
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid detection and quantification of paracetamol and its major metabolites using surface enhanced Raman scattering†
Najla
AlMasoud
ab,
Taghrid S.
Alomar
ab,
Yun
Xu
 b,
Cassio
Lima
b and
Royston
Goodacre
*b
b,
Cassio
Lima
b and
Royston
Goodacre
*b
aDepartment of Chemistry, College of Science, Princess Nourah bint Abdulrahman University, Riyadh 11671, Saudi Arabia
bCentre for Metabolomics Research, Department of Biochemistry and Systems Biology, Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, UK. E-mail: roy.goodacre@liverpool.ac.uk
First published on 14th March 2023
Abstract
Paracetamol (also known as acetaminophen) is an over-the-counter (OTC) drug that is commonly used as an analgesic for mild pain, headache, cold and flu. While in the short term it is a safe and effective medicine, it is sometimes used for attempted suicides particularly in young adults. In such circumstances it is important for rapid diagnosis of overdoses as antidotes can be given to limit liver damage from one of its primary metabolites N-acetyl-p-benzoquinone imine (NAPQI). Unfortunately, the demand for rapid and sensitive analytical techniques to accurately monitor the abuse of OTC drugs has significantly risen. Ideally these techniques would be highly specific, sensitive, reproducible, portable and rapid. In addition, an ideal point of care (PoC) test would enable quantitative detection of drugs and their metabolites present in body fluids. While Raman spectroscopy meets these specifications, there is a need for enhancement of the signal because the Raman effect is weak. In this study, we developed a surface-enhanced Raman scattering (SERS) methodology in conjunction with chemometrics to quantify the amount of paracetamol and its main primary metabolites (viz., paracetamol sulfate, p-acetamidophenyl β-D-glucuronide and NAPQI) in water and artificial urine. The enhancement of the SERS signals was achieved by mixing the drug or xenometabolites with a gold nanoparticle followed by aggregation with 0.045 M NaCl. We found that the SERS data could be collected directly, due to immediate analyte association with the Au surface and colloid aggregation. Accurate and precise measurements were generated, with a limit of detection (LoD) of paracetamol in water and artificial urine at 7.18 × 10−6 M and 2.11 × 10−5 M, respectively, which is well below the limit needed for overdose and indeed normal levels of paracetamol in serum after taking 1 g orally. The predictive values obtained from the analysis of paracetamol in water and artificial urine were also excellent, with the coefficient of determination (Q2) being 0.995 and 0.996, respectively (1 suggests a perfect model). It was noteworthy that when artificial urine was spiked with paracetamol, no aggregating agent was required due to the salt rich medium, which led to spontaneous aggregation. Moreover, for the xenometabolites of paracetamol excellent LoDs were obtained and these ranged from 2.6 × 10−4 M to 5 × 10−5 M with paracetamol sulfate and NAPQI having Q2 values of 0.934 and 0.892 and for p-acetamidophenyl β-D-glucuronide this was slightly lower at 0.6437.
1. Introduction
Paracetamol, also known as acetaminophen, N-acetyl-p-aminophenol or APAP, was first manufactured in 1878.1 It is an active pharmaceutical ingredient (API) that has been commonly used as an analgesic for mild pain, headache, cold and flu.2 Paracetamol can be purchased over-the-counter in the form of tablets, capsules, suppositories, suspensions and solutions. The recommended daily dosage of this medication is 1–4 g per day and 50–75 mg per kg per day for adults and children, respectively.3,4 It is widely accepted that this drug is not associated with the most frequently reported side effects of nonsteroidal anti-inflammatory drugs (NSAIDS).Despite the wide use of paracetamol, exceeding the recommended maximum single dose of 7 g per day in adults and 150 mg kg−1 in children may have life threatening implications and cause long-term damage and failure of vital organs, such as the liver. The normal serum level after taking 1 g paracetamol orally is 30 mg L−1, while for acute overdose victims this rises to 50–400 mg L−1 serum.5 In addition, other side effects of paracetamol misuse are skin rashes and suppression of blood sugar levels.6–8 Paracetamol is converted into three major metabolites in the human body (Fig. 1) including an inactive glucuronide (52–57% of urinary metabolites), sulfate conjugates (30–44%) and N-acetyl-p-benzoquinone imine (NAPQI) (5–15%).9,10 In case of acute overdose, the paracetamol toxicity is mainly due to NAPQI and this is due to depletion of glutathione which normally conjugates to NAPQI making it harmless.11 The liver and to a less extent the kidney and intestines, are the major organs that are damaged severely by the metabolites of paracetamol.12
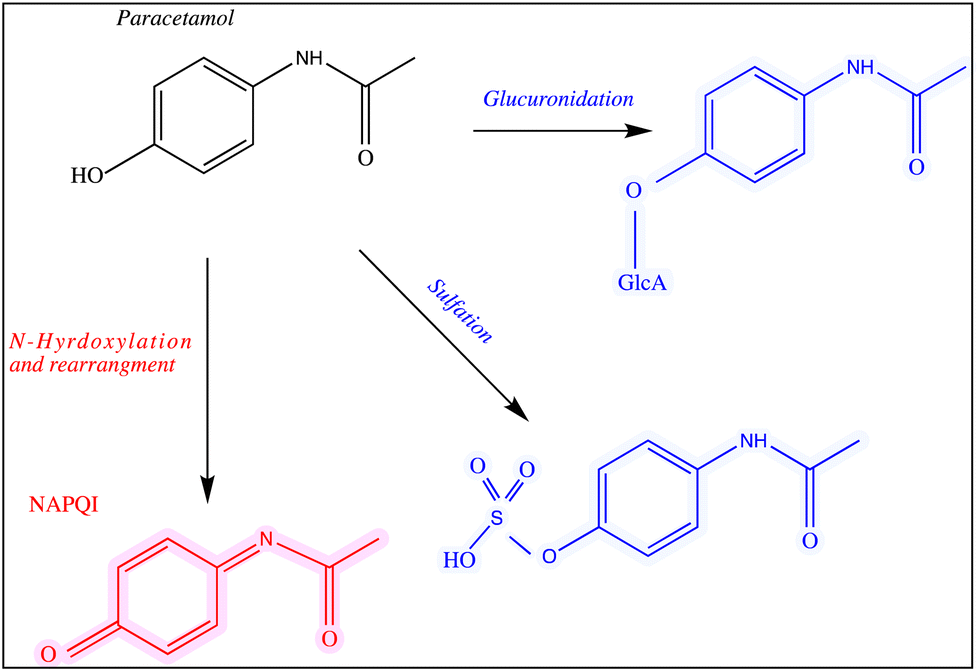 | ||
| Fig. 1 The main pathways of paracetamol metabolism. The structures of the drug and its metabolites are shown and all of these analytes were used in this study. | ||
In order to minimise minor and major side effects of medications, it is vital to identify and quantify over-the-counter drugs in human biofluids to ensure whether compliance is met by users and providers. Many research studies use different methods for the quantification and characterisation of drugs. The routine clinical use of paracetamol for a number of mild health conditions, has resulted in the application of many analytical tools for drug identification and quantification. These include chromatography,13 spectrophotometry14 and electrochemical analysis.15 Chromatography, in particular liquid chromatography, is widely used in the pharmaceutical industry to characterise drugs and this is due to its high resolution, precision and accuracy.16 However, chromatography is destructive, pre-treatment of samples is time-consuming and large quantities of organic/inorganic solvents are consumed. As a result, there is a need for alternative methods to characterise drugs and their metabolites that are faster, more reliable and sustainable.17–20 The ideal technique should have these features as well as being portable/handheld to enable point of care (PoC) use.
Raman spectroscopy is a vibrational analytical technique that has the advantage of providing a specific molecular pattern of a compound to enable identification and quantification. This analytical technique is non-invasive, increasingly available as portable devices and has been shown to be useful in the analysis of biofluids.21–23 However, this technique suffers from a weak signal and fluorescence from biological samples; in combination, these affect the quality of the data and limit its application in biological samples, such as urine.24 One way to enhance the Raman signal significantly is to apply surface-enhanced Raman scattering (SERS). This enhancement requires the analyte to be in close contact with a roughened metal surface25–28 such as gold/silver in colloidal suspensions and highly quantitative analysis has been readily reported.29–32
We have conducted extensive research in order to optimise SERS substrates for the detection of traces of drugs in human samples,33 both illicit and legal.34,35 Previous research has focused on the identification of both single and multiple analytes to detect various illicit drugs in body fluids using SERS. While there are reported of SERS on paracetamol,36,37 most are qualitative and do not include the quantitative analyses of the primary metabolites of paracetamol. Therefore, the aim of this study was to develop a SERS based portable assay to quantify paracetamol and its major metabolites.
2. Materials and method
2.1 Chemicals
The chemicals used for sample preparations included silver nitrate (purity >99.99%), trisodium citrate (99%), sodium tetrahydridoborate, hydroxylamine hydrochloride (99%), chloroauric acid (99.9%), sodium chloride, artificial urine, paracetamol and its metabolites paracetamol sulfate potassium, N-acetyl-p-benzoquinone imine and p-acetamidophenyl β-D-glucuronide sodium salts (99%). All of these materials were purchased from Sigma-Aldrich (Dorset, UK). The chemical structure of paracetamol and its major primary metabolites are illustrated in Fig. 1.2.2 Preparation of glassware
The glassware used for the preparation of gold and silver nanoparticles was cleaned using aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, HCl/HNO3). Next, double distilled water was used to rinse the glassware to remove any remaining residues, and it was then allowed to dry at 50 °C.
:1, HCl/HNO3). Next, double distilled water was used to rinse the glassware to remove any remaining residues, and it was then allowed to dry at 50 °C.
2.3 Synthesis of metal nanoparticles
The resultant colloids from the three methods were stable when poured into glass flasks. They were covered in foil and stored in a dark cupboard at room temperature for several weeks.
2.4 Nanoparticles characterisation
Different colloids were characterised by a UV-Vis spectrophotometer on a Jenway 7200 Visible 72 Series Diode Array Scanning Spectrophotometer (Cole-Parmer Ltd, Staffordshire, UK) (with a range of 335–800 nm). The UV-Vis absorbance spectra of gold nanoparticles are shown in Fig. S1.† In addition, scanning electron microscopy (SEM) was used to visualize the colloids using a Hitachi S-4800 microscope operating at a voltage of 1 kV: images of the same gold colloid have been reported elsewhere,40 and as the silver colloids were not optimal for SERS (see below) the micrographs are not reported here.2.5 Instrumentation
To collect SERS data, we used a DeltaNu Advantage portable Raman spectroscopy (DeltaNu, Laramie, WY, USA), equipped with a 785 nm HeNe laser and powered with an output of ∼60 mW on the sample. This Raman device was calibrated daily using a polystyrene internal standard supplied by the instrument manufacturer. The spectral range used was 200–1800 cm−1 and the laser exposure time was set to 30 s.2.6 Sample preparation for SERS measurements
The following protocol was used for measuring paracetamol and its major metabolites: 250 μL of each drug sample was mixed with 250 μL of the colloid, followed by the addition of 50 μL NaCl (0.5 M) or KNO3 (0.5 M) as the aggregating agents. The mixtures were then immediately placed in the DeltaNu Raman system and the spectral data were collected.Initial experiments were carried out to optimize the SERS conditions. These involved the assessment of the three different colloids described above: gold citrate colloid, silver hydroxylamine colloid or silver borohydride. In addition, tests were conducted on whether NaCl or KNO3 was the best aggregating agent. Finally, after the analyte, colloid and aggregating agent were mixed spectra were collected after aggregation for 0 to 40 min in intervals of 5 min, to ascertain at which point the signal was strongest.
For quantitative SERS analysis paracetamol and its metabolites were dissolved in water individually at a series of concentrations ranging from 3 mM to 0.003 mM. In addition, the same range was used for paracetamol spiked into artificial urine, and for this experiment SERS measurements were conducted in both the presence or absence of the aggregating agent (NaCl).
2.7 Chemometrics analysis
After exporting the data off the DeltaNu spectrometers as ASCII files, MATLAB software R2021b (The Math Works Inc., Natick, MA, USA) was used to analyze the SERS spectral data. The SERS spectra were baseline corrected by using asymmetric least squares,41 after which these SERS spectra were smoothed via a Savitzky–Golay filter using a 2nd order polynomial and a window width of 11 points. Finally the spectra were normalized by vector norm so that the sum of square of each spectrum equals 1 and subjected to unsupervised principal components analysis (PCA)42,43 to visualize the natural patterns within these SERS data. Supervised partial least-squares regression (PLSR)44,45 was used to quantify the concentration of paracetamol and its metabolites in the mixtures as described previously.20,31 As the concentration ranges of these analytes spanned over two orders of magnitude the natural log (ln) of the output was taken to normalise the data and to stabilize the variance. The PLSR models were trained and validated using a k-fold double cross-validation procedure where k is the number of different concentrations within each analyte dilution range.46 Finally, the limit of detection (LoD) of paracetamol and its metabolites were also estimated based on the predictions PLSR models on the test sets only using the method described in ref. 47. For PLSR modelling and tests, the SERS spectra were pre-processed by standard normal variate (SNV) normalization only so that each spectrum had a mean of 0 and standard deviation of 1. This is to minimize steps of pre-processing and reduce risk of over-fitting associated to pre-processing methods and their parameters.3. Results and discussion
3.1 SERS optimisation experiments
SERS is a technique that uses nanoscale roughened metal surfaces, such as gold and silver, in order to boost the rather weak Raman signal. This study started with the optimisation of SERS in order to generate not only enhanced signals but spectra that were reproducible. The optimisation of SERS was carried out as follows: (a) the most suitable colloid was determined from three different nanoparticle preparations; (b) the most compatible aggregating agent between NaCl and KNO3 was determined; and (c) the time taken for the analyte (paracetamol in this case for the optimisation experiments) to associate with the metal surface and aggregation agent was recorded.Step (a) involved the synthesis of three different colloids including – silver borohydride, silver hydroxylamine and gold citrate – in order to investigate the optimal metal surface for SERS along with the assessment of the two aggregating agents (step (b)). Raman bands were observed using all three SERS substrates when detecting paracetamol (Fig. S2†). It is noteworthy that the SERS signal from paracetamol was noticeably different for each of the three colloids, and for silver hydroxylamine and silver borohydride colloids the spectra were non-reproducible and thus unreliable. By contrast, it can be seen that the gold nanoparticles gave highly reproducible signals as the three replicate spectra overlapped when either NaCl or KNO3 was used as the aggregating agent (Fig. S2†). Both combinations gave similar SERS spectra indicating that the effect is nanoparticle dependent rather than being contingent on the aggregating agent. Close inspection of the spectra in Fig. S2† showed that when NaCl was used as the aggregating agent the most reproducible signals were observed. As a result, gold nanoparticles were utilized in subsequent investigations of paracetamol and its metabolites, along with employing 0.045 M NaCl (total within the sample) as the aggregating agent.
After choosing the colloid-aggregating agent pair, the final step (c) involved investigating the effect of analyte, colloid and aggregating agent association on the time required to observe SERS signals. Paracetamol (3 mM) was mixed with the gold colloid, followed by the addition of 50 μL of 0.5 M NaCl as the aggregating agent under ambient conditions (i.e. RT) for association to occur. Analysis of each mixture was carried at 5-minute intervals for a duration of 0 to 40 min. Plots of the raw SERS spectra during this time frame for paracetamol are shown in Fig. S3.† All spectra in this time series show very similar bands and it was found that in the first 10 min of association, there was no obvious change in the SERS signal intensity and this implies that the analyte (paracetamol) associated rapidly with the gold surface. After 10 min there was a slight decrease in the signal intensity which suggests that upon aggregation larger aggregates were precipitating out of suspension. Thus, for all consequent analyses, the association of samples took place at 0 min, hence the sample was analyzed immediately.
3.2 Limit of detections for paracetamol and its major metabolites
As determined and reported above the optimal SERS protocol to quantify the amount of paracetamol in a given sample was determined using gold colloid prepared by reduction of chloroauric acid with citrate ions and 0.045 M NaCl as the aggregating agent. The resultant SERS data were collected immediately; i.e., immediately after 0 min of preparation for each sample. Using this SERS protocol, we generated a dilution series of paracetamol and then each of its metabolites to determining the limit of detection (LoD) for each analyte.For each of the four analytes a combination of visible inspection of the dilution series along with PCA was used to assess whether there was an obvious trend in PCA scores space that was related to the concentration of the analyte under investigation. The corresponding PCA loadings plots were then used to assess the most diagnostic peaks. This was then followed by PLSR modelling to predict the level of the drug or its xenometabolites and in these plots only the test sets are plotted from the k-fold cross validation procedure used. These PLSR models were then used to estimate the LoDs for each analyte.
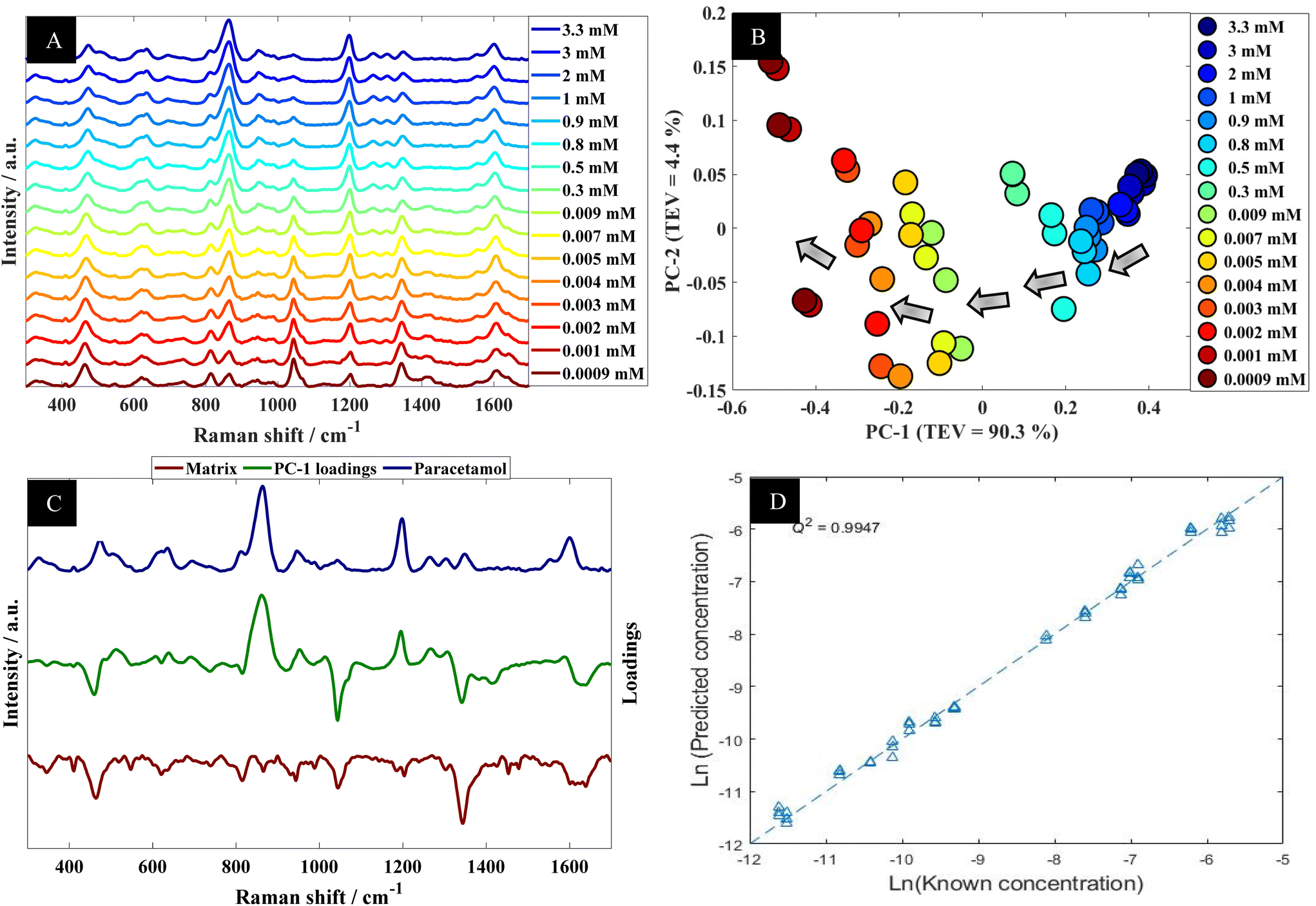 | ||
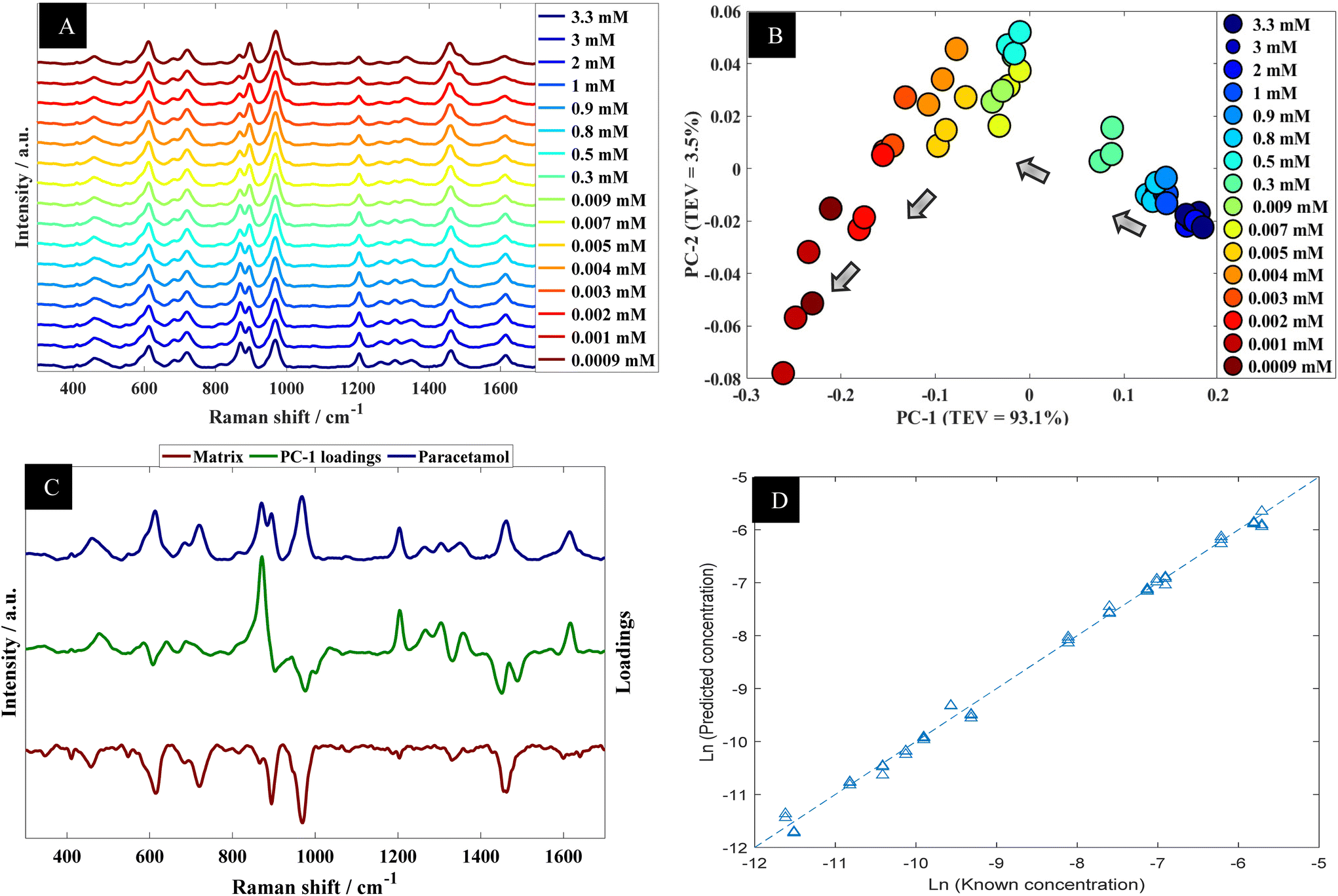
| Fig. 2 (A) A series of SERS spectra of paracetamol in water at different concentration; spectra are offset for clarity. (B) PCA scores plot of SERS data acquired from paracetamol in water at different concentrations; TEV refers to total explained variance. (C) The corresponding PC-1 loadings plot (green line), with SERS spectra of paracetamol in water (blue) and the matrix (brown line; this has been multiplied by (−1) for clarity). (D) PLSR quantitative output generated using SERS data from paracetamol in water; data shown are the test set outputs only from the k-fold validation procedure. | ||
Further analysis using PCA was carried out to analyze SERS spectra of paracetamol in water (Fig. 2B). The PCA scores plot shows a clear trend along the PC-1 axis from positive to negative scores which correlates with decreasing concentrations of paracetamol. In the PCA plot, the first principal component (PC-1) explains the most variance within the data, so this is highly significant and accounts for 90.3% of the TEV (total explained variance). By contrast, the variance associated with PC-2 is relatively low (4.4%). Note that the replicate spectra cluster very closely together in the PCA scores plot which shows that using our optimal SERS protocol we achieved high reproducibility of signals. Furthermore, inspection of the loadings plot (Fig. 2C) showed two peaks were highly characteristic for paracetamol and these were at around 860 cm−1 and 1202 cm−1. The peak at 860 cm−1 corresponds to the aromatic ring bend (C–H out-of-plane skeletal deformation) in paracetamol.48 In accordance with the electromagnetic selection rules the C–H stretching mode should be relatively enhanced when the C–H bond is perpendicular to the metal surface plane, as compared with the case in which the C–H bond lies parallel to the surface.49
Next the whole range of the SERS spectra (i.e., 200–1800 cm−1) were used to construct PLSR models in order to test the ability of SERS to quantify paracetamol at low concentration in water. The PLSR predictions for paracetamol concentrations are shown in Fig. 2D. This figure illustrates excellent predictive values, with Q2 of 0.99 in predictions in a natural logarithm scaled concentration and Q2 of 0.95 when these predictions were reverted back to original scale (in moles). This shows an excellent agreement between predicted and corresponding known concentrations of test set samples as the closer the Q2 value to 1, the better the predictions. Furthermore, the root means square error of cross validation (RMSECV) was 2.12 × 10−4 M, and the LoD was found to be 7.18 × 10−6 M. This LoD is lower than the LoD that was reported in a previous study on paracetamol using SERS.50 Note as mentioned above the normal serum level after taking 1 g paracetamol orally is 30 mg L−1 (∼2 × 10−4 M), while for acute overdose victims this rises to 50–400 mg L−1 (3.3 × 10−4 M to 2.6 × 10−3 M) serum,5 so these detection limits are clinically useful and much faster than liquid chromatography.
The optimised SERS protocol was further used to detect metabolites of paracetamol in water. For this purpose, three different metabolites were investigated including: p-acetamidophenyl β-D-glucuronide, paracetamol sulfate as well as NAPQI. Fig. 3 illustrates SERS spectra for all three paracetamol metabolites along with paracetamol and the matrix. Despite the similarities regarding their chemical structure, these SERS spectra from all four analytes displayed distinct spectral features, which could be associated with the different chemical conjugates due to glucuronidation or sulfation and for NAPQI hydroxylation and structural rearrangement. By contrast, these differences could also be due to orientation effects resulting in different molecule adsorption onto the metal surface as reported by Lombardi and Birke;52 note, there is no direct evidence of this.
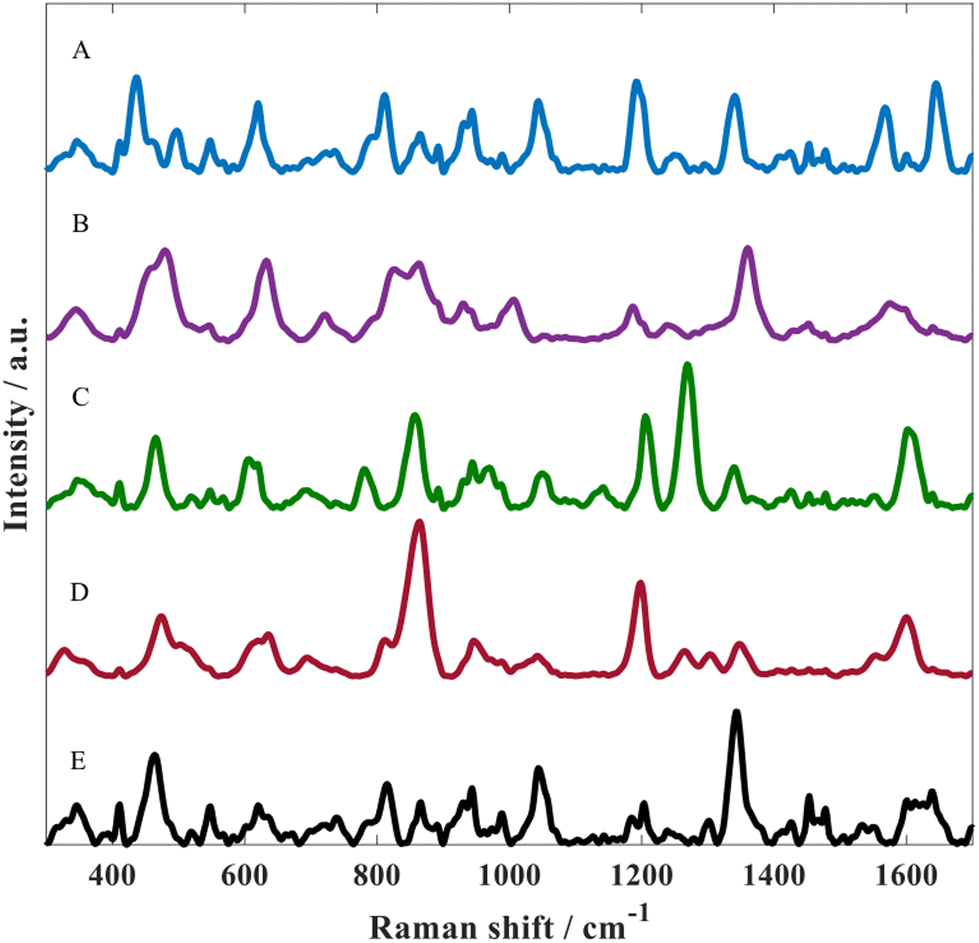 | ||
| Fig. 3 SERS spectra of (A) paracetamol sulfate potassium, (B) N-acetyl-p-benzoquinone imine, (C) p-acetamidophenyl β-D-glucuronide, (D) paracetamol and (E) matrix (i.e., the nanoparticles and aggregating agent alone without any analyte). SERS conditions used: gold nanoparticles with 0.045 M NaCl as the aggregating agent, and the collection time was 30 s. | ||
In all of these experiments we include the matrix. This is the colloid after aggregation without the addition of any analyte. Thus, the peaks seen in the spectrum in Fig. 3E are not analyte but belong to citrate which is the capping agent that remains on the gold surface after metal reduction during nanoparticle formation.
Using p-acetamidophenyl β-D-glucuronide in water as an example the next stage of the analysis was to determine quantitative information that could be extracted from the SERS spectra of this metabolite. Fig. 4A shows the multiple peaks at 606, 860, 1202 and 1270 cm−1 in the baseline-corrected SERS spectra of p-acetamidophenyl β-D-glucuronide decreased in intensity with respect to concentrations of the analyte in the solutions. The data obtained from SERS was further investigated by PCA (Fig. 4B). The PCA score plot for p-acetamidophenyl β-D-glucuronide demonstrated a clear concentration-dependent trend, which is reflected on PC1 with TEV of 83.8%. Concentrations of p-acetamidophenyl β-D-glucuronide less than 0.06 mM were separated slightly on PC2 (TEV of 8.5%) indicating at lower concentrations the SERS spectra were less reproducible than those >0.06 mM. PC-1 loadings plot (Fig. 4C) showed that peaks at 1202 and 1270 cm−1 were the most significant vibrational bands contributing to the separation.
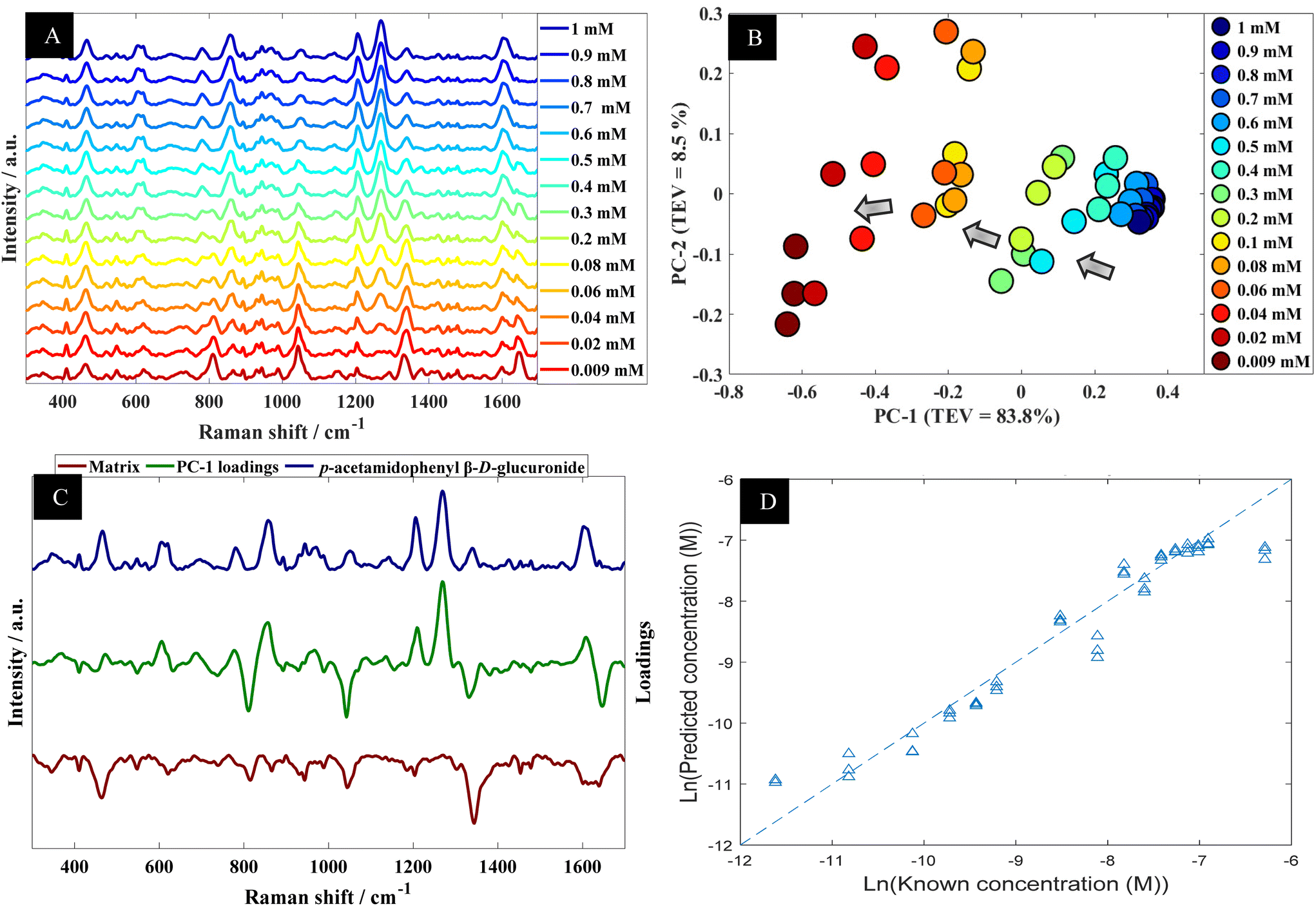 | ||
| Fig. 4 (A) A series of SERS spectra of p-acetamidophenyl β-D-glucuronide in water at different concentration; spectra are offset for clarity. (B) PCA scores plot of SERS data acquired from p-acetamidophenyl β-D-glucuronide in water at different concentrations; TEV refers to total explained variance. (C) Corresponding PC-1 loadings plot (green line), with SERS spectra of p-acetamidophenyl β-D-glucuronide in water (blue) and matrix (brown line; this has been multiplied by (−1) for clarity). (D) PLSR quantitative output generated using SERS data from p-acetamidophenyl β-D-glucuronide in water; data shown are the test set outputs only from the k-fold validation procedure. | ||
Furthermore, other bands were observed and were possibly specific to the matrix because it is anticipated that p-acetamidophenyl β-D-glucuronide interacts more strongly with the gold nanoparticles. In light of the PCA, next PLSR modelling was carried out for the quantification of p-acetamidophenyl β-D-glucuronide using the SERS spectral data. The PLSR predictions for the glucuronide conjugated metabolite concentrations are shown in Fig. 4D. These predictions showed excellent predictive accuracies, with Q2 of 0.9379, RMSECV of 2.88 × 10−4 M and a LoD at 4.97 × 10−5 M.
The two other metabolites were also analyzed as described above. Briefly, Fig. S4A and D† show a concentrations profile and the effects on the SERS spectra of paracetamol sulfate and NAPQI. After PCA (not shown) the corresponding PCA loading plots were inspected (Fig. S4B and E†) for these two metabolites in order to detect significant peaks. The PCA loading plots correlated to the wavenumber shifts from the analytes as they replaced the citrate on the gold surface. PLSR models were also employed and predictions were generated as shown in Fig. S4C and F.† It was apparent from these observations that there was an abundance of information within the SERS spectra, allowing excellent predictions of the concentrations of metabolites in the samples. In general, very low root means square error (RMSE) of cross validation (RMSECV) confirmed a high prediction accuracies for paracetamol and its three metabolites (Table 1). For paracetamol and p-acetamidophenyl β-D-glucuronide, the concentration ranged over three orders of magnitude, therefore log-scaling was needed to prevent model being dominated by high concentration samples. By contrast, for paracetamol sulfate potassium and NAPQI, the range of concentration varied within one order of magnitude and a log-scaling pre-process was not necessary. In fact, log-scaling had adverse effect when applied to these two sets of data on the accuracies in predicting test set, resulted in much higher RMSECV and lower Q2 (Table 1). The results showed that the concentrations of samples in both low and high concentrations had been over estimated, and this suggests that when variance stabilization was not necessary, a forced log-linear modelling had resulted in extra error in predictions.
| Analyte | Q 2(ln) | Q 2 | RMSECV | LoD |
|---|---|---|---|---|
| Q 2(ln) is the coefficient of determination of natural log-scaled PLSR model predictions; Q2 is the coefficient of determination calculated after the predictions of log-scaled PLSR model predictions had been scaled back to their original scale using exponential function, RMSECV is the root-mean-squared error of cross-validation, calculated in the original scale (M); LoD is the estimated limit of detection using the corresponding PLSR model as described in the text. | ||||
| Paracetamol | 0.9947 | 0.9590 | 2.12 × 10−4 M | 7.18 × 10−6 M |
| p-Acetamidophenyl β-D-glucuronide | 0.9379 | 0.6437 | 2.88 × 10−4 M | 4.97 × 10−5 M |
| Paracetamol sulfate potassium | 0.8538 | 0.2475 | 3.90 × 10−4 M | 3.28 × 10−4 M |
| NAPQI | 0.7263 | 0.0253 | 4.21 × 10−4 M | 3.91 × 10−4 M |
| Paracetamol sulfate potassium (without log-scaling) | — | 0.9339 | 1.16 × 10−4 M | 2.56 × 10−4 M |
| NAPQI (without log-scaling) | — | 0.8920 | 1.40 × 10−4 M | 2.81 × 10−4 M |
 | ||
| Fig. 5 (A) A series of SERS spectra of paracetamol in artificial urine at different concentration; spectra are offset for clarity. (B) PCA scores plot of SERS data acquired from paracetamol in artificial urine at different concentrations. (C) Corresponding PC-1 loadings plot (green line), with SERS spectra of paracetamol (blue) and matrix (brown line; this has been multiplied by (−1) for clarity). Spectra are offset for clarity. (D) PLSR quantitative output generated using SERS data from paracetamol in artificial urine; data shown are the test set outputs only from the k-fold validation procedure. | ||
The data collected from the spiked urine samples, were analyzed using PCA (Fig. 5B) where there was a clear trend in PCA space where PC-1 correlated with the concentration of paracetamol. The corresponding PC-1 loading plot (Fig. 5C) also highlighted 860 and 1202 cm−1 as being important contributors to this model. Furthermore, PLSR model predictions of the collected data were plotted against corresponding known concentrations as shown in Fig. 5D, which showed excellent agreement between predicted concentrations and corresponding known concentrations. The figures of merit from PLS in terms of Q2, RMSECV and LoD are reported in Table 2.
| Analytes | Q 2(ln) | Q 2 | RMSECV | LoD |
|---|---|---|---|---|
| Q 2(ln) is the coefficient of determination of natural log-scaled PLSR model predictions; Q2 is the coefficient of determination calculated after the predictions of natural log-scaled PLSR models had been scaled back to their original scale using exponential functions; RMSECV is the root-mean-squared error of cross-validation, calculated in the original scale (M); LoD is the estimated limit of detection using the corresponding PLSR model as described in the text. | ||||
| Paracetamol in urine with aggregation agent | 0.9956 | 0.9801 | 1.48 × 10−4 M | 2.24 × 10−5 M |
| Paracetamol in urine without aggregation agent | 0.9954 | 0.9897 | 1.06 × 10−4 M | 2.11 × 10−5 M |
We also collected a series of SERS spectra from urine alone, along with a spike of paracetamol (with and without NaCl as the aggregating agent), along with paracetamol in water as a comparison (Fig. S5†). It is clear from these spectra that there are SERS peaks that are present in urine which are also observed when paracetamol is present. Many of these peaks have been described previously19,53–55 and many can be assigned to uric acid (∼640 cm−1: ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–N deformation; ∼890 cm−1: N–H bending), hypoxanthine (∼720 cm−1: C–H stretching) and tryptophan (∼680 cm−1; ∼1360 cm−1 (also ascribed to guanine)). It is noteworthy that despite these SERS urine peaks being present when paracetamol is analysed, they are not use in quantitative modelling as illustrated in the PCA loading plots in Fig. 5C (green line).
O–N deformation; ∼890 cm−1: N–H bending), hypoxanthine (∼720 cm−1: C–H stretching) and tryptophan (∼680 cm−1; ∼1360 cm−1 (also ascribed to guanine)). It is noteworthy that despite these SERS urine peaks being present when paracetamol is analysed, they are not use in quantitative modelling as illustrated in the PCA loading plots in Fig. 5C (green line).
During these experiments we observed that spontaneous aggregation occurred which is most likely due to the presence of salts in artificial urine. Therefore, further experiments were carried out on these spiked samples without the addition of the NaCl aggregating agent. The same results as reported above in terms of visible inspection of the SERS spectra (Fig. S6A†), use of PCA (Fig. S6B and C†) and PLSR (Fig. S6D†).
Table 2 compares the values that were generated from the PLS models for paracetamol in urine with and without an aggregation agent. Surprisingly, the Q2(ln) and Q2 values for paracetamol in artificial urine were slightly higher than in water. In addition, both the RMSECV and LoDs were lower for artificial urine. These results are likely due to be due to an increase in colloid aggregation, in addition to an enhanced association of paracetamol with a gold surface, which is most likely due to the presence of salts found in artificial urine samples. The artificial urine used in this study was rich in salts, such as NaCl as well as many other salts (Table S1†). These observations lead to the interesting prospect of using a combination of salts in future experiments to further enhance SERs signals through optimal aggregation.
4. Conclusion
The use of rapid and sensitive portable/handheld techniques to measure the concentration of drugs and their metabolites is vital, especially in drug overdoses where administration of the most appropriate antidote is essential and time limited. Such technology would reduce unwanted side effects would be quickly detected and prevented. Quantification of drugs and their metabolites is typically conducted by using LC-MS. However, this is costly, labor and time intense and samples generally need to be transported to a dedicated laboratory which means that results are retrospective. Thus, alternative portable techniques are in urgent demand for the rapid quantification of drugs in biofluids. For this purpose, we chose paracetamol as the model drug and its three main metabolites including NAPQI which can cause drug-induced liver injury. These analytes were subsequently analysed using Raman spectroscopy. However, despite this method being reproducible, portable, robust, less laborious and time efficient, the scattering process of Raman is very weak, therefore we enhanced the Raman signal using a gold colloid to effect SERS. After optimisation of the SERS conductions multivariate data analysis was conducted using PCA and PLSR to look at trends in the SERS data and to quantify paracetamol and its major metabolites in a series of samples where the target analyte was diluted typically from 3 mM to 0.003 mM. The predictions showed excellent predicative values of paracetamol in water as well as artificial urine, and all three metabolites could also be readily quantified in water. To illustrate that all four analytes can be observed in a single mixture we also generated a SERS spectrum of all four analytes together in water. It is clear from Fig. S7† that the multiplexed spectra of all four analytes contains clear features (peaks) that can be assigned to the pure drug and drug metabolite spectra. It was noted that there was no need to add an aggregating agent to the spiked artificial urine with paracetamol, which is likely because of the salt rich medium in the urine and we shall be exploring mixed salts in the future for enhanced aggregation and SERS. Future suggested studies will also focus on developing suitable pre-sample treatment methods on the different compounds found in blood.Finally, while there is some discussion in the field as to the reproducibility of SERS for generating quantitative data from target analytes in mixtures, there are several excellent reviews have highlighted the use of internal standards that can be used to compensate for any variations due to concentration of analytes-nanoparticles and laser power within the system.30,56–58 These strategies have been used within clinical sciences and the interested reader will find details within these reviews.
Author contributions
Conceptualization, RG; methodology, RG, NA and TSA; formal analysis, CL and YX; resources, RG; data curation, CL; writing—original draft preparation, NA; writing—review and editing, CL, YX, TSA and RG; funding acquisition, Princess Nourah Bint Abdulrahman University.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
NA and TSA thank Princess Nourah bint Abdulrahman University for supporting this research through scholarship program for funding and support.References
- H. N. Morse, Ber. Dtsch. Chem. Ges., 1878, 11, 232–233 CrossRef.
- C. Thompson, M. C. Davies, C. J. Roberts, S. J. Tendler and M. J. Wilkinson, Int. J. Pharm., 2004, 280, 137–150 CrossRef CAS PubMed.
- L. L. Mazaleuskaya, K. Sangkuhl, C. F. Thorn, G. A. FitzGerald, R. B. Altman and T. E. Klein, Pharmacogenet. Genomics, 2015, 25, 416 CrossRef CAS PubMed.
- A. R. Temple, B. R. Temple and E. K. Kuffner, Clin. Ther., 2013, 35, 1361–1375 CrossRef CAS PubMed .e1345.
- J. A. Marx, R. Hockberger and R. Walls, Rosen's Emergency Medicine: Concepts and Clinical Practice, Elsevier, 2010, vol. 2 Search PubMed.
- S. R. Rose, Am. J. Health-Syst. Pharm., 1994, 51, 3065–3068 CrossRef CAS.
- J. D. Perkins, Liver Transplant., 2006, 12, 682–686 CrossRef PubMed.
- N. AlRuwaili, F. Zaidan, A. AlFattani and S. Alenazi, Int. J. Pediatr. Adolesc. Med., 2022, 9, 62–65 CrossRef PubMed.
- L. Prescott, Drugs, 1983, 25, 290–314 CrossRef CAS PubMed.
- M. R. McGill and H. Jaeschke, Pharm. Res., 2013, 30, 2174–2187 CrossRef CAS PubMed.
- M. J. Hodgman and A. R. Garrard, Crit. Care Clin., 2012, 28, 499–516 CrossRef PubMed.
- J. G. Bessems and N. P. Vermeulen, Crit. Rev. Toxicol., 2001, 31, 55–138 CrossRef CAS PubMed.
- A. F. El-Yazbi, K. M. Guirguis, M. M. Bedair and T. S. Belal, Drug Dev. Ind. Pharm., 2020, 46, 1853–1861 CrossRef CAS PubMed.
- E. A. Abdelaleem, I. A. Naguib, E. S. Hassan and N. W. Ali, Anal. Chem. Lett., 2016, 6, 13–23 CrossRef CAS.
- S. J. Saleem and M. Guler, Electroanalysis, 2019, 31, 2187–2198 CrossRef CAS.
- M. Gumustas, S. Kurbanoglu, B. Uslu and S. A. Ozkan, Chromatographia, 2013, 76, 1365–1427 CrossRef CAS.
- M. A. Bakkar, H. Nawaz, M. I. Majeed, A. Naseem, A. Ditta, N. Rashid, S. Ali, J. Bajwa, S. Bashir and S. Ahmad, Spectrochim. Acta, Part A, 2021, 245, 118900 CrossRef CAS PubMed.
- T. De Beer, W. Baeyens, A. Vermeire, D. Broes, J. P. Remon and C. Vervaet, Anal. Chim. Acta, 2007, 589, 192–199 CrossRef CAS PubMed.
- A. Subaihi, L. Almanqur, H. Muhamadali, N. AlMasoud, D. I. Ellis, D. K. Trivedi, K. A. Hollywood, Y. Xu and R. Goodacre, Anal. Chem., 2016, 88, 10884–10892 CrossRef CAS PubMed.
- H. Muhamadali, M. Chisanga, A. Subaihi and R. Goodacre, Anal. Chem., 2015, 87, 4578–4586 CrossRef CAS PubMed.
- K. J. Ember, M. A. Hoeve, S. L. McAughtrie, M. S. Bergholt, B. J. Dwyer, M. M. Stevens, K. Faulds, S. J. Forbes and C. J. Campbell, npj Regener. Med., 2017, 2, 1–10 CrossRef PubMed.
- D. I. Ellis, D. P. Cowcher, L. Ashton, S. O'Hagan and R. Goodacre, Analyst, 2013, 138, 3871–3884 RSC.
- C. A. F. de Oliveira Penido, M. T. T. Pacheco, I. K. Lednev and L. Silveira Jr., J. Raman Spectrosc., 2016, 47, 28–38 CrossRef.
- J. Cailletaud, C. De Bleye, E. Dumont, P.-Y. Sacré, L. Netchacovitch, Y. Gut, M. Boiret, Y.-M. Ginot, P. Hubert and E. Ziemons, J. Pharm. Biomed. Anal., 2018, 147, 458–472 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- Y. Lu, L.-W. Wu, W. Cao and Y.-F. Huang, Anal. Chem., 2022, 15, 6011–6016 CrossRef PubMed.
- W. Smith, Chem. Soc. Rev., 2008, 37, 955–964 RSC.
- C. De Bleye, E. Dumont, E. Rozet, P.-Y. Sacré, P.-F. Chavez, L. Netchacovitch, G. Piel, P. Hubert and E. Ziemons, Talanta, 2013, 116, 899–905 CrossRef CAS PubMed.
- L. Gozdzialski, A. Rowley, S. A. Borden, A. Saatchi, C. G. Gill, B. Wallace and D. K. Hore, Int. J. Drug Policy, 2022, 102, 103611 CrossRef PubMed.
- R. Goodacre, D. Graham and K. Faulds, TrAC, Trends Anal. Chem., 2018, 102, 359–368 CrossRef CAS.
- D. P. Cowcher, Y. Xu and R. Goodacre, Anal. Chem., 2013, 85, 3297–3302 CrossRef CAS PubMed.
- S. Adomavičiūtė, M. Velička and V. Šablinskas, Plasmonics in Biology and Medicine XVI, Proc. SPIE, 2019, vol. 10894, p. 108940E Search PubMed.
- C. Levene, E. Correa, E. W. Blanch and R. Goodacre, Anal. Chem., 2012, 84, 7899–7905 CrossRef CAS PubMed.
- S. Mabbott, A. Eckmann, C. Casiraghi and R. Goodacre, Analyst, 2013, 138, 118–122 RSC.
- S. Mabbott, E. Correa, D. P. Cowcher, J. W. Allwood and R. Goodacre, Anal. Chem., 2013, 85, 923–931 CrossRef CAS PubMed.
- E. de Barros Santos, E. C. N. L. Lima, C. S. de Oliveira, F. A. Sigoli and I. O. Mazali, Anal. Methods, 2014, 6, 3564–3568 RSC.
- S. Farquharson, C. Brouillette, W. Smith and C. Shende, Front. Chem., 2019, 7, 706 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- N. Leopold and B. Lendl, J. Phys. Chem. B, 2003, 107, 5723–5727 CrossRef CAS.
- T. S. Alomar, N. AlMasoud, Y. Xu, C. Lima, B. Akbali, S. Maher and R. Goodacre, Sensors, 2022, 22, 7832 CrossRef PubMed.
- P. H. Eilers, Anal. Chem., 2004, 76, 404–411 CrossRef CAS PubMed.
- R. Vidal, Y. Ma and S. S. Sastry, Generalized Principal Component Analysis, Springer, 2016, pp. 25–62 Search PubMed.
- H.-P. Wang, P. Chen, J.-W. Dai, D. Liu, J.-Y. Li, Y.-P. Xu and X.-L. Chu, TrAC, Trends Anal. Chem., 2022, 116648 CrossRef CAS.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17 CrossRef CAS.
- X. Zhao, N. Wang, M. Zhu, X. Qiu, S. Sun, Y. Liu, T. Zhao, J. Yao and G. Shan, Molecules, 2022, 27, 1707 CrossRef CAS PubMed.
- K. Roy and P. Ambure, Chemom. Intell. Lab. Syst., 2016, 159, 108–126 CrossRef CAS.
- A. C. Olivieri, H. C. Goicoechea and F. A. Iñón, Chemom. Intell. Lab. Syst., 2004, 73, 189–197 CrossRef CAS.
- M. Elbagerma, H. Edwards, T. Munshi and I. Scowen, CrystEngComm, 2011, 13, 1877–1884 RSC.
- L. Andronie, S. Cîntă Pînzaru, N. Peica, N. Leopold and O. Cozar, Stud. Univ. Babes-Bolyai, Phys., 2009, 2, 51–57 Search PubMed.
- Z. Mukanova, K. Gudun, Z. Elemessova, L. Khamkhash, E. Ralchenko and R. Bukasov, Anal. Sci., 2018, 34, 183–187 CrossRef CAS PubMed.
- E. Piperno and D. Berssenbruegge, Lancet, 1976, 308, 738–739 CrossRef PubMed.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2008, 112, 5605–5617 CrossRef CAS.
- A. Bonifacio, S. Cervo and V. Sergo, Anal. Bioanal. Chem., 2015, 407, 8265–8277 CrossRef CAS PubMed.
- C. Westley, Y. Xu, B. Thilaganathan, A. J. Carnell, N. J. Turner and R. Goodacre, Anal. Chem., 2017, 89, 2472–2477 CrossRef CAS PubMed.
- J. Lin, Z. Huang, X. Lin, Q. Wu, K. Quan, Y. Cheng, M. Zheng, J. Xu, Y. Dai and H. Qiu, Biomed. Opt. Express, 2020, 11, 7109–7119 CrossRef CAS PubMed.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. G. de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- R. D. Norton, H. T. Phan, S. N. Gibbons and A. J. Haes, Annu. Rev. Phys. Chem., 2022, 73, 141–162 CrossRef PubMed.
- S. E. Bell, G. Charron, E. Cortés, J. Kneipp, M. L. de la Chapelle, J. Langer, M. Procházka, V. Tran and S. Schlücker, Angew. Chem., Int. Ed., 2020, 59, 5454–5462 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00249g |
| This journal is © The Royal Society of Chemistry 2023 |