
Fabrication of a tunable photothermal actuator via in situ oxidative polymerization of polydopamine nanoparticles in hydrogel bilayers†
Chaewon
Lee
a,
Jin Hyeok
Park
b,
Mina
Kim‡
a,
Jong Sik
Kim
 a and
Tae Soup
Shim
*ab
a and
Tae Soup
Shim
*ab
aDepartment of Energy Systems Research, Ajou University, Suwon 16499, Republic of Korea. E-mail: tsshim@ajou.ac.kr
bDepartment of Chemical Engineering, Ajou University, Suwon 16499, Republic of Korea
First published on 7th June 2022
Abstract
Photothermally triggered actuation enables the remote and local control of a material. The complex actuation can be achieved by controlling the photothermal efficiency of the material, which is crucial for the development of soft actuators. In this study, the photothermal efficiency of a hydrogel bilayer actuator consisting of a passive agarose/alginate double-network hydrogel layer and an active poly(N-isopropylacrylamide) (PNIPAm) layer was controlled via in situ oxidative polymerization of polydopamine nanoparticles (PDA NPs). Highly concentrated PDA NPs were successfully incorporated into the hydrogel bilayer without interrupting or weakening the polymer network during polymerization. The photothermal efficiency of the actuator was controlled using the number of polymerization cycles. Upon light irradiation, the heat generated by the photothermal effect of PDA NPs caused the shrinkage of the PNIPAm layer, resulting in the shape-morphing of the bilayer. The broad light absorption properties of PDA NPs allowed the bilayer to actuate under sunlight or visible light. Finally, we demonstrated controlled photothermal actuation using a pinwheel-shaped actuator consisting of four panels with different photothermal efficiencies.
Introduction
A soft actuator is a physicochemically powered robot inspired by the actuation mechanism in plants. Mimicking the anisotropic volume phase transition of multilayered cellular structures in natural plants, such as the leaves of a Venus flytrap,1 the seed capsule of an ice plant,2 or a pine cone,3 stimuli-responsive actuators have been actively developed since the early 2000s.4 Stimuli-responsive materials that can change their shape in response to specific environmental conditions, such as temperature,5 humidity,6 pressure,7 pH,8 chemical gradients,9 DNA,10 and light,11,12 have been designed to perform programmed mechanical actuation. Among the various stimuli mentioned above, light is a distinctive stimulus for the remote and selective control of actuators. While other physical stimuli, such as temperature, humidity, and pressure, are applied isotropically to the global system, light can be applied locally by controlling the direction, shape, intensity, wavelength, and polarization of light sources. As a result, light can be used for the remotely controlled local actuation of a soft actuator.Light-responsive actuators have been developed by introducing either light-responsive polymers (e.g., azopolymers13,14 or azocompound-functionalized liquid crystal elastomers)15 or temperature-responsive polymers (e.g., hydrogels16 and liquid crystal elastomers17) into photothermal materials that can convert light into thermal energy through the photothermal effect. In the case of an azopolymer-based actuator, the actuation condition is solely dependent on the intrinsic photoisomerization property of the azocompound. In the case of photothermally controlled actuators or photothermal actuators, a versatile design has been proposed that combines photothermal materials and temperature-responsive polymers. For example, liquid crystal elastomer-based and hydrogel-based actuators have been designed for photothermal actuation by introducing plasmonic gold nanoparticles18,19 or light-absorbing materials, such as dyes,20 carbon nanotubes,21 graphene oxide,22,23 and magnetic nanoparticles,11 which enable remotely and locally controlled actuation. In photothermal actuation, the degree of actuation can be controlled using the photothermal efficiency of photothermal materials because the rate of photothermal energy conversion determines the temperature of the actuator. Therefore, the control of photothermal materials in a polymer matrix affects the photothermal actuation performance.
Photothermal actuators were prepared by mixing photothermal materials with a prepolymer solution followed by polymerization. In such cases, the increase in the concentration of photothermal materials may cause nonuniform dispersion, which hinders the formation of homogeneous polymeric networks after polymerization, leading to the deterioration of mechanical properties. The fabrication of hydrogel actuators is more challenging owing to the poor dispersion stability of photothermal materials in water. In addition, the micropatterning of an actuator through photopolymerization is limited because the high light absorption ability of the photothermal material hinders the photolysis of the photoinitiator. Therefore, the development of a photothermal actuator that can control its photothermal characteristics while maintaining its mechanical properties is required.
Herein, we address the issues via the control of the concentration of photothermal materials in a hydrogel bilayer through an in-situ polymerization strategy. We chose polydopamine nanoparticles (PDA NPs) as a photothermal material because of their excellent light absorption in the ultraviolet (UV) to the near-infrared range, good water dispersibility, and biocompatibility. PDA is a material having a similar chemical structure to natural melanin widely observed in nature. Although the synthesis mechanism and the structure of PDA have not been fully elucidated,24 research on its application is being conducted in many fields due to the ease of coating on the surface of various materials and the easy synthesis of nanoparticles in aqueous media. Similar to how natural melanin is utilized for colorization of a living creature,25 antioxidant,26 protection of living organisms through absorption of harmful UV rays,27 and thermoregulation28 due to its excellent optical properties, PDA has been applied to develop coloration material,29,30 UV shielding,31 antioxidant,32 and photothermal agent for biomedical application.33 These examples show the potential of PDA as a photonic application.
To fabricate the photothermal actuator, a neat hydrogel bilayer consisting of a passive agarose/alginate double-network hydrogel (DN gel) layer, and an active temperature-responsive poly(N-isopropylacrylamide) (PNIPAm) layer was prepared prior to incorporate the PDA NPs. For the passive layer, agarose/alginate DN gel was selected as a material with high thermal and mechanical stability to cope with repeated mechanical deformation by the photothermal effect. Recently, due to the high elasticity and elasticity of DN gel, it has been applied to many flexible materials.34 In addition, the agarose/alginate DN gel used in this study shows good thermal stability that can overcome the thermo-gelation characteristic of a neat agarose gel at high temperatures (Fig. S1, ESI†). Subsequently, PDA NPs were incorporated into the hydrogel matrix via in situ oxidative polymerization. The photothermal efficiency and thereby the equilibrium temperature of the hydrogel was controlled according to the concentration of PDA NPs in the hydrogel matrix. The broadband light absorption property of PDA NPs enabled the continuous light-driven local control of hydrogel grippers. Finally, the tunable photothermal actuation of a pinwheel actuator was demonstrated by designing a pinwheel with four panels of hydrogel bilayers having different PDA NP concentrations.
Results and discussion
The broad absorption spectrum of agarose/alginate DN gels can be engineered via the in situ oxidative polymerization of PDA NPs in a hydrogel matrix. The comprehensive process for the preparation of PDA NP-incorporated agarose/alginate DN gels is illustrated in Fig. 1a. Briefly, a thin film of agarose gel consisting of 2 wt% agarose in water was prepared via physical gelation. The film was immersed in a 0.5 wt% alginate precursor solution for 3 h, followed by immersion in an aqueous 5 wt% calcium chloride solution for another 3 h to complete the ionic polymerization of alginate within the agarose matrix. The formation of interpenetrated networks between the physical agarose gel and ionic alginate gel has a synergistic effect that enhances the mechanical strength (i.e., stiffness and elastic modulus),35 enabling the application of these materials to soft actuators. The as-prepared agarose/alginate DN gel was then alternately immersed in a 10 wt% dopamine hydrochloride solution and 10 mM NaOH solution for the polymerization of PDA NPs in the hydrogel matrix. PDA NPs synthesized through the in situ polymerization formed uniform particles with a size of about 120 nm as in the batch polymerization method (Fig. S2, ESI†). In the in situ oxidative polymerization of PDA NPs, the rate-determining factor can be considered as the diffusion rate of the PDA NP precursor and alkali ions through the porous gel network. Because PDA NP polymerization occurs within the gel, it can be considered that the larger the gel network, the faster the diffusion; hence, the PDA NP polymerization process becomes more efficient. Although the agarose/alginate DN gel has a smaller pore size than the neat agarose gel, as shown in Fig. 1b, no difference was observed in the in situ polymerization of PDA NP (Fig. S3, ESI†). This is because both hydrogels have sufficiently large pores for the diffusion of dopamine precursors and alkali ions. | ||
| Fig. 1 (a) Schematic of the fabrication of polydopamine nanoparticle (PDA NP)-incorporated agarose/alginate double-network hydrogel (DN gel). (b) Scanning electron microscopy images of freeze-dried 2 wt% agarose gel (left) and agarose/alginate DN gel (right). (c) Optical microscopy images of PDA NP-incorporated agarose/alginate DN gels with various PDA NP concentrations via in situ oxidative polymerization cycles. The number of polymerization cycles of each sample including the control sample was indicated at the bottom of the image. (d) Transmittance spectra of PDA NP-incorporated agarose/alginate DN gels presented in (c). (e) Transmittance peaks at 550 nm of PDA NP-incorporated agarose/alginate DN gels presented in (c). (f) Concentration-dependent absorption intensity at 550 nm of the aqueous PDA NP dispersion. Linear regression equation of the absorption data is A = 1.65 × 10−2CPdNP,dispersion + 5.45 × 10−3. (g) Calculation of the PDA NP concentration in agarose/alginate DN gels presented in (c) based on the extrapolated linear regression line in (f). | ||
The concentration of PDA NPs and thereby the light absorption properties of the agarose/alginate DN gel were controlled through repeated polymerization cycles. The optical microscope image in Fig. 1c shows that the color of the agarose/alginate DN gel darkened with increasing PDA NP concentration. The qualitative analysis of light absorption was conducted by measuring the transmittance spectrum of a 1 mm thick agarose/alginate DN gel. Fig. 1d shows that the transmittance of the agarose/alginate DN gel significantly decreased during the initial dipping cycle, followed by a gradual decrease. After eight polymerization cycles, the transmittance of the agarose/alginate DN gel fall below the detection limit. The change in transmittance at a fixed wavelength of 550 nm with respect to the number of dipping cycles is also confirmed in Fig. 1e, which shows the precise control of the light absorption property through the repeated in situ oxidative polymerization of PDA NPs is possible.
Further analysis was performed to determine the concentration of PDA NPs in the agarose/alginate DN gel. Although a direct measurement of the weight of PDA NPs in the agarose/alginate DN gel is difficult, its value can be determined by comparing the light absorption spectrum of the PDA NP-incorporated agarose/alginate DN gel with that of the aqueous PDA NP dispersion. We synthesized uniform PDA NPs via oxidative polymerization following the method used in a previous study.36 Thereafter, the absorption spectrum of the known concentration of the PDA NP dispersion was measured using a UV-visible spectrometer. The absorption intensity of the PDA NP dispersion in the visible light range was proportional to the PDA NP concentration (Fig. S4, ESI†). This was confirmed by plotting the absorption intensity at 550 nm, as shown in Fig. 1f. It can be comprehended by the Beer–Lambert law as follows:
| A = εlc | (1) |
 | (2) |
The photothermal effect of PDA NP-incorporated agarose/alginate DN gels under visible light was qualitatively investigated. The 1 mm thick PDA NP-incorporated agarose/alginate DN gels were prepared to analyze the photothermal effect of PDA NPs. Four samples with transmittances of 100% (negative control), 70%, 35%, and 5% at 550 nm were prepared and labeled as 1, 2, 3, and 4, respectively, as shown in Fig. 2a. Mimicking sunlight, visible light from a 300 W xenon arc lamp was used to illuminate the agarose/alginate DN gels to benefit from the broad absorption property of PDA NPs. Fig. 2b shows the infrared thermal images taken 1 min after the illumination of the agarose/alginate DN gels prepared in air (dried) and water (wetted) environments. In contrast to the neat agarose/alginate DN gel, which showed almost no temperature change in water and air environments, other samples exhibited an increase in temperature owing to the photothermal effect proportional to the PDA NP concentration. Local heating by the photothermal effect was higher in air than in a water environment for all samples. This is because the samples immersed in water lost heat energy more easily through heat conduction, and the irradiated light was reflected by the water surface, which reduced its intensity. The temperature gradually increased during light irradiation and then dropped to room temperature within 1 min when the light was turned off (Fig. 2c and d). The increase in temperature due to the photothermal effect was mainly observed during the first 10 s of light irradiation. The rate of change of temperature after the initial photothermal effect remained the same regardless of the PDA NP concentration (Fig. 2e and f). Similarly, the temperature of all samples rapidly decreased as soon as the light was turned off. These observations suggest that the photothermal effect of PDA NPs is an immediate and key phenomenon for the local and remote control of the temperature of agarose/alginate DN gels.
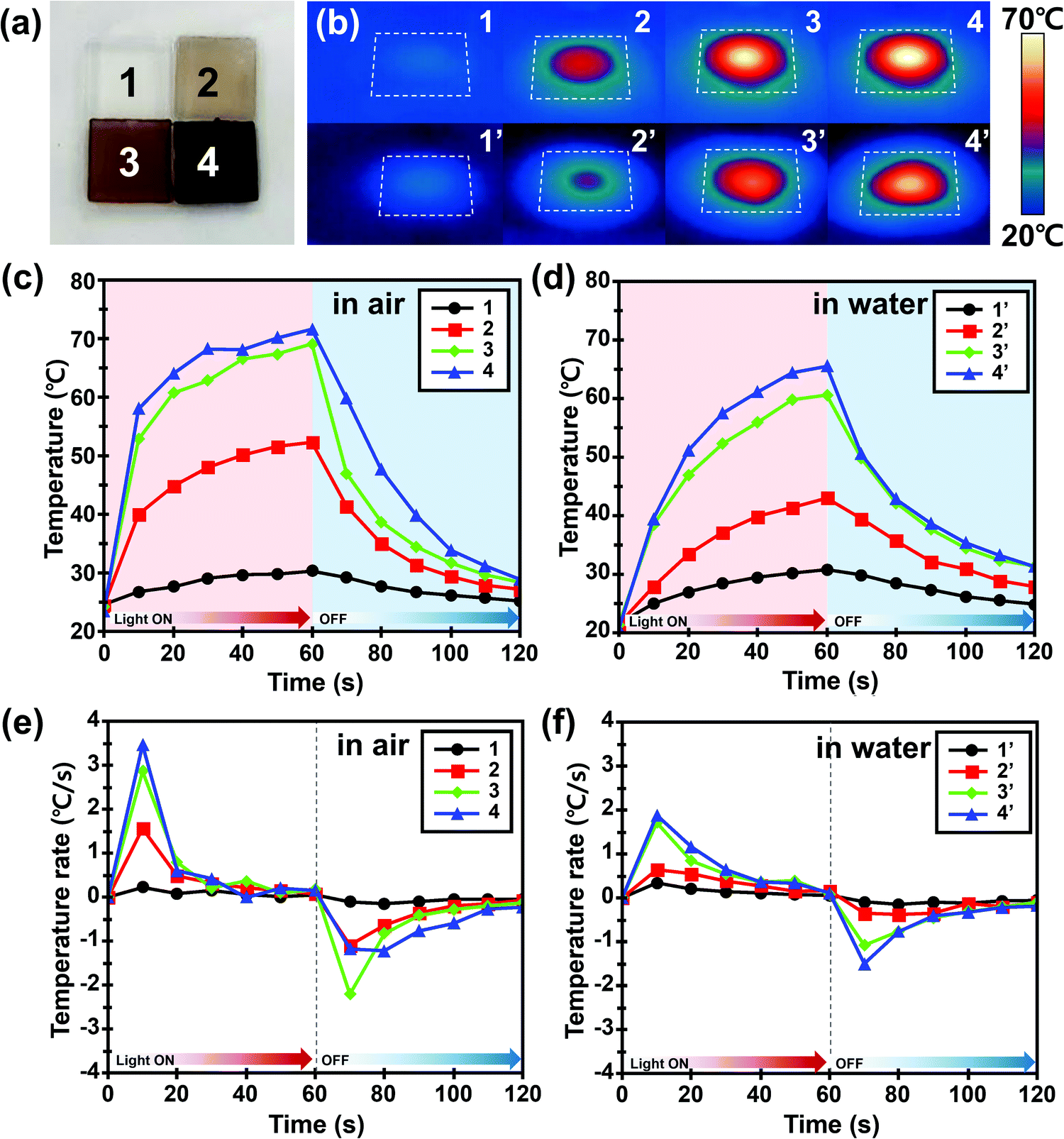 | ||
| Fig. 2 (a) Digital photograph of PDA NP-incorporated agarose/alginate DN gels with four different PDA NP concentrations. Transmittance at 550 nm was 100%, 70%, 35%, and 5% for samples 1, 2, 3, and 4, respectively. (b) Infrared thermal images of four different agarose/alginate DN gels introduced in (a) after visible light irradiation by a 300 W xenon arc lamp for 1 min. Each sample was measured in air (top) and water (bottom) environments, respectively. (c and d) Temperature change of four different hydrogels in (b) air and (c) water environments during visible light irradiation. Light was turned off after 1 min and remained off for an additional 1 min. (e and f) Rate of temperature change of four different agarose/alginate DN gels in (e) air and (f) water environments during visible light irradiation. Light was turned off after 1 min and remained off for an additional 1 min. Scale bar in (a) indicates 1 mm. | ||
We introduced a PDA NP-incorporated bilayer system consisting of an agarose/alginate DN gel as the passive layer and PNIPAm as the temperature-responsive active layer, as shown in Fig. 3a. Briefly, a thin film of agarose/alginate DN gel was prepared via the physical gelation of agarose, followed by the ionic crosslinking of sodium alginate. Afterward, the PNIPAm was polymerized on the agarose/alginate DN gel through photopolymerization. Finally, PDA NPs were incorporated into the neat bilayer via in situ oxidative polymerization. As mentioned in the Introduction, the post-incorporation process enables the facile control of the nanoparticle concentration in the hydrogel matrix and the homogeneous distribution of nanoparticles. Moreover, the photopolymerized PNIPAm forms a physicochemically intact interface with the agarose/alginate DN gel, enabling the formation of a mechanically robust bilayer structure, as shown in the cross-sectional scanning electron microscope (SEM) image in Fig. 3a. It is noted that the formation of mechanically robust interface is particularly important in bilayer actuator design. This is because a mechanical strain is applied during shape morphing of the film due to anisotropic volume change of material. When the PDA NPs were introduced prior to the photopolymerization of PNIPAm layer, incomplete photopolymerization near the interface results in delamination between bilayers upon actuation (Fig. S5, ESI†).
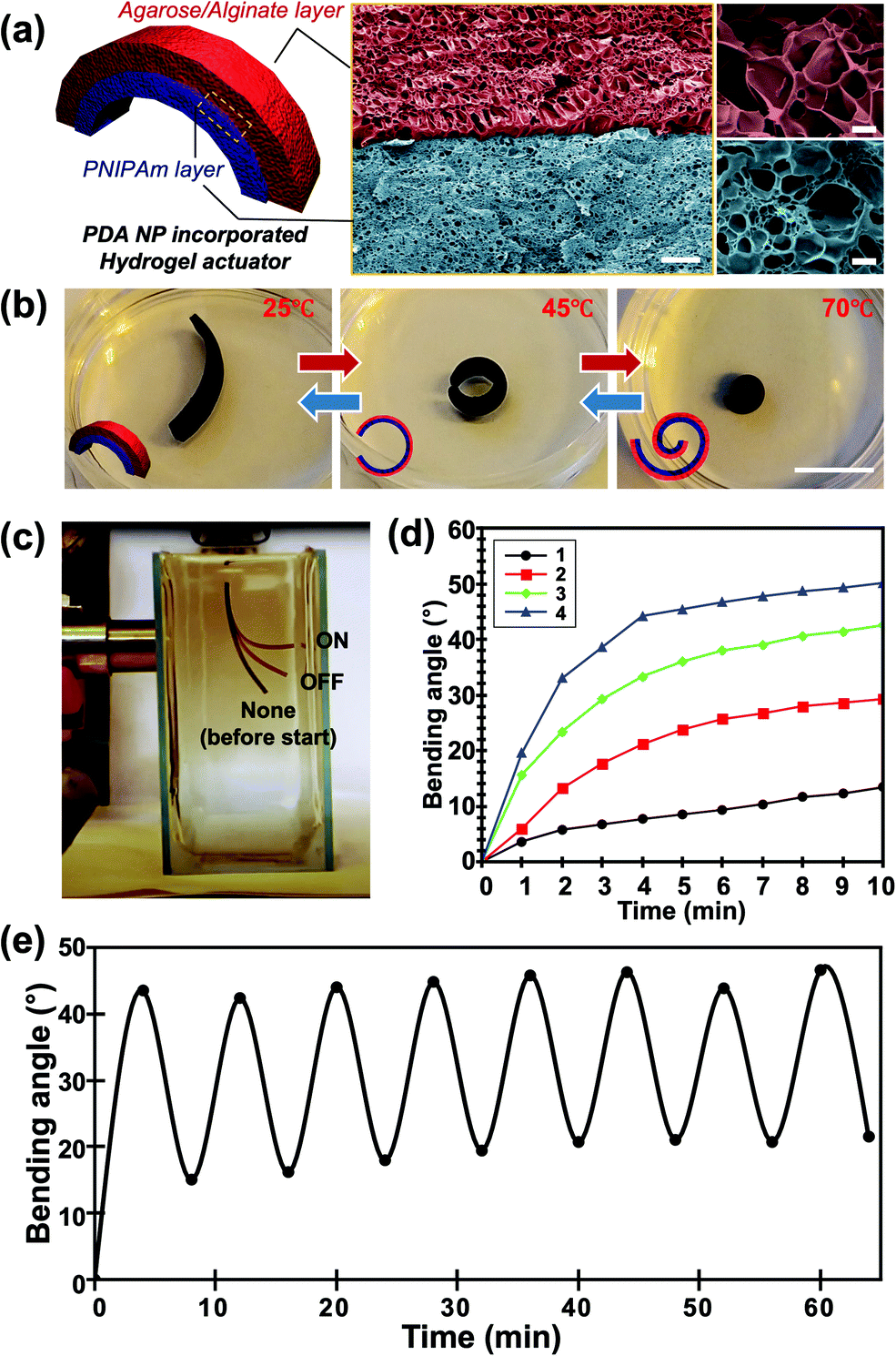 | ||
| Fig. 3 (a) Schematic illustration (left panel) and false-colored scanning electron microscopy images (right panel) of the PDA NP-incorporated hydrogel actuator consisting of an agarose/alginate DN gel layer and a PNIPAm layer. Red and blue layers indicate the agarose/alginate DN gel layer and PNIPAm layer, respectively, with a clear boundary. (b) Photographs of the temperature-dependent actuation of the PDA NP-incorporated hydrogel actuator taken at 25 °C (left), 45 °C (center), and 70 °C (right), respectively. Insets indicate that the outer layer is the agar/alginate DN gel layer (red), while the inner layer is the PNIPAm layer (blue). (c) Photograph showing the photothermal actuation of the hydrogel actuator. Film images before and after light irradiation were superimposed for demonstration. Bending angle (θ) of film was measured based on the image analysis. (d) Temperature-dependent bending angles of four different films with different PDA NP concentrations (shown in Fig. 2) via photothermal actuation. (e) Bending angle of the actuator film upon repetitive photothermal actuation showing reversibility. Scale bars in (a) indicate 10 μm (center) and 1 μm (right), respectively. Scale bar in (b) indicates 1 mm. | ||
The PDA NP-incorporated hydrogel bilayer exhibited controlled shape-morphing actuation in response to temperature and light. We prepared a 2 cm (length) × 0.5 cm (width) agarose/alginate DN gel/PNIPAm bilayer consisting of 750 μm thick layers. The initial bilayer structure was slightly curled toward the PNIPAm in room temperature water because of the difference in water uptake between the agarose/alginate DN gel and PNIPAm, which can be discerned from the different pore sizes of the freeze-dried sample in Fig. 3a. The hydrogen bonds between the amide groups of the PNIPAm layer are stronger than those between the amide and water molecules above the low critical solution temperature of 32 °C. Therefore, the bilayer curled toward the PNIPAm that shrunk as the temperature increased, as shown in Fig. 3b. To fully utilize the photothermal ability of PDA NPs, a light-driven actuation experiment was conducted with visible light irradiation of the PDA NP-incorporated hydrogel bilayer using a xenon arc lamp. As shown in Fig. 3c, the bilayer immersed in water was curled toward the PNIPAm layer owing to photothermal actuation and recovered slowly when the light was turned off. We prepared four different bilayers with different photothermal responses by controlling the in situ oxidative polymerization of PDA NPs. The photothermal responses of each sample were compared by measuring the bending angles at 1 min intervals during light irradiation. The bending angle were obtained by continuously taking photos of bilayer actuator during photothermal actuation. Then, we drew an imaginary line segment connecting both ends of the film, and the angle between the line segments were measured. As shown in Fig. 3d, the bending angle of all samples continuously increased until an equilibrium was reached. Time-resolved bending behavior reflected the photothermal heating behavior of the gel, as shown in Fig. 2d. This implies that the photothermal effect of PDA NPs is directly influenced by the actuation of the bilayer. The actuation time was longer than the time required for temperature equilibration for all samples. This is a reasonable result because light energy is directly converted into heat, whereas bilayer actuation requires the mass transfer of water inside the gel. In addition, we conducted an experiment on the reversible photothermal actuation of the bilayer. The bending angle of the bilayer was measured in each actuation cycle by turning the light on and off every 4 min. We found that the bilayer was not fully restored to its original position during the initial actuation when the heating and cooling times were equally controlled, as shown in Fig. 3e. However, the bending angle consistently changed when the light was turned on and off after the second actuation. This is because the expansion and shrinkage kinetics of the PNIPAm layer are affected by the heat generation and dissipation followed by water diffusion. Owing to the photothermal effect of PDA NPs, heat was rapidly generated, while cooling occurred via convection, which explains the slower shape recovery of the bilayer following the actuation cycle.
To emphasize the advantages of tunable and locally controlled photothermal actuation, we designed several actuation systems for investigation. First, remotely controlled local actuation was demonstrated using four independent microgrippers. The microgrippers were prepared by attaching plastic wires to the straight-shaped bilayers and placing them on a polydimethylsiloxane (PDMS) substrate with four-hole cups, as shown in Fig. 4a. The capture, delivery, and release of the target glass beads were repeatedly performed via the photothermal actuation of the microgrippers as shown in Fig. 4b (see Movie M1, ESI†). The weights of the hydrated microgripper and glass bead were 620 mg and 840 mg, respectively; the microgrippers consisting of agarose/alginate DN gel and PNIPAm showed sufficient strength to grip and deliver the heavier target.
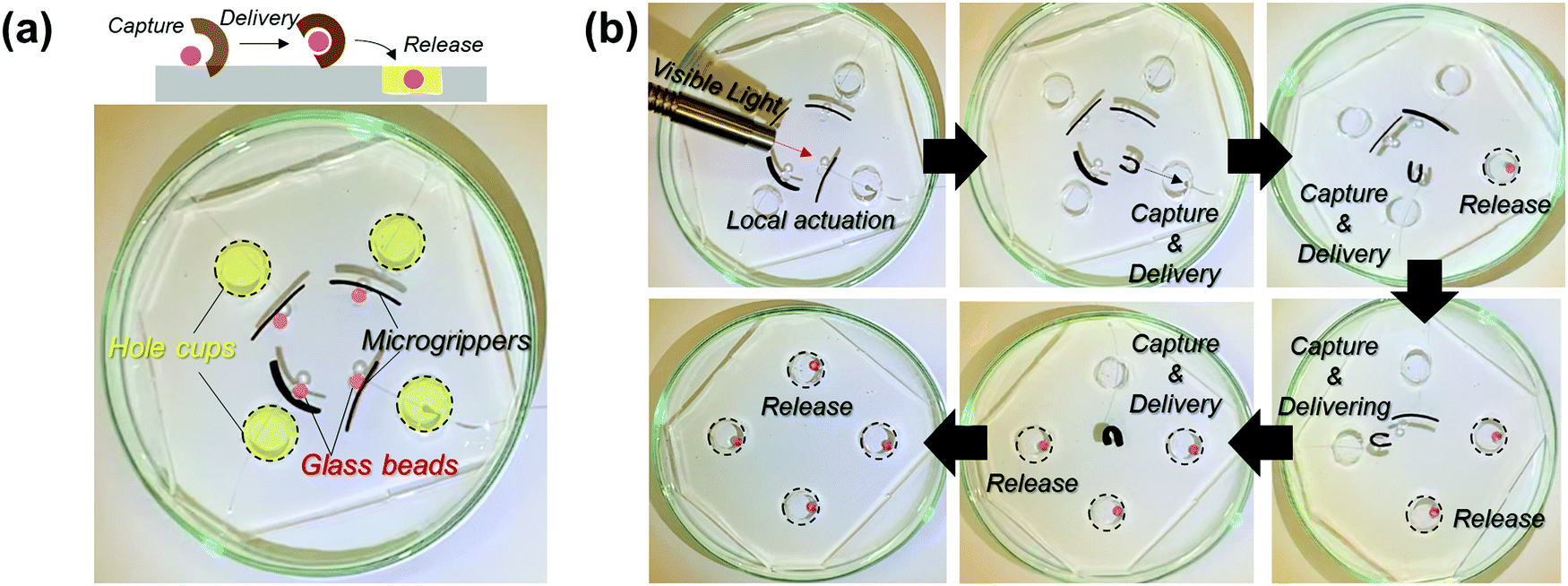 | ||
| Fig. 4 (a) Demonstration of the remotely controlled local actuation of bilayer microgrippers. (b) Snapshots of the remotely controlled sequential actuation of four bilayer microgrippers showing the capture, delivery, and release of targets. | ||
A pinwheel-shaped actuator consisting of four isosceles triangular panels with different PDA NP concentrations was fabricated for tunable photothermal actuation. We prepared four triangular hydrogel bilayer panels, and the transmittance of each bilayer was controlled by changing the polymerization cycles of the PDA NPs (Fig. S6, ESI†). As shown in Fig. 5a, the bilayers were arranged into the shape of a pinwheel according to the order of transmittance. The anchor point at the center was fixed via polymerization of the epoxy resin by UV light. For photothermal actuation, the sample was irradiated with visible light for 5 min using xenon arc lamp. The different shape morphing of each bilayer panel was observed according to the transmittance of the bilayer (i.e., the PDA NP concentration), as shown in Fig. 5b, which corresponds to Fig. 3d. From the top-view image of the pinwheel-shaped actuator, we measured the change in the end-to-end distances before and after irradiation for further analysis. As shown in Fig. 5c, the end-to-end distance of the bilayer panel decreased the most for the bilayer with the lowest transmittance in the visible light range; this is reasonable considering the photothermal efficiency. The degree of shape morphing was not linearly proportional to the PDA NP concentration. When the actuation of the bilayer occurred in a water environment, the heat generated by the photothermal reaction dissipated to the surroundings, which interrupts local heating. Therefore, the rate of heat generation must be sufficient to allow the local temperature of the bilayer film to exceed the low critical solution temperature (LCST) of PNIPAm.
 | ||
| Fig. 5 (a) Schematic of the tunable photothermal actuation of the pinwheel-shaped actuator. (b) Top-view (bottom panel) and side-view (top panel) photographs of the pinwheel-shaped actuator before (left panel) and after (right panel) visible light illumination. (c) Normalized end-to-end distance (L/L0) of four bilayer panels after the photothermal actuation (black) and transmittance of four bilayer panels at 550 nm (diagonal line). | ||
The strong light absorption property of PDA NPs enables the sunlight-driven photothermal actuation of agarose/alginate/PNIPAm bilayers. We immersed the PDA NP-incorporated and neat flower-shaped bilayers in water and exposed them outdoors to sunlight for 20 min. As shown in Fig. 6, the PDA NP-incorporated bilayer exhibited notable shape morphing after 5 min, while the neat bilayer exhibited only a slight shape change after 20 min. After the 20 min of sunlight exposure, the water temperature of the PDA NP-incorporated and neat flower-shaped bilayers increased to 32.6 °C and 30.4 °C, respectively, which were lower than the LCST of PNIPAm. The above results imply that the shape morphing of the PDA NP-incorporated bilayer is driven by the local photothermal effect of PDA NPs instead of solar thermal energy.
 | ||
| Fig. 6 Sunlight-driven photothermal actuation of flower-shaped bilayers over time. PDA NP-incorporated (top panel) and control (bottom panel) agarose/alginate DN gel/PNIPAm bilayer films were compared. Surface temperature of water was measured by an infrared thermal camera (insets). | ||
Summary
In summary, we developed a tunable photothermal actuator by fabricating an anisotropic hydrogel bilayer incorporated with PDA NPs. Utilizing the photothermal effect of PDA NPs, excellent absorption characteristics over a broad range of wavelengths of light was achieved, and visible light-driven photothermal actuation was demonstrated. In particular, the photothermal reaction was controlled using the in situ oxidative polymerization of PDA NPs in the hydrogel matrix, thereby controlling bilayer actuation. The strategy of in situ oxidative polymerization of PDA NPs in the hydrogel matrix resolved several issues in the development of visible-light- or sunlight-driven soft actuators. It enabled the homogeneous distribution of nanoparticles within the hydrogel matrix. As materials with a broad range of visible light absorption (e.g., carbons and iron oxide) have poor dispersibility in aqueous media, the use of in situ oxidative polymerization strategy enables the fabrication of homogeneous hydrogel films. In addition, post-incorporation of PDA NPs facilitated the fabrication of actuators by solving photopolymerization inhibition caused by light absorption of photothermal materials. Although we did not present it in this paper, the in situ polymerization strategy may enable the fabrication of materials with higher complexity. For example, if PDA NPs are selectively polymerized at the desired location, photothermal materials with different concentrations can be patterned on a monolithic film (Fig. S7, ESI†). In addition, it will be possible to fabricate a monolithic film in which the photothermal material has a concentration gradient by engineering the diffusion kinetics. Such approaches are thought to be difficult to make with the conventional methods. The in situ oxidative polymerization strategy could be an effective approach to mitigate the challenges of film production, in which nanomaterials with high light absorption abilities are employed.Experimental section
Materials
Agarose powder (extra pure) was purchased from Samchun Pure Chemicals. Dopamine hydrochloride and N,N′-methylenebisacrylamide (MBAA) were purchased from Sigma-Aldrich. Sodium alginate and calcium chloride dihydrate were purchased from Junsei Chemical. The photoinitiator, Omnirad 2959, was obtained from IGM Resins. PDMS (Sylgard A,B) was purchased from Dow Corning. The 1 N NaOH standard solution was purchased from Daejung Chemical & Metals. N-Isopropylacrylamide (NIPAm) was purchased from Tokyo Chemical Industry. All chemicals were used as received without further treatment.Fabrication of the agarose/alginate DN gel film
To fabricate the agarose/alginate DN gel film, the agarose film was first prepared, followed by an additional crosslinking process by alginate. For the agarose film, an aqueous 2 wt% agarose solution was prepared by dissolving agarose powder in deionized water. The agarose solution was melted in a 90 °C thermostat water bath prior to use. Thereafter, glass or acrylic spacers of various thicknesses were placed on the glass substrate to control the thickness of the agarose film. Finally, the hot agarose solution was poured onto the substrate and covered with another glass substrate without leaving trapped air bubbles in the middle of the film. The film was then cooled in a refrigerator for complete gelation. To fabricate the DN gel film, agarose film was immersed in a 0.5% sodium alginate solution for 3 h and then transferred to a 5% calcium chloride solution for 3 h for ionic polymerization. The final DN gel film was thoroughly washed and stored in deionized water.Author contributions
T. S. S. designed the study. C. L. and M. K. prepared PDA NPs and PDA NP-incorporated hydrogel bilayers. C. L. and M. K. conducted qualitative studies on the photothermal actuation of hydrogel bilayers. C. L., J. H. P., and J. S. K. performed the robotic actuation of the bilayer applications. C. L., J. H. P., and T. S. S. interpreted the results and wrote the manuscript. C. L., J. H. P., and T. S. S. prepared the figures. All the authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Government of Korea (MSIT) (no. NRF-2017R1A5A1015365; NRF-2021R1F1A1048628).References
- Y. Forterre, J. M. Skotheim, J. Dumais and L. Mahadevan, Nature, 2005, 433, 421–425 CrossRef CAS PubMed.
- M. J. Harrington, K. Razghandi, F. Ditsch, L. Guiducci, M. Rueggeberg, J. W. C. Dunlop, P. Fratzl, C. Neinhuis and I. Burgert, Nat. Commun., 2011, 2, 337 CrossRef PubMed.
- A. L. Duigou and M. Castro, Sci. Rep., 2016, 6, 18105 CrossRef PubMed.
- T. S. Shim and J. M. Kim, Korean J. Chem. Eng., 2017, 34, 2355–2365 CrossRef CAS.
- S. Jiang, F. Liu, A. Lerch, L. Ionov and S. Agarwal, Adv. Mater., 2015, 27, 4865–4870 CrossRef CAS PubMed.
- A. S. Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef PubMed.
- S. A. Morin, R. F. Shepherd, S. W. Kwok, A. A. Stokes, A. Nemiroski and G. M. Whitesides, Science, 2012, 337, 828–832 CrossRef CAS PubMed.
- T. S. Shim, S. Kim, C. Heo, H. C. Jeon and S. Yang, Angew. Chem., Int. Ed., 2012, 51, 1420–1423 CrossRef CAS PubMed.
- S. Maeda, Y. Hara, T. Sakai, R. Yoshida and S. Hashimoto, Adv. Mater., 2007, 19, 3480–3484 CrossRef CAS.
- T. S. Shim, Z. G. Estephan, Z. Qian, J. H. Prosser, S. Y. Lee, D. M. Chenoweth, D. Lee, S.-J. Park and J. C. Crocker, Nat. Nanotechnol., 2017, 12, 41–47 CrossRef CAS PubMed.
- E. Lee, D. Kim, H. Kim and J. Yoon, Sci. Rep., 2015, 5, 15124 CrossRef CAS PubMed.
- D. Kim, H. S. Lee and J. Yoon, Sci. Rep., 2016, 6, 20921 CrossRef CAS PubMed.
- K. Iwaso, Y. Takashima and A. Harada, Nat. Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- J. G. Kim, J. Jeon, R. Sivakumar, J. Lee, Y. H. Kim, M. Cho, J. H. Youk and J. J. Wie, Adv. Intell. Syst., 2021, 2100148 Search PubMed.
- S. Ahn, T. H. Ware, K. M. Lee, V. P. Tondiglia and T. J. White, Adv. Funct. Mater., 2016, 26, 5819–5826 CrossRef CAS.
- X. Le, W. Lu, J. Zhang and T. Chen, Adv. Sci., 2019, 6, 1801584 CrossRef PubMed.
- L. Dong and Y. Zhao, Mater. Chem. Front., 2018, 2, 1932–1943 RSC.
- S. R. Mishra and J. B. Tracy, ACS Appl. Nano Mater., 2018, 1, 3063–3067 CrossRef CAS.
- H. Yang, J.-J. Liu, Z.-F. Wang, L.-X. Guo, P. Keller, B.-P. Lin, Y. Sun and X.-Q. Zhang, Chem. Commun., 2015, 51, 12126–12129 RSC.
- M. Wang, X.-B. Hu, B. Zuo, S. Huang, X.-M. Chen and H. Yang, Chem. Commun., 2020, 56, 7597–7600 RSC.
- C. Li, Y. Liu, C. Lo and H. Jiang, Soft Matter, 2011, 7, 7511–7516 RSC.
- C. Wu, J. Feng, L. Peng, Y. Ni, H. Liang, L. He and Y. Xie, J. Mater. Chem., 2011, 21, 18584–18591 RSC.
- Y. Yang, W. Zhan, R. Peng, C. He, X. Pang, D. Shi, T. Jiang and Z. Lin, Adv. Mater., 2015, 27, 6376–6381 CrossRef CAS PubMed.
- J. Liebscher, R. Mrowczynski, H. A. Scheidt, C. Filip, N. D. Hadade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed.
- B. D. Wilts, K. Michielsen, H. D. Raedt and D. G. Stavenga, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4363–4368 CrossRef CAS PubMed.
- L. Panzella, G. Gentile, G. D’Errico, N. F. Della Vecchia, M. E. Errico, A. Napolitano, C. Carfagna and M. d’Ischia, Angew. Chem., Int. Ed., 2013, 52, 12684–12687 CrossRef CAS PubMed.
- M. Brenner and V. J. Hearing, Photochem. Photobiol., 2008, 84, 539–549 CrossRef CAS PubMed.
- S. Clusella-Trullas, J. H. Wyk and J. R. Spotila, Ecology, 2009, 90, 2297–2312 CrossRef PubMed.
- S. Cho, T. S. Shim, J. H. Kim, D.-H. Kim and S.-H. Kim, Adv. Mater., 2017, 29, 1700256 CrossRef PubMed.
- Y.-S. Lim, J. S. Kim, J. H. Choi, J. M. Kim and T. S. Shim, Colloid Interface Sci. Commun., 2022, 48, 100624 CrossRef.
- Y. Huang, Y. Li, Z. Hu, X. Yue, M. T. Proetto, Y. Jones and N. C. Gianneschi, ACS Cent. Sci., 2017, 3, 564–569 CrossRef CAS PubMed.
- S. E. Yakhlifi, M.-L. Alfieri, Y. Arntz, M. Eredia, A. Ciesielski, P. Samorì, M. d’Ischia and V. Ball, Colloids Surf. Physicochem. Eng. Asp., 2021, 614, 126134 CrossRef.
- Y. Xing, J. Zhang, F. Chen, J. Liu and K. Cai, Nanoscale, 2017, 9, 8781–8790 RSC.
- Q. Chen, H. Chen, L. Zhu and J. Zheng, J. Mater. Chem. B, 2015, 3, 3654–3676 RSC.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- S. Cho and S.-H. Kim, J. Colloid Interface Sci., 2015, 458, 87–93 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00420h |
| ‡ Current address: KU-KIST Graduate School of Converging Science and Technology, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2022 |