
Unraveling the evolution of exsolved Fe–Ni alloy nanoparticles in Ni-doped La0.3Ca0.7Fe0.7Cr0.3O3−δ and their role in enhancing CO2–CO electrocatalysis†
Haris Masood
Ansari
 a,
Adam Stuart
Bass
a,
Nabeel
Ahmad
b and
Viola I.
Birss
*a
a,
Adam Stuart
Bass
a,
Nabeel
Ahmad
b and
Viola I.
Birss
*a
aDepartment of Chemistry, University of Calgary, 2500 University Dr NW, Calgary, AB T2N1N4, Canada. E-mail: birss@ucalgary.ca
bRosalind Franklin Institute, Harwell Campus, Didcot, OX11 0FA, UK
First published on 13th October 2021
Abstract
The growth and phase evolution characteristics of exsolved metal nanoparticles (NPs) in a Ni-doped La0.3Ca0.70Fe0.7Cr0.3O3−δ (LCFCrN) perovskite is investigated in H2–N2 and CO–CO2 environments. Exsolution kinetics are rapid in H2–N2 while those in CO–CO2 atmospheres are sluggish, possibly due to a combination of pO2 difference in excess of three orders of magnitude as compared to that in H2–N2, and a different reaction pathway in the two atmospheres. NPs grown in H2–N2 exhibit a compositional and structural progression from an initially Ni-rich phase to an Fe-rich phase at short and long heat treatment durations, respectively. Once the subsurface Ni depletes, the NPs seem to coarsen via a combination of addition of Fe from the parent perovskite and Ostwald ripening. For longer heat treatment durations, CaO particles are observed to be appended to the Fe–Ni NPs. Exsolution also occurred in CO2–CO atmospheres exhibiting similar trends, although the composition of the NPs was Ni-rich even after a 25 h reduction treatment in 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 CO:CO2 at 800 °C, indicating that the NPs are resistant to coarsening and stable for use in highly reducing CO–CO2 environments. In reversible solid oxide cells (RSOC) applications, the CO oxidation kinetics are typically sluggish on single phase perovskite electrodes. However, for Fe–Ni alloy NP-decorated LCFCrN (Fe–Ni@LCFCrN), the NPs are shown to enhance the CO oxidation kinetics (by ca. 75%) and the CO2 reduction reaction (CO2-RR) kinetics (by ca. 15%) as compared to the parent material, LCFCr. This makes the Fe–Ni@LCFCrN catalyst equally active for both reactions, hence significantly enhancing its potential for use in reversible solid oxide cell applications.
:30 CO:CO2 at 800 °C, indicating that the NPs are resistant to coarsening and stable for use in highly reducing CO–CO2 environments. In reversible solid oxide cells (RSOC) applications, the CO oxidation kinetics are typically sluggish on single phase perovskite electrodes. However, for Fe–Ni alloy NP-decorated LCFCrN (Fe–Ni@LCFCrN), the NPs are shown to enhance the CO oxidation kinetics (by ca. 75%) and the CO2 reduction reaction (CO2-RR) kinetics (by ca. 15%) as compared to the parent material, LCFCr. This makes the Fe–Ni@LCFCrN catalyst equally active for both reactions, hence significantly enhancing its potential for use in reversible solid oxide cell applications.
1. Introduction
Mixed ion-electron conductors (MIEC) belonging to the perovskite oxide class, with a chemical formula ABO3, are a very promising class of inorganic materials that have gained significant traction in the last decade or so for use as the cathode material in solid oxide electrolysis cells (SOEC) to replace conventional Ni-YSZ cathodes, due to their propensity for loss of activity due to coking,1,2 as well as Ni oxidation, especially under pure CO2.3–5 One of the main advantages of the use of MIECs is that they can operate as a single phase in the electrode layers, without the need for the presence of a second ionically conducting phase (typically YSZ), as MIECs typically have both good ionic and electronic conducting properties. This then can simplify the microstructural requirements of these electrodes, removing the requirement for maximization of the triple phase boundary length, as is the case in Ni-YSZ.Of the perovskite oxides that have been examined to date, single phase La0.3M0.7Fe0.7Cr0.3O3−δ (LMFCr, M = Sr, Ca) has emerged as a very promising material, showing exceptionally active and stable performance characteristics at both the cathode (fuel electrode) during CO2 reduction as well as during oxygen evolution at the anode (oxygen electrode).6–8 What is remarkable is the fact that LMFCr exhibits high activity for CO2 reduction as well as oxygen evolution in the absence of a second phase, such as doped ceria, which is generally added to perovskites to increase the ionic conductivity and generate a triple phase boundary (TPB) length, that, in turn, enhances performance. Efforts are still underway to further improve the achievable current densities at the lowest overpotentials by using symmetrical SOECs that employ LMFCr at both electrodes.
Furthermore, there is growing interest in RSOCs that can run reversibly in both the fuel cell (SOFC) mode (CO oxidation) as well as the SOEC mode (CO2 reduction). This could enable using off-peak renewable electricity for electrolysis of CO2 (and possibly H2O) to produce fuel (CO and H2), and, vice versa, converting the fuel back to electricity during peak demand, all using the same RSOC. However, although single phase LMFCr shows a reasonable performance for CO oxidation, much like other similar perovskites, the CO oxidation activity is sluggish as compared to that for the CO2 reduction reaction (CO2-RR).7,9,10 Therefore, it is imperative to explore means to improve the CO oxidation activity of LMFCr and make it equivalent to the activity for CO2-RR to enhance its applicability in RSOC applications.
In the last few years, there has been a growing interest in employing nanomaterials and nano-engineering approaches to enhance the stability and performance of perovskite electrode materials. Metal nanoparticle (NP) infiltration and in situ metal NP exsolution are two such methods by which electrode performance can be improved.11–15 Of these two, exsolution offers more benefits in terms of exercising control over the size and areal density of the NPs and their stability towards coking and agglomeration during operation.16–18 Perovskite oxides, such as La0.3Ca0.7Fe0.7Cr0.3O3−δ (LCFCr), are a particularly versatile class of materials with properties that can be tailored by doping a variety of cations into either the A-site or the B-site.19 By placing reducible/redox active cations on the B-site, there is a possibility of producing metal catalyst NPs on the perovskite backbone by exsolution.20
In material systems that do not readily exsolve B-site cations, an A-site deficiency helps to facilitate exsolution from the B-site, and in turn, inhibits the formation of undesirable reaction products, such as phases containing A-site cations.21 The NPs are strongly anchored to the parent perovskite backbone, which is the defining factor in their excellent long-term stability and coking resistance.22 The NPs not only provide active sites for CO2/CO adsorption but also increase the catalytically active surface area.23,24In situ NP exsolution has been demonstrated in a variety of perovskite systems, such as La0.52Sr0.28Ni0.06Ti0.94O3,21 La0.43Ca0.37Ni0.03Fe0.03Ti0.94O3−γ,25 La0.6Sr0.4Fe0.8Ni0.2O3−δ,9 La0.6Ca0.4Fe0.8Ni0.2O3−δ,26 La0.3Sr0.7Cr0.3Fe0.6Co0.1O3−δ,27 Sr1.95Fe1.35Mo0.45Ni0.2O6−δ,24 and, Pr0.5Ba0.5Mn0.9Co0.1O3,28 to name a few.
Employment of Ni or Co-doped perovskites as cathodes for the electrolysis (or oxidation) of CO2 (CO) requires exposure of the cathode (or fuel electrode) to a reducing atmosphere, typically 5:95 H2:N2 (or Ar), for a few hours at the SOC operating temperature, typically 800–900 °C. This ensures that the exposed surfaces of the porous cathode are decorated with a reasonable population of metal NPs with sub-100 nm sizes. During CO2 reduction and CO oxidation, the fuel electrode is exposed to CO2–CO atmospheres having a pO2 ranging from 10−7 to 10−27 atm (assuming 0.01% CO in pure CO2 and 0.01% CO2 in pure CO, respectively). As will be shown later in this paper, exsolution of metal NPs from Ni-doped LCFCr can occur in 5:95 CO:CO2 having a pO2 that is several orders of magnitude lower (∼10−16 atm) than that in 5:95 H2:N2 (pO2 < 10−23 atm). However, the vast majority of the literature on exsolution is based on reduction in H2 prior to CO2 electrolysis, even though the fuel electrode is mostly exposed to CO2–CO atmospheres during CO2 reduction and CO oxidation. According to our knowledge, there are no reports in literature on the formation, evolution, and stability of NPs exsolved in CO2–CO atmospheres and the similarities and differences with those formed in H2.
In this paper, we have employed 5 mol% A-site deficient compositions of LCFCr ((La0.3Ca0.70)0.95Fe0.7Cr0.25Ni0.05O3−δ) with a 5 mol% B-site doping of Ni to demonstrate the robustness of the system toward in situ NP exsolution and the excellent NP size tunability it affords for relatively short reduction times at 750–800 °C. We also compare exsolution trends in two different gas environments, namely H2–N2 and CO2–CO, with pO2 ranging from ca. 10−16 to < 10−23 atm, to highlight the difference in NPs characteristics in the two gas environments. Finally, we demonstrate the enhancement of CO2 reduction kinetics and especially CO oxidation activity due to the NPs anchored on the parent perovskite surface.
2. Experimental methods
(La0.3Ca0.7)0.95Fe0.7Cr0.25Ni0.05O3−δ (LCFCrN) powder was prepared by dissolving La(NO3)3·6H2O, Ca(NO3)2·4H2O, Fe(NO3)3·9H2O, Cr(NO3)3·9H2O and Ni(NO3)2·6H2O (Alfa Aesar, USA) in DI water (100 mL) according to the desired stoichiometry. Glycine was added in a 2:1 mole ratio versus the total metal content and the mixture was heated at 375 °C on a hot plate until auto ignition. The ash was calcined in a high temperature furnace at 1200 °C for 2 h and then ball milled in an isopropanol medium and zirconia balls at 150 rpm for 4 h. The milled powder was uniaxially pressed into pellets at 200 MPa, and then sintered in air at 1200 °C for 8 h. Both powder and pellet samples were exposed to flowing 5:95 H2:N2 or X CO:(100 − X) CO2 (X = 5–95) in a tube furnace at 750–800 °C for 1–50 h (the pO2 of the gas mixtures used in this work is listed in Table S1†). The furnace was ramped up at 5 °C min−1 in N2 (20 sccm) and allowed to stabilize for 15 minutes upon reaching the temperature set point before introducing the reducing gases at a total flow rate of 20 sccm. Upon completion of the reduction treatment, N2 was reintroduced for 15 min at temperature after which the furnace was ramped down at 5 °C min to room temperature in flowing N2.
Field emission scanning electron microscopy was performed on a Zeiss Σigma FEG-SEM. X-ray diffraction was carried out over a 2θ range of 20–80° (step size = 0.02°, time per step = 1 s) on Bruker D8 and Rigaku Multiflex-1200 XRD spectrometers with Cu Kα radiation. High Angle Annular Dark Field (HAADF) Scanning Transmission Electron microscopy (STEM) and Energy Dispersive Spectroscopy (EDS) were performed using a FEI Titan Themis STEM.
The electrochemical performance of LCFCrN was evaluated using symmetrical cells, where single phase LCFCrN powder was mixed with an ink vehicle in a 1:1 ratio (by weight) and screen printed on an area of ca. 0.53 cm2 on both sides of 130 μm thick (1 inch in diameter) samaria-doped ceria (SDC)-buffered scandia-stabilized zirconia (ScSZ) electrolytes (Hionic™, Nexceris, USA) followed by firing at 1100 °C for 2 h. The current collectors were comprised of Au paste, which was painted on both electrodes followed by firing at 825 °C for 1 h, while Au wires were used as the contact leads.
The cells were mounted on an alumina tube and sealed using Ceramabond VFG-552 sealant (Aremco Products, USA) to isolate the fuel and the air sides. Electrochemical performance measurements were carried out at 800 °C under various CO2–CO atmospheres (pure CO2, 90:10, 70:30, and 50:50 CO2:CO). The gas mixtures were chosen to simulate the various environments the fuel electrode could be exposed to under a typical SOC operation. A Solartron 1287/1255 potentiostat/impedance analyzer was used to acquire electrochemical impedance spectra (EIS) in the frequency range of 0.01–65000 Hz at an AC amplitude of 50 mV. Cyclic voltammograms (CVs) were acquired at a sweep rate of 5 mV s−1 using a Solartron 1287 interface and Corrware software. Short-term electrochemical stability of the cells was evaluated by 15 minute potentiostatic tests at cell voltages of 1.0, 1.2, 1.4, and 1.6 V, respectively, in pure CO2, 90:10 and 70:30 CO2:CO atmospheres. Medium-term stability was evaluated via a 10 h potentiostatic test at a cell voltage of 1.3 V in a 70:30 CO2:CO atmosphere.
3. Results and discussion
3.1 Exsolution of Fe–Ni alloy NPs from LCFCrN in H2–N2 environment
Fig. 1(a) shows an XRD pattern of the as-prepared LCFCrN powder. While the parent perovskite (LCFCr) is a pure orthorhombic phase in the space group Pnma, the XRD pattern of A-site deficient LCFCrN shows the presence of Fe2O3 and NiO. These impurities have also been observed by Lv et al. in A-site deficient Ni-doped SFMN (Sr1.95Fe1.35Mo0.45Ni0.2O6−δ and Sr1.90Fe1.35Mo0.45Ni0.2O6−δ).24 Lai et al. doped a similar material, namely La0.3Sr0.7Fe0.7Cr0.3O3−δ, with Co to attain the composition (La0.3Sr0.7)0.95Fe0.6Cr0.3Co0.1O3−δ and found (Fe,Cr)3O4 impurities in their powder.27 They argued that a pure phase cannot be attained with 5 mol% A-site deficiency in this material because of the high Sr content and low Cr content. From an electroneutrality viewpoint, the A-site deficiency can either be compensated for by the creation of built-in oxygen vacancies or by an increase in the oxidation state of the B-site cations (since the A-site cations, La3+ and Ca2+, have only one oxidation state). Xiao et al. have shown that the oxygen non-stoichiometry (δ) in La0.3Ca0.7Fe0.7Cr0.3O3−δ is ca. 0.05,29 since Cr prevents oxygen loss due to a strong preference for 6-fold coordination. Charge compensation in these perovskites is achieved by Cr3+ changing its oxidation state to Cr4+, which has a limit set by the amount of Ca2+ and Cr3+ present.27,30 In LCFCrN, the amount of Ca2+ is high and that of Cr3+ is low and hence Fe2O3 and NiO impurities are inevitable along with a near-stoichiometric perovskite.27 Furthermore, a close look at the phase diagrams of the La–Fe–O and La–Cr–O systems shows that the equilibrium A/B ratio in LaFeO3 and LaCrO3 in air is close to 1. Since LCFCrN is based on these aforementioned compounds, any deviation from A/B = 1 could very well result in impurity phases.31,32The SEM micrograph in Fig. 1(b) shows the surface of the LCFCrN pellet before reduction, while that in Fig. 1(c) shows the early stages of NP formation when the pellet is reduced in 5:95 H2:N2 for 1 h at 800 °C. At this stage, the smaller particles are hemispherical, while the bigger particles begin to facet, such as the large particle with a square base in the SEM image of Fig. 1(d). The NPs are 25–70 nm in size with an average of ca. 46 nm and an areal density of ca. 18 NPs/μm2, as depicted in Fig. 1(e). The robustness of the system to NP exsolution in a H2 environment is apparent in that a sizeable NP population with sub-100 nm sizes is observed after only 1 h of reduction treatment at 800 °C, the typical operating temperature of SOCs. The practicality of catalyst systems with exsolved NPs depends on (1) how quickly a reasonable NP population is established on the catalyst surface, (2) the average size of the NPs, and (3) the thermal stability of the NPs. Many SOC catalyst systems have suffered from this practical limitation, whereby the exsolution kinetics were either too sluggish33 or required temperatures as high as 1400 °C to achieve a reasonable NP population.34 In some systems, such as La0.52Sr0.28Ni0.06Ti0.94O3,21 the A-site deficiency had to be significantly increased to force NP exsolution. However, these time and temperature requirements to achieve a reasonable NP population still render the system impractical for SOC application.
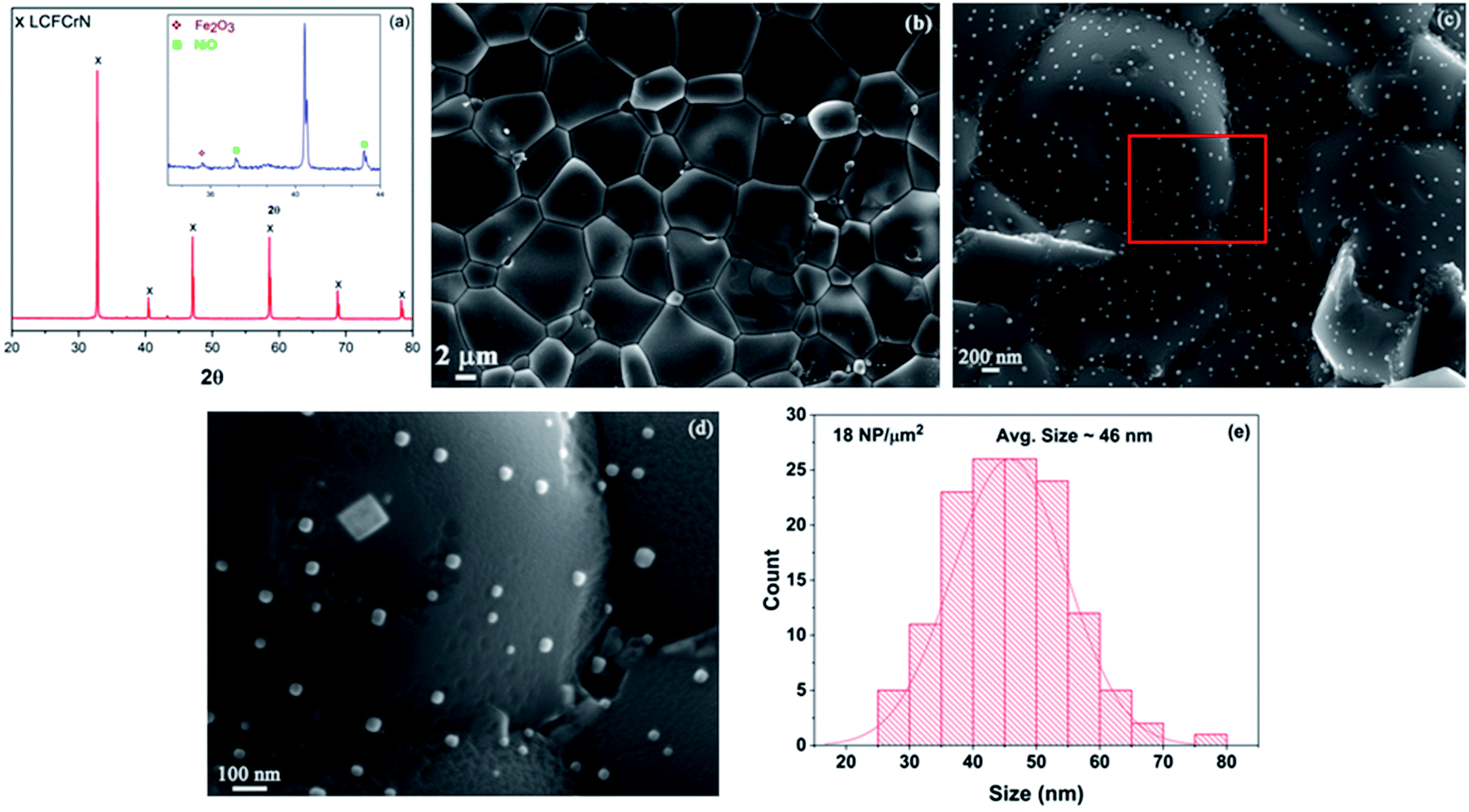 | ||
| Fig. 1 (a) XRD pattern of the as-prepared LCFCrN powder (inset: narrower 2θ range showing the Fe2O3 and NiO peaks). (b) SEM micrograph showing the surface of an LCFCrN pellet before the reduction treatment, (c) SEM micrograph showing NPs on the surface of an LCFCrN pellet upon reduction in 5:95 H2:N2 atmosphere for 1 h at 800 °C, with (d) showing a higher magnification image of the area highlighted by the red box in (c), and (e) a histogram depicting the NP size distribution. | ||
In the case of LCFCrN, two factors that could determine its strong exsolution capability in H2 are the A-site deficiency and the presence of other redox active cations on the B-site, namely Fe and Cr. Under a reducing environment, the loss of lattice oxygen reduces the 6-fold coordination of the B-site cations. Oxygen stripping during exposure to a reducing environment has been identified as a vital factor in the surface segregation of reducible species and their eventual exsolution.35,36 Haag et al. have shown that, in a reducing environment, the oxygen stoichiometry for La0.3Sr0.7Fe0.7Cr0.3O3−δ, a perovskite similar to LCFCr, is ∼2.65 (δ ∼0.35) when B-site cations reduce fully to an oxidation state of +3.37 This, in turn, destabilizes the perovskite lattice by lowering the co-ordination number of B-site cations and could have a strong influence on forcing B-site exsolution. Sun et al. have argued that lattice coordination has a strong influence on the rate of exsolution.13 Through experiments and density functional theory (DFT) calculations on Pr0.5Ba0.5Mn0.9Co0.1O3, they have shown that crystal reconstruction and subsequent lowering of the coordination number of B-site cations favors Co NP exsolution in a reducing environment. Since the A-site deficiency in LCFCrN is low in our case, the loss of lattice oxygen and lowering of co-ordination number could be the dominant factor in the speed of exsolution.35
It has been shown via high temperature AFM studies that the initial stage of NP formation involves the appearance of pits on perovskite surfaces exposed to a reducing atmosphere from where the NPs subsequently emerge.38 A direct consequence of this is that the NPs are firmly anchored to the parent perovskite, with as much as 30% of the NP residing below the surface.17 The existence of surface pits is clearly visible in Fig. 1(d) and many NPs seem to have a trench around them. A recent in situ TEM study on a La0.43Ca0.37Ni0.06Ti0.94O3 perovskite has shown that the Ni NPs nucleate on the perovskite surface rather than in the bulk and that anchoring is achieved when the expanding NPs push the perovskite lattice around them, causing it to rise around the particles to create a volcano-shaped socket.39 However, it is unclear from our experiments whether pit formation is a precursor to exsolution and eventual emergence of NPs from them, or whether the pits are formed later when the NPs mound up as they grow and draw material from their vicinity.
To understand the phase evolution of the NPs and the perovskite, first in a H2 atmosphere, the as-prepared LCFCrN powder was reduced at two different temperatures, namely 750 and 800 °C, in 5:95 H2:N2 for 1–50 h Fig. 2 shows a series of XRD patterns of LCFCrN after exposure to 5:95 H2:N2 at 750 and 800 °C for 1, 2, and 5 h. Although slightly suppressed peaks of Fe2O3 and NiO are still visible after a 1 h reduction at 750 °C, these peaks disappear after a 5 h reduction at 750 °C (Fig. 2(a) and (b)). At the same time, peaks for FeNi3 (JCPDS 38-0419) appear at 2θ values of 44.33° and 51.7°. A weak peak at 2θ = 43.64° is also visible, which belongs to the Fe-rich Fe0.64Ni0.36 (JCPDS 47-1405) Fe–Ni alloy. Lv et al. reduced Sr2Fe1.35Mo0.45Ni0.2O6−δ (SFMN) perovskites in 5:95 H2:Ar at 700 °C for 2 h and found the surface of the SFMN powder particles decorated with FeNi3 NPs,24 while Liu et al. reduced La0.6Sr0.4Fe0.8Ni0.2O3−δ (LSFN) in 5:95 H2:N2 at 850 °C for 2 h and observed Fe0.64Ni0.36 NPs on the surface of LSFN.9 That we observe both FeNi3 and Fe0.64Ni0.36 NPs could be due to the combination of a lower temperature as compared to that employed by Liu et al. and a longer reduction time compared to that used by Lv et al.
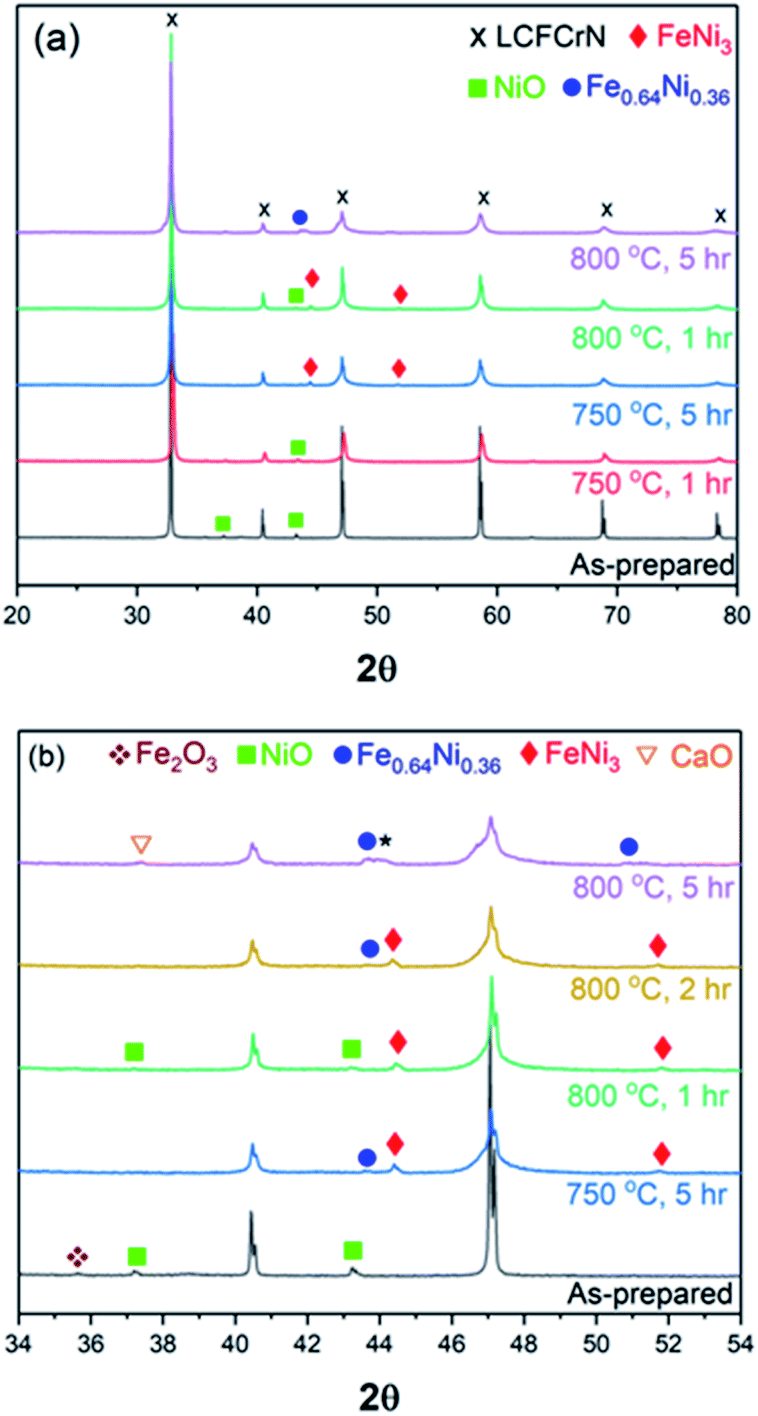 | ||
| Fig. 2 X-ray diffraction patterns of (a) as-prepared LCFCrN, LCFCrN exposed to 5:95 H2:N2 at 750 °C for 1 and 5 h, and 800 °C for 1 and 5 h, respectively, and (b) patterns over a narrower 2θ range, showing the phase evolution upon reduction of LCFCrN at 750 °C for 5 h and 800 °C for 1, 2, and 5 h, respectively. | ||
Another factor that could be at play in determining the NP composition/stoichiometry is the amount of dopant present, which in our case is 5 mol%, while it was 10–20 mol% in the two aforementioned cases.9,24 Our results suggest that the first generation NPs that were initially FeNi3 coarsened and formed Fe0.64Ni0.36 at a later stage, while the next generation of NPs with a FeNi3 composition nucleated at the same time. This also suggests that the NP size and composition can be tuned in LCFCrN to suit the application by varying the reduction temperature and duration. It is hardly surprising that the NPs formed at lower temperatures or short reduction durations are Ni-rich, since the Gibb's free energy change for Ni2+ → Ni0 reduction (ΔGr) is highly negative at 800 °C, while that for Fe2+ → Fe0 is less negative.40 All the other cations in LCFCrN have a positive ΔGr value and hence are stable in reducing environments and thus do not exsolve.
Increasing the reduction temperature to 800 °C for 1 h results in an XRD pattern similar to the one obtained after a 5 h reduction at 750 °C, although weak NiO peaks are still visible (Fig. 2). These NiO peaks disappear when the reduction duration was increased to 2 h, while a weak Fe0.64Ni0.36 peak appears at a 2θ value of 43.65°. The NPs formed under these conditions are composed predominantly of FeNi3, while the parent perovskite peaks shift to higher 2θ, suggesting a lattice contraction. A longer reduction duration at 800 °C (5 h) resulted in NPs with a mixed Fe0.64Ni0.36 and FeNi3 composition, although the NP stoichiometry possibly deviates from Fe0.64Ni0.36 and FeNi3, which causes peak shifts such as indicated by * in Fig. 2(b).
Exsolution is often accompanied by the formation of AO type oxides9,21 and, in this case too, CaO peaks appear after a 5 h reduction at 800 °C, possibly as a consequence of loss of Fe3+ from the B-site and a strong affinity of the system to revert to ABO3 stoichiometry. Neagu et al. have suggested that increasing the perovskite A-site deficiency (≥0.1) could circumvent the formation of AO byproduct and also produce a stable residual perovskite.41 In this work, we have employed an A-site deficiency of 0.05 and have used native perovskite surfaces rather than cleaved bulk surfaces. It is shown in literature that native surfaces undergo A-cation surface segregation21,27, which could interfere with the exsolution process and also promote the formation of the AO byproduct. However, for real SOC catalysts, their native surfaces, rather than cleaved bulks, are exposed to the process gases. Also, as we have already shown (Fig. 1(c)), the exsolution process in LCFCrN does not seem to be inhibited in any way by the use of native surfaces and hence we did not use cleaved bulk surfaces in this work. Fig. 1(c) shows that the entire surface of the LCFCrN pellet is uniformly covered with NPs even after 1 h exposure to 5:95 H2:N2 at 800 °C. The system is so robust that lower growth temperatures can also be employed and the size and areal density of the NPs can be tuned by varying the time of reduction treatment.
Fig. 3 shows a series of SEM micrographs, displaying the surfaces of LCFCrN pellet samples, reduced at 750 and 800 °C for 5 and 25 h, respectively. The dark phase adjoining the NPs in Fig. 3(b–d) appears to be CaO, since it was evident in the XRD pattern in Fig. 2. Similar oxide growth attached to the NPs has also been observed by Neagu et al. in La0.8Ce0.1Ti0.6Ni0.4O3,39 as well as Thalinger et al. in La0.6Sr0.4FeO3−δ.42 Notably, CaO formation during exsolution may not necessarily have a deleterious effect on the electrochemical performance of the material, as it has been shown to promote CO2 adsorption and enhance the electrochemical activity in synergy with exsolved NPs.26 One feature that is common to all of the micrographs shown in this work is the surface roughness that develops on the surfaces of the LCFCrN grains upon exposure to 5:95 H2:N2 environment. Although others27,43 have also shown the development of a similar degree of surface roughness, it is unclear whether this is a result of exposure to the reducing atmosphere or the exsolution process, since the grain surfaces are smooth in the as-prepared pellets in air as seen in Fig. 1(b).
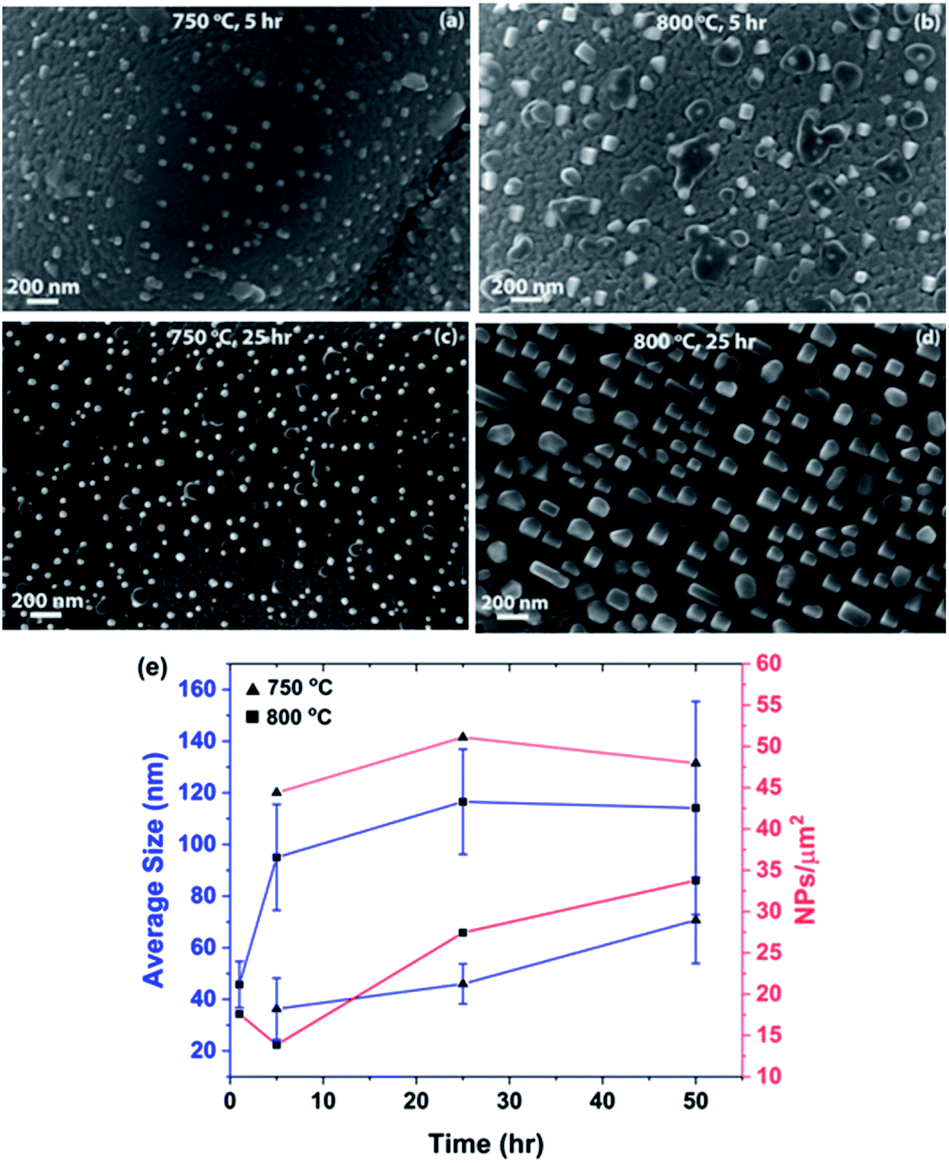 | ||
| Fig. 3 SEM micrographs showing the metal NPs on the surface of LCFCrN pellets, formed upon exposure to 5:95 H2:N2 for (a) 5 h at 750 °C, (b) 5 h at 800 °C, (c) 25 h at 750 °C, and (d) 25 h at 800 °C. (e) Plots of NP average size and areal density vs. heat treatment duration. | ||
The NPs that form at 750 °C in 5:95 H2:N2 are smaller in size and appear to be hemispherical in shape as compared to those formed at 800 °C for the same time duration. Fig. 3(e) shows plots of the NP average size and areal density vs. heat treatment duration. For a 5 h reduction treatment, the NPs formed at 750 °C are 36 nm in size on average with an areal density of ca. 44 NPs/μm2, while those formed at 800 °C are 95 nm in size with an areal density of ca. 14 NPs/μm2. The average size of the NPs at both 750 and 800 °C exhibits an increasing trend with the heat treatment duration and the size distribution becomes broader with time (Fig. S1†).
The NPs formed at 800 °C have an initially steep growth rate (Fig. 3(e)), nearly doubling their average size after 5 h, but saturating beyond that point, leading to average sizes of ca. 115 nm after 50 h. It is pertinent to mention here that the NP size distribution becomes broader with heat treatment time in the 5:95 H2:N2 environment (Fig. S1†), suggesting the presence of two competing processes, namely nucleation and growth. It seems that, after an initial nucleation burst, growth is the dominant process. This could explain the slight decrease in NP areal density between 1 and 5 h of reduction treatment (Fig. 3(e)) as the NPs double in size. At this point, although the subsurface Ni is depleted, more Ni diffuses to the surface from the bulk, initiating new nucleation events and hence causing the areal density to double thereafter, from 14 NPs/μm2 after 5 h to 27 NPs/μm2 after 25 h.
The results at 750 °C paint a slightly different picture. Fig. 3(e) shows that the NP growth rate in 5:95 H2:N2 is sluggish at all times at the lower temperature and requires 50 h for a twofold increase in size (from ca. 36 nm after 5 h to ca. 70 nm after 50 h). The slower growth rate leads to a significantly narrower NP size distribution for all heat treatment durations at 750 °C as compared to those formed at 800 °C (Fig. S1†). The NP areal density is also 2–3 times higher at the lower temperature than at 800 °C. This suggests that the conditions at 800 °C favor NP growth, which is suppressed at 750 °C, likely due to smaller diffusivities of Fe and Ni although the pO2 is lower at 750 °C.
Overall, our results suggest that the kinetics of nucleation and growth, the two competing processes in NP formation, are faster at 800 °C than at 750 °C in 5:95 H2:N2, as expected. Hence, the average particle size shifts from 36 nm (750 °C 5 h) to 95 nm (800 °C 5 h), because the growth rate is much faster for only a 50 °C increase in temperature. As a consequence, the areal density drops from ca. 44 to 14 NPs/μm2. However, the areal density of NPs after 25 h at 750 °C does not change significantly (51 NPs/μm2) while that at 800 °C almost doubles to 27 NPs/μm2. This could be attributed to the fact that, at the higher temperature (800 °C), the Ni and Fe required to form NPs can diffuse from deeper in the bulk to nucleate new surface NPs, while once the sub-surface Ni concentration is depleted at 750 °C, nucleation of new NPs is restricted while the existing ones coarsen to an average size of 46 nm, possibly by the addition of Fe. A typical NP population consists of a variety of shapes ranging from hemispherical for small particles to cuboidal and faceted for larger ones.
To understand whether NP coarsening in LCFCrN is a result of particle coalescence or Ostwald ripening, an experiment was carried out in which the NP evolution in a particular area on a pellet sample was tracked with time at 800 °C in 5:95 H2:N2. Fig. 4(a) shows an SEM image after a 5 h exposure, in which cuboidal NPs ca. 55 nm in size are clearly visible, while much smaller NPs can also be seen emerging from the surface (indicated by the yellow arrow in the inset in Fig. 4(a)). The same sample was placed in the furnace again and reduced for another 5 h for a total of 10 h exposure to 5:95 H2:N2 at 800 °C. Fig. 4(b) shows that after a 10 h exposure, the dark phase completely disappeared from the surface while the grain surface appears to have roughened as the particles grew in size. A few individual particles were tracked for changes in shape and size (marked by red oval and blue box). The tracked NPs appear to be anchored to the same spot on the sample in the same orientation. Their shapes, however, changed from cuboidal to irregular and their size doubled on average (from ca. 55 nm to ca. 104 nm). The smaller NPs that were seen emerging after 5 h have completely disappeared, possibly merging with the larger ones, which suggests that the dominant coarsening mechanism in this case is Ostwald ripening, although the possibility of the contribution of NP coalescence to the overall growth process cannot be ruled out. Lai et al. also suggested Ostwald ripening as the dominant NP growth mechanism for a similar perovskite with Co doping (La0.3Sr0.7Cr0.3Fe0.6Co0.1O3−δ), although they based their argument on particle size and areal density measurements rather than corroborating it with a particle tracking experiment.27
 | ||
| Fig. 4 SEM micrographs showing NPs on the surface of an LCFCrN pellet upon exposure to 5:95 H2:N2 for (a) 5 h, and (b) 10 h at 800 °C. The same area was imaged for both (a) and (b). | ||
HAADF STEM imaging was performed to determine the morphology and composition of the NPs formed after the reduction treatment in H2–N2. Fig. 5(a) shows a STEM image of a NP protruding from the perovskite particle surface. This sample was exposed to 5:95 H2:N2 for 1 h at 800 °C. The EDS elemental color maps in Fig. 5(b–e) reveal that the NP is an Fe–Ni alloy, while the EDS line scan across the center of the particle reveals that the stoichiometry is close to Fe0.5Ni0.5 (f). Similar results were obtained for the sample that was exposed to 5:95 H2:N2 for 5 h at 750 °C. Fig. S2(a–e)† shows a NP with a square base that is composed of only Fe and Ni, also with a stoichiometry close to Fe0.55Ni0.45 (Fig. S2(f)†). Longer exposures to the reducing environment appear to allow the NPs to shift from a Ni-rich to Fe-rich stoichiometry. These STEM results, in conjunction with XRD, suggest that there is a series of Fe–Ni alloys that can form by exsolution from LCFCrN at these temperatures and that the transition from FeNi3 to Fe0.64Ni0.36 and beyond is not stepwise. Instead, this process likely proceeds through intermediate compositions, such as Fe0.5Ni0.5, by the addition of Fe to the growing NPs.
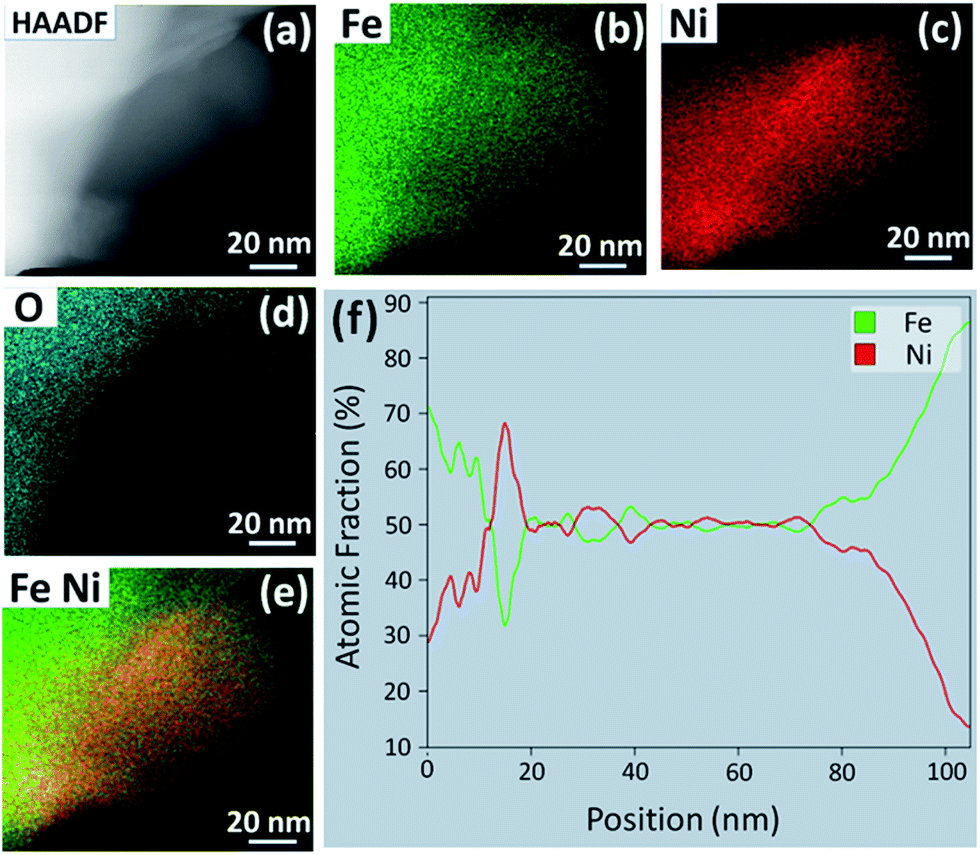 | ||
| Fig. 5 (a) HAADF STEM image of a NP exsolved upon reduction of LCFCrN in 5:95 H2:N2 for 1 h at 800 °C. EDS elemental maps of (b) Fe, (c) Ni, (d) O, and (e) Fe and Ni overlayed, for the NP seen in (a). (f) EDS line scan across the middle of the NP in (a). | ||
3.2 Exsolution of Fe–Ni alloy NPs from LCFCrN in CO–CO2 environments
In SOC conditions, the fuel electrode is exposed to CO–CO2 atmospheres having a pO2 ranging from 10−7 to 10−27 atm (assuming 0.01% CO in pure CO2 and 0.01% CO2 in pure CO). Even if the NPs are first formed in 5:95 H2:N2 before being used as catalysts in CO–CO2 atmospheres, the atmosphere could have enough reducing power to influence the NP stability during SOC operation. Although there are many reports in which cell post-mortem analysis after exposure to CO–CO2 has been reported, there are no reports of systematic studies on the nucleation and growth of exsolved Fe–Ni alloy NPs in CO–CO2, to the best of our knowledge. For this reason, herein we compare the Fe–Ni alloy NP nucleation and growth characteristics during exsolution from LCFCrN perovskites in 5:95 H2:N2 and CO–CO2 atmospheres.
Fig. 6(a–d) show SEM micrographs of the surfaces of LCFCrN pellets exposed to 10:90, 30:70, 70:30 and 90:10 CO:CO2 gas mixtures, respectively. Small hemispherical NPs are visible in all of these gas mixtures as well as for gas mixtures with 5:95 and 95:5 CO:CO2 ratios after 5 h exposure at 800 °C. One important observation made from the SEM micrographs in Fig. 6 is that the dark phase, possibly CaO, which was visible in H2–N2 atmospheres, does not appear in all atmospheres up to 90:10 CO:CO2 after a 5 h exposure. In 90:10 CO:CO2, the NPs appear to be enveloped by a second phase with a morphology that is reminiscent of a red blood cell (see inset of Fig. 6(d)). It is unclear at present as to what this phase may be but it is possible that it is the incipient CaO that was seen to append to the NPs previously, although further characterization is needed for accurate determination.
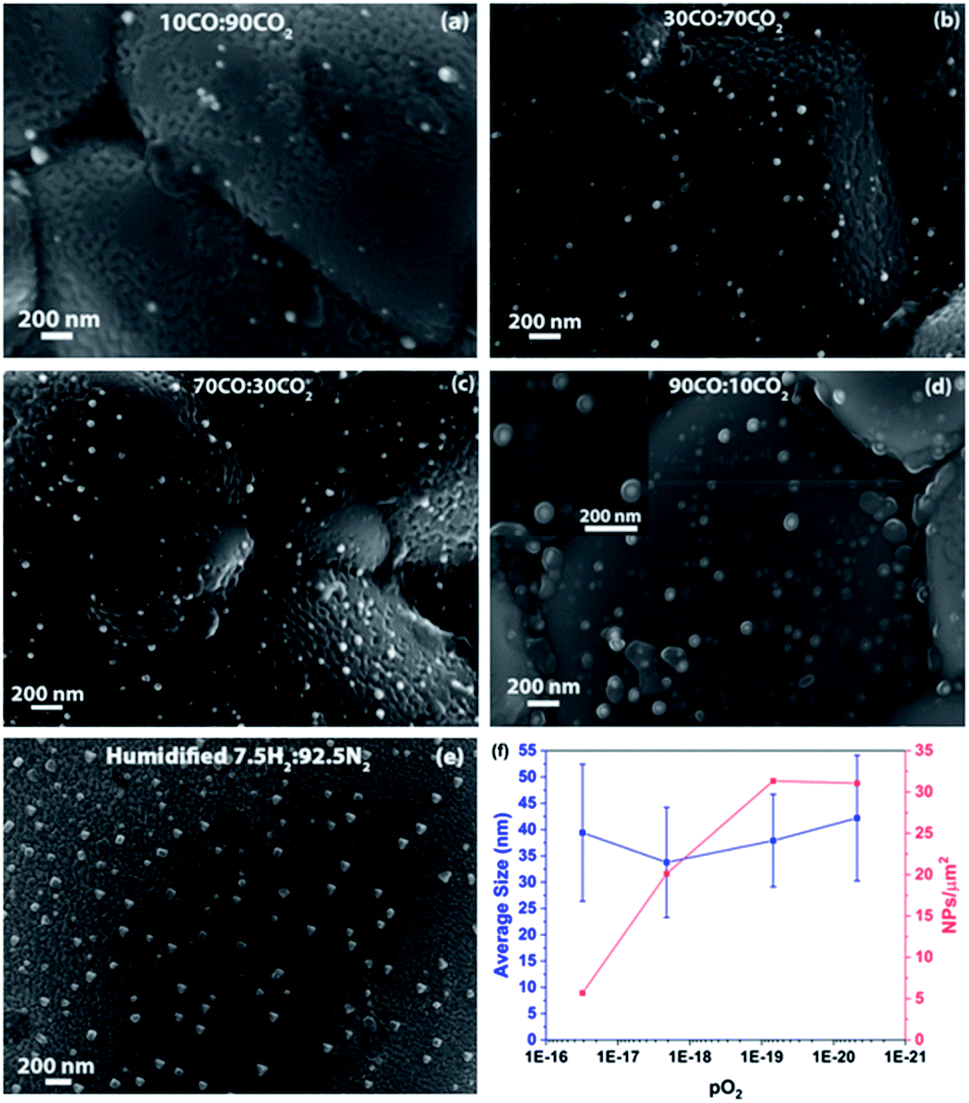 | ||
| Fig. 6 SEM micrographs showing NPs on the surface of LCFCrN pellets upon exposure to (a) 10:90, (b) 30:70, (c) 70:30, (d) 90:10 CO:CO2 gas mixtures (inset shows the SEM image demonstrating the Fe–Ni NPs encapsulated by a second phase), and (e) humidified 7.5:92.5 H2:N2 for 5 h at 800 °C. (f) Plots of NP average size and areal density vs. pO2 in various CO–CO2 gas mixtures. | ||
Fig. 7 shows XRD patterns for LCFCrN powder exposed to various CO–CO2 mixtures, indicating that CaO only begins to appear after exposures to >90% CO, with distinct peaks visible for 95% CO after 5 and 25 h exposures. It is worth mentioning that the pellets coked excessively after exposure to 95:5 CO:CO2 for 25 h (see graphite peak at 2θ value of 26.38° in the XRD pattern in Fig. 7) and pulverized completely after exposure to 100% CO. Another interesting observation from the XRD results is that the NPs are composed of Ni-rich FeNi3 up to at least 70% CO, while distinct peaks for Fe0.64Ni0.36 alloy NPs (clearly visible in 5:95 H2:N2) only begin to appear after exposure to >90% CO. Also, the Ni-rich alloy peak disappears after a 25 h exposure to 95% CO and a peak at a 2θ value of 44.94° appears, possibly reflecting α-Fe.
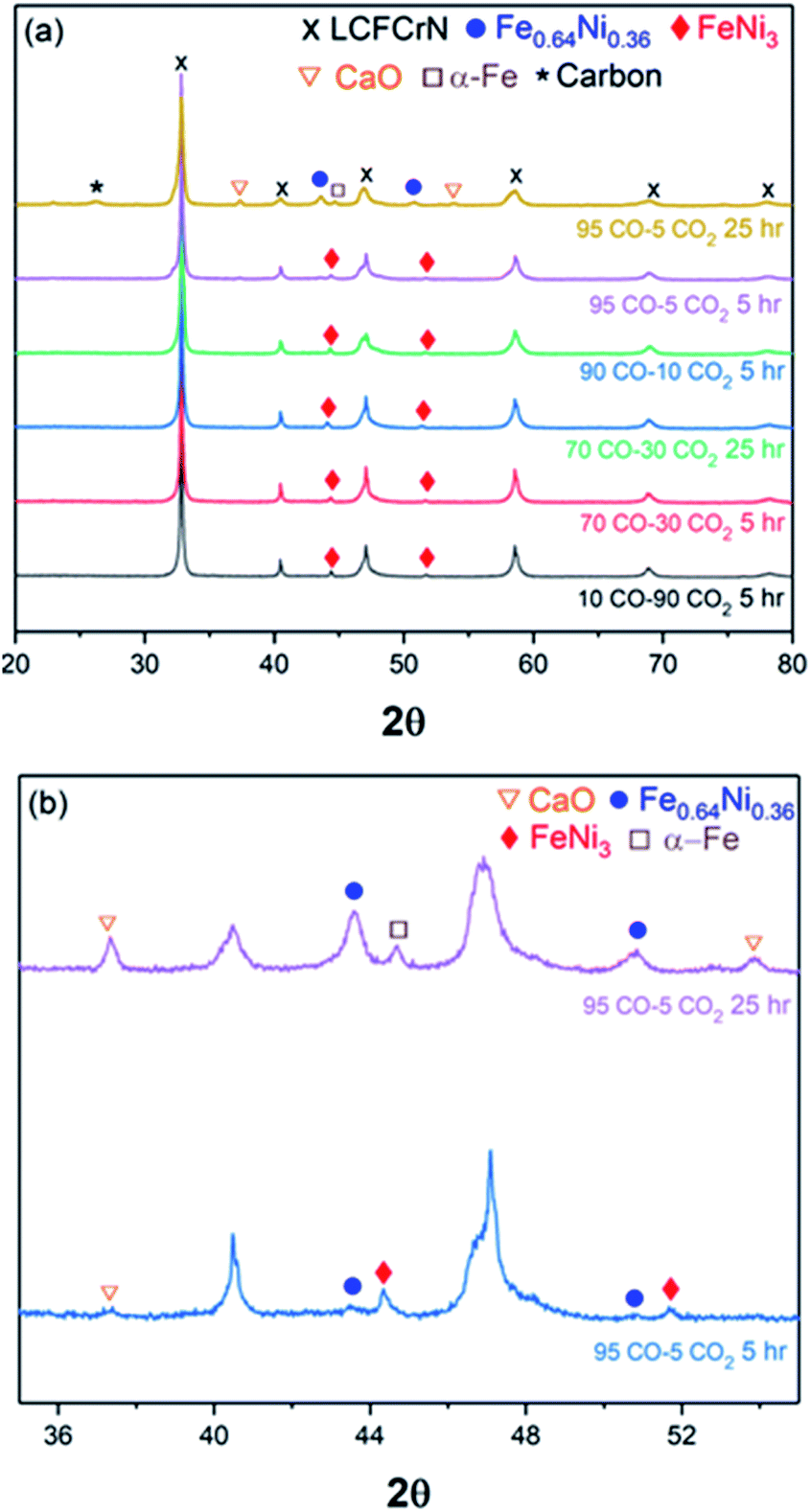 | ||
| Fig. 7 (a) Room temperature X-ray diffraction patterns showing phase evolution of LCFCrN exposed to varying CO–CO2 ratios at 800 °C. (b) X-ray diffraction patterns showing the emergence of CaO and Fe–Ni alloy NP peaks for LCFCrN exposed to 95:5 CO:CO2 at 800 °C for 5 and 25 h. | ||
Significant surface roughness was observed on the LCFCrN grains upon exposure to CO2–CO atmospheres, similar to what was observed in 5:95 H2:N2 atmospheres. Fig. 6(f) shows plots for the variation in NP size and areal density with pO2 (which varies with the CO2:CO gas composition) for a 5 h exposure at 800 °C. The average NP size does not change significantly, increasing only by ca. 10% over a 3 orders of magnitude decrease in pO2. On the other hand, a decrease in pO2 has a marked effect on the Ni NP areal density, which increases by a factor of ca. 5.5 when the pO2 was reduced from 3.1 × 10−17 atm to 6.9 × 10−20 atm. This suggests that, although the decrease in pO2 promotes NP nucleation, it does not significantly impact NP growth in CO–CO2 environments.
3.3 Comparison of nanoparticles grown in CO–CO2vs. H2–N2 environments
To verify whether the NP composition, size, areal density, and shape, are a function of pO2 alone, pellet samples were reduced in humidified 7.5:92.5 H2:N2 at 800 °C for 5 h. This gas composition was chosen because it has a pO2 almost identical to that of 70:30 CO:CO2 at 800 °C (Table S1†). Fig. 6(e) shows an SEM image of the surface of the pellet, depicting the NP distribution and morphology in humidified 7.5:92.5 H2:N2. The NPs have a composition close to FeNi3, an average size of 67 nm, and an areal density of 14 NPs/μm2 (Fig. S4†). Although the NP areal density is identical, the average NP size is almost 2/3rd that in dry 5:95 H2:N2, while the appended CaO particles visible in Fig. 3 are also less abundant. These results suggest that the nucleation kinetics are similar in both the atmospheres (dry vs. humidified H2) while the coarsening kinetics are slower in humidified 7.5:92.5 H2:N2.
In comparison, while the Ni-rich NP composition (FeNi3) in humidified 7.5:92.5 H2:N2 is similar, the average size is almost twice, and the areal density is almost half that in 70:30 CO:CO2, although the pO2for both atmospheres is similar. To maintain the composition close to Ni-rich FeNi3 at a bigger size, the rate of Ni diffusion to the growing NP needs to keep up to prevent Fe enrichment. That this composition is maintained in humidified 7.5:92.5 H2:N2 at an average NP size of 67 nm (as opposed to 38 nm in 70:30 CO:CO2) suggests that the H2–N2 environment is more potent for NP exsolution than CO–CO2 environment and that the nature of the reducing gas in conjunction with the pO2 dictates the NP characteristics. With regards to the NP shape, those formed in humidified 7.5:92.5 H2:N2 are strongly faceted with cuboidal or pyramidal shapes (Fig. 6(e)), while those formed in 70:30 CO:CO2 are more or less hemispherical (Fig. 6(c)). The stark differences in the shapes of the NPs in both the environments also lend credence to the assertion that the nature of the gas, not just the pO2, plays an important role in determining NP characteristics.
Since the two gas environments, humidified 7.5:92.5 H2:N2 and 70:30 CO:CO2, have a similar pO2 but exhibit starkly different NP characteristics, the nature of the gas and consequently the role of the reaction pathway for reduction in H2–N2vs. CO–CO2 are clearly very important. For exsolution to proceed, a complex series of steps are required, i.e., the reactant gas must diffuse to the NP/perovskite interface and then undergo many individual reaction steps, including multiple solid-state processes, as shown as net reactions (1) and (2) in H2–N2:
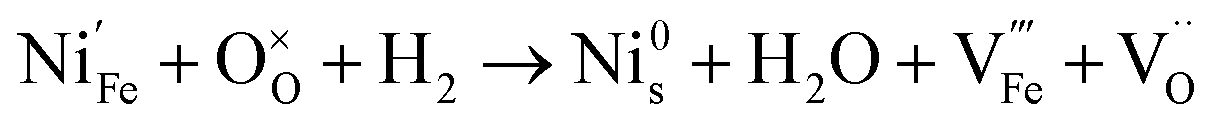 | (1) |
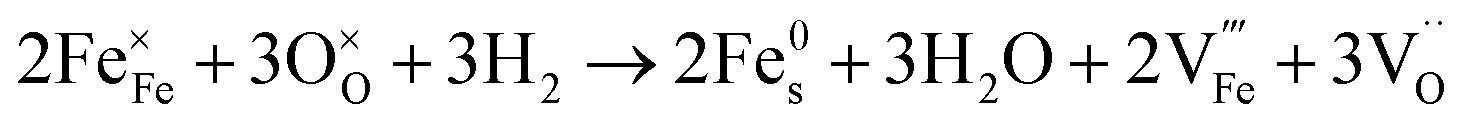 | (2) |
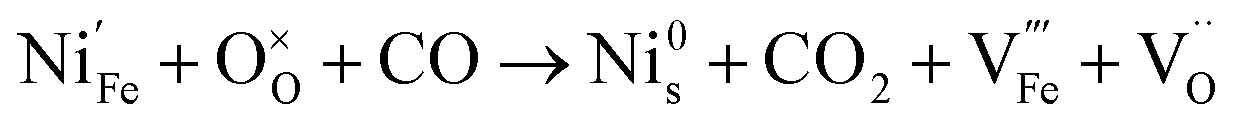 | (3) |
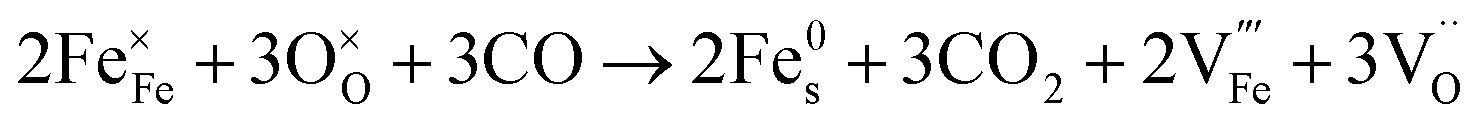 | (4) |
It is possible that the rate limiting step in both the H2 and CO environments is the diffusion of the H2 or CO to the NP/perovskite interface, where the reaction is most likely to occur, once Ni2+ is depleted from the perovskite lattice in the surface region, forming Ni. H2 is a small molecule and thus can diffuse faster to that interface rather than the larger CO molecule and hence reactions (1) and (2) would likely have faster kinetics than reactions (3) and (4). Since the CO–CO2 gas mixture contains a significant amount of CO2 and as it has been shown in the literature that the NPs enhance CO2 adsorption, it is possible that the ease of access of the gas to the NP/perovskite interface becomes increasing difficult for CO. Thus a lower coarsening rate is seen in CO–CO2 atmospheres as compared to that in H2–N2. Furthermore, as stated above, reactions (2) and (4) will each likely involve multiple individual steps, any of which could be rate limiting.
Although not a fair comparison, some interesting contrasts can be drawn between the characteristics of the NPs formed in 5:95 H2:N2 (at 750 and 800 °C) versus those formed in 70–30 CO:CO2 atmospheres at 800 °C for the same heat treatment duration (5 h). This CO:CO2 ratio was chosen assuming that the CO2 to CO conversion efficiency of a typical SOC fuel electrode is 70%. In terms of their size and areal density, the NPs exsolved under 5:95 H2:N2 at 800 °C are bigger by a factor of ca. 2.5, while their areal density is roughly one half of those formed under 70–30 CO:CO2 atmospheres. This corroborates our earlier assertion that the NP growth kinetics are faster at 800 °C in 5:95 H2:N2 than in 70:30 CO:CO2. Slower NP growth kinetics also mean that the NP shape is not as well developed (hemispherical) versus those formed in 5:95 H2:N2 (faceted). Also, since NP coarsening is restrained, the size distribution is narrow in 70:30 CO:CO2 (Fig. 6(e) and S3†). On the other hand, the NPs formed under 5:95 H2:N2 at 750 °C have a similar average size (36 nm), while their areal density is roughly 1.5 times those formed under 70:30 CO:CO2 atmospheres. This suggests that the NP nucleation kinetics are also faster in 5:95 H2:N2 than in 70:30 CO:CO2 even though the temperature is lower. This could be advantageous, as this suggests that NP coarsening would be restrained during exposure to highly reducing CO–CO2 environments that are prevalent in typical SOC conditions.
However, it would be misleading to suggest that the NP size and composition would remain the same after long-term exposure of LCFCrN to a reducing environment under cathodic polarization. While studies focused on CO2 electrolysis using exsolved NPs have reported a particular NP size distribution and initial composition, there are few studies that show the composition after long-term exposure to the reducing environment. As we have shown in the present study, the NP size and composition are transient and depend on the reducible species, reduction temperature and duration, the local pO2, and the nature of the reducing gas. In the case of material systems containing Ni and Fe, such as LCFCrN, once Ni in the surface/sub-surface is depleted and the diffusion of Ni from deeper within the lattice is restrained, the NPs would continue to grow in size with the addition of Fe, while the composition would continuously shift from Ni-rich to Fe-rich alloys, such as the transition from FeNi3 to Fe0.64Ni0.36 in the current case.
However, as we have shown here, this transition can be accelerated or suppressed by changing the pO2 of the reducing environment, the reducing atmosphere (CO vs. H2), and the reduction temperature and duration, among other factors. Zhu et al. have shown that slight changes in pO2 can have a significant impact on the NP composition.44 They showed that the Fe–Ni alloy NPs formed at 850 °C in Ni-substituted Sr(Ti,Fe)O3 shifted from a stoichiometry of Ni0.51Fe0.49 to Ni0.72Fe0.28 with an order of magnitude increase in pO2. Since higher pO2 seems to favor Ni-rich (Fe-lean) alloys, the NPs formed in CO–CO2 and humidified H2–N2 in the present work retain their Ni-rich stoichiometry as opposed to those formed in dry H2–N2, as the pO2 is at least 3 orders of magnitude higher.
HAADF STEM imaging was performed to determine the morphology and composition of the NPs formed in the 70:30 CO:CO2 environment. Fig. 8(a) shows a STEM image of a NP protruding from the perovskite particle surface, where the sample was exposed to 70:30 CO:CO2 for 5 h at 800 °C. The particle is ca. 30 nm in size and appear to be spherical in shape with slightly faceted corners. A significant volume of the NPs seems to be embedded in the perovskite base, suggesting strong anchoring. EDS elemental mapping and line scans across the center of the particle in Fig. 8(b–e) show that the NPs are Ni-rich with a stoichiometry of close to that of FeNi3 (f). Fig. 8(d) shows an EDS elemental map for oxygen which reveals a faint oxygen shell around the NP. It is pertinent to mention here that although very faint, this shell was seen across many particles which led us to believe that this is likely an artefact due to exposure to air during sample retrieval and handling.
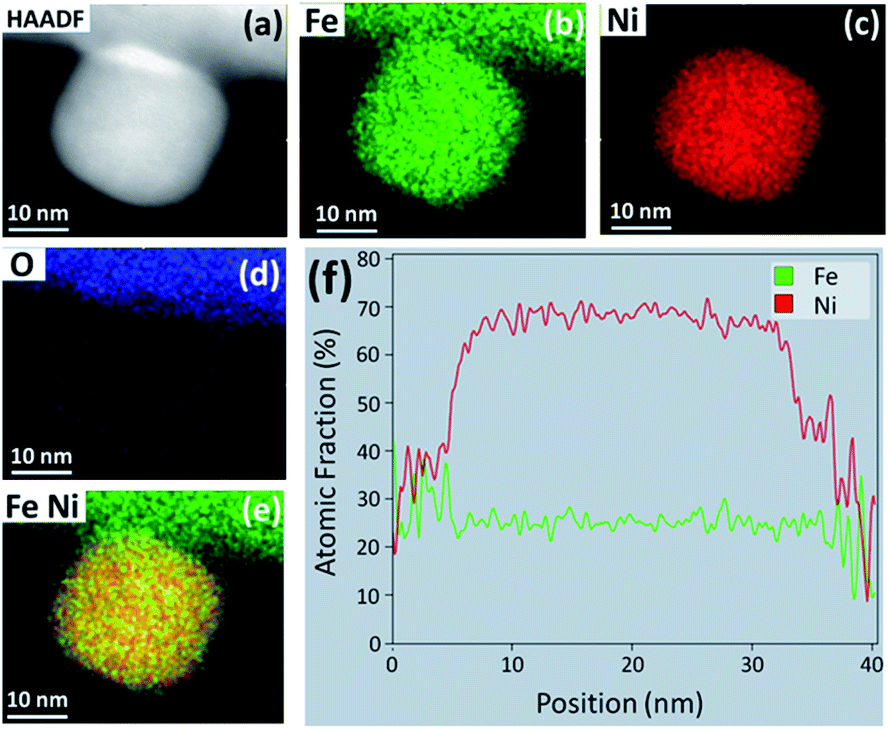 | ||
| Fig. 8 (a) HAADF STEM image of a NP protruding from the surface of the LCFCrN perovskite reduced in 70:30 CO:CO2 at 800 °C for 5 h. EDS elemental maps of (b) Fe, (c) Ni, (d) O, and (e) Fe and Ni overlayed, for the NP seen in (a). (f) EDS line scan across the middle of the NP in (a). | ||
Even after exposure to the same atmosphere for 25 h (Fig. 9), the NPs do not coarsen significantly (Fig. 9(a)) and retain the Ni-rich composition, as revealed by the STEM/EDS analysis presented in Fig. 9. However, the NP shown in Fig. 9 has a slightly lower Ni:Fe ratio (Fig. 9(f)), which suggests that NP coarsening is a result of the addition of Fe, possibly due to the sub-surface depletion of Ni and the abundance of Fe in the vicinity of the NP.
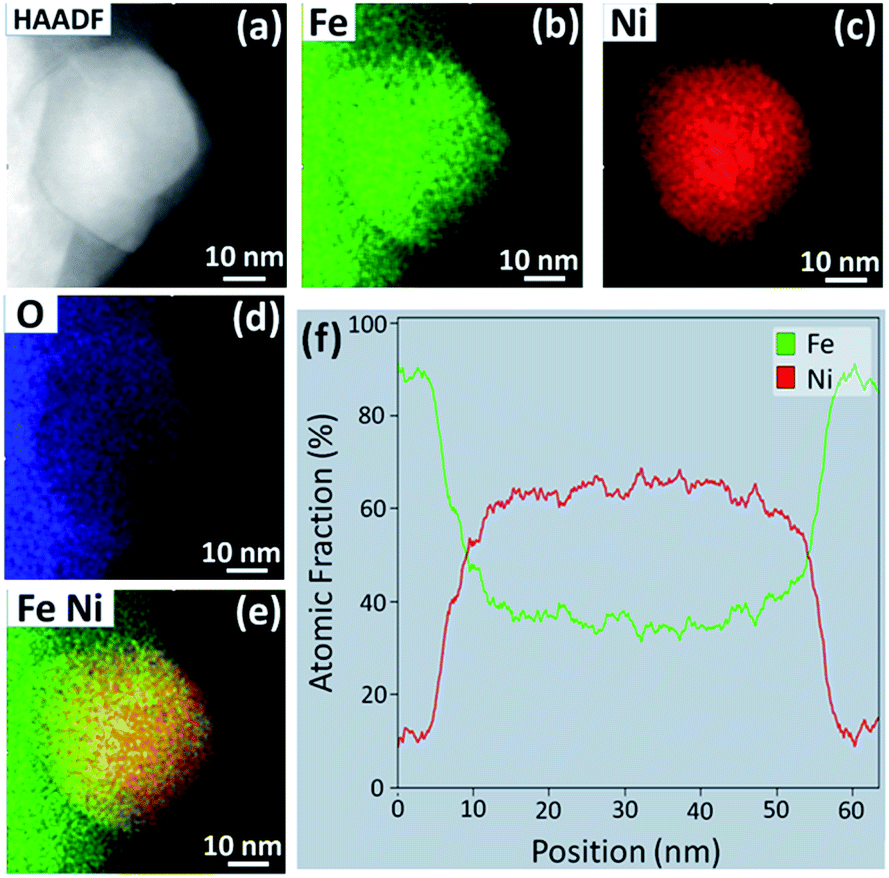 | ||
| Fig. 9 (a) HAADF STEM image of a NP exsolved upon reduction in 70:30 CO:CO2 for 25 h at 800 °C. EDS elemental maps of (b) Fe, (c) Ni, (d) O, and (e) Fe and Ni overlayed, for the NP in (a). (c) EDS line scan across the middle of the NP in (a). | ||
To summarize, we have found a clear indication of faster nucleation and growth kinetics in 5:95 H2:N2 than in all of the CO–CO2 environments employed in this work but the mechanism causing this difference is not clear at this time, although it can be argued that both pO2 and the nature of the reducing gas play a role in the exsolution kinetics and outcomes. An important conclusion reached in the previous discussion is that, once the NPs are formed in either gas environment, they are likely to remain stable when they are employed in CO2 reduction/CO oxidation catalysis. For the system in question, we suggest a short reduction treatment (1–2 h) in 5:95 H2:N2 at 800 °C for the fuel electrode to achieve a densely populated NP distribution with average sizes of sub-50 nm, before exposure to CO2–CO environments for CO2 electrolysis or CO oxidation studies.
3.4 Electrochemical performance of LCFCrN symmetrical cells in CO2–CO environments
To assess the electrochemical performance of Fe–Ni NP-decorated LCFCrN (Fe–Ni@LCFCrN), symmetrical cells were prepared on samaria-doped ceria (SDC)-buffered scandia-stabilized zirconia (ScSZ) electrolytes, with the buffer layer added to prevent reaction of La and Ca with YSZ.7,8 The fuel electrode was reduced in 5:95 H2:N2 at 800 °C for 2 h, as discussed above, to produce the NPs on the electrode surface, followed by exposure to various CO2:CO ratios, while the oxygen electrode was supplied with air.
Fig. 10(a) shows the cyclic voltammograms (CVs) acquired when the fuel electrode was supplied with pure CO2, 90:10 CO2:CO, 70:30 CO2:CO, and 50:50 CO2:CO, respectively. The measured open circuit voltages (OCV) for each gas mixture were 0.134 V, 0.847 V, 0.905 V, and 0.943 V, respectively, all at 800 °C, in agreement with predicted thermodynamic values.
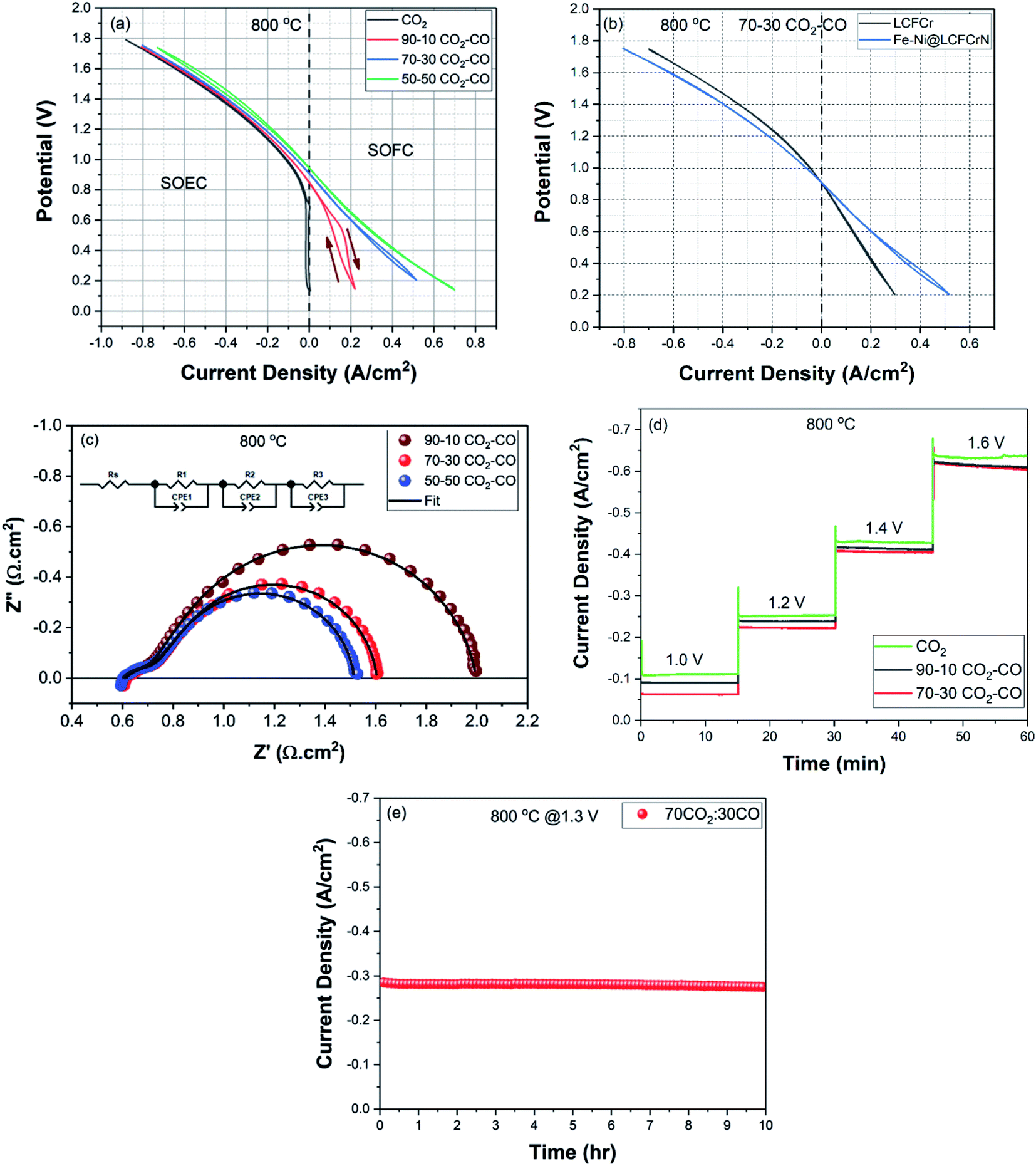 | ||
| Fig. 10 Electrochemical results for a symmetrical cell with the configuration Fe–Ni@LCFCrN/SDC/SSZ/SDC/LCFCrN. (a) CVs acquired at 800 °C (scan direction is indicated by arrows) in pure CO2, and in 90:10, 70:30, and 50:50 CO2:CO gas mixtures, (b) CVs showing the enhancement in current density vs. LCFCr in a 70:30 CO2:CO gas mixture due to the exsolved NPs, (c) Nyquist plots acquired at open circuit in 90:10, 70:30, and 50:50 CO2:CO gas mixtures, respectively, (d) 15 minute stability tests between 1.2–1.6 V for the electrolysis of pure CO2, and 90:10 and 70:30 CO2:CO gas mixtures, respectively, and (e) medium term stability test at a cell potential of 1.3 V in a 70:30 CO2:CO gas mixture. | ||
The CVs reveal the excellent activity of Fe–Ni@LCFCrN for the CO2-RR, with current densities of −0.649, −0.634, −0.618, and −0.583 A cm−2, respectively, at a cell voltage of 1.6 V. The current densities achieved in this work for the electrolysis of pure CO2 are comparable to or better than many other electrode materials reported in the literature (Table S2†), noting that we have not added a second phase, normally an ionic conductor, to the catalyst layer and are thus reporting the excellent activity of the mixed conducting LCFCrN material on its own. Interestingly, the CO2-RR current densities at a constant overpotential (Fig. 10(d)) are almost independent of the pCO2 of the gas feed. Although an in-depth study of this observation has not been carried out as yet, we speculate that this could be due to two possible reasons; (i) during CO2 electrolysis, a redox transition step, such as the reduction of Fe and Cr, is the slow step before CO2 adsorption and activation occurs, or (ii) the surface coverage of CO2 is saturated at a lower pCO2, hence making the CO2 reduction rate essentially independent of pCO2.
Fig. 10(a) also shows the excellent activity of Fe–Ni@LCFCrN in the SOFC mode (CO oxidation), especially as the CO content in the gas phase is increased. In 90:10 CO2:CO, the CV shows significant hysteresis in the SOFC mode, possibly due to reactant (CO) starvation at the fuel electrode. However, when the amount of CO fed to the fuel electrode is increased, the hysteresis progressively disappears and high current densities for CO oxidation are also achieved. For example, from the iR corrected CV in 50:50 CO2:CO shown in Fig. S5,† at an absolute current density of 0.4 A cm−2, the overpotential for CO2RR is 241 mV, while for CO oxidation, it is 273 mV, showing that the CO2RR is only slightly faster than CO oxidation under these conditions.
To reveal the impact of the exsolved Fe–Ni NPs, Fig. 10(b) compares the electrochemical response of symmetrical LCFCr and Fe–Ni@LCFCrN cells when the fuel electrodes were supplied with 70:30 CO2:CO and air was fed to the oxygen electrodes. It is evident that, while there is only a ca. 15% enhancement in current density for CO2-RR in Fe–Ni@LCFCrN vs. LCFCr at a cell potential of 1.6 V, the current density for CO oxidation shows a ca. 75% enhancement at 0.2 V, with these potentials being equivalent to a similar overpotential of ∼0.7 V. These results are very important, especially from the point of view of developing electrode materials for symmetrical SOCs. Generally, in perovskite-based fuel electrodes, the CO2-RR is significantly more efficient than is CO oxidation,9,10,45 which limits the applicability of these materials for RSOC applications. However, in the present work, the Fe–Ni NPs in Fe–Ni@LCFCrN greatly increase the kinetics of the CO oxidation reaction and make the material essentially equally active for both CO2-RR and CO oxidation, thus significantly improving the material for use in RSOC applications.
Our results are contrary to what was observed by Liu et al., who used La0.6Sr0.4Fe0.8Ni0.2O3−δ as the fuel electrode. Their cells exhibited a greater enhancement in CO2-RR activity (ca. 35%) when tested at 850 °C due to exsolved Fe–Ni alloy NPs, while the CO oxidation activity showed a significantly lower (ca. 20%) extent of improvement.9 It should also be kept in mind that the fuel electrodes in Liu et al.'s work and those in most published literature are composite electrodes, where the active material is mixed with an ion-conducting material, such as gadolinia- or samaria-doped ceria.9,22,24,46 Similarly, their air electrode consisted of LSCF or LSM composite electrodes containing doped cerias or YSZ.9,22,24,46,47 It is well known that the addition of an ion-conducting phase increases the TPB length that, in turn, enhances the catalytic activity. In our case, the material for both the fuel and the oxygen electrodes was intentionally used as a single phase material, and as has been shown previously, the parent perovskite (LCFCr) is also highly active for the oxygen reduction (ORR) and oxygen evolution (OER) reactions.6,8 Importantly, as our electrodes are not composited with doped-cerias, this means that the electrochemical data can be interpreted in a straightforward manner and any enhancement in activity can be unambiguously ascribed to the presence of the exsolved NPs.
The impedance spectra of the Fe–Ni@LCFCrN full cell at OCP in 90:10, 70:30, and 50:50 CO2:CO gas mixtures are presented in Fig. 10(c). The corresponding resistance and capacitance values based on a best-fit to an equivalent circuit model (ECM) [Rs(R1//CPE1)(R2//CPE2)(R3//CPE3)] are given in Table 1. The series resistance, Rs, was found to be ∼0.60 Ω cm2, invariant with the CO2:CO ratio, as expected. The Rp in 90:10 CO2:CO was 1.39 Ω cm2, which decreased to 1.0 Ω cm2 when the gas composition was changed to 70:30 CO2:CO, and to 0.93 Ω cm2 when the gas composition was changed to 50:50 CO2:CO.
| Gas composition | R 1 (Ω cm2) | C 1 (F cm−2) | n 1 | R 2 (Ω cm2) | C 2 (F cm−2) | n 2 | R 3 (Ω cm2) | C 3 (F cm−2) | n 3 | R p (Ω cm−2) |
|---|---|---|---|---|---|---|---|---|---|---|
| 90% CO2–10% CO | 0.15 | 1.0 × 10−2 | 0.52 | 0.06 | 5.2 ×10−1 | 0.88 | 1.18 | 2.3 × 10−1 | 0.91 | 1.39 |
| 70% CO2–30% CO | 0.16 | 8.5 × 10−3 | 0.53 | 0.06 | 5.2 × 10−1 | 0.88 | 0.78 | 3.0 × 10−1 | 0.94 | 1.0 |
| 50% CO2–50% CO | 0.16 | 8.3 × 10−3 | 0.53 | 0.06 | 5.2 × 10−1 | 0.88 | 0.71 | 4.0 × 10−1 | 0.94 | 0.93 |
The Rp value obtained in two-electrode studies represents the electrochemical processes occurring at both electrodes. Table 1 shows that R1 and R2 (high and medium frequencies, respectively) seem to be independent of the gas composition supplied at the fuel electrode, possibly attributable to underlying interfaces but perhaps more likely to processes occurring at the oxygen electrode. In order to clarify this, half-cell impedance spectra were acquired in air, which yielded a relatively low Rp of 0.46 Ω cm2 (0.23 Ω cm2 per electrode). Comparison of the time constants obtained from equivalent circuit fitting for the half cell in air with those observed at the full cell suggests that R2 (0.06 Ω cm2) in Table 1 arises primarily from the oxygen electrode. In terms of the high frequency R1 (ca. 0.15–0.16 Ω cm2) (Table 1) for the full cell, our results suggest that it is due to the combination of processes occurring within the porous structures of both electrodes.
The comparison of the EIS-determined R values in the full cell versus in an air half-cell shows that LCFCrN, like its undoped analogue (LCFCr), is a still better catalyst for the oxygen reduction and evolution reactions (ORR and OER)6,8 than for CO2 reduction/CO oxidation, and that the processes occurring at the fuel electrode make the dominant contribution to the full cell Rp in Fig. 10(c). Also, the decrease in Rp with increasing amounts of CO in Fig. 10(c) means that CO oxidation dominates the low frequency response at these open circuit conditions.
Table 1 also shows the capacitance values measured in our full cells, revealing that the low frequency capacitor (C3) is very large, with an associated n3 value close to 1. As this arc is very responsive to the CO2:CO ratio, it is attributed either to gas phase mass transport limitations of CO (which is present in lower concentrations, at least in 90:10 and 70:30 CO2:CO gas mixtures) or to the chemical capacitance of the LCFCrN electrode in the CO2–CO environment,48–51 with a small increase in C3 noted as the CO content of the gas increases. As stated above, C2 likely has similar origins but is generated at the air electrode, as it is independent of the CO2:CO ratio. C1 and its characteristic n1 value of ca. 0.5 suggests a Gerischer element, arising from a distribution of potentials within both of the porous electrodes under high frequency perturbations.52,53
Fig. 10(d) shows the results of 15 min potentiostatic stability tests for the CO2-RR reaction (and oxygen evolution) at a cell potential in the range of 1.0–1.6 V when the fuel electrode was exposed to pure CO2 and 90:10 and 70:30 CO2:CO gas mixtures. It is seen that the current densities are stable in pure CO2 at all voltages, while the current densities for the 90:10, and 70:30 CO2:CO gas mixtures are also stable up to a cell voltage of 1.4 V, with a slight degradation seen only at the relatively high voltage of 1.6 V. However, this drop in current density can be rapidly reversed by retracting the voltage and running the cell at the OCP for a few minutes.
Liu et al. attributed a similar drop in current density for La0.6Sr0.4Fe0.8Ni0.2O3−δ electrodes at high cell voltages to concentration polarization arising from starvation of CO2 in the gas mixture and/or restrained oxygen evolution at the air electrode,9 while Optiz et al. attributed it to coke formation by CO disproportionation or electrochemical reduction.54 As our cells can be so easily re-activated, it seems more likely that this is due to mass transport limitations in our case.
Chronoamperometry was also carried out to examine the medium-term stability of the Fe–Ni@LCFCrN cells in a 70:30 CO2:CO atmosphere. Fig. 10(e) shows that at a cell potential of 1.3 V, the cell exhibits a very stable current density of ca. 0.28 A cm−2 for 10 h for the electrolysis of CO2, without appreciable degradation.
Although the growth and phase evolution of exsolved Fe–Ni alloy NPs presented herein focus on one system, namely LCFCrN, we believe that this could be generalized to other Fe and Ni containing perovskites. This could apply especially to perovskites with abundant Fe and low Ni doping levels on the B-site, such as SrFe1.35Mo0.45Ni0.2O6−δ,24 La0.6Sr0.4Fe0.8Ni0.2O3−δ,9 La0.6Ca0.4Fe0.8Ni0.2O3−δ,26 (La0.75Sr0.25)(Cr0.5Fe0.5−xNix)O3,55etc. The insights presented in this paper should also be very helpful in controlling the NP characteristics, such as size, areal density, and composition, to enable stable and durable electrochemical performance of catalyst systems with exsolved NPs.
4. Conclusions
Ni-doped La0.3Ca0.7Fe0.7Cr0.3O3−δ (LCFCrN) is a robust material system in which a uniform dispersion of metal nanoparticles can be exsolved onto the parent perovskite surface via a short reduction treatment under typical SOC operating temperatures (750–800 °C). The nanoparticle size, composition and areal density can be tuned by controlling the reduction treatment duration and temperature in a particular reducing atmosphere. Although XRD reveals that CaO also forms as a by-product of exsolution, its formation can be mitigated by using a lower growth temperature, short reduction duration, or reducing in CO–CO2 environments (lower pO2). Also, the NPs produced in the CO–CO2 atmospheres are composed of Ni-rich Fe–Ni alloys, while those produced in 5:95 H2:N2 quickly transition from the composition of FeNi3 to Fe0.64Ni0.36. However, the nucleation and growth kinetics are slower in CO–CO2 atmospheres as compared to those in H2–N2. This makes the LCFCrN system a promising fuel electrode material for RSOC applications as the NP coarsening can be restrained during exposure to CO–CO2 atmospheres under typical SOC conditions.
Overall, Fe–Ni@LCFCrN exhibits a remarkable electrochemical performance, even without the addition of an ionically conducting phase, such as GDC. Short-term stability tests reveal a very high and stable performance with a maximum current density of 0.65 A cm−2 at a cell voltage of 1.6 V in pure CO2 at 800 °C. As compared to the parent perovskite LCFCr, the exsolved NPs in Fe–Ni@LCFCrN greatly enhance the CO oxidation kinetics (by ca. 75%) more notably than the CO2-RR kinetics (ca. 15%), which makes the exsolved catalyst equally active for both CO2-RR and CO oxidation, showing great promise for future use as an SOFC anode as well as in RSOCs.
Author contributions
Haris Masood Ansari: conceptualization, formal analysis, investigation, writing – original draft. Adam Stuart Bass: investigation, formal analysis, writing – review & editing. Nabeel Ahmad: investigation, formal analysis, writing – review & editing. Viola I. Birss: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Canada First Research Excellence Fund (CFREF) and Alberta Innovates-Technology Futures (AITF, Grant No. G2016000655) for the support of this work. A. S. B. would like to acknowledge the Program for Undergraduate Research Experience (PURE) for the award to support his work. We would also like to acknowledge Mykhailo Pidburtnyi for helpful discussions related to the interpretation of the electrochemical data.References
- M. S. Khan, S.-B. Lee, R.-H. Song, J.-W. Lee, T.-H. Lim and S.-J. Park, Fundamental Mechanisms Involved in the Degradation of Nickel-Yttria Stabilized Zirconia (Ni-YSZ) Anode During Solid Oxide Fuel Cells Operation: A Review, Ceram. Int., 2016, 42, 35–48 CrossRef CAS.
- J. Masek, B. Novosel and M. Marinsek, Ni-YSZ SOFC Anodes-Minimization of Carbon Deposition, J. Eur. Ceram. Soc., 2007, 27, 487–491 CrossRef.
- S. P. Jiang, X. J. Chen, S. H. Chan, J. T. Kwok and K. A. Khor, (La0.75Sr0.25)(Cr0.5Mn0.5)O3/YSZ Composite Anodes for Methane Oxidation Reaction in Solid Oxide Fuel Cells, Solid State Ionics, 2006, 177, 149–157 CrossRef CAS.
- X. Yue and J. T. S. Irvine, Alternative Cathode Material for Co2 Reduction by High Temperature Solid Oxide Electrolysis Cells, J. Electrochem. Soc., 2012, 159, F442–F448 CrossRef CAS.
- Y. Song, Z. Zhou, X. Zhang, Y. Zhou, H. Gong, H. Lv, Q. Liu, G. Wang and X. Bao, Pure Co2 Electrolysis over an Ni/YSZ Cathode in a Solid Oxide Electrolysis Cell, J. Mater. Chem. A, 2018, 6, 13661–13667 RSC.
- B. Molero-Sanchez, J. Prado-Gonjal, D. Avila-Brande, M. Chen, E. Moran and V. Birss, High Performance La0.3Ca0.7Cr0.3Fe0.7O3−δ Air Electrode for Reversible Solid Oxide Fuel Cell Applications, Int. J. Hydrogen Energy, 2015, 40, 1902–1910 CrossRef CAS.
- P. K. Addo, B. Molero-Sanchez, M. Chen, S. Paulson and V. Birss, CO/CO2 Study of High Performance La0.3Sr0.7Fe0.7Cr0.3O3−δ Reversible SOFC Electrodes, Fuel Cells, 2015, 15, 689–696 CrossRef CAS.
- B. Molero-Sanchez, P. Addo, A. Buyukaksoy, S. Paulson and V. Birss, Electrochemistry of La0.3Sr0.7Fe0.7Cr0.3O3−δ as an Oxygen and Fuel Electrode for RSOFCs, Faraday Discuss., 2015, 182, 159–175 RSC.
- S. Liu, Q. Liu and J.-L. Luo, Highly Stable and Efficient Catalyst with in Situ Exsolved Fe-Ni Alloy Nanospheres Socketed on an Oxygen Deficient Perovskite for Direct CO2 Electrolysis, ACS Catal., 2016, 6, 6219–6228 CrossRef CAS.
- S. Park, Y. Kim, H. Han, Y. S. Chung, W. Yoon, J. Choi and W. B. Kim, In Situ Exsolved Co Nanoparticles on Ruddlesden-Popper Material as Highly Active Catalyst for CO2 Electrolysis to CO, Appl. Catal., B, 2019, 248, 147–156 CrossRef CAS.
- K. Chen, N. Ai and S. P. Jiang, Enhanced Electrochemical Performance and Stability of (La,Sr)MnO3-(Gd,Ce)O2 Oxygen Electrodes of Solid Oxide Electrolysis Cells by Palladium Infiltration, Int. J. Hydrogen Energy, 2012, 37, 1301–1310 CrossRef CAS.
- K. Chen, N. Ai and S. P. Jiang, Performance and Structural Stability of Gd0.2Ce0.8O1.9 Infiltrated La0.8Sr0.2MnO3 Nano-Structured Oxygen Electrodes of Solid Oxide Electrolysis Cells, Int. J. Hydrogen Energy, 2014, 39, 10349–10358 CrossRef CAS.
- Y.-F. Sun, Y.-Q. Zhang, J. Chen, J.-H. Li, Y.-T. Zhu, Y.-M. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J.-L. Luo, New Opportunity for in Situ Exsolution of Metallic Nanoparticles on Perovskite Parent, Nano Lett., 2016, 16, 5303–5309 CrossRef CAS.
- Y. Li, Y. Wang, W. Doherty, K. Xie and Y. Wu, Perovskite Chromates Cathode with Exsolved Iron Nanoparticles for Direct High-Temperature Steam Electrolysis, ACS Appl. Mater. Interfaces, 2013, 5, 8553–8562 CrossRef CAS.
- G. Yang, W. Zhou, M. Liu and Z. Shao, Enhancing Electrode Performance by Exsolved Nanoparticles: A Superior Cobalt-Free Perovskite Electrocatalyst for Solid Oxide Fuel Cells, ACS Appl. Mater. Interfaces, 2016, 8, 35308–35314 CrossRef CAS.
- T. Z. Sholklapper, C. P. Jacobson, S. J. Visco and L. C. De Jonghe, Synthesis of Dispersed and Contiguous Nanoparticles in Solid Oxide Fuel Cell Electrodes, Fuel Cells, 2008, 8, 303–312 CrossRef CAS.
- D. Neagu, T.-S. Oh, D. N. Miller, H. Menard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nano-Socketed Nickel Particles with Enhanced Coking Resistance Grown in Situ by Redox Exsolution, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y.-W. Ju, J. W. Han and G. Kim, Exsolution Trends and Co-Segregation Aspects of Self-Grown Catalyst Nanoparticles in Perovskites, Nat. Commun., 2017, 8, 15967 CrossRef CAS PubMed.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, Perovskite Oxides: Preparation, Characterizations, and Applications in Heterogeneous Catalysis, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- G. Tsekouras, D. Neagu and J. T. S. Irvine, Step-Change in High Temperature Steam Electrolysis Performance of Perovskite Oxide Cathodes with Exsolution of B-Site Dopants, Energy Environ. Sci., 2013, 6, 256–266 RSC.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Menard and J. T. S. Irvine, In Situ Growth of Nanoparticles through Control of Non-Stoichiometry, Nat. Chem., 2013, 5, 916 CrossRef CAS PubMed.
- L. Ye, M. Zhang, P. Huang, G. Guo, M. Hong, C. Li, J. T. S. Irvine and K. Xie, Enhancing CO2 Electrolysis through Synergistic Control of Non-Stoichiometry and Doping to Tune Cathode Surface Structures, Nat. Commun., 2017, 8, 14785 CrossRef CAS PubMed.
- Y. Zhu, J. Dai, W. Zhou, Y. Zhong, H. Wang and Z. Shao, Synergistically Enhanced Hydrogen Evolution Electrocatalysis by in Situ Exsolution of Metallic Nanoparticles on Perovskites, J. Mater. Chem. A, 2018, 6, 13582–13587 RSC.
- H. Lv, L. Lin, X. Zhang, D. Gao, Y. Song, Y. Zhou, Q. Liu, G. Wang and X. Bao, In Situ Exsolved FeNi3 Nanoparticles on Nickel Doped Sr2Fe1.5Mo0.5O6−δ Perovskite for Efficient Electrochemical CO2 Reduction Reaction, J. Mater. Chem. A, 2019, 7, 11967–11975 RSC.
- M. Chanthanumataporn, J. Hui, X. Yue, K. Kakinuma, J. T. S. Irvine and K. Hanamura, Electrical Reduction of Perovskite Electrodes for Accelerating Exsolution of Nanoparticles, Electrochim. Acta, 2019, 306, 159–166 CrossRef CAS.
- Y. Tian, Y. Liu, A. Naden, L. Jia, M. Xu, W. Cui, B. Chi, J. Pu, J. T. S. Irvine and J. Li, Boosting CO2 Electrolysis Performance Via Calcium-Oxide-Looping Combined with in Situ Exsolved Ni-Fe Nanoparticles in a Symmetrical Solid Oxide Electrolysis Cell, J. Mater. Chem. A, 2020, 8, 14895–14899 RSC.
- K.-Y. Lai and A. Manthiram, Self-Regenerating Co-Fe Nanoparticles on Perovskite Oxides as a Hydrocarbon Fuel Oxidation Catalyst in Solid Oxide Fuel Cells, Chem. Mater., 2018, 30, 2515–2525 CrossRef CAS.
- Y.-F. Sun, J.-H. Li, Y.-Q. Zhang, B. Hua and J.-L. Luo, Bifunctional Catalyst of Core-Shell Nanoparticles Socketed on Oxygen-Deficient Layered Perovskite for Soot Combustion: In Situ Observation of Synergistic Dual Active Sites, ACS Catal., 2016, 6, 2710–2714 CrossRef CAS.
- J. Xiao, Q. Xu, D.-P. Huang, M. Chen, K. Zhao and B.-H. Kim, Evaluation of La0.3Ca0.7Fe1−YCryO3−δ (Y=0.1-0.3) Cathodes for Intermediate Temperature Solid Oxide Fuel Cells, Mater. Res. Bull., 2017, 90, 104–110 CrossRef CAS.
- M. F. Lu, E. V. Tsipis, J. C. Waerenborgh, A. A. Yaremchenko, V. A. Kolotygin, S. Bredikhin and V. V. Kharton, Thermomechanical, Transport and Anodic Properties of Perovskite-Type (La0.75Sr0.25)0.95Cr1−XFexO3−δ, J. Power Sources, 2012, 206, 59–69 CrossRef.
- E. Povoden, M. Chen, A. N. Grundy, T. Ivas and L. J. Gauckler, Thermodynamic Assessment of the La-Cr-O System, J. Phase Equilib. Diff., 2009, 30, 12–27 CrossRef CAS.
- E. Povoden-Karadeniz, A. N. Grundy, M. Chen, T. Ivas and L. J. Gauckler, Thermodynamic Assessment of the La-Fe-O System, J. Phase Equilib. Diff., 2009, 30, 351–366 CrossRef CAS.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, M. Shah, L. D. Marks and S. A. Barnett, Nickel- and Ruthenium-Doped Lanthanum Chromite Anodes: Effects of Nanoscale Metal Precipitation on Solid Oxide Fuel Cell Performance, J. Electrochem. Soc., 2010, 157, B279–B284 CrossRef CAS.
- S.-H. Cui, J.-H. Li, X.-W. Zhou, G.-Y. Wang, J.-L. Luo, K. T. Chuang, Y. Bai and L.-J. Qiao, Cobalt Doped LaSrTiO3−δ as an Anode Catalyst: Effect of Co Nanoparticle Precipitation on SOFCs Operating on H2S-Containing Hydrogen, J. Mater. Chem. A, 2013, 1, 9689–9696 RSC.
- J.-H. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Switching on Electrocatalytic Activity in Solid Oxide Cells, Nature, 2016, 537, 528 CrossRef CAS PubMed.
- Y. Gao, Z. Lu, T. L. You, J. Wang, L. Xie, J. He and F. Ciucci, Energetics of Nanoparticle Exsolution from Perovskite Oxides, J. Phys. Chem. Lett., 2018, 9, 3772–3778 CrossRef CAS PubMed.
- J. M. Haag, S. A. Barnett, J. W. Richardson and K. R. Poeppelmeier, Structural and Chemical Evolution of the SOFC Anode La0.30Sr0.70Fe0.70Cr0.30O3−δ Upon Reduction and Oxidation: An in Situ Neutron Diffraction Study, Chem. Mater., 2010, 22, 3283–3289 CrossRef CAS.
- T.-S. Oh, E. K. Rahani, D. Neagu, J. T. S. Irvine, V. B. Shenoy, R. J. Gorte and J. M. Vohs, Evidence and Model for Strain-Driven Release of Metal Nanocatalysts from Perovskites During Exsolution, J. Phys. Chem. Lett., 2015, 6, 5106–5110 CrossRef CAS.
- D. Neagu, V. Kyriakou, I.-L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, In Situ Observation of Nanoparticle Exsolution from Perovskite Oxides: From Atomic Scale Mechanistic Insight to Nanostructure Tailoring, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, From Material Design to Mechanism Study: Nanoscale Ni Exsolution on a Highly Active A-Site Deficient Anode Material for Solid Oxide Fuel Cells, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- D. Neagu, E. I. Papaioannou, W. K. W. Ramli, D. N. Miller, B. J. Murdoch, H. Menard, A. Umar, A. J. Barlow, P. J. Cumpson, J. T. S. Irvine and I. S. Metcalfe, Demonstration of Chemistry at a Point through Restructuring and Catalytic Activation at Anchored Nanoparticles, Nat. Commun., 2017, 8, 1855 CrossRef PubMed.
- R. Thalinger, M. Gocyla, M. Heggen, B. Klotzer and S. Penner, Exsolution of Fe and SrO Nanorods and Nanoparticles from Lanthanum Strontium Ferrite La0.6Sr0.4FeO3−δ Materials by Hydrogen Reduction, J. Phys. Chem. C, 2015, 119, 22050–22056 CrossRef CAS.
- H. Lv, L. Lin, X. Zhang, Y. Song, H. Matsumoto, C. Zeng, N. Ta, W. Liu, D. Gao, G. Wang and X. Bao, In Situ Investigation of Reversible Exsolution/Dissolution of CoFe Alloy Nanoparticles in a Co-Doped Sr2Fe1.5Mo0.5O6−δ Cathode for CO2 Electrolysis, Adv. Mater., 2020, 32, 1906193 CrossRef CAS PubMed.
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Ni-Substituted Sr(Ti,Fe)O3 SOFC Anodes: Achieving High Performance Via Metal Alloy Nanoparticle Exsolution, Joule, 2018, 2, 478–496 CrossRef CAS.
- P. K. Addo, B. Molero-Sanchez, A. Buyukaksoy, S. Paulson and V. Birss, Sulfur Tolerance of La0.3M0.7Fe0.7Cr0.3O3−δ (M = Sr, Ca) Solid Oxide Fuel Cell Anodes, ECS Trans., 2015, 66, 219–228 CrossRef CAS.
- S. Liu, Q. Liu and J.-L. Luo, The Excellence of La(Sr)Fe(Ni)O3 as an Active and Efficient Cathode for Direct CO2 Electrochemical Reduction at Elevated Temperatures, J. Mater. Chem. A, 2017, 5, 2673–2680 RSC.
- Y. Zhou, Z. Zhou, Y. Song, X. Zhang, F. Guan, H. Lv, Q. Liu, S. Miao, G. Wang and X. Bao, Enhancing CO2 Electrolysis Performance with Vanadium-Doped Perovskite Cathode in Solid Oxide Electrolysis Cell, Nano Energy, 2018, 50, 43–51 CrossRef CAS.
- S. Primdahl and M. Mogensen, Gas Conversion Impedance: A Test Geometry Effect in Characterization of Solid Oxide Fuel Cell Anodes, J. Electrochem. Soc., 1998, 145, 2431–2438 CrossRef CAS.
- S. Primdahl and M. Mogensen, Gas Diffusion Impedance in Characterization of Solid Oxide Fuel Cell Anodes, J. Electrochem. Soc., 1999, 146, 2827–2833 CrossRef CAS.
- S. B. Adler, Factors Governing Oxygen Reduction in Solid Oxide Fuel Cell Cathodes, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- Q. X. Fu, F. Tietz and D. Stover, La0.4Sr0.6Ti1−XMnxO3−δ Perovskites as Anode Materials for Solid Oxide Fuel Cells, J. Electrochem. Soc., 2006, 153, D74 CrossRef CAS.
- B. A. Boukamp and H. J. M. Bouwmeester, Interpretation of the Gerischer Impedance in Solid State Ionics, Solid State Ionics, 2003, 157, 29–33 CrossRef CAS.
- X. Yue and J. T. S. Irvine, Impedance Studies on LSCM/GDC Cathode for High Temperature CO2 Electrolysis, Electrochem. Solid-State Lett., 2012, 15, B31 CrossRef CAS.
- A. K. Opitz, A. Nenning, C. Rameshan, M. Kubicek, T. Gotsch, R. Blume, M. Havecker, A. Knop-Gericke, G. Rupprechter, B. Klotzer and J. Fleig, Surface Chemistry of Perovskite-Type Electrodes During High Temperature CO2 Electrolysis Investigated by Operando Photoelectron Spectroscopy, ACS Appl. Mater. Interfaces, 2017, 9, 35847–35860 CrossRef CAS PubMed.
- D. Papargyriou, D. N. Miller and J. T. S. Irvine, Exsolution of Fe-Ni Alloy Nanoparticles from (La,Sr)(Cr,Fe,Ni)O3 Perovskites as Potential Oxygen Transport Membrane Catalysts for Methane Reforming, J. Mater. Chem. A, 2019, 7, 15812–15822 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07552g |
| This journal is © The Royal Society of Chemistry 2022 |