
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of formamidinium lead iodide-based perovskite solar cells: efficiency and stability
Ziwei
Zheng†
,
Shiyu
Wang†
,
Yue
Hu
 ,
Yaoguang
Rong
,
Anyi
Mei
and
Hongwei
Han
*
,
Yaoguang
Rong
,
Anyi
Mei
and
Hongwei
Han
*
Michael Grätzel Center for Mesoscopic Solar Cells, Wuhan National Laboratory for Optoelectronics, Key Laboratory of Materials Chemistry for Energy Conversion and Storage of Ministry of Education, Huazhong University of Science and Technology, Wuhan, 430074, Hubei, PR China. E-mail: hongwei.han@mail.hust.edu.cn
First published on 28th December 2021
Abstract
Perovskite materials have been particularly eye-catching by virtue of their excellent properties such as high light absorption coefficient, long carrier lifetime, low exciton binding energy and ambipolar transmission (perovskites have the characteristics of transporting both electrons and holes). Limited by the wider band gap (1.55 eV), worse thermal stability and more defect states, the first widely used methylammonium lead iodide has been gradually replaced by formamidinium lead iodide (FAPbI3) with a narrower band gap of 1.48 eV and better thermal stability. However, FAPbI3 is stabilized as the yellow non-perovskite active phase at low temperatures, and the required black phase (α-FAPbI3) can only be obtained at high temperatures. In this perspective, we summarize the current efforts to stabilize α-FAPbI3, and propose that pure α-FAPbI3 is an ideal material for single-junction cells, and a triple-layer mesoporous architecture could help to stabilize pure α-FAPbI3. Furthermore, reducing the band gap and using tandem solar cells may ulteriorly approach the Shockley–Queisser limit efficiency. We also make a prospect that the enhancement of industrial applications as well as the lifetime of devices may help achieve commercialization of PSCs in the future.
1. Introduction
As one of the most promising emerging photovoltaics, perovskite solar cells (PSCs) have attracted extensive attention and developed rapidly in the past decade for renewable and clean energy.1–5 Miyasaka et al. introduced methylammonium-based halide perovskites, namely, methylammonium lead iodide (MAPbI3) and methylammonium lead bromine (MAPbBr3), into dye-sensitized solar cells as light absorbers for the first time in 2009 and achieved an efficiency of 3.8% at that moment.6 With the optimization of deposition technology and device configuration,7–11 PSCs with MAPbI3 perovskites as the light-absorbing layer reached the highest efficiency of 22.28% in 2020.12 MAPbI3 faces several problems such as poor thermal stability and wider band gap (1.55 eV).13,14 Formamidinium lead iodide (FAPbI3) perovskites are considered to exhibit higher thermal stability and a more ideal band gap of 1.48 eV.15,16 The efficiency of FAPbI3-based PSCs prepared for the first time is not high.17 Snaith et al. found that FAPbI3 has sufficiently long electron and hole diffusion lengths for planar heterojunction solar cells to be a suitable configuration.18 M. Grätzel et al. first used FA/MA mixed cationic perovskites, but MA is still the main ingredient.19 Park et al. formed a thin MAPbI3 overlayer on FAPbI3 to enhance the short-circuit current density.20 Seok et al. stabilized the black phase of FAPbI3 (α-FAPbI3) by introducing MAPbBr3 and obtained a breakthrough for FA-based PSCs.21 Park et al. found that cesium (Cs) can stabilize the black phase of FAPbI3.22 Furthermore, mixed Cs-MA-FA cation-based devices can achieve a high efficiency of more than 22%.23–28 Since then, an increasing number of devices have been using FA-based perovskites as the light-absorbing layer and have achieved a higher efficiency.29,30 FAPbI3-based PSCs have recently obtained a certified efficiency of 25.21%.31 These results indicate that FAPbI3 shows excellent potential for efficient PSCs.We have sorted out the annual efficiency growth trends of PSCs with MA-based perovskites (Table 1) and FA-based perovskites (Table 2). The current focus of PSCs is shifting from MA-based ones to FA-based ones (Fig. 1). As the most ideal single-junction light-absorbing material, FAPbI3 with 1.48 eV band gap has displayed the impressive potential. In this work, we focused on the future development direction of PSCs. For single-junction cells, we compared the basic properties of MAPbI3 and FAPbI3. Herein, we summarize the relevant methods for stabilizing α-FAPbI3, and speculate that nano-localization effects may be one of the potential methods to stabilize the pure α-FAPbI3 phase. For other aspects, further narrowing the band gap and developing tandem cells may be another way to achieve or even break the Shockley–Queisser (S–Q) limit. Besides, we simply summarize the development of Sn–Pb and tandem solar cells and discuss the current problems. In order to realize the industrialization of PSCs, it is very important to realize the preparation of perovskite solar cells in the air. We summarize some successful cases of preparation in the air and put forward several prospects.
| Year | Main composition | V oc (mV) | J sc (mA cm−2) | FF | PCE (%) | Ref. |
|---|---|---|---|---|---|---|
| 2009 | MAPbI3 | 610 | 11.00 | 0.570 | 3.81 | 6 |
| 2011 | MAPbI3 | 706 | 15.82 | 0.586 | 6.54 | 32 |
| 2012 | MAPbI3 | 888 | 17.60 | 0.620 | 9.70 | 1 |
| 2013 | MAPbI3 | 980 | 17.80 | 0.630 | 10.90 | 33 |
| 2013 | MAPbI3 | 1020 | 18.00 | 0.670 | 12.30 | 2 |
| 2014 | MAPbI3 | 1130 | 22.75 | 0.751 | 19.30 | 7 |
| 2015 | MAPbI3 | 1086 | 23.83 | 0.762 | 19.71 | 8 |
| 2016 | MAPbI3 | 1113 | 23.69 | 0.773 | 20.40 | 9 |
| 2017 | MAPbI3 | 1120 | 23.40 | 0.813 | 21.30 | 10 |
| 2018 | MAPbI3 | 1100 | 22.70 | 0.810 | 20.20 | 11 |
| 2019 | MAPbI3 | 1120 | 23.23 | 0.814 | 21.17 | 34 |
| 2020 | MAPbI3 | 1159 | 24.11 | 0.797 | 22.28 (certified) | 12 |
| Year | Main composition | V oc (mV) | J sc (mA cm−2) | FF | PCE (%) | Ref. |
|---|---|---|---|---|---|---|
| 2013 | FAPbI3 | 970 | 6.45 | 0.687 | 4.30 | 17 |
| 2014 | FAPbI3 | 1032 | 20.97 | 0.740 | 16.01 | 20 |
| 2015 | FAPbI3 | 1059 | 24.65 | 0.770 | 20.11 (certified) | 35 |
| 2016 | FAPbI3 | 1134 | 23.70 | 0.780 | 21.02 (certified) | 36 |
| 2017 | FAPbI3 | 1100 | 25.00 | 0.803 | 22.10 (certified) | 37 |
| 2018 | FAPbI3 | 1080 | 25.06 | 0.755 | 20.35 | 38 |
| 2019 | FAPbI3 | 1144 | 26.70 | 0.776 | 23.69 (certified) | 27 |
| 2020 | FAPbI3 | 1181 | 26.18 | 0.796 | 24.64 (certified) | 39 |
| 2021 | FAPbI3 | 1174 | 26.25 | 0.818 | 25.21 (certified) | 31 |
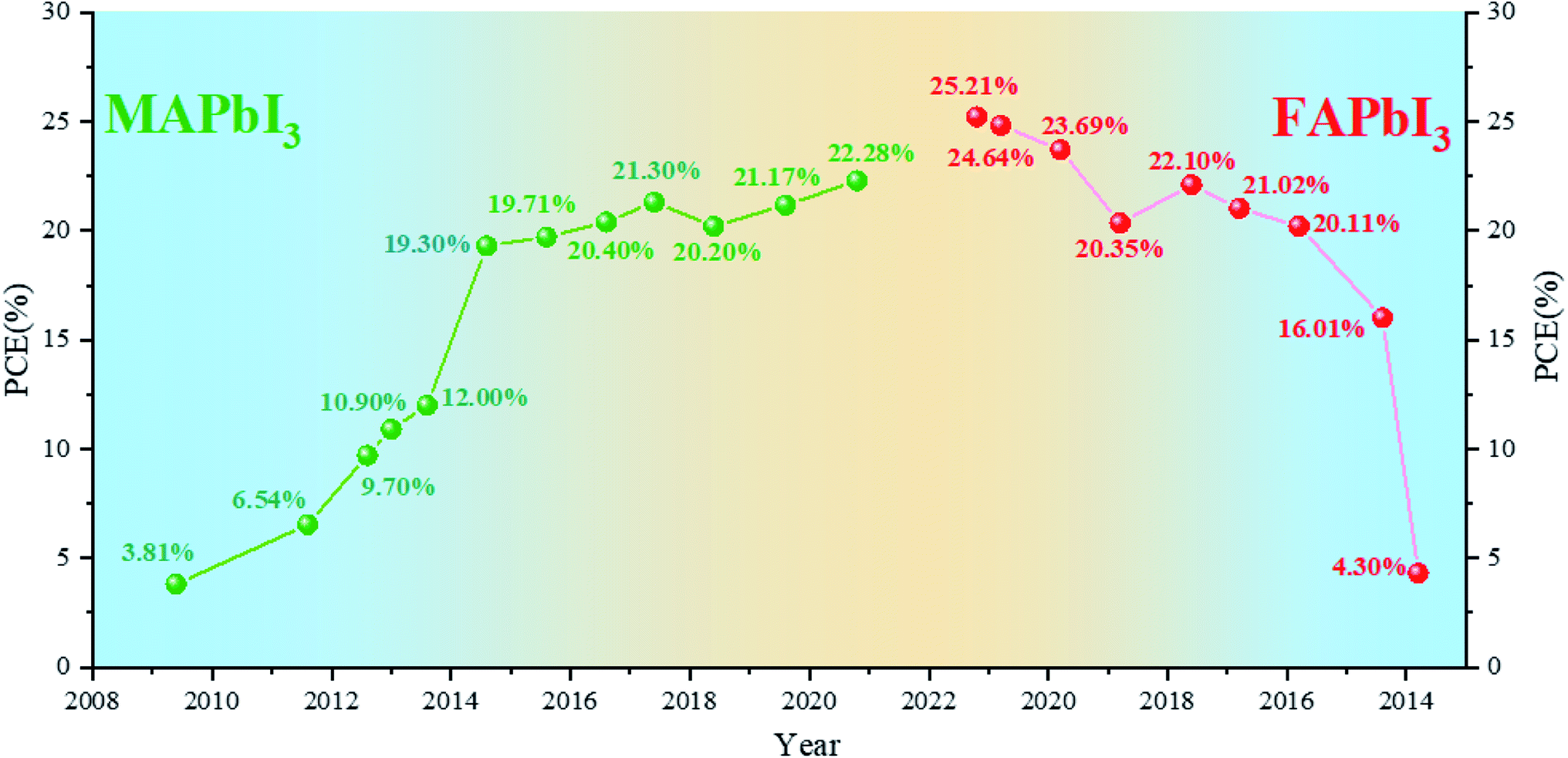 | ||
| Fig. 1 Development of MA-based and FA-based perovskite solar cells. | ||
2. A promising photovoltaic material of FAPbI3
2.1 Inherent properties
Perovskite crystals have a general ABX3 chemical formula. The ions at the A site are MA+, FA+ or Cs+; the ions at the B site are Pb2+ or Sn2+; and the ions at the X site are I−, Br− or Cl−, as shown in Fig. 2a. The A, B and X ions satisfying the Goldschmidt equation are a prerequisite to form a perovskite structure.40 As the first used material in PSCs, MAPbI3 has been widely employed due to its excellent photovoltaic performance. However, it has been gradually discovered that the larger band gap, lower charge-carrier mobility and more defect state density of MAPbI3 limit its further improvement in efficiency.7,41–43 At the same time, since MA cations are thermally unstable, the stability of the MAPbI3 perovskite has always been a problem.44 In 2013, Tom et al. introduced FA into PSCs for the first time, allowing FA cations to enter researchers' field of vision.17 Compared with the lower tolerance factor of 0.91 for MAPbI3, FAPbI3 has a higher tolerance factor of 0.99,45 which is more close to the upper limit of the tolerance factor, and this indicates a perfect fit (Fig. 2b).46 Meanwhile, while maintaining the same band structure and absorption coefficient, FAPbI3 has a narrower band gap than that of MAPbI3 (Fig. 2c), and it is more likely to achieve a more efficient solar cell.18 Starting from the physical properties of the material itself, we systematically compared the physical properties of the two materials. The effective masses of holes and electrons of MAPbI3 are 0.19me and 0.25mh, while the effective masses of holes and electrons of FAPbI3 are slightly smaller than 0.18 me and 0.23 mh.47 Generally, the smaller the effective mass is, the faster the carrier mobility will be. J. Kubicki et al. used 14N, 2H, 13C, and 1H solid-state MAS NMR magnetic resonance (NMR) to elucidate the cation reorientation dynamics of (MA)x(FA)1−xPbI3 between 100 and 330 K. The reorientation rate of the FA cation in the lattice was found to be much higher (8.7 ± 0.5 ps) than that of the MA cation (108 ± 18 ps). Faster rotation results in a better orbital overlap, and it is easier to form polarons, so electrons overcome recombination more effectively.48,49 The carrier mobility and diffusion length of FAPbI3 are similar to those of MAPbI3.50 The hydrogen vacancy total capture coefficient of MAPbI3 and α-FAPbI3 is 3.2 × 10−8 cm3 s−1 and 0.8 × 10−4 cm3 s−1, and H vacancy in FAPbI3 has a higher formation energy than that of MAPbI3. Compared with MA cations, the use of organic FA cations has greater chemical thermal stability and evaporation resistance.16Table 3 presents the summary of the inherent properties of α-FAPbI3 and MAPbI3.51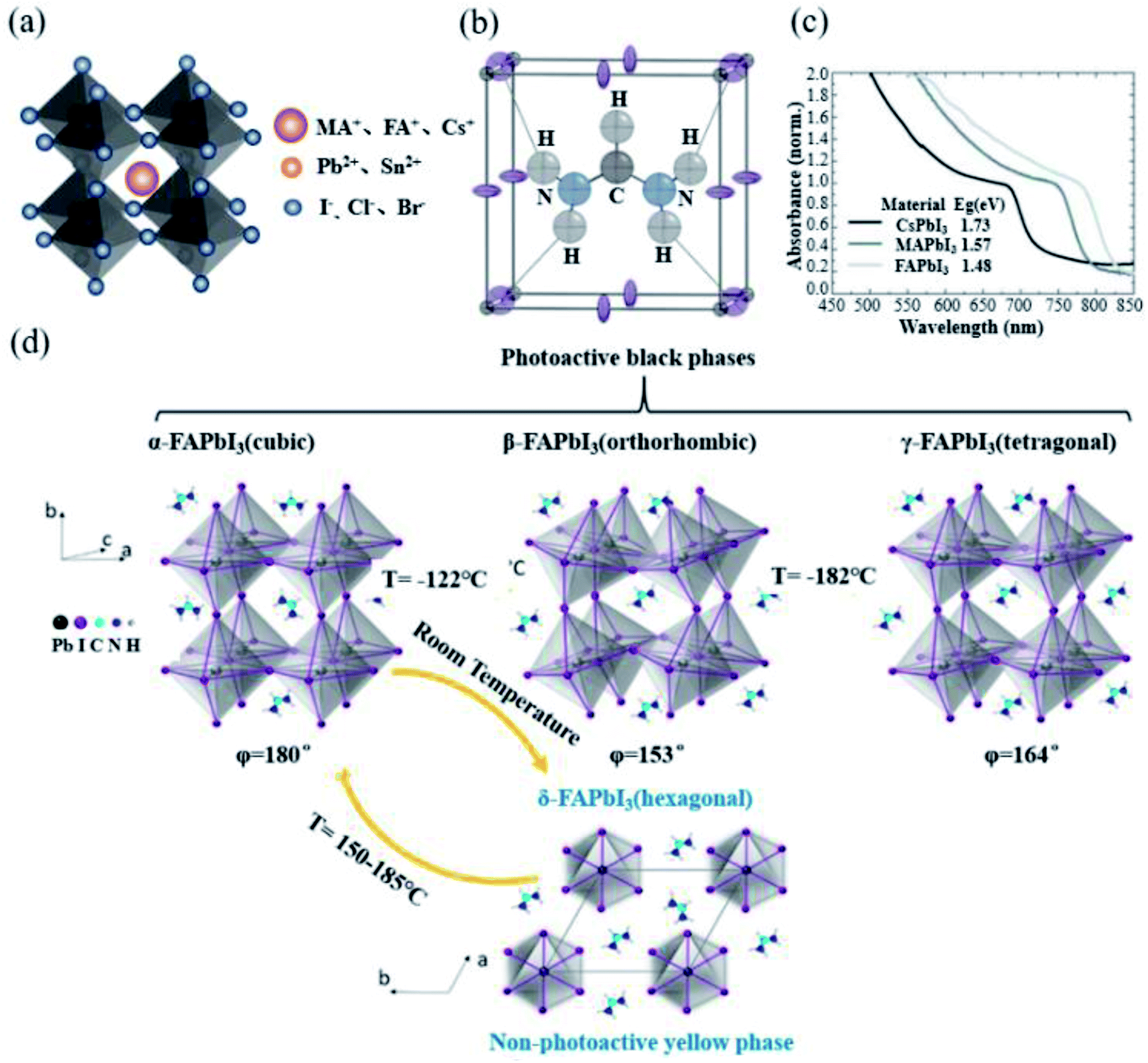 | ||
| Fig. 2 (a) Schematic diagram of the crystal structure of perovskites. (b) Unit cell structure of α-FAPbI3 at 298 K. The dark gray, purple, black, blue, and pale gray spheres correspond to the Pb, I, C, N, and H atoms. NH–I hydrogen bonds are shown as dotted lines. Reprinted with permission from ref. 53, copyright 2015, American Chemical Society. (c) UV-Vis spectra for the APbI3 perovskites formed, where A is either cesium (Cs), MA or FA. Reprinted with permission from ref. 18, copyright 2014, The Royal Society of Chemistry. (d) Crystal structure of the FAPbI3 perovskite and phase transition between each crystal form. The common crystal structure of FAPbI3 involves mainly α and δ phases, and β and γ phases are formed at extremely low temperatures. Reprinted with permission from ref. 54, copyright 2020, American Chemical Society. | ||
| Name | Band gap (eV)7,15 | Cation radius of A (pm)15,45 | Tolerance factor45 | Effective mass47 | Carriers mobility (cm2 V−1 s−1)50 | Diffusion length (μm)50 | Cation reorientation rate (ps)48,49 | Hydrogen vacancies total capture coefficient (cm3 s−1)52 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| e | h | e | h | e | h | ||||||
| MAPbI3 | 1.55 | 217 | 0.91 | 0.19me | 0.25mh | 2–40 | 10–44 | 2–8 | 1.8–12 | 108 ± 18 (300 K) | 3.2 × 10−8 |
| α-FAPbI3 | 1.48 | 253 | 0.99 | 0.18me | 0.23mh | — | 35 | 6.6 | — | 8.7 ± 0.5 (294 K) | 0.8 × 10−4 |
The combination of polar materials, high masses, and therefore, low phonon energies and fairly decent densities of deep defects is a desirable condition for slow non-radiative recombination. For α-FAPbI3 perovskite materials, fairly large defect densities that are not midgap should be tolerable as long as there is no trap-to-trap transition possible. In 2015, Derezis et al. found that perovskites would also decompose due to the molecular defects in the absence of water and heating in MAPbI3. They speculated that the hydrogen iodide (HI) vacancies caused by volatile HI on the surface broke the thermodynamic equilibrium of the structure.55 Later, researchers believed that the I-rich environment is a key factor in achieving high-efficiency solar cells.37 However, in an iodine-rich environment, it was discovered that iodine interstitial is a non-radiative recombination center.56 In order to understand how far the defect tolerance range of the lead halide perovskite is, Yavari et al. deliberately added BiI3 to the precursor solution. They attributed the significant drop in the device performance to the introduction of effective recombination centers by Bi.57 In 2020, Zhang et al. used rigorous first-principles calculations to demonstrate that Bi acts as a donor, and pushes the Fermi level closer to the conduction band; this shift promotes the formation of iodine interstitials, which are the actual nonradiative recombination centers.58 In 2021, Zhang et al. found that the H vacancy has been ignored. They explicitly reveal that the non-radiative capture coefficient of hydrogen vacancy is 10−4 cm3 s−1 by first-principles calculations, four orders of magnitude higher than that of I. Further research found that the H vacancy in FAPbI3 has a higher formation energy compared with MAPbI3. The dissociation energy of the N–H (C–H) bond in MA is lower than that of the N–H (C–H) bond in FA by 0.9 eV (0.4 eV), which implies that FA is intrinsically more stable than MA.52
However, FAPbI3 is inferior for its phase stability.21,23,27,31,38,59 FAPbI3 mainly has four different crystal structures, of which the common crystal forms are α-FAPbI3 and non-perovskite phase (δ-FAPbI3). Distortions with the [PbI6]4− octahedra promote the formation of lowered-symmetry β-FAPbI3 and γ-FAPbI3 black phases, as the temperature decreases (Fig. 2d).54 For α-FAPbI3, it corresponds to the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.60 The C–H in the FA cation points directly to the surface of the cube. The position of a planar formamidinium cation within this cubic framework was energy-minimized via density functional theory (DFT, PBEsol functional) within the regular 6.36 Å cube and found to adopt an orientation with the N–CH–N molecular ion fragment lying in the unit cell midplane, the (200) plane, with the C–H bond pointing directly toward the cube face (thereby minimizing CH⋯I interactions) and the C–N bonds directed toward adjacent cube faces. This allows the –NH2 groups to be orientated toward the unit cell edges with the potential formation of NH⋯I hydrogen bonds.53 For δ-FAPbI3, it corresponds to the P63/mmc space group at room temperature. Compared with α-FAPbI3, the yellow non-perovskite δ-FAPbI3 has a larger band gap and poor charge transport, which has an adverse effect on PSCs.61 Unfortunately, α-FAPbI3 can only be obtained at a high temperature (160 °C). The energy barrier for the conversion of α-FAPbI3 to δ-FAPbI3 obtained at high temperatures is 0.6 eV.62 However, due to the presence of highly polar ionic bonds, water will cause a large number of defects on the surface of the device, thereby greatly reducing the transformation energy barrier and accelerating the transition rate to δ-FAPbI3.63 The irreversible degradation product from the thermal degradation of FA was sym-triazine, but it was only probed by the mass spectrometry technique above 95 °C.16 Recently, it has been discovered that light induced FA+ degradation process in the perovskite precursor solution, and the –CH
m space group.60 The C–H in the FA cation points directly to the surface of the cube. The position of a planar formamidinium cation within this cubic framework was energy-minimized via density functional theory (DFT, PBEsol functional) within the regular 6.36 Å cube and found to adopt an orientation with the N–CH–N molecular ion fragment lying in the unit cell midplane, the (200) plane, with the C–H bond pointing directly toward the cube face (thereby minimizing CH⋯I interactions) and the C–N bonds directed toward adjacent cube faces. This allows the –NH2 groups to be orientated toward the unit cell edges with the potential formation of NH⋯I hydrogen bonds.53 For δ-FAPbI3, it corresponds to the P63/mmc space group at room temperature. Compared with α-FAPbI3, the yellow non-perovskite δ-FAPbI3 has a larger band gap and poor charge transport, which has an adverse effect on PSCs.61 Unfortunately, α-FAPbI3 can only be obtained at a high temperature (160 °C). The energy barrier for the conversion of α-FAPbI3 to δ-FAPbI3 obtained at high temperatures is 0.6 eV.62 However, due to the presence of highly polar ionic bonds, water will cause a large number of defects on the surface of the device, thereby greatly reducing the transformation energy barrier and accelerating the transition rate to δ-FAPbI3.63 The irreversible degradation product from the thermal degradation of FA was sym-triazine, but it was only probed by the mass spectrometry technique above 95 °C.16 Recently, it has been discovered that light induced FA+ degradation process in the perovskite precursor solution, and the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH double bonds of FA+ ions are oxidized to –CHO double bonds as an irreversible degradation process.64 Deng et al. showed that a slight excess of FAI cannot further improve the efficiency, but can greatly alleviate the expansion process to improve the photostability of FA-based perovskites.65
NH double bonds of FA+ ions are oxidized to –CHO double bonds as an irreversible degradation process.64 Deng et al. showed that a slight excess of FAI cannot further improve the efficiency, but can greatly alleviate the expansion process to improve the photostability of FA-based perovskites.65
2.2 Strategies to stabilize α-FAPbI3
As we have seen, FAPbI3 perovskite materials have been mainly used in high-efficiency devices in recent years. The essential idea is to generate α-FAPbI3 and avoid the generation of δ-FAPbI3.Grätzel et al. introduced FA+ in MAPbI3, which makes the absorption edge redshift to increase the absorption range, resulting in a higher short circuit current density.19 However, the device performance is still inferior due to the component dominated by MAPbI3. Seok et al. found that addition of a small amount of MAPbBr3 to FAPbI3 can stabilize the α-FAPbI3 phase (Fig. 3a), contributing to a more uniform morphology and better crystallization.21 The introduction of Cs also leads to the dominance of FAPbI3, achieving a efficiency of 16.5%.22 In addition, from the term of the tolerance factor, it has also been found that the mixed cation structure increases the energy and mixed entropy contribution, resulting in a decrease in free energy, thereby improving the stability.66 Based on this, Grätzel et al. began to use ternary mixed perovskite materials (Cs0.5(MA0.17FA0.83)95Pb(I0.83Br0.17)3).24 Soon after, rubidium (Rb) belonging to the same group of Cs was also introduced into the mixed perovskite.
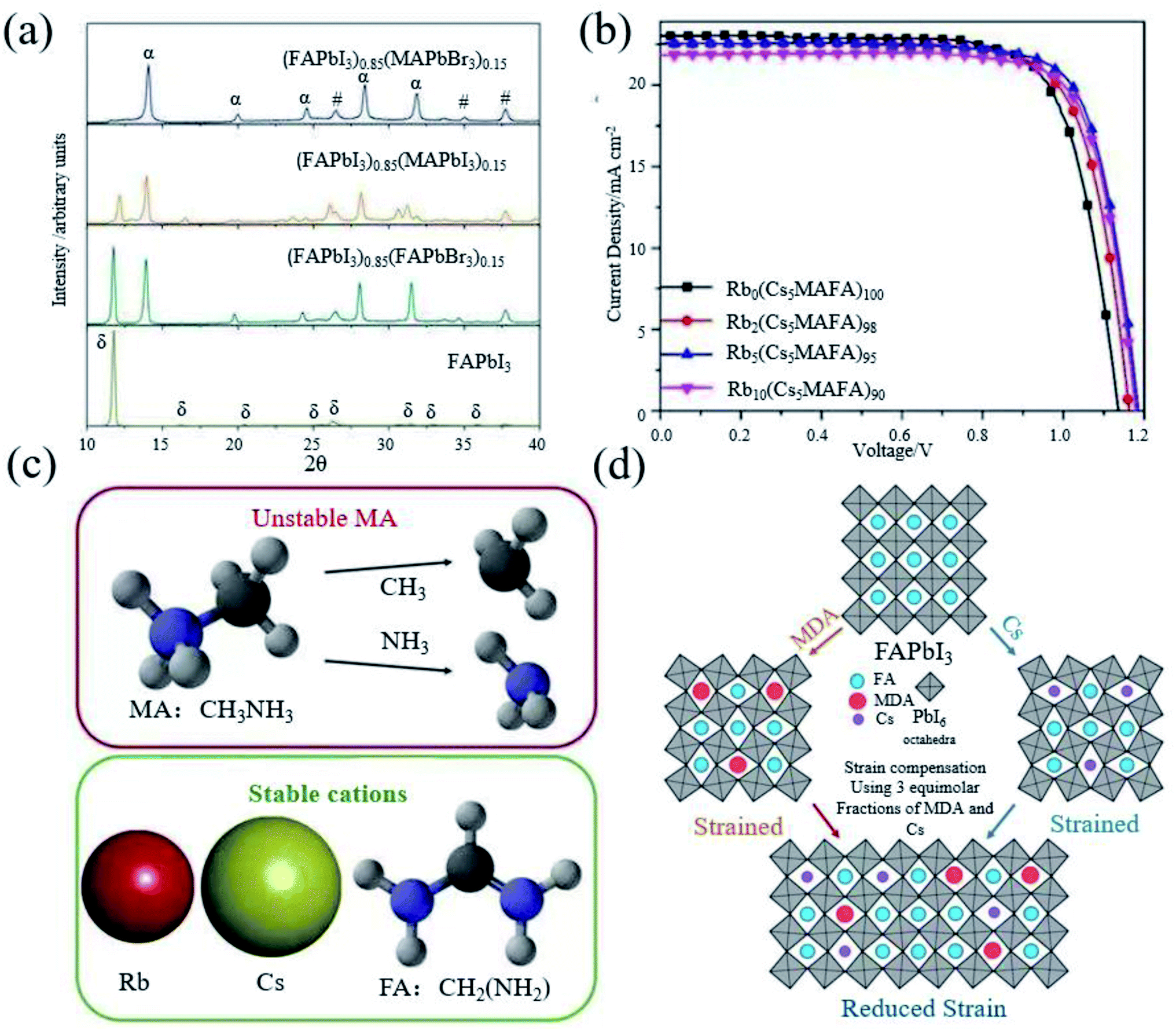 | ||
Fig. 3 (a) XRD spectra of solvent-engineering-processed FAPbI3, (FAPbI3)1−x(MAPbI3)x, (FAPbI3)1−x(FAPbBr3)x, and (FAPbI3)1−x(MAPbBr3)x, respectively. Reprinted with permission from ref. 21, copyright 2015, Nature Publishing Group. (b) J–V test of Rb0(Cs5MAFA)100PbI3, Rb2(Cs5MAFA)98PbI3, Rb5(Cs5MAFA)95PbI3, and Rb10(Cs5MAFA)90PbI3, respectively. Reprinted with permission from ref. 23, copyright 2016, Science Publishing Group. (c) Illustration of the volatile nature of the MA molecule (the I atom is omitted for simplicity), by contrast, Rb, Cs, and FA are thermally more stable cations. Reprinted with permission from ref. 38, copyright 2018, Science Publishing Group. (d) Schematic diagram of the distribution of FA, MDA and Cs in the perovskite crystal lattice, by adding 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 equimolar fractions of MDA and Cs cations to the perovskite crystal to compensate for the lattice strain. Reprinted with permission from ref. 28, copyright 2020, Science Publishing Group. :3 equimolar fractions of MDA and Cs cations to the perovskite crystal to compensate for the lattice strain. Reprinted with permission from ref. 28, copyright 2020, Science Publishing Group. | ||
Although RbPbI3 cannot form a black perovskite phase, a small amount of Rb can stabilize α-FAPbI3 and improve the performance to 21.6% (Fig. 3b).23 It has been found that all the devices containing MA severely degraded after the perovskite devices were annealed for 3 h, although the doping of MA can effectively stabilize α-FAPbI3. It has also been found that the doping of Br will obviously lead to a larger band gap, which reduces the photovoltaic performance of the perovskite.66 Therefore, it is imperative to make perovskite materials free from MA and Br. Therefore, Saliba et al. prepared a Rb0.05Cs0.1FA0.85PbI3 perovskite, without using MA and Br.38 At the same time, PMMA was used to passivate the surface of the perovskite, so as to obtain an efficiency of 20.35% (Fig. 3c). Methylenediammonium (MDA) has also been introduced to stabilize α-FAPbI3 in recent years due to its ionic radius similar to FA+. As it has more H atoms and can form more hydrogen bonds with I, only a very small amount of MDA is required. The MDA can achieve the effect of stabilizing the perovskite.27 Cs was introduced to reduce the local tensile and compressive strains in the perovskite lattice (Fig. 3d), and a certified efficiency of 24.4% was obtained.28 However, for mixed cationic systems, FA/MA, due to the thermal instability of MA of multi-element cationic mixed perovskites, phase separation will inevitably occur due to the tilt, deformation, expansion and contraction of the octahedral network, local strain may increase consequently, and the short circuit current density will reduce due to the narrowing down of the light-absorption range.44,66–68 Even though PSCs based on FA0.9Cs0.1PbI3 exhibit reasonable thermal stability, exposure to 1 sun light can cause significant degradation after hundreds of hours of operation.69
A small amount of HI added to the perovskite precursor solution with a stoichiometric ratio of FAI to PbI2 of 1:1 can assist to form a very uniform and continuous film with high phase purity.18 Yang et al. also introduced thiocyanate (SCN−) into the FAPbI3 system by adding ammonium thiocyanate (NH4SCN) into the precursor solution. They found that addition of 30% NH4SCN can not only improve the water stability, but also promote the generation of α-FAPbI3 and inhibit the generation of δ-FAPbI3 simultaneously. It was found that there was still a small amount of SCN− in the perovskite, which is attributed to the strong interaction between SCN− and Pb2+ (Fig. 4a). In addition, NH4SCN mostly evaporates by the end of the annealing process.70 Xu et al. invented a facile bi-additive method (BA method) using HI and Pb(SCN)2 as dual additives to form a new FAI–Pb(SCN)2–HI–N,N-dimethylformamide (DMF) intermediate to induce the transition of δ-FAPbI3 to α-FAPbI3.73 The Lewis acid–base addition method has been widely used to form a uniform perovskite film, which provides a methodological basis for the development of high-performance PSCs. Dimethyl sulfoxide (DMSO) is commonly used as an additive for preparing FAPbI3 and the resulting film is mainly δ-FAPbI3. N-Methylpyrrolidone (NMP) avoids the formation of the δ-FAPbI3 phase by forming a more stable mesophase than DMSO, thereby obtaining a pure α-FAPbI3 phase film (Fig. 4b and c).71,74 The ionic liquid can be used to stabilize α-FAPbI3 by adjusting the crystallization kinetics of the perovskite active layer. 1-Hexyl-3-methylimidazolium iodide ionic liquid (IL) doping is beneficial to FAPbI3 (Fig. 4d). The high polarity and high boiling point of the crystal grains are coarsened, which yields liquid domains between neighbouring grains to reduce the activation energy of the grain-boundary migration.72
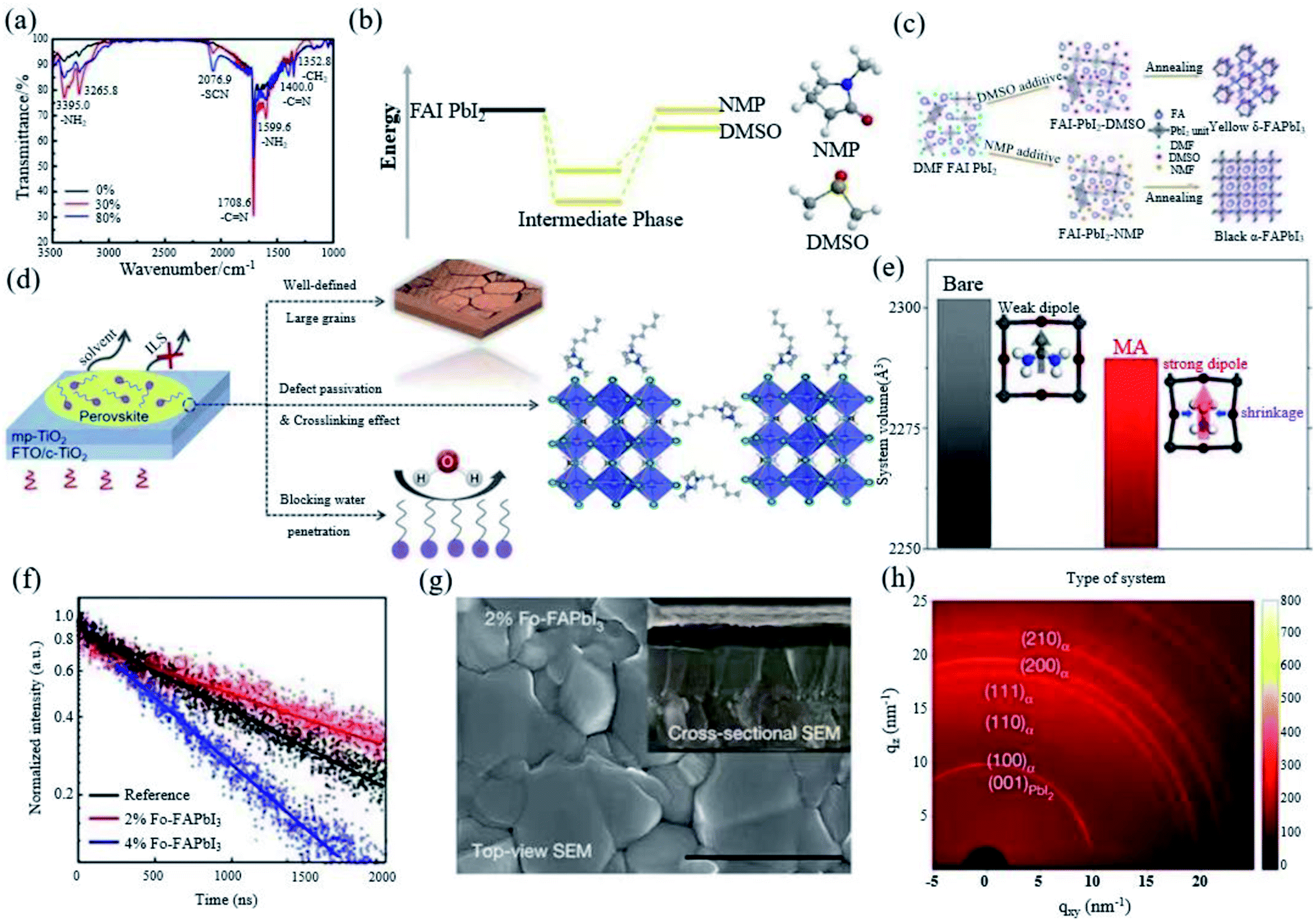 | ||
| Fig. 4 (a) Fourier transform infrared spectra of FAPbI3 perovskite films with and without SCN−. Reprinted with permission from ref. 70, copyright 2016, The Royal Society of Chemistry. (b) Comparison of the interaction energy of an adduct formed between FAI, PbI2 and DMSO as the Lewis base and that of the related adduct formed with NMP as the Lewis base. (c) Schematic of a blade-coated perovskite film with DMSO or NMP additives. Reprinted with permission from ref. 71, copyright 2020, Advanced Energy Materials. (d) Schematic illustration showing the effects of HMII IL in FAPbI3 active layer. Reprinted with permission from ref. 72, copyright 2020, Advanced Functional Materials. (e) Total volume and cubic-octahedral structure of the bare α-FAPbI3 perovskite structure and that prepared with MA. Reprinted with permission from ref. 29, copyright 2019, Joule. (f) Time-resolved photoluminescence of FAPbI3, 2% Fo-FAPbI3, and 4% Fo-FAPbI3 films, respectively. (g) SEM images of 2% Fo-FAPbI3. (h) Two-dimensional grazing-incidence XRD patterns of 2% Fo-FAPbI3 films. Reprinted with permission from ref. 31, copyright 2021, Nature Publishing Group. | ||
Cl− has been demonstrated to improve the crystallization and reduce the non-radiative recombination of perovskites.59,71,75–77 Methylenediammonium dichloride (MDACl2),27,28 MACl29 and FACl78 have already been introduced into α-FAPbI3. MACl is considered to be a transitional “stabilizer” that does not affect the crystal structure, and can induce the formation of α-FAPbI3.79 In addition, it has a good synergistic effect with the commonly used solvent DMSO.80 In order to reveal the specific mechanism of MACl effected on FAPbI3, Kim et al. conducted that the grain size of the perovskite film is directly related to the amount of MACl and the grain size is as high as 1.5 μm with 40% MACl added.
Moreover, compared with the control device without MACl, the roughness of the film reduced from 64.7 nm to 24.7 nm with the addition of 40% MACl. MA has a certain substitution for FA (Fig. 4e), but no Cl is detected, indicating that Cl does not affect the composition of the perovskite, which avoids the discontinuity of perovskite crystallization. The addition of 40% MACl can affect the formation of the α-FAPbI3 structure before the annealing step, and the stable mesophase can be induced to ultra-pure α-phase perovskite after the annealing step. The DFT calculation further proves that Cl can enhance the interaction between FA and I, which helps to improve the stability of the FAPbI3 perovskite. The FAPbI3 device with 40% MACl obtained a certified efficiency of 23.48%.29 Recently, Jeong et al. have introduced 35% MACl and 2% FAHCOO into the precursor solution. HCOO− is small enough to fill the I vacancies, thereby reducing the defects. The resulting non-radiative composite (Fig. 4f) increases the voltage and fill factor of the device, and a FAPbI3 perovskite film (Fig. 4g) with improved crystallinity and a larger grain size was obtained. δ-FAPbI3 was not found in synchrotron-based two-dimensional grazing-incidence XRD measurements, which proves that FAHCOO can stabilize the α phase (Fig. 4h), and the device has a certification efficiency of 25.2%.31
2.3 From efficiency to stability
The stabilization of α-FAPbI3via cation doping or using additives will more or less cause the perovskite lattice parameters to produce an octahedral tilt, resulting in an increase in the band gap and phase separation, which seriously affects the photovoltaic performance and stability of the PSCs.69 Therefore, what we need to consider is to achieve the intrinsic stability of FAPbI3 PSCs, while gaining high efficiency, quantum dot passivation, interface processing and preparation engineering has been tried.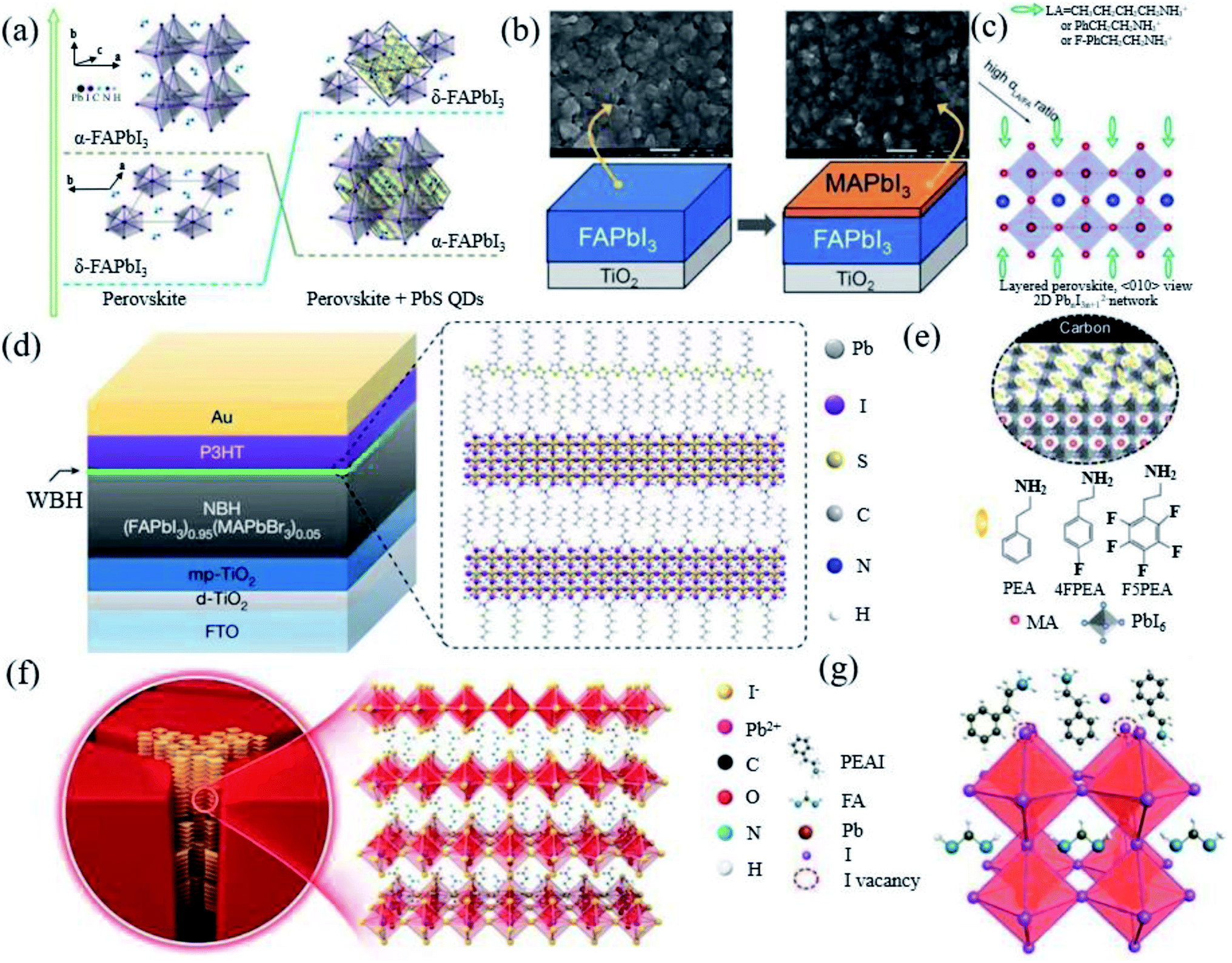 | ||
| Fig. 5 (a) Interaction process diagram between PbS and FAPbI3. Reprinted with permission from ref. 81, copyright 2020, American Chemical Society. (b) Diagram of MAPbI3 deposited on FAPbI3. Reprinted with permission from ref. 20, copyright 2014, Advanced Materials. (c) Schematic diagram of the long-chain alkyl or aromatic ammonium (LA) cations bound to the surface of the perovskite structure. Reprinted with permission from ref. 82, copyright 2014, Advanced Materials. (d) Diagram of the ultrathin wide-bandgap halide (WBH) stacked onto a narrow-bandgap halide (NBH). Reprinted with permission from ref. 83, copyright 2019, Nature Publishing Group. (e) Interface scheme of F5PEAI at the interface between the 3D perovskite and carbon. Reprinted with permission from ref. 84, copyright 2021, Advanced Energy Materials. (f) Diagram illustrating the crystal structure of the 2D perovskite with n = 1. Reprinted with permission from ref. 30, copyright 2020, Advanced Materials. (g) Schematic diagram of the possible passivation mechanism of the PEAI layer of the perovskite film. Reprinted with permission from ref. 26, copyright 2019, Nature Publishing Group. | ||
Macromolecular polymer polymethyl methacrylate (PMMA) has been reported as a buffer layer to improve the air stability of the device.38 Long-chain alkyl or aromatic ammonium (LA)-like n-butylammonium (n-C4H9NH3+, BA), phenylethylammonium (C6H5CH2CH2NH3+, PEA), and 4-fluorophenylethylammonium (4-FC6H5CH2CH2NH3+, FPEA) iodide have proved that surface functionalization can enable the stabilization of the metastable perovskite phase in pure FAPbI3 without cation or anion alloying (Fig. 5c).82 The longer alkyl ammonium chain promotes the hydrogen bond with the octahedron and self-assembles to form a new perovskite structure product, thereby stabilizing the FA perovskite. The appropriate amount of LA ions will not enter the perovskite lattice. Surface passivation or the construction of 2D/3D perovskite heterostructures via post-processing has also been proven to effectively increase the photovoltage of PSCs. Jung et al. used the in situ reaction of n-hexyl trimethyl ammonium bromide (HTAB) on the surface of the perovskite to form a thin layer of wide-bandgap halide perovskite on top of the narrow-band-gap light absorption layer (Fig. 5d).83 Jiang et al. formed phenethylammonium iodide (PEAI) on the surface of a 3D perovskite (Fig. 5g).26 Yao et al. used β-guanidinopropionic acid molecules to embed in the grain boundaries of 3D perovskite grains, distributed in half the thickness of the film (Fig. 5f).30 Chen et al. applied pentafluorophenylethylammonium iodide (F5PEAI) to post treat the perovskite/carbon interface (Fig. 5e).84 These 2D perovskites are all on the surface and grain boundaries of the 3D FAPbI3 crystal lattice. The resulting longer and hydrophobic carbon chain improves the water stability of the perovskite. This may be an effective structural stabilizer (inhibiting lattice deformation) and chemical stabilizer (enhancing the interaction between 3D grains) in FA-based perovskites. Recently, an in situ crosslinking-enabled strain-regulating crystallization (CSRC) method with trimethylolpropane triacrylate has been introduced to precisely regulate the top section of the perovskite film, where the largest lattice distortion occurs by Zhang et al.77
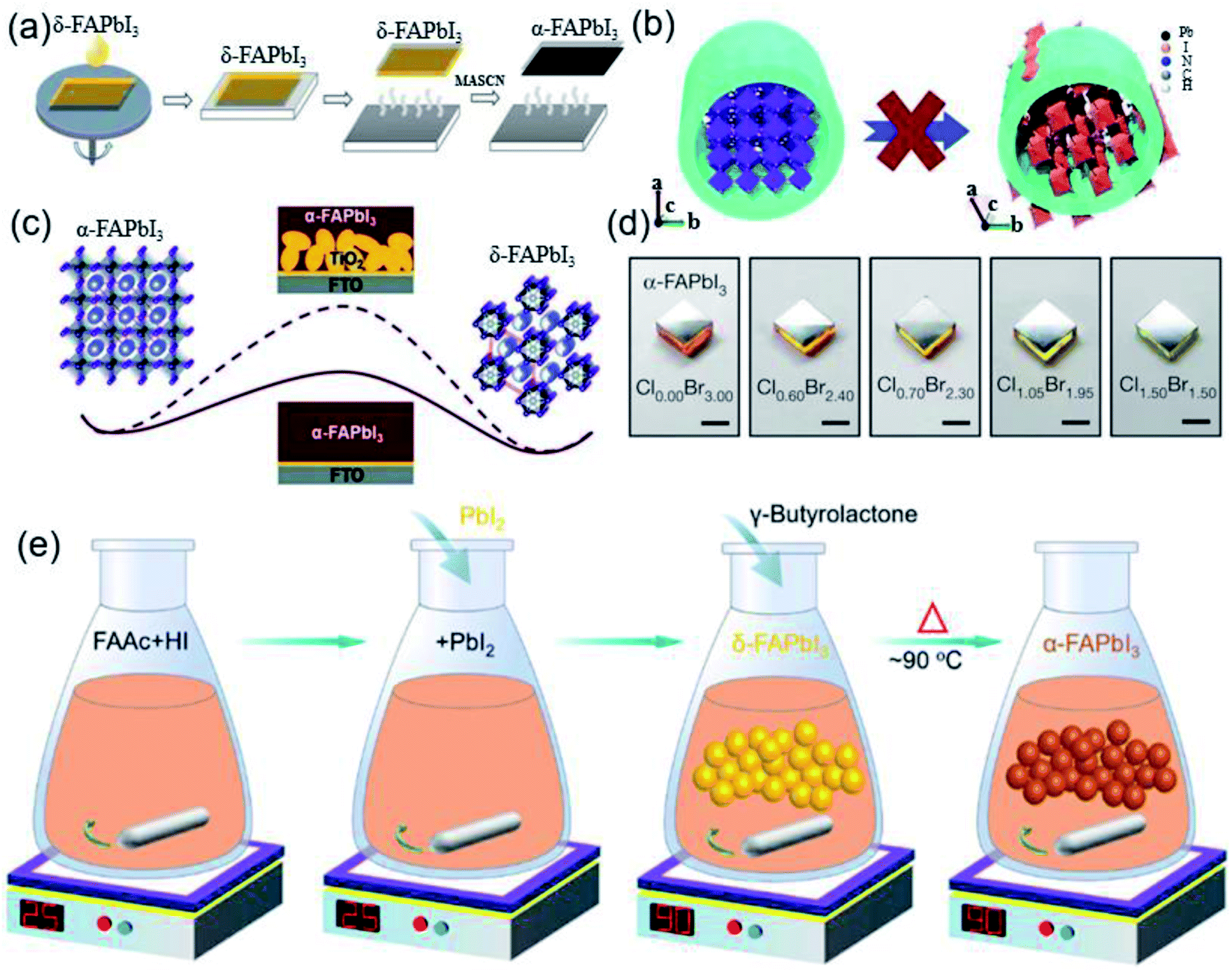 | ||
| Fig. 6 (a) Simplified scheme of the MASCN vapor treatment process for the pure black-phase FAPbI3 perovskite film. Reprinted with permission from ref. 80, copyright 2020, Science Publishing Group. (b) Schematic of the mechanism of the anodized alumina membrane (AAM) prohibiting the α-to-δ phase transition of FAPbI3 NWs via spatial confinement. Reprinted with permission from ref. 81, copyright 2018, The Royal Society of Chemistry. (c) Barrier diagram increment of the α-FAPbI3-to-δ-FAPbI3 transition with the increase in FAPbI3/TiO2 interface area. Reprinted with permission from ref. 82, copyright 2016, The Royal Society of Chemistry. (d) Optical images of α-FAPbI3 epitaxial films grown on different substrates. Reprinted with permission from ref. 84, copyright 2020, Nature Publishing Group. (e) Diagram of a simple method for the synthesis of high-purity FAPbI3 powder to prepare high-efficiency devices. Reprinted with permission from ref. 86, copyright 2019, American Chemical Society. | ||
3. Perspective and outlook
In order to achieve higher efficiency to approach or exceed the S–Q limit, and to look for feasible industry-academia cooperation, we make a perspective and outlook in the future development of FAPbI3 perovskite materials after solving the α-FAPbI3 phase problem.3.1 Theoretical efficiency
At present, the four parameters of the highest efficiency FAPbI3-based single-junction PSCs (Fo-FAPbI3, Eg = 1.53 eV) are as follows: Voc = 1.174 V, Jsc = 26.25 mA cm−2, FF = 0.818, efficiency = 25.21%.31 The four parameters of solar cells with light absorbers corresponding to a band gap of 1.53 eV calculated by the S–Q limit are as follows: Voc = 1.243 eV, Jsc = 27.99 mA cm−2, FF = 0.901, efficiency = 31.34%.13 The four parameters of FAPbI3-based PSCs account for 94.4% (Voc), 93.8% (Jsc), 90.8% (FF), and 80.4% (efficiency) of the S–Q limit, respectively. We can see that the lowest fill factor (FF) may be responsible for the efficiency loss, and the improvement of FF is usually the most difficult. In PSCs, the resistance element is usually composed of series resistance (Rs) and parallel resistance (Rsh). In order to obtain a higher FF, Rs as small as possible and Rsh as large as possible are usually required. Rs is related to the resistance of the device materials and resistive contacts, and Rsh is caused by the current leakage through the device, such as the body and the interface, the carrier recombination, the contact between the ETL and the HTL, and the leakage around the edge of the device.94 The improvement of the device efficiency is to further solve the interface contact problem of the device to obtain a higher Rsh, thereby obtaining a higher FF. Due to the reduction in the interface recombination, a higher Voc can theoretically be obtained. At present, the band gap of FA-based perovskites is more or less widened due to doping or additive use.In order to further improve the performance of the device, obtaining pure α-FAPbI3 and obtaining a lower band gap perovskite may be a potential means.
3.2 Band gap adjustment
The band gap of FAPbI3 is 1.48 eV, which has a certain distance with the S–Q limit of 1.34 eV.95 Therefore, band gap tuning, for example, by mixing halides or making Sn–Pb compounds (Fig. 7a and b) is one of the main ways to improve the efficiency.40 Both pure Sn and Sn–Pb show better charge transport performance than that of Pb-based perovskites,96–98 and the highest efficiency reaches 21.74%.99 Moreover, compared to the voltage loss of Pb-based perovskites, the voltage loss of the Sn–Pb perovskite can now be 0.33 V, which is comparable to that of FAPbI3-based devices.100 The carrier lifetime has reached more than 1 μs.101 There are reasons to believe that in the future development, Sn–Pb perovskites can reach a new height.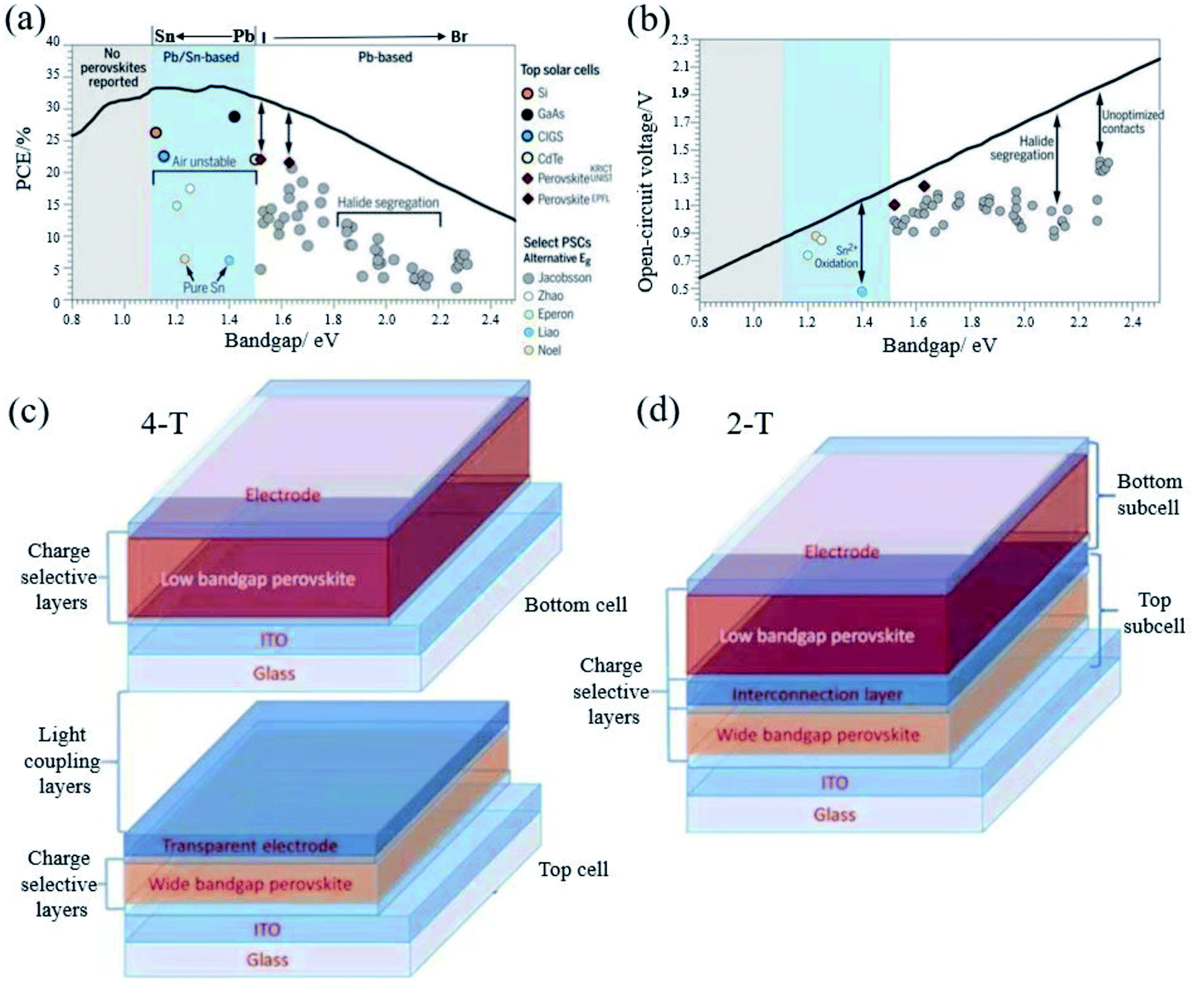 | ||
| Fig. 7 (a) Shockley–Queisser efficiency and (b) calculated maximum VOC (radiative limit). Reprinted with permission from ref. 32, copyright 2017, Science Publishing Group. Structure diagram of (c) 4-T and (d) 2-T solar cells. Reprinted with permission from ref. 96, copyright 2019, Advanced Functional Materials. | ||
However, so far, Sn–Pb perovskites have a certain distance from their theoretical efficiency. This is mainly because Sn2+ is easily oxidized to Sn4+, which leads to the generation of Sn vacancies. At the same time, the Sn–Pb mixed perovskite also has the problem of phase separation. In the future, there may be better ways to solve this problem and move closer to the S–Q limit.
3.3 Tandem solar cells
In 1961, Shockley and Queisser reported that the theoretical efficiency of a single junction solar cell was 33% via calculation.13 The tandem solar cell is considered to be the most likely means to break the S–Q limit. PSCs exhibit excellent photovoltaic performance, band gap tunability, low temperature and low-cost solution processability, which make them ideal candidates for the preparation of tandem solar cells. The maximum PCE is expected to exceed 40%.102 There are many types of tandem solar cells, and PSCs have important applications in tandem solar cells because of their adjustable band gap and low cost.Tandem cells mainly have two-terminal (2-T) and four-terminal (4-T) stacking methods (Fig. 7c and d).102 The 4-T series unit consists of independent wide band gap and narrow band gap devices. 4-T devices are mechanically stacked together, and the two sub-units are manufactured separately and connected by an external circuit. Although the production and operation process of 4-T is relatively simple, the additional two electrodes and corresponding optical loss usually result in high cost.101 In contrast to 4-T devices, 2-T cells are sequentially fabricated on a single substrate with a transparent front electrode and an opaque back electrode, where the front cell and the back cell are connected by an interconnect layer (ICL). As shown in Fig. 7d, ICL is where the recombination of photo-generated carriers from the front and back sub-cells occurs to maintain charge neutrality in the metal composite layer or the inter-band tunnel junction.103 2-T tandem solar cells can avoid additional manufacturing costs and spectrum loss, which make them more promising than 4-T tandem solar cells in practical applications. In 2-T tandem solar cells, because the narrow band gap and wide band gap solar cells are connected in series, the voltage generated by each sub-cell is added. Kirchhoff's law stipulates that the current flowing through each sub-cell must match, which means that the photocurrent is limited by the lower sub-cell current, and the two sets of cells collect different regions of the solar spectrum. In order to obtain a higher efficiency, it is necessary to achieve photocurrent matching under the condition of the maximum power point of each sub-cell, which requires that the band gaps of the bottom cell and the top cell are extremely matched. Due to the limitations of the manufacturing process and electrical coupling operation, manufacturing 2-T cells is practically more challenging than manufacturing 4-T cells and 4-T tandem solar cells. At present, PSCs are mainly composed of perovskite/perovskite tandem solar cells and perovskite/silicon tandem solar cells. Silicon and low band gap perovskites are generally used as bottom cells. For perovskites, Sn is generally doped to narrow the band gap of perovskites. Recently, the highest efficiency of perovskite/perovskite tandem solar cells has reached 25.6%.104 Perovskite/silicon tandem solar cells even reached 29.15%.105 A breakthrough of 30% is just around the corner. Long-term stability is an issue that must be paid attention to for tandem PSCs in order to achieve commercialization. Although 4-T is more expensive than 2-T in terms of production process, the stability of PSCs is still a big problem at present. 4-T tandem solar cells can be replaced with new cells after the perovskite efficiency has decayed. From this perspective, 4-T tandem solar cells are more suitable for commercialization than the 2-T ones.106 However, for actual operation, the glass, encapsulation materials and junction box in the 4-T can easily account for half or more of the cost of the panel. At the same time, further realizing the large-scale production of PSCs is also the only way to realize the commercialization of tandem cells. Amplifying the laboratory process and realizing the industry-university-research collaboration are vital.
3.4 Lifetime estimation
Limited by the problem of phase stability, there are still big gaps towards the actual work stability referred to International Electrotechnical Commission (IEC)-61265 standards, especially for the pure-phase α-FAPbI3. Consequently, several modification methods were employed to help the device endure the aging tests.Monica et al. first introduced a FAPbI3(0.85)MAPbBr3(0.15)-based traditional mesoporous n-i-p device, and the device operated for >1000 h under real outdoor conditions following the ISOS-1 procedures.107,108 Park et al. further incorporated an ultrathin two-dimensional (2D) perovskite (5-AVA)2PbI4 (5-AVA = 5-ammoniumvaleric acid) layer, acting as a passivation layer between (FAPbI3)0.88(CsPbBr3)0.12 and the hole transporting CuSCN layer in the mesoporous n-i-p structure device by forming 2D/3D heterostructures. The encapsulated device maintains 98% of the PCE after 63 days under moisture exposure of about 10% in the darkness.109 Dai et al. found that unsealed (Cs0.1FA0.9PbI3)0.9(FAPbBr3)0.1-based devices retain about 80% and 90% of the initial PCE at 85 °C with 20% relative humidity (RH) after 260 h and at room temperature with 45 ± 5% RH after 1440 h, respectively.110 Zhao et al. also prepared a 2D/3D-based device using (FAPbI3)n(BA2PbX4) (n = 60) as an absorber. They delivered excellent ambient stability with a t80 lifetime exceeding 2880 hours without encapsulation and a PCE as high as 20.62%.111 Sang et al. reported that an edge-encapsulated FAPbI3−XBrX-based PSC exhibits only 5.20% degradation during 1000 h maximum power point tracking under simultaneous damp heat (85 °C, 85% RH) and 1 sun light soaking test conditions.112
Seok et al. found that PSCs based on FAPbI3 doped with MMDACl2 maintained remarkable stability. As shown in Fig. 8a, the target device retained >90% of the initial PCE after 70 h under high humidity (85% RH, 25 °C), and the device maintained >90% of its initial PCE and exhibited greatly improved thermal stability monitored at 150 °C and ∼25% RH (Fig. 8b). The target device also exhibits high photostability, maintaining ∼90% of its initial PCE (>23.0%) over 600 h of irradiation (Fig. 8c).27
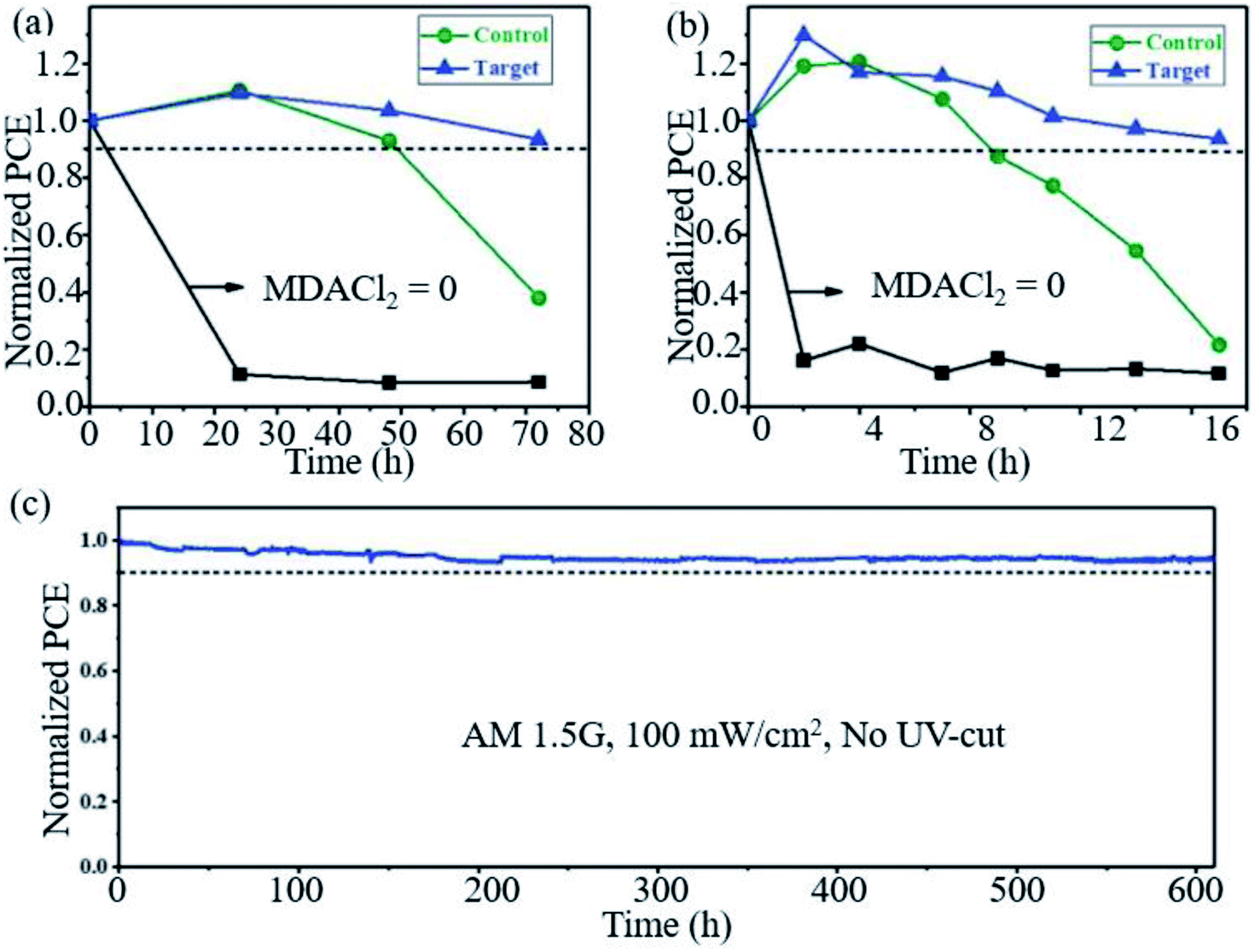 | ||
| Fig. 8 Long-term stability test. Comparison of (a) humidity (85% RH, 25 °C) and (b) thermal (150 °C, ∼25% RH) stability performances of unencapsulated control and target. (c) Maximum power point tracking measured with the encapsulated target device under full solar illumination (AM 1.5G, 100 mW cm−2 under ambient conditions) without a UV filter. Reprinted with permission from ref. 27, copyright 2019, Science Publishing Group. | ||
Hao et al. introduced a DMSO molecule process for improving the quality of Cs-(FAPbI3)0.85(MAPbBr3)0.15, and 1 cm2 devices retained 90% of the initial PCE after aging for 50 days in ambient air.113 Zhao et al. used a high-energy metastable 2D intermediate of MAFAPbI3Cl to prepare highly crystallized α-FAPbI3 with uniaxial-oriented nature at low temperatures. The unencapsulated device could retain over 95% of its initial PCE after 700 h of illumination, and the champion device could maintain ∼19% PCE after storage in a desiccator for 30 days.114 As shown in Fig. 9a, the 2D/3D IBA2FAPb2I7/α-FAPbI3-based PSCs reached a PCE close to 23%. The devices retained over 95% and 85% of its initial efficiency under simultaneous exposure with maximum power point tracking (MPPT) to full-simulated sunlight at room temperature and 80 °C over a period of 700 h and 500 h, respectively.76Fig. 9c shows that thiocyanate (SCN−)-based vapor-treated α-FAPbI3 PSCs retained 90% of the initial value (21.4%) after 500 hours of MPP measurements, and the PCE partially recovered to 20.2%, which is 94.4% of the initial value after 12 h of rest under open-circuit conditions in the darkness.86 Li et al. used a hydrazinium cation (HA+: NH2NH3+) to obtain novel 1D/3D-based PSCs, and the device retained 90% at room temperature in ambient environment for 2520 h.115 Kim et al. introduced FAPbI3 perovskite films with improved crystallinity and larger grain size by introducing 2% FAHCOO into the precursor solution, and they display that the PCE of the FAPbI3 cells declined by ∼35% after 1000 h of aging, whereas FAPbI3 with 2% FAHCOO additive-based cells (target) showed a degradation of only 10% (Fig. 10a). Fig. 10b shows that the target cell retained around 80% of its initial efficiency after 1000 h of aging, whereas the reference cell retained only about 40% at 60 °C under 20% relative humidity. Fig. 10c shows the PCE of the PSCs under continuous light soaking using a xenon lamp. The PCE of the target cell remained above 24% after 10 h MPP tracking, whereas that of the reference cell decreased to 22.8%. Fig. 10d shows that the PCE of the reference cell decreased by about 30%, whereas the target cell lost only around 15% of its initial efficiency.31 Yan et al. further prepared PSCs with remarkable stability. The PSCs triple retained 100% of the initial PCE performance over 104 days under ambient conditions (room temperature, RH = 50 ± 5%) without encapsulation. Han et al. innovatively presents that the triple-layer mesoporous structure may provide the nano-confine effect to stabilize pure MA perovskites, Cs/FA perovskites and inorganic pure Cs-based perovskites.5,116,117 We think that these confined nano structures may also help the stabilization of pure FA-based perovskites.
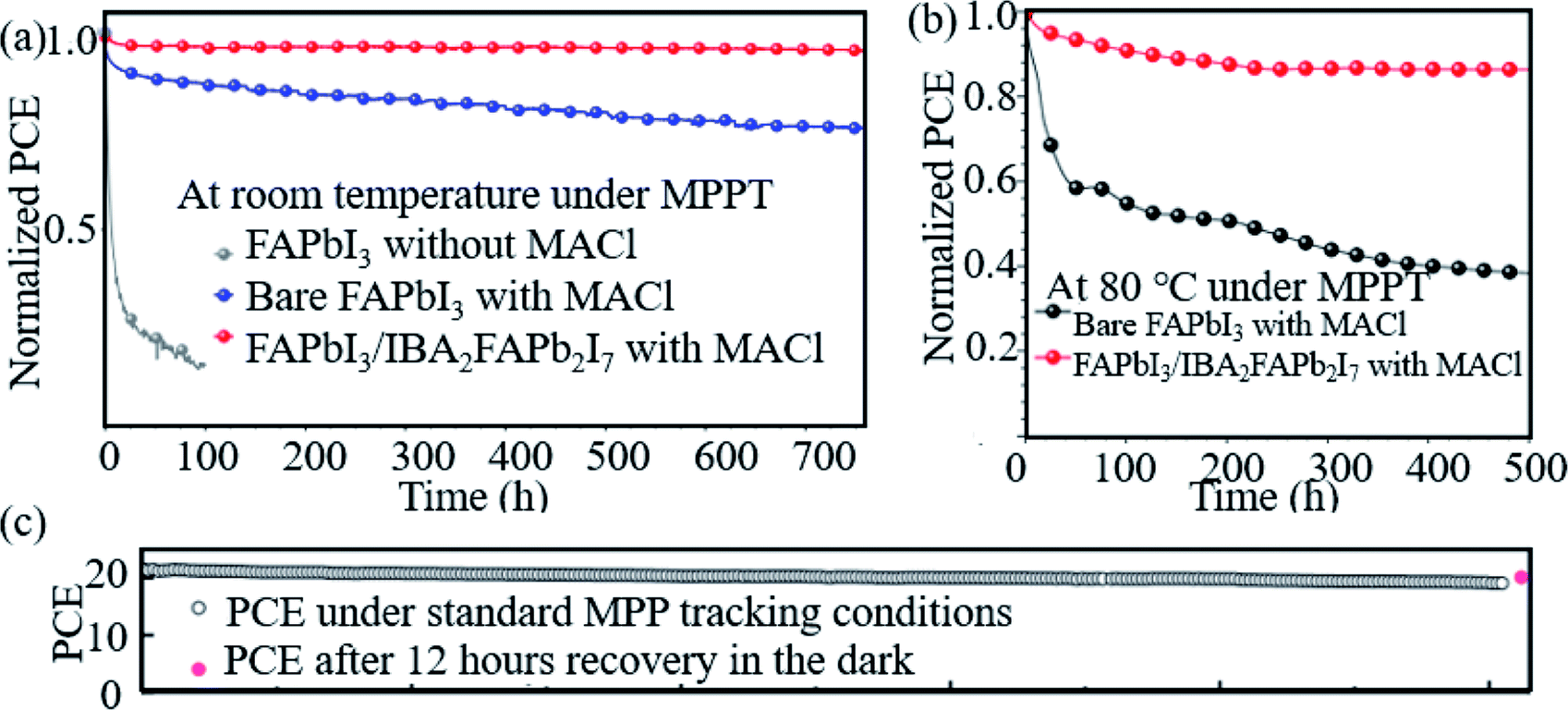 | ||
| Fig. 9 Aging results (a) and (b) of PSCs based on bare FAPbI3 (black dotted line) and α-FAPbI3/IBA2FAPb2I7 perovskites (red dotted line). Reprinted with permission from ref. 70, copyright 2020, Wiley. (c) Operational stability test of the vapor-treated FAPbI3-based PSCs under 500 hours of MPP-tracking conditions. Reprinted with permission from ref. 80, copyright 2020, Science Publishing Group. | ||
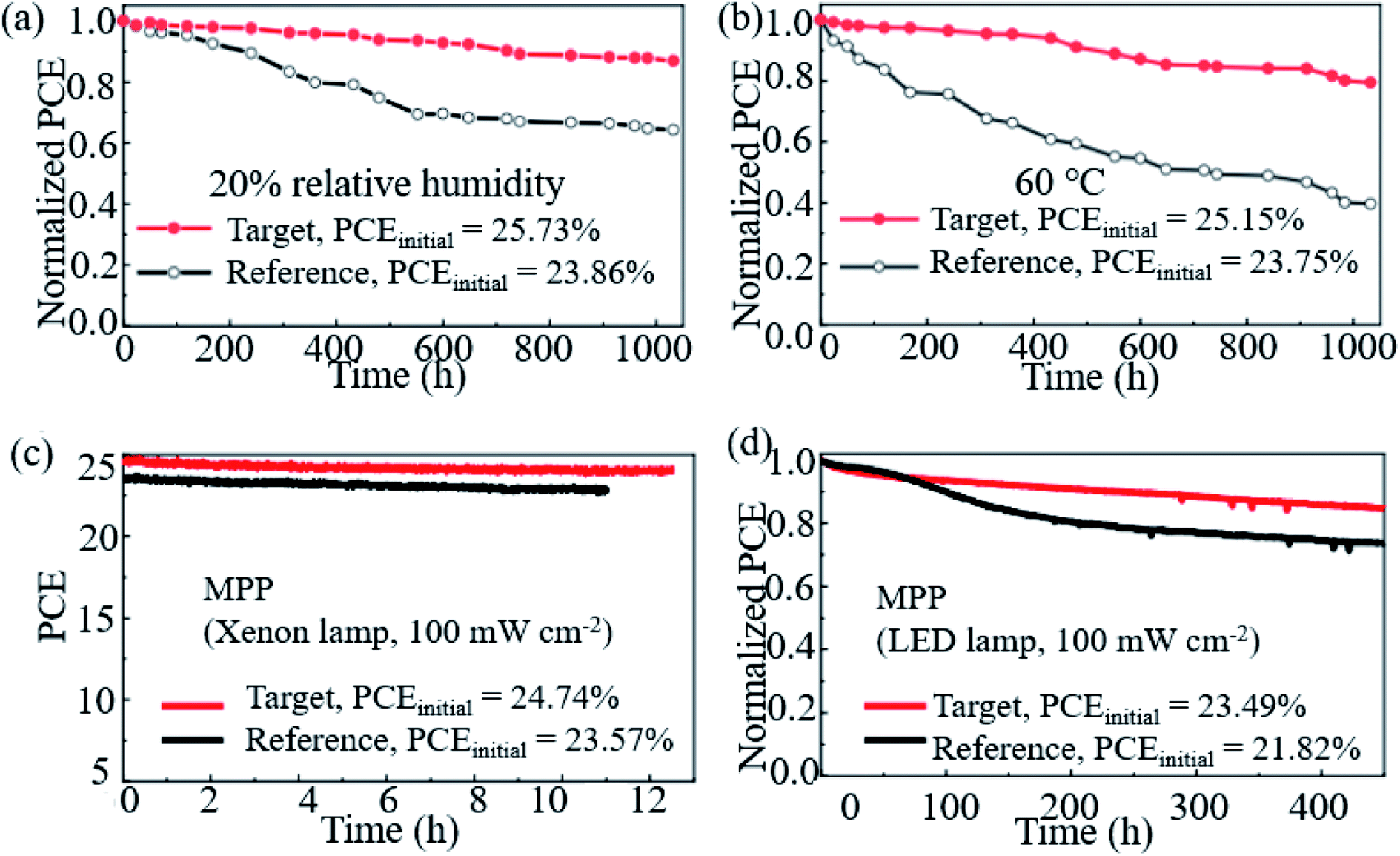 | ||
| Fig. 10 Stability of the FAPbI3 PSCs. (a) Shelf-life stability of the reference (FAPbI3) and target (FAPbI3 with 2% FAHCOO additive) PSCs. (b) Heat stability of the reference and target PSCs. (c) Operational stability of the reference and target PSCs. (d) Long-term operational stability of the reference and target PSCs. Reprinted with permission from ref. 31, copyright 2021, Nature Publishing Group. | ||
3.5 Possibility of preparation in ambient air
To further push the advancement to commercialize PSCs, the preparation of PSCs under ambient air conditions even at moderate/high RH will be top priority undoubtedly.Zhao et al. used two-dimensional Ruddlesden–Popper phases to passivate the FAPbI3 perovskite and achieved a PCE as high as 20.62% and remarkable long-term ambient stability with a t80 lifetime exceeding 2880 hours without encapsulation (30–40% relative humidity and 25 °C).111 Jiang et al. introduced an N-methyl pyrrolidone (NMP) additive-based method to prepare FAPbI3 layers in ambient air under a RH of ∼40%, demonstrating the reliability for pure FA-perovskite ambient-air fabrication.118 Huang et al. used methylamine formate to synthesize stable α-FAPbI3 in ambient air regardless of the humidity and temperature, and the unencapsulated cells retain 80 and 90% of their initial efficiencies for 500 hours at 85 °C and continuous light stress.119 Salim et al. also further showed that the long-term stability of NMP additive-pure FAPbI3 PSCs shows a dramatic increase when prepared under ambient air compared to PSCs made under nitrogen.120 Ma et al. incorporated methylamine thiocyanate as a stabilizer, and high-quality and stable α-phase FA-based perovskites in ambient 50 ± 5% RH even at room temperature.121 Only Huang et al.119 really prepared the FAPbI3 PSCs at high humidity in the atmospheric air and obtained excellent stability. Actually, there are lots of difficulties in the preparation of FAPbI3 perovskites in ambient air. Maybe the combination of nano-confine effects and ion-liquid additive/solvent methods will do the positive effect for the preparation of FAPbI3 in ambient air, and further to advance the industry.
4. Conclusion
Pure α-FAPbI3 will become the main research focus in the field of perovskite in the future. The key issue is how to eliminate lattice stress and stabilize the pure α-FAPbI3 phase. To further improve the performance and stability of PSCs, we think that a triple-layer mesoporous structure may assist in achieving the stability of perovskites. At the same time, we can continue to explore applications in lower band gaps, such as Sn–Pb PSCs, and also study tandem solar cells to further approach the S–Q limit efficiency. In the future scientific research work, it is necessary to try to use more convenient technical methods, which are easy to scale and make industrialization possible.Author contributions
Anyi Mei and Hongwei Han designed and directed this paper. Ziwei Zheng and Shiyu Wang wrote this perspective. Yue Hu and Yaoguang Rong revised this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 91733301, 21702069, 51572102 and 51972133), the Fundamental Research Funds for the Central Universities, the Science and Technology Department of Hubei Province (No. 2017AAA190), the 111 Project (No. B07038) and the Program for HUST Academic Frontier Youth Team (2016QYTD06).References
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Gratzel and N. G. Park, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- J. M. Ball, M. M. Lee, A. Hey and H. J. Snaith, Energy Environ. Sci., 2013, 6, 1739–1743 RSC.
- J.-Y. Jeng, Y.-F. Chiang, M.-H. Lee, S.-R. Peng, T.-F. Guo, P. Chen and T.-C. Wen, Adv. Mater., 2013, 25, 3727–3732 CrossRef CAS PubMed.
- K.-C. Wang, J.-Y. Jeng, P.-S. Shen, Y.-C. Chang, E. W.-G. Diau, C.-H. Tsai, T.-Y. Chao, H.-C. Hsu, P.-Y. Lin, P. Chen, T.-F. Guo and T.-C. Wen, Sci. Rep., 2014, 4, 4756 CrossRef PubMed.
- A. Mei, X. Li, L. Liu, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Gratzel and H. Han, Science, 2014, 345, 295–298 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-b. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- N. Ahn, D.-Y. Son, I.-H. Jang, S. M. Kang, M. Choi and N.-G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef CAS PubMed.
- D.-Y. Son, J.-W. Lee, Y. J. Choi, I.-H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N.-G. Park, Nat. Energy, 2016, 1, 16081 CrossRef CAS.
- S. S. Shin, E. J. Yeom, W. S. Yang, S. Hur, M. G. Kim, J. Im, J. Seo, J. H. Noh and S. I. Seok, Science, 2017, 356, 167–171 CrossRef CAS PubMed.
- W.-Q. Wu, Q. Wang, Y. Fang, Y. Shao, S. Tang, Y. Deng, H. Lu, Y. Liu, T. Li and Z. Yang, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- K. Wang, C. Wu, Y. Hou, D. Yang, T. Ye, J. Yoon, M. Sanghadasa and S. Priya, Energy Environ. Sci., 2020, 13, 3412–3422 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- S. Rühle, Sol. Energy, 2016, 130, 139–147 CrossRef.
- F. Ma, J. Li, W. Li, N. Lin, L. Wang and J. Qiao, Chem. Sci., 2017, 8, 800–805 RSC.
- E. J. Juarez-Perez, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 16912–16919 RSC.
- T. M. Koh, K. Fu, Y. Fang, S. Chen, T. C. Sum, N. Mathews, S. G. Mhaisalkar, P. P. Boix and T. Baikie, J. Phys. Chem. C, 2013, 118, 16458–16462 CrossRef.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 RSC.
- N. Pellet, P. Gao, G. Gregori, T. Y. Yang, M. K. Nazeeruddin, J. Maier and M. Gratzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- J. W. Lee, D. J. Seol, A. N. Cho and N. G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- J. W. Lee, D. H. Kim, H. S. Kim, S. W. Seo, S. M. Cho and N. G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- M. Saliba, T. Matsui, K. Domanski, J. Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J. P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- L. Xie, K. Lin, J. Lu, W. Feng, P. Song, C. Yan, K. Liu, L. Shen, C. Tian and Z. Wei, J. Am. Chem. Soc., 2019, 141, 20537–20546 CrossRef CAS PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Science, 2019, 366, 749–753 CrossRef CAS PubMed.
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. I. Seok, Science, 2020, 370, 108–112 CrossRef CAS PubMed.
- M. Kim, G.-H. Kim, T. K. Lee, I. W. Choi, H. W. Choi, Y. Jo, Y. J. Yoon, J. W. Kim, J. Lee and D. Huh, Joule, 2019, 3, 2179–2192 CrossRef CAS.
- Q. Yao, Q. Xue, Z. Li, K. Zhang, T. Zhang, N. Li, S. Yang, C. J. Brabec, H. L. Yip and Y. Cao, Adv. Mater., 2020, 32, 2000571 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer and M. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- J. H. Im, C. R. Lee, J. W. Lee, S. W. Park and N. G. Park, Nanoscale, 2011, 3, 4088–4093 RSC.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- Y. Wang, W. Chen, L. Wang, B. Tu, T. Chen, B. Liu, K. Yang, C. W. Koh, X. Zhang and H. Sun, Adv. Mater., 2019, 31, 1902781 CrossRef PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim and J. H. Noh, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS PubMed.
- M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J.-H. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 369, 1615–1620 CrossRef CAS PubMed.
- J.-P. Correa-Baena, M. Saliba, T. Buonassisi, M. Grätzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T.-W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582–587 Search PubMed.
- L. M. Herz, ACS Energy Lett., 2017, 2, 1539–1548 Search PubMed.
- G. Giorgi, J.-I. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. Lett., 2013, 4, 4213–4216 Search PubMed.
- E. Smecca, Y. Numata, I. Deretzis, G. Pellegrino, S. Boninelli, T. Miyasaka, A. La Magna and A. Alberti, Phys. Chem. Chem. Phys., 2016, 18, 13413–13422 RSC.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- W. Travis, E. Glover, H. Bronstein, D. Scanlon and R. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Gratzel and F. De Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. t. Péchy, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, M. Saski, P. Yadav, D. Bi, N. Pellet, J. Lewiński, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2018, 140, 3345–3351 CrossRef CAS PubMed.
- O. Gunawan, S. R. Pae, D. M. Bishop, Y. Virgus, J. H. Noh, N. J. Jeon, Y. S. Lee, X. Shao, T. Todorov and D. B. Mitzi, Nature, 2019, 575, 151–155 CrossRef CAS PubMed.
- J. Y. Kim, J. W. Lee, H. S. Jung, H. Shin and N. G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- X. Zhang, J.-X. Shen, M. E. Turiansky and C. G. Van de Walle, Nat. Mater., 2021, 1–6 Search PubMed.
- M. T. Weller, O. J. Weber, J. M. Frost and A. Walsh, J. Phys. Chem. Lett., 2015, 6, 3209–3212 CrossRef CAS.
- S. Masi, A. F. Gualdrón-Reyes and I. Mora-Sero, ACS Energy Lett., 2020, 5, 1974–1985 CrossRef CAS.
- I. Deretzis, A. Alberti, G. Pellegrino, E. Smecca, F. Giannazzo, N. Sakai, T. Miyasaka and A. J. A. P. L. La Magna, Appl. Phys. Lett., 2015, 106, 131904 CrossRef.
- X. Zhang, M. E. Turiansky, J.-X. Shen and C. G. Van de Walle, Phys. Rev. B, 2020, 101, 140101 CrossRef CAS.
- M. Yavari, F. Ebadi, S. Meloni, Z. S. Wang, T. C.-J. Yang, S. Sun, H. Schwartz, Z. Wang, B. Niesen, J. Durantini, P. Rieder, K. Tvingstedt, T. Buonassisi, W. C. H. Choy, A. Filippetti, T. Dittrich, S. Olthof, J.-P. Correa-Baena and W. Tress, J. Mater. Chem. A, 2019, 7, 23838–23853 RSC.
- X. Zhang, J.-X. Shen, M. E. Turiansky and C. G. Van de Walle, J. Mater. Chem. A, 2020, 8, 12964–12967 RSC.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulovic, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590 Search PubMed.
- O. J. Weber, D. Ghosh, S. Gaines, P. F. Henry, A. B. Walker, M. S. Islam and M. T. Weller, Chem. Mater., 2018, 30, 3768–3778 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- T. Chen, B. J. Foley, C. Park, C. M. Brown, L. W. Harriger, J. Lee, J. Ruff, M. Yoon, J. J. Choi and S.-H. Lee, Sci. Adv., 2016, 2, e1601650 CrossRef PubMed.
- J. Lin, M. Lai, L. Dou, C. S. Kley, H. Chen, F. Peng, J. Sun, D. Lu, S. A. Hawks and C. Xie, Nat. Mater., 2018, 17, 261–267 CrossRef CAS PubMed.
- H. Wei, S. Chen, J. Zhao, Z. Yu and J. Huang, Chem. Mater., 2020, 32, 2501–2507 CrossRef CAS.
- Y. Deng, S. Xu, S. Chen, X. Xiao, J. Zhao and J. Huang, Nat. Energy, 2021, 6, 633–641 CrossRef CAS.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Grätzel, S. M. Zakeeruddin, U. Röthlisberger and M. Grätzel, Energy Environ. Sci., 2016, 9, 656–662 RSC.
- N. Rolston, K. A. Bush, A. D. Printz, A. Gold-Parker, Y. Ding, M. F. Toney, M. D. McGehee and R. H. Dauskardt, Adv. Energy Mater., 2018, 8, 1802139 CrossRef.
- L. Zhang, W. Geng, C.-j. Tong, X. Chen, T. Cao and M. Chen, Sci. Rep., 2018, 8, 1–9 Search PubMed.
- N. Li, Y. Luo, Z. Chen, X. Niu, X. Zhang, J. Lu, R. Kumar, J. Jiang, H. Liu and X. Guo, Joule, 2020, 4, 1743–1758 CrossRef CAS.
- S. Yang, W. Liu, L. Zuo, X. Zhang, T. Ye, J. Chen, C.-Z. Li, G. Wu and H. Chen, J. Mater. Chem. A, 2016, 4, 9430–9436 RSC.
- F. Yang, L. Dong, D. Jang, K. C. Tam, K. Zhang, N. Li, F. Guo, C. Li, C. Arrive and M. Bertrand, Adv. Energy Mater., 2020, 10, 2001869 CrossRef CAS.
- S. Akin, E. Akman and S. Sonmezoglu, Adv. Funct. Mater., 2020, 30, 2002964 CrossRef CAS.
- X. Xu, H. Zheng, G. Liu, L. Zhu, D. He, S. Xu, H. Xu, L. Zhang, X. Zhang and X. Pan, ChemSusChem, 2020, 13, 956–963 CrossRef CAS PubMed.
- J.-W. Lee, Z. Dai, C. Lee, H. M. Lee, T.-H. Han, N. De Marco, O. Lin, C. S. Choi, B. Dunn and J. Koh, J. Am. Chem. Soc., 2018, 140, 6317–6324 CrossRef CAS PubMed.
- Y. Rong, X. Hou, Y. Hu, A. Mei, L. Liu, P. Wang and H. J. N. c. Han, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- Y. Liu, S. Akin, A. Hinderhofer, F. T. Eickemeyer, H. Zhu, J. Y. Seo, J. Zhang, F. Schreiber, H. Zhang and S. M. Zakeeruddin, Angew. Chem., Int. Ed., 2020, 59, 15688–15694 CrossRef CAS PubMed.
- H. Zhang, Z. Chen, M. Qin, Z. Ren, K. Liu, J. Huang, D. Shen, Z. Wu, Y. Zhang, J. Hao, C.-s. Lee, X. Lu, Z. Zheng, W. Yu and G. Li, Adv. Mater., 2021, 2008487 CrossRef CAS PubMed.
- M. Lyu and N.-G. Park, Sol. RRL, 2020, 4, 2000331 CrossRef CAS.
- C. Mu, J. Pan, S. Feng, Q. Li and D. Xu, Adv. Energy Mater., 2017, 7, 1601297 CrossRef.
- J. Qing, X.-K. Liu, M. Li, F. Liu, Z. Yuan, E. Tiukalova, Z. Yan, M. Duchamp, S. Chen, Y. Wang, S. Bai, J.-M. Liu, H. J. Snaith, C.-S. Lee, T. C. Sum and F. Gao, Adv. Energy Mater., 2018, 8, 1800185 CrossRef.
- S. Masi, C. Echeverría-Arrondo, K. M. Salim, T. T. Ngo, P. F. Mendez, E. López-Fraguas, D. F. Macias-Pinilla, J. Planelles, J. I. Climente and I. Mora-Sero, ACS Energy Lett., 2020, 5, 418–427 CrossRef CAS.
- Y. Fu, T. Wu, J. Wang, J. Zhai, M. J. Shearer, Y. Zhao, R. J. Hamers, E. Kan, K. Deng and X.-Y. Zhu, Nano Lett., 2017, 17, 4405–4414 CrossRef CAS PubMed.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T. Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS PubMed.
- X. Chen, Y. Xia, Q. Huang, Z. Li, A. Mei, Y. Hu, T. Wang, R. Cheacharoen, Y. Rong and H. J. A. E. M. Han, Adv. Energy Mater., 2021, 2100292 CrossRef CAS.
- M. Que, Z. Dai, H. Yang, H. Zhu, Y. Zong, W. Que, N. P. Padture, Y. Zhou and O. Chen, ACS Energy Lett., 2019, 4, 1970–1975 CrossRef CAS.
- H. Lu, Y. Liu, P. Ahlawat, A. Mishra, W. R. Tress, F. T. Eickemeyer, Y. Yang, F. Fu, Z. Wang and C. E. Avalos, Science, 2020, 370 Search PubMed.
- L. Gu, D. Zhang, M. Kam, Q. Zhang, S. Poddar, Y. Fu, X. Mo and Z. Fan, Nanoscale, 2018, 10, 15164–15172 RSC.
- Y. Y. Zhou, J. Kwun, H. F. Garces, S. P. Pang and N. P. Padture, Chem. Commun., 2016, 52, 7273–7275 RSC.
- A. Mei, Y. Sheng, Y. Ming, Y. Hu, Y. Rong, W. Zhang, S. Luo, G. Na, C. Tian, X. Hou, Y. Xiong, Z. Zhang, S. Liu, S. Uchida, T.-W. Kim, Y. Yuan, L. Zhang, Y. Zhou and H. Han, Joule, 2020, 4, 2646–2660 CrossRef CAS.
- Y. Chen, Y. Lei, Y. Li, Y. Yu, J. Cai, M.-H. Chiu, R. Rao, Y. Gu, C. Wang and W. Choi, Nature, 2020, 577, 209–215 CrossRef CAS PubMed.
- V. L. Pool, B. Dou, D. G. Van Campen, T. R. Klein-Stockert, F. S. Barnes, S. E. Shaheen, M. I. Ahmad, M. F. Van Hest and M. F. Toney, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- G. Tong, D.-Y. Son, L. K. Ono, H.-B. Kang, S. He, L. Qiu, H. Zhang, Y. Liu, J. Hieulle and Y. Qi, Nano Energy, 2021, 87, 106152 CrossRef CAS.
- Y. Zhang, S. Seo, S. Y. Lim, Y. Kim, S.-G. Kim, D.-K. Lee, S.-H. Lee, H. Shin, H. Cheong and N.-G. Park, ACS Energy Lett., 2019, 5, 360–366 CrossRef.
- C. Ma and N.-G. Park, Chem, 2020, 6, 1254–1264 CAS.
- W. Tress, N. Marinova, O. Inganäs, M. K. Nazeeruddin, S. M. Zakeeruddin and M. Graetzel, Adv. Energy Mater., 2015, 5, 1400812 CrossRef.
- D. Zhao, C. Chen, C. Wang, M. M. Junda, Z. Song, C. R. Grice, Y. Yu, C. Li, B. Subedi, N. J. Podraza, X. Zhao, G. Fang, R.-G. Xiong, K. Zhu and Y. Yan, Nat. Energy, 2018, 3, 1093–1100 CrossRef CAS.
- M. Y. Wei, K. Xiao, G. Walters, R. X. Lin, Y. B. Zhao, M. I. Saidaminov, P. Todorovic, A. Johnston, Z. R. Huang, H. J. Chen, A. D. Li, J. Zhu, Z. Y. Yang, Y. K. Wang, A. H. Proppe, S. O. Kelley, Y. Hou, O. Voznyy, H. R. Tan and E. H. Sargent, Adv. Mater., 2020, 32, 8 Search PubMed.
- X. Jiang, H. Li, Q. Zhou, Q. Wei, M. Wei, L. Jiang, Z. Wang, Z. Peng, F. Wang, Z. Zang, K. Xu, Y. Hou, S. Teale, W. Zhou, R. Si, X. Gao, E. H. Sargent and Z. Ning, J. Am. Chem. Soc., 2021, 143, 10970–10976 CrossRef CAS PubMed.
- G. Kapil, T. Bessho, T. Maekawa, A. K. Baranwal, Y. Zhang, M. A. Kamarudin, D. Hirotani, Q. Shen, H. Segawa and S. Hayase, Adv. Energy Mater., 2021, 2101069 CrossRef CAS.
- X. Y. Zhou, L. Z. Zhang, X. Z. Wang, C. Liu, S. Chen, M. Q. Zhang, X. N. Li, W. D. Yi and B. M. Xu, Adv. Mater., 2020, 32, 8 Search PubMed.
- J. Tong, Z. Song, D. H. Kim, X. Chen, C. Chen, A. F. Palmstrom, P. F. Ndione, M. O. Reese, S. P. Dunfield and O. G. Reid, Science, 2019, 364, 475–479 CrossRef CAS PubMed.
- C. L. Wang, Z. N. Song, C. W. Li, D. W. Zhao and Y. F. Yan, Adv. Funct. Mater., 2019, 29, 30 Search PubMed.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang, M. Wei, M. I. Saidaminov, Y. Gao, J. Xu and M. Xiao, Nat. Energy, 2019, 4, 864–873 CrossRef CAS.
- K. Xiao, R. Lin, Q. Han, Y. Hou, Z. Qin, H. T. Nguyen, J. Wen, M. Wei, V. Yeddu and M. I. Saidaminov, Nat. Energy, 2020, 5, 870–880 CrossRef CAS.
- A. Al-Ashouri, E. Köhnen, B. Li, A. Magomedov, H. Hempel, P. Caprioglio, J. A. Márquez, A. B. M. Vilches, E. Kasparavicius and J. A. Smith, Science, 2020, 370, 1300–1309 CrossRef CAS PubMed.
- R. Wang, T. Huang, J. Xue, J. Tong, K. Zhu and Y. Yang, Nat. Photonics, 2021, 15, 411–425 CrossRef CAS.
- Y. Reyna, M. Salado, S. Kazim, A. Pérez-Tomas, S. Ahmad and M. Lira-Cantu, Nano Energy, 2016, 30, 570–579 CrossRef CAS.
- M. O. Reese, S. A. Gevorgyan, M. Jørgensen, E. Bundgaard, S. R. Kurtz, D. S. Ginley, D. C. Olson, G. Yaman-Uzunoglu, J.-B. Bonekamp, A. J. J. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2011, 95, 1253–1267 CrossRef CAS.
- J. Chen, J.-Y. Seo and N.-G. Park, Adv. Energy Mater., 2018, 8, 1702714 CrossRef.
- G. Liu, H. Zheng, L. Zhu, A. Alsaedi, T. Hayat, X. Pan, L. e. Mo and S. Dai, ChemSusChem, 2018, 11, 2436–2443 CrossRef CAS PubMed.
- T. Niu, J. Lu, M.-C. Tang, D. Barrit, D.-M. Smilgies, Z. Yang, J. Li, Y. Fan, T. Luo, I. McCulloch, A. Amassian, S. Liu and K. Zhao, Energy Environ. Sci., 2018, 11, 3358–3366 RSC.
- J. H. Heo, Y. K. Choi, C. W. Koh, H. Y. Woo and S. H. Im, Adv. Mater. Technol., 2019, 4, 1800390 CrossRef.
- X. Liu, L. Shi, J. Huang, Z. Liu, P. Zhang, J. S. Yun, A. M. Soufiani, J. Seidel, K. Sun, Z. Hameiri, J. A. Stride, Y. Zhang, M. A. Green, H. Lin and X. Hao, Sol. RRL, 2019, 3, 1800338 CrossRef.
- T. Zhang, Q. Xu, F. Xu, Y. Fu, Y. Wang, Y. Yan, L. Zhang and Y. Zhao, Sci. Bull., 2019, 64, 1608–1616 CrossRef CAS.
- S. Yu, H. Liu, S. Wang, H. Zhu, X. Dong and X. Li, Chem. Eng. J., 2021, 403, 125724 CrossRef CAS.
- X. Hou, M. Xu, C. Tong, W. Ji, Z. Fu, Z. Wan, F. Hao, Y. Ming, S. Liu, Y. Hu, H. Han, Y. Rong and Y. Yao, J. Power Sources, 2019, 415, 105–111 CrossRef CAS.
- S. Wang, W. Shen, Y. Chu, W. Zhang, L. Hong, A. Mei, Y. Rong, Y. Tang, Y. Hu and H. Han, J. Phys. Chem. Lett., 2020, 11, 9689–9695 CrossRef CAS PubMed.
- G. Wang, L. Wang, J. Qiu, Z. Yan, K. Tai, W. Yu and X. Jiang, Sol. Energy, 2019, 187, 147–155 CrossRef CAS.
- W. Hui, L. Chao, H. Lu, F. Xia, Q. Wei, Z. Su, T. Niu, L. Tao, B. Du, D. Li, Y. Wang, H. Dong, S. Zuo, B. Li, W. Shi, X. Ran, P. Li, H. Zhang, Z. Wu, C. Ran, L. Song, G. Xing, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Science, 2021, 371, 1359–1364 CrossRef CAS PubMed.
- K. M. Salim, S. Masi, A. F. Gualdrón-Reyes, R. S. Sánchez, E. M. Barea, M. Kreĉmarová, J. F. Sánchez-Royo and I. Mora-Seró, ACS Energy Lett., 2021, 6, 3511–3521 CrossRef CAS PubMed.
- J. Zhao, G. He, D. Yang, D. Guo, L. Yang, J. Chen and D. Ma, Sustainable Energy Fuels, 2021, 5, 4268–4276 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |