
Enhancing the polymer electrolyte–Li metal interface on high-voltage solid-state batteries with Li-based additives inspired by the surface chemistry of Li7La3Zr2O12†
Ander
Orue
 a,
Mikel
Arrese-Igor
ab,
Rosalia
Cid
a,
Xabier
Júdez
a,
Nuria
Gómez
a,
Juan Miguel
López del Amo
a,
William
Manalastas
c,
Madhavi
Srinivasan
cd,
Catleya
Rojviriya
e,
Michel
Armand
a,
Frédéric
Aguesse
a and
Pedro
López-Aranguren
*a
a,
Mikel
Arrese-Igor
ab,
Rosalia
Cid
a,
Xabier
Júdez
a,
Nuria
Gómez
a,
Juan Miguel
López del Amo
a,
William
Manalastas
c,
Madhavi
Srinivasan
cd,
Catleya
Rojviriya
e,
Michel
Armand
a,
Frédéric
Aguesse
a and
Pedro
López-Aranguren
*a
aCenter for Cooperative Research on Alternative Energies (CIC EnergiGUNE), Basque Research and Technology Alliance (BRTA), Parque Tecnológico de Álava, Albert Einstein, 48, 01510, Vitoria-Gasteiz, Spain. E-mail: plopez@cicenergigune.com
bUniversity of the Basque Country (EHU/UPV), Barrio Sarriena, s/n, 48940 Leioa, Spain
cSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Ave, 639798 Singapore, Singapore
dEnergy Research Institute a Nanyang Technological University, Research Techno Plaza, 50 Nanyang Drive, Singapore 637553, Singapore
eSynchrotron Light Research Institute (SLRI), Nakhon Ratchasima, 30000, Thailand
First published on 15th December 2021
Abstract
High-voltage Li metal solid-state batteries are in the spotlight as high energy and power density devices for the next generation of batteries. However, the lack of robust solid-electrolyte interfaces (SEIs) and the propagation of Li dendrites still need to be addressed for practical application with extended cyclability. In the present work, high-voltage Li metal cells with LiNi0.6Mn0.2Co0.2O2 active material were assembled with a polyethylene(oxide) based electrolyte mixed with lithium bis(fluorosulfonyl)imide (LiFSI) salt. The addition of Li7La3Zr2O12 garnet to form a composite electrolyte demonstrated a beneficial effect for cell cycling stability. Inspired by the improved interface of ceramic Li7La3Zr2O12 garnet and Li metal, as well as by previous knowledge of favorable SEI forming species, various additive candidates were selected to optimize its electrolyte composition. Among them, lithium hydroxide (LiOH) is a key favorable species that shows a relevant improvement in the cyclability of the cells. X-ray photoelectron spectroscopy showed that the SEI layer is composed mainly of chemical species arising from the reduction of the Li salt, with lithium fluoride (LiF) being the main product. In addition, solid-state nuclear magnetic resonance proved that LiOH induces the cleavage of the labile S–F bond, increasing the concentration of LiF. Herein, we highlight that SEI-forming additives need to be considered for the interfacial engineering design of stable SEIs to expand the performance boundary of SSBs.
Introduction
Solid-state batteries (SSBs) consisting of high-voltage active materials such as LiNixMnyCo1−x−yO2 (NMC), thin polymer electrolytes and Li metal anodes should allow high energy density over 400 W h kg−1 and 1000 W h L−1 to be reached, demanded by the automotive industry.1,2 In addition, the thermal stability of solid electrolytes improves the safety of these devices with respect to conventional lithium ion batteries (LIBs).3–6 Amongst the organic and inorganic solid electrolytes, solid polymer electrolytes (SPEs) such as polyethylene oxide (PEO) with imide-based salts have been extensively used due to their low density, excellent processability, flexibility, reduced interfacial resistance with the electrodes, and reasonable ionic conductivity of ∼10−4 S cm−1 at 40–60 °C.7–11The lithium metal anode enables high energy density SSBs, but low resistance solid electrolyte interfaces (SEIs) with good mechanical integrity need to be developed in order to suppress the formation of Li dendrites and to extend the cyclability of the devices.12 The cycling performance of Li metal PEO-based SSBs with a high-voltage NMC positive electrode displays a sudden failure during the charging step (delithiation of the NMC).13,14 Homann et al.14 ascribed it to the propagation of Li from the Li|SPE interface towards the positive electrode during the charging process. It is expected that a robust and favorable SEI will prevent such dendritic behavior. For example, Esthetu et al.15 have highlighted the importance of the choice of the imide-based Li salt, by showing that lithium bis(fluorosulfonyl)imide (LiFSI) in PEO-based SPEs forms a more favorable lithium fluoride (LiF)-rich SEI than lithium bis(trifluoromethanesulfonyl) imide salt, leading to less dendritic, more dense and compact Li metal plating.
The addition of ceramic Li7La3Zr2O12 (LLZO) garnet to composite polymer electrolytes (CPEs) has recently been a subject of study with respect to the Li-ion dynamic properties of the polymer|LLZO interface owing to the improvement of the conductivity.16–18 In addition, CPEs have shown a higher interfacial stability with lithium,19,20 likely ascribed to the complex surface chemistry of the LLZO ceramic garnet, which includes lithium carbonate (Li2CO3), lithium hydroxide (LiOH) and other adventitious carbon species that may take part in the formation of the SEI.21–24 Likewise, LiF is amongst the chemical species that contribute to a robust SEI on conventional LIBs.25,26 Despite the extensive work on additives for LIBs, only a few studies have reported the improvement of the SEI on polymer SSBs upon using salt additives.27,28
In the present work, we initially demonstrate the improved performance of high-voltage NMC Li metal with a CPE containing LLZO imbedded in a SPE. Inspired by the surface chemistry of LLZO, we selected various additives that could trigger similar effects, including Li2CO3, LiOH and LiF, and assembled SPE-based cells. This enabled us to evaluate the effect of each additive on the electrochemical performance of SSBs. Amongst the Li-based additives, LiOH shows a relevant improvement in the cycling performance, with the cells showing a smooth voltage during the charging step and delivering the theoretical discharge capacity. After the addition of LiOH to the solid electrolyte, the interfacial resistance with the Li metal anode is reduced and an enhancement in electrochemical stability is observed in the plating/stripping test. These facts suggest that the SEI layer formed in the presence of LiOH is beneficial for obtaining a less resistive and more robust SEI layer. X-ray photoelectron spectroscopy (XPS) analysis showed that the SEI layer formed in the presence of LiOH is rich in inorganic products, especially in LiF, which could provide a less resistive layer. Besides, solid-state nuclear magnetic resonance (ssNMR) demonstrated that LiFSI molecules react with LiOH at 70 °C leading to the formation of LiF. These results pave the way towards the rational selection of SPE additives as well as the design of robust and favorable SEIs for high-voltage Li metal SPE batteries.
Experimental
Preparation of solid polymer electrolytes
The SPEs were prepared from a mixture of LiFSI (Provisco, 99.9%) and polyethylene oxide (PEO, Sigma Aldrich, Mw = 5 × 106). To limit the presence of water, PEO and LiFSI were dried at 50 and 80 °C, respectively, in a vacuum oven for 12 h before being transferred to an argon glovebox. To prepare the self-standing membranes, PEO and LiFSI were stirred in anhydrous acetonitrile (ACN, Sigma Aldrich, 99.8%) for 12 h. The as-milled slurry was cast onto a Mylar sheet using a Hohsen MC-20 Minicoater with a blade gap of 1500 μm and a speed of 10 mm s−1. Finally, the cast membrane was dried at 50 °C for 12 h under a dynamic vacuum.For the preparation of the CPE containing 10 vol% LLZO ceramic garnet, Li6.55Ga0.15La3Zr2O12 was firstly synthesized using a chelate-gel route as previously reported.16,29,30 X-ray diffraction (XRD) of the powders confirmed the cubic-garnet structure with no impurities (Fig. S1†). The CPE was prepared following a similar procedure to that for the SPEs. However, to ensure a homogeneously dispersed composite, the mixture of the components was ball-milled under soft conditions. A detailed procedure for the preparation of the CPE can be found elsewhere.13
Additive-containing SPEs (SPE-X, where X refers to LiOH, Li2CO3 and LiF) with a content of 5 vol% of Li-based additive were also prepared. LiOH (Sigma Aldrich, >98%), Li2CO3 (Sigma Aldrich, >99%) and LiF (Sigma Aldrich, >99%) were used for this purpose and were also thoroughly dried under vacuum prior to their addition to the PEO–LiFSI mixture. In order to prepare different SPE-X membranes, the same preparation procedure described previously for the preparation of a CPE was followed.13 For all the solid electrolytes, the Li salt concentration was fixed at an optimized molar ratio of EO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Li = 20:1,15 while the thickness of solid electrolytes was fixed at ∼50 μm.
:Li = 20:1,15 while the thickness of solid electrolytes was fixed at ∼50 μm.
Cathode laminate preparation
The cathode laminate was prepared by mixing LiNi0.6Mn0.2Co0.2O2 (NMC622), C65 carbon black (Imerys, France) and PEO–LiFSI catholyte, the last component having the same composition as the SPE. The cathode laminate was prepared with the NMC622:C65:catholyte ratio fixed at 74:7:19 wt% (45:10:45 vol%). To prepare the positive electrode, NMC622 and C65 were dried overnight under vacuum and transferred to an argon glovebox (H2O, O2 ≤ 1 ppm) where the preparation of the cathode laminate was carried out. For that, NMC622 and C65 were mixed in an agate mortar with a small amount of ACN and the resulting mixture was then left overnight in order to evaporate all the solvent. This mixture was subsequently added to the PEO–LiFSI catholyte slurry and homogenized with an IKA® ULTRA-TURRAX® disperser at 16000 rpm for 25 min. The resulting slurry was cast onto an Al foil (thickness of 20 μm) in a coater with a blade gap of 750 μm and a speed of 10 mm s−1. Finally, the cathode laminate was dried at 50 °C for 12 h under a dynamic vacuum. The areal capacity of the NMC622 electrodes was fixed to 1 mA h cm−2 (6 mg cm−2). A composite cathode laminate containing 10 vol% of LLZO on the catholyte was also prepared for X-ray tomography analysis following a procedure described previously.13
Microstructure and composition
XRD patterns of the powders of NMC622 and Ga-doped LLZ were recorded on a Bruker D8 Discover diffractometer using Cu-Kα1,2 radiation. To avoid air contamination, the XRD analysis of both powders was performed under an argon atmosphere in an airtight sample holder.The morphological characterization of the NMC622 cathode electrode was conducted via scanning electron microscopy (SEM) with an FEI Quanta 200F SEM station operated at 20 keV collecting either secondary or backscattered electrons. The NMC622 cathode electrodes were hardened using liquid nitrogen and then cut with a blade, thus obtaining a sharp and neat cross section. The composition of the cathode was determined by energy dispersive X-ray spectroscopy (EDX) using an Oxford Instruments detector.
Synchrotron Radiation X-ray Tomographic Microscopy (SRXTM) of the cathode was conducted at the high-flux 2.4 Tesla multipole wiggler beamline (BL1.2W) of the Synchrotron Light Research Institute (Thailand). For the sample preparation, cathode laminates (including the Al current collector) were cut into longitudinal strips measuring 0.5 to 1 mm in width, and meticulously aligned to stand vertically on 90° Al pin stubs. For this step, the field-of-view was adjusted such that a vertical clearance of at least 5 mm from the metallic pin stubs was achieved, to minimize fringe effects from any stray radiation. The cathode laminates were scanned using a filtered white beam at a mean X-ray energy of 6 keV. The X-ray projections were acquired at a pixel size of 0.72 μm with the detection system comprising a YAG-Ce scintillator, 10× objective lens-coupled microscope, and PCO.edge 5.5. For each cathode sample, a total of 2001 projections were collected in a 180° angular range, corresponding to a step size of 0.100°. Sinogram processing and filtered back projection algorithms were iteratively applied using the Octopus Reconstruction software package31 and Drishti project software32 to finally achieve the optimal noise speckling–smoothing–compiling–thresholding segmentation of computed tomography (CT) slices for 3D visual exploration.
Electrochemical characterization and interfacial analysis
Solid-state full cells were assembled in CR2032 coin cells with two 500 μm stainless steel spacers. For cell assembly, a 16 mm diameter disk of SPE, CPE or SPE-X was sandwiched between 12 mm and 14 mm diameter discs of the NMC622 cathode laminate and a 500 μm thickness Li metal, respectively. The cycling performance of the cells was evaluated on a VMP3 (BioLogic, France) at C/10 between 4.2 and 2.7 V vs. Li/Li+ at 70 °C.Electrochemical impedance spectroscopy (EIS) of the solid-state full cells was evaluated on the same device in the frequency range from 1 MHz to 1 Hz applying a 50 mV polarization amplitude. On the other hand, EIS was also applied to determine the ionic conductivity of the SPEs on symmetric cells using stainless steel (SS) blocking electrodes in an SS|SPE|SS configuration. These measurements were performed using a Solartron 1260A impedance analyzer in the frequency range from 32 MHz to 1 Hz applying a 50 mV polarization amplitude. The Nyquist plots were recorded every 5 °C in the temperature range from 80 °C to 0 °C and then fitted to an equivalent circuit to extract the resistance of the electrolyte. The ionic conductivity (σ) of SPEs was calculated using the equation below:
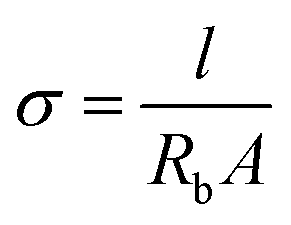
Cyclic Voltammetry (CV) measurements of the solid electrolytes were performed at 70 °C using stainless steel as the working electrode and Li metal was used as both the counter and the reference electrode in an SS|SPE|Li cell configuration. Cyclic voltammograms were obtained by performing a CV test from −0.5 V to 5 V vs. Li/Li+, at a scan rate of 0.5 mV s−1.
Li metal symmetric coin cells (Li|SPE|Li) were assembled to study the electrochemical stability of the electrolyte/Li metal interface. The galvanostatic cycling of the Li metal symmetric cells was evaluated using a Maccor battery tester at a current density of 0.1 mA cm−2, wherein the duration of each half cycle was 2.5 h.
X-ray photoelectron spectroscopy (XPS)
The analysis of the surface composition on the Li metal deposits was examined by XPS. 1,2-Dimethoxyethane (DME) was selected as an archetypal liquid electrolyte due to its analogous chemical structure to PEO to carry out the Li plating on Cu foil. First, Li metal was electrochemically plated on Cu substrates either in Li|LiFSI/DME|Cu or Li|LiFSI–LiOH/DME|Cu cells at a current density of 0.1 mA cm−2 for 20 h. After the galvanostatic deposition, the samples were gently rinsed in DME and thoroughly dried under vacuum before being transferred into the XPS spectrometer. They were transported from the glovebox to the XPS chamber in an Ar-filled airtight transfer vessel. The surface composition was examined using a Phoibos 150 XPS spectrometer (SPECS Surface Nano Analysis) installed in an ultra-high-vacuum (UHV) chamber with a base pressure of 1 × 10−10 mbar with a non-monochromatic Mg Kα source (hν = 1253.6 eV). The high-resolution spectra were recorded in fixed analyzer transmission (FAT) mode with a field of view on the sample of 2 mm, operating the source at low power (100 W) and employing a 30 eV pass energy and 0.1 eV energy step. The binding energy scale was calibrated by setting the aliphatic C–C bond at 284.8 eV. A Shirley background was subtracted to remove the inelastically scattered photoelectron signal, and Voigt profiles (30% Gaussian, 70% Lorentzian) were employed as line shapes. Tabulated Scofield cross sections,33 together with correction factors accounting for the energy dependent analyzer transmission and differences in effective attenuation length (EAL) of the collected photoelectrons,34 were applied to the integrated intensities for quantifying the atomic surface species.Solid-state nuclear magnetic resonance (ssNMR)
Magic Angle Spinning Nuclear Magnetic Resonance (MAS NMR) was applied to determine the chemical local environments of 19F and 7Li. SPE and SPE-LiOH membranes were packed into 2.5 mm rotors. MAS NMR measurements were performed using a Bruker Advance III 500 spectrometer. Spectra were recorded at spinning frequencies of 20 kHz. The spectra were recorded using simple single pulse experiments for 7Li and an echo sequence for 19F without decoupling. 32 scans were acquired for 7Li and 256 scans for the 19F spectra. Samples were referenced to a 0.1 M LiOH solution and externally to solid LiF resonating at −204 ppm for 19F.Results and discussion
Morphological characterization of NMC622 positive electrodes
The diffractogram of NMC622 powder from Fig. S2a† confirmed a hexagonal α-NaFeO2 type structure with an R1m space group. Homogeneous composite electrodes with the catholyte fully wetting the active material particles and an optimized ionic and electronic conductive network are required for high-performance SSBs.35,36 The microstructure of the cathode laminate electrodes was observed by SEM and SRXTM. The micrographs of the surface and cross section of the positive electrode laminate are shown in Fig. 1a and b, respectively. Top-view micrographs confirmed the smooth surface of the electrodes with sparse pores and micron-sized NMC622 particles (Fig. S2b†). The absence of pores evidences the high density of the composites with active material loading of 1 mA h cm−2. The cross-section of the electrode in Fig. 1b, cast on 20 mm thick Al foil, indicates an electrode thickness of ∼25 μm. As shown in Fig. S3,† EDX analysis of the NMC622 electrode confirmed that the amount of NMC622 active material is around 74%. SRXTM is a powerful tool to visualize the geometry of the electrode pore space.37,38 The reconstruction at the electrode level enables the inspection of macroscopic design parameters and defects. The 3D computed images of the composite electrodes rely on the attenuation contrast, where greyscale values represent the attenuation coefficients on the different materials in the sample.37 The reconstruction of NMC622 particles evidenced that the active material particles are well dispersed throughout the laminate and homogeneously coated by the composite PEO–LLZO catholyte (Fig. 1c and Video S1†). The estimated porosity from SRXTM with a resolution of 0.72 μm × 0.72 μm × 0.72 μm is about 2.4%. | ||
| Fig. 1 Morphological characterization of the NMC622 electrode. (a) Top and (b) cross-section view SEM micrographs and (c) X-ray absorption contrast computed tomography image. | ||
Electrochemical characterization of solid-state cells
The voltage profiles for the cells assembled with the pristine SPE, CPE and SPEs containing LiOH, LiF and Li2CO3 are shown in Fig. 2. The cell assembled with the SPE evidences voltage instability above 3.7 V during the charging step and the cell does not reach the upper cut-off voltage (4.2 V). In addition, the cell delivers a poor discharge capacity of 85 mA h g−1. In contrast, the cell with the CPE displays an improved performance. The cell reaches the upper cut-off voltage and shows a high discharge capacity close to the theoretical one. We ascribed the improvement in the cell with the CPE to the SEI layer formed from the inorganic species present at the surface of LLZO. LLZO particles readily react with the moisture present in the air, leading to a contamination layer mainly composed of Li2CO3 and LiOH. In order to trigger a similar effect, Li2CO3 and LiOH have been used as additives on SPEs. Besides, LiF is commonly found at the SEI of LIBs and has therefore also been selected as an additive.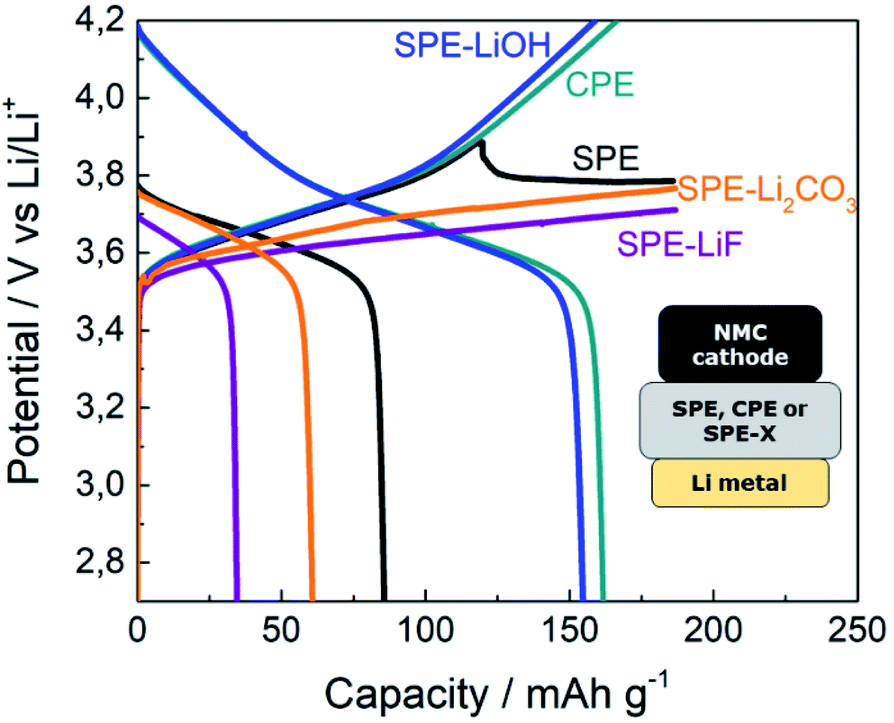 | ||
| Fig. 2 5th cycle galvanostatic cycling voltage profiles of NMC622|SPE|Li metal cells at C/20 and 70 °C with different additives in the SPE separator. | ||
The addition of neither Li2CO3 nor LiF to the SPE improves the cycling of the cells, as evidenced by the low voltage during the charging step and the poor discharge capacity below 61 mA h g−1. However, the cell with SPE-LiOH shows a good electrochemical performance, with the cell smoothly reaching 4.2 V during charging and delivering the theoretical capacity on discharge (164 mA h g−1).
The galvanostatic cycling performance of the cell assembled with SPE-LiOH at a constant current is shown in Fig. 3a. The cell initially delivers the theoretical capacity which decreases down to 145 mA h g−1 in the 20th cycle, while the coulombic efficiency (CE) of the cell remains over 90%. The observed capacity fading has previously been ascribed to the oxidation of the PEO in SPEs.13 The inset in Fig. 3a shows the EIS profiles of the cell before and after 20 cycles. Both Nyquist plots fit well with an equivalent circuit (EC) consisting of various resistances (R), constant-phase-elements (CPEs) and a Warburg element (Wd). The resistances estimated from the different elements in the EC are summarized in Table S1.† Before cycling, the cell displays a high frequency semicircle and a straight line at low frequency, ascribed to an interfacial resistance and a Warburg element, respectively. According to the fitting displayed in Table S1,† a bulk solid electrolyte resistance (Rb) of 19 Ω is obtained while the resistance of the electrode interfaces (Rint) is 25 Ω with a capacitance of 10−6 F. After 20 cycles, Rb slightly increases to 26 Ω, likely due to the lower conductivity of the electrolyte owing to the oxidation of the SPE-LiOH. The trend observed in the resistance of the cell after cycling is in good agreement with the voltage profiles observed in Fig. S4a,† where a low-capacity fade is accompanied by an increase in the overpotential of the discharge curve upon cycling.
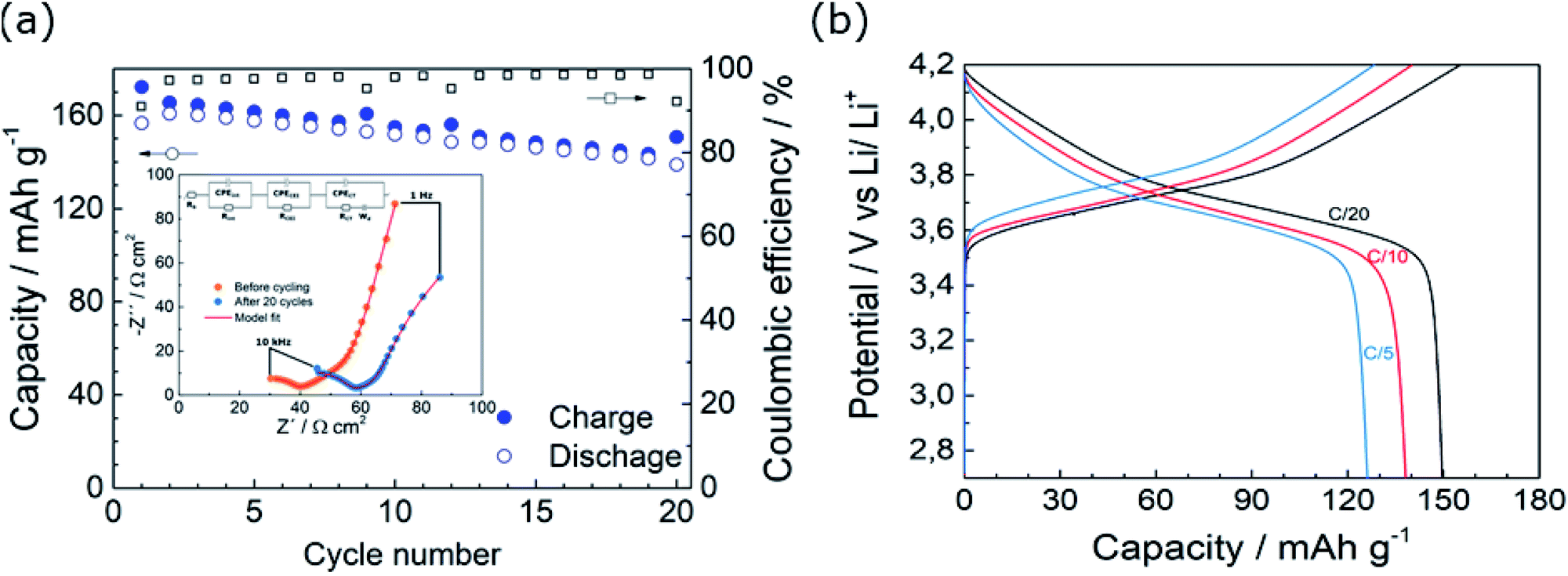 | ||
| Fig. 3 Galvanostatic cycling of NMC622|SPE-LiOH|Li metal cells at 70 °C with 1 mA h cm−2 loading. (a) Capacity evolution upon cycling at C/10 and EIS of the cell before cycling and after 20 cycles and (b) galvanostatic voltage profiles at different C-rates. | ||
Fig. 3b shows the galvanostatic voltage profile for the rate capability test from C/20 up to C/5 (Fig. S4b†). In spite of the increasing polarization of the cell at higher currents, the cell shows a capacity retention of 94 and 83% at C/10 and C/5, respectively.
Electrochemical properties and surface analysis
The LSVs at oxidative potentials of both electrolytes exhibit good electrochemical stability below 4.2 V (Fig. 4a). However, at higher voltage, the low anodic current of 10 μA cm−2 evidences the oxidative degradation of PEO in both electrolytes.39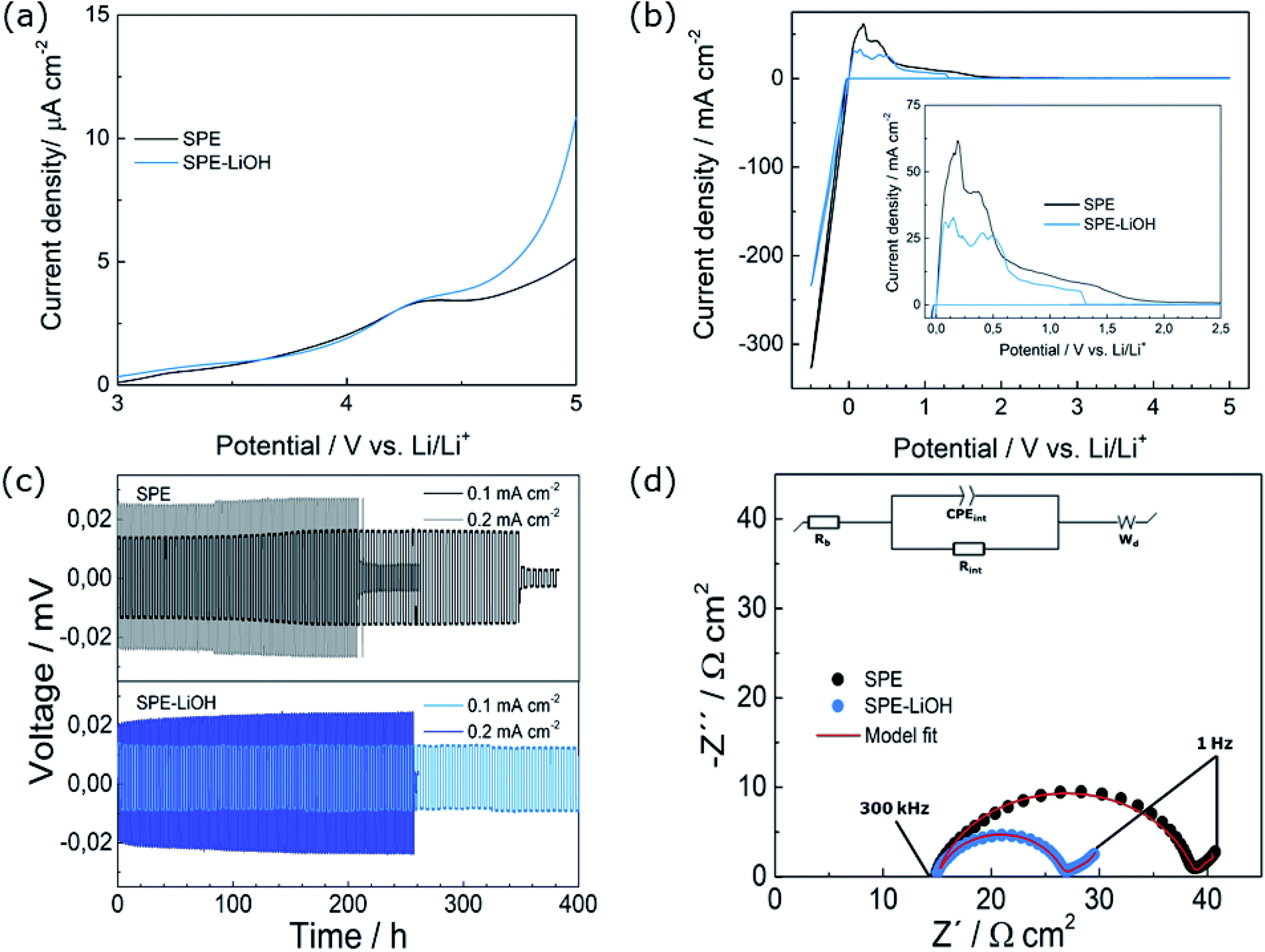 | ||
| Fig. 4 Electrochemical characterization of SPE and SPE-LiOH through (a) LSV voltammograms at oxidation potentials and at a scan rate of 0.5 mV s−1, (b) CV voltammograms at a scan rate of 0.5 mV s−1 in the range of −0.5 to +5 V, (c) lithium stripping/plating at 0.1 and 0.2 mA·cm−2, fixing the areal capacity at 0.25 mA h cm−2 and (d) EIS measurement of Li symmetric cells with two different electrolytes after conditioning for 24 h at 70 °C. | ||
The CVs recorded in the −0.5–5 V range display anodic and cathodic currents (Fig. 4b) at −0.5 and +0.5 V, respectively, corresponding to the lithium stripping/plating on the working electrode,40 and show a lower current for SPE-LiOH. The electrochemical stability against Li metal and ionic conductivity were assessed for the SPE-LiOH electrolyte in order to gain understanding on the improved properties of the full cell. As shown in the Arrhenius plot (Fig. S5†), the SPE and SPE-LiOH display similar ionic conductivities in the 0–80 °C range, indicating that the addition of LiOH does not have an impact on the ionic conductivity. These results indicate that the improvement of cycling upon the addition of LiOH likely arises from the interface formed between Li metal and the SPE.
Therefore, galvanostatic cycling of the SPEs in symmetric Li cells has been performed at two different current densities to investigate the interfacial stability (Fig. 4c). At 0.1 mA cm−2, the SPE shows an overpotential of 16 mV and the cell experiences a short-circuit after 350 h (70 cycles). With the addition of LiOH to the solid electrolyte, the overpotential of SPE-LiOH decreases to 12 mV owing to the lower interfacial resistance between the Li metal and electrolyte, and the cell cycles for 400 h without short-circuiting. At a higher current density of 0.2 mA cm−2, the SPE-LiOH also displays an improved performance over the SPE, with a lower overpotential of 23 mV, withstanding almost 50 hours more than the SPE before the cell experiences internal short-circuiting. The EIS measurement from Fig. 4d evidences the lower interfacial resistance obtained for SPE-LiOH. Table S2† summarizes the estimated bulk (Rb) and interfacial (Rint) resistance after fitting to an EC consisting of two resistances, constant-phase-elements and a Warburg element. Both cells show similar Rb while the SPE-LiOH displays an Rint 47% lower than that of the SPE, highlighting the improvement upon the addition of LiOH.
Therefore, in order to study possible variations in the chemical composition of the formed SEI, a detailed analysis of the species derived from the addition of LiOH at the Li metal interface was performed by XPS. After plating Li metal with pristine and LiOH-containing electrolytes, the outermost surface SEI layer was analyzed to identify spectral features in the F 1s, O 1s, C 1s (Fig. 5a and b) and S 2p (Fig. S6†) spectra. In order to investigate the composition of the SEI layer at different depths, all samples were etched using 1 keV focused Ar+ ions with different etching times. The distribution of different surface elements at different depths can be found in Fig. S7.†
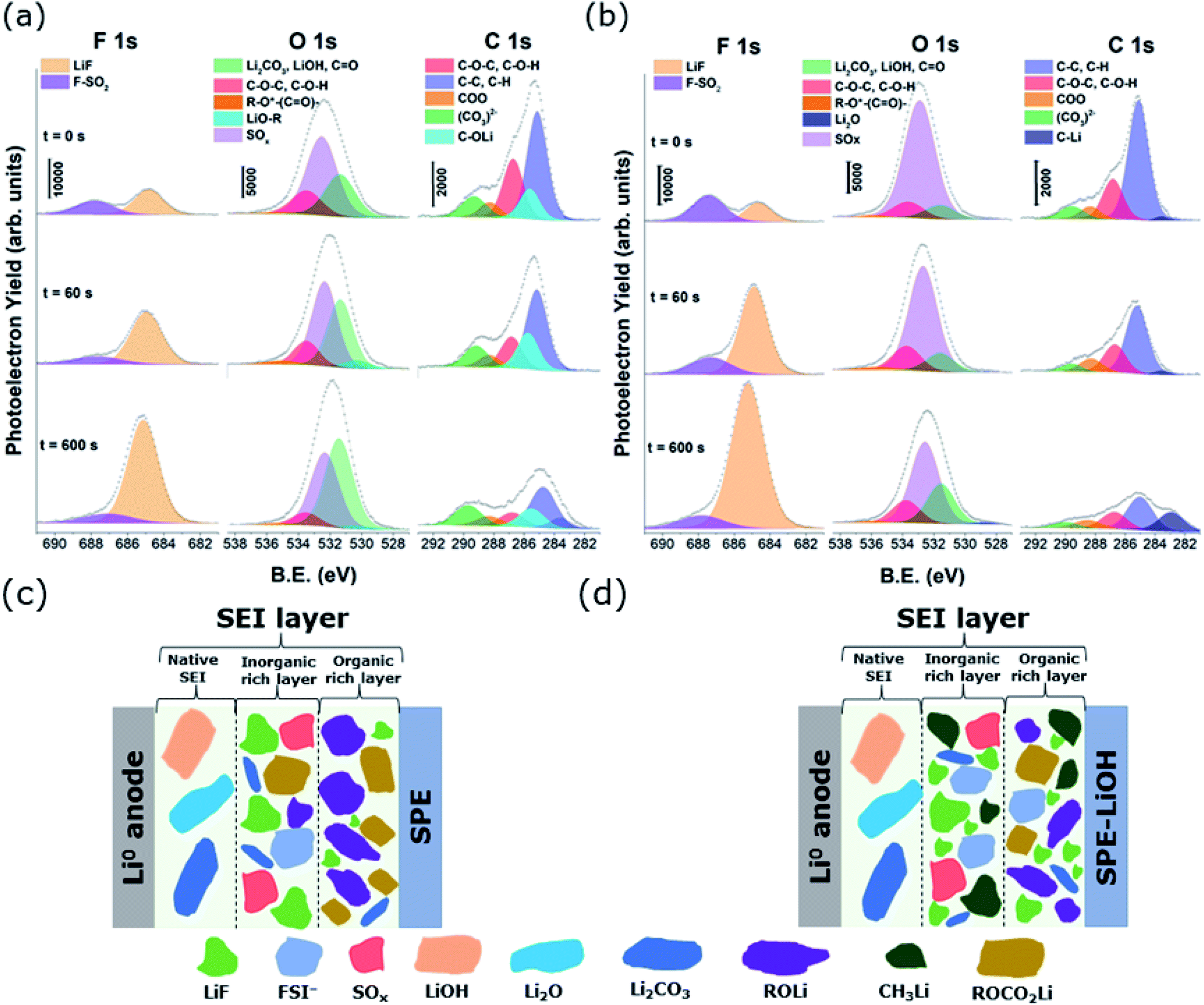 | ||
| Fig. 5 F 1s, O 1s, and C 1s spectra of the outermost surface of the SEI layer formed on plated Li metal with (a) pristine and (b) LiOH-containing electrolyte. Schematic illustration of the main species of the SEI layer formed on the Li metal anode electrode with (c) pristine and (d) LiOH-containing electrolyte. | ||
Fig. 5a and b show the F 1s, O 1s and C 1s XPS spectra of 2 mA h cm−2 lithium deposited on Cu electrodes. The overall spectra of both electrodes indicate that the SEI layer is formed by the decomposition products of both solvent and salt, consisting mainly of inorganic compounds including Li2CO3, LiOH, LiF, Li2O, and other Li salt derivatives, as well as organic compounds.41–43 A deeper analysis reveals that the C 1s peaks observed in Fig. 5a and b are characteristic of different carbon environments corresponding to species formed in the SEI layer. The signal at 284.8 eV is related to C atoms bound to H or other C atoms, corresponding usually to aliphatic chains. The peak observed at 286.8 eV is associated with the C atoms bound to one oxygen atom characteristic of ethylene oxide oligomers while the peak located at 288.3 eV corresponds to O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O environments, confirming the presence of lithium carboxylates. In addition, the signal at 289.5 eV is linked to carbonate species such as lithium carbonate, lithium alkyl carbonate (ROCO2Li) and/or semicarbonate species ((CH2OCO2Li)2).44–46 Comparing C 1s spectra obtained with the pristine electrolyte, a carbon signal at 282.9 eV appears in the spectra of the electrodeposits with LiOH containing electrolyte, which is more apparent upon sputtering. Provided that this peak does not reflect sputtering-induced reactions, it can be concluded that close to the Li surface, a portion of the surface species are further reduced to form C–Li bonds.47 On the other hand, a peak at 285.5 eV can be seen in the spectra of the electrodeposits with the pristine electrolyte, which is absent when LiOH is added, pointing to the formation of alkoxides (ROLi) in the SEI layer of the pristine electrolyte upon reduction.
O environments, confirming the presence of lithium carboxylates. In addition, the signal at 289.5 eV is linked to carbonate species such as lithium carbonate, lithium alkyl carbonate (ROCO2Li) and/or semicarbonate species ((CH2OCO2Li)2).44–46 Comparing C 1s spectra obtained with the pristine electrolyte, a carbon signal at 282.9 eV appears in the spectra of the electrodeposits with LiOH containing electrolyte, which is more apparent upon sputtering. Provided that this peak does not reflect sputtering-induced reactions, it can be concluded that close to the Li surface, a portion of the surface species are further reduced to form C–Li bonds.47 On the other hand, a peak at 285.5 eV can be seen in the spectra of the electrodeposits with the pristine electrolyte, which is absent when LiOH is added, pointing to the formation of alkoxides (ROLi) in the SEI layer of the pristine electrolyte upon reduction.
Small differences are found in the detailed F 1s spectra of both electrolytes (Fig. 5a and b). Both spectra reveal two peak contributions at 688 eV and 685 eV, corresponding to the residual salt and LiF, respectively, which is formed as a result of the reduction/nucleophilic attack of LiFSI salt.48,49 Notably, the electrode cycled with the electrolyte containing LiOH shows more F (Fig. 5b) than the pristine electrolyte at all depths, which is consistent with the formation of a LiF rich SEI layer. This suggests that the addition of LiOH may induce the cleavage of S–F labile bonds in LiFSI, therefore leading to a larger amount of LiF at the SEI. Considering the reported reactivity between LiFSI and CH3CO2Li49 and the results from XPS obtained in this work, herein we propose a reaction mechanism in which the nucleophilic attack of OH− leads to the cleavage of the S–F bond in N(SO2F)2− (Scheme 1), releasing LiF and leading to a modified salt which, in addition to LiF, may be part of the SEI. The improvement of the modified SEI is likely due to the synergy from the formation of LiF, unreacted LiOH, and LiH2NO6S2 and LiSO2N(Li)SO2Li salts resulting from the chemical reaction with LiOH and electrochemical reduction of LiFSI, respectively.
 | ||
| Scheme 1 | ||
Considering the detailed spectra of O 1s (Fig. 5a and b), the LiOH containing electrolyte induces a slight formation of Li2O, as revealed by the small peak at ∼528 eV. The amount of Li2O seems to be rather low on the surface, while it is not detectable when the pristine electrolyte is reduced. This is not surprising as Li2O should be found mostly in the inner part of the SEI.44 In addition, both spectra show a signal at 531.5 eV related to lithium carbonate, lithium carboxylate or lithium hydroxide.45,46,50 Interestingly, and in correlation with the corresponding peaks in C 1s, the intensity of this contribution in the LiOH containing electrolyte shows a much less pronounced signal than in the pristine electrolyte. Moreover, an extra component appears at 530.4 eV in the case of the pristine electrolyte, accounting for the oxygens in lithium alkoxide environments (ROLi).46,51 In addition, part of its contribution is probably included in the bigger peak at 531.5 eV, since they partly overlap. These findings, together with the smaller amount of C found in the SEI layer when LiOH additive is present (Fig. S7†), suggest a poor SEI in organic species and, since those bonds (COLi, COO, and CO3) result from the decomposition of the ethylene oxide, probably less electrolyte degradation occurs in the presence of LiOH. The peak observed at 532.6 eV is assigned to the sulfonyl group of the pristine LiFSI salt and other possible SOx products resulting from the salt decomposition.52 In agreement with the higher S content in the SEI layer, when LiOH is added to the electrolyte (Fig. S7†), this O–S contribution is also much more pronounced than in the case of the pristine electrolyte.
For the S 2p spectra of both electrodes (Fig. S6†), the doublet with an S 2p3/2 peak at 170 eV indicates the presence of the N–SO2–F product in the LiFSI salt, as observed previously in the F 1s spectra.44,52 Other S 2p3/2 peaks observed in the range of 166–173 eV could be attributed to different decomposition products of the LiFSI salt with SO groups.44,52 Concretely, the doublet with the 2p3/2 component at 169.1 eV may be attributed to N–SO2− units after the cleavage of the S–F bond and the other one (2p3/2 peak) at 167.3 eV may be attributed to the formation of Li2SO3 upon further reduction of the LiFSI salt. Interestingly, and in agreement with the higher LiF formation, the amount of F cleaved –SO2− units is also much higher when LiOH is added to the electrolyte. In contrast, more (SO3)2− reduced species are found in the electrode cycled with the pristine formulation.
Based on the XPS results, the SEI layers formed in the two solid electrolytes are schematically illustrated in Fig. 5c and d. According to our findings, the electrochemical decomposition of LiFSI salt is much lower in the LiOH containing electrolyte than in the pristine electrolyte. In addition, the amount of fluorine at the different depths of the SEI layer is higher, leading to the formation of a larger amount of LiF in the presence of LiOH. The use of LiFSI in conventional LIB technology results in a low resistance and robust SEI layer, which contains a substantial amount of LiF upon the electrochemical reduction of the salt. This compound passivates and protects the surface of the cathode and anode from further electrolyte decomposition, and promotes Li-ion transport.15,48,53–56 Due to the unexpected augmented presence of LiF in the inner part of the SEI layer with the LiOH containing electrolyte, ssNMR measurements of both SPEs have been performed prior to applying any current or voltage.
Solid-state nuclear magnetic resonance
19F ssNMR spectra were recorded for the SPE and SPE-LiOH samples, in order to assess the chemical stability of the FSI− anions. The resulting spectra are shown in Fig. 6a. Both spectra are characterized by the main isotropic 19F NMR signals of FSI− anions centered at around 50 ppm.49 In addition to the isotropic lines, the related sets of rotational sidebands are visible in the spectra (marked by “*” in Fig. 6a). These signals are characteristic of the solid-state NMR spectra of samples under MAS rotation and are located in the spectra at distances from the isotropic lines that are multiples of the MAS frequency (20 kHz in this case). It is interesting to note that in both spectra, two signals are observed for FSI− anions with slightly different chemical shifts and linewidths (inset of Fig. 6a). Broader lines in solid-state NMR are generally related to slower local motions. We therefore ascribe the broad components in the spectra to the LiFSI salt present in the microcrystalline regions of the PEO material, and the narrower components to LiFSI in the amorphous polymer regions. From an inspection of Fig. 6a, it is clearly observed that the proportion of FSI− anions at crystalline positions is higher in SPE-LiOH. In addition to the FSI− signals, a broad component is clearly observed in the spectrum of SPE-LiOH at −204 ppm, which is not present in that of SPE. This signal is characteristic of LiF,57 and is therefore an indication of the chemical decomposition of LiFSI molecules upon reaction with LiOH at 70 °C, without inducing any current or potential. Also, some minor signals are observed at around −78 ppm that are tentatively ascribed to intermediate degradation products of the FSI− anions which could be Li[HOSO2NSO2F] from the nucleophilic attack of SO2F by LiOH. As mentioned earlier, the amount of FSI− at amorphous positions of the polymer material is observed to be smaller in SPE-LiOH. This can be explained by a higher reactivity of LiFSI with LiOH at the amorphous regions of the material. Similar to the SPE-LiOH, the 19F ssNMR spectrum of CPE also shows the signal of LiF at −204 ppm, evidencing the reaction between the LiFSI and LiOH present at the surface of LLZO (Fig. S8†).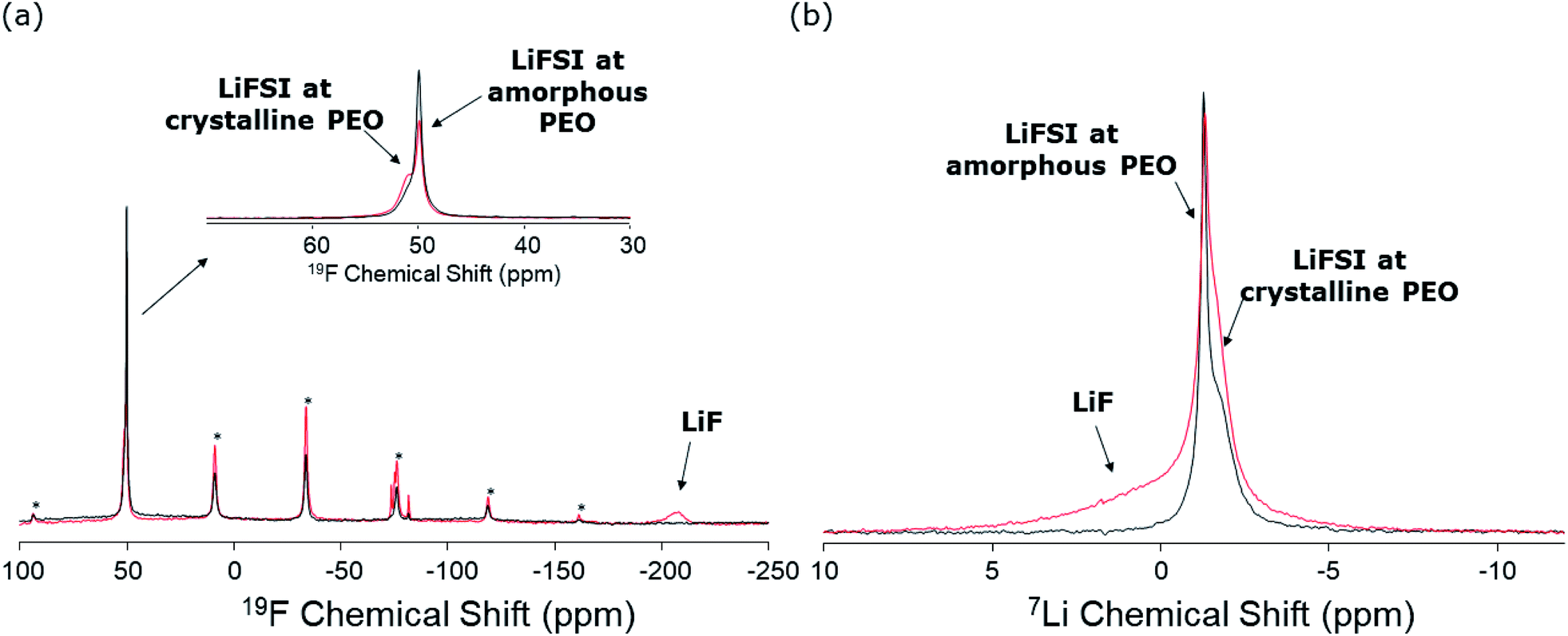 | ||
| Fig. 6 (a) 19F and (b) 7Li solid-state NMR spectra of SPE (black) and SPE-LiOH (red). Signals marked by “*” correspond to rotational sidebands of the FSI isotropic lines. | ||
The 7Li NMR spectra of both samples are shown in Fig. 6b. The spectra of both phases are characterized by two signals at around −1.4 ppm with slightly different chemical shifts and line broadenings, in agreement with the observations made in Fig. 6a. These signals are therefore assigned to the Li-ions of the LiFSI salt, at the amorphous and crystalline positions of the PEO. Also in this case, the ratio of LiFSI observed at the crystalline positions is larger in the SPE-LiOH sample. Finally, a broad component is observed in the spectrum of SPE-LiOH, which is not present in that of SPE. This signal is assigned to LiF, as observed also by 19F NMR (Fig. 6a). The broader character of this LiF signal in the 7Li NMR spectrum also matches the broader width of the corresponding signal in the 19F spectrum.
Conclusions
Based on the surface chemistry of LLZO ceramic garnet, it is shown that some chemical compounds are beneficial for the performance of high-voltage Li metal cells. In particular, we demonstrate that LiOH, used as an additive, has a significant effect on the stability during cycling of high-voltage cells with loading of 1 mA h cm−2, owing to an improvement of the SPE|Li metal anode interface. The addition of LiOH to the SPE promotes the cleavage of the S–F bond in the LiFSI salt, leading to the spontaneous formation of LiF. Consequently, a less resistive and more robust SEI is formed, withstanding Li plating and stripping of around 5 μm thickness. The present work provides further understanding of the role of additives in polymer electrolytes, which can be used to improve high-voltage SSBs.Author contributions
Conceptualization, P. L.-A., M. A. and F. A.; methodology, A. O., M. A.-I., N. G., J. M. L. d. A., R. C., X. J., W. M. and C. R.; validation, P. L.-A., A. O., and M. A.-I.; formal analysis, A. O., M. A.-I., C. R, W. M. and P. L.-A.; investigation, A. O., M. A.-I, N. G., J. M. L. d. A., R. C., X. J., W. M., C. R., M. S. and P. L.-A.; resources, P. L.-A. and F. A.; data curation, A. O., M. A.-I., and P. L.-A.; writing – original draft preparation, A. O., M. A.-I., and P. L.-A.; writing – review and editing, A. O., M. A.-I., C. R., W. M., M. S. and P. L.-A.; visualization, P. L.-A.; supervision, P. L.-A. and F. A.; project administration, P. L.-A.; funding acquisition, P. L.-A. and F. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Synchrotron Light Research Institute (BL1.2W: X-ray Imaging & Microtomography), Nakhon Ratchasima, Thailand, and especially Phakkhananan Pakawanit and Chalermluck Phoovasawat for performing the SRXTM scanning acquisition. W. M. and M. S. are thankful for the financial support from the National Research Foundation of Singapore (NRF) under Investigatorship Award Number NRFI2017-08.Notes and references
- J. Deng, C. Bae, A. Denlinger and T. Miller, Joule, 2020, 4, 511–515 CrossRef.
- J. A. Sanguesa, V. Torres-Sanz, P. Garrido, F. J. Martinez and J. M. Marquez-Barja, Smart Cities, 2021, 4, 372–404 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- F. Zheng, M. Kotobuki, S. Song, M. O. Lai and L. Lu, J. Power Sources, 2018, 389, 198–213 CrossRef CAS.
- C. Yu, S. Ganapathy, E. R. H. van Eck, H. Wang, S. Basak, Z. Li and M. Wagemaker, Nat. Commun., 2017, 8, 1086 CrossRef PubMed.
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- W. Zhao, J. Yi, P. He and H. Zhou, Electrochem. Energy Rev., 2019, 2, 574–605 CrossRef CAS.
- M. B. Armand, Fast Ion Transport in Solids, ed. P. Vashishta, J. N. Mundy and G. K. Shenoy, North-Holland, Amsterdam, 1979 Search PubMed.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- P. López-Aranguren, X. Judez, M. Chakir, M. Armand and L. Buannic, J. Electrochem. Soc., 2020, 167, 020548 CrossRef.
- G. Homann, L. Stolz, J. Nair, I. C. Laskovic, M. Winter and J. Kasnatscheew, Sci. Rep., 2020, 10, 4390 CrossRef PubMed.
- G. G. Eshetu, X. Judez, C. Li, M. Martinez-Ibañez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, J. Am. Chem. Soc., 2018, 140, 9921–9933 CrossRef CAS PubMed.
- J. Zagórski, B. Silván, D. Saurel, F. Aguesse and A. Llordés, ACS Appl. Energy Mater., 2020, 3, 8344–8355 CrossRef.
- M. R. Bonilla, F. A. García Daza, P. Ranque, F. Aguesse, J. Carrasco and E. Akhmatskaya, ACS Appl. Mater. Interfaces, 2021, 13, 30653–30667 CrossRef CAS PubMed.
- P. Ranque, J. Zagórski, D. Shanmukaraj, F. Aguesse and J. Lopez del Amo, J. Mater. Chem. A, 2021, 9, 17812–17820 RSC.
- S. Wang, Q. Sun, W. Peng, Y. Ma, Y. Zhou, D. Song, H. Zhang, X. Shi, C. Li and L. Zhang, J. Energy Chem., 2021, 58, 85–93 CrossRef.
- C.-Z. Zhao, X.-Q. Zhang, X.-B. Cheng, R. Zhang, R. Xu, P.-Y. Chen, H.-J. Peng, J.-Q. Huang and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11069–11074 CrossRef CAS PubMed.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- M. M. Besli, C. Usubelli, M. Metzger, V. Pande, K. Harry, D. Nordlund, S. Sainio, J. Christensen, M. M. Doeff and S. Kuppan, ACS Appl. Mater. Interfaces, 2020, 12, 20605–20612 CrossRef CAS PubMed.
- L. Cheng, M. Liu, A. Mehta, H. Xin, F. Lin, K. Persson, G. Chen, E. J. Crumlin and M. Doeff, ACS Appl. Energy Mater., 2018, 1, 7244–7252 CrossRef CAS.
- J. Wang, G. Huang and X. Zhang, Batteries Supercaps, 2020, 3, 1006–1015 CrossRef CAS.
- M. He, R. Guo, G. M. Hobold, H. Gao and B. M. Gallant, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 73–79 CrossRef CAS.
- S. Jurng, Z. L. Brown, J. Kim and B. L. Lucht, Energy Environ. Sci., 2018, 11, 2600–2608 RSC.
- A. Santiago, J. Castillo, I. Garbayo, A. Saenz de Buruaga, J. A. Coca Clemente, L. Qiao, R. Cid Barreno, M. Martinez-Ibañez, M. Armand, H. Zhang and C. Li, ACS Appl. Energy Mater., 2021, 4, 4459–4464 CrossRef CAS.
- R. Fang, B. Xu, N. S. Grundish, Y. Xia, Y. Li, C. Lu, Y. Liu, N. Wu and J. B. Goodenough, Angew. Chem., Int. Ed., 2021, 60, 17701–17706 CrossRef CAS PubMed.
- J. Zagórski, J. M. López del Amo, M. J. Cordill, F. Aguesse, L. Buannic and A. Llordés, ACS Appl. Energy Mater., 2019, 2, 1734–1746 CrossRef.
- W. Manalastas, J. Rikarte, R. J. Chater, R. Brugge, A. Aguadero, L. Buannic, A. Llordés, F. Aguesse and J. Kilner, J. Power Sources, 2019, 412, 287–293 CrossRef CAS.
- M. Dierick, B. Masschaele and L. V. Hoorebeke, Meas. Sci. Technol., 2004, 15, 1366–1370 CrossRef CAS.
- A. Limaye, Drishti: a volume exploration and presentation tool, in Proceedings Volume 8506, Developments in X-ray Tomography VIII, 85060X (2012), ed. S. R. Stock, San Diego, California, USA, 2012, DOI:10.1117/12.935640.
- J. H. Scofield, Theoretical photoionization cross sections from 1 to 1500 keV, 1973, p. 375 Search PubMed.
- M. P. Seah, Surf. Interface Anal., 2012, 44, 1353–1359 CrossRef CAS.
- A. Orue Mendizabal, N. Gomez, F. Aguesse and P. López-Aranguren, Materials, 2021, 14, 1213 CrossRef PubMed.
- A. I. Pitillas Martinez, F. Aguesse, L. Otaegui, M. Schneider, A. Roters, A. Llordés and L. Buannic, J. Phys. Chem. C, 2019, 123, 3270–3278 CrossRef CAS.
- V. Wood, Nat. Rev. Mater., 2018, 3, 293–295 CrossRef.
- C. Villevieille, in Rechargeable Lithium Batteries, Elsevier, 2015, pp. 183–232 Search PubMed.
- S. Li, Y.-M. Chen, W. Liang, Y. Shao, K. Liu, Z. Nikolov and Y. Zhu, Joule, 2018, 2, 1838–1856 CrossRef CAS.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed.
- X. Wu and Z. Du, Electrochem. Commun., 2021, 129, 107088 CrossRef CAS.
- S.-J. Kang, K. Park, S.-H. Park and H. Lee, Electrochim. Acta, 2018, 259, 949–954 CrossRef CAS.
- X. Wu, T. Liu, Y. Bai, X. Feng, M. M. Rahman, C.-J. Sun, F. Lin, K. Zhao and Z. Du, Electrochim. Acta, 2020, 353, 136453 CrossRef CAS.
- G. G. Eshetu, T. Diemant, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, ACS Appl. Mater. Interfaces, 2016, 8, 16087–16100 CrossRef CAS PubMed.
- A. M. Andersson, M. Herstedt, A. G. Bishop and K. Edström, Electrochim. Acta, 2002, 47, 1885–1898 CrossRef CAS.
- N. Schulz, R. Hausbrand, L. Dimesso and W. Jaegermann, J. Electrochem. Soc., 2018, 165, A819–A832 CrossRef CAS.
- D. Aurbach, I. Weissman, A. Schechter and H. Cohen, Langmuir, 1996, 12, 3991–4007 CrossRef CAS.
- A. Santiago, X. Judez, J. Castillo, I. Garbayo, A. Sáenz de Buruaga, L. Qiao, G. Baraldi, J. A. Coca-Clemente, M. Armand, C. Li and H. Zhang, J. Phys. Chem. Lett., 2020, 11, 6133–6138 CrossRef CAS PubMed.
- G. G. Eshetu, S. Grugeon, G. Gachot, D. Mathiron, M. Armand and S. Laruelle, Electrochim. Acta, 2013, 102, 133–141 CrossRef CAS.
- D. Bar-Tow, E. Peled and L. Burstein, J. Electrochem. Soc., 1999, 146, 824–832 CrossRef CAS.
- C. Xu, B. Sun, T. Gustafsson, K. Edström, D. Brandell and M. Hahlin, J. Mater. Chem. A, 2014, 2, 7256–7264 RSC.
- B. Philippe, R. Dedryvère, M. Gorgoi, H. Rensmo, D. Gonbeau and K. Edström, J. Am. Chem. Soc., 2013, 135, 9829–9842 CrossRef CAS PubMed.
- V. Sharova, A. Moretti, T. Diemant, A. Varzi, R. J. Behm and S. Passerini, J. Power Sources, 2018, 375, 43–52 CrossRef CAS.
- S. Han, S. Liu, J. Gao, J. Wu, Y. Yang, T. Yan, Y. Sun, L. Bao and W. Tang, ChemistrySelect, 2020, 5, 14008–14016 CrossRef CAS.
- S.-J. Cho, D.-E. Yu, T. P. Pollard, H. Moon, M. Jang, O. Borodin and S.-Y. Lee, iScience, 2020, 23, 100844 CrossRef CAS PubMed.
- H. Q. Pham, G. J. Chung, J. Han, E.-H. Hwang, Y.-G. Kwon and S.-W. Song, J. Chem. Phys., 2020, 152, 094709 CrossRef CAS PubMed.
- M. Ménétrier, J. Bains, L. Croguennec, A. Flambard, E. Bekaert, C. Jordy, Ph. Biensan and C. Delmas, J. Solid State Chem., 2008, 181, 3303–3307 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08362g |
| This journal is © The Royal Society of Chemistry 2022 |