
Pyrolysis-free synthesis of single-atom cobalt catalysts for efficient oxygen reduction†
Rui
Ma
a,
Xun
Cui
 *ab,
Yonglin
Wang
a,
Zongying
Xiao
a,
Rui
Luo
a,
Likun
Gao
d,
Zhengnan
Wei
e and
Yingkui
Yang
*ac
*ab,
Yonglin
Wang
a,
Zongying
Xiao
a,
Rui
Luo
a,
Likun
Gao
d,
Zhengnan
Wei
e and
Yingkui
Yang
*ac
aKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education, Hubei Key Laboratory of Catalysis and Materials Science, South-Central University for Nationalities, Wuhan 430074, China. E-mail: xcui@scuec.edu.cn; ykyang@mail.scuec.edu.cn
bEngineering Research Centre of Nano-Geomaterials of Ministry of Education, China University of Geosciences, Wuhan 430074, China
cHubei Engineering Technology Research Centre of Energy Polymer Materials, School of Chemistry and Materials Science, South-Central University for Nationalities, Wuhan 430074, China
dKey Laboratory of Bio-based Material Science and Technology of Ministry of Education, Northeast Forestry University, Harbin 150040, China
eNew Energy Development Centre, Shengli Petroleum Administration Co., Ltd, SINOPEC, China
First published on 16th December 2021
Abstract
Nitrogen-coordinated single-atom catalysts (SACs) have emerged as one of the most promising alternatives to noble metal-containing benchmarks for highly efficient oxygen reduction reaction (ORR). However, the commonly required high-temperature pyrolysis usually results in undesirable structural changes and randomly produced active sites which gives rise to great challenges to the structure–property relationships, inevitably hindering the understanding of the reaction mechanisms. Herein, we demonstrate a simple yet robust pyrolysis-free route to craft single-atom cobalt catalysts with high electrocatalytic ORR activity via judiciously in situ wrapping an electrocatalytically active porphyrin-based thiophene-sulfur site-containing covalent organic polymer (PTS-COP) shell around a highly conductive multiwalled CNT (MWCNT) core, followed by accurately anchoring single-atom Co–N4 sites onto the macrocyclic porphyrin structure. The resulting Co-PTS-COPs@MWCNTs was exploited as an ORR electrocatalyst and displayed outstanding performance as a direct consequence of the advantageous architecture (i.e., 1D core@shell heterostructures with few-layer-thick PTS-COP shells) and unique active site configurations (i.e., atomically anchored Co–N4 sites and homogeneously dispersed thiophene-sulfur sites). Remarkably, an alkaline electrolyte capitalizing on Co-PTS-COPs@MWCNTs achieved excellent ORR activity (Eonset, 0.930 V; E1/2, 0.835 V), and favourable long-term durability, comparable to that of state-of-the-art carbon-based electrocatalysts.
Introduction
Oxygen electrocatalysis plays a critical role in the practical operation of clean and sustainable electrochemical energy conversion technologies.1–5 In particular, as the most important electrode process in fuel cells and metal–air batteries, the oxygen reduction reaction (ORR) has been the research hotspot in the past few decades.6–10 To date, platinum (Pt)-based materials have been widely recognised as state-of-the-art ORR electrocatalysts.11–14 However, the inherent drawbacks of prohibitive cost, small reserves and poor stability have greatly hampered their widespread applications. Consequently, the development of cost-efficient non-noble metal-based ORR electrocatalysts with improved activity and enhanced long-term durability is vital for the large-scale implementation of these sustainable electrochemical energy technologies.The last few years have witnessed continuous endeavours and enormous achievements in the development of low-cost and high-performance carbon-based single-atom catalysts (SACs) towards the ORR.6,7,15 As demonstrated by both experimental and theoretical studies, the nitrogen-coordinated SACs offer versatile active sites, while the carbon matrix mainly provides channels for electron transfer and mass transport, ensuring a high-efficiency gas/liquid/solid tri-phase electrocatalytic system.16–18 Currently, almost all of the reported carbon-based SACs commonly require a rigorous pyrolysis procedure (usually 700–1100 °C) of the precursor mixtures with highly precise elemental ratio to realize the construction of single-atom active sites and improvement of electrical conductivity as well as corrosion resistance. Nevertheless, the requisite high-temperature pyrolysis treatment always inevitably results in undesirable structural changes and/or even reconstruction of the original fine structure, and usually slight changes in pyrolysis temperature will cause remarkable differences in electrocatalytic ORR activity.19,20 Notably, the high-temperature pyrolysis treatment also results in the generation of unpredictable and poorly defined electrocatalytically active sites as the thermodynamically unstable metal atoms tend to agglomerate, especially at relatively high metal concentrations, which gives rise to great challenges to the structure–property relationships, severely hindering the understanding of reaction mechanisms.21,22 To this end, the development of the pyrolysis-free strategy towards the synthesis of carbon-based SACs can definitely eliminate the aforementioned disadvantages. Note that the pyrolysis-free approach can also significantly reduce the energy consumption, improve the repeatability of material preparation and electrocatalytic performance, and realize the controllable construction of well-defined active sites, thereby providing model catalysts in order to gain deep insight into the structure–property relationships and electrocatalytic mechanisms.
Recently, well-defined pyrolysis-free covalent organic polymer (COP)-based materials have been proposed as oxygen electrocatalysts due to their precisely controllable capabilities with regard to active site creation and channel/porosity tailoring as well as their high thermodynamic stability.23–25 On the one hand, the pyrolysis-free COPs prepared by polymerization of small monomers maintain the correct active site structure with desired activity. On the other hand, pyrolysis-free COPs with clear structure–property relationships offer an inherent advantage for mechanism studies and performance optimization. However, the electrical conductivity of pyrolysis-free COPs is generally inferior to that of other typical inorganic electrocatalytic materials due to the absence of sufficient free electrons and low charge carrier mobility, leading to low electrocatalytic performance.23,25,26 In addition, the densely stacked COP layers of the generally obtained bulk COP powder could also lead to poor mass transport and low utilization of active sites.23 In this regard, the synthesis of few-layer-thick pyrolysis-free COPs and construction of interfacial electron transfer channels via compositing with highly conductive supporting materials are of particular interest towards the electrocatalytic ORR.
Herein, to overcome the aforementioned challenges while maintaining the atomically precisely controllable capabilities, we report a simple yet robust pyrolysis-free approach to craft a hybrid material (i.e., Co-PTS-COPs@MWCNTs) via judiciously in situ wrapping an electrocatalytically active porphyrin-based thiophene-sulfur site-containing covalent organic polymer (PTS-COP) shell around a highly conductive multiwalled CNT (MWCNT) core, followed by accurately anchoring single-atom Co–N4 active sites onto the macrocyclic porphyrin structure. As a highly conductive 1D material, MWCNTs not only possess a large specific surface area and highly conjugated structure, but also exhibit high charge mobility and strong thermal/chemical stability, which could significantly improve electron transfer from the current collector to the intrinsically active COPs.23,25 To achieve abundant active sites, the macrocyclic porphyrin analogues with four pyrrole groups which could offer a suitable region for anchoring the single-atom cobalt to produce abundant well-defined Co–N4 active sites were selected to prepare COP-based electrocatalysts. As expected, the as-synthesized Co-PTS-COPs@MWCNTs demonstrated excellent electrocatalytic ORR activity (onset potential (Eonset) of 0.930 V; half-wave potential (E1/2) of 0.835 V) and outstanding long-term durability, comparable to that of state-of-the-art carbon-based electrocatalysts under alkaline conditions owing to the favourable configuration of active sites (i.e., atomically anchored Co–N4 sites and homogeneously dispersed thiophene-sulfur sites) and advantageous 1D core@shell heterostructures. As such, the carefully crafted pyrolysis-free Co-PTS-COPs@MWCNTs emerges as a promising candidate for highly efficient ORR.
Results and discussion
As illustrated in Fig. 1, the general synthetic route to the pyrolysis-free Co-PTS-COPs@MWCNT hybrid involves in situ structural crafting to form 1D core@shell heterostructures with a few-layer-thick COP shell (i.e., PTS-COPs@MWCNTs) followed by accurately anchoring single-atom Co–N4 active sites onto the macrocyclic porphyrin structure to obtain the target Co-PTS-COPs@MWCNTs. Specifically, the PTS-COPs@MWCNTs was first synthesized through a Schiff-base polycondensation between planar tetragonal linkers (i.e., tetraamine porphyrin (TPP)) and linear thiophene-sulfur site-containing building units (i.e., thieno/thiophene-2,5-dicarboxaldehyde (bTDA)) in the presence of MWCNTs, leading to the formation of 1D core@shell heterostructures which favour high electrical conductivity. Subsequently, abundant well-defined Co–N4 active sites can be accurately anchored onto the optimal PTS-COPs@MWCNTs due to the presence of abundant quadridentate chelating ligands (i.e., four pyrrole groups bonded together by methine bridges in the macrocyclic porphyrin structure). As a result, the resulting Co-PTS-COPs@MWCNT hybrid features a 1D core@shell heterostructure with a few-layer-thick COP shell which contains abundant Co–N4 and thiophene-sulfur active sites, favouring high electrical conductivity and fast mass transport of ORR-relevant species.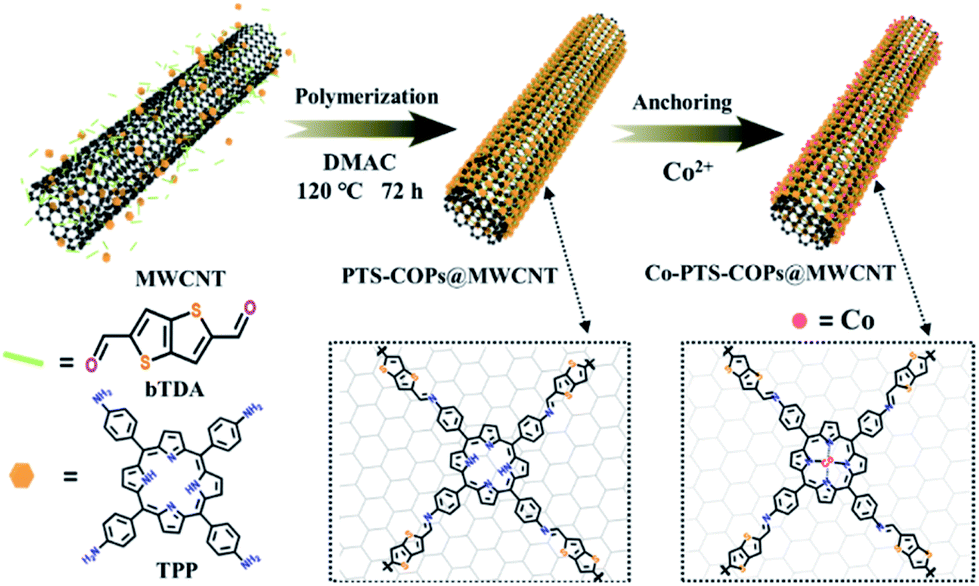 | ||
| Fig. 1 Schematic illustration of the pyrolysis-free synthesis of the Co-PTS-COPs@MWCNT hybrid. | ||
As shown in Fig. S1 and S2,† to verify the positive electrocatalytic capability of thiophene-sulfur sites towards the ORR, the same planar tetragonal linkers (TPP) and three similar linear molecules (i.e., bTDA, 2,5-thiophenedicarboxaldehyde (TDA) and p-phthalaldehyde (PPA), respectively) with different contents of thiophene-sulfur sites were employed as monomers. The Schiff-base polycondensation between TPP and bTDA, and TDA and PPA, respectively, in the absence of MWCNTs resulted in the corresponding PTS-COPs, PTS-COPs-1 and P-COPs with similar morphologies of densely stacked powders (Fig. S3†). Fourier transform infrared spectra (FTIR) exhibited the disappearance of the peaks at 1668 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band for bTDA, TDA and PPA) and around 2850 cm−1 (H–CO band for bTDA, TDA and PPA) (Fig. S4†) and the appearance of a new peak located at 1625 cm−1 (CN band for PTS-COPs, PTS-COPs-1 and P-COPs) (Fig. S5†), validating the complete transformation of the amine and aldehyde groups and the successful formation of PTS-COPs, PTS-COPs-1 and P-COPs. Note that the peaks in the range of 3200–3500 cm−1 (Fig. S5†) can be attributed to the stretching of the residual –NH2 groups located at the edge of the COPs after polymerization.
O band for bTDA, TDA and PPA) and around 2850 cm−1 (H–CO band for bTDA, TDA and PPA) (Fig. S4†) and the appearance of a new peak located at 1625 cm−1 (CN band for PTS-COPs, PTS-COPs-1 and P-COPs) (Fig. S5†), validating the complete transformation of the amine and aldehyde groups and the successful formation of PTS-COPs, PTS-COPs-1 and P-COPs. Note that the peaks in the range of 3200–3500 cm−1 (Fig. S5†) can be attributed to the stretching of the residual –NH2 groups located at the edge of the COPs after polymerization.
To investigate the electrocatalytic ORR activity of the three as-synthesized COPs, cyclic voltammetry (CV) and linear sweep voltammetry (LSV) curves were obtained in an alkaline medium based on a rotating disk electrode (RDE). The CV curves exhibit a much more positive ORR peak for PTS-COPs (0.64 V) than for PTS-COPs-1 (0.63 V) and P-COPs (0.61 V) in O2-saturated electrolyte, demonstrating the better electrocatalytic ORR activity of PTS-COPs (Fig. S6†). Fig. S7† depicts the LSV curves at a rotation rate of 1600 rpm, where PTS-COPs and PTS-COPs-1 display much more efficient electrocatalytic ORR activity than thiophene-sulfur site-free P-COPs in terms of the more positive E1/2 and Eonset and larger limited current density (Jd), further supporting the view that the thiophene-sulfur sites provided the positive ORR electrocatalytic capability, which is in good agreement with previously published work.27,28 Based on the theoretical calculation results reported previously, the C–S region at the thiophene moiety can be specifically recognised as the active sites.28 In fact, the initial O2 molecular adsorption preferentially occurs at the carbon atoms right next to the thiophene-sulfur atom in the thiophene structure. After accepting a proton–electron pair, the OOH* intermediate diffuses to the neighbouring thiophene-sulfur atom and proceeds following the dissociative ORR pathway. Besides, the thiophene-sulfur structures are also believed to be capable of reducing the band gap of the COPs, thereby lowering the overpotential for the ORR.27 Note that although the as-prepared PTS-COPs with higher content of thiophene-sulfur sites exhibit the best ORR performance, it is still not satisfactory and cannot compete with the reported carbon-based electrocatalysts produced by high-temperature pyrolysis. The most probable reasons are the low electrical conductivity of the as-synthesised COPs and the poor mass transport at the gas/liquid/solid tri-phase interface during the electrocatalytic ORR process caused by the densely stacked COP layers.
To this end, bTDA with higher content of thiophene-sulfur sites was chosen as the linear monomer to build the 1D core@shell structured PTS-COPs@MWCNT hybrid to further improve the electrical conductivity, enhance the density of active sites and prevent excessive stacking of polymer layers. The morphology and microstructure of the as-obtained PTS-COPs@MWCNT hybrid were determined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Compared with the pristine MWCNTs with smooth and clean surfaces (Fig. S8†), the PTS-COPs@MWCNTs shows that seamless PTS-COP shells are uniformly wrapped onto the MWCNTs (Fig. S9†). Notably, the thickness of in situ coated PTS-COP shells can be readily tuned by adjusting the mass ratio between COP monomers and MWCNTs (mmonomers/mMWCNTs = 0.5, 1, or 2) to obtain a set of PTS-COPs@MWCNTs (denoted as PTS-COPs@MWCNTs-x, where x refers to the mass ratio of 0.5, 1, or 2) (Fig. S10†). The FTIR (Fig. S11†) results demonstrated the successful polycondensation between TPP and bTDA in the presence of MWCNTs.
As expected, all of the as-synthesized PTS-COPs@MWCNTs-x (x = 0.5, 1, or 2) hybrids exhibited significantly improved electrocatalytic ORR activity. As shown in Fig. S12,† the CV curves of PTS-COPs@MWCNTs-x demonstrate much more positive ORR peaks than that of the PTS-COPs (Fig. S6†). Besides, the LSV curves (Fig. S7 and S13†) also support the more efficient electrocatalytic ORR activity of PTS-COPs@MWCNTs-x in terms of the positively shifted E1/2 and Eonset and largely enhanced Jd. Interestingly, among the three samples, PTS-COPs@MWCNTs-1 with a shell thickness of 2 ± 0.5 nm exhibited the best electrocatalytic ORR activity, mainly due to the uniform 1D core@shell heterostructures with an appropriate shell thickness of PTS-COPs that can remarkably improve the electrical conductivity, enhance the specific surface area (Fig. S14†), expose plenty of active sites, and facilitate mass transport to promote the ORR process. It is worth noting that the MWCNT substrate may inject abundant delocalized electrons into the thiophene moieties of the COP shell, leading to an n-doping effect. The injected delocalized electrons that serve as excessive carriers could further modify the electronic properties of the COP shell and lower the COP shell's bandgap and work function, thereby reducing the charge transfer barrier between the COP shell and adsorbed oxygen intermediates and improving the electrochemical activity.29,30
Based on the above results, the optimal PTS-COPs@MWCNTs-1 was further employed to anchor abundant well-defined Co–N4 active sites by taking full advantage of the strong chelation between Co2+ cations and quadridentate chelating ligands (i.e., four pyrrole groups bonded together by methine bridges in the macrocyclic porphyrin structure). The morphology of the resulting pyrolysis-free Co-PTS-COPs@MWCNTs was studied by SEM (Fig. S15†), TEM and high-resolution TEM (HRTEM) (Fig. 2a and b). The Co-PTS-COPs@MWCNTs shows that seamless polymer shells (Fig. 2a and b) with a thickness of 2 ± 0.5 nm are uniformly coated on MWCNTs, similar to what is observed for the PTS-COPs@MWCNTs-1 (Fig. S10b†). The energy dispersive X-ray (EDX) elemental mapping analysis (Fig. 2c) of Co-PTS-COPs@MWCNTs confirms the high-level and uniform doping of N and S elements in the polymer shells. Notably, elemental Co was also found uniformly anchored to the polymer shells and partly overlapped with the N element signal, implying the presence of highly dispersed Co–Nx sites. To further validate the existence of atomically dispersed Co–Nx sites, the atomically scaled high-angle annular dark-field scanning TEM (HAADF-STEM) measurement was conducted to gain detailed morphological insight into Co-PTS-COPs@MWCNTs. As displayed in Fig. 2d, numerous bright spots corresponding to the atomically dispersed Co–Nx sites are observed, which is in good agreement with the XRD results (Fig. S16†). The Co content in the Co-PTS-COPs@MWCNTs sample was determined to be 0.57 at% by X-ray photoelectron spectroscopy (XPS) and 1.33 wt% by inductively coupled plasma mass spectrometry (ICP-MS). High-resolution XPS was used to further scrutinize the surface chemical and Co–N bonding information of Co-PTS-COPs@MWCNTs, as shown in Fig. 2e–g. The high-resolution Co 2p spectrum shows signals centered at 796.5 and 781.3 eV, as well as related satellite peaks (787.5 and 802.4 eV), which can be well attributed to the 2p3/2 and 2p1/2 of Co–N bonds, respectively. Note that the absence of significant signals corresponding to Co0 (typically at about 778.5 eV) excludes the existence of Co–Co bonds. Meanwhile, in addition to the stronger Co2+ peaks (781.7 and 796.8 eV, respectively), the relatively weaker signals corresponding to Co3+ (779.4 and 795.2 eV, respectively) can also be deconvoluted, implying that the average valence state of the positively charged cobalt species is between Co2+ and Co3+ but closer to Co2+.31 This result suggests the presence of charge transfer from the cobalt centre to the macrocyclic porphyrin structure owing to the strong electronegativity of N, which is in good agreement with previous observations.31 The N 1s spectrum reveals the coexistence of pyrrolic N and Co–Nx.32 The binding energy of Co–Nx (399.1 eV) is close to that of pyrrolic N (400.6 eV), indicating that pyrrolic N in the macrocyclic porphyrin structure is the main site to anchor the Co atom, which is consistent with our assumption. Notably, the binding energy of the deconvoluted Co–Nx was a little bit lower than that of pyrrolic N, further supporting the partial charge transfer from the cobalt centre to the macrocyclic porphyrin structure. Besides, the S 2p spectrum was deconvoluted into two peaks centred at 165.8 eV and 164.6 eV, corresponding to C–S–C bonds.33
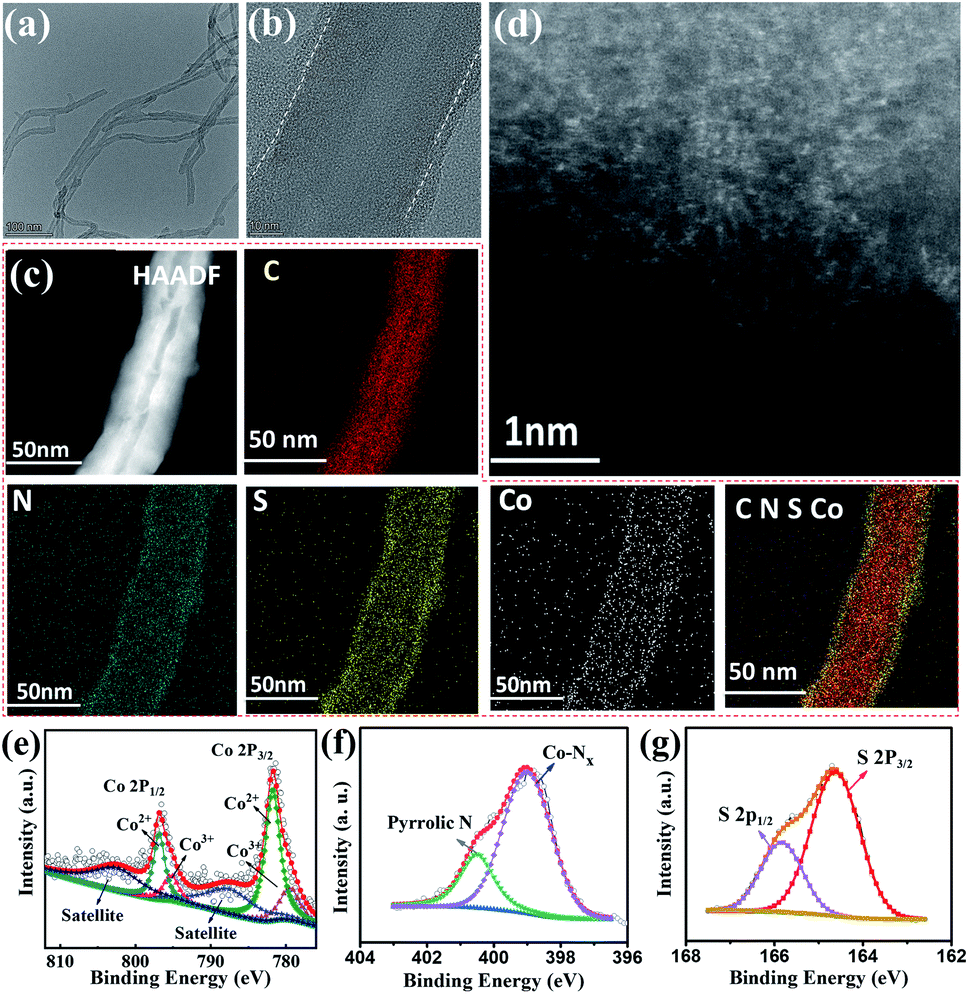 | ||
| Fig. 2 (a) TEM, (b) HRTEM, (c) HAADF-STEM and EDX elemental mapping, and (d) HAADF-STEM images of Co-PTS-COPs@MWCNTs. High-resolution XPS spectra of (e) Co 2p, (f) N 1s and (g) S 2p of Co-PTS-COPs@MWCNTs. | ||
Furthermore, Co K-edge X-ray absorption fine structure (XAFS) of Co-PTS-COPs@MWCNTs was used to analyse the coordination environment of cobalt species. As shown in Fig. 3a, compared with the references of Co foil, CoO, Co3O4 and cobalt phthalocyanine (CoPc), the absorption edge position of Co-PTS-COPs@MWCNTs is close to that of CoO, suggesting that the valence state of Co is close to +2, consistent with the XPS results. The Fourier transform plot of the Co K-edge extended X-ray absorption fine structure (FT-EXAFS) spectra for Co-PTS-COPs@MWCNTs (Fig. 3b) only presents one prominent peak at 1.45 Å, which could be ascribed to the Co–Nx coordination of Co at the first shell. Note that the Co–O coordination is located at slightly higher R values in the CoO (1.70 Å) and Co3O4 (1.55 Å) samples. Besides, it can also be found that the FT-EXAFS curve of Co-PTS-COPs@MWCNTs indicates similar characteristic peaks to those of CoPc but different peak-distributions to those of Co foil, CoO, and Co3O4. This result suggests that the coordination environment of Co-PTS-COPs@MWCNTs is closer to that of CoPc, where the Co atoms are coordinated with nitrogen to form Co–Nx bonds. Meanwhile, no obvious Co–Co peak, typically at 2.17 Å, is found in Co-PTS-COPs@MWCNTs, which eliminates the possibility of aggregated Co nanoparticles, consistent with the HAADF-STEM observation. Moreover, the EXAFS fitting was conducted to extract the quantitative structural parameters of Co sites in Co-PTS-COPs@MWCNTs. The EXAFS R-space fitting curve of Co-PTS-COPs@MWCNTs (Fig. 3c) and the fitted parameters summarized in Table S1† clearly suggested that the atomically dispersed cobalt sites existed in the Co–N4 configuration with a bond distance of 1.95 Å. The wavelet transform (WT) of Co-PTS-COPs@MWCNTs in Fig. 3d exhibits a maximum peak at around 4 Å−1, which is similar to that of CoPc and can be assigned to the Co–N coordination. No intensity maximum corresponding to Co–Co coordination can be observed, in sharp contrast with those of Co foil, CoO, and Co3O4, demonstrating the complete absence of Co-derived crystalline structures in Co-PTS-COPs@MWCNTs.
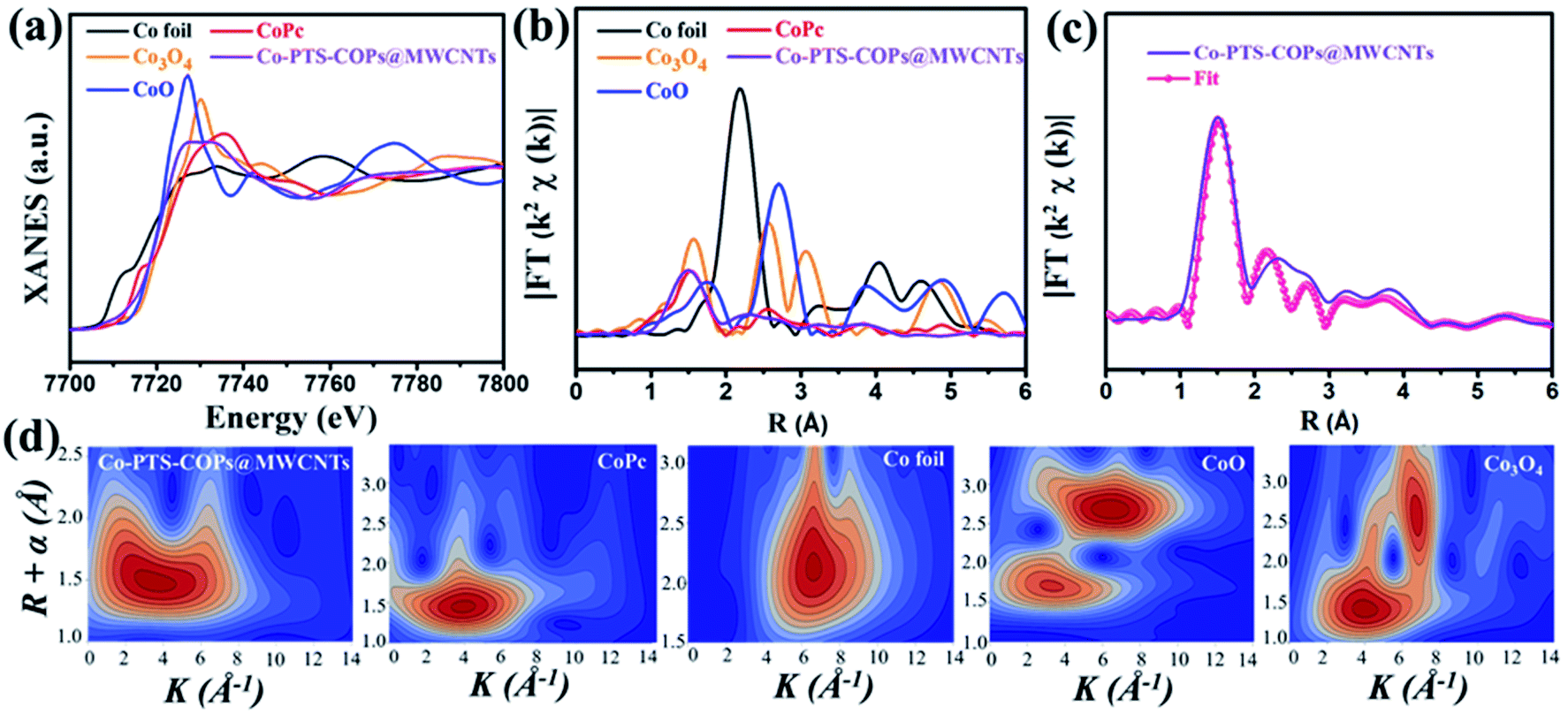 | ||
| Fig. 3 (a) Co K edge XANES spectra and (b) Fourier-transformed EXAFS (FT-EXAFS) spectra of the Co-PTS-COPs@MWCNTs and standard samples (Co foil, CoO, Co3O4 and CoPc), (c) FT-EXAFS fitting result of Co-PTS-COPs@MWCNTs in R space and (d) WT for the k3-weighted EXAFS signals. | ||
The electrocatalytic ORR performance of the as-synthesized pyrolysis-free Co-PTS-COPs@MWCNTs was then scrutinized under alkaline conditions. To reveal the electrochemical role of Co–N4 sites in Co-PTS-COPs@MWCNTs, PTS-COPs@MWCNTs and commercial Pt/C were selected as control samples. Fig. 4a displays the CV curves for Co-PTS-COPs@MWCNTs, PTS-COPs@MWCNTs and commercial Pt/C catalysts in O2- or N2-saturated 0.1 M KOH solution. Clearly, the Co-PTS-COPs@MWCNTs exhibits a more positive ORR peak (0.859 V) than that of PTS-COPs@MWCNTs (0.730 V), implying a superior electrocatalytic ORR activity of Co-PTS-COPs@MWCNTs. Fig. 4b depicts the LSV curves at a rotation rate of 1600 rpm, where PTS-COPs@MWCNTs shows the worst electrocatalytic ORR performance in terms of the most negative Eonset of 0.836 V and E1/2 of 0.670 V and lowest Jd of about −4.0 mA cm−2 as summarized in Fig. 4c, suggesting that the activity of Co-PTS-COPs@MWCNTs mainly derives from Co–N4 sites despite the thiophene-sulfur sites definitely having a positive electrocatalytic capability towards the ORR as discussed above. This result is further supported by anchoring the Co–N4 active sites on the three pristine COPs (i.e., PTS-COPs-1, PTS-COPs, and P-COPs) to obtain Co-PTS-COPs-1, Co-PTS-COPs, and Co–P-COPs (Fig. S17–S22†). Compared with the three pristine COPs (Fig. S7†), the as-synthesized Co-PTS-COPs-1, Co-PTS-COPs, and Co–P-COPs exhibit significantly improved electrocatalytic ORR performance (Fig. S22†), signifying that the metallic Co–N4 sites play a more critical role than the non-metallic thiophene-sulfur sites. To further evaluate the effect of Co–N4 content on the electrocatalytic ORR activity, the mass ratio of Co precursor/PTS-COPs@MWCNTs-1 was adjusted to explore the relationship between the catalytic activity and the Co content, as shown in Fig. S23–S25.† Obviously, with the increase of the mass ratio, the Co content gradually reaches saturation (i.e., Co-PTS-COPs@MWCNTs obtained at a mass ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Fig. S23a†). No significant increase of Co content can be observed with the further increase of the mass ratio due to the limited volume of the macrocyclic porphyrin structures exposed on the surface. Notably, TEM, HRTEM and XRD results (Fig. S24 and S25†) exclude the possibility of aggregation of Co sites. The LSV curves (Fig. S23b†) confirm that the increase of Co–N4 content is indeed beneficial to the improvement of electrocatalytic ORR activity. Consequently, with the incorporation of Co–N4 sites, the Co-PTS-COPs@MWCNTs delivers significantly improved ORR performance (Eonset = 0.933 V; E1/2 = 0.835 V; Jd = −5.5 mA cm−2). The corresponding Tafel curves (Fig. S26†) further signify that the Co-PTS-COPs@MWCNTs with the smallest Tafel slope (71 mV dec−1) is an efficient electrocatalyst. In fact, the more critical role of metallic Co–N4 sites than the non-metallic thiophene-sulfur sites can be mainly attributed to the unique d orbitals of the metal Co atoms that can interact with the p electrons of the oxygen feedstock and intermediates during the ORR, resulting in the adsorption of oxygen molecules and consequent electron transfer.34 In addition, the favourable charge density penetration from the isolated Co atoms to the adjacent N atoms as mentioned above could markedly improve the adsorption ability of the oxygen feedstock and reduce the limiting barrier for the ORR.35 Although it is still slightly lower than that of the commercial Pt/C catalyst (Eonset = 0.958 V; E1/2 = 0.850 V; Jd = −5.2 mA cm−2), to the best of our knowledge, the electrocatalytic ORR activity of pyrolysis-free Co-PTS-COPs@MWCNTs is among the best reported performances of various carbon-based electrocatalysts prepared by pyrolysis (Table S2†).
:1) (Fig. S23a†). No significant increase of Co content can be observed with the further increase of the mass ratio due to the limited volume of the macrocyclic porphyrin structures exposed on the surface. Notably, TEM, HRTEM and XRD results (Fig. S24 and S25†) exclude the possibility of aggregation of Co sites. The LSV curves (Fig. S23b†) confirm that the increase of Co–N4 content is indeed beneficial to the improvement of electrocatalytic ORR activity. Consequently, with the incorporation of Co–N4 sites, the Co-PTS-COPs@MWCNTs delivers significantly improved ORR performance (Eonset = 0.933 V; E1/2 = 0.835 V; Jd = −5.5 mA cm−2). The corresponding Tafel curves (Fig. S26†) further signify that the Co-PTS-COPs@MWCNTs with the smallest Tafel slope (71 mV dec−1) is an efficient electrocatalyst. In fact, the more critical role of metallic Co–N4 sites than the non-metallic thiophene-sulfur sites can be mainly attributed to the unique d orbitals of the metal Co atoms that can interact with the p electrons of the oxygen feedstock and intermediates during the ORR, resulting in the adsorption of oxygen molecules and consequent electron transfer.34 In addition, the favourable charge density penetration from the isolated Co atoms to the adjacent N atoms as mentioned above could markedly improve the adsorption ability of the oxygen feedstock and reduce the limiting barrier for the ORR.35 Although it is still slightly lower than that of the commercial Pt/C catalyst (Eonset = 0.958 V; E1/2 = 0.850 V; Jd = −5.2 mA cm−2), to the best of our knowledge, the electrocatalytic ORR activity of pyrolysis-free Co-PTS-COPs@MWCNTs is among the best reported performances of various carbon-based electrocatalysts prepared by pyrolysis (Table S2†).
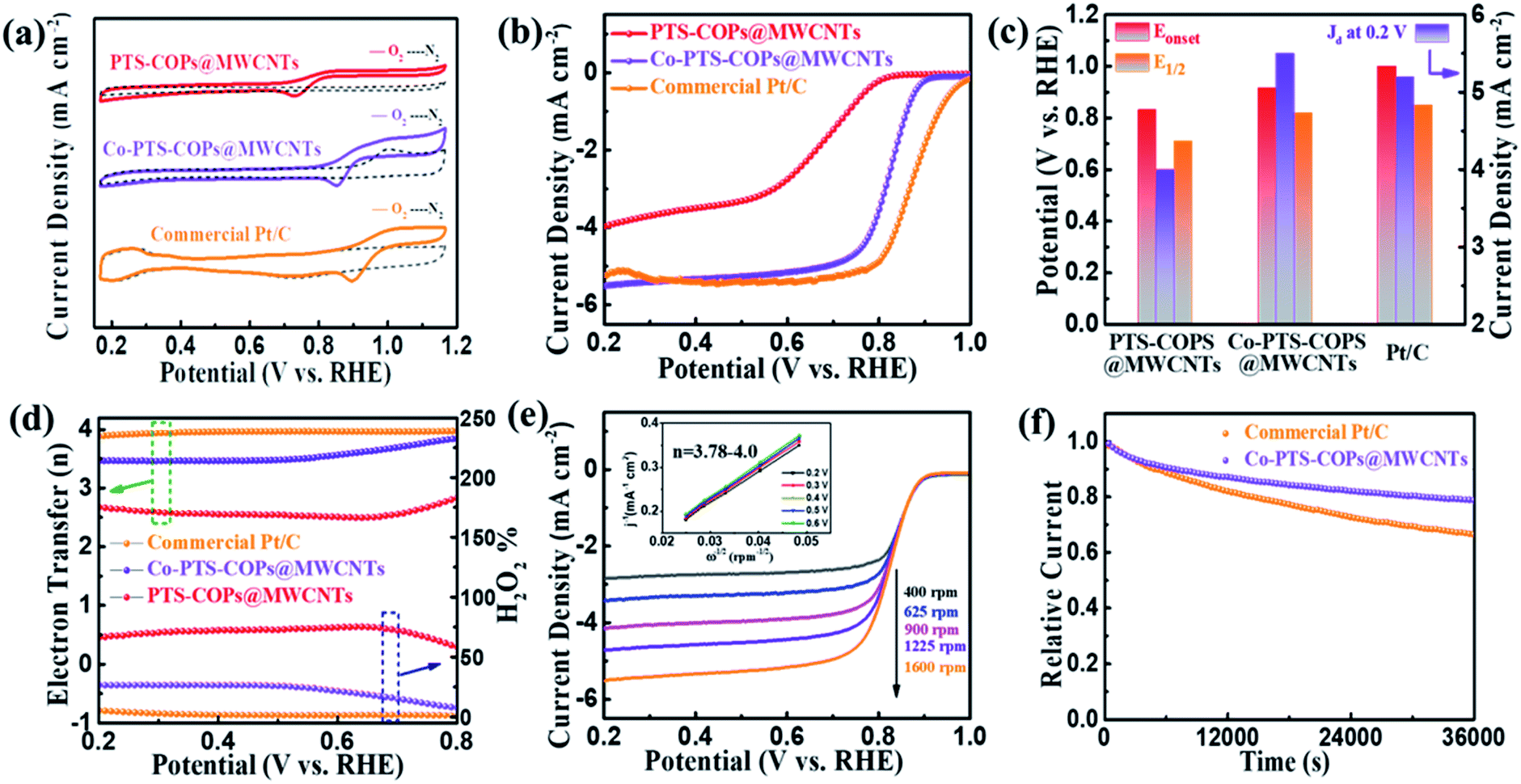 | ||
| Fig. 4 (a) CV and (b) LSV curves, (c) E1/2, Eonset and Jd, and (d) H2O2 yields and electron transfer number of Co-PTS-COPs@MWCNTs, PTS-COPs@MWCNTs and Pt/C. (e) LSV curves of Co-PTS-COPs@MWCNTs at different rotation speeds. Inset: the corresponding K–L plots and electron transfer number. (f) Chronoamperometric response of Co-PTS-COPs@MWCNTs and Pt/C. | ||
To further quantify the ORR pathway, both Koutecky–Levich (K–L) plots at various rotation rates and rotating ring disk electrode (RRDE) measurements were employed to track the formation of H2O2 during the ORR process. As revealed in Fig. 4e, the K–L plot of Co-PTS-COPs@MWCNTs presents parallel fitting lines, implying the first-order reaction kinetics.36 The kinetic electron transfer number (n) of Co-PTS-COPs@MWCNTs was calculated to be 3.78–4.00 (Fig. S27†), similar to that of the Pt/C catalyst (3.92–3.96) (Fig. S28†), suggesting a highly efficient four-electron (4e−) ORR pathway and direct electrocatalytic reduction of O2 to OH− species without the production of OOH− species in alkaline electrolyte. Furthermore, the n values of 3.51–3.85 with a low peroxide yield of 6.8–21.4% (Fig. 4d) for the Co-PTS-COPs@MWCNTs obtained by RRDE measurements also evidence a desired 4e− ORR pathway towards the formation of H2O.
Besides the electrocatalytic activity, the long-term durability of Co-PTS-COPs@MWCNTs was further evaluated via chronoamperometric measurements. After a continuous 36000 s (10 h) chronoamperometric test, the commercial Pt/C catalyst suffered from 35% loss of its initial ORR current, while Co-PTS-COPs@MWCNTs only lost 18.5% of its initial ORR current (Fig. 4f), indicating a superior long-term durability of Co-PTS-COPs@MWCNTs. The high-resolution Co and N XPS spectra of Co-PTS-COPs@MWCNTs after the long-term durability test (Fig. S29a and b†) contained peaks identical to those of the as-prepared catalyst (Fig. 2e and f). The XRD pattern (Fig. S29c†), HAADF-STEM image and EDX elemental mapping analysis (Fig. S30†) suggest that no aggregation of Co species can be observed. In addition, the slightly reduced Co content of Co-PTS-COPs@MWCNTs (0.45 at% determined by XPS) compared with that of the as-prepared catalyst (0.57 at%) excluded the apparent dissolution of Co species. The above results confirm that the highly active Co–N4 sites were well retained after the stability test. The outstanding long-term durability of Co-PTS-COPs@MWCNTs can be attributed to the unique 1D core@shell architecture, which prevents the aggregation and dissolution of Co species, boosts the mass transport, and maintains a continuous 4e− transfer pathway during long-term testing.
Finally, considering the similar chelation between the macrocyclic porphyrin structures and Fe2+ and Ni2+ cations, the cobalt precursor was replaced by Fe and Ni precursors in the synthetic procedure to obtain Fe-PTS-COPs@MWCNTs and Ni-PTS-COPs@MWCNTs, respectively. Both the as-synthesized Fe-PTS-COPs@MWCNTs and Ni-PTS-COPs@MWCNTs show similar morphology to that of Co-PTS-COPs@MWCNTs (Fig. S15 and S31†). HAADF-STEM, EDX elemental mapping, XPS, and XRD characterizations were carried out to confirm the atomically dispersed metal sites in both cases (Fig. S32–34†). Compared with Co-PTS-COPs@MWCNTs, both Fe-PTS-COPs@MWCNTs and Ni-PTS-COPs@MWCNTs exhibit slightly reduced ORR performance (Fig. S35†) in terms of the negatively shifted E1/2 (0.83 V and 0.93 V, respectively), which is consistent with previously reported experimental and theoretical results.37,38
Conclusions
In summary, instead of randomly creating single-atom sites on carbon through a rigorous pyrolysis procedure, we developed a simple yet robust pyrolysis-free approach for crafting single-atom cobalt catalysts with high electrocatalytic ORR activity via judiciously in situ wrapping an electrocatalytically active porphyrin-based thiophene-sulfur site-containing PTS-COP shell around a highly conductive MWCNT core, followed by accurately anchoring single-atom Co–N4 active sites onto the macrocyclic porphyrin structure. This route eliminates the high-temperature pyrolysis treatment that usually results in undesirable structural changes of the COPs, therefore enabling atomically accurate control of the electrocatalyst structure. Impressively, the optimal Co-PTS-COPs@MWCNTs confers excellent electrocatalytic ORR activity (Eonset of 0.930 V and E1/2 of 0.835 V) and outstanding long-term durability, comparable to that of state-of-the-art carbon-based electrocatalysts in alkaline media. Such excellent performance is collectively ascribed to the favorable configuration of active sites (i.e., atomically anchored Co–N4 sites and homogeneously dispersed thiophene-sulfur sites) in conjunction with the advantageous well-designed 1D core@shell heterostructures. As such, the carefully crafted pyrolysis-free Co-PTS-COPs@MWCNTs emerges as a promising candidate for highly efficient ORR. This work offers a promising pyrolysis-free approach for rational design and controllable synthesis of high-performance ORR electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52003300, 51973235 and 52173091), Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan), Program for Leading Talents of National Ethnic Affairs Commission of China (MZR21001), Hubei Provincial Natural Science Foundation of China (2021CFA022), Wuhan Science and Technology Bureau (2020010601012198), and the Fundamental Research Funds for the Central Universities (CZP19001).Notes and references
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- C. D. Sewell, Z. Wang, Y.-W. Harn, S. Liang, L. Gao, X. Cui and Z. Lin, J. Mater. Chem. A, 2021, 9, 20375–20384 RSC.
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- L. Gao, X. Cui, Z. Wang, C. D. Sewell, Z. Li, S. Liang, M. Zhang, J. Li, Y. Hu and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023421118 CrossRef CAS PubMed.
- X. Cui, W. Guo, M. Zhou, Y. Yang, Y. Li, P. Xiao, Y. Zhang and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 493–503 CrossRef CAS PubMed.
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C. D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS.
- W. Xue, Q. Zhou, X. Cui, S. Jia, J. Zhang and Z. Lin, Nano Energy, 2021, 86, 106073 CrossRef CAS.
- X. Cui, Y. Yang, Y. Li, F. Liu, H. Peng, Y. Zhang and P. Xiao, J. Electrochem. Soc., 2015, 162, F1415–F1424 CrossRef CAS.
- Y.-W. Harn, S. Liang, S. Liu, Y. Yan, Z. Wang, J. Jiang, J. Zhang, Q. Li, Y. He, Z. Li, L. Zhu, H.-P. Cheng and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2014086118 CrossRef CAS PubMed.
- Y. Yan, S. Liang, X. Wang, M. Zhang, S.-M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110036118 CrossRef CAS PubMed.
- H. Wang, R. Liu, Y. Li, X. Lü, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, R. Zhang, M. Lei and Z. Lin, Joule, 2018, 2, 337–348 CrossRef CAS.
- T. Wang, C. Yang, Y. Liu, S. M. Yang, X. Li, M. Yang, Y. He, H. Li, H. Chen and Z. Lin, Nano Lett., 2020, 20, 5639–5645 CrossRef CAS PubMed.
- T. Wang, Y. He, Y. Liu, F. Guo, X. Li, H. Chen, H. Li and Z. Lin, Nano Energy, 2020, 79, 105487 CrossRef.
- C. Yang, R. Gao and H. Yang, EnergyChem, 2021, 3, 100062 CrossRef CAS.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 2–18 CrossRef.
- H. L. Fei, J. C. Dong, Y. X. Feng, C. S. Allen, C. Z. Wan, B. Volosskiy, M. F. Li, Z. P. Zhao, Y. L. Wang, H. T. Sun, P. F. An, W. X. Chen, Z. Y. Guo, C. Lee, D. L. Chen, I. Shakir, M. J. Liu, T. D. Hu, Y. D. Li, A. I. Kirkland, X. F. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- H. Zhou, T. Yang, Z. Kou, L. Shen, Y. Zhao, Z. Wang, X. Wang, Z. Yang, J. Du, J. Xu, M. Chen, L. Tian, W. Guo, Q. Wang, H. Lv, W. Chen, X. Hong, J. Luo, D. He and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 20465–20469 CrossRef CAS PubMed.
- T. Sun, S. Zhao, W. Chen, D. Zhai, J. Dong, Y. Wang, S. Zhang, A. Han, L. Gu, R. Yu, X. Wen, H. Ren, L. Xu, C. Chen, Q. Peng, D. Wang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12692–12697 CrossRef CAS PubMed.
- J. Guo, C.-Y. Lin, Z. Xia and Z. Xiang, Angew. Chem., Int. Ed., 2018, 57, 12567–12572 CrossRef CAS PubMed.
- P. Peng, L. Shi, F. Huo, C. Mi, X. Wu, S. Zhang and Z. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef CAS PubMed.
- S. Tao and D. Jiang, CCS Chem., 2020, 2, 2003–2024 Search PubMed.
- C. Mi, P. Peng and Z. Xiang, Chin. Sci. Bull., 2020, 65, 1348–1357 CrossRef.
- X. Cui, L. Gao, R. Ma, Z. Wei, C.-H. Lu, Z. Li and Y. Yang, J. Mater. Chem. A, 2021, 9, 20985–21004 RSC.
- B. Huang, L. Li, X. Tang, W. Zhai, Y. Hong, T. Hu, K. Yuan and Y. Chen, Energy Environ. Sci., 2021, 14, 2789–2808 RSC.
- X. Cui, S. Lei, A. C. Wang, L. Gao, Q. Zhang, Yi. Yang and Z. Lin, Nano Energy, 2020, 70, 104525 CrossRef CAS.
- X. Cui, Y. Chen, M. Zhang, Y. W. Harn, J. Qi, L. Gao, Z. L. Wang, J. Huang, Y. Yang and Z. Lin, Energy Environ. Sci., 2020, 13, 1743–1752 RSC.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- C. Liu, F. Liu, H. Li, J. Chen, J. Fei, Z. Yu, Z. Yuan, C. Wang, H. Zheng, Z. Liu, M. Xu, G. Henkelman, L. Wei and Y. Chen, ACS Nano, 2021, 15, 3309–3319 CrossRef CAS PubMed.
- Z. Wang, B. Yang, Y. Wang, Y. Zhao, X.-M. Cao and P. Hu, Phys. Chem. Chem. Phys., 2013, 15, 9498–9502 RSC.
- J. Y. Cheon, J. H. Kim, J. H. Kim, K. C. Goddeti, J. Y. Park and S. H. Joo, J. Am. Chem. Soc., 2014, 136, 8875–8878 CrossRef CAS PubMed.
- Q. Zhao, Y. Wang, W.-H. Lai, F. Xiao, Y. Lyu, C. Liao and M. Shao, Energy Environ. Sci., 2021, 14, 5444–5456 RSC.
- X. X. Wang, D. A. Cullen, Y. T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- Y. Kim, D. Kim, J. Lee, L. Y. S. Lee and D. K. P. Ng, Adv. Funct. Mater., 2021, 2103290 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- H. Zhang, W. Zhou, T. Chen, B. Y. Guan, Z. Lia and X. W. Lou, Energy Environ. Sci., 2018, 11, 1980–1984 RSC.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- S. Wannakao, T. Maihom, K. Kongpatpanich, J. Limtrakul and V. Promarak, Phys. Chem. Chem. Phys., 2017, 19, 29540–29548 RSC.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu, J.-Q. Huang and Q. Zhang, Chin. Chem. Lett., 2019, 30, 911–914 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08412g |
| This journal is © The Royal Society of Chemistry 2022 |