic mixer. We established that the size and zeta potential of mRNA LNP formulations were similar regardless of their PEG-lipid composition. All formulations were measured with dynamic light scattering (DLS) to have ∼70 nm hydrodynamic diameters, low polydispersity indices under 0.06 (Fig. 1b and Fig. S2†), and near-neutral surface potentials (Fig. 1c). As with mRNA formulations, size and zeta potential of siRNA LNP formulations were similar regardless of their PEG-lipid composition (Fig. S3a and b†). Size and zeta potential are physicochemical measurements known to affect LNP pharmacokinetics.43,44 The similar size and zeta potential measured for each formulation allowed us to compare LNP pharmacokinetic and activity differences based on their PEG-lipid composition. The mRNA and siRNA encapsulation measured was also similar for all three formulations (Fig. 1d and Fig. S3c†), and therefore administrations matched by cargo also contained equal amounts of lipid.
:DSPC:cholesterol:PEG-lipid at mole ratios of 50:10:38.5:1.5 (Fig. S1†). DLin-MC3-DMA (MC3) is the ionizable lipid essential for endosomal escape, DSPC is the helper lipid, and cholesterol and the PEG-lipid enhance the stability of the LNP.61 With the goal to study whether the PEG-lipid anchor length impacts brain accumulation and LNP pharmacokinetics, we compared LNPs formulated with a fixed total mole percent of 1.5% PEG-lipid but with three ratios of DSPE-PEG:DMG-PEG (0.1:1.4, 0.5:1, 1:0.5) (Fig. 1a Table). Each formulation included 0.1% of DSPE-PEG-Cy7 in order to quantify between formulations. Small and uniform LNPs were formulated through nanoprecipitation in a microfluidic mixer with staggered herringbone features, as described previously (Fig. 1a).66,67 For each of the LNP formulations, lipids were combined in ethanol before formulation in the microfluidic mixer. We established that the size and zeta potential of mRNA LNP formulations were similar regardless of their PEG-lipid composition. All formulations were measured with dynamic light scattering (DLS) to have ∼70 nm hydrodynamic diameters, low polydispersity indices under 0.06 (Fig. 1b and Fig. S2†), and near-neutral surface potentials (Fig. 1c). As with mRNA formulations, size and zeta potential of siRNA LNP formulations were similar regardless of their PEG-lipid composition (Fig. S3a and b†). Size and zeta potential are physicochemical measurements known to affect LNP pharmacokinetics.43,44 The similar size and zeta potential measured for each formulation allowed us to compare LNP pharmacokinetic and activity differences based on their PEG-lipid composition. The mRNA and siRNA encapsulation measured was also similar for all three formulations (Fig. 1d and Fig. S3c†), and therefore administrations matched by cargo also contained equal amounts of lipid.
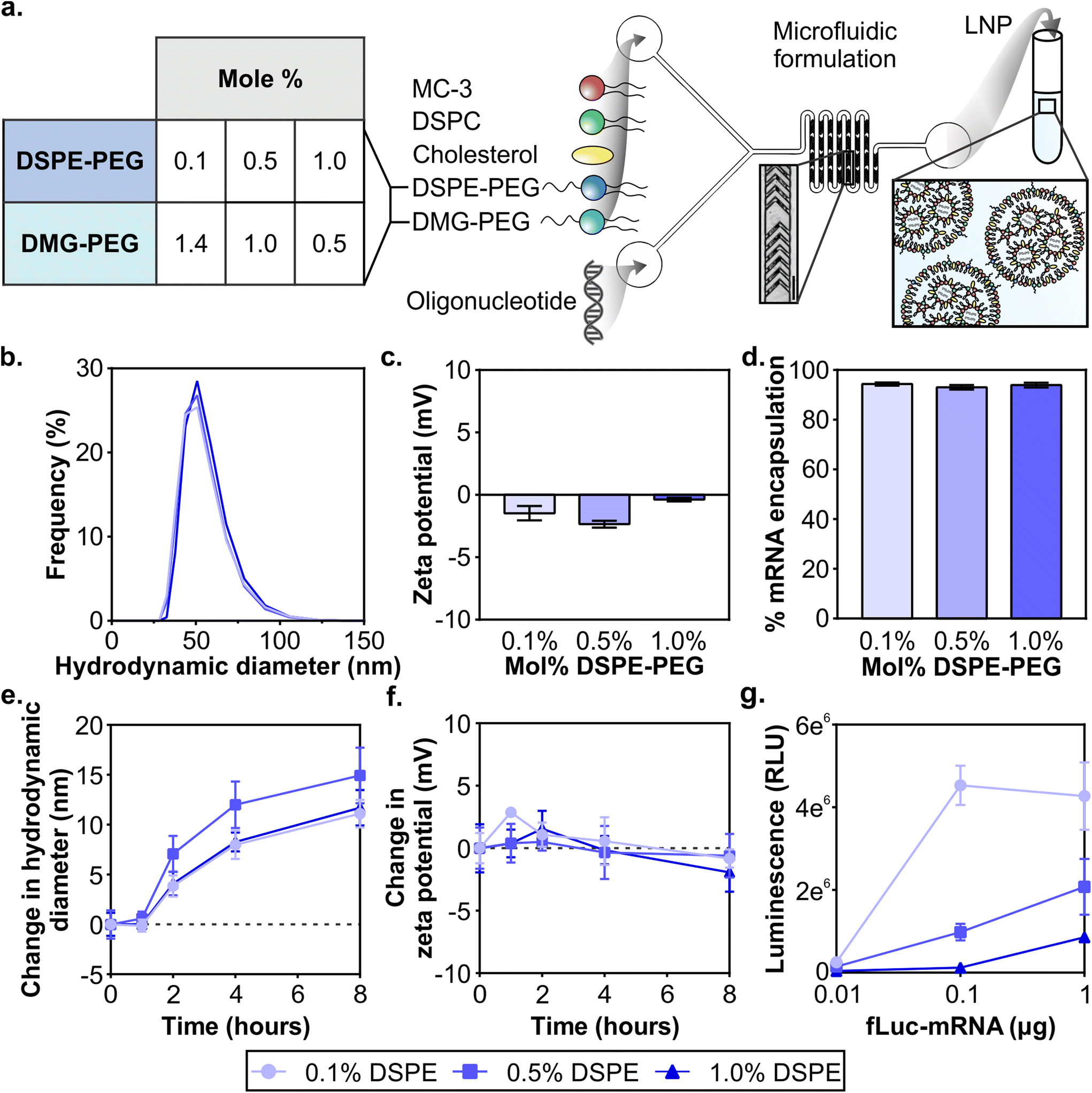 | ||
| Fig. 1 Formulation of nucleic acid LNPs with varying PEG-lipid compositions. (a) Schematic of LNP formulation and the PEG-lipid compositions evaluated listing the mol% of DSPE and DMG-PEG in the formulation (scale bar = 200 μm). DLS analysis of (b) size distributions and (c) zeta potentials (n = 2) of LNP formulations. (d) Percent encapsulation of mRNA in LNPs (n = 2). (e) Average sizes and (f) zeta potentials of LNPs when incubated in serum over time (n = 3). (g) Luciferase activity in 293T cells after 24 hours of treatment with LNPs at various mRNA doses (n = 3) (mean ± SD). | ||
It is known that protein coronas form on the surface of nanoparticles, including LNPs. The surface adsorption of serum-derived ApoE onto the LNP surface is how the clinical formulation patisiran traffics to the liver after systemic delivery.20,62 Therefore, in order to understand the impact of protein adsorption on the LNP formulations, we incubated LNPs with 55% exosome-free serum in PBS at 37 °C to emulate physiological protein concentrations in blood,68 and measured their size and zeta potential at 1, 2, 4, and 8 hours. The formation of a protein corona on PEGylated and non-PEGylated nanoparticles exposed to serum is well-studied, with PEGylated nanoparticles typically adsorbing less proteins due to the steric hindrance of the PEG layer.69–71 Over the incubation period, we observed that all LNP formulations increase in size, culminating in a 10–15 nm increase at 8 hours, suggesting the formation of a protein corona (Fig. 1e). We also observed a slight negative shift in zeta potential for all 3 formulations at 8 hours (Fig. 1f), consistent with the abundance of negatively charged serum proteins.72 Increases in size up to 30 nm and negative shifts in zeta potential have been previously observed after protein corona formation on PEGylated liposomes after incubation in serum.73 The results of this study indicated that serum adsorption occurred equally in all three formulations at least in vitro, although there may be differences in protein corona composition which may impact in vivo functionality.74
Next, we validated that mRNA LNP formulations displayed in vitro transfection activity in the highly transfectable cell line 293T. We formulated LNPs with mRNA encoding firefly luciferase (fLuc-mRNA) and applied them to 293T cells in media supplemented with serum (Fig. 1g). When cells were measured for luciferase activity at 24 hours, we observed that all formulations had measurable dose-dependent transfection efficiency. LNP formulations with the least amount of DSPE-PEG content demonstrated more activity at all tested doses, consistent with previous reports.75–77 Similar results were obtained in an analysis of the in vitro activity of siRNA LNP formulations (Fig. S4†). Prior studies have established the fast desorption kinetics of short C14 anchor PEG-lipids in the presence of plasma proteins, which reduces the steric barrier and allows LNPs to interact with the cell.51,53,78 Having established baseline in vitro activity of all LNP formulations, we subsequently compared LNP formulations in vivo.
PEG-lipid anchor length impacts LNP pharmacokinetics after systemic administration in a mouse model of TBI
Due to the complex interactions of nanoparticles on the whole organism level, in vivo activity is difficult to predict with in vitro measurements.79 Therefore, in subsequent studies formulations were evaluated in vivo. Having confirmed that LNPs formulated with varying ratios of DSPE-PEG:DMG-PEG had similar physicochemical properties (Fig. 1 and Fig. S2–S4†), we next evaluated their pharmacokinetics after systemic administration in a mouse model of TBI. LNPs were formulated with Dy677-labeled siRNA and Cy7-labeled DSPE-PEG to monitor the distribution of both siRNA cargo and PEG-lipid. All three LNP formulations included 0.1 mol% Cy7-labeled DSPE-PEG to minimize the influence of dye molecules on the physicochemical properties of the LNPs. Controlled cortical impact (CCI) is a well-studied and reproducible model of TBI that models several important clinical outcomes in human TBI, including inflammation, oxidative stress, cell death, and transient BBB permeability due to vascular damage in the injured tissue.30,31,80,81 However, biological variables including sex, age, and race still remain understudied across TBI models and further studies are required to understand how models can recapitulate the heterogeneity of human TBI.82 Mice were injured on the right hemisphere of the brain with a CCI and 0.75 mg kg−1 of siRNA LNPs were administered via the tail-vein 1 hour post-injury (Fig. 2a). We and others have previously established that nanoparticles can passively transport across the damaged BBB into the injured brain after systemic administration 1–24 hours post-injury for a variety of nanoparticle types, although maximal accumulation is typically observed when administration occurs within 6 hours post-injury.33,35,39,83 Blood signal over 4 hours and organ biodistribution at 4 hours were measured by fluorescence of siRNA cargo and the Cy7-labeled DSPE-PEG (Fig. 2a).
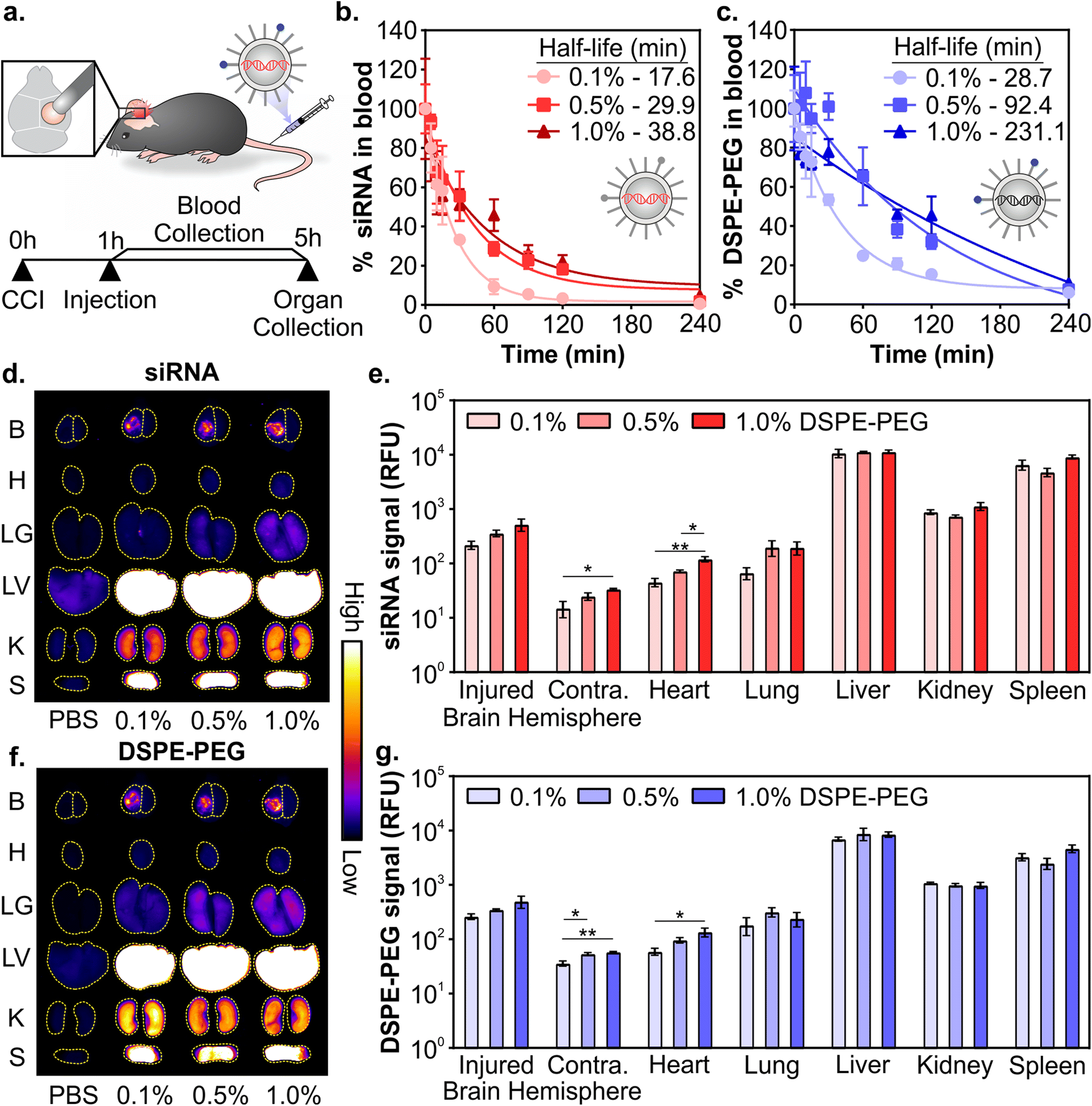 | ||
| Fig. 2 Pharmacokinetics of siRNA LNPs in a mouse model of TBI as a function of PEG-lipid composition. (a) Schematic of experiment. Blood half-lives of siRNA LNPs as measured by fluorescently labeled (b) siRNA and (c) DSPE-PEG. Representative whole organ fluorescent surface images and their quantification measured by labeled (d and e) siRNA and (f and g) DSPE-PEG. Brains were analyzed by integrating signal from the injured vs. uninjured contralateral hemispheres. LNP organ accumulation is compared with a one-way ANOVA with Tukey's post-test within each organ type (mean ± SEM, n = 3, *p < 0.05, **p < 0.01). | ||
Composition of PEG-lipid anchor lengths affected the blood half-lives of LNPs when measured both by siRNA (Fig. 2b) and the PEG-lipid (Fig. 2c). LNPs formulated with high proportions of DSPE-PEG had longer blood half-lives, consistent with previous literature that reported that longer anchor PEG-lipids increase the circulation time of LNPs after intravenous administration.55,59,84,85 The blood half-lives of LNPs measured by the fluorescence of the PEG-lipid (Fig. 2c) are extended compared to the blood half-lives measured by the fluorescence of the siRNA (Fig. 2b). Mui et al. also observed longer blood half-lives of LNPs when measured by radiolabeled PEG-lipids compared to MC3.55 The longer blood half-lives calculated from measuring labeled PEG-lipid over cargo is likely due to the association of PEG-lipids with lipid rich and/or hydrophobic domains on extracellular vesicles, apolipoproteins, and albumin, leading to a perceived extended blood circulation.50
Organ biodistribution of both siRNA (Fig. 2d and e) and PEG-lipid (Fig. 2f and g) were evaluated by quantifying the fluorescence intensity of the organs in surface scans. Relative signal between labeled siRNA and DSPE-PEG were largely consistent across organs. We observed that LNPs accumulate in the injured hemisphere of the brain ∼10-fold over the uninjured contralateral hemisphere (Fig. 2e and g), similar to what has been observed with the passive accumulation of other nanomaterials in the injured brain tissue after TBI due to the dysregulation of the BBB.33–36 LNPs formulated with more DSPE-PEG demonstrate slightly greater accumulation in the injured brain hemisphere (Fig. 2e and g), likely due to the longer blood half-lives of these LNP formulations (Fig. 2b and c). Previous research has observed increased tumor41,48,86 and injured brain39 passive accumulation with polymeric, metallic, and lipid-based nanomaterials that have longer blood half-lives. We also observe greater LNP accumulation in the contralateral brain hemisphere and heart that is statistically significant when LNPs are formulated with more DSPE-PEG, likely also due to their longer blood half-lives, although these organs have the least overall accumulation compared to other organs. Lee et al. has established that PEGylated liposomes have longer blood half-lives and greater heart accumulation compared to conventional liposomes after intravenous administration,87 supporting our observation of increased accumulation in the heart when mice were treated with LNPs with more DSPE-PEG. In line with our previous work studying the pharmacokinetics of various nanomaterials after intravenous administration in a mouse model of TBI,39,88,89 the RES organs have greater overall LNP accumulation than the injured brain. However, there are no notable differences in accumulation between the formulations evaluated. The similarity in LNP organ distribution when measured by labeled siRNA or DSPE-PEG is likely due to the relatively slow desorption of DSPE-PEG from the LNP (0.2% desorbed per hour for C18 PEG-lipid),55 indicating that the accumulation of the labeled PEG-lipid in organs is a reasonable proxy for intact LNPs. Supporting our observation, correlative biodistribution trends have similarly been established with LNPs tracked by radiolabeled C18 PEG-lipid and MC3.55
Next, we determined how the pharmacokinetic profiles of LNPs changed when formulated with mRNA cargo. siRNA and mRNA LNPs were formulated with N/P ratios (the mole ratio of ionizable lipid amine (N) and nucleotide phosphate (P)) of 3 and 5.6, respectively, which were chosen based on the N/P ratios of the siRNA LNP therapeutic patisiran (N/P 3) and the mRNA LNP COVID vaccines (N/P 5.6).61,90,91 Similar to the siRNA LNPs, the mRNA LNPs were formulated with 0.1 mol% Cy7-labeled DSPE-PEG and LNP signal was measured in blood and organs using fluorescence. After intravenous administration (0.5 mg kg−1) in a mouse model of TBI, we observed that mRNA LNP circulation times were longer with higher proportions of DSPE-PEG in the formulation (Fig. 3a). Overall, mRNA LNPs exhibited shorter blood half-lives than siRNA LNPs (Fig. 2c), likely due to the higher N/P ratio of mRNA LNPs. Positively charged nanoparticles are known to have shorter blood half-lives.41,45,92 Previous research has also demonstrated that N/P ratios impact LNP biodistribution, with lower N/P ratios leading to greater spleen accumulation and higher N/P ratios leading to greater lung accumulation.93 We observed that the content of DSPE-PEG had more impact on mRNA LNP than siRNA LNP accumulation; statistically significant differences were observed in the liver, kidney, and spleen (Fig. 3b and Fig. S5†). In the liver and kidney, the LNP formulations with less DSPE-PEG demonstrate greater accumulation, consistent with previous studies that observed higher liver accumulation in formulations with short anchor PEG-lipids.55,59 However, LNP accumulation in the spleen increases when formulations have more DSPE-PEG. In previous studies, PEGylation of nanoparticles has been shown to reduce uptake by the phagocytic Kupffer cells of the liver,56 leaving a larger blood pool of nanoparticles that are available to accumulate in the spleen.94 We similarly observed LNPs increased spleen accumulation concomitant with decreased liver accumulation. Having established that the pharmacokinetics of LNPs formulated with either siRNA or mRNA cargos were impacted similarly by PEG-lipid compositions, we focused our subsequent analysis on mRNA delivery.
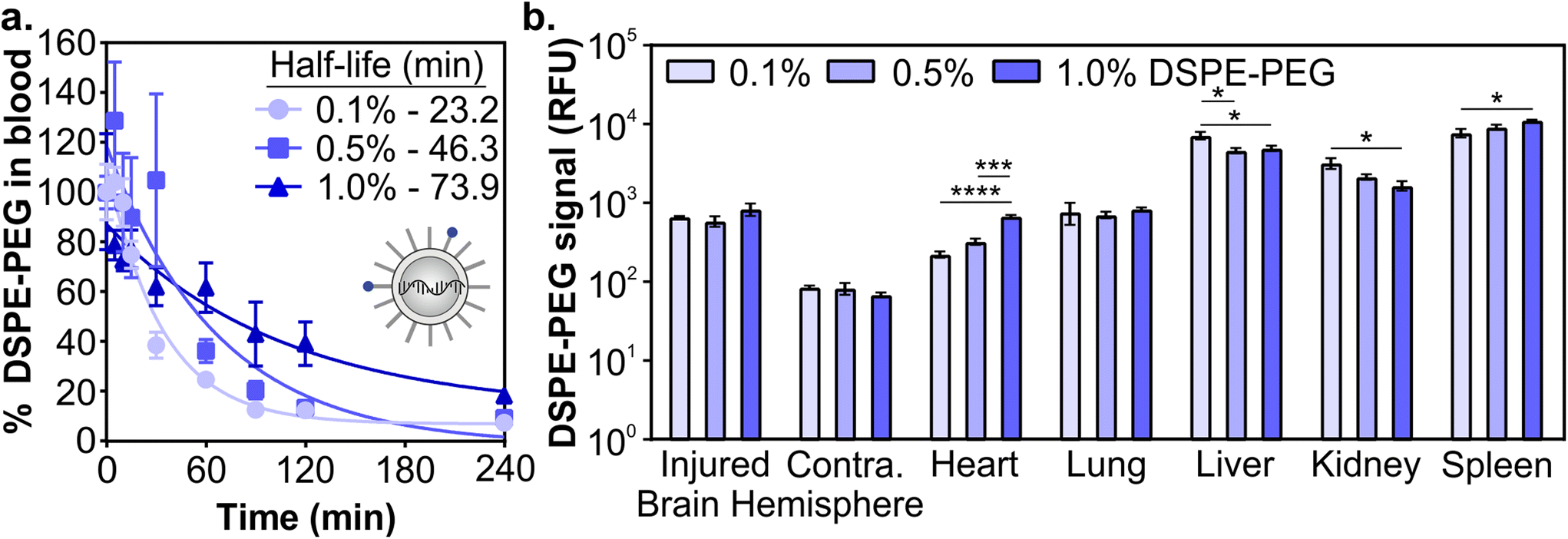 | ||
| Fig. 3 Pharmacokinetics of mRNA LNPs in a mouse model of TBI as a function of PEG-lipid composition. (a) Blood half-lives and (b) organ biodistribution of mRNA LNPs by Cy7-labeled DSPE-PEG. LNP organ accumulation is compared with a one-way ANOVA with Tukey's post-test within each organ type (mean ± SEM, n = 3, *p < 0.05, ***p < 0.001, ****p < 0.0001). | ||
PEG-lipid anchor length influences mRNA LNP activity after systemic administration in a mouse model of TBI
After determining how PEG-lipid anchor length affects mRNA LNP pharmacokinetics in a mouse model of TBI, we evaluated LNP activity. To do so, we used LNPs formulated with mRNA encoding firefly luciferase. We intravenously administered LNPs at 0.5 mg kg−1 1 hour post-CCI and quantified the expression of luciferase at 4 and 24 hours post-administration by measuring luciferase activity in homogenized organs. LNPs mediated higher transfection efficiency in the injured brain hemisphere compared to the uninjured contralateral brain hemisphere (Fig. 4a and b). Larger relative differences in transfection efficiency in injured vs. contralateral brain tissue were observed when LNPs were formulated with lower amounts of DSPE-PEG at both time points. In off-target organs, we observed a significant decrease in transfection efficiency in the heart, lung, liver, and spleen at 4 hours as DSPE-PEG content increased (Fig. 4a). Transfection efficiency of LNPs in off-target organs differed from accumulation at 4 hours (Fig. 3b); the heart and spleen had increased LNP accumulation as DSPE-PEG content increased, but transfection efficiency decreased. This may be due to the decreased cellular interaction of LNPs with more DSPE-PEG at the cellular level, which we observed in the transfection of cultured 293T cells (Fig. 1g). Mui et al. previously established that the in vivo desorption rates of C14 PEG-lipid and C18 PEG-lipid were 45% and 0.2% per hour respectively,55 suggesting that the DMG-PEG is largely desorbed from LNPs while the majority of DSPE-PEG is still associated with LNPs 4 hours post-administration.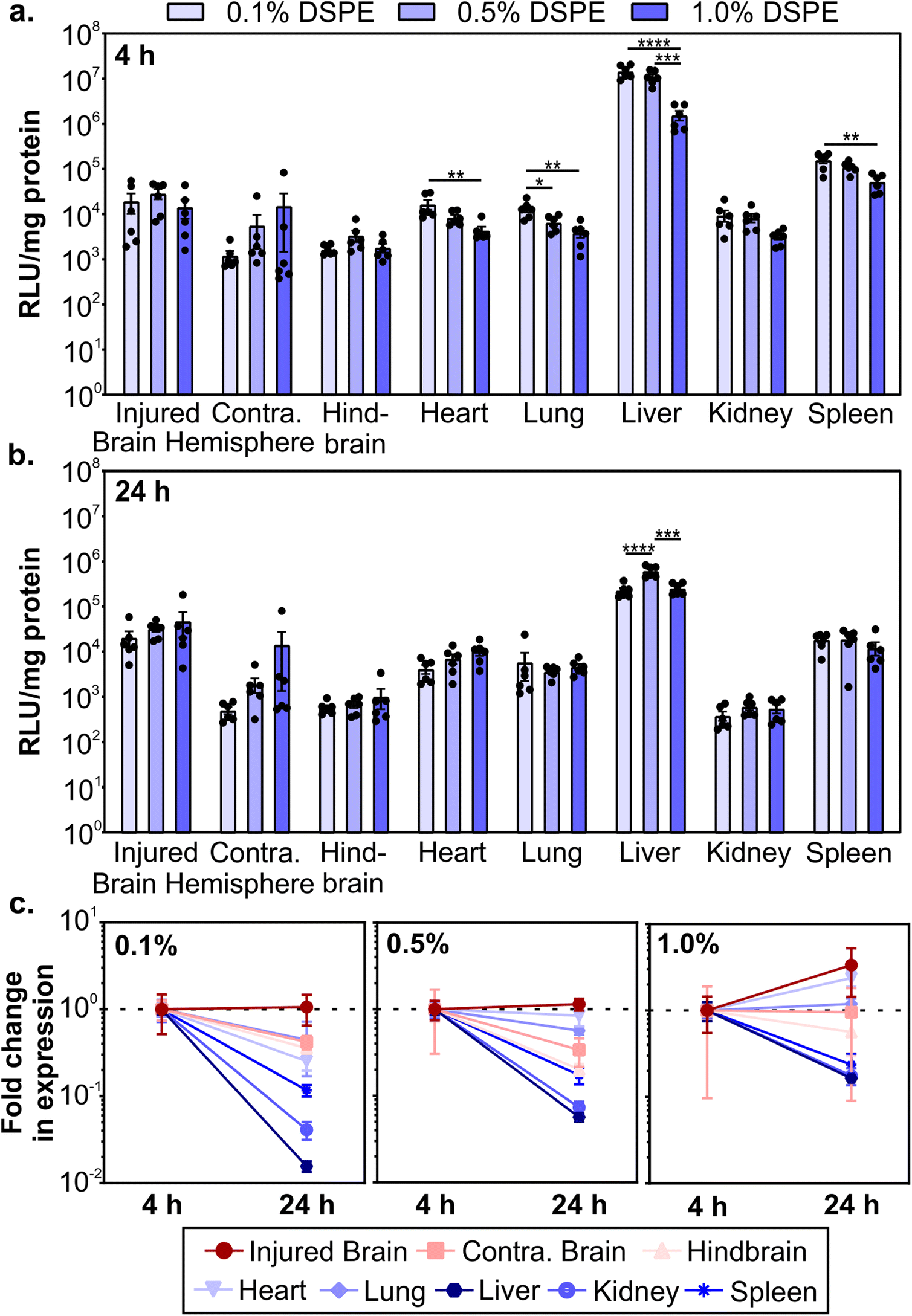 | ||
| Fig. 4 Transgene expression of mRNA LNPs in a mouse model of TBI as a function of PEG-lipid composition. Luciferase activity in organ homogenates normalized by protein content after the injection of fLuc-mRNA LNPs at (a) 4 and (b) 24 hours post-injection, presented as relative luminescence units per mg of protein (RLU per mg). Luciferase expression in organ homogenate is compared with a one-way ANOVA with Tukey's post-test within each organ type (mean ± SEM, n = 6, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). (c) Fold change in RLU per mg protein from 4 to 24 hours (normalized by the average of each organ at 4 hours) for 0.1%, 0.5%, and 1.0% DSPE-PEG LNPs (mean ± SEM, n = 6). | ||
Luciferase expression could still be detected 24 hours post-administration (Fig. 4b). Comparing the expression changes from 4 to 24 hours, we observe that luciferase expression levels in the injured brain hemisphere remain constant or increase, in contrast to the rapid loss of expression in the liver, kidney, and spleen (Fig. 4c). Loss of expression over time in off-target organs is the most dramatic for formulations made with the lowest proportion of DSPE-PEG. Similar reductions in RES organ expression have previously been observed after the systemic administration of LNPs. Pardi et al. observed a faster reduction in liver expression over 24 hours compared to other organs and determined that the translation half-life of mRNA was 6.8 hours after LNP intravenous administration, postulating that the liver turns over LNPs and/or mRNA more rapidly than other organs.12 In mammalian cells, luciferase is known to have a short half-life of 2–3 hours,95,96 so the expression of luciferase at 4 and 24 hours is expected to be due to active translation of mRNA from LNPs. Importantly, expression in the injured brain hemisphere has the least signal attenuation from 4 to 24 hours, indicating that LNPs could achieve greater expression duration in the brain compared to off-target organs. Tissue-dependent differences in translation half-lives after LNP administration have been demonstrated previously.13 Interestingly, expression levels between 4 and 24 hours increased with increasing proportions of DSPE-PEG in the LNP formulation (Fig. 4c), possibly due to the known extended desorption time of C18 PEG-lipids compared to C14 PEG-lipids causing delayed cellular interactions.55 To further investigate the PEG-lipid dependent LNP cellular interactions as a function of time, we treated 293T cells with fLuc-mRNA LNPs and measured luciferase expression at time points over 6–72 hours (Fig. S6†). As we observed previously (Fig. 1g), higher contents of DSPE-PEG in the formulation decreased overall transfection levels (Fig. S6a†). When we analyze luciferase expression across time, LNPs with higher DSPE-PEG content mediated longer delays in expression, with relative expression increasing substantially over time (Fig. S6b†). Therefore, we hypothesize that the extended expression mediated by LNPs with higher content of DSPE-PEG we observe in vivo is likely contributed to by delayed cellular interactions.
mRNA LNP distribution after convection enhanced delivery in a healthy brain
Once systemically administered LNPs enter the brain across injured vasculature, they must diffuse through the tissue to transfect cells relevant to injury repair. Although the healthy and injured brain microenvironment are distinct in both molecular and cellular compositions, we evaluated the impact of LNP PEG-lipid anchor length on the diffusion and cellular interactions of LNPs in the healthy brain microenvironment. In order to do so, we used CED to administer Cy5-labeled mRNA LNP formulations to the striatum of a healthy brain and imaged LNP distribution and cell association in the striatum of coronal brain slices collected at 0, 0.5, and 1 mm away from the injection site (Fig. 5a). In tiled images of brain hemispheres, we detected LNPs at least 1 mm away from the injection site on the anterior-posterior axis and 1–1.5 mm away on the lateral axis, demonstrating LNP diffusion away from the injection site through the brain (Fig. 5b and Fig. S7a–c†). Nance et al. previously established that model polystyrene nanoparticles with a dense surface modification of PEG can diffuse through the brain extracellular space more readily than unmodified polystyrene nanoparticles due to their neutral and non-interactive surface.97 Although previous work has shown that multiple properties impact nanoparticle diffusion in brain tissue, including surface chemistry,39,98 we did not observe significant PEG-lipid composition dependent differences in LNP distribution in our study (Fig. S7d†). In higher magnification images of brain slices 0.5 mm away from the injection site stained with cell type markers, we observed that all LNP formulations appeared to associate more with neurons and microglia than astrocytes (Fig. 5c and Fig. S8†). Consistent with our observations, Rungta et al. found that siRNA LNPs formulated with 1.5 mol% DMG-PEG associated robustly with neurons.99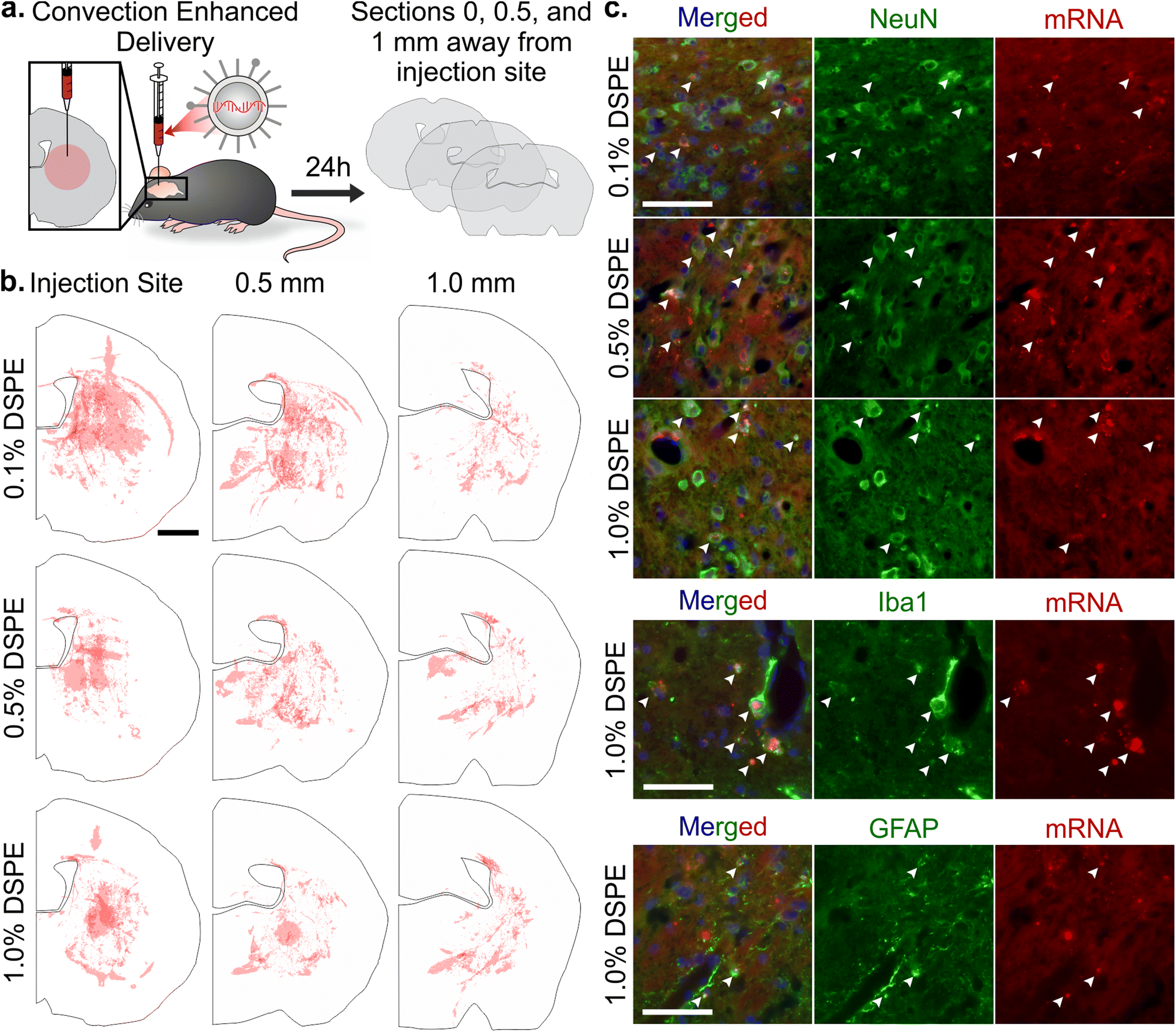 | ||
| Fig. 5 Analysis of mRNA LNP distribution and cell uptake after convection enhanced delivery. (a) Schematic of CED experiment and serial histological analysis. (b) Distribution of Cy5-labeled mRNA LNPs after CED administration in brain sections 0, 0.5, and 1 mm away from the injection site. Each hemisphere is an overlay of three injections (scale bar = 1 mm). (c) Imaging of LNPs in the striatum in brain sections 0.5 mm away from the injection site after CED. Colocalization, indicated by white arrows, of Cy5-labeled mRNA LNPs with neurons (NeuN), microglia (Iba1), and astrocytes (GFAP) (nuclei, blue; scale bar = 50 μm). | ||
Conclusion
LNPs are a promising platform for non-viral gene therapy to treat TBI. Systemically administered LNPs offer a minimally invasive administration route for gene therapy in the injured brain tissue. While the access of nanomaterials to the injured brain via the injured BBB is transient on the order of hours,35 it is known that early invention is important in management of TBI outcomes.100–102 In addition, the transient action of LNPs has the potential to match the timescale of the secondary injury caused by TBI.6 Gene therapy could address pathogenic transcriptional pathways during secondary injury, such as dampening the production of pro-inflammatory cytokines103,104 and reactive oxygen species,105 or providing regenerative cues, such as the production of neuroprotective/neurotrophic growth factors.106–109 Although LNP formulations have been established for delivery to the liver after systemic administration,62 LNP design has yet to be explored for delivery in the context of the injured brain. In this study, we observed that LNPs demonstrated greater accumulation and mediated greater transgene expression in the injured brain hemisphere compared to the contralateral uninjured brain hemisphere, indicating that LNPs can passively accumulate into injured brain tissue by exploiting the temporary permeability of the BBB created by the injury. We engineered LNPs with different compositions of DSPE(C18)-PEG and DMG(C14)-PEG and demonstrated that increasing the content of long anchor PEG-lipid extended circulation time after intravenous administration in a mouse model of TBI. Importantly, mRNA expression in injured brain tissue remained constant or increased when measured between 4 and 24 hours after administration, while off-target RES organ expression decreased ∼10-fold, indicating that gene expression mediated by LNPs have greater duration in the brain compared to off-target organs. This effect was further pronounced with increasing content of DSPE-PEG in the formulation. Our work suggests that PEG-lipid anchor length is an important design parameter for LNPs and can be used to tune the pharmacokinetics and activity of LNPs in a mouse model of TBI. In future work, we plan to evaluate the levels and duration of expression that can be achieved for therapeutic proteins for the preservation and/or regeneration of brain tissue after injury.Materials and methods
Lipid nanoparticle formulation and characterization
(6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate (DLin-MC3-DMA) was purchased from BioFine International Inc. 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG-2000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG-2000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]-N-(Cyanine 7) (DSPE PEG(2000)-N-Cy7), and cholesterol were purchased from Avanti Polar Lipids. mRNA encoding firefly luciferase was purchased from TriLink BioTechnologies. Cy5 labeled mRNA encoding EGFP was purchased from APExBIO. siRNA against luciferase with label was purchased from Dharmacon and siRNA against luciferase without label was purchased from IDT (siLuc sequence: CUUACGCUGAGUACUUCGAdTdT).mRNA LNPs were formulated with DLin-MC3-DMA:DSPC : cholesterol:PEG-lipid at a mole ratio of 50:10:38.5:1.5 and N/P of 5.6 (weight ratio of 20.7). siRNA LNPs were similarly formulated, with N/P of 3 (weight ratio of 11). To formulate LNPs with three compositions of DSPE-PEG, the 1.5 mol% of total PEG-lipid was prepared at 0.1:1.4, 0.5:1, and 1:0.5 ratios of DSPE-PEG:DMG-PEG. To prepare LNPs, lipids in ethanol and oligonucleotides in 25 mM acetate buffer, pH 4.0 were combined at a flow rate of 1:3 in a PDMS staggered herringbone mixer.94,95 Mixer channels were 200 by 100 μm, with herringbone structures 30 μm high and 50 μm wide. Immediately after formulation, 3-fold volume of PBS was added and LNPs were purified in 100 kDa MWCO centrifugal filters to less than 0.5% ethanol and sterile filtered. LNPs were stored in PBS at 4 °C for up to 2 weeks or cryoprotected in 20 mM Tris with 10% sucrose (pH 7.4) and stored at −80 °C for up to 1 month before use. Cryoprotectant buffer was exchanged into PBS after storage at −80 °C. LNP hydrodynamic diameter, polydispersity index, and zeta potential were measured by dynamic light scattering (Malvern NanoZS Zetasizer) in PBS and after incubation in 55% exosome free NCS in PBS at 37 °C. The RNA content and percent encapsulation were measured with and without Triton X-100 using a Quant-it RiboGreen RNA Assay (Invitrogen) according to the manufacturer's protocol.
Lipid nanoparticle activity in vitro
293T cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (Corning) supplemented with 10% fetal bovine serum and 2 mM GlutaMAX (Thermo Fisher Scientific). Cells were cultured to 30–50% confluency before use in experiments. For characterizing in vitro transfection efficiency, cells were plated at 25000 cells per cm2 in 96 well plates treated with 0.05 mg mL−1 poly-D-lysine overnight. LNPs formulated with fLuc-mRNA at 1, 0.1, and 0.01 μg mL−1 were added to 293T cell culture through a media change and cultured for an additional 24 hours. For analysis of luciferase activity, cells were lysed with Cell Culture Lysis Reagent (Promega) and luciferase expression was quantified with the Luciferase Assay System (Promega).
Controlled cortical impact model of TBI
All animal experiments were performed in accordance with the University of California’s Policy on the Use of Animals in Research and Teaching and are approved by the University of California San Diego Institutional Animal Care and Use Committee (IACUC), PHS Assurance Number D16-00020. 8-week-old C57BL/6J female mice (Jackson Labs) were used in these experiments. Mice were put in a stereotaxic frame under 2.5% isoflurane anesthesia. A 5 mm diameter craniotomy was performed over the right cortex midway between lambda and bregma. The right cortex was injured with a 2 mm diameter stainless steel piston tip at a depth of 2 mm and a rate of 3 m s−1 using an ImpactOne (Leica Biosystems).Pharmacokinetics of LNPs after systemic administration in a mouse model of TBI
One hour post-CCI, 0.75 mg kg−1 siRNA LNPs or 0.5 mg kg−1 mRNA LNPs formulated with Cy5-labeled EGFP mRNA were injected via the tail-vein and allowed to circulate for 4 hours. Blood was collected from the tail at 0, 5, 10, 15, 30, 60, 90, 120, and 240 minutes for blood half-life analysis. Four hours post-administration, mice were perfused with fixative and organs were harvested for imaging analysis. Fluorescent signals from Dy677-labeled siRNA and Cy7-labeled DSPE-PEG in the blood and organ samples were imaged with a LiCor Odyssey fluorescence scanner and quantified in ImageJ. Injured and contralateral brain signal were determined by integrating signal over the right and left brain hemispheres, respectively.Expression of LNPs after systemic administration in a mouse model of TBI
One hour post-CCI, 0.5 mg kg−1 fLuc-mRNA LNPs were injected via the tail-vein and allowed to circulate for 4 hours or 24 hours. Organs were harvested at each experimental endpoint and the brain was separated into the injured hemisphere, contralateral hemisphere, and hindbrain. Organ tissue was lysed through 3 freeze/thaw cycles and homogenized with a Tissue-Tearor handheld homogenizer (BioSpec) in Reporter Lysis Buffer (Promega) until homogenous. Supernatants of homogenates were measured with a Luciferase Assay System (Promega) following manufacturer protocols. Protein content of supernatants was determined with a BCA assay following standard protocols.Diffusion of LNPs through the healthy brain after CED administration
8-week-old female C57BL/6J mice (Jackson Labs) were secured in a stereotaxic frame under 2.5% isoflurane anesthesia. A 24-gauge needle was inserted through a 0.5 mm hole at a depth of 3 mm, 0.5 mm rostral and 1.75 mm right of bregma, and allowed to equilibrate for 1 minute. 1 μg of Cy5-labeled mRNA LNPs in 3.5 μL of PBS was injected at 0.5 μL min−1. The needle was left for 1 minute after injection to reduce backflow. 24 hours after injection, mice were perfused in 4% paraformaldehyde in PBS and brains were harvested for analysis by histology.Histology
Triplicate 10 μm thick brain sections 0, 0.5, and 1 mm away from the needle tract were counterstained with Hoechst using standard protocols. Additional sections 0.5 mm from the needle tract were stained with mouse anti-NeuN (1:800, Millipore MAB377), rabbit anti-Iba1 (1:500, Wako 019-19741), and chicken anti-GFAP (1:500, Abcam ab4674) using standard protocols. Fluorescent images were obtained on a Nikon Eclipse Ti2 (Nikon Instruments Inc.) at 10x magnification for distribution studies in whole brain slices. Distribution images were thresholded in ImageJ to obtain particle distribution, which was then quantified by area and represented as a schematic image with overlapping replicates. Cell localization images were obtained at 40x magnification in the striatum.
Statistical analysis
Statistical analysis was performed on GraphPad Prism 8 software, version 9.1.2. LNP accumulation and activity was analyzed by a one-way ANOVA with Tukey's post-test within each organ p < 0.05.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (NIH) Director's New Innovator Award (Number DP2 NS111507). K. F. M. was supported by a training grant from the National Heart, Lung, and Blood Institute of the National Institutes of Health (Number 1 T32 HL 160507-1 A1). Experiments were performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-2025752).The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- D. G. Stein, Embracing Failure: What the Phase III Progesterone Studies Can Teach about TBI Clinical Trials, Brain Inj., 2015, 29(11), 1259–1272, DOI:10.3109/02699052.2015.1065344.
- S. V. Kabadi and A. I. Faden, Neuroprotective Strategies for Traumatic Brain Injury: Improving Clinical Translation, Int. J. Mol. Sci., 2014, 15(1), 1216–1236, DOI:10.3390/ijms15011216.
- D. J. Loane and A. I. Faden, Neuroprotection for Traumatic Brain Injury: Translational Challenges and Emerging Therapeutic Strategies, Trends Pharmacol. Sci., 2010, 31(12), 596–604, DOI:10.1016/j.tips.2010.09.005.
- A. W. Selassie, E. Zaloshnja, J. A. Langlois, T. Miller, P. Jones and C. Steiner, Incidence of Long-Term Disability Following Traumatic Brain Injury Hospitalization, United States, 2003, J.Head Trauma Rehabil., 2008, 23(2), 123–131, DOI:10.1097/01.HTR.0000314531.30401.39.
- N. Andelic, S. Sigurdardottir, A.-K. Schanke, L. Sandvik, U. Sveen and C. Roe, Disability, Physical Health and Mental Health 1 Year after Traumatic Brain Injury, Disabil. Rehabil., 2010, 32(13), 1122–1131, DOI:10.3109/09638280903410722.
- C. Werner and K. Engelhard, Pathophysiology of Traumatic Brain Injury, Br. J. Anaesth., 2007, 99(1), 4–9, DOI:10.1093/bja/aem131.
- E. Kenjo, H. Hozumi, Y. Makita, K. A. Iwabuchi, N. Fujimoto, S. Matsumoto, M. Kimura, Y. Amano, M. Ifuku, Y. Naoe, N. Inukai and A. Hotta, Low Immunogenicity of LNP Allows Repeated Administrations of CRISPR-Cas9 MRNA into Skeletal Muscle in Mice, Nat. Commun., 2021, 12(1), 7101, DOI:10.1038/s41467-021-26714-w.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, The Current Landscape of Nucleic Acid Therapeutics, Nat. Nanotechnol., 2021, 16(6), 630–643, DOI:10.1038/s41565-021-00898-0.
- M. Herrera-Barrera, R. C. Ryals, M. Gautam, A. Jozic, M. Landry, T. Korzun, M. Gupta, C. Acosta, J. Stoddard, R. Reynaga, W. Tschetter, N. Jacomino, O. Taratula, C. Sun, A. K. Lauer, M. Neuringer and G. Sahay, Peptide-Guided Lipid Nanoparticles Deliver MRNA to the Neural Retina of Rodents and Nonhuman Primates, Sci. Adv., 2023, 9(2), eadd4623, DOI:10.1126/sciadv.add4623.
- P. D. Kessler, G. M. Podsakoff, X. Chen, S. A. McQuiston, P. C. Colosi, L. A. Matelis, G. J. Kurtzman and B. J. Byrne, Gene Delivery to Skeletal Muscle Results in Sustained Expression and Systemic Delivery of a Therapeutic Protein, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(24), 14082–14087, DOI:10.1073/pnas.93.24.14082.
- X. Xiao, J. Li and R. J. Samulski, Efficient Long-Term Gene Transfer into Muscle Tissue of Immunocompetent Mice by Adeno-Associated Virus Vector, J. Virol., 1996, 70(11), 8098–8108, DOI:10.1128/jvi.70.11.8098-8108.1996.
- N. Pardi, S. Tuyishime, H. Muramatsu, K. Kariko, B. L. Mui, Y. K. Tam, T. D. Madden, M. J. Hope and D. Weissman, Expression Kinetics of Nucleoside-Modified MRNA Delivered in Lipid Nanoparticles to Mice by Various Routes, J. Controlled Release, 2015, 217, 345–351, DOI:10.1016/j.jconrel.2015.08.007.
- J. Di, Z. Du, K. Wu, S. Jin, X. Wang, T. Li and Y. Xu, Biodistribution and Non-Linear Gene Expression of MRNA LNPs Affected by Delivery Route and Particle Size, Pharm. Res., 2022, 39(1), 105–114, DOI:10.1007/s11095-022-03166-5.
- F. Shen, L. Wen, X. Yang and W. Liu, The Potential Application of Gene Therapy in the Treatment of Traumatic Brain Injury, Neurosurg. Rev., 2007, 30(4), 291–298, DOI:10.1007/s10143-007-0094-4.
- Y. Long, L. Zou, H. Liu, H. Lu, X. Yuan, C. S. Robertson and K. Yang, Altered Expression of Randomly Selected Genes in Mouse Hippocampus after Traumatic Brain Injury, J. Neurosci. Res., 2003, 71(5), 710–720, DOI:10.1002/jnr.10524.
- R. N. Munji, A. L. Soung, G. A. Weiner, F. Sohet, B. D. Semple, A. Trivedi, K. Gimlin, M. Kotoda, M. Korai, S. Aydin, A. Batugal, A. C. Cabangcala, P. G. Schupp, M. C. Oldham, T. Hashimoto, L. J. Noble-Haeusslein and R. Daneman, Profiling the Mouse Brain Endothelial Transcriptome in Health and Disease Models Reveals a Core Blood–Brain Barrier Dysfunction Module, Nat. Neurosci., 2019, 22(11), 1892–1902, DOI:10.1038/s41593-019-0497-x.
- S. M. Hoy, Patisiran: First Global Approval, Drugs, 2018, 78(15), 1625–1631, DOI:10.1007/s40265-018-0983-6.
- Commissioner, O. of the. FDA Approves First COVID-19 Vaccine. FDA, https://www.fda.gov/news-events/press-announcements/fda-approves-first-covid-19-vaccine, (accessed 2022-09-19).
- Commissioner, O. of the. Coronavirus (COVID-19) Update: FDA Takes Key Action by Approving Second COVID-19 Vaccine. FDA, https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-takes-key-action-approving-second-covid-19-vaccine, (accessed 2022-09-19).
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs, Nat. Nanotechnol., 2019, 14(12), 1084–1087, DOI:10.1038/s41565-019-0591-y.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Maximizing the Potency of SiRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo**, Angew. Chem., 2012, 124(34), 8657–8661, DOI:10.1002/ange.201203263.
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey, R. Novak, D. Diemert, S. A. Spector, N. Rouphael, C. B. Creech, J. McGettigan, S. Khetan, N. Segall, J. Solis, A. Brosz, C. Fierro, H. Schwartz, K. Neuzil, L. Corey, P. Gilbert, H. Janes, D. Follmann, M. Marovich, J. Mascola, L. Polakowski, J. Ledgerwood, B. S. Graham, H. Bennett, R. Pajon, C. Knightly, B. Leav, W. Deng, H. Zhou, S. Han, M. Ivarsson, J. Miller and T. Zaks, Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine, N. Engl. J. Med., 2021, 384(5), 403–416, DOI:10.1056/NEJMoa2035389.
- F. Wang, R. M. Kream and G. B. Stefano, An Evidence Based Perspective on MRNA-SARS-CoV-2 Vaccine Development, Med. Sci. Monit., 2020, 26, e924700-1–e924700-8, DOI:10.12659/MSM.924700.
- E. J. Anderson, N. G. Rouphael, A. T. Widge, L. A. Jackson, P. C. Roberts, M. Makhene, J. D. Chappell, M. R. Denison, L. J. Stevens, A. J. Pruijssers, A. B. McDermott, B. Flach, B. C. Lin, N. A. Doria-Rose, S. O'Dell, S. D. Schmidt, K. S. Corbett, P. A. Swanson, M. Padilla, K. M. Neuzil, H. Bennett, B. Leav, M. Makowski, J. Albert, K. Cross, V. V. Edara, K. Floyd, M. S. Suthar, D. R. Martinez, R. Baric, W. Buchanan, C. J. Luke, V. K. Phadke, C. A. Rostad, J. E. Ledgerwood, B. S. Graham and J. H. Beigel, Safety and Immunogenicity of SARS-CoV-2 MRNA-1273 Vaccine in Older Adults, N. Engl. J. Med., 2020, 383(25), 2427–2438, DOI:10.1056/NEJMoa2028436.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Lipid Nanoparticles for MRNA Delivery, Nat. Rev. Mater., 2021, 6(12), 1078–1094, DOI:10.1038/s41578-021-00358-0.
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines, Vaccines, 2021, 9(4), 359, DOI:10.3390/vaccines9040359.
- P. R. Cullis and M. J. Hope, Lipid Nanoparticle Systems for Enabling Gene Therapies, Mol. Ther., 2017, 25(7), 1467–1475, DOI:10.1016/j.ymthe.2017.03.013.
- A. Chodobski, B. J. Zink and J. Szmydynger-Chodobska, Blood–Brain Barrier Pathophysiology in Traumatic Brain Injury, Transl. Stroke Res., 2011, 2(4), 492–516, DOI:10.1007/s12975-011-0125-x.
- S. L. Smith, P. K. Andrus, J.-R. Zhang and E. D. Hall, Direct Measurement of Hydroxyl Radicals, Lipid Peroxidation, and Blood–Brain Barrier Disruption Following Unilateral Cortical Impact Head Injury in the Rat, J. Neurotrauma, 1994, 11(4), 393–404, DOI:10.1089/neu.1994.11.393.
- H. Alluri, C. A. Shaji, M. L. Davis and B. Tharakan, A Mouse Controlled Cortical Impact Model of Traumatic Brain Injury for Studying Blood–Brain Barrier Dysfunctions, in Traumatic and Ischemic Injury: Methods and Protocols, ed. Tharakan, B., Methods in Molecular Biology, Springer, New York, NY, 2018, pp. 37–52. DOI:10.1007/978-1-4939-7526-6_4.
- M. J. Whalen, T. M. Carlos, P. M. Kochanek and S. Heineman, Blood-Brain Barrier Permeability, Neutrophil Accumulation and Vascular Adhesion Molecule Expression after Controlled Cortical Impact in Rats: A Preliminary Study, in Intracranial Pressure and Neuromonitoring in Brain Injury, ed. A. Marmarou, R. Bullock, C. Avezaat, A. Baethmann, D. Becker, M. Brock, J. Hoff, H. Nagai, H.-J. Reulen and G. Teasdale, Acta Neurochirurgica Supplements, Springer, Vienna, 1998, pp. 212–214. DOI:10.1007/978-3-7091-6475-4_61.
- V. N. Bharadwaj, D. T. Nguyen, V. D. Kodibagkar and S. E. Stabenfeldt, Nanoparticle-Based Therapeutics for Brain Injury, Adv. Healthcare Mater., 2018, 7(1), 1700668, DOI:10.1002/adhm.201700668.
- V. N. Bharadwaj, J. Lifshitz, P. D. Adelson, V. D. Kodibagkar and S. E. Stabenfeldt, Temporal Assessment of Nanoparticle Accumulation after Experimental Brain Injury: Effect of Particle Size, Sci. Rep., 2016, 6(1), 29988, DOI:10.1038/srep29988.
- V. N. Bharadwaj, R. K. Rowe, J. Harrison, C. Wu, T. R. Anderson, J. Lifshitz, P. D. Adelson, V. D. Kodibagkar and S. E. Stabenfeldt, Blood–Brainbarrier Disruption Dictates Nanoparticle Accumulation Following Experimental Brain Injury, Nanomedicine, 2018, 14(7), 2155–2166, DOI:10.1016/j.nano.2018.06.004.
- E. J. Kwon, M. Skalak, R. Lo Bu and S. N. Bhatia, Neuron-Targeted Nanoparticle for SiRNA Delivery to Traumatic Brain Injuries, ACS Nano, 2016, 10(8), 7926–7933, DOI:10.1021/acsnano.6b03858.
- B. J. Boyd, A. Galle, M. Daglas, J. V. Rosenfeld and R. Medcalf, Traumatic Brain Injury Opens Blood–Brain Barrier to Stealth Liposomes via an Enhanced Permeability and Retention (EPR)-like Effect, J. Drug Targeting, 2015, 23(9), 847–853, DOI:10.3109/1061186X.2015.1034280.
- R. M. Kandell, L. E. Waggoner and E. J. Kwon, Nanomedicine for Acute Brain Injuries: Insight from Decades of Cancer Nanomedicine, Mol. Pharmaceutics, 2021, 18(2), 522–538, DOI:10.1021/acs.molpharmaceut.0c00287.
- E. A. Sykes, J. Chen, G. Zheng and W. C. W. Chan, Investigating the Impact of Nanoparticle Size on Active and Passive Tumor Targeting Efficiency, ACS Nano, 2014, 8(6), 5696–5706, DOI:10.1021/nn500299p.
- L. E. Waggoner, M. I. Madias, A. A. Hurtado and E. J. Kwon, Pharmacokinetic Analysis of Peptide-Modified Nanoparticles with Engineered Physicochemical Properties in a Mouse Model of Traumatic Brain Injury, AAPS J., 2021, 23(5), 100, DOI:10.1208/s12248-021-00626-5.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, The Effect of Surface Charge on in Vivo Biodistribution of PEG-Oligocholic Acid Based Micellar Nanoparticles, Biomaterials, 2011, 32(13), 3435–3446, DOI:10.1016/j.biomaterials.2011.01.021.
- R. R. Arvizo, O. R. Miranda, D. F. Moyano, C. A. Walden, K. Giri, R. Bhattacharya, J. D. Robertson, V. M. Rotello, J. M. Reid and P. Mukherjee, Modulating Pharmacokinetics, Tumor Uptake and Biodistribution by Engineered Nanoparticles, PLoS One, 2011, 6(9), e24374, DOI:10.1371/journal.pone.0024374.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Mediating Tumor Targeting Efficiency of Nanoparticles Through Design, Nano Lett., 2009, 9(5), 1909–1915, DOI:10.1021/nl900031y.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR–Cas Gene Editing, Nat. Nanotechnol., 2020, 15(4), 313–320, DOI:10.1038/s41565-020-0669-6.
- S. Chen, Y. Y. C. Tam, P. J. C. Lin, M. M. H. Sung, Y. K. Tam and P. R. Cullis, Influence of Particle Size on the in Vivo Potency of Lipid Nanoparticle Formulations of SiRNA, J. Controlled Release, 2016, 235, 236–244, DOI:10.1016/j.jconrel.2016.05.059.
- S.-D. Li and L. Huang, Pharmacokinetics and Biodistribution of Nanoparticles, Mol. Pharmaceutics, 2008, 5(4), 496–504, DOI:10.1021/mp800049w.
- K. Maruyama, Intracellular Targeting Delivery of Liposomal Drugs to Solid Tumors Based on EPR Effects, Adv. Drug Delivery Rev., 2011, 63(3), 161–169, DOI:10.1016/j.addr.2010.09.003.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Analysis of Nanoparticle Delivery to Tumours, Nat. Rev. Mater., 2016, 1(5), 16014, DOI:10.1038/natrevmats.2016.14.
- T. Yang, F.-D. Cui, M.-K. Choi, J.-W. Cho, S.-J. Chung, C.-K. Shim and D.-D. Kim, Enhanced Solubility and Stability of PEGylated Liposomal Paclitaxel: In Vitro and in Vivo Evaluation, Int. J. Pharm., 2007, 338(1), 317–326, DOI:10.1016/j.ijpharm.2007.02.011.
- J. A. Kulkarni, D. Witzigmann, J. Leung, Y. Y. C. Tam and P. R. Cullis, On the Role of Helper Lipids in Lipid Nanoparticle Formulations of SiRNA, Nanoscale, 2019, 11(45), 21733–21739, 10.1039/C9NR09347H.
- C. Hald Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, The Role of Lipid Components in Lipid Nanoparticles for Vaccines and Gene Therapy, Adv. Drug Delivery Rev., 2022, 188, 114416, DOI:10.1016/j.addr.2022.114416.
- H. Hatakeyama, H. Akita and H. Harashima, A Multifunctional Envelope Type Nano Device (MEND) for Gene Delivery to Tumours Based on the EPR Effect: A Strategy for Overcoming the PEG Dilemma, Adv. Drug Delivery Rev., 2011, 63(3), 152–160, DOI:10.1016/j.addr.2010.09.001.
- S. Mishra, P. Webster and M. E. Davis, PEGylation Significantly Affects Cellular Uptake and Intracellular Trafficking of Non-Viral Gene Delivery Particles, Eur. J. Cell Biol., 2004, 83(3), 97–111, DOI:10.1078/0171-9335-00363.
- K. Remaut, B. Lucas, K. Braeckmans, J. Demeester and S. C. De Smedt, Pegylation of Liposomes Favours the Endosomal Degradation of the Delivered Phosphodiester Oligonucleotides, J. Controlled Release, 2007, 117(2), 256–266, DOI:10.1016/j.jconrel.2006.10.029.
- D. Zukancic, E. J. A. Suys, E. H. Pilkington, A. Algarni, H. Al-Wassiti and N. P. Truong, The Importance of Poly(Ethylene Glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles, Pharmaceutics, 2020, 12(11), 1068, DOI:10.3390/pharmaceutics12111068.
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Du, Y. Y. C. Tam, P. J. Lin, S. Chen, J. K. Narayanannair, K. G. Rajeev, M. Manoharan, A. Akinc, M. A. Maier, P. Cullis, T. D. Madden and M. J. Hope, Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of SiRNA Lipid Nanoparticles, Mol. Ther. – Nucleic Acids, 2013, 2, e139, DOI:10.1038/mtna.2013.66.
- Y. Bao, Y. Jin, P. Chivukula, J. Zhang, Y. Liu, J. Liu, J.-P. Clamme, R. I. Mahato, D. Ng, W. Ying, Y. Wang and L. Yu, Effect of PEGylation on Biodistribution and Gene Silencing of SiRNA/Lipid Nanoparticle Complexes, Pharm. Res., 2013, 30(2), 342–351, DOI:10.1007/s11095-012-0874-6.
- K. J. Kauffman, J. R. Dorkin, J. H. Yang, M. W. Heartlein, F. DeRosa, F. F. Mir, O. S. Fenton and D. G. Anderson, Optimization of Lipid Nanoparticle Formulations for MRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs, Nano Lett., 2015, 15(11), 7300–7306, DOI:10.1021/acs.nanolett.5b02497.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Effect of Polyethyleneglycol (PEG) Chain Length on the Bio–Nano-Interactions between PEGylated Lipid Nanoparticles and Biological Fluids: From Nanostructure to Uptake in Cancer Cells, Nanoscale, 2014, 6(5), 2782, 10.1039/c3nr05559k.
- E. Ambegia, S. Ansell, P. Cullis, J. Heyes, L. Palmer and I. MacLachlan, Stabilized Plasmid–Lipid Particles Containing PEG-Diacylglycerols Exhibit Extended Circulation Lifetimes and Tumor Selective Gene Expression, Biochim. Biophys. Acta, Biomembr., 2005, 1669(2), 155–163, DOI:10.1016/j.bbamem.2005.02.001.
- S. C. Wilson, J. L. Baryza, A. J. Reynolds, K. Bowman, M. E. Keegan, S. M. Standley, N. P. Gardner, P. Parmar, V. O. Agir, S. Yadav, A. Zunic, C. Vargeese, C. C. Lee and S. Rajan, Real Time Measurement of PEG Shedding from Lipid Nanoparticles in Serum via NMR Spectroscopy, Mol. Pharmaceutics, 2015, 12(2), 386–392, DOI:10.1021/mp500400k.
- Y. Suzuki and H. Ishihara, Difference in the Lipid Nanoparticle Technology Employed in Three Approved SiRNA (Patisiran) and MRNA (COVID-19 Vaccine) Drugs, Drug Metab. Pharmacokinet., 2021, 41, 100424, DOI:10.1016/j.dmpk.2021.100424.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley, J. R. Dorkin, J. S. Butler, L. Qin, T. Racie, A. Sprague, E. Fava, A. Zeigerer, M. J. Hope, M. Zerial, D. W. Sah, K. Fitzgerald, M. A. Tracy, M. Manoharan, V. Koteliansky, A. de Fougerolles and M. A. Maier, Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms, Mol. Ther., 2010, 18(7), 1357–1364, DOI:10.1038/mt.2010.85.
- J. B. Lee, K. Zhang, Y. Y. C. Tam, Y. K. Tam, N. M. Belliveau, V. Y. C. Sung, P. J. C. Lin, E. LeBlanc, M. A. Ciufolini, P. S. Rennie and P. R. Cullis, Lipid Nanoparticle SiRNA Systems for Silencing the Androgen Receptor in Human Prostate Cancer in Vivo, Int. J. Cancer, 2012, 131(5), E781–E790, DOI:10.1002/ijc.27361.
- J. B. Lee, K. Zhang, Y. Y. C. Tam, J. Quick, Y. K. Tam, P. J. Lin, S. Chen, Y. Liu, J. K. Nair, I. Zlatev, K. G. Rajeev, M. Manoharan, P. S. Rennie and P. R. Cullis, A Glu-Urea-Lys Ligand-Conjugated Lipid Nanoparticle/SiRNA System Inhibits Androgen Receptor Expression In Vivo, Mol. Ther. – Nucleic Acids, 2016, 5, e348, DOI:10.1038/mtna.2016.43.
- Y. Yamamoto, P. J. C. Lin, E. Beraldi, F. Zhang, Y. Kawai, J. Leong, H. Katsumi, L. Fazli, R. Fraser, P. R. Cullis and M. Gleave, SiRNA Lipid Nanoparticle Potently Silences Clusterin and Delays Progression When Combined with Androgen Receptor Cotargeting in Enzalutamide-Resistant Prostate Cancer, Clin. Cancer Res., 2015, 21(21), 4845–4855, DOI:10.1158/1078-0432.CCR-15-0866.
- N. M. Belliveau, J. Huft, P. J. Lin, S. Chen, A. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Microfluidic Synthesis of Highly Potent Limit-Size Lipid Nanoparticles for In Vivo Delivery of SiRNA, Mol. Ther. – Nucleic Acids, 2012, 1(8), e37, DOI:10.1038/mtna.2012.28.
- D. Chen, K. T. Love, Y. Chen, A. A. Eltoukhy, C. Kastrup, G. Sahay, A. Jeon, Y. Dong, K. A. Whitehead and D. G. Anderson, Rapid Discovery of Potent SiRNA-Containing Lipid Nanoparticles Enabled by Controlled Microfluidic Formulation, J. Am. Chem. Soc., 2012, 134(16), 6948–6951, DOI:10.1021/ja301621z.
- L. L. Aylward, Biomarkers of Environmental Exposures in Blood, in Encyclopedia of Environmental Health, ed. J. Nriagu, Elsevier, Oxford, 2nd edn, 2019, pp. 376–385. DOI:10.1016/B978-0-12-409548-9.10658-X.
- K. Partikel, R. Korte, N. C. Stein, D. Mulac, F. C. Herrmann, H.-U. Humpf and K. Langer, Effect of Nanoparticle Size and PEGylation on the Protein Corona of PLGA Nanoparticles, Eur. J. Pharm. Biopharm., 2019, 141, 70–80, DOI:10.1016/j.ejpb.2019.05.006.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Müller, ‘Stealth’ Corona-Core Nanoparticles Surface Modified by Polyethylene Glycol (PEG): Influences of the Corona (PEG Chain Length and Surface Density) and of the Core Composition on Phagocytic Uptake and Plasma Protein Adsorption, Colloids Surf., B, 2000, 18(3), 301–313, DOI:10.1016/S0927-7765(99)00156-3.
- H. Li, Y. Wang, Q. Tang, D. Yin, C. Tang, E. He, L. Zou and Q. Peng, The Protein Corona and Its Effects on Nanoparticle-Based Drug Delivery Systems, Acta Biomater., 2021, 129, 57–72, DOI:10.1016/j.actbio.2021.05.019.
- J. T. Busher, Serum Albumin and Globulin, in Clinical Methods: The History, Physical, and Laboratory Examinations, ed. H. K. Walker, W. D. Hall and J. W. Hurst, Butterworths, Boston, 1990 Search PubMed.
- S. Palchetti, V. Colapicchioni, L. Digiacomo, G. Caracciolo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, The Protein Corona of Circulating PEGylated Liposomes, Biochim. Biophys. Acta, Biomembr., 2016, 1858(2), 189–196, DOI:10.1016/j.bbamem.2015.11.012.
- D. Chen, N. Parayath, S. Ganesh, W. Wang and M. Amiji, The Role of Apolipoprotein- and Vitronectin-Enriched Protein Corona on Lipid Nanoparticles for in Vivo Targeted Delivery and Transfection of Oligonucleotides in Murine Tumor Models, Nanoscale, 2019, 11(40), 18806–18824, 10.1039/C9NR05788A.
- K. W. C. Mok, A. M. I. Lam and P. R. Cullis, Stabilized Plasmid-Lipid Particles: Factors Influencing Plasmid Entrapment and Transfection Properties, Biochim. Biophys. Acta, Biomembr., 1999, 1419(2), 137–150, DOI:10.1016/S0005-2736(99)00059-0.
- A. Sarode, Y. Fan, A. E. Byrnes, M. Hammel, G. L. Hura, Y. Fu, P. Kou, C. Hu, F. I. Hinz, J. Roberts, S. G. Koenig, K. Nagapudi, C. C. Hoogenraad, T. Chen, D. Leung and C.-W. Yen, Predictive High-Throughput Screening of PEGylated Lipids in Oligonucleotide-Loaded Lipid Nanoparticles for Neuronal Gene Silencing, Nanoscale Adv., 2022, 4(9), 2107–2123, 10.1039/D1NA00712B.
- L. Y. Song, Q. F. Ahkong, Q. Rong, Z. Wang, S. Ansell, M. J. Hope and B. Mui, Characterization of the Inhibitory Effect of PEG-Lipid Conjugates on the Intracellular Delivery of Plasmid and Antisense DNA Mediated by Cationic Lipid Liposomes, Biochim. Biophys. Acta, Biomembr., 2002, 1558(1), 1–13, DOI:10.1016/S0005-2736(01)00399-6.
- J. J. Wheeler, L. Palmer, M. Ossanlou, I. MacLachlan, R. W. Graham, Y. P. Zhang, M. J. Hope, P. Scherrer and P. R. Cullis, Stabilized Plasmid-Lipid Particles: Construction and Characterization, Gene Ther., 1999, 6(2), 271–281, DOI:10.1038/sj.gt.3300821.
- Y. Zhu, R. Shen, I. Vuong, R. A. Reynolds, M. J. Shears, Z.-C. Yao, Y. Hu, W. J. Cho, J. Kong, S. K. Reddy, S. C. Murphy and H.-Q. Mao, Multi-Step Screening of DNA/Lipid Nanoparticles and Co-Delivery with SiRNA to Enhance and Prolong Gene Expression, Nat. Commun., 2022, 13(1), 4282, DOI:10.1038/s41467-022-31993-y.
- Y. Xiong, A. Mahmood and M. Chopp, Animal Models of Traumatic Brain Injury, Nat. Rev. Neurosci., 2013, 14(2), 128–142, DOI:10.1038/nrn3407.
- N. D. Osier, J. R. Korpon and C. E. Dixon, Controlled Cortical Impact Model, in Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects, ed. F. H. Kobeissy, Frontiers in Neuroengineering, CRC Press/Taylor & Francis, Boca Raton (FL), 2015 Search PubMed.
- T. Wood and E. Nance, Disease-Directed Engineering for Physiology-Driven Treatment Interventions in Neurological Disorders, APL Bioeng., 2019, 3(4), 040901, DOI:10.1063/1.5117299.
- L. E. Waggoner, J. Kang, J. M. Zuidema, S. Vijayakumar, A. A. Hurtado, M. J. Sailor and E. J. Kwon, Porous Silicon Nanoparticles Targeted to the Extracellular Matrix for Therapeutic Protein Delivery in Traumatic Brain Injury, Bioconjugate Chem., 2022, 33(9), 1685–1697, DOI:10.1021/acs.bioconjchem.2c00305.
- Y. Xu, M. Ou, E. Keough, J. Roberts, K. Koeplinger, M. Lyman, S. Fauty, E. Carlini, M. Stern, R. Zhang, S. Yeh, E. Mahan, Y. Wang, D. Slaughter, M. Gindy, C. Raab, C. Thompson and J. Hochman, Quantitation of Physiological and Biochemical Barriers to SiRNA Liver Delivery via Lipid Nanoparticle Platform, Mol. Pharmaceutics, 2014, 11(5), 1424–1434, DOI:10.1021/mp400584h.
- M. A. Monck, A. Mori, D. Lee, P. Tam, J. J. Wheeler, P. R. Cullis and P. Scherrer, Stabilized Plasmid–Lipid Particles: Pharmacokinetics and Plasmid Delivery to Distal Tumors Following Intravenous Injection, J. Drug Targeting, 2000, 7(6), 439–452, DOI:10.3109/10611860009102218.
- C. Yao, P. Wang, L. Zhou, R. Wang, X. Li, D. Zhao and F. Zhang, Highly Biocompatible Zwitterionic Phospholipids Coated Upconversion Nanoparticles for Efficient Bioimaging, Anal. Chem., 2014, 86(19), 9749–9757, DOI:10.1021/ac5023259.
- C.-M. Lee, Y. Choi, E. J. Huh, K. Y. Lee, H.-C. Song, M. J. Sun, H.-J. Jeong, C.-S. Cho and H.-S. Bom, Polyethylene Glycol (PEG) Modified 99mTc-HMPAOLiposome for Improving Blood Circulation and Biodistribution: The Effect of the Extent of PEGylation, Cancer Biother.Radiopharm., 2005, 20(6), 620–628, DOI:10.1089/cbr.2005.20.620.
- R. M. Kandell, J. A. Kudryashev and E. J. Kwon, Targeting the Extracellular Matrix in Traumatic Brain Injury Increases Signal Generation from an Activity-Based Nanosensor, ACS Nano, 2021, 15(12), 20504–20516, DOI:10.1021/acsnano.1c09064.
- J. A. Kudryashev, L. E. Waggoner, H. T. Leng, N. H. Mininni and E. J. Kwon, An Activity-Based Nanosensor for Traumatic Brain Injury, ACS Sens., 2020, 5(3), 686–692, DOI:10.1021/acssensors.9b01812.
- K. J. Hassett, K. E. Benenato, E. Jacquinet, A. Lee, A. Woods, O. Yuzhakov, S. Himansu, J. Deterling, B. M. Geilich, T. Ketova, C. Mihai, A. Lynn, I. McFadyen, M. J. Moore, J. J. Senn, M. G. Stanton, Ö. Almarsson, G. Ciaramella and L. A. Brito, Optimization of Lipid Nanoparticles for Intramuscular Administration of MRNA Vaccines, Mol. Ther. – Nucleic Acids, 2019, 15, 1–11, DOI:10.1016/j.omtn.2019.01.013.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, MRNA-Lipid Nanoparticle COVID-19 Vaccines: Structure and Stability, Int. J. Pharm., 2021, 601, 120586, DOI:10.1016/j.ijpharm.2021.120586.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Effects of Particle Size and Surface Charge on Cellular Uptake and Biodistribution of Polymeric Nanoparticles, Biomaterials, 2010, 31(13), 3657–3666, DOI:10.1016/j.biomaterials.2010.01.065.
- L. M. Kranz, M. Diken, H. Haas, S. Kreiter, C. Loquai, K. C. Reuter, M. Meng, D. Fritz, F. Vascotto, H. Hefesha, C. Grunwitz, M. Vormehr, Y. Hüsemann, A. Selmi, A. N. Kuhn, J. Buck, E. Derhovanessian, R. Rae, S. Attig, J. Diekmann, R. A. Jabulowsky, S. Heesch, J. Hassel, P. Langguth, S. Grabbe, C. Huber, Ö. Türeci and U. Sahin, Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy, Nature, 2016, 534(7607), 396–401, DOI:10.1038/nature18300.
- I. A. Khalil, M. A. Younis, S. Kimura and H. Harashima, Lipid Nanoparticles for Cell-Specific in Vivo Targeted Delivery of Nucleic Acids, Biol. Pharm. Bull., 2020, 43(4), 584–595, DOI:10.1248/bpb.b19-00743.
- J. F. Thompson, L. S. Hayes and D. B. Lloyd, Modulation of Firefly Luciferase Stability and Impact on Studies of Gene Regulation, Gene, 1991, 103(2), 171–177, DOI:10.1016/0378-1119(91)90270l.
- J. M. Ignowski and D. V. Schaffer, Kinetic Analysis and Modeling of Firefly Luciferase as a Quantitative Reporter Gene in Live Mammalian Cells, Biotechnol. Bioeng., 2004, 86(7), 827–834, DOI:10.1002/bit.20059.
- E. A. Nance, G. F. Woodworth, K. A. Sailor, T.-Y. Shih, Q. Xu, G. Swaminathan, D. Xiang, C. Eberhart and J. Hanes, A Dense Poly(Ethylene Glycol) Coating Improves Penetration of Large Polymeric Nanoparticles Within Brain Tissue, Sci. Transl. Med., 2012, 4(149), 149ra119–149ra119, DOI:10.1126/scitranslmed.3003594.
- E. Song, A. Gaudin, A. R. King, Y. E. Seo, H. W. Suh, Y. Deng, J. Cui, G. T. Tietjen, A. Huttner and W. M. Saltzman, Surface Chemistry Governs Cellular Tropism of Nanoparticles in the Brain, Nat. Commun., 2017, 8(1), 15322, DOI:10.1038/ncomms15322.
- R. L. Rungta, H. B. Choi, P. J. Lin, R. W. Ko, D. Ashby, J. Nair, M. Manoharan, P. R. Cullis and B. A. MacVicar, Lipid Nanoparticle Delivery of SiRNA to Silence Neuronal Gene Expression in the Brain, Mol. Ther. – Nucleic Acids, 2013, 2(12), e136, DOI:10.1038/mtna.2013.65.
- M. Mohamadpour, K. Whitney and P. J. Bergold, The Importance of Therapeutic Time Window in the Treatment of Traumatic Brain Injury, Front. Neurosci., 2019, 13, 7, DOI:10.3389/fnins.2019.00007.
- J. Ponsford, C. Willmott, A. Rothwell, P. Cameron, G. Ayton, R. Nelms, C. Curran and K. Ng, Impact of Early Intervention on Outcome After Mild Traumatic Brain Injury in Children, Pediatrics, 2001, 108(6), 1297–1303, DOI:10.1542/peds.108.6.1297.
- J. Ponsford, C. Willmott, A. Rothwell, P. Cameron, A. Kelly, R. Nelms and C. Curran, Impact of Early Intervention on Outcome Following Mild Head Injury in Adults, J. Neurol., Neurosurg. Psychiatry, 2002, 73(3), 330–332, DOI:10.1136/jnnp.73.3.330.
- M. L. Block and J.-S. Hong, Microglia and Inflammation-Mediated Neurodegeneration: Multiple Triggers with a Common Mechanism, Prog. Neurobiol., 2005, 76(2), 77–98, DOI:10.1016/j.pneurobio.2005.06.004.
- S. Kalra, R. Malik, G. Singh, S. Bhatia, A. Al-Harrasi, S. Mohan, M. Albratty, A. Albarrati and M. M. Tambuwala, Pathogenesis and Management of Traumatic Brain Injury (TBI): Role of Neuroinflammation and Anti-Inflammatory Drugs, Inflammopharmacol, 2022, 30(4), 1153–1166, DOI:10.1007/s10787-022-01017-8.
- D. R. Boone, J. M. Leek, M. T. Falduto, K. E. O. Torres, S. L. Sell, M. A. Parsley, J. C. Cowart, T. Uchida, M.-A. Micci, D. S. DeWitt, D. S. Prough and H. L. Hellmich, Effects of AAV-Mediated Knockdown of NNOS and GPx-1 Gene Expression in Rat Hippocampus after Traumatic Brain Injury, PLoS One, 2017, 12(10), e0185943, DOI:10.1371/journal.pone.0185943.
- L. Longhi, D. J. Watson, K. E. Saatman, H. J. Thompson, C. Zhang, S. Fujimoto, N. Royo, D. Castelbuono, R. Raghupathi, J. Q. Trojanowski, V. M.-Y. Lee, J. H. Wolfe, N. Stocchetti and T. K. McIntosh, Ex Vivo Gene Therapy Using Targeted Engraftment of NGF-Expressing Human NT2N Neurons Attenuates Cognitive Deficits Following Traumatic Brain Injury in Mice, J. Neurotrauma, 2004, 21(12), 1723–1736, DOI:10.1089/neu.2004.21.1723.
- L. L. Zou, L. Huang, R. L. Hayes, C. Black, Y. H. Qiu, J. R. Perez-Polo, W. Le, G. L. Clifton and K. Yang, Liposome-Mediated NGF Gene Transfection Following Neuronal Injury: Potential Therapeutic Applications, Gene Ther., 1999, 6(6), 994–1005, DOI:10.1038/sj.gt.3300936.
- M. F. Philips, G. Mattiasson, T. Wieloch, A. Björklund, B. B. Johansson, G. Tomasevic, A. Martínez-Serrano, P. M. Lenzlinger, G. Sinson, M. S. Grady and T. K. McIntosh, Neuroprotective and Behavioral Efficacy of Nerve Growth Factor—Transfected Hippocampal Progenitor Cell Transplants after Experimental Traumatic Brain Injury, J. Neurosurg., 2001, 94(5), 765–774, DOI:10.3171/jns.2001.94.5.0765.
- H. Ma, B. Yu, L. Kong, Y. Zhang and Y. Shi, Neural Stem Cells Over-Expressing Brain-Derived Neurotrophic Factor (BDNF) Stimulate Synaptic Protein Expression and Promote Functional Recovery Following Transplantation in Rat Model of Traumatic Brain Injury, Neurochem. Res., 2012, 37(1), 69–83, DOI:10.1007/s11064-011-0584-1.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01846b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |