
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReview on fluorinated nucleoside/non-nucleoside FDA-approved antiviral drugs
Magda M. F. Ismaila and
Mohammed Salah Ayoup *b
*b
aDepartment of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo 11754, Egypt
bDepartment of Chemistry, Faculty of Science, Alexandria University, P. O. Box 426, Alexandria 21321, Egypt. E-mail: mohammedsalahayoup@gmail.com
First published on 31st October 2022
Abstract
FDA-approved antiviral agents represent an important class that has attracted attention in recent years to combat current and future threats of viral pandemics. Fluorine ameliorates the electronic, lipophilic and steric problems of drugs. Additionally, fluorine can prolong drug activity and improve metabolic stability, thereby, modifying their pharmacodynamic and pharmacokinetic character. Herein, we summarized the fluorinated FDA-approved antiviral agents, dealing with biological aspects, mechanisms of action, and synthetic pathways.
1. Introduction
Viruses are amongst the dominant pathogenic organisms that cause a lot of severe diseases.1 Advancing approaches for the treatment of viral infections focuses on either the viruses or the infected host cell. Some antiviral agents act directly as inhibitors of viral attachment, uncoating, entry, or fusion,2 and some inhibit RNA polymerase,3 protease,4 integrase,5 nucleoside, or nonnucleoside reverse transcriptase.6,7During the 2010s, the protease viral DNA polymerase and integrase inhibitors were indexed among the top 200 marketed drugs. To date, there is no effectual antiviral agent for numerous viral diseases. Nevertheless, a pair of drugs has been discovered for herpes viruses, many drugs for influenza and a number of therapeutics for hepatitis C infection and human immunodeficiency virus type 1 (HIV-1).
Antiviral drugs are primarily transformed to their triphosphate then inhibit viral DNA synthesis. Mechanisms of famous antiviral drugs have been analyzed revealing their ability to improve the body's resistance to a virus,8 repress adsorption or diffusion of the virus into the cell and denature viral protein. The inhibition of nucleic acid synthesis is another mechanism for antimetabolites.9
The modifications brought to the ribose or 2′-deoxyribose moiety of a nucleoside comprise changes in the sugar moiety, oxygen replacement with an isostere, ring size changes, or replacement with an open-chain moiety. These modifications may lead to astonishing discrepancies in toxicity and biological assets.10
It is well known that the high electronegativity of fluorine results in the polarization of adjacent bonds resulting in acidity enhancement and a decrease in the basicity of the substituents in close proximity. The electronegativity gives rise to a more polarizable C–F bond with increased electrostatic character. Conventionally, C–F bonds can bind with their receptors by either dipole or electrostatic interaction.11,12 The pKa is increased, which can affect the potency, selectivity, and toxicity.13,14 The hydroxyl group is always replaced by fluorine,15 which is characterized by the same polarity and ability to form a hydrogen bond.16 The introduction of a fluorine atom or trifluoromethyl group (–CF3) changes both the pharmacodynamics and pharmacokinetics (absorption, distribution, metabolism, and excretion) of the molecule.17–30 Fluorine is a metabolic blocker; the blocking occurs when the C–H bond is replaced by a C–F bond which has a high impact on the biological half-life and bioavailability enhancement.17 Fluorine is small compared to the hydrogen atom, therefore, replacing the hydrogen atom with fluorine can considerably increase the binding affinity of a drug with receptors via the formation of noncovalent bonds. On the other hand, cell penetration is increased due to less plasma protein binding and a higher free fraction of the drug. These outcomes lead to a greatly improved therapeutic activity.31
Among various antiviral drugs presently used, this review will address the mechanisms of action, synthetic pathways, and therapeutic uses of the fluorinated U.S. Food and Drug Administration (FDA)-approved antiviral agents over the last four decades. Furthermore, the biological value of the fluorine substituent will be clarified for the antiviral activity against HIV type-1, COVID-19, influenza, smallpox, acute asthma, and hepatitis C.
2. Treatment of smallpox
Tecovirimat (ST-246) has been endorsed in the United States and approved in 2018 as a therapy for smallpox, monkeypox, and orthopoxvirus diseases for which it showed potent antiviral activity. This drug blocks the viral escape from an infected cell, burdening the dissemination of the virus within the body. The para-trifluoromethyl group has an important role in the optimal metabolic stability and pharmacokinetic profile of this drug.32 Tecovirimat (7) was synthesized in 2014 by Dai using versatile methods, starting from the reaction of maleic anhydride (1) with monoBoc hydrazine to afford the imide derivative 2. Boc deprotection of 2 using 4 M HCl afforded N-amino maleimide hydrochloride 3 which can be acylated by p-trifluoromethyl benzoyl chloride 4 to give the hydrazide derivative 5. Then, cyclo-addition of 5 was achieved with cycloheptatriene in xylene to produce tecovirimat, 7, in excellent yield as shown in Scheme 1.33 | ||
| Scheme 1 Synthesis of tecovirimat (7) from maleic anhydride. | ||
Another modified protocol was employed to prepare tecovirimat starting from the cyclo-addition reaction of maleic anhydride, 1, with cycloheptatriene, 6, to yield the adduct 8, which after reaction with BocNHNH2 provided the fused imide derivative, 9. Deprotection of 9 in 4 M HCl gave rise to the free hydrazine derivative, 10, which is finally acylated by p-trifluoromethylbenzoyl chloride, 4, to afford 7 (Scheme 2).34
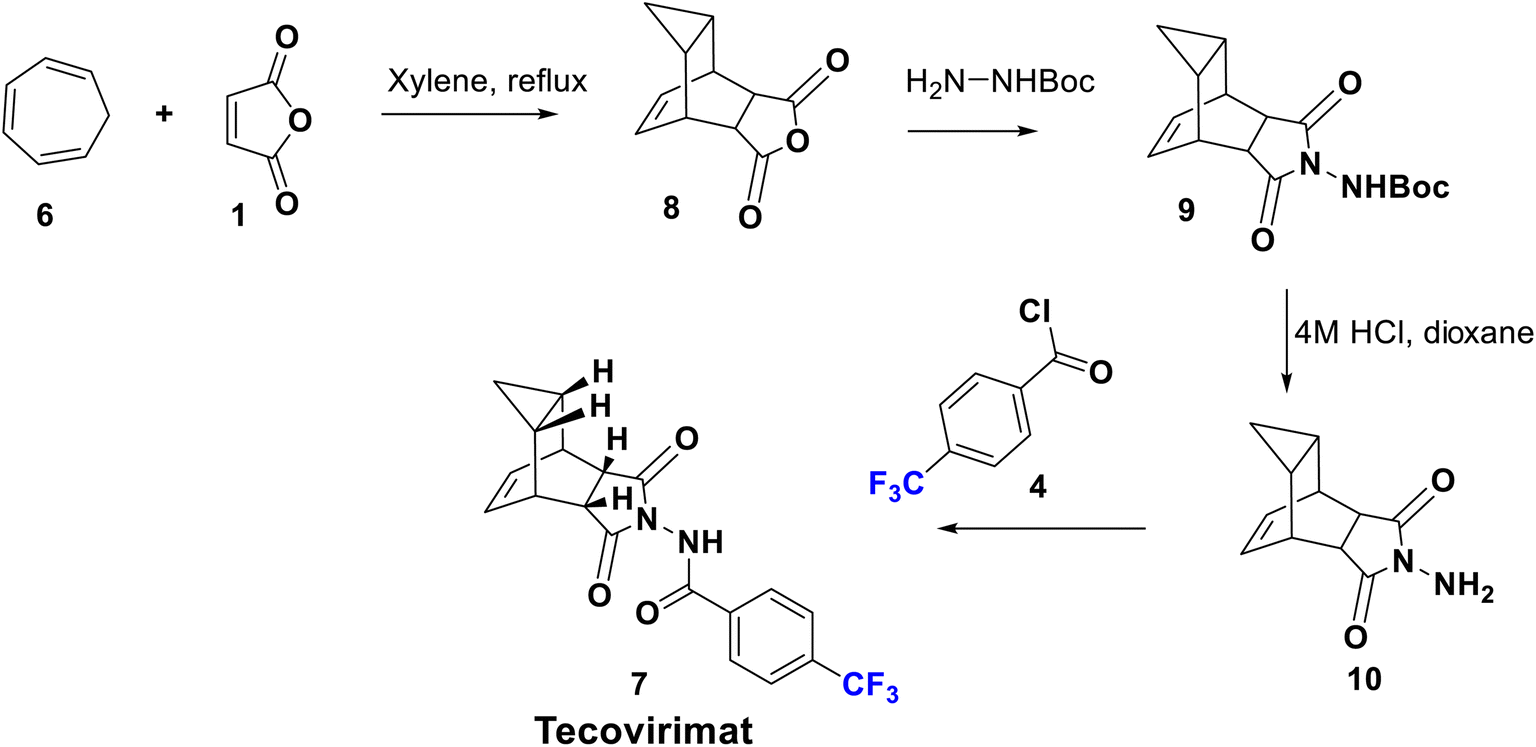 | ||
| Scheme 2 Synthesis of tecovirimat (7) from heptatriene. | ||
3. Treatment of acute asthma
Pleconaril (Picovir) was originally manufactured by Schering-Plough, who made an agreement with ViroPharma in 2004 to license intranasal pleconaril for the treatment of the common cold in the US and Canada. Pleconaril is active against viruses in the picornavirus group; it is accommodated in a hydrophobic pocket of the virion, which leads to its rigidification and viral capsid compression in the capsid structural protein. Consequently, it prevents the viral attachment and uncoating process in the host cell, a crucial step in the life cycle of the virus.35 Pleconaril is employed for the prevention of asthma exacerbation and currently, its intranasal spray formulation is in clinical stages for treatment of rhinovirus infections, while the oral medication is only formerly used due to reported aftereffects. A remarkable enhancement of its activity was observed due to the combination of two factors: the bulky trifluoromethyl group which interferes with the enzyme binding, and its effect as a metabolic blocker, especially against cytochrome P450 enzymes to protect the oxadiazole ring from hydroxylation.35 The U.S. Food and Drug Administration rejected pleconaril in 2002 due to the fact that it has been found to induce CYP3A enzyme activity, therefore increasing the risk of serious drug interactions.The synthesis of pleconaril (18) by Diana et al. in 1995 is shown in Scheme 3, starting from the reaction of dimethyl isoxazole, 11, with oxirane (12) via carbon–carbon bond formation using BuLi to afford the primary alcohol, 13, which provided the corresponding chloro derivative, 14, upon treatment with thionyl chloride. Then, Williamson synthesis was carried out via reaction of 14 with the phenol 15 via SN2 reaction, to yield the ether 16 which upon addition of hydroxylamine to the nitrile functionality gave the corresponding amidoxime, 17. Subsequently, the latter was cyclized using TFAA to afford pleconaril (18).36–39
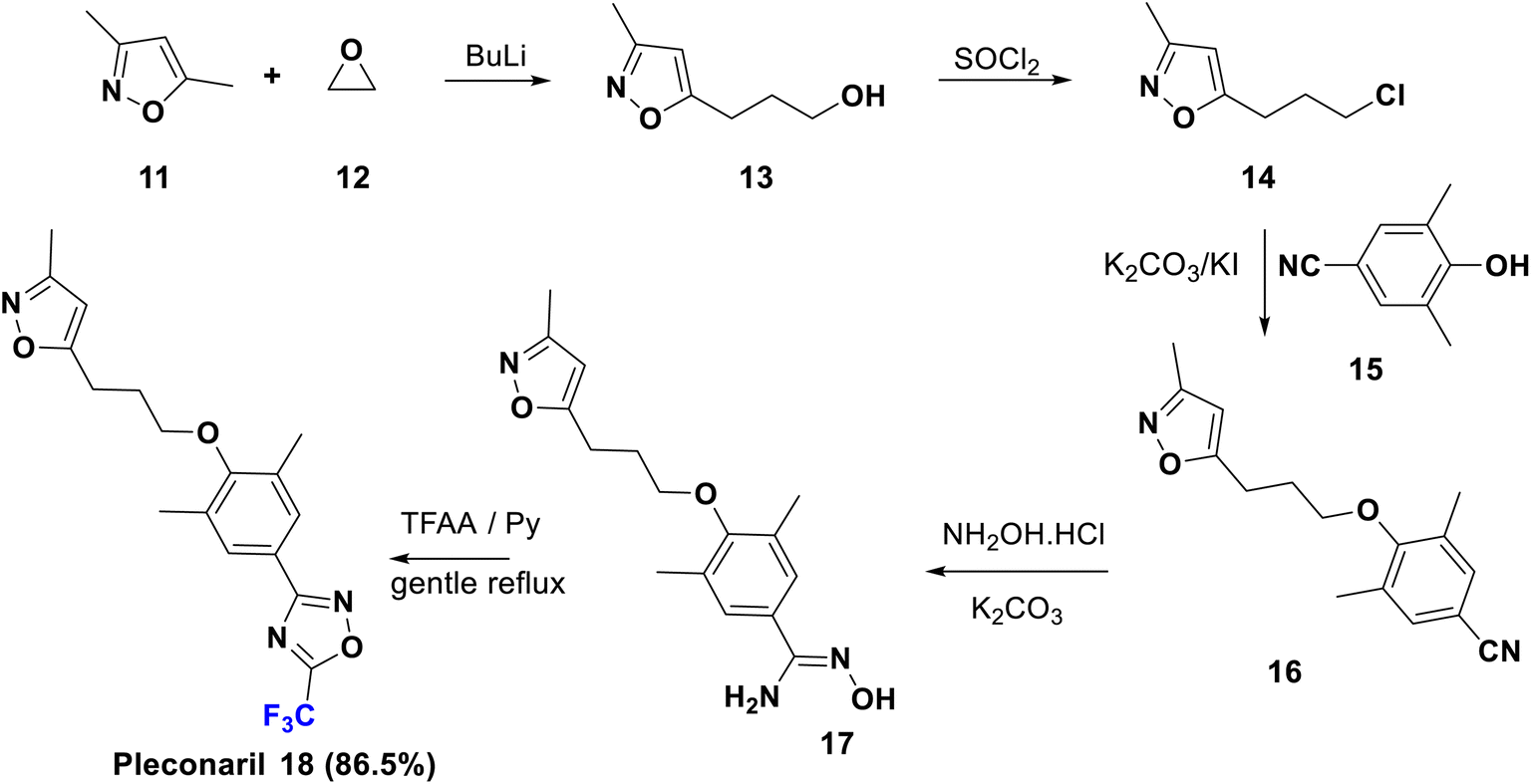 | ||
| Scheme 3 Synthesis of pleconaril (18) from dimethyl isoxazole 11 and oxirane 12. | ||
4. Treatment of HIV type-1
When the immune system is attacked by HIV without treatment, severe acquired immunodeficiency syndrome (AIDS) will result, and unfortunately, there is no effective cure yet.1,40 The presently accepted anti-HIV agents can be categorized as inhibitors of reverse transcriptase, integrase, protease, fusion, and co-receptor. Combination therapy using antiretroviral agents greatly suppresses viral growth and replenishes the immune system.41,42 Development of drug resistance, in addition to their toxicity, limits the use of these drugs.8,43,44Maraviroc (MVC) has commercial names such as Selzentry and Celsentri and was approved in 2007; it acts as a selective HIV entry and fusion inhibitor for C–C chemokine receptor type-5 (CCR5), and is accepted for the treatment of advanced patients infected with CCR5-tropic HIV-1. Much effort have been applied to decrease the affinity of these agents towards human receptors closely correlated with CCR5 e.g. the potassium channel HERG, responsible for the cardiotoxicity that appears with this type of medicament.45 Combination therapy of MVC with other medications is advised to cure a certain type of human immunodeficiency virus (HIV) infection. The complete synthesis of maraviroc 33 was described in a US patent46 by Igor et al. in 2014 via the formation of tropinone, 22, by Robinson synthesis using benzylamine, 2-oxomalonic acid, and a furan derivative. Condensation of 22 with hydroxylamine affords the corresponding oxime 23, which upon subsequent reduction via reflux with sodium in penthanol afforded the exo-amine 24, which reacted with 25 to yield the corresponding amide, 26. Cyclocondensation of 26 with acid hydrazide 27 afforded the 1,2,4-triazole derivative, 28, which is then deprotected to 29 under transfer-hydrogenolysis conditions. Next, the addition of the tropane-triazole adduct 29 to the aldehyde 30 occurred followed by deprotection of the Boc group to yield 31. Peptide coupling of 31 with the difluorinated acid, 32, afforded the target antiviral drug maraviroc (33) (Scheme 4).47,48
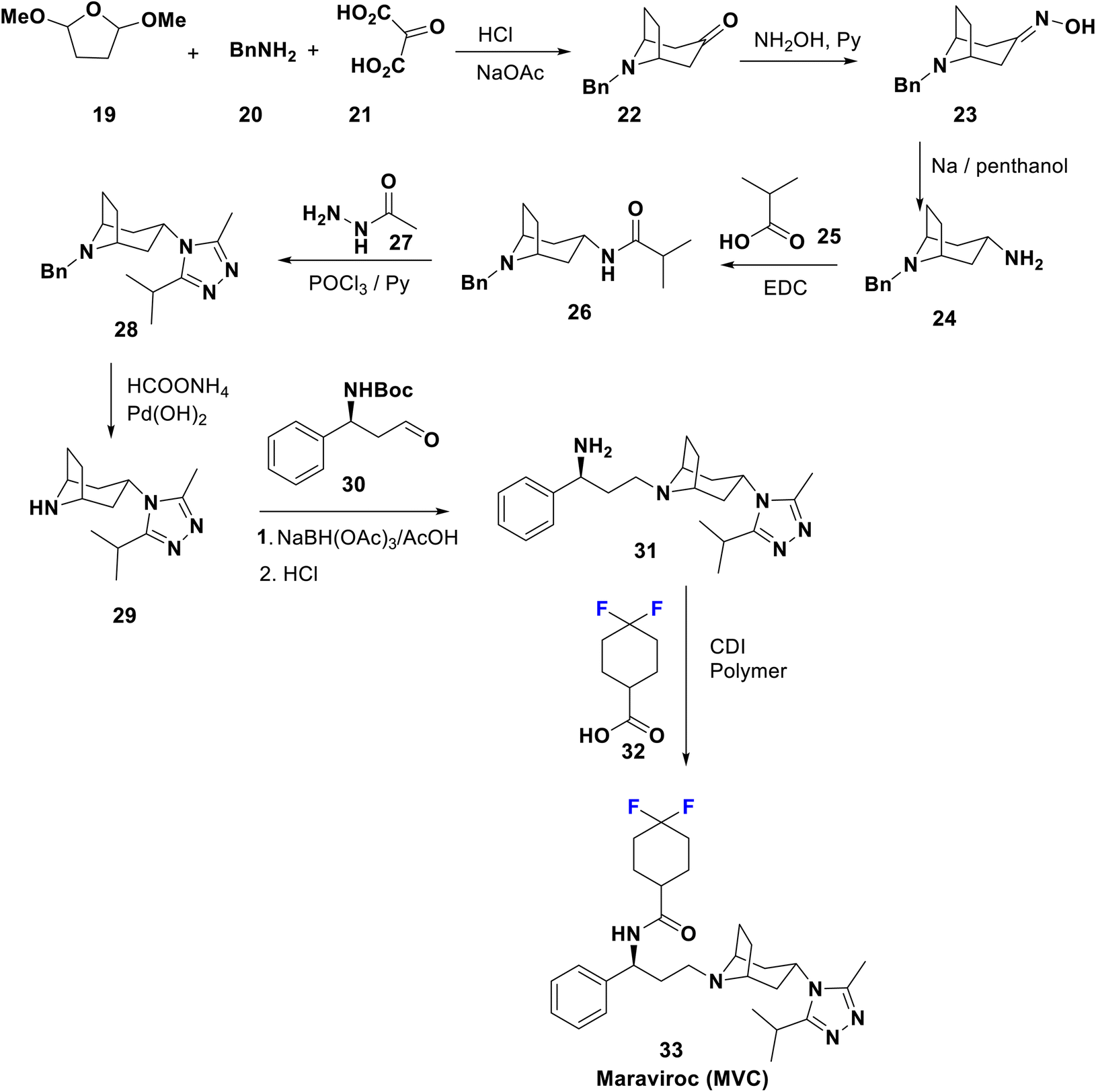 | ||
| Scheme 4 Synthesis of maraviroc 33 from tropinone 22. | ||
Integration of the viral DNA into the host genome required an essential enzyme, HIV integrase, which is a well-executed target in combating HIV where there is no human homolog. Integrase inhibitors are composed of two distinctive constituents. The first is a diketo acid moiety, which is capable of forming covalent bonds with DNA, with the aid of integrase enzyme; then, the sealing of the viral DNA into the chromosome occurs by cellular repair activities. The second moiety is the benzyl group with lipophilic nature which permits binding to an important hydrophobic pocket of integrase.49 The fluorine attached to the benzyl moiety results in a higher affinity between the drug and the protein via binding to numerous nonpolar residues within the hydrophobic pocket of the integrase enzyme. In the first generation of HIV integrase inhibitors, the para-fluorophenyl is in a favored position e.g. in raltegravir (RAL), or the ortho-fluorophenyl e.g. in elvitegravir, while second-generation inhibitors like dolutegravir have the ortho/para bis-fluoro phenyl as an essential moiety.
Raltegravir is a first generation HIV integrase strand transfer inhibitor (INSTI), FDA-approved in 2007 and sold under the trade name Isentress. It inhibits the role of HIV integrase, required for viral reproduction. The para-fluoro group is capable of binding with the former viral nucleotide of the enzyme. RAL can be utilized as a prophylactic or as combined therapy with other drugs to treat HIV/AIDS. It is worth mentioning that RAL and other integrase strand transfer inhibitors are well-tolerated antiretroviral drugs (ARVs).50 Raltegravir 44 was synthesized in 2013 by Gurjar et al., starting from acylation of 2-amino-2-methyl propanenitrile, 35, by oxadiazole acid chloride, 34, in the presence of NMM, 36, to afford the nitrile, 37. Then, the addition of hydroxylamine to compound 37 yielded the corresponding amidoxime, 38, which upon cyclization with dimethylacetylene-dicarboxylate, 39, provided the dihydropyrimidine ester derivative, 40. Amidation of the latter by p-fluorobenzylamine, 41, gave the norraltegravir, 42, which undergoes N-methylation using trimethylsulphoxide iodide, 43, to afford raltegravir (44) (Scheme 5).51
 | ||
| Scheme 5 Synthesis of raltegravir 44 from oxadiazole acid chloride 34. | ||
Elvitegravir (EVG) 56 was first approved in 2012 and is utilized for HIV therapy; it acts via inhibition of the HIV-1 integrase enzyme and it also inhibits the integrase enzyme of HIV-2 to a lesser degree.52 Phase II clinical trials showed that patients taking elvitegravir once a day enhanced by ritonavir (protease inhibitor, PI) had a more significant decline in viral burden when compared to patients receiving a ritonavir-boosted protease inhibitor after the same time period. Clearly, ortho-fluoro in EVG has a favorable role where it contributes to van der Waals interactions with glutamate residues in the enzymatic pocket. The synthetic procedure for EVG was achieved by Stanislav et al. in 2014 via the reaction of difluoro-iodobenzoic acid 45 with thionyl chloride to give the corresponding acid chloride 46. Treatment of the latter with the ester 47 allowed the transformation to benzoyl acrylate 48; then, a nucleophilic substitution reaction with S-(+)-valinol 49 provided the enamine 50, which was then condensed to quinolin-4-one 51. Activation of the primary alcohol of 51 was carried out to produce the carbonate ester 52 using methyl chloroformate; then Negishi cross-coupling with an organo-zinc reagent 53 in the presence of a palladium catalyst provided the benzyl quinolone 54. This was followed by alkaline deprotection to yield the corresponding free primary alcohol 55, which was subjected to reaction with sodium methoxide to provide elvitegravir (56) (Scheme 6).53
 | ||
| Scheme 6 Synthesis of elvitegravir (56) from difluoro-iodobenzoic acid 45. | ||
Dolutegravir (DTG) 69 was FDA approved in 2013 and is marketed with the commercial name Tivicay; it is an antiretroviral therapy used, combined with other drugs, to treat HIV/AIDS. A combination of dolutegravir/abacavir/lamivudine is also available. Notably, DTG shows longer duration of action than raltegravir; this may be attributed to the amide linker attaching the diketo acid to the benzyl group which allows the difluorobenzyl moiety to interact more strongly with the lipophilic pocket of the integrase enzyme.54 Since 2019, the WHO has advocated DTG as the first- and second-line treatment for everyone with HIV. The synthesis of DTG proceeds as mentioned in the US patent by Sumino et al. in 2012 and Maras et al. in 2016, starting from the condensation of ethyl chloro-acetoacetate 57 with the acetal 58 to afford the enaminone 59 which was subsequently cyclized with chloro-oxalate 60 to give 4-pyrone derivative 61. This was transformed to the corresponding 4-pyridone 63 via reaction with acetalamine, 62. Then, acidic hydrolysis of 63 gave the corresponding aldehyde 64; treatment of the latter with butanolamine 65 afforded the fused tricyclic ester 66. Acidic hydrolysis of 66 gave 67 which underwent coupling with difluoro-benzylamine 68 to provide the target antiviral 69 (Scheme 7).55,56
 | ||
| Scheme 7 Synthesis of dolutegravir 69 from ethyl chloroacetoacetate 57. | ||
Tipranavir (TPV) 78 was approved by the FDA in 2005 for pediatric use to treat HIV infection. Only combination therapy of tipranavir/ritonavir is allowed to be administered. Among protease inhibitors, tipranavir is capable of inhibiting viral replication which is resistant to other medications. Two mechanisms may elucidate the potency of tipranavir against C-resistant HIV. First, it may bind to protease's active site with a smaller number of hydrogen bonds than peptidic PIs, leading to more flexibility to tune amino acid substitutions at the active site.57 Second, the presence of a trifluoromethyl pyridyl-sulfonamide moiety enhances the pharmacodynamic and pharmacokinetic profile.58 Some side effects of tipranavir include intracranial hemorrhage, hepatitis, hyperglycemia and diabetes mellitus, in addition to an increase in total cholesterol and triglycerides.
Tipranavir (78) was prepared by Romines et al. in 1998 via an efficient and short enantio-selective synthesis starting with allylic alkylation of 70 with methyl sodiomalonate using molybdenum complex Mo(CO)3(C3H8) and the ligand 71 as a catalyst. The key compound 73 was obtained in an excellent yield from the malonate derivative 72. Next, aldol coupling of the ester derivative 73 with the aldehyde 74, followed by Dess–Martin oxidation afforded β-ketoester 75 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture). The pyrone 76 was obtained after cleavage of the PMB group, and subsequent reduction of the NO2 functionality followed by treatment of the resulting amine with trifluoromethyl pyridine sulfonyl chloride 77 to provide tipranavir (78) (Scheme 8).59,60
:1 mixture). The pyrone 76 was obtained after cleavage of the PMB group, and subsequent reduction of the NO2 functionality followed by treatment of the resulting amine with trifluoromethyl pyridine sulfonyl chloride 77 to provide tipranavir (78) (Scheme 8).59,60
 | ||
| Scheme 8 Synthesis of tipranavir (78) using molybdenum-catalyzed allylic alkylation. | ||
Viral infections are classified into three basic classes; the first one encompasses lethal viruses such as HIV, hepatitis B, and C viruses. The second class encompasses acute infections like influenza and the third class includes viral infections e.g. the common cold which is nonlethal.61,62 Fluorinated nucleoside reverse transcriptase inhibitors (NRTI) such as emtricitabine (FTC) and racivir (RCV) are a notable category of FDA-approved medications for the treatment of HIV type-1.
Emtricitabine (88) is a 5-fluoro deoxycytidine approved in 2003 and is in the index of important drugs assembled by the WHO.63 It is therapeutically employed for treatment of HIV infection in both adults and children via reverse transcriptase inhibition.64,65 The FDA in 2006 accepted a three-drug combination (Atripla), emtricitabine, tenofovir and efavirenz. Additionally, the WHO registered two other combinations: emtricitabine/tenofovir and emtricitabine/efavirenz as indispensable drugs. Emtricitabine consists of a cytosine base with a fluorine atom at the C-5 position and an uncommon oxathiolane ring as a sugar moiety. It was realized that the activated form of FTC (triphosphate metabolite) was integrated faster into growing viral DNA than that of lamivudine. Furthermore, the introduction of fluorine improves its bio-accessibility parameters and duration compared to lamivudine.66 A stereoselective synthetic route of emtricitabine (88) was accomplished by Mansour et al. in 1997, starting from the formation of trans-oxathiolane derivative 81 from the reaction of 2,5-dihydroxy-1,4-dithiane (79) and glyoxylic acid (80). Acetylation of 81 afforded 82 followed by esterification with a chiral auxiliary 1-menthol (83) which led to a mixture of diastereoisomers 84 and 85. The desired diastereoisomer 84 was isolated by fractional crystallization and directly coupled with silylated 5-fluorocytosine (86), to provide the N-nucleoside derivative 87. Finally, reductive removal of the chiral auxiliary from 87 gives emtricitabine 88, represented with inversion of configuration in position 5 of oxathiolane67–70 (Scheme 9).
 | ||
| Scheme 9 Synthesis of emtricitabine (88) from trans oxathiolane derivative 81. | ||
Racivir is an enantiomer of emtricitabine. It is a widely used nucleoside reverse transcriptase inhibitor advanced by Pharmasset Company for HIV treatment and is currently in phase 2b clinical trials; it blocks a further increase in the amount of HIV virus in the body.71 Racivir is a racemic mixture of the two β-enantiomers of emtricitabine (88), and a neat synthetic method has been patented by Glaxo Wellcome Inc. Its synthesis was achieved by reaction of 2,4-dichloro-5-fluoropyrimidine (89) with NaOEt to yield 90; which is then reacted with the anion of 2,2-dimethoxyethanol (91) to produce the pyrimidine derivative 92. Deprotection of the latter gave the corresponding aldehyde 93. Then, the formation of 1,3-oxathiolane 94 occurred via reaction of 93 and 2,5-dihydroxy-1,4-dithiane 79. 1,3-Oxathiolane 94 was later acetylated to give 95 which upon treatment with TMSOTf afforded racemic 96. Eventually, the reaction of 96 with ammonia led to the racemic mixture 97 (racivir) (Scheme 10).72–74
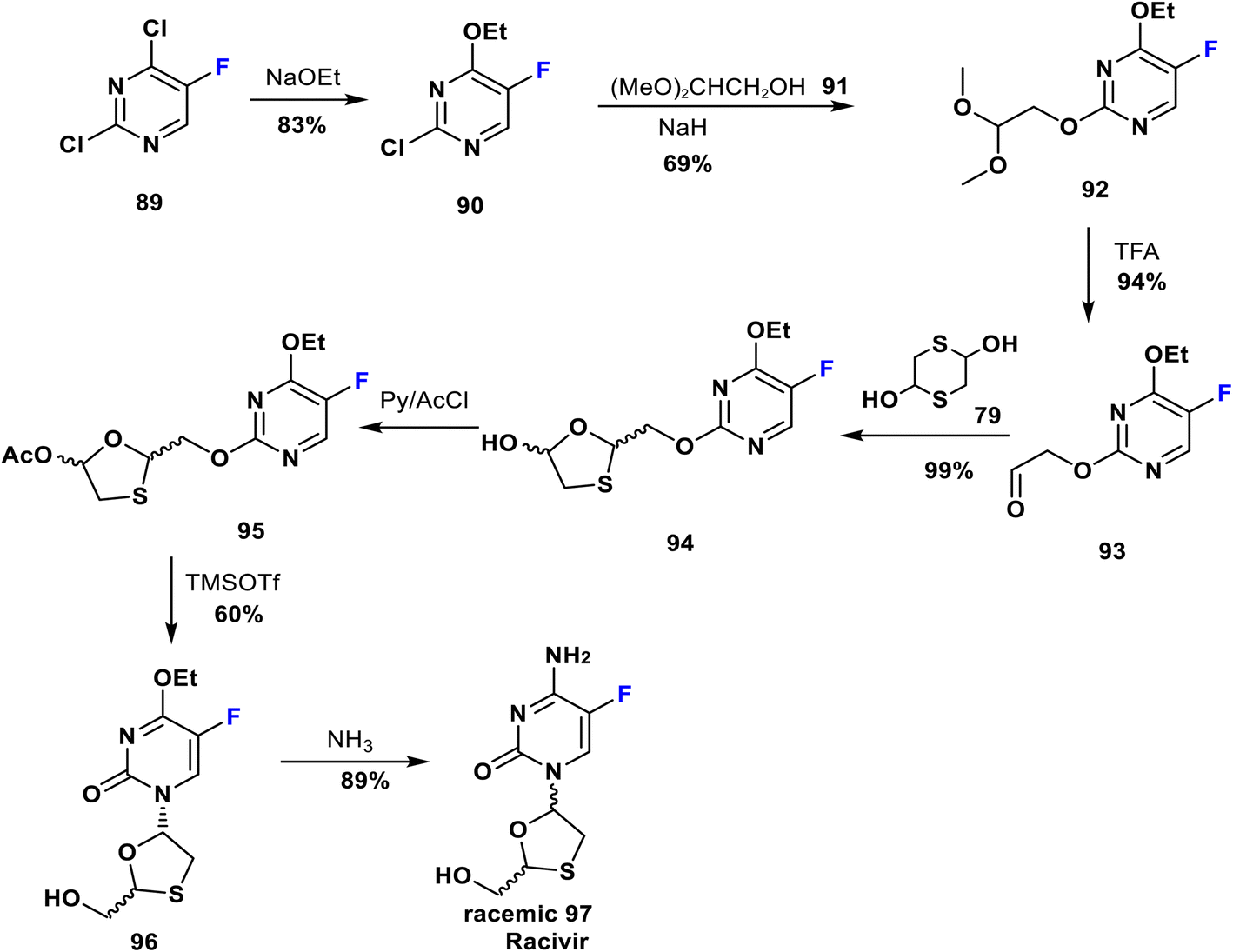 | ||
| Scheme 10 Synthesis of racivir from 2,4-dichloro-5-fluoropyrimidine (89). | ||
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are dissimilar to NRTIs where metabolic activation is not required. Alternatively, they act as noncompetitive blockers targeting the nearby allosteric site of HIV-1 RT,75–77 which leads to conformational changes and impairment of the viral replication.78
The FDA approved efavirenz (Sustiva, EFV) in 1998; it is considered one of the vital medicines by the WHO, and since 2016 it has been available as a generic medication. EFV is an essential antiretroviral drug for the prevention and therapy of HIV/AIDS. It is marketed as either a single drug or a combined form with other antiviral agents such as efavirenz, emtricitabine, and tenofovir for the treatment of HIV infection. Efavirenz is characterized by a trifluoromethyl group at the 4-position which reduces the pKa of the cyclic carbamate. Therefore, the drug potency is improved by hydrogen bond formation between EFV and lysine residue inside the allosteric binding site of the enzyme.79–81
Udit et al. reported in 1999 the synthetic pathway of efavirenz 108. The complete synthesis starts from the acylation of p-chloroaniline 98 by t-butylcarbonyl chloride to afford the amide 99, followed by acylation of the aromatic ring using BuLi and trifluoroacetate to give the trifluoromethyl ketone derivative 100. Acidic hydrolysis of 100 afforded the O-trifluoroacetyl aniline derivative 101, and then N-alkylation of 101 using p-methoxybenzyl chloride (102) provided the corresponding N-PMB aniline derivative 103. Asymmetric addition of the lithium salt of cyclopropyl acetylide, 105, to the ketone group of 103 in the presence of the chiral catalyst 104 afforded the pure tertiary alcohol 106 in excellent enantiomeric excess. Cyclization of 106 was achieved by treatment with phosgene gas to afford the protected N-PMB efavirenz 107 which was deprotected by a cerium nitrate and ammonium nitrate protocol to provide efavirenz 108 (Scheme 11).82,83
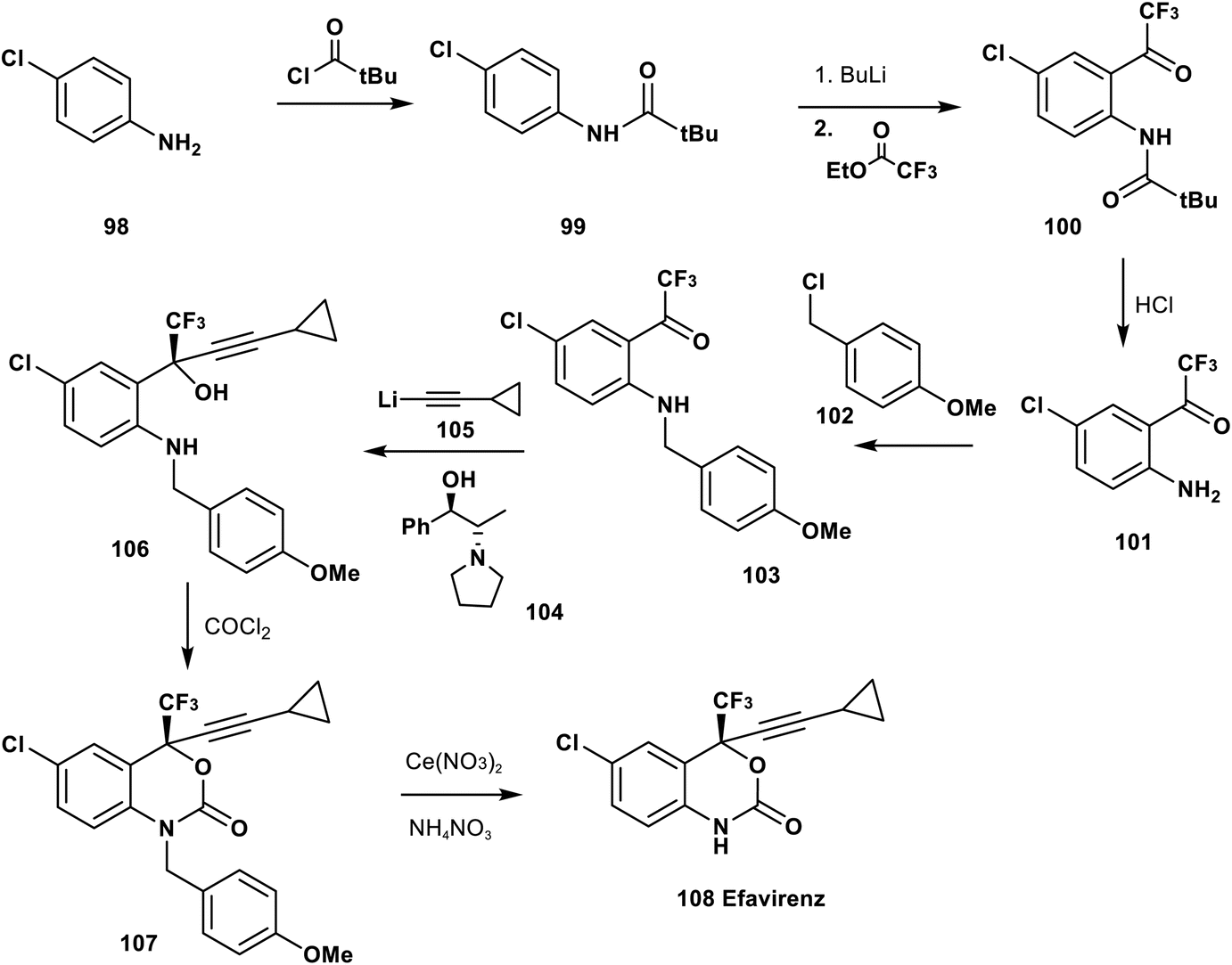 | ||
| Scheme 11 Synthesis of efavirenz 108 from p-chloroaniline (98). | ||
5. Treatment of hepatitis C
Sofosbuvir (Sovaldi®, GS7977) 113 is an RNA-dependant RNA polymerase inhibitor approved by the FDA in 2013, manufactured by Gilead Sciences, Inc. It is a prodrug and has the advantage that the active phosphorylated nucleotide enhances both cell permeability and oral bioavailability. It is the nucleotide analog that interferes with the HCV lifecycle, curtailing viral replication. It can be integrated with HCV RNA by the NS5B polymerase which leads to chain termination. This prodrug is utilized along with ribavirin and sometimes another medication to treat certain types of chronic hepatitis C in adults. Sofosbuvir appears to have a high threshold to the development of resistance. Among sugar-fluorinated nucleosides, sofosbuvir is authorized by the FDA as a therapy for chronic HCV infections including for patients who suffer from hepatocellular carcinoma and others with HCV/HIV-1 co-infection.84 Its preparation was accomplished by Kaushik et al. in 2015 via the reaction of fluorinated deoxynucleoside 110 with the phosphonate 109 which contains a good leaving group (X) such as tosylate, camphorsulfonate, mesylate, trifluoroacetate, trifluorosulfonate, an aryloxide, or heteroaryl oxide substituted with at least one electron-withdrawing group to afford the key compound phosphonucleoside 111. Then, debenzoylation was carried out via reaction of the latter with aqueous acetic acid 70% to produce 112. Subsequently, hydrolysis in the last step was performed using an aqueous acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, formic acid, acetic acid, fumaric acid, oxalic acid, or mixtures thereof to provide sofosbuvir 113 (Scheme 12).85 | ||
| Scheme 12 Synthesis of sofosbuvir (113) from fluorinated deoxynucleoside 110. | ||
Ledipasvir was developed by Gilead Sciences. It inhibits an important hepatitis C virus protein (NS5A), which is incorporated into viral assembly, secretion, and replication.86 Interestingly, patients with genotypes 1a or 1b recover upon using a combination of ledipasvir and sofosbuvir (approved in 2014 by the FDA), as a direct-acting antiviral agent without PEG-interferon or ribavirin.87 Metabolism of sofosbuvir generates active uridine triphosphate which causes RNA chain termination, when integrated in RNA by NS5B polymerase. The addition of ribavirin to this combination will result in an effective therapy for chronic hepatitis C in both children and adults. Ledipasvir was prepared by Scott et al. in 2013 via acylation of 2-bromo-9,9-difluoro-7-iodo-9H-fluorene (114) with 2-chloro-N-methoxy-N-methylacetamide (115) in THF with a catalytic amount of i-PrMgCl at −10 to 0 °C to afford the chloroacetyl-fluorene derivative 116. Then, the nucleophilic substitution of 116 was carried out with the potassium salt of the spiro compound 117 in acetone to produce the corresponding ester 118. Subsequently, reflux of the latter in methoxyethanol with ammonium acetate was performed to provide the imidazole derivative 119 via a cyclization mechanism. New C–C bond formation occurred via the treatment of 119 with borono compound 120 using a palladium acetate, Mephos, and potassium acetate protocol to produce 121. Then, Boc deprotection of 121 was achieved using HCl to give the tetrahydrochloride salt 122. Ultimately, bis-peptide coupling of 122 with (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (Moc-valine) (123) using an EDC-HC1/NMM/HOBt/DMF protocol afforded the target ledipasvir (124) (Scheme 13).88,89
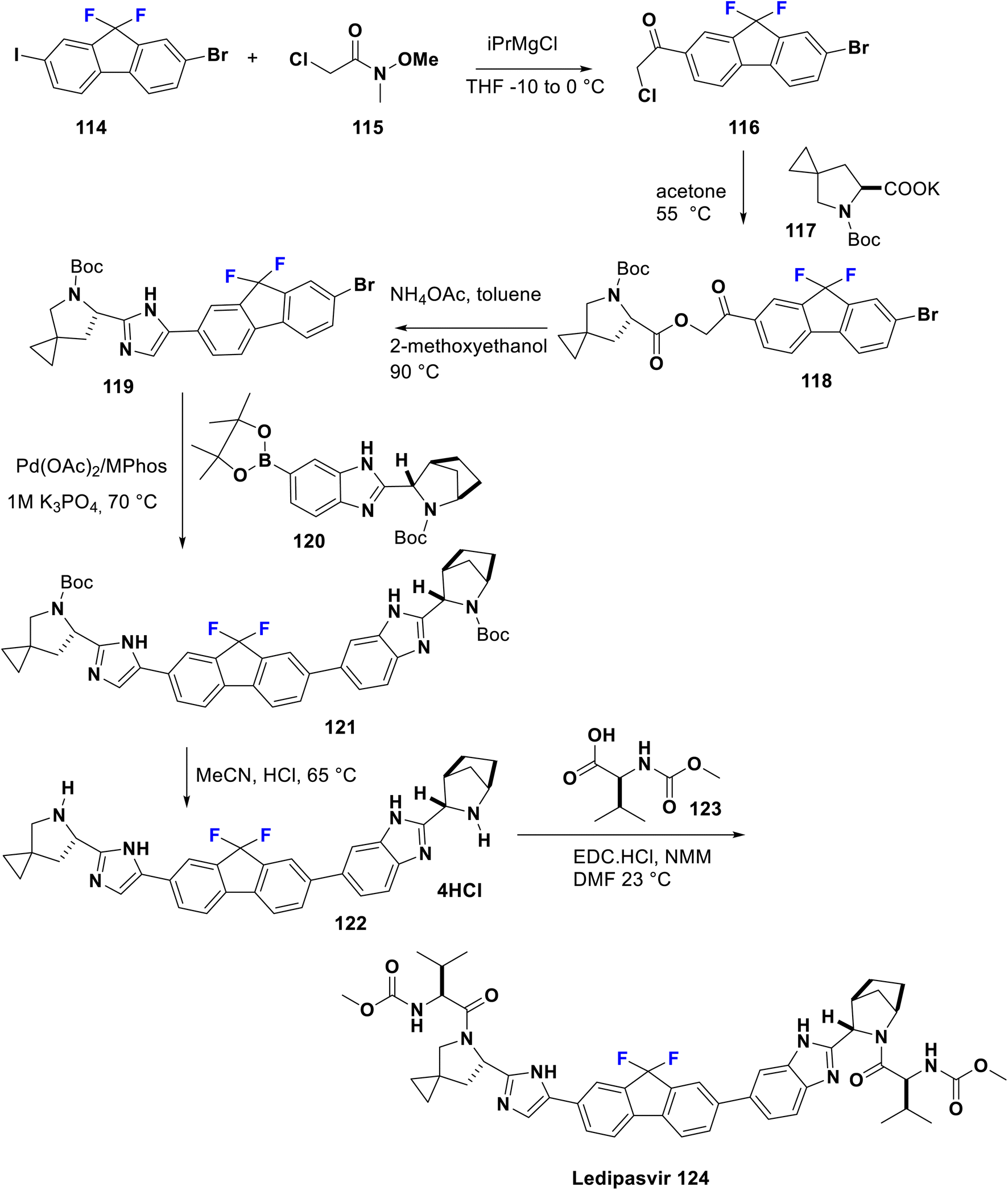 | ||
| Scheme 13 Synthesis of ledipasvir (124) from 2-bromo-9,9-difluoro-7-iodo-9H-fluorene (114). | ||
6. Treatment of eye infections
Trifluridine 127 (trifluorothymidine or TFT) was FDA approved in 1980. It was sold under the trade name Viroptic by Glaxo Wellcome; now it is merged into GlaxoSmithKline as an ophthalmic drug. It is utilized for viral eye infections caused by the herpes simplex virus in particular, and does not affect bacteria or fungi.90 Furthermore, combined therapy of trifluridine and tipiracil is beneficial as an oral anticancer agent for colon or rectal cancers. Trifluridine is a thymidylate synthetase and DNA polymerase inhibitor. It is a suicide substrate that is converted to an irreversible inhibitor at the active site of the enzyme, preventing the conversion of deoxyuridine monophosphate (dUMP) into thymidine monophosphate (TMP), a crucial step in DNA biosynthesis. This will lead to apoptosis, and alter viral or cancer cell replication.81The synthetic procedure of trifluridine, 127, was achieved by Heidelberger et al. in 1962 via a free radical reaction of uridine (125) with Togni's reagent (126) in the presence of catalytic amounts of cuprous salt. Alternatively, trifluridine 127 could be prepared directly by reaction of 5-iodouridine (128) with CF3Cu (that generated from the reaction in situ of CF3I and copper powder)91,92 as shown in Scheme 14.
 | ||
| Scheme 14 Synthesis of trifluridine (127) from uridine. | ||
7. Treatment of influenza and corona viruses
Favipiravir (T-705) (134) was approved for medical use in Japan by Toyama Chemical (a subsidiary of Fujifilm) in 2014. Then, in 2016, it became a generic drug and was sold under the brand name Avigan in 2019. T-705 has a selective and broad-spectrum antiviral effect towards the RNA-dependent RNA polymerase (RdRp) of RNA viruses.93 It is a fluorinated pyrazine carboxamide derivative, used to treat influenza in Japan.94 Concerning the oral formulation, the addition of a 6-fluoro group at the pyrazine ring enhanced both the biological potency and pharmacokinetic character of T-705 in comparison to other derivatives.95 In addition, patients of COVID-19 suffering from a mild-to-moderate illness treated with T-705 show viral clearance and clinical improvement.96 The synthesis of favipiravir (135) was achieved by Furuta et al. in 2000 as shown in Scheme 15,97–99 starting from pyrazine-2-carboxylate derivative 129 which was transformed to its methoxy derivative 130 via diazotization in methanol. Next, 130 was subjected to Buchwald–Hartwig amination to give 131; followed by amidation utilizing aq. ammonia to afford the corresponding amide 132. Diazotization of 132 was carried out in the presence of pyridine hydrofluoride (Olah's reagent) to produce the fluorinated pyrazine derivative 133. Then, deprotection of the methyl ether 133 occurred to give favipiravir (134).97 | ||
| Scheme 15 Synthesis of favipiravir (134) from pyrazine-2-carboxylate derivative 129. | ||
8. Conclusion
Most of the extraordinary potential of fluorine-containing molecules, as FDA-approved antiviral agents in medicinal chemistry and drug discovery, has been covered and clarified in this review. Clearly, the introduction of a fluorine atom or trifluoromethyl group to these drugs enhances both their pharmacodynamic and pharmacokinetic properties. Future directions must include larger clinical trials to assess the safety, tolerability, and resistance of the antiviral drugs. The antiviral combination strategy is highly beneficial to solve problems with these drugs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. S. A. express sincere gratitude to Academy of Scientific Research and Technology (Egypt) for funding project no 7207.References
- E. De Clercq, Biochim. Biophys. Acta, Mol. Basis Dis., 2002, 1587, 258–275 CrossRef CAS.
- D. R. Kuritzkes, Curr. Opin. HIV AIDS, 2009, 4, 82–87 CrossRef.
- E. P. Tchesnokov, C. Gilbert, G. Boivin and M. Götte, J. Virol., 2006, 80, 1440–1450 CrossRef CAS.
- E. De Clercq, J. Clin. Virol., 2004, 30, 115–133 CrossRef CAS PubMed.
- D. J. Hazuda, P. Felock, M. Witmer, A. Wolfe, K. Stillmock, J. A. Grobler, A. Espeseth, L. Gabryelski, W. Schleif and C. Blau, Science, 2000, 287, 646–650 CrossRef CAS PubMed.
- P. A. Furman, J. A. Fyfe, M. H. St Clair, K. Weinhold, J. L. Rideout, G. A. Freeman, S. N. Lehrman, D. P. Bolognesi, S. Broder and H. Mitsuya, PNAS, 1986, 83, 8333–8337 CrossRef CAS.
- E. Tuaillon, M. Gueudin, V. Lemée, I. Gueit, P. Roques, G. E. Corrigan, J.-C. Plantier, F. Simon and J. Braun, J. Acquired Immune Defic. Syndr., 2004, 37, 1543–1549 CrossRef CAS.
- J. Martinez-Picado, M. P. DePasquale, N. Kartsonis, G. J. Hanna, J. Wong, D. Finzi, E. Rosenberg, H. F. Günthard, L. Sutton and A. Savara, PNAS, 2000, 97, 10948–10953 CrossRef CAS.
- E. De Clercq, Nat. Rev. Drug Discovery, 2006, 5, 1015–1025 CrossRef CAS PubMed.
- S. Mahmoud, S. Hasabelnaby, S. Hammad and T. Sakr, J. Adv. Pharm. Res., 2018, 2, 73–88 CrossRef.
- P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS.
- T. Al-Harthy, W. Zoghaib and R. Abdel-Jalil, Molecules, 2020, 25, 4677 CrossRef CAS.
- K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS.
- M. Derudas and S. Meneghesso, in Fluorinated Pharmaceuticals: Advances in Medicinal Chemistry, Future Science Ltd, 2015, pp. 30–42 Search PubMed.
- A. V. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613–12622 CrossRef CAS.
- H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- M. S. T. Makki, D. A. Bakhotmah, R. M. Abdel-Rahman and M. S. El-Shahawy, Int. J. Org. Chem., 2012, 2, 311–320 CrossRef CAS.
- R. M. Abdel-Rahman, M. S. I. T. Makki and W. A. Bawazir, E-J. Chem., 2011, 8, 405–414 CrossRef CAS.
- L. F. Awad and M. S. Ayoup, Beilstein J. Org. Chem., 2020, 16, 1022–1050 CrossRef CAS.
- D.-J. Wang, L. Fan, C.-Y. Zheng and Z.-D. Fang, J. Fluorine Chem., 2010, 131, 584–586 CrossRef CAS.
- R. Filler and R. Saha, Future Med. Chem., 2009, 1, 777–791 CrossRef CAS.
- G. Sandford, J. Fluorine Chem., 2007, 128, 90–104 CrossRef CAS.
- C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
- S. Liu, X. Qian, G. Song, J. Chen and W. Chen, J. Fluorine Chem., 2000, 105, 111–115 CrossRef CAS.
- R. M. Abdel-Rahman, Pharmazie, 1999, 54, 791–803 CAS.
- A. A. Ramadan, R. M. Abdel-Rahman and M. H. Seada, Asian J. Chem., 1992, 4, 569–578 CAS.
- A. A. T. Ramadan, R. M. Abdel-Rahman, M. A. El-Behairy, A. I. Ismail and M. M. Mahmoud, Thermochim. Acta, 1993, 222, 291–303 CrossRef CAS.
- F. M. Foss, Best Pract. Res., Clin. Haematol., 2004, 17, 573–584 CrossRef CAS.
- M. S. I. Makki, R. M. Abdel-Rahman and K. A. Khan, J. Chem., 2014, 2014, 1–14 CrossRef.
- D. W. Grosenbach, R. Jordan and D. E. Hruby, Future Virol., 2011, 6, 653–671 CrossRef CAS.
- D. Dai, WO2014028545A1, 2014.
- D. Dai, US Pat., 9546137B2, 2018.
- A. M. De Palma, I. Vliegen, E. De Clercq and J. Neyts, Med. Res. Rev., 2008, 28, 823–884 CrossRef CAS.
- G. D. Diana, P. Rudewicz, D. C. Pevear, T. J. Nitz, S. C. Aldous, D. J. Aldous, D. T. Robinson, T. Draper, F. J. Dutko, C. Aldi, G. Gendron, R. C. Oglesby, D. L. Volkots, M. Reuman, T. R. Bailey, R. Czerniak, T. Block, R. Roland and J. Oppermann, J. Med. Chem., 1995, 38, 1355–1371 CrossRef CAS PubMed.
- N. R. Florea, D. Maglio and D. P. Nicolau, Pharmacotherapy, 2003, 23, 339–348 CrossRef CAS.
- D. Pevear, G. Rhodes, T. Nitz, M. McKinlay and M. Collett, US Pat., 20060167109A1, 2006.
- J. S. S. W. Staudinger, WO2010009288A1, 2010.
- M. S. Serna-Arbeláez, L. Florez-Sampedro, L. P. Orozco, K. Ramírez, E. Galeano and W. Zapata, Adv. Virol., 2021, 2021, 1–22 CrossRef PubMed.
- C. M. Kitchen, S. G. Kitchen, J. A. Dubin and M. S. Gottlieb, Clin. Infect. Dis., 2001, 33, 466–472 CrossRef CAS PubMed.
- W. M. Valenti, The AIDS Reader, 2001, vol. 11, pp. 260–262 Search PubMed.
- S. J. Little, S. Holte, J.-P. Routy, E. S. Daar, M. Markowitz, A. C. Collier, R. A. Koup, J. W. Mellors, E. Connick and B. Conway, N. Engl. J. Med., 2002, 347, 385–394 CrossRef CAS.
- A. Carr, Nat. Rev. Drug Discovery, 2003, 2, 624–634 CrossRef CAS.
- A. Lazzarin, J. Reynes, J.-M. Molina, S. Valluri, G. Mukwaya, J. Heera, C. Craig, E. Van Der Ryst and J. G. Sierra-Madero, HIV Clin. Trials, 2015, 16, 10–21 CrossRef.
- J. Hajicek and I. Cerna, WO2014173375A1, 2014.
- S. J. Haycock-Lewandowski, A. Wilder and J. Åhman, Org. Process Res. Dev., 2008, 12, 1094–1103 CrossRef CAS.
- Y. Zhu, H. Li, K. Lin, B. Wang and W. Zhou, Synth. Commun., 2019, 49, 1721–1728 CrossRef CAS.
- E. Serrao, S. Odde, K. Ramkumar and N. Neamati, Retrovirology, 2009, 6, 1–14 CrossRef.
- P. Krogstad, P. Samson, E. P. Acosta, J. Moye, E. Townley, S. Bradford, E. Brown, K. Denson, B. Graham and L. Hovind, J. Pediatr. Infect. Dis. Soc., 2021, 10, 201–204 CrossRef CAS.
- M. K. Gurjar, S. P. Sonawane, G. S. Maikap, G. D. Patil, S. B. Shinde, P. S. Patil and S. S. Mehta, WO2013098854A2, 2014.
- H. Haberfeld, Austria-Codex (in German)Vienna: Österreichischer Apothekerverlag, Beč, 2015.
- S. Radl, WO2014056465A1, 2014.
- S. Hare, S. J. Smith, M. Metifiot, A. Jaxa-Chamiec, Y. Pommier, S. H. Hughes and P. Cherepanov, Mol. Pharmacol., 2011, 80, 565–572 CrossRef CAS.
- Y. Sumino, M. Masui, D. Yamada, F. Ikarashi and K. Okamoto, WO2012018065A1, 2012.
- N. Maras, L. Selic and A. Cusak, WO2016113372A1, 2016.
- B. Vergani and S. Rusconi, Drugs R&D, 2011, 11, 291–293 CrossRef.
- S. R. Turner, J. W. Strohbach, R. A. Tommasi, P. A. Aristoff, P. D. Johnson, H. I. Skulnick, L. A. Dolak, E. P. Seest, P. K. Tomich and M. J. Bohanon, J. Med. Chem., 1998, 41, 3467–3476 CrossRef CAS PubMed.
- M. Kraft and D. Mayers, US Pat., 20060135562A1, 2006.
- K. R. Romines, G. L. Bundy, T. M. Schwartz, R. A. Tommasi, J. W. Strohbach, S. R. Turner, S. Thaisrivongs, P. A. Aristoff, P. D. Johnson and H. I. Skulnick, WO1995030670A3, 1998.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS.
- A. S. Ray, R. F. Schinazi, E. Murakami, A. Basavapathruni, J. Shi, S. M. Zorca, C. K. Chu and K. S. Anderson, Antiviral Chem. Chemother., 2003, 14, 115–125 CrossRef CAS.
- https://list.essentialmeds.org/, retrieved 12 October 2022.
- A. Cavaliere, K. C. Probst, A. D. Westwell and M. Slusarczyk, Future Med. Chem., 2017, 9, 1809–1833 CrossRef CAS PubMed.
- P. Liu, A. Sharon and C. K. Chu, J. Fluorine Chem., 2008, 129, 743–766 CrossRef CAS PubMed.
- J. E. Frampton and C. M. Perry, Drugs, 2005, 65, 1427–1448 CrossRef CAS PubMed.
- T. Mansour, H. Jin, H. L. Allan and M. A. Siddiqui, EP0515157A1, 1997.
- D. Mandala and P. Watts, ChemistrySelect, 2017, 2, 1102–1105 CrossRef CAS.
- P. B. Kshirsagar, S. M. Bhoge, S. Richhariya and K. Singh, CA2792044A1, 2014.
- S. Simhadri, V. S. K. Indukuri, S. R. Gorantla, V. R. Gollapalli and N. Arikatla, WO2014174532A2, 2015.
- https://clinicaltrials.gov/ct2/show/NCT00121979.
- E. De Clercq and G. Li, Clin. Microbiol. Rev., 2016, 29, 695–747 CrossRef PubMed.
- E. De Clercq, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 1–24 CrossRef CAS.
- D. M. Volochnyuk, O. O. Grygorenko and A. O. Gorlova, Fluorine in Heterocyclic Chemistry, 2014, vol. 2, pp. 577–672 Search PubMed.
- C. Tantillo, J. Ding, A. Jacobo-Molina, R. G. Nanni, P. L. Boyer, S. H. Hughes, R. Pauwels, K. Andries, P. A. J. Janssen and E. Arnold, J. Mol. Biol., 1994, 243, 369–387 CrossRef CAS PubMed.
- J. Ding, K. Das, H. Moereels, L. Koymans, K. Andries, P. A. J. Janssen, S. H. Hughes and E. Arnold, Nat. Struct. Biol., 1995, 2, 407–415 CrossRef CAS PubMed.
- K. Das, J. Ding, Y. Hsiou, A. D. Clark Jr, H. Moereels, L. Koymans, K. Andries, R. Pauwels, P. A. J. Janssen and P. L. Boyer, J. Mol. Biol., 1996, 264, 1085–1100 CrossRef CAS.
- O. J. D'Cruz and F. M. Uckun, J. Antimicrob. Chemother., 2006, 57, 411–423 CrossRef PubMed.
- S. R. Rabel, S. Sun, M. B. Maurin and M. Patel, AAPS PharmSci, 2001, 3, 26–29 CrossRef PubMed.
- M. R. P. Rao and L. S. Babrekar, Indian J. Pharm. Sci., 2018, 80, 1115–1124 CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- M. K. Gurjar, A. A. Deshmukh, S. S. Deshmukh and S. R. Mehta, US Pat., 8604189B2, 2013.
- U. Batra, R. J. Higgins, K. C. Thompson and A. V. Katdare, WO1999061026A1, 1999.
- A. Cha and A. Budovich, Pharmacol. Ther., 2014, 39, 345 Search PubMed.
- V. K. Kaushik, V. K. Ravi, S. Vakiti and B. Tirumalaraju, WO2015097605A1, 2015.
- https://www.hepatitisc.uw.edu, accessed 23rd August 2022.
- K. M. Bullard-Feibelman, J. Govero, Z. Zhu, V. Salazar, M. Veselinovic, M. S. Diamond and B. J. Geiss, Antiviral Res., 2017, 137, 134–140 CrossRef CAS PubMed.
- R. W. Scott, J. P. Vitale, K. S. Matthews, M. G. Teresk, A. Formella and J. W. Evans, US Pat., 9056860B2, 2013.
- R. W. Scott, F. Wang, B. Shi and E. Mogalian, WO2013184698A1, 2015.
- D. V. Santi and T. T. Sakai, Biochemistry, 1971, 10, 3598–3607 CrossRef CAS.
- C. Heidelberger, D. G. Parsons and D. C. Remy, J. Med. Chem., 1964, 7, 1–5 CrossRef CAS PubMed.
- C. Heidelberger, D. Parsons and D. C. Remy, J. Am. Chem. Soc., 1962, 84, 3597–3598 CrossRef CAS.
- E. Vanderlinden, B. Vrancken, J. Van Houdt, V. K. Rajwanshi, S. Gillemot, G. Andrei, P. Lemey and L. Naesens, Antimicrob. Agents Chemother., 2016, 60, 6679–6691 CrossRef CAS.
- Y. Furuta, K. Takahashi, K. Shiraki, K. Sakamoto, D. F. Smee, D. L. Barnard, B. B. Gowen, J. G. Julander and J. D. Morrey, Antiviral Res., 2009, 82, 95–102 CrossRef CAS PubMed.
- E. De Clercq, Curr. Med. Chem., 2015, 22, 3866–3880 CrossRef CAS.
- T. Manabe, D. Kambayashi, H. Akatsu and K. Kudo, BMC Infect. Dis., 2021, 21, 1–13 CrossRef.
- Q. Guo, M. Xu, S. Guo, F. Zhu, Y. Xie and J. Shen, Chem. Pap., 2019, 73, 1043–1051 CrossRef CAS.
- Y. Furuta and H. Egawa, WO2000010569A1, 2000.
- E. J. Lane, Fujifilm in Avigan API license with Zhejiang Hisun Pharmaceuticals, Fierce Pharma, 22 June 2016, retrieved 22 September 2022 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |