
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEight new phenolic acids from the leaves of Illicium dunnianum and their osteoprotective activities†
Hai-bo Li‡
 b,
Sen-ju Ma‡ac,
Ying-xin Shana,
Ting Lia,
Zhen-zhong Wang*b,
Wei Xiaob,
Zuo-cheng Qiu*a and
Yang Yu*a
b,
Sen-ju Ma‡ac,
Ying-xin Shana,
Ting Lia,
Zhen-zhong Wang*b,
Wei Xiaob,
Zuo-cheng Qiu*a and
Yang Yu*a
aInstitute of Traditional Chinese Medicine & Natural Products, Guangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM and New Drugs Research, Jinan University, Guangzhou 510632, P. R. China. E-mail: 1018yuyang@163.com; zuocq@jun.edu.cn; Fax: +86-20-85221559; Tel: +86-20-85221559
bJiangsu Kanion Pharmaceutical Co., Ltd, State Key Laboratory of New-tech for Chinese Medicine Pharmaceutical Process, Jiangsu, Lianyungang, 222001, China. E-mail: wzhzh-nj@163.com
cShenzhen Institute for Drug Control (Shenzhen Testing Center of Medical Devices), Shenzhen 518057, China
First published on 4th August 2022
Abstract
Eight previously unreported phenolic acids (1–8), including three new phenylpropenoid glycosides (1–3), and five undescribed shikimic acid derivatives (4–8), together with six known analogues (9–14), were obtained from the dried leaves of Illicium dunnianum. The structures of these new compounds were elucidated by extensive spectroscopic analyses (1D, 2D-NMR, HRESIMS, IR, UV) and chemical methods. Compounds 1, 2, 4, and 5 were tested for their promotion effect on osteoblastogenesis of pre-osteoblastic MC3T3-E1 cells and inhibitory effect on osteoclastogenesis of RANKL-induced RAW264.7 cells. As a result, 1 and 4 exerted a promotion effect on osteoblastogenesis, but without activity on osteoclastogenesis. Our studies not only enrich the structural diversity of phenolic acids in nature, but also discover new lead compounds from folk plants with activities on osteoblastogenesis or osteoclastogenesis.
1 Introduction
Illicium dunnianum Tutcher, belonging to the genus Illicium of Magnoliaceae, is a folk plant found throughout Southern China and used as a medicine for bone injury and fractures, relieving pain, and treating rheumatism.1,2 Previous phytochemical investigations of I. dunnianum indicated the presence of phenylpropanoids,3–7 sesquiterpenes,3,8–11 phenolic glycosides,12 flavonoids,1,13,14 triterpenes1,6 and other chemical components.1,3,13 Modern pharmacological studies have shown that the extracts of I. dunnianum and some of these types of compounds possess multiple biological activities, such as anti-inflammatory15,16 and analgesic,2,17 relieving gastrointestinal smooth muscle spasm,16 regulation immune,16 repairing bone fracture2 and other pharmacological activities. In our previous research, 24 lignans (including nine previously uncharacterized compounds), one new phenolic glycoside, and one new benzofuran derivative with anti-inflammatory activities were isolated from the leaves of I. dunnianum.18,19 As a part of further investigation of constituents with activities on osteoblastogenesis or osteoclastogenesis from the leaves of I. dunnianum has led to the isolation and structural elucidation of eight previously unreported phenolic acids (1–8), including three new phenylpropenoid glycosides (1–3), and five undescribed shikimic acid derivatives (4–8), together with six known analogues (9–14) (Fig. 1). In addition, compounds 1, 2, 4, and 5 were tested for their promotion effect on osteoblastogenesis of pre-osteoblastic MC3T3-E1 cells and inhibitory effect on osteoclastogenesis of RANKL-induced RAW264.7 cell. Herein, the isolation, structural identification, and bioactivity of these new compounds are discussed.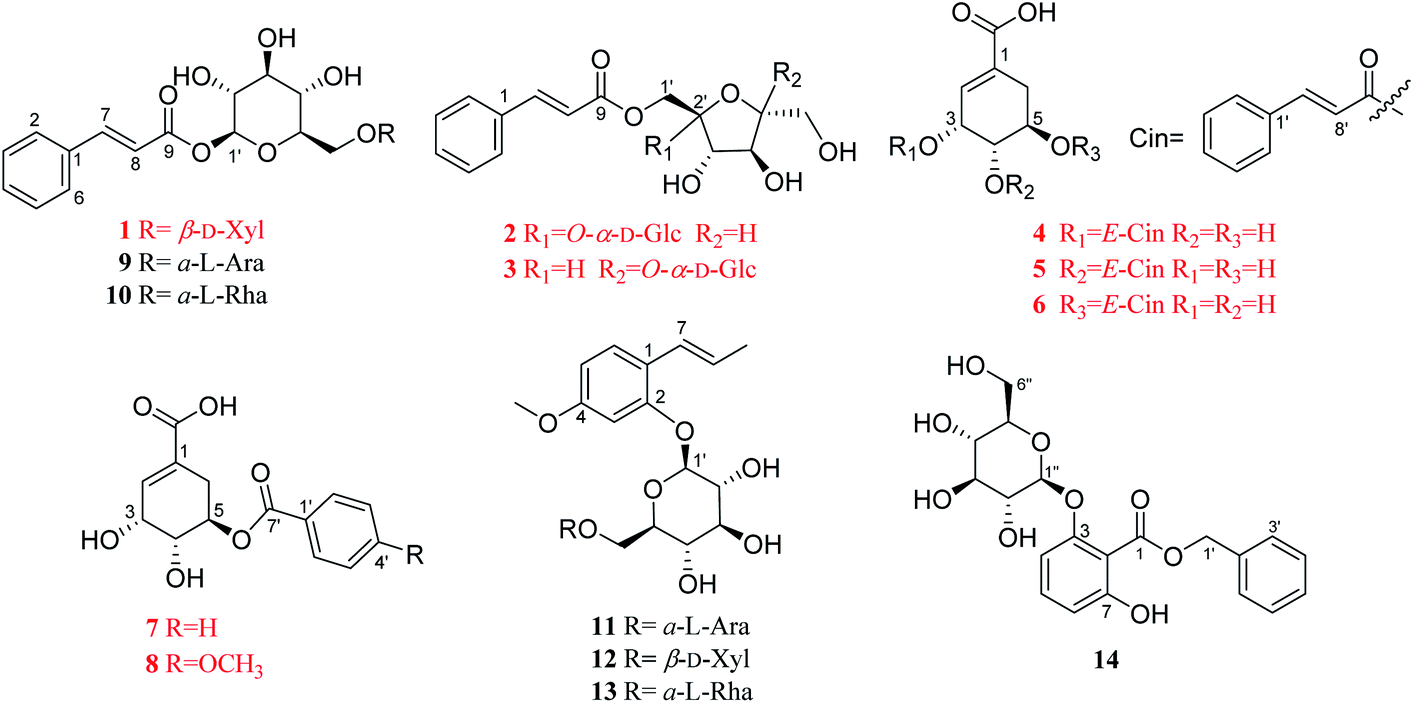 | ||
| Fig. 1 Chemical structures of compounds 1–14. | ||
2 Results and discussion
2.1 Structural elucidation
Compound 1 was obtained as a brown amorphous powder. The sodium adduct ion at m/z 465.1373 [M + Na]+ (calcd. for 465.1373) by HR-ESI-MS demonstrated that the molecular formula of 1 was C20H26O11. The 1H NMR (Table 1) spectrum showed five aromatic proton signals at [δH 7.63 (2H, m, H-2, 6), 7.42 (3H, m, H-3, 4, 5)], a trans-substituted double bond [δH 7.81 (1H, d, J = 16.0 Hz, H-7), 6.58 (1H, d, J = 16.0 Hz, H-8)] and two anomeric proton signals [δH 5.58 (1H, d, J = 7.4 Hz, H-1′), 4.29 (1H, d, J = 7.5 Hz, H-1′′)]; The 13C NMR spectrum displayed eight sp2 carbon resonances (δC 147.8, 135.6, 131.8, 130.1 × 2, 129.4 × 2, 118.2) and a carbonyl carbon signal (δC 167.1), combined with the 1H–1H COSY correlations of H-2/H-3/H-4/H-5/H-6, H-7/H-8 and the key HMBC correlations (Fig. 2) of H-7/C-1,2,6,8,9; H-8/C-1, suggested the presence of a trans-cinnamoyl. In addition, two saccharide groups were determined to be a β-D-glucopyranose and β-D-xylopyranose by HPLC analysis after acid hydrolysis and glycosyl derivatization, as well as the J value of the anomeric protons mentioned above. Furthermore, the HMBC correlations from H-1′ to C-9 and H-1′′ to C-6′ (Fig. 2) suggested that the glucose group was connected to C-9 of the cinnamoyl group, and the xylosyl was linked to C-6′ of glucose. Therefore, compound 1 was identified as (E)-1-O-cinnamoyl-β-D-xylopyranosyl-(1→6)-β-D-glucopyranoside.| Pos. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a Multiplets and or overlapped signals are reported without designating multiplicity [the scale for 1H-NMR (0–10 ppm), and 13C-NMR (0–200 ppm)]. | ||||||
| 1 | 135.6 | 135.7 | 135.7 | |||
| 2 | 129.4 | 7.63 | 129.3 | 7.62 | 129.3 | 7.62 |
| 3 | 130.1 | 7.42 | 130.0 | 7.41 | 130.0 | 7.41 |
| 4 | 131.8 | 7.42 | 131.6 | 7.41 | 131.6 | 7.41 |
| 5 | 130.1 | 7.42 | 130.0 | 7.41 | 130.0 | 7.41 |
| 6 | 129.4 | 7.63 | 129.3 | 7.62 | 129.3 | 7.62 |
| 7 | 147.8 | 7.81, d (16.0) | 146.9 | 7.74, d (16.0) | 146.6 | 7.73, d (16.1) |
| 8 | 118.2 | 6.58, d (16.0) | 118.4 | 6.57, d (16.0) | 118.7 | 6.56, d (16.1) |
| 9 | 167.1 | 167.9 | 168.5 | |||
| 1′ | 95.9 | 5.58, d (7.4) | 64.2 | 4.47, d (12.1) | 63.8 | 3.64, s |
| 4.30, d (12.1) | ||||||
| 2′ | 74.0 | 3.46 | 104.1 | 105.6 | ||
| 3′ | 77.8 | 3.46 | 78.9 | 4.16, d (8.5) | 78.9 | 4.13, d (8.2) |
| 4′ | 71.0 | 3.46 | 75.1 | 4.07, t (8.0) | 76.8 | 4.08, t (7.9) |
| 5′ | 77.8 | 3.59 | 83.8 | 3.80, d (2.1) | 80.7 | 4.0, td (7.5, 3.3) |
| 6′ | 69.5 | 4.12, dd (11.1, 1.2) | 63.3 | 3.78 | 67.0 | 4.52, dd (11.7, 7.4) |
| 3.75, dd (11.1, 5.3) | 4.45, dd (11.7, 3.3) | |||||
| 1′′ | 105.2 | 4.29, d (7.5) | 94.2 | 5.45, d (3.8) | 93.5 | 5.40, d (3.7) |
| 2′′ | 74.9 | 3.21, d (9.2) | 73.1 | 3.44, dd (9.8, 3.8) | 73.3 | 3.41, dd (9.8, 3.8) |
| 3′′ | 77.7 | 3.31 | 74.6 | 3.7, d (9.5) | 74.8 | 3.70, d (9.4) |
| 4′′ | 71.1 | 3.46 | 71.4 | 3.39, t (9.4) | 71.6 | 3.35, d (9.4) |
| 5′′ | 66.9 | 3.84, dd (11.4, 5.1) | 74.4 | 3.87 | 74.3 | 3.87 |
| 3.17, t (7.5) | ||||||
| 6′′ | 62.2 | 3.74, d (4.4) | 62.5 | 3.75, d (5.1) | ||
| 3.84 | 3.84 | |||||
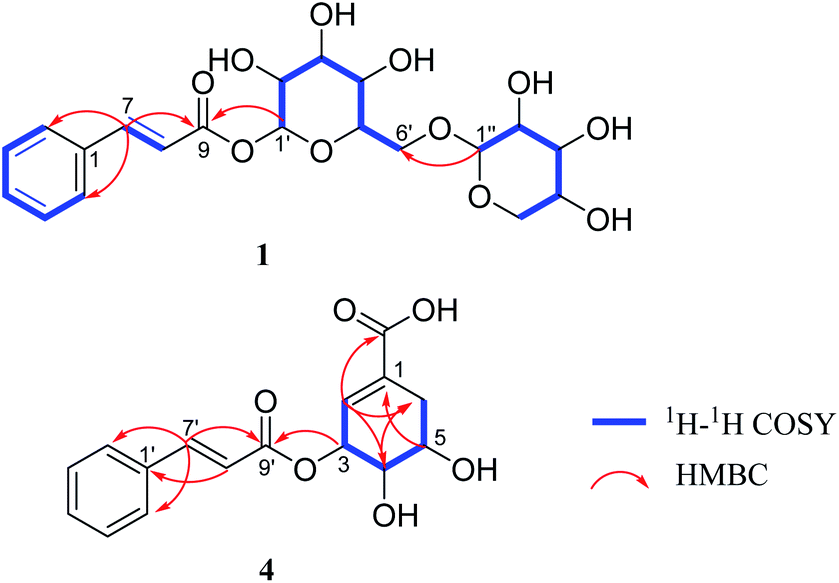 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1 and 4. | ||
2 was obtained as a yellowish oil. The molecular formula of 2 was determined as C21H28O12 by HR-ESI-MS (m/z 495.1471 [M + Na]+; calcd for C21H28O12Na, 495.1478). The detailed analyses of 1D/2D NMR data of 2 determined a planar structure consisting of trans-cinnamoyl and a sucrose unit. 1H-NMR (Table 1) spectrum displayed the presence of an anomeric proton signals [δH 5.45 (1H, d, J = 3.8 Hz, H-1′′)]. The 13C NMR spectrum displayed 12 oxygen carbon signals, including two anomeric carbon signals (δC 104.1, 94.2), three methylene carbon signals (δC 64.2, 63.3, 62.2) and seven methine carbon signals (δC 83.8, 78.9, 75.1, 74.6, 74.4, 73.1, 71.4). Comparing the above data with the literature,20 indicated the presence a sucrose unit, which was confirmed by acid hydrolysis and TLC analysis. Furthermore, the HMBC correlations from H-1′ to C-9 suggested that the sucrose unit was connected to C-9 of the trans-cinnamoyl. Thus, the structure of 2 was assigned as (E)-1′-O-cinnamoyl sucrose glycoside.
Compound 3, a yellowish oil, its molecular formula was consistent with 2 by the molecular ion at m/z 495.1476 [M + Na]+ (calcd. for 495.1478). The NMR spectra (Table 1) of 3 were highly similar to 2, both have a trans-cinnamoyl and a sucrose unit. In the HMBC spectrum, the correlations from H-6′ to C-9 suggesting that the hydroxyl group at C-6′ of sucrose forms an ester bond with the cinnamoyl group. Therefore, the structure was named as (E)-6′-O-cinnamyl sucrose glycoside.
Compound 4 was isolated as a yellowish oil, gave the molecular formula of C16H16O6 based on HR-ESI-MS (m/z 305.1024 [M + H]+, calcd. for 305.1025). Its NMR data (Table 2) indicated the presence of a trans-cinnamoyl [δH 7.62 (2H, m, H-2′,6′), 7.41 (3H, m, H-3′,4′,5′), 7.77 (1H, d, J = 16.0 Hz, H-7′), 6.60 (1H, d, J = 16.0 Hz, H-8′); δC 135.8 (C-1′), 129.3 (C-2′, 6′), 130.0 (C-3′, 5′), 131.6 (C-4′), 146.7 (C-7′), 118.8 (C-8′), 168.0 (C-9′)]. The remaining olefnic proton signal at δH 6.73 (s, H-2), the signals of a CH2-group [δH 2.30 (1H, dd, J = 18.4, 5.1 Hz, H-6a), 2.78 (1H, dt, J = 18.4, 2.3 Hz, H-6b)] and the three methine protons appearing at [δH 3.95 (1H, dd, J = 7.1, 4.1 Hz, H-4), 4.09 (1H, dd, J = 12.1, 5.1 Hz, H-5) and 5.70 (s, H-3)], were attributed to a shikimic acid residue,21 which was further confirmed by 1H–1H COSY correlations of H-2/H-3/H-4/H-5/H2-6 and HMBC cross peaks from H-2 to C-4, 6, 7; H-4 to C-2, 6; H-5 to C-1; H-6 to C-2, 4 (Fig. 2). In addition, the ester linkage of both units via the hydroxyl group at C-3 of the shikimic acid unit could be deduced by the key HMBC correlation from H-3 to C-9′. Accordingly, the structure of 4 could be established as 3-O-cinnamoyl shikimic acid.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c
c
| Pos. | 4a | 5b | 6a | 7b | 8b | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| a Measured at 600 MHz for 1H and 150 MHz for 13C in CD3OD.b Measured at 400 MHz for 1H and 100 MHz for 13C in CD3OD.c Multiplets and or overlapped signals are reported without designating multiplicity [the scale for 1H-NMR (0–10 ppm), and 13C-NMR (0–200 ppm)]. | ||||||||||
| 1 | 134.0 | 131.2 | 131.5 | 130.2 | 130.7 | |||||
| 2 | 133.6 | 6.73 | 138.0 | 6.85, d (3.1) | 137.6 | 6.82 | 139.0 | 6.90, d (1.5) | 138.6 | 6.87 |
| 3 | 71.0 | 5.70, s | 65.6 | 4.63 | 67.4 | 4.41 | 67.4 | 4.45, s | 67.5 | 4.44, s |
| 4 | 70.8 | 3.95, dd (7.1, 4.1) | 75.3 | 5.04, dd (7.8, 4.2) | 70.3 | 3.91, dd (7.2, 3.9) | 69.9 | 4.0, dd (7.4, 4.2) | 70.0 | 3.98, dd (7.4, 4.1) |
| 5 | 68.7 | 4.09, dd (12.1, 5.1) | 65.7 | 4.22, dd (13.2, 5.6) | 71.8 | 5.27, dd (12.2, 6.0) | 72.1 | 5.40, dd (12.5, 5.5) | 71.8 | 5.36, dd (12.6, 5.2) |
| 6 | 31.8 | 2.78, dt (18.4, 2.3) | 32.2 | 2.75, dd (18.4, 5.0) | 29.7 | 2.90, dd (18.4, 3.7) | 29.1 | 2.92, dd (18.3, 4.7) | 29.2 | 2.90, dd (18.4, 5.0) |
| 2.30, dd (18.4, 5.1) | 2.32, dd (18.4, 5.8) | 2.33, dd (18.4, 5.1) | 2.41, dd (18.3, 5.2) | 2.38, dd (18.4, 5.3) | ||||||
| 7 | 170.5 | 169.9 | 170.1 | 169.6 | 170.1 | |||||
| 1′ | 135.8 | 135.8 | 135.8 | 131.4 | 123.5 | |||||
| 2′ | 129.3 | 7.62 | 129.3 | 7.60 | 129.3 | 7.61 | 130.6 | 8.02, d (7.9) | 132.7 | 7.96, d (8.9) |
| 3′ | 130.0 | 7.41 | 130.0 | 7.40 | 130.0 | 7.41 | 129.6 | 7.48, t (7.6) | 114.8 | 6.99, d (8.9) |
| 4′ | 131.6 | 7.41 | 131.5 | 7.40 | 131.6 | 7.41 | 134.4 | 7.61, t (7.4) | 165.3 | |
| 5′ | 130.0 | 7.41 | 130.0 | 7.40 | 130.0 | 7.41 | 129.6 | 7.48, t (7.6) | 114.8 | 6.99, d (8.9) |
| 6′ | 129.3 | 7.62 | 129.3 | 7.60 | 129.3 | 7.61 | 130.6 | 8.02, d (7.9) | 132.7 | 7.96, d (8.9) |
| 7′ | 146.7 | 7.77, d (16.0) | 146.5 | 7.74, d (16.0) | 146.6 | 7.72, d (16.0) | 167.4 | 167.3 | ||
| 8′ | 118.8 | 6.60, d (16.0) | 118.9 | 6.60, d (16.0) | 118.9 | 6.56, d (16.0) | ||||
| 9′ | 168.0 | 168.3 | 168.1 | |||||||
| 4′-OCH3 | 56.0 | 3.86, s | ||||||||
Compounds 5 and 6 were yield as a yellow amorphous powder, their molecular formulas were consistent with 4 by HRESIMS at m/z 305.1029 [M + H]+, and 305.1024 [M + H]+ (calcd. for 305.1025), respectively. The NMR spectroscopic data of 5 and 6 were very similar to those of 4, except for the substitute position of a trans-cinnamoyl. Furthermore, the key HMBC correlation from H-4 to C-9′ in 5, suggested that the trans-cinnamoyl was connected to C-4 of the shikimic acid unit; the key HMBC correlation between H-5 and C-9′ in 6, indicated that the trans-cinnamoyl was connected to C-5 of the shikimic acid unit. Thus, the structures of 5 and 6 were determined and named as 4-O-cinnamoyl shikimic acid and 5-O-cinnamoyl shikimic acid, respectively.
7 was obtained as a yellow needle crystal. The HRESIMS of 7 showed a quasimolecular peak at m/z 279.0870 [M + H]+ (calcd for C14H14O6, 279.0869), corresponding to the molecular formula of C14H14O6. Its NMR data (Table 2) possessed similar signals to those of 6 except for the loss of the trans-substituted double bond. The benzoyl group was located at C-5 of the shikimic acid unit, which was evidenced by HMBC correlations from H-5 (δH 5.40) to C-7 (δC 167.4). Therefore, the structure of 7 was assigned, and named 5-O-benzoyl shikimic acid.
Compound 8 was isolated as a yellow needle crystal. The molecular formula of 8 was determined as C15H16O7 by HRESIMS (m/z 309.0975 [M + H]+; calcd. 309.0974). A comparison of the spectral data (Table 2) of 8 and 7 revealed that an additional methoxyl group (δH 3.86) was substituted at C-4′, which was confirmed by HMBC correlation from OCH3-4′ (δH 3.86, s) to C-4′ (δC 165.3). To sum up, 8 was identified as 5-O-p-methoxybenzoyl shikimic acid.
The other six known compounds were identified as (E)-2-hydroxy-4-methoxyphenylpropene-2-O-α-L-arabifuranosyl-(1→6)-β-D-glucopyranoside (9),22 (E)-2-hydroxy-4-methoxyphenylpropene-2-O-β-D-xylopyranosyl-(1→6)-β-D-glucopyranoside (10).22 (E)-2-Hydroxy-4-methoxybenzene-propene-2-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside23 (11), (E)-1-O-cin-namoyl-α-L-arabifuranosyl-(1→6)-β-D-glucopyranoside24,25 (12), gleditschiaside A26 (13) and benzyl-3-O-β-D-glucopyranosyl-7-hydroxybenzoate27 (14). Their structures were established by comparison of their spectroscopic data recorded in the literature. Among them, compounds 12 and 13 were firstly isolated from the genus Illicium.
2.2 Osteoclastogenesis inhibitory effects of compounds 1, 2, 4, 5
Compounds 1, 2, 4, and 5 were evaluated for their osteoclastogenesis effect on the pre-osteoclastic RAW264.7 cells. As shown in Fig. 3A, 2 increased the cell proliferation of RAW 264.7 cells about 20% at the test concentration, although no statistic significant was indicated at the concentration of 1 μM. For the osteoclastogenesis activity, all the tested compounds exerted no effect on the number of multinucleated osteoclasts induced by RANKL (Fig. 3B and C). | ||
| Fig. 3 The osteoblastogenesis and osteoclastogenesis activities of compounds 1, 2, 4, and 5. (A) Cell proliferation activity of RAW264.7 cells; (B) quantitative of osteoclasts (TRAP positive multi-nucleated cells (MNCs), more than 3 nuclei) induced from RAW264.7 cells by osteoclast differentiation medium for 4 days (n = 3). (C) Representative images of TRAP staining osteoclast at day 4 in each well of 96-well plate. (D) Cell proliferation activity of pre-osteoblast MC3T3-E1 cells; (F) representative morphological pictures of ALP staining on the MC3T3-E1 cells induced by osteoblast differentiation medium at day 6. NC: negative control, 0.1% DMSO without osteoclast differentiation medium (OCM); C: control, 0.1% DMSO with OCM; results were obtained from three independent experiments in triplicate and expressed are means ± SD; *P < 0.05, **P < 0.01, ***P < 0.001 significantly different from the control group. | ||
2.3 Osteoblastogenesis promoting effects of compounds 1, 2, 4, and 5
Compounds 1, 2, 4, and 5 were also evaluated for their osteoblastogenesis activities using pre-osteoblastic MC3T3-E1 cells. As shown in Fig. 3D, all the tested compounds exhibited no cytotoxicity or cell proliferation effect on the MC3T3-E1 cells. However, 1 (1 and 10 μM) and 4 (1 μM) could significantly deepen the staining colour of ALP staining, which indicated that 1 and 4 could promote the osteoblastogenesis activity of pre-osteoblast MC-3T3 E1 cells.3 Conclusions
In summary, eight previously unreported phenolic acids, including three new phenylpropenoid glycosides (1–3), and five undescribed shikimic acid derivatives (4–8), together with six known analogues (9–14) were isolated and elucidated via varies of chromatographic and spectroscopic methods from the dried leaves of Illicium dunnianum. In addition, compounds 1, 2, 4, and 5 were tested for their promotion effect on osteoblastogenesis of pre-oteoblastic MC3T3-E1 cells and inhibitory effect on osteoclastogenesis of RANKL-induced RAW264.7 cell. As a result, 1 and 4 could promote osteoblastogenesis activity without affect osteoclastogenesis activity, which will be helpful for gaining a deep insight into the biological role of such phenolic acids in bone fracture repair.4 Experimental
4.1 General experimental procedures
Optical rotations were measured on a JASCO P-1020 polarimeter with a 1 cm cell at room temperature. IR spectra were obtained using a JASCO FT/IR-480 plus spectrometer. UV spectra were recorded on a JASCO V-550 UV/Vis spectrometer. HRESIMS spectra were acquired using a Waters Synapt G2 mass spectrometer. 1D and 2D NMR spectra were acquired with Bruker AV 600/400 using solvent signals (CD3OD: δH 3.30/δC 49.0) as the internal standard. Analytical HPLC was conducted on a Shimadzu HPLC system with an LC-20AB solvent delivery system and an SPD-20A UV/vis detector using a Phenomenex Gemini C18 column (5 μm, Φ 4.6 × 250 mm; Phenomenex Inc., Los Angeles, USA). Semi-preparative HPLC was carried out on a Shimadzu LC-6AD liquid chromatography system equipped with a SPD-20A detector on a Phenomenex Gemini C18 column (5 μm, Φ 10.0 × 250 mm; Phenomenex Inc., Los Angeles, USA) and Preparative HPLC using a Cosmosil Packed C18 column (5 μm, Φ 20.0 × 250 mm, Nacalai Tesque Inc., Kyoto, Japan).Silica gel (200–300 mesh, Qingdao Marine Chemical Ltd, China), Diaion HP-20 (Mitsubishi Chemical Co., Tokyo, Japan) and ODS (YMC Ltd, Japan) were used for column chromatography (CC). TLC was performed on precoated silica gel plate (SGF254, 0.2 mm, Yantai Chemical Industry Research Institute, China). HPLC-grade methanol and acetonitrile were bought from Oceanpack Alexative Chemicals Co. Ltd (Gothenburg, Sweden). All analytical grade reagents were from Concord Chemicals Co. Ltd, (Tianjin, China). Modified eagle medium alpha (α-MEM), fetal bovine serum (FBS), penicillin and streptomycin, trypsin–EDTA were purchased from Gibco (Gaithersburg, MD, USA). Murine sRANKL and MCSF were the products of Peprotech (Rocky Hill, NJ, USA). CCK-8 assay kit was purchased from Dojindo (Kumamoto, Japan). Leucocyte acid phosphatase staining kit was purchased from Sigma (St Louis, MO, USA).
4.2 Plant material
The leaves of I. dunnianum were purchased from Ji'an County, Jiangxi, China, in 2018, and were identified by Prof. Zhou Wu (Jiangsu Kanion Pharmaceutical Co. Ltd). A sample (2018ID101) was deposited in Institute of Traditional Chinese Medicine & Natural Products, college of pharmacy, Jinan University, Guangzhou, China.4.3 Extraction and isolation
The air-dried leaves of I. dunnianum (ID, 15.5 kg) were extracted with 50% EtOH by heat reflux for 3 times (2 h each). Total extracts (IDEs, 2 kg) were yielded by evaporation under reduced pressure. IDEs were separated by HP-20 resin column chromatography (CC) eluted with EtOH–H2O (0:100, 30:70, 50:50, 95:5) gradient to afford 4 fractions (ID-1 to ID-4). Fr. ID-3 (180 g) was separated over a silica gel column eluting with a CH2Cl2–MeOH gradient (100:0, 98:2, 95:5, 90:10, 85:15, 80:20, 70:30, 60:40, 50:50, 0:100) to afford 15 fractions (Fr. 3A to 3O). Fr. 3 L (17.4 g) was chromatographed by ODS CC using a CH3OH–H2O gradient elution (30:70–70:30, 100:0) to give 9 subfractions (Fr. 3L1–3L9). Fr. 3L3 (3.1 g) was chromatographed by silica gel column eluting with a CH2Cl2–MeOH gradient (95:5–1:1, 0:100) to give 9 subfractions (Fr. 3L3A–3L3I). Fr. 3L3D and Fr. 3L3E was isolated using semipreparative HPLC [30% CH3OH–H2O (0.1% HCOOH), 3 mL min−1] to yield 12 (7.5 mg), 1 (12.3 mg) and 13 (14 mg). Fr. 3L4 was further chromatographed by polyamide CC using a EtOH –H2O gradient elution (10:90–40:60, 95:5) to give 7 subfractions (Fr. 3L4A-3L4G). Fr. 3L4A was isolated using preparative HPLC [35% CH3OH–H2O (containing 0.1% HCOOH), 8 mL min−1] to give fractions 3L4A2–3L4A9. Fr. 3 L4A2 was isolated using semipreparative HPLC [15% CH3CN–H2O (0.1% HCOOH), 3 mL min−1] to yield 2 (14.9 mg). Fr. 3 L4A4 was isolated using semipreparative HPLC [19% CH3CN–H2O (0.1% HCOOH), 3 mL min−1] to yield 9 (10 mg), 10 (14.6 mg) and 3 (4.4 mg). Fr. 3L4A6 was isolated using semipreparative HPLC [16% CH3CN–H2O (0.1% HCOOH), 3 mL min−1] to yield 11 (7.1 mg). Fr. 3G (7.1 g) was chromatographed by ODS CC using a CH3OH–H2O gradient elution (25:75–100:0) to give 9 subfractions (Fr. 3G1–3G9). Fr. 3G9 was isolated using semipreparative HPLC [20% CH3CN–H2O (0.1% HCOOH), 3 mL min−1] to yield 4 (148.7 mg) and 5 (73.2 mg). Fr. 3G7 was chromatographed by Sephadex LH-20 eluting with a CH3OH to give six subfractions (Fr. 3G7A–3G7F). Fr. 3G7F was isolated using semipreparative HPLC [27% CH3CN–H2O (0.1% HCOOH), 3 mL min−1] to yield 6 (3.5 mg), 7 (3.7 mg) and 8 (5.5 mg). Fr. 3G7E was isolated using semipreparative HPLC [18% CH3CN–H2O (0.1% HCOOH), 3 mL min−1] to yield 14 (47.9 mg).
4.4 Structural characterization of undescribed compounds
ε): 205 (4.23), 217 (4.07), 223 (4.0), 280 (4.08) nm; IR (KBr) νmax 3281, 2927, 2890, 1716, 1637, 1509, 1455, 1374, 1333, 1271, 1172, 1068 cm−1; The 1H and 13C NMR spectra data are shown in Table 1; HR-ESI-MS m/z: 465.1373 [M + Na]+ (calcd for C20H26O11Na, 465.1373).ε): 205 (4.23), 217 (4.0), 222 (4.16), 279 (4.36) nm; IR (KBr) νmax 3317, 2927, 2887, 1710, 1637, 1450, 1381, 1333, 1160, 1051 cm−1; the 1H and 13C NMR spectra data are shown in Table 1; HR-ESI-MS m/z: 495.1471 [M + Na]+ (calcd for C21H28O12Na, 495.1478).ε): 205 (4.39), 216 (4.3), 223 (4.2), 278 (4.31) nm; IR (KBr) νmax 3312, 2933, 2882, 1707, 1634, 1596, 1498, 1453, 1320, 1283, 1329, 1065 cm−1; The 1H and 13C NMR spectra data are shown in Table 1; HR-ESI-MS m/z: 495.1476 [M + Na]+ (calcd for C21H28O12Na, 495.1478).ε): 206 (4.34), 217 (4.33), 222 (4.25), 278 (4.32) nm; IR (KBr) νmax 3258, 2902, 1704, 1639, 1554, 1402, 1175 cm−1; the 1H and 13C NMR spectra data are shown in Table 2; HR-ESI-MS m/z: 305.1024 [M + H]+ (calcd for C16H17O6, 305.1025).ε): 205 (4.27), 216 (4.28), 278 (4.26) nm; IR (KBr) νmax 3383, 3069, 1719, 1696, 1424, 1277, 1065 cm−1; the 1H and 13C NMR spectra data are shown in Table 2; HR-ESI-MS m/z: 305.1029 [M + H]+ (calcd for C16H17O6, 305.1025).ε): 206 (4.78), 217 (4.76), 278 (4.73) nm; IR (KBr) νmax 3394, 2921, 1704, 1599, 1402, 1172, 1045 cm−1; the 1H and 13C NMR spectra data are shown in Table 2; HR-ESI-MS m/z: 305.1024 [M + H]+ (calcd for C16H17O6, 305.1025).ε): 207 (5.19), 257 (5.04) nm; IR (KBr) νmax 3300, 2964, 2910, 2842, 2627, 2497, 2208, 2044, 1707, 1608, 1511, 1271, 1102 cm−1; the 1H and 13C NMR spectra data are shown in Table 2; HR-ESI-MS m/z: 309.0975 [M + H]+ (calcd for C15H17O7, 309.0974).4.5 Acid hydrolysis and sugar analysis
The compounds (1.0 mg) were hydrolyzed with 2 mL of 2 M HCl for 2 h at 90 °C. The hydrolysates were extracted with equal volume of ethyl acetate twice. The aqueous layer was dried, and then reacted with 2.5 mg L-cysteine methyl ester hydrochloride in 1 mL of pyridine for 1 h at 60 °C. After 1 h, a total of 5 μL of o-tolyl isothiocyanate was added to the reaction mixture and further reacted at 60 °C for 1 h. The reaction products were filtered by a 0.45 μm filter membrane for HPLC analysis, detected by a UV detector at 250 nm. Authentic samples of D-Glc, L-Glc, D-Xyl, L-Xyl, D-Rha, L-Rha and D-Fuc, L-Fuc were treated following same procedure.4.6 Cell culture and viability
Murine pre-osteoblastic MC3T3-E1 cells (ATCC, USA) and RAW 264.7 cells (ATCC, USA) were routinely cultured in modified Eagle medium alpha (α-MEM, Gibco, USA), containing 10% fetal bovine serum (Gibco, USA), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. They were cultured at 37 °C in a humidified atmosphere of 5% CO2. Cells were digested with 0.05% trypsin–EDTA (Gibco, USA) and culture in a new culture dish every three days. MC3T3-E1 cells and RAW264.7 cells were seeded in 96-well plates at a density of 5 × 103 or 3 × 103 cells per well, respectively. After drug treatment for 48 h, CCK-8 assay was used as an indirect colorimetric measurement of cell viability.4.7 Effect of compounds 1, 2, 4, and 5 on the osteoblastogenesis of MC3T3-E1 cells
Alkaline phosphatase (ALP) activity was commonly used as a marker of osteoblast differentiation. Cells were seeded in 12-well plates with a cell density of 5 × 104 cells per well. After 2 days, the medium was changed to osteoblast differentiation medium (α-MEM with 10% FBS and 50 μg mL−1 of ascorbic acid and 10 mM of β-glycerophosphate) in the present or absent of compound 1, 2, 4, and 5 (1, 10 μM). Upon treatment for 7 days, ALP activity was measured by staining with the BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime, China) following the instruction of manufacturer. The cells were rinsed with PBS, and images of each well were captured.4.8 Effect of compounds 1, 2, 4, and 5 on the osteoclastogenesis of RANKL-induced RAW264.7 cells
RAW264.7 cells were seeded at 3000 cells per well in 96 well plates. After 24 h of incubation, cells were treated with different concentrations of 1, 2, 4, and 5 in osteoclast differentiation medium (α-MEM with 10% FBS and 100 ng mL−1 of RANKL and 10 ng mL−1 of M-CSF). After 4 days, TRAP staining was performed according to our previous method.28 The number of multinucleated osteoclasts were quantified under microscopy (Olympus CKX53).4.9 Data analysis
The data obtained are presented as the means ± SD of three independent experiments. A one-way analysis of variance (ANOVA) test was used for statistical analysis, followed by a Dunnett's post hoc test for multiple comparisons. GraphPad Prism 5.02 (GraphPad Software Inc., San Diego, CA, USA) was used to perform the analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants from the Programs Foundation for leading Talents in National Administration of Traditional Chinese Medicine of China “Qihuang scholars” project; National Natural Science Foundation of China (Program No. 81903426) and Guangdong Basic and Applied Basic Research Foundation (2022A1515010034).Notes and references
- J. W. Zhang, Chemical constituents of the stems and leaves of Illicium dunnianum Tutch, Zhongguo Zhongyao Zazhi, 1989, 14(1), 36–37 CAS.
- Q. Lin, Medicinal plant resources of Illicium L, Chin. Tradit. Herb. Drugs, 2002, 33(07), 81–84 Search PubMed.
- J. Bai, H. Chen, Z. F. Fang, S. S. Yu, S. G. Ma, Y. Li, S. Xu, J. H. Ren, H. N. Lv and X. Chen, Sesquiterpenes and neolignans from the roots of Illicium dunnianum, J. Asian Nat. Prod. Res., 2012, 14(10), 940–949 CrossRef CAS PubMed.
- J. Li, D. Geng, J. Xu, L. J. Weng, Q. Liu and L. T. Yi, Antidepressant-like effect of macranthol isolated from Illicium dunnianum tutch in mice, Eur. J. Pharmacol., 2013, 707(1–3), 112–119 CrossRef CAS PubMed.
- L. Sy and G. Brown, A sesquilignan from Illicium dunnianum, Phytochemistry, 1996, 43(6), 1417–1419 CrossRef CAS.
- L. Sy, R. Saunders and G. Brown, Phytochemistry of Illicium dunnianum and the systematic position of the Illiciaceae, Phytochemistry, 1997, 44(6), 1099–1108 CrossRef CAS.
- C. Y. IsaoKouno, Two new Sesquineolignans from the bark of Illicium dunnianum, Chem. Pharm. Bull., 1991, 39(10), 2606–2608 CrossRef.
- J. Bai, H. Chen, Z. F. Fang, S. S. Yu, W. J. Weng, Y. Liu, S. G. Ma, Y. Li, J. Qu, S. Xu, J. H. Liu, F. Zhao and N. Zhao, Sesquiterpenes from the roots of Illicium dunnianum, Phytochemistry, 2012, 80(80), 137–147 CrossRef CAS PubMed.
- L. Sy and G. Brown, A sesquiterpene class from Illicium dunnianum, Phytochemistry, 1998, 47(47), 301–302 CAS.
- J. Huang, J. Wang and C. Yang, Sesquiterpene lactones from the pericarp of Illicium dunnianum, Phytochemistry, 1997, 46(4), 777–780 CrossRef CAS.
- I. Kouno, N. Kawano and C. S. Yang, New pseudoanisatin-like sesquiterpene lactones from the bark of Illicium dunnianum, J. Chem. Soc., Perkin Trans. 1, 1988,(6), 1537–1539 RSC.
- J. Bai, Z. F. Fang, H. Chen, S. S. Yu, D. Zhang, W. J. Weng, S. G. Ma, Y. Li, J. Qu, S. Xu, J. H. Ren, F. Zhao, N. Zhao and J. H. Liu, Antioxidant phenolic glycosides from the roots of Illicium dunnianum, Carbohydr. Res., 2012, 361(11), 206–211 CrossRef CAS PubMed.
- D. Geng, L. J. Weng, Y. Y. Han and X. Yang, Chemical Constituents from Illicium dunnianum, Adv. Mater. Res., 2012, 1914(1103), 1586–1589 Search PubMed.
- D. Geng, L. J. Weng, Y. Y. Han and X. Yang, Flavonoids from Illicium dunnianum, Adv. Mater. Res., 2013, 2115(1223), 3375–3377 Search PubMed.
- L. F. Zhang, K. Z. Lin, W. D. Yang, J. L. Wei, G. H. Yang and X. W. Lv, Identification of Illicium dunnianum and its Confusing products, China J. Chin. Mater. Med., 1995, 20(12), 717–718 Search PubMed.
- W. L. Zeng, J. Y. Song and Y. F. Cen, Effect of the alcohol extract of Illicium dunnianum on the immune function of mice, Journal of Guiyang College of Traditional Chinese Medicine, 1992, 14(02), 60–62 Search PubMed.
- Y. Z. Fang, J. Y. Song, Y. F. Cen, W. L. Zeng and D. S. l Xie, Study on analgesic effect of shihimic acid, Journal of Guiyang College of Traditional Chinese Medicine, 1989, 11(01), 59–62 Search PubMed.
- S. J. Ma, H. B. Li, T. Li, Z. Z. Su, Z. Z. Wang, X. S. Yao, W. Xiao and Y. Yu, Illiciumlignans G–O from the leaves of Illicium dunnianum and their anti-inflammatory activities, RSC Adv., 2021, 11, 30725–30733 RSC.
- J. R. Shao, S. J. Ma, T. Li, X. Q. He, Z. Z. Wang, W. Xiao, X. S. Yao, H. B. Li and Y. Yu, Two new chemical constituents from the leaves of Illicium dunnianum, Nat. Prod. Res., 2022 DOI:10.1080/14786419.2021.2004599.
- N. Wang, X. Yao and R. Ishii, et al., Bioactive sucrose esters from Bidens parviflora, Phytochemistry, 2003, 62(5), 741–746 CrossRef CAS PubMed.
- K. P. Adam, Phenolic constituents of the fern Phegopteris connectilis, Phytochemistry, 1999, 52(5), 929–934 CrossRef CAS.
- Y. L. Wang, H. Li, L. Song, Y. H. Gao and T. F. Chen, Two new phenylpropenoid glycosides from the roots of Illicium dunnianum, Nat. Prod. Res., 2021 DOI:10.1080/14786419.2021.2019736.
- S. W. Lee, G. Li, K. S. Lee, D. K. Song and J. K. Son, A new phenylpropanoid glucoside from the fruits of Illicium verum, Arch. Pharmacal Res., 2003, 26(8), 591–593 CrossRef CAS PubMed.
- S. Latza, D. Ganßer and R. G. Berger, Carbohydrate esters of cinnamic acid from fruits of Physalis peruviana, Psidium guajava and Vaccinium vitisidaea, . Phytochemistry, 1996, 43(2), 481–485 CrossRef CAS.
- E. Selenge, G. Odontuya, T. Murata, K. Sasaki, K. Kobayashi, J. Batkhuu and F. Yoshizaki, Phytochemical constituents of Mongolian traditional medicinal plants, Chamaerhodos erecta and C. altaica, and its constituents prevents the extracellular matrix degradation factors, J. Nat. Med., 2013, 67(4), 867–875 CrossRef CAS PubMed.
- W. Q. Chen, Z. J. Song and H. H. Xu, A new antifungal and cytotoxic C-methylated flavone glycoside from Picea neoveitchii, Bioorg. Med. Chem. Lett., 2012, 22(18), 5819–5822 CrossRef CAS PubMed.
- T. Tanaka, T. Nakashima, T. Ueda, K. Tomii and I. Kouno, Facile discrimination of aldose enantiomers by reversedphase HPLC, Chem. Pharm. Bull., 2007, 55(6), 899–901 CrossRef CAS PubMed.
- Z. C. Qiu, X. L. Dong, Y. Dai, G. K. Xiao, X. L. Wang, K. C. Wong, M. S. Wong and X. S. Yao, Discovery of a New Class of Cathepsin K Inhibitors in Rhizoma Drynariae as Potential Candidates for the Treatment of Osteoporosis, Int. J. Mol. Sci., 2016, 17(12), 2116 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of all new compounds. See https://doi.org/10.1039/d2ra03589h |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |