
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, biological and computational studies of flavonoid acetamide derivatives†
Daniel K. Isika a,
Fatma Nur Özkömeçb,
Mustafa Çeşme*c and
Omowunmi A. Sadik*a
a,
Fatma Nur Özkömeçb,
Mustafa Çeşme*c and
Omowunmi A. Sadik*a
aDepartment of Chemistry and Environmental Science, BioSensor Materials for Advanced Research & Technology (BioSMART Center), New Jersey Institute of Technology, University Heights, 161 Warren Street, Newark, NJ 07102, USA. E-mail: dki3@njit.edu; sadik@njit.edu
bDepartment of Biology, Faculty of Art and Sciences, Kahramanmaras Sutcu Imam University, 46040, Kahramanmaras, Turkey. E-mail: fatmanurozkomec9@gmail.com
cDepartment of Chemistry, Faculty of Art and Sciences, Kahramanmaras Sutcu Imam University, 46040, Kahramanmaras, Turkey. E-mail: mustafacesme@msn.com
First published on 30th March 2022
Abstract
This study reports the synthesis and characterization of a novel class of flavonoid acetamide derivatives (FA) of quercetin, apigenin, fisetin, kaempferol, and luteolin. Flavonoids display numerous biological properties but are limited by aqueous insolubility, enzymatic degradation, instability, and low bioavailability. FAs were synthesized, with 80–82% yields, through the sequential modification of the flavonoid hydroxyl groups into the acetamide moieties. Bioavailability, antioxidant, and ADMET are structure–activity-dependent properties that vary across different classes of flavonoids and dictate the prevalent biological applications of the flavonoids. Thus, the FAs were evaluated for their bioavailability, antioxidant, and ADMET toxicity properties versus the unmodified flavonoids (UFs). In vitro bioavailability analysis shows that the UFs have bio-availabilities in the range of 10.78–19.29% against that of the FAs in the range of 20.70–34.87%. The antioxidant capacity was measured using the 2,2-diphenyl-1-picrylhydrazyl (DPPH·) assay with recorded IC50 values of 2.19–13.03 μM for the UFs. Conversely, the FAs had high DPPH IC50 values ranging from 33.83 to 67.10 μM and corresponding to lower antioxidant activity. The FAs showed favorable ADMET properties. The modification of flavonoids into FAs significantly improves the bioavailability and the ADMET toxicity properties, albeit with decreased antioxidant activity. This work highlights the effect of the global modification of the flavonoids with the acetamide groups on the bioavailability, antioxidant, and ADMET toxicity properties which are critical determinants in the biological applications of the flavonoids.
1. Introduction
Flavonoids are polyphenolic compounds abundant in vegetables and fruits. Their fundamental chemical structure reveals benzene rings interlinked by a linear three-carbon chain forming a C6–C3–C6 carbon skeleton. They possess abundant biological applications, such as anticancer and antioxidant, and can inhibit enzymes such as cyclooxygenase, prostaglandin synthase, and lipoxygenase.1,2 The daily intake of flavonoids depends on the prevalent diet, age, culture, geographic location, and other factors. In the United States, the daily intake of flavonoids is estimated at 500–1000 mg.3The extensive biological applications of flavonoids are restricted by rapid body clearance, rapid body metabolism, enzymatic degradation, aqueous insolubility, and low oral bioavailability.4 These limitations can be addressed through chemical modifications of the flavonoids to achieve flavonoid derivatives with satisfactory properties which maintain and improve the desired biological applications.1
Chemical modifications of flavonoids alter the structure–activity-dependent properties of the flavonoids and determine the biological applications of the new flavonoid derivatives. Therefore, it is indispensable to modify the flavonoids with chemical groups that preserve and improve the prevalent favorable properties. This can be achieved by using bioisosteric groups to replace some chemical groups in the molecule. Whereas there exist lots of structure–activity-based flavonoid properties, the key ones are antimicrobial, antioxidant, ADMET, anticancer, and bioavailability properties. Therefore, it is imperative to look at these properties in the new flavonoid derivatives to assess the effect of chemical modifications on the overall properties of the flavonoids.
The low bioavailability of flavonoids can be attributed to the fact that flavonoids are unstable and prone to (enzymatic) degradation.4
Flavonoids undergo high rates of metabolism, which result in biologically futile metabolites that are eliminated from the body, thus resulting in reduced overall bioavailability.5 During oral administration, the low bioavailability of flavonoids can be attributed to low aqueous solubility, enzymatic degradation, and minimal absorption. Owing to the polar hydroxyl groups in the flavonoids, they suffer from high polarity, thus reducing the ability of these molecules to cross the phospholipid layer of the cell membranes. Flavonoids readily undergo different chemical and enzymatic reactions such as methylation,6 hydroxylation7 and glycosylation.8 The unstable flavonoids degrade to form flavonoid-based compounds, such as quinones, that cannot revert to the original flavonoid forms, hence losing the intended biological activities.9 Flavonoid degradation and metabolism products depend on the class of the flavonoid. The global effect of degradation is the reduced overall bioavailability of these flavonoids.
Several strategies have been proposed to improve the bioavailability of flavonoids. Among these are the chemical modifications of the flavonoids and the use of delivery vehicles such as nanoparticles and polymers. Chemical modifications to reduce the polarity of the flavonoids and improve both the lipophilicity and flexibility of the flavonoids are some viable ways to improve the bioavailability.10 The molecule's polarity can be altered by replacing the hydroxyl groups with isosteres with reduced polarity to achieve low overall hydrophilicity hence facilitating the ability of the new derivatives to cross the biological membranes.1 The use of lipophilic groups, such as methylene groups, between polar groups may help to reduce the polarity and improve the overall bioavailability of a molecule.2
The antioxidant capacity of the flavonoids can be explained based on electron delocalization and the formation of radicals. Taking this into perspective, a closer look at a flavonol such as quercetin (1S0) shows that the antioxidant capacity will be determined by the presence of hydroxyl groups, the carbonyl group at position C-4, and the presence of double bonds in rings A, B, and C. The lone pair of electrons on the carbonyl oxygen in C-4 can trigger electron delocalization, which is facilitated by the double bonds in rings. The formation of radicals from any of the 5 hydroxyl groups can induce the formation of radicals which are further stabilized across rings A, B, and C.11 It is therefore projected that the modification of flavonoids into derivatives that have chemical groups which break the free flow of electrons and the general delocalization may reduce the overall antioxidant capacity of the flavonoids. The reduced antioxidant capacity can be remedied by using isosteres that allow electron delocalization. The antioxidant capacity of flavonoids can be determined using the 2,2-diphenyl-1-picrylhydrazyl (DPPH˙·) assay.12
The objective of this study was to synthesize flavonoid acetamide derivatives and evaluate their bioavailability, antioxidant, and ADMET properties. The flavonoid acetamide derivatives in this study are: 2,2′,2′′-((2-(3,4-bis(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetamide (1S3);2 2,2′-((2-(4-(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-5,7-diyl)bis(oxy))diacetamide (2S3);2 2,2′-((4-(3,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (3S3); 2,2′,2′′-((2-(4-(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetamide (4S3) and 2,2′-((4-(5,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (5S3). The antioxidant capacity was evaluated using the DPPH· radical assay; in vitro bioavailability was determined using the dialysis tubing procedure while the ADMET properties of these compounds were determined using computational tools.
2. Materials and methods
High purity flavonoids, quercetin, apigenin, luteolin, fisetin, kaempferol (Indofine Chemical Company, Hillsborough, NJ, USA), and ethyl chloroacetate, dimethylformamide, anhydrous potassium carbonate, lithium hydroxide, hydrochloric acid, ethyl acetate, thionyl chloride, acetone, ammonia solution, hexane, silica gel, sodium bicarbonate, sodium chloride, sodium sulfate, sodium cholate, porcine pepsin, dialysis tubing, amylase, esterase, porcine pancreatin, dimethyl sulfoxide, and phosphate-buffered saline were purchased from Millipore Sigma (Burlington, MA, USA).2.1 Synthesis of flavonoid acetamide derivatives
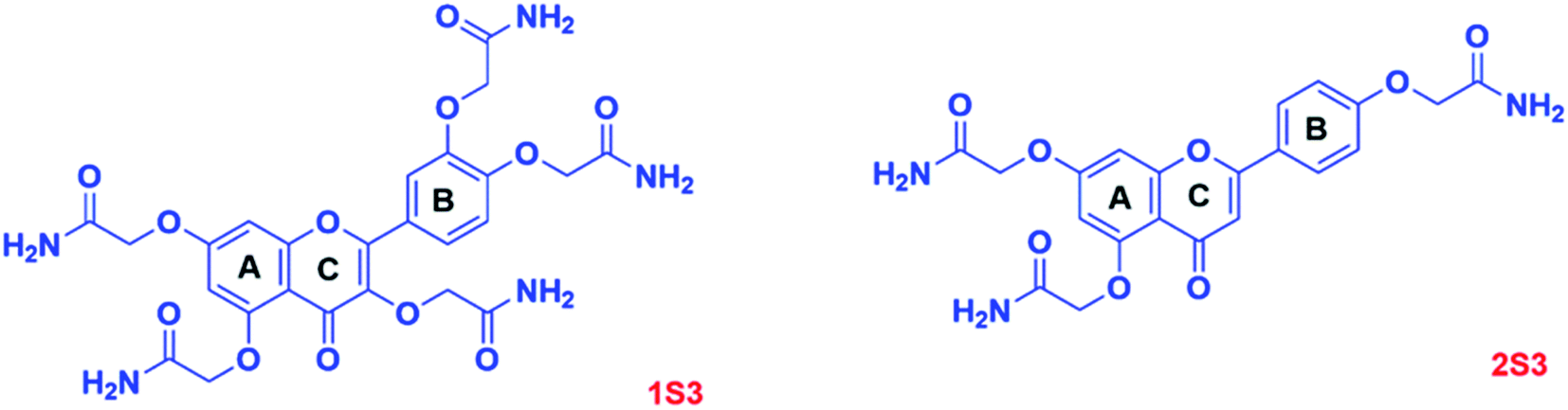 | ||
| Fig. 1 Structures of 1S3 and 2S3 derivatives. | ||
The synthesis of 3S3, 4S3, and 5S3 derivatives was achieved using our optimized procedure as described below.2 The structures of the flavonoids (S0 series), the flavonoid intermediates (S1 & S2 series), and the final flavonoid acetamide derivatives (S3 series) are shown in the scheme below. The IUPAC names of the compounds are shown below, under the procedure section.
2.1.1.1 Synthesis of diethyl 2,2′-((4-(3,7-bis(2-ethoxy-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetate (3S1); triethyl 2,2′,2′′-((2-(4-(2-ethoxy-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetate (4S1) and diethyl 2,2′-((4-(5,7-bis(2-ethoxy-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetate (5S1). Fisetin, 3S0, (0.686 g, 2.396 mmol, 1 eq.) was dissolved in dry DMF (12 mL) in an oven-dried round-bottomed flask. Anhydrous K2CO3 (6 eq.) was added, and the mixture was stirred for 45 minutes. Ethyl chloroacetate (1.409 g, 11.5 mmol, 4.8 eq.) was added to the reaction mixture, which was stirred under nitrogen gas at room temperature. The reaction progress was monitored by TLC for 21 hours. The resultant reaction mixture was suction filtered; the filtrate was concentrated in a rotavapor to obtain a yellow crude product. The solid product was re-dissolved and washed with 5% sodium bicarbonate, brine, and water. The organic layer was dried in anhydrous Na2SO4 before purification by flash column chromatography using silica gel in (hexane/ethyl acetate, 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) to afford 3S1. The final 3S1 product was a white solid, 1.254 g, 83% yield, Rf = 0.21 silica gel in (hexane/ethyl acetate, 1:1, v/v). The intermediates 4S1 and 5S1 were similarly synthesized to obtain 4S1; a white solid, 84% yield, Rf = 0.10 and 5S1; a white solid, 80% yield, Rf = 0.06 silica gel in (hexane/ethyl acetate, 1:1, v/v). The structures of these compounds were verified by 1H and 13C NMR characterizations.
:1 v/v) to afford 3S1. The final 3S1 product was a white solid, 1.254 g, 83% yield, Rf = 0.21 silica gel in (hexane/ethyl acetate, 1:1, v/v). The intermediates 4S1 and 5S1 were similarly synthesized to obtain 4S1; a white solid, 84% yield, Rf = 0.10 and 5S1; a white solid, 80% yield, Rf = 0.06 silica gel in (hexane/ethyl acetate, 1:1, v/v). The structures of these compounds were verified by 1H and 13C NMR characterizations.3S1: δ1H (400 MHz, DMSO-d6): 7.98 (1H, d, J = 8.9 Hz), 7.83–7.75 (2H, m), 7.35–7.27 (1H, m), 7.18–7.08 (2H, m), 5.12–4.70 (8H, m), 4.27–4.06 (8H, m), 1.29–1.12 (12H, m). δ13C (400 MHz, DMSO-d6): 173.41, 169.27, 169.22, 169.04, 168.66, 167.99, 162.71, 156.67, 154.02, 150.12, 147.23, 138.83, 127.04, 124.09, 123.44, 118.18, 115.54, 115.37, 114.22, 102.23, 68.39, 66.30, 65.82, 65.73, 61.64, 61.52, 61.39, 61.29, 61.12, 14.64, 14.59, 14.56.
4S1: δ1H (400 MHz, DMSO-d6): 8.16–8.07 (2H, m), 7.13–7.05 (2H, m), 6.90 (1H, d, J = 2.3 Hz), 6.46 (1H, d, J = 2.3 Hz), 4.92 (6H, d, J = 14.1 Hz), 4.78 (2H, s), 4.26–4.14 (6H, m), 4.10 (2H, q, J = 7.1 Hz), 1.27–1.11 (12H, m). δ13C (400 MHz, DMSO-d6): 171.75, 168.61, 168.43, 168.19, 167.97, 161.69, 159.29, 158.57, 157.78, 151.45, 138.48, 129.97, 123.06, 114.48, 108.98, 97.89, 94.83, 67.67, 65.69, 65.03, 64.67, 60.84, 60.73, 60.39, 14.02, 13.94.
5S1: δ1H (400 MHz, DMSO-d6): 7.65 (1H, dt, J = 8.5, 2.0 Hz), 7.58 (1H, t, J = 2.2 Hz), 7.10 (1H, dd, J = 9.9, 8.7 Hz), 6.90 (1H, dd, J = 5.8, 2.3 Hz), 6.83 (1H, d, J = 6.9 Hz), 6.46 (1H, d, J = 2.3 Hz), 5.15–4.77 (8H, m), 4.19 (8H, pd, J = 7.2, 2.0 Hz), 1.23 (12H, tdd, J = 7.2, 5.1, 2.4 Hz). δ13C (400 MHz, DMSO-d6): 175.97, 169.09, 168.88, 168.78, 168.49, 162.04, 159.92, 159.33, 159.05, 150.52, 147.87, 124.46, 120.52, 114.51, 112.34, 109.53, 107.89, 98.54, 95.75, 66.13, 66.03, 65.77, 65.52, 61.35, 61.20, 61.11, 14.55, 14.51.
2.1.1.2 Synthesis of 2,2′-((4-(3,7-bis(carboxymethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetic acid (3S2); 2,2′,2′′-((2-(4-(carboxymethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetic acid (4S2) and 2,2′-((4-(5,7-bis(carboxymethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetic acid (5S2). 3S1 (0.914 g, 1.449 mmol, 1 eq.) was dissolved in THF/H2O (1
:2). LiOH·H2O (486.5 mg, 11.6 mmol, 8 eq.) was added. The reaction mixture was stirred, at room temperature and the progress of the reaction was monitored by TLC for 23 hours. The reaction was quenched with 1 N HCl, the product was concentrated and purified through a repeated cycle of washes with ethyl acetate, acetone and hexane. The final product, 3S2, was a white solid, 0.713 g, 95% yield. The final product, 3S2, was a white solid, 95% yield. The intermediates 4S2 and 5S2 were similarly synthesized to obtain 4S2, as a white solid, 95% yield, and 5S2, as a white solid, 94% yield. The structures of the products were confirmed using 1H NMR characterization.3S2: δ1H (400 MHz, DMSO-d6): 7.95 (1H, d, J = 8.7 Hz), 7.83 (1H, s), 7.76 (1H, d, J = 8.7 Hz), 7.23 (1H, s), 7.07 (2H, d, J = 7.6 Hz), 4.83 (2H, s), 4.75 (6H, s).
4S2: δ1H (400 MHz, deuterium oxide): 7.91 (2H, d, J = 8.5 Hz), 6.91 (2H, d, J = 8.5 Hz), 6.54 (1H, s), 6.28 (1H, s), 4.58 (2H, s), 4.52 (4H, d, J = 7.1 Hz), 4.37 (2H, s).
5S2: δ1H (400 MHz, DMSO-d6): 7.68–7.59 (1H, m), 7.59–7.53 (1H, m), 7.06 (1H, d, J = 8.7 Hz), 6.90 (1H, d, J = 2.3 Hz), 6.83 (1H, s), 6.49 (1H, d, J = 2.3 Hz), 4.85 (8H, dd, J = 10.9, 7.7 Hz).
2.1.1.3 Synthesis of 2,2′-((4-(3,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (3S3); 2,2′,2′′-((2-(4-(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetamide (4S3) and 2,2′-((4-(5,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (5S3). 3S2 (0.620 g, 1.196 mmol, 1 eq.) was suspended in excess thionyl chloride in an oven-dried round-bottomed flask. The reaction mixture was refluxed amidst stirring and the progress of the reaction was monitored by TLC for 8 hours. The reaction mixture was filtered off to obtain the acetyl chloride intermediate product which was slowly transferred into ice-cold ammonium hydroxide (excess). The reaction mixture was stirred at room temperature for 3 hours. The excess solvent was removed by rotavapor. The product was concentrated in a vacuum and purified in flash column chromatography using silica gel in methanol/water, 7
:1, v/v to achieve the final product, 3S3; yellow solid, 0.498 g, 81% yield. The derivatives 4S3 and 5S3 were similarly synthesized to obtain 4S3; faint yellow solid, 80% yield and 5S3; yellow-brown solid, 82% yield. The structures of the products were confirmed using 1H, 13C NMR and FTIR characterizations.3S3: δH (400 MHz, DMSO-d6): 8.02 (1H, d, J = 8.8 Hz), 7.83–7.73 (3H, m), 7.66 (1H, s), 7.52 (2H, s), 7.47 (3H, s), 7.40 (1H, s), 7.26 (1H, d, J = 2.3 Hz), 7.19–7.11 (2H, m), 4.66–4.57 (6H, m), 4.37 (2H, s). δ13C (400 MHz, DMSO-d6): 173.60, 170.64, 170.40, 170.05, 169.48, 162.77, 156.69, 154.76, 150.42, 147.68, 139.39, 126.85, 123.74, 123.37, 117.89, 115.59, 115.19, 114.18, 102.08, 70.98, 68.58, 67.93, 67.50.
4S3: δH (400 MHz, DMSO-d6): 8.11 (2H, d, J = 9.0 Hz), 8.09 (1H, s), 7.73 (1H, s), 7.66 (1H, s), 7.63–7.58 (2H, m), 7.47 (1H, s), 7.41 (2H, d, J = 9.6 Hz), 7.13 (2H, d, J = 8.9 Hz), 6.88 (1H, d, J = 2.2 Hz), 6.66 (1H, d, J = 2.2 Hz), 4.62 (2H, s), 4.55 (4H, s), 4.30 (2H, s). 13C NMR (400 MHz, DMSO-d6) δ 173.39, 170.53, 170.15, 169.95, 169.42, 165.23, 162.92, 160.18, 158.19, 158.15, 153.57, 139.41, 133.99, 130.47, 130.05, 123.07, 115.36, 108.93, 98.83, 95.33, 70.66, 68.26, 67.49, 67.16.
5S3: δH (400 MHz, DMSO-d6): 8.21 (1H, dd, J = 18.5, 3.0 Hz), 7.82–7.67 (2H, m), 7.67–7.57 (2H, m), 7.49 (4H, d, J = 12.6 Hz), 7.25 (1H, d, J = 2.4 Hz), 7.13 (1H, dd, J = 8.7, 4.3 Hz), 6.83 (1H, s), 6.65 (1H, dd, J = 14.4, 2.3 Hz), 6.59–6.50 (1H, m), 4.71 (1H, s), 4.67–4.57 (4H, m), 4.57–4.49 (2H, m), 4.42 (1H, d, J = 14.9 Hz). 13C NMR (400 MHz, DMSO-d6) δ 187.76, 170.65, 170.41, 170.29, 170.00, 169.66, 169.45, 169.35, 162.78, 160.77, 159.10, 158.14, 151.10, 148.18, 124.30, 120.92, 114.46, 112.68, 108.91, 107.72, 99.10, 95.82, 68.55, 68.37, 67.96, 67.47.
2.2 In vitro bioavailability
The in vitro bioavailability was determined using a slightly modified dialysis tubing procedure available in the literature.13–15Each of the samples collected after the stage 1 and stage 2 experiments was characterized using UV-Vis. The UV-Vis vis spectra of the recovered samples were compared with those of the original compounds of the study. The concentrations of the recovered samples were evaluated from the calibration curves of the standard samples. The calibration curves for each standard study compound were made at both pH 1.0 and pH 7.0. The equations obtained from these calibration curves were used to calculate the concentration of each sample after each stage of digestion.
In this study, percentage bioavailability was defined as the fraction of the standard compound recovered in the unchanged form expressed as a percentage.
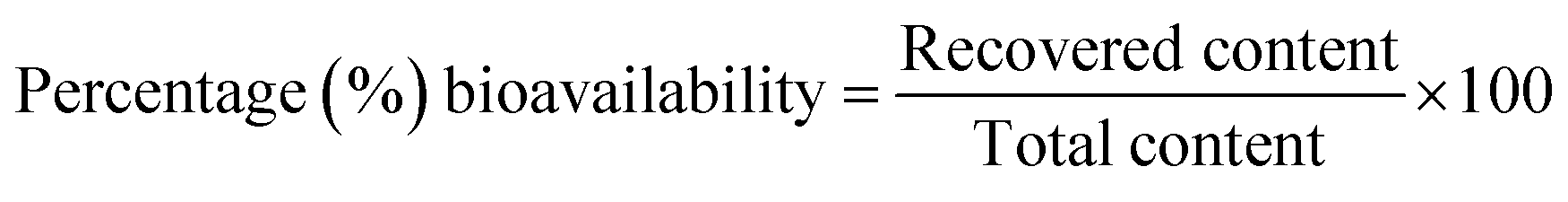 | (1) |
2.3 2,2-Diphenyl-1-picrylhydrazyl (DPPH˙) assay
The DPPH˙ radical solution was prepared for immediate use at a concentration of 50 μM, in methanol. Stock solutions of the S0 and S3 series of compounds were prepared in DMSO. The DPPH· radical scavenging activities of the compounds were determined using a slightly modified literature procedure.16 Briefly, 100 μL of each sample was mixed with 3.0 mL of a freshly prepared DPPH· solution in methanol. The mixture was incubated in the dark for 30 minutes, and the absorbance was read off at 517 nm wavelength using a UV-Vis spectrophotometer. The stability of the DPPH˙ radical at different concentrations were determined in both the methanol and the DMSO solvents. A concentration-based study was done by repeating each experiment across different sample concentrations. The %DPPH˙ radical scavenging activity of the samples was calculated against vehicle control (DMSO) using the formula below.
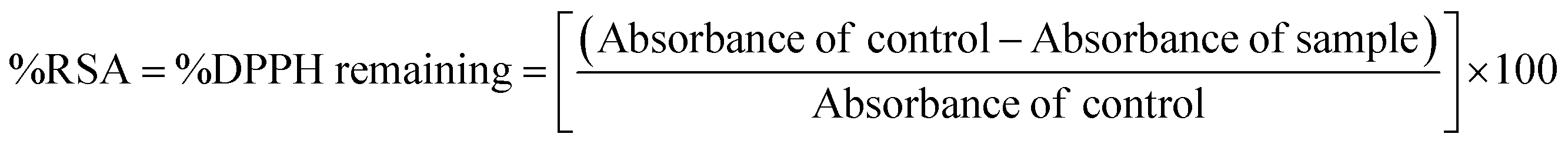 | (2) |
The %RSA data was plotted and used to calculate the half-maximal inhibitory concentration (IC50) values. The IC50 values from the plots were further double-checked using verified available online tools.16,17 The final antioxidant activity of each compound was expressed as the IC50 value.
2.4 Drug relevant and toxicity properties prediction
Property Explorer is a free prediction tool used to calculate physicochemical and toxicological molecular properties, which must be perfected when designing pharmaceutically active compounds. It was initially developed by T. Sander and used at Actelion Pharmaceutical Ltd as an integrated part of the compound registration system in the drug discovery department. Whenever a structure is valid, the OSIRIS Property Explorer allows one to draw chemical structures and determine on-the-fly various drug-relevant properties (clogP, solubility, molecular weight, toxicity risk assessment, overall drug score, etc.).18
The results from the estimation are valued and color-coded. For example, properties with a high probability of adverse effects are described in red; such include mutagenicity or low intestinal absorption. Similarly, a green color implies that the properties conform to desirable drug properties. The drug-relevant properties and toxicity risk assessment by OSIRIS property explorer were computed and represented in Tables 3 and 4.
In silico toxicity prediction has tremendous importance in the early stages of drug discovery, considering that ∼30% of drug candidates fail owing to these issues. We tried to develop a reliable model which can readily classify mutagenicity of Ames Salmonella TA100, TA1535 species, and rodent carcinogenicity 2 year assay of rat and mouse by backward elimination and Rprop neural net method.19
Ames test uses several strains of the bacterium Salmonella typhimurium that carry mutations in genes involved in histidine synthesis to require histidine for growth. The variable being tested is the mutagen's ability to cause a reversion to growth on a histidine-free medium. PreADMET predicts toxicity to TA98, TA100, and TA1535, often used in the Ames test. Moreover, the result can be calculated using consideration of metabolite (metabolic activation by rat liver 10% homogenate, +S9) and without consideration of metabolite (no metabolic activation, −S9). The actual value of the prediction result is “positive” or “negative” (https://preadmet.bmdrc.kr/toxicity/)'.
3. Results and discussion
3.1 Characterization of flavonoid acetamide derivatives
The global flavonoid acetamide derivatives for this study were synthesized following our optimized procedure.2 The flavonoid acetamide derivatives were synthesized through a series of steps, illustrated in Scheme 1, to achieve global flavonoids acetamide derivatives in which all the hydroxyl groups attached to 2-phenylchromen-4-one structure of the flavonoids were converted into acetamide groups. This synthesis was achieved consecutively by first converting the flavonoids (S0 series) into esters (S1 series) which were hydrolyzed into carboxylic acids (S2 series) through the ester saponification reaction. The carboxylic acid intermediates (S2 series) were then converted into the final acetamide derivatives (S3 series) through the amidification process. The intermediates and the final products were characterized by 1H & 13C NMR confirming the formation of the flavonoid derivatives.2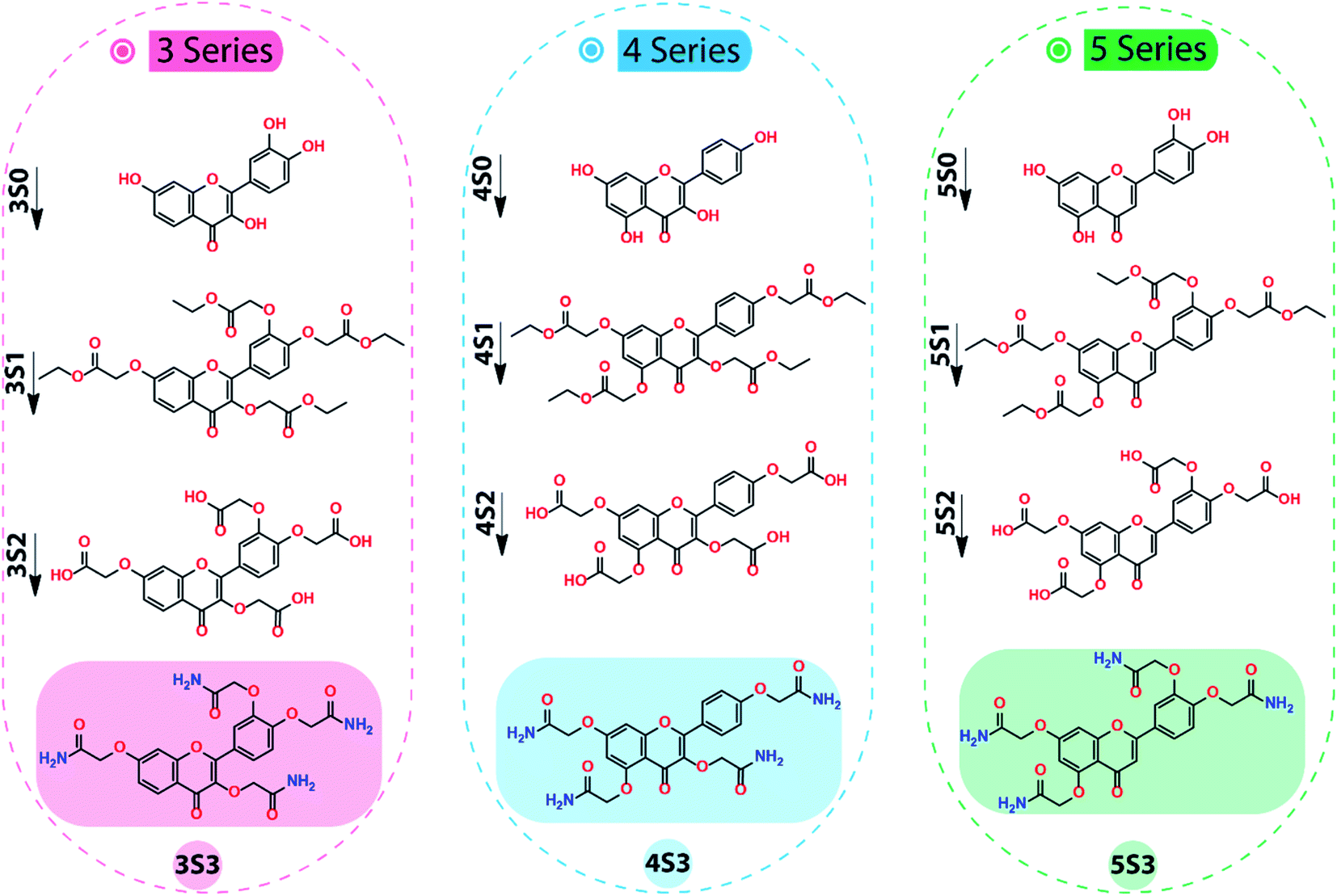 | ||
| Scheme 1 Steps in the synthesis of 2,2′-((4-(3,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (3S3), 2,2′,2′′-((2-(4-(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetamide (4S3) and 2,2′-((4-(5,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (5S3) derivatives. (Reagents and reaction conditions: Step1 (S0 → S1) ethyl chloroacetate (4.8 eq.), K2CO3 (excess), DMF, Rt. Step 2 (S1 → S2) LiOH·H2O (8 eq.), THF/H2O, Rt. Step 3 (S2 → S3) SOCl2 (excess), reflux, 75 °C; NH4OH (excess), 0 °C-Rt.). | ||
1H and 13C NMR characterizations were used to confirm the formation of the S1 derivatives. The 1H NMR characterizations of 3S1, 4S1 and 5S1 confirmed 6 aromatic, 8 methylene (ether), 8 methylene (ester), and 12 methyl (ester) hydrogens for each intermediate. The 1H NMR spectra of the 3S1, 4S1 and 5S1 are shown in Fig. S1–S3,† respectively.
The conversion of the S1 derivatives into the S2 intermediates was confirmed using the 1H NMR characterizations. The 1H NMR characterization confirmed the cleavage of the –OCH2CH3 group in the S1 intermediate, the integration confirmed the expected number of the aromatic and the methylene (ether) protons and the formation of the carboxylic acid, S2 derivative. The 3S2, 4S2 and 5S2 derivatives each had 6 aromatic protons and 8 methylene protons as confirmed by the 1H NMR. The representative 1H NMR spectra of 4S2 derivative are shown in Fig. S4.† In the final step, the S2 intermediate was converted into the acetamide, S3 derivative. These products were confirmed using 1H & 13C NMR and FT-IR characterizations. The 1H NMR confirmed the amide, aromatic, and methylene protons. Using the 1H NMR characterization, 8-CONH2 (amide) protons, 6 aromatic and 8 methylene protons were confirmed. The 1H NMR spectra of the 3S3, 4S3 & 5S3 derivatives are shown in Fig. S5–S7,† respectively. Further characterization of these derivatives was done using the 13C NMR characterization, which further confirms the formation of the 3S3, 4S3, and 5S3 derivatives.
The flavonoid acetamide derivatives have the methylene group as well as the iconic carbonyl and amide functional groups. These functional groups were confirmed using FT-IR characterization. The FT-IR data obtained from these spectra conform to the structures of these compounds. The FT-IR spectra of the flavonoid acetamide derivatives 3S3, 4S3, and 5S3 are shown in Fig. S8–S10.† The corresponding FT-IR peak assignments for all the compounds are shown in Table S1.†
3.2 Bioavailability studies results
The biological applications of flavonoids are highly limited by their low bioavailability.2 Flavonoids can be chemically modified to achieve derivatives which are more resistant to (enzymatic) degradation and metabolic activities. Flavonoid derivatives which are more resistant to enzymatic degradation and metabolic activities can reach their target sites after oral administration hence improving their overall bioavailability. The chemical modification of flavonoids with groups that alter the pH enables the efficient delivery of the flavonoids by inducing the ‘proton sponge effect’ consequently facilitating endosomal escape in the cells. In this study, the flavonoids were modified to achieve flavonoid acetamide derivatives in which the latter is more lipophilic, has reduced polarity, and enhanced flexibility.2 These three properties are related to improved bioavailability in flavonoids.In vitro bioavailability studies were carried out using the dialysis tubing procedure adopted from the literature.13,14 The dialysis tubing procedure involves two stages of digestion, the pepsin digestion (stage 1) and pancreatin digestion (stage 2). These two stages of digestion are carried out sequentially. The dialysis tubing procedure is based on an in vitro stimulation of gastric and intestinal digestive fluids and conditions. The pH was adjusted to match that of the digestive chambers.
For the purposes of this study, the test samples were dissolved in dimethyl sulfoxide. After every step of the digestion, aliquot samples were collected for characterization. The calibration curves for the standards in each study compound were made, at the two pH values and media, using UV-Vis characterizations. The equations developed from these calibration curves were used to calculate the concentrations of the study samples after the pepsin and pancreatin digestion. The total in vitro bioavailability was the sum of the pepsin digestion (2 hours, stage 1) bioavailability and pancreatin digestion (4 hours, stage 2) bioavailability. The in vitro bioavailability was expressed as the average of three studies for each sample.
Grande et al. evaluated the in vitro bioavailability of quercetin (1S0) using the same dialysis tubing procedure adopted in this study.13 Based on their studies, they reported that quercetin (1S0) had a bioavailability of 4.4 ± 0.9% after the pepsin digestion (2 hours, stage 1) and 6.3 ± 0.8% after the pancreatin digestion (4 hours, stage 2); the corresponding total in vitro bioavailability of quercetin (1S0) was found to be 10.7 ± 1.3%.13 The in vitro bioavailability studies of the flavonoids and flavonoid acetamide derivatives in this study were carried out using the dialysis tubing procedure reported by Grande et al.13 Based on our studies, quercetin (1S0) had an in vitro bioavailability of 4.68 ± 0.19% after pepsin digestion (stage 1) and 6.10 ± 0.64% after pancreatin digestion (stage 2) amounting to 10.78 ± 0.82% in total in vitro bioavailability. This data was statistically similar to that reported in the adopted procedure.13
The in vitro bioavailability data obtained for the flavonoids were reported as the mean of the 3 trials ± the standard deviation. The in vitro bioavailability of the other flavonoids after the pepsin and pancreatic digestions was 2S0 (4.77%, 11.83%); 3S0 (8.25%, 7.94%); 4S0 (4.33%,11.07%) and 5S0 (8.48%, 10.81%). The corresponding total bioavailability for these compounds was 2S0 (16.59%); 3S0 (16.19%); 4S0 (15.40%) and 5S0 (19.29%). The in vitro bioavailabilities after both the pepsin digestion and the pancreatic digestions and the total in vitro bioavailability for the flavonoids are illustrated in Fig. 2.
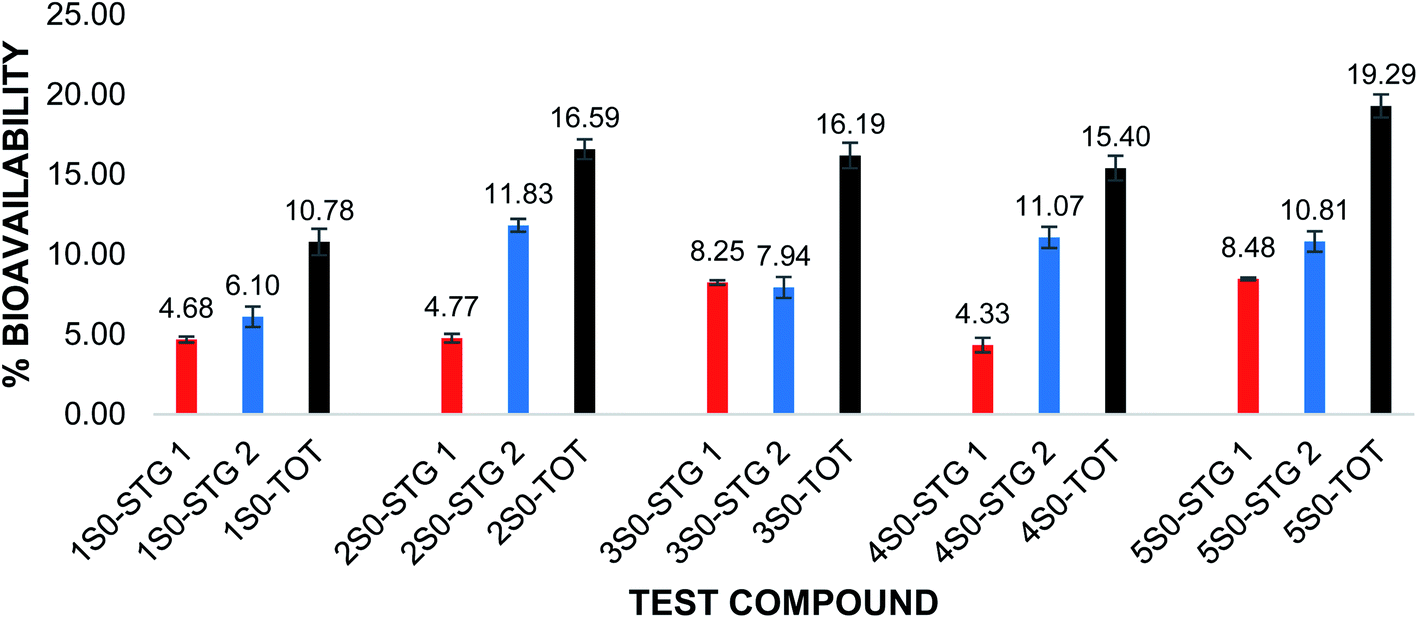 | ||
| Fig. 2 Percentage (%) bioavailability of UFs after pepsin and pancreatin digestion and total in vitro bioavailability. Red: %bioavailability after pepsin digestion (STG 1); blue: %bioavailability after pancreatin digestion (STG 2); black: total %bioavailability after both pepsin and pancreatin digestions. Compounds 1S0–5S0. The error bars show ± standard deviation. | ||
The in vitro bioavailability of quercetin penta acetamide (1S3) was 11.86 ± 0.03% after the pepsin digestion and 8.84 ± 0.32% after the pancreatin digestion. The total in vitro bioavailability for the quercetin penta acetamide derivative was 20.70 ± 0.29%. The total in vitro bioavailability of the apigenin triacetamide (2S3), fisetin tetra-acetamide (3S3), kaempferol tetra acetamide (4S3) and luteolin tetra acetamide (5S3) was 22.20 ± 0.82%, 34.81 ± 0.85%, 21.34 ± 0.50% and 34.87 ± 0.87%, respectively. The bioavailability data detailing the pepsin and pancreatin digestions as well as the total in vitro bioavailability of the flavonoid acetamide derivatives, is illustrated in Fig. 3.
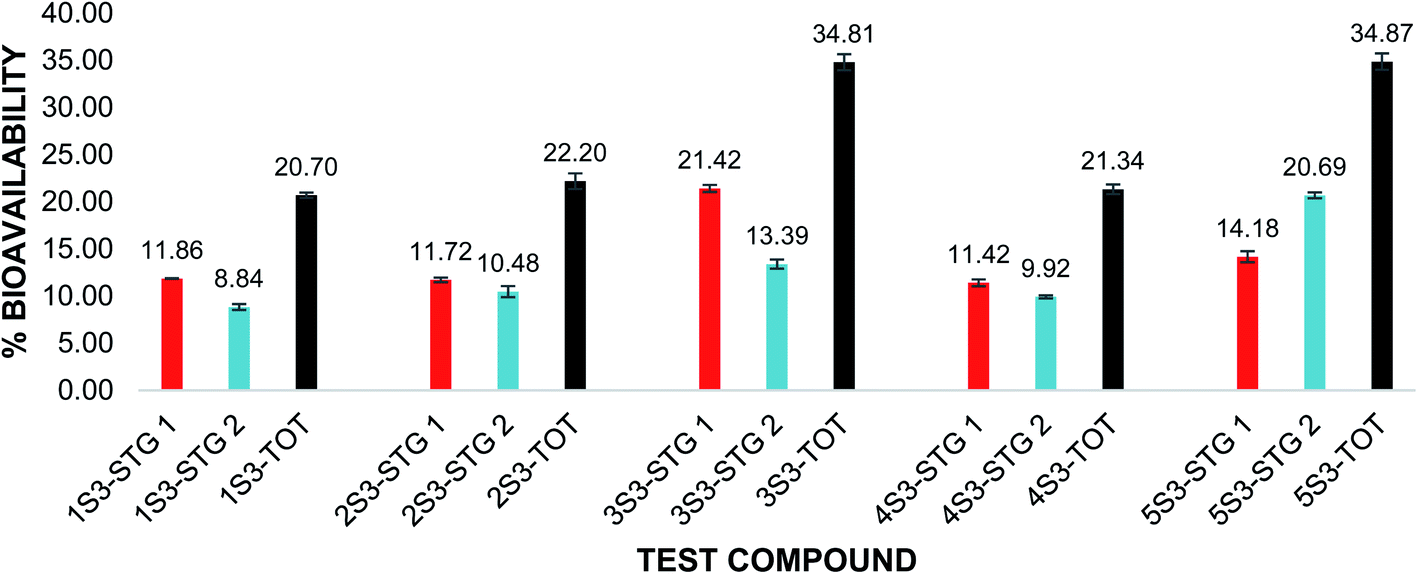 | ||
| Fig. 3 Percentage (%) bioavailability of FAs after pepsin and pancreatin digestion and total in vitro bioavailability. Red: %bioavailability after pepsin digestion (STG 1); blue: %bioavailability after pancreatin digestion (STG 2); black: total %bioavailability after both pepsin and pancreatin digestions. Compounds 1S3–5S3. The error bars show ± standard deviation. | ||
The flavonoid acetamide derivatives showed significantly higher bioavailability than the corresponding unmodified flavonoids. The total in vitro bioavailabilities of the flavonoids relative to the flavonoid acetamide derivatives are shown in Fig. 4. The differences in the bioavailability between the unmodified flavonoids and the flavonoid acetamide derivatives can be explained by the presence of the methylene and amide groups (collectively forming the acetamide group) in the flavonoid acetamide derivatives. The flavonoid acetamide derivatives have different flexibility, lipophilicity and polarity compared to the unmodified flavonoids, which are key factors affecting the bioavailability of flavonoids.20
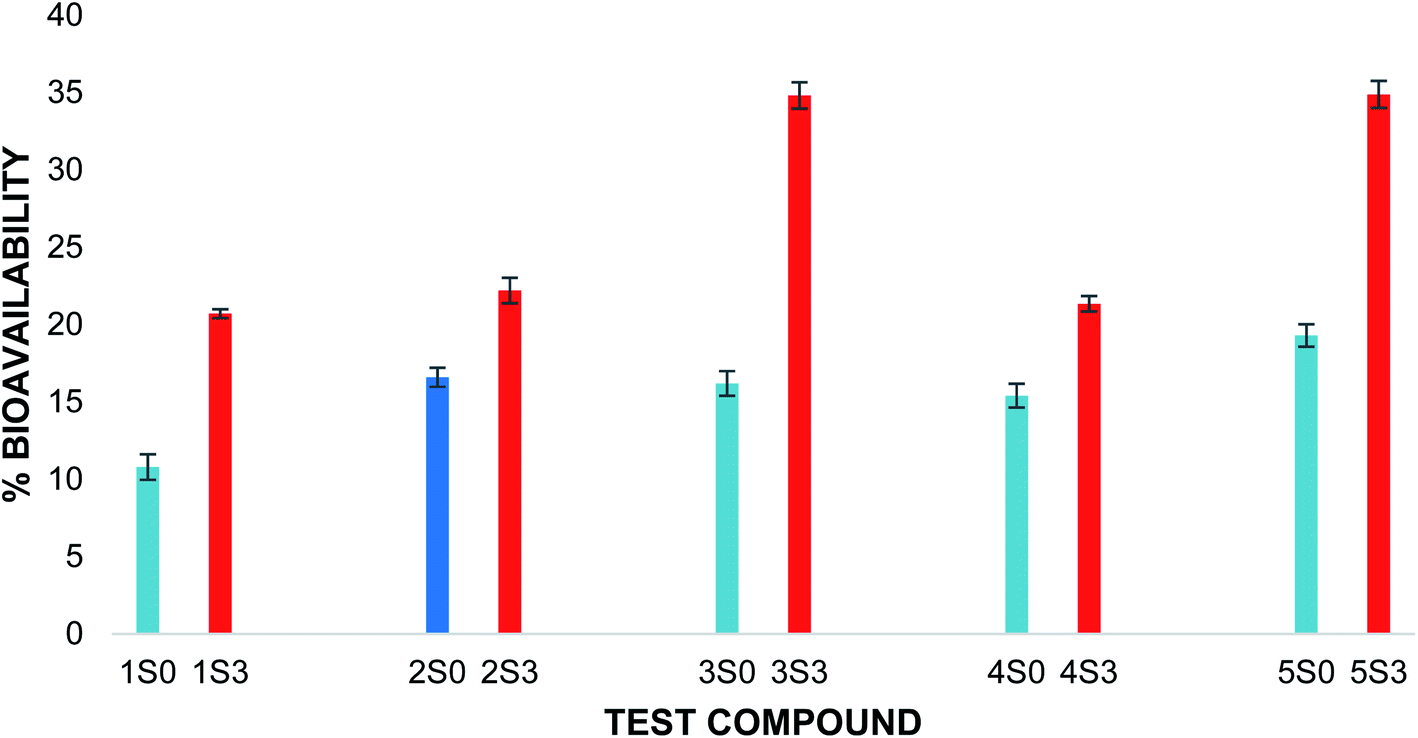 | ||
| Fig. 4 Total percentage bioavailability of S0 & S3 series. Blue: unmodified flavonoid, red: flavonoid acetamide derivative. The error bars show ± standard deviation. | ||
3.3 2,2-Diphenyl-1-picrylhydrazyl (DPPH˙) assay results
The 2,2-diphenyl-1-picrylhydrazyl (DPPH˙) assay was used to determine the antioxidant capacity of the flavonoids and the flavonoid acetamide derivatives. The DPPH free radical is stable and violet in color; it is discolored towards yellow upon adding a significant amount of free radical scavenging agents. This assay measures the ability of the flavonoids to scavenge the free radicals. The color changes correspond to the amount of stable DPPH free radicals remaining. Thus the antioxidant capacity of the study samples can be quantified spectrophotometrically from the absorbance of the remaining DPPH free radicals.The stability of DPPH free radicals may vary across different reaction media. The stability of different concentrations of the DPPH free radicals in methanol was monitored. The UV-Vis absorption spectra of different concentrations of DPPH free radicals in methanol across different wavelengths are shown in Fig. 5. The absorbance of the DPPH free radical is proportional to the DPPH free radical concentration, as monitored at 517 nm, which is highlighted in Fig. 5. A plot of the absorbance of the DPPH free radical at 517 nm in wavelength versus the concentration of the DPPH free radicals shows a linear relationship. A linear regression equation was obtained from the calibration curve; thus, the exact initial DPPH free radical concentration in the reaction medium was calculated using the equation below.
| A517 = 0.0171 × 10−6 × [DPPH] + 0.0191, R2 = 0.999 | (3) |
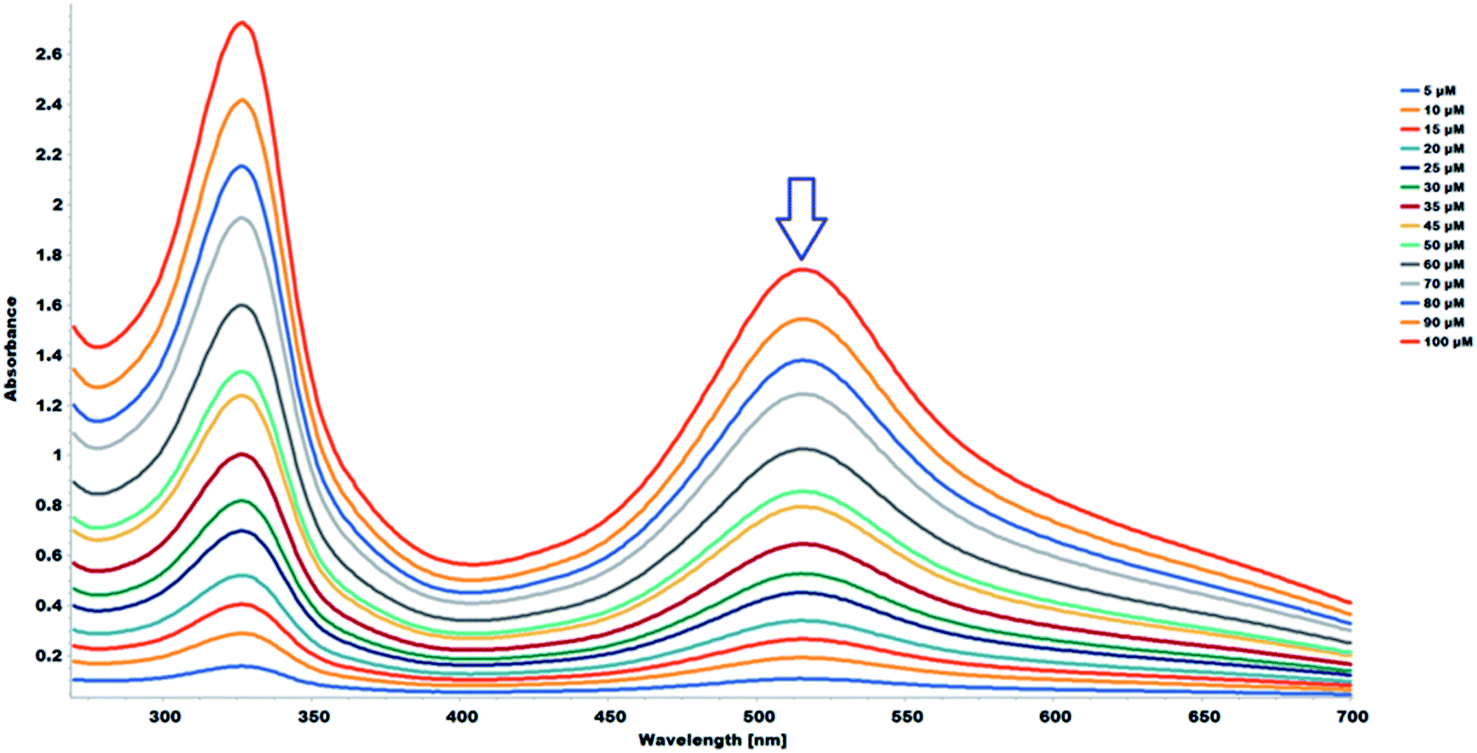 | ||
| Fig. 5 UV-Vis absorption spectra (268–700 nm) in methanol of DPPH (5.0–100 μM). | ||
The study samples were prepared in DMSO. The stability of the methanolic DPPH in DMSO was evaluated across different concentrations with 100 μL of DMSO, the same amount used for the antioxidant capacity evaluation. The methanolic DPPH free radicals remained stable across the different DPPH free radical concentrations. The UV-Vis absorption spectra of different concentrations of DPPH free radicals in methanol, in the presence of 100 μL of DMSO, across different wavelengths is shown in Fig. 6. The absorbance of the DPPH free radical is proportional to concentration at 517 nm in wavelength, which is highlighted in Fig. 6. A linear regression equation was obtained from the calibration curve; thus, the exact initial concentration of the DPPH free radicals in the reaction medium, in the presence of the sample solvent, DMSO, was calculated using the equation below:
| A517 = 0.0164 × 10−6 × [DPPH] + 0.0213, R2 = 0.999 | (4) |
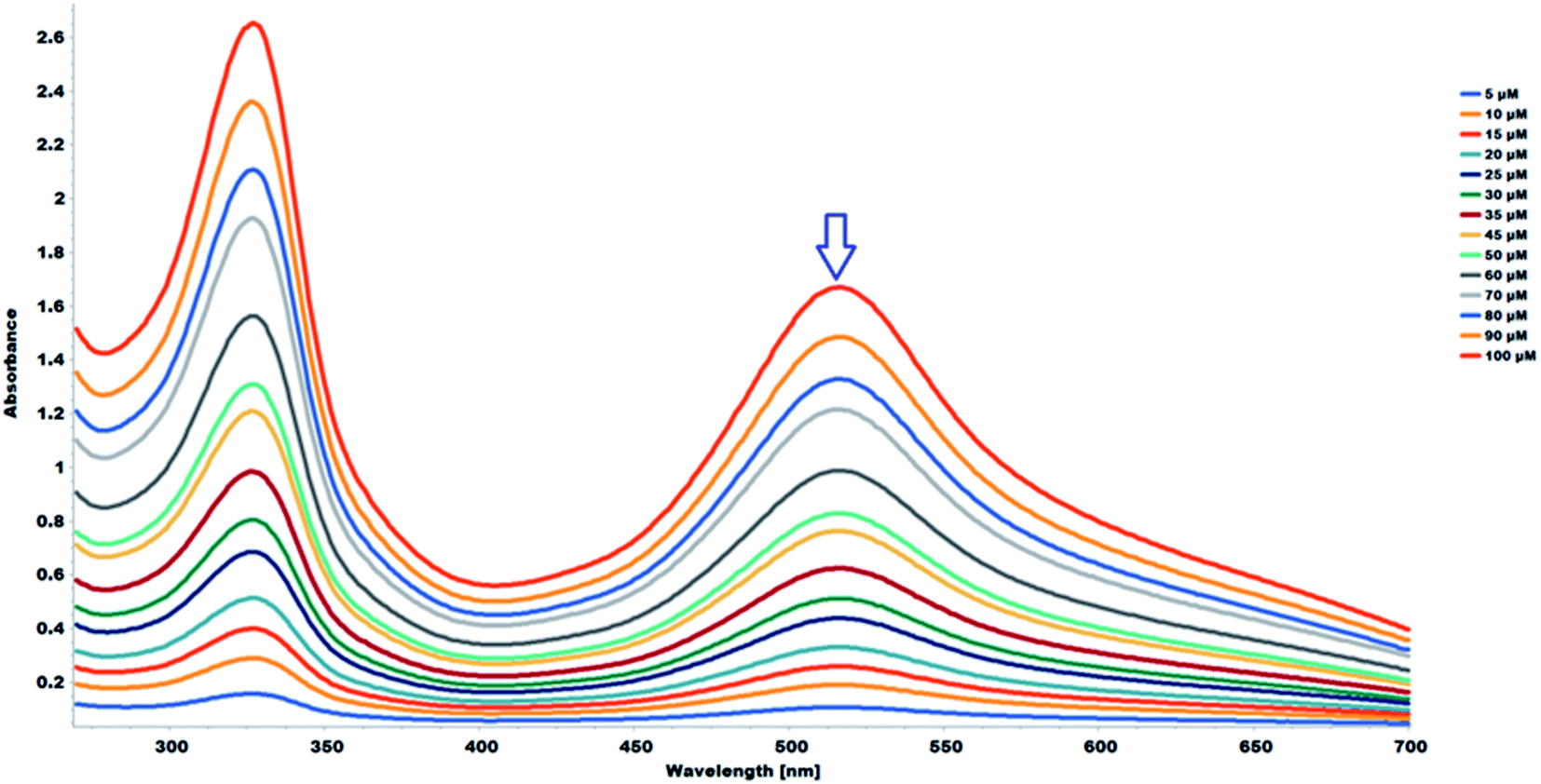 | ||
| Fig. 6 UV-Vis absorption spectra (268–700 nm) in methanol of DPPH (5.0–100 μM) with 100 μL of DMSO. | ||
The data obtained from the methanolic DPPH in DMSO more accurately factor the effect of the DMSO in the calculation of the antioxidant activity and was therefore used as the blank for the quantitative antioxidant capacity analysis.
The interaction between the DPPH˙ radical and flavonoids can be monitored using the decay curve. The reaction between the DPPH and the flavonoids follows two successive steps as seen from the DPPH decay curves. The first step is a quick decay of the DPPH free radicals in the solvent medium within ∼120 seconds. The second step is a slow decay which represents the interaction between the DPPH free radicals and the antiradical agent and typically occurs within ∼60 minutes.20,21 Considering the DPPH decay curves, the choice of optimal reaction time for the study is consequential in determining the antioxidant capacity of the samples. The decay curve of the reaction shows a concentration-dependent kinetic behavior. The decay curves for the reactions were recorded for 30 minutes. The decay curves for the interaction between the DPPH and the 1S0 samples are shown in Fig. 7. The corresponding decay curves for the other study samples are shown in Fig. S11–S17.†
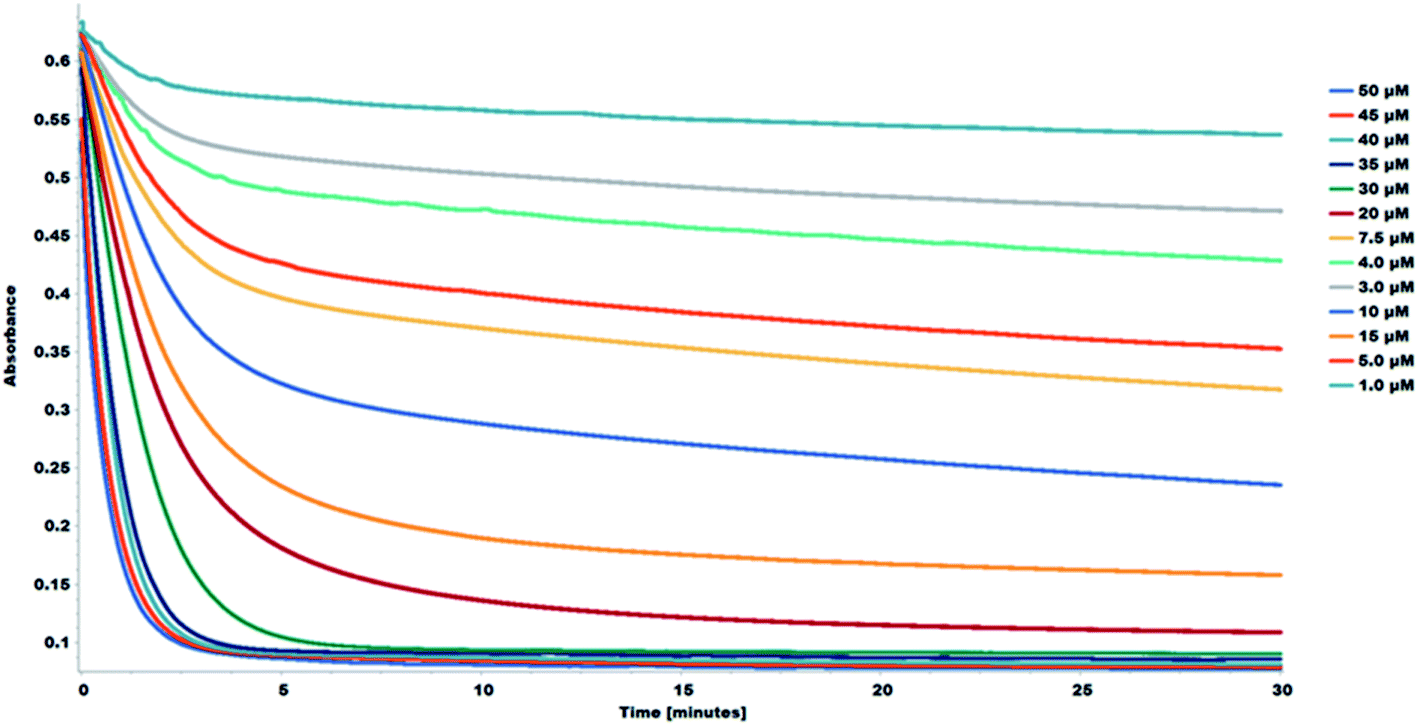 | ||
| Fig. 7 Decay curve for the reaction between DPPH and 1S0. Decrease in absorbance, at 517 nm of DPPH in methanol in the presence of various concentrations of 1S0 (1.0–50 μM). | ||
The effect of the addition of an antiradical agent can be determined spectrophotometrically by monitoring the change in the DPPH free radical concentration. The amount of the stable DPPH free radicals remaining was expressed as the %RSA which was used to calculate the IC50 values. The IC50 values indicate 50% of the maximal inhibitory concentration. Thus, the IC50 is inversely proportional to the antioxidant activity; a compound with a low IC50 value has a high antioxidant capacity, the vice versa holds.
The S0 series of compounds showed the lowest IC50 values and thus the best antioxidant capacities. The IC50 values of 2.19–13.03 μM were recorded. The S3 series of compounds had very high IC50 values corresponding to low antioxidant activities. The IC50 values for the S3 series of compounds ranged from 33.83–67.10 μM. The order of the antioxidant activity among the S0 series of compounds was 3S0 ≥ 1S0 > 5S0 > 4S0 > 2S0 and 3S3 > 1S3 > 4S3 > 5S3 > 2S3, among the S3 series. A similar order in the flavonoid (S0 series) antioxidant activity was reported by Tian Chunlian, et al. as quercetin (1S0) > luteolin (5S0) > kaempferol (4S0) > apigenin (2S0), from their DPPH free radical antioxidant studies.22 Similarly, Zheng Cheng-Dong, et al. reported the following trend in flavonoid (S0) antioxidant capacity: quercetin (1S0) > luteolin (5S0) > kaempferol (4S0) > apigenin (2S0), as determined by the DPPH free radical scavenging activity studies.23 On the contrary, Hirano Reiko, et al. reported the following order in the flavonoid (S0) antioxidant capacity as determined using the DPPH free radical scavenging assay: quercetin (1S0) > kaempferol (4S0) > luteolin (5S0).24 The DPPH free radical scavenging activity of the apigenin (2S0) and the corresponding apigenin triacetamide derivative (2S3) were very low. The %RSA of these compounds was very low, and so were the corresponding maximum DPPH free radical inhibitory values. The individual DPPH IC50 values calculated for the S0 and S3 series of compounds are for the study samples are shown in Fig. 8.
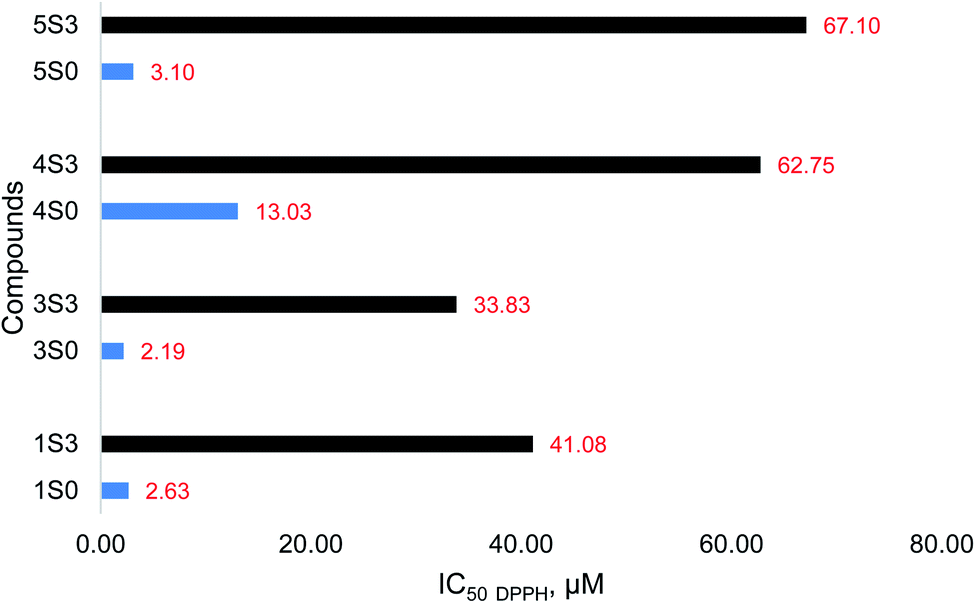 | ||
| Fig. 8 DPPH free radical scavenging activity (value reported as IC50 DPPH) of the S0 (blue) and S3 (black) compounds. | ||
The flavonoid acetamide derivatives (S3) showed substantially reduced antioxidant capacities relative to the unmodified flavonoids (S0). The DPPH˙ accepts an electron or hydrogen radical to create a stable diamagnetic molecule. Therefore, this assay can be used to quantify the extent of compounds acting as free radical scavengers or hydrogen donors and thus evaluate the antioxidant capacities of compounds that can act as electron or hydrogen donors.25 The presence, nature, number, and location of hydrogen atoms and the ability of the compound to donate electrons and hydrogen atoms play a critical role in determining the antioxidant capacity of the compounds.26–28 In the flavonoid acetamide derivatives, there exists sp3 hybridized carbon groups which imperatively reduce the antioxidant capacities of these flavonoids. This effect is slightly counterbalanced by the amide group. Despite electron delocalization in the amide group of the flavonoid acetamide derivatives, the resultant effect is not strong enough to offset the impact of the methylene groups in these derivatives. Thus, the overall antioxidant capacities of the S3 series are lower than those of the S0 series.
3.4 ADMET results
Molinspiration and swissADME online tools were used to analyze the molecular identifiers and drug similarity properties of the compounds used in this study, according to Lipinski's RO5. Pharmaceutical chemists widely use Lipinski's five drug design and development rules to anticipate the oral bioavailability of potential drug candidates or drug molecules. Molinspiration, which we have used, offers a wide range of software tools that can predict intestinal absorption following Lipinski's five rules. swissADME and Molinspiration, web-based tools were used to obtain parameters such as logP, TPSA, drug-likeness. logP (octanol–water partition coefficient) is essential in quantitative structure–activity relationship (QSAR) studies and rational drug design as a factor of molecular hydrophobicity. Hydrophobicity affects drug absorption, bioavailability, hydrophobic drug–receptor interactions, metabolism of molecules, as well as their toxicity. logP (octanol/water partition coefficient) is calculated by the methodology developed by Molinspiration as a sum of fragment-based contributions and correction factors used to predict the permeability of molecules across the cell membrane. Topological polar surface area (TPSA) is a handy parameter for the prediction of drug transport properties. Polar surface area is defined as a sum of surfaces of polar atoms (usually oxygen, nitrogen, and attached hydrogen) in a molecule. The absorption percentage (%Ab.) was calculated by using the following formula: %Ab. = 109 − [0.345 × TPSA].29–31
Physicochemical properties of the compounds according to RO5 are given in Table 1. Molecular weight is an important consideration in therapeutic drug effect accordingly; if it increases, it will affect the effect of the drug. Although these values are very close to 500, they violate the RO5 parameters in this respect; but it is seen that this rule is violated in terms of molecular mass in some commercially used drugs. Considering a slight deviation above 500 in the synthesized molecules, this difference is negligible. Therefore, based on this property, we can say that flavonoid acetamide derivatives will have comparable drug similarities to the starting materials. The number of hydrogen bond acceptors (O and N atoms) and hydrogen bond donors (NH and OH) of compounds other than 1S3 in the studied compounds were found to be in the Lipinski range of 2 to 7 and less. This property further presents flavonoid acetamide derivatives as promising drug candidates. The physicochemical properties of the flavonoids (S0) and the flavonoid acetamide derivatives (S3) are shown in Table 1.
| Compound | MW | %Ab. | TPSA (Å2) | RB | HB | HB | logP (o/w) |
LV |
|---|---|---|---|---|---|---|---|---|
| a Key: MW-molecular weight, TPSA-topological polar surface area, RB-number of rotatable bonds, HBA-number of hydrogen bond acceptors, HBD-number of hydrogen bond donors, LV-number of Lipinski rule of 5 violations, logP (o/w) (ilogP)-lipophilicity, %Ab.-percentage absorbance. |
||||||||
| 1S3 | 587.49 | 8.33 | 291.81 | 16 | 12 | 5 | −0.32 | 2 |
| 2S3 | 441.39 | 44.43 | 187.17 | 10 | 8 | 3 | 0.78 | 1 |
| 3S3 | 514.44 | 26.38 | 239.49 | 13 | 10 | 4 | 0.57 | 2 |
| 4S3 | 514.44 | 26.38 | 239.49 | 13 | 10 | 4 | 0.35 | 2 |
| 5S3 | 514.44 | 26.38 | 239.49 | 13 | 10 | 4 | −0.09 | 2 |
| Apigenin (2S0) | 270.24 | 77.64 | 90.90 | 1 | 5 | 1 | 1.89 | 0 |
| Fisetin (3S0) | 286.24 | 70.66 | 111.13 | 1 | 6 | 4 | 1.50 | 0 |
| Kaempferol (4S0) | 286.24 | 70.66 | 111.13 | 1 | 6 | 4 | 1.70 | 0 |
| Luteolin (5S0) | 286.24 | 70.66 | 111.13 | 1 | 6 | 4 | 1.86 | 0 |
| Quercetin (1S0) | 302.24 | 63.68 | 131.36 | 1 | 7 | 5 | 1.63 | 0 |
TPSA was calculated and found within 90.90 and 291.81 range for all of the compounds. TPSA parameter has been shown to be a perfect signifier and characterization for drug absorption, including intestinal absorption, bioavailability, BBB penetration, and Caco-2 permeability. TPSA was calculated from the surface areas occupied by the oxygen and nitrogen atoms and the hydrogen atoms attached to them. Therefore, TPSA is closely related to any compound's hydrogen bonding potential. In this study, all study compounds exhibited a TPSA of 187.17–291.81 Å2, showing better oral bioavailability compared to the parent compounds. Good bioavailability correlates closely with the 10 number of rotatable ligaments and TPSA. As the number of rotatable bonds increases, the molecule becomes more flexible and more adaptable for efficient interaction with a particular binding site. The S3-series (1S3–5S3) have 10–16 rotatable bonds and are flexible. The lipophilicity was calculated for all compounds as an indicator of good lipid solubility that will help the drug to interact with membranes. The enhanced lipophilicity in the flavonoid acetamide derivatives, compared to the unmodified flavonoids, can be attributed to the presence of the sp3 hybridized carbon from the acetamide groups. There exist three and five sp3 hybridized carbons in the 2S3 and 1S3 derivatives, respectively. The 3S3, 4S3, and 5S3 each have four sp3 hybridized carbons. This explains why 1S3 has the best lipophilicity out of the study compounds. Interestingly, 3S3, 4S3, and 5S3 have the same number of sp3 hybridized carbons, but varying lipophilicity indicates that the location of the acetamide group plays a role.
The compounds were also inspected for bioactivity by calculating activity scores for different targets such as GPCR (G-protein-bound receptor) ligand, ion channel modulator, a kinase inhibitor, nuclear receptor ligand. All parameters were checked with the help of the drug similarity score of Molinspiration (Table 2). All compounds showed good GPCR ligand affinity. A molecule with a bioactivity score above 0.00 is likely to exhibit significant biological activity, while values between −0.50 and 0.00 are expected to be moderately active and are assumed to be inactive if the score is lower than −0.50.28,32,33
| Compound | 1S3 | 2S3 | 3S3 | 4S3 | 5S3 | 2S0 | 3S0 | 4S0 | 5S0 | 1S0 |
|---|---|---|---|---|---|---|---|---|---|---|
| 3D structure |  |
 |
 |
 |
 |
 |
 |
 |
 |
 |
| GPCR ligand | −0.16 | −0.07 | −0.14 | −0.11 | −0.08 | −0.07 | −0.11 | −0.10 | −0.02 | −0.06 |
| Ion channel modulator | −0.63 | −0.27 | −0.38 | −0.34 | −0.31 | −0.09 | −0.27 | −0.21 | −0.07 | −0.19 |
| Kinase inhibitor | −0.19 | −0.02 | −0.08 | −0.05 | 0.00 | 0.18 | 0.18 | 0.21 | 0.26 | 0.28 |
| Nuclear receptor ligand | −0.23 | 0.04 | −0.14 | −0.05 | 0.01 | 0.34 | 0.2 | 0.32 | 0.39 | 0.36 |
| Protease inhibitor | −0.13 | −0.07 | −0.17 | −0.11 | −0.09 | −0.25 | −0.36 | −0.27 | −0.22 | −0.25 |
| Enzyme inhibitor | −0.17 | −0.01 | −0.04 | −0.01 | −0.01 | 0.26 | 0.2 | 0.26 | 0.28 | 0.28 |
The results clarify that the physiological effects of synthetic compounds (S3 series) may involve multiple mechanisms and may be caused by interactions with GPCR ligands, protease and ion channel nuclear receptor ligands, and other enzymes. The flavonoid acetamide derivatives, 1S3–5S3, showed moderate biological activity. The 1S3, 2S3, 5S3, fisetin, kaempferol, quercetin, and apigenin all showed moderate to significant biological activities. Both 3S3 and 4S3 showed moderate biological activities. The bioactivity scores of the compounds suggest moderate interaction with all drug targets and present the flavonoid acetamide derivatives as promising drug candidates. The specific bioactivity scores of the molecules are shown in Table 2.
Various biological activating properties of the compounds were examined with the OSIRIS Property Explorer. Osiris property explorer is a cheminformatics tool used to determine the pharmacokinetics, such as toxicity potential, solubility, overall drug similarity, and drug score of compounds.34,35
The results of the toxicity risks and drug score assessment of the synthetic compounds (1S3–5S3) were estimated by the OSIRIS Property Explorer and presented in Table 3. This software estimates the studied compound based on functional group similarity with the extensively studied compounds. Results are coded in colors such as red, green, and yellow. The green indicates low toxic potential, the yellow indicates mild toxicity, and the red indicates high toxicity probability.18 According to the results, it is predicted that 1S3–5S3 are not toxic in terms of tumorigenic effect.
| Compd. | Toxicity | Drug-relevant properties | |||||
|---|---|---|---|---|---|---|---|
| Tumorigenic | Reproductive effect | Irritant effect | Mutagenicity | Solubility | Drug-likeness | Drug score | |
| a Green indicates no risk or low risk, red indicates more toxicity risk. | |||||||
| 1S3 | Green | Green | Green | Red | −3.13 | 1.41 | 0.32 |
| 2S3 | Green | Red | Green | Green | −3.35 | 2.55 | 0.44 |
| 3S3 | Green | Green | Green | Green | −3.26 | 1.54 | 0.61 |
| 4S3 | Green | Red | Green | Red | −3.26 | 2.16 | 0.23 |
| 5S3 | Green | Green | Green | Green | −3.22 | 1.8 | 0.63 |
| 2S0 | Green | Green | Green | Red | −2.86 | 1.21 | 0.47 |
| 3S0 | Green | Green | Green | Red | −2.79 | −0.07 | 0.4 |
| 4S0 | Green | Green | Green | Red | −2.79 | 0.9 | 0.46 |
| 5S0 | Green | Green | Green | Green | −2.56 | 1.9 | 0.84 |
| 1S0 | Red | Green | Green | Red | −2.49 | 1.6 | 0.3 |
The toxicity was estimated through the tumorigenic, reproductive effect, irritant effect, and mutagenicity toxicity tests. Amongst the 10 compounds tested, only 3S3, 5S3, had all green indicators. All other molecules had at least one high-risk (red indicator) toxicity parameter associated with them. Both 1S3 and 2S3 had only one red indicator, while 4S3 had two. A recent study on the mutagenicity study on the 1S3 and 2S3 proved that these compounds were nontoxic, and the in silico data hereof conforms to such findings.2 Apigenin (2S0), fisetin (3S0), and kaempferol (4S0) all had one red indicator, while quercetin (1S0) had two. The less toxicity in the flavonoid acetamide derivatives further presents these molecules as potential drug candidates.
ADMET data can be used to estimate the drug-likeness of the molecule in question. The drug scores of the flavonoid and the flavonoid acetamide derivatives were estimated using OSIRIS property explorer. The flavonoid acetamide derivatives had drug score values of 0.32, 0.44, 0.61, 0.23 and 0.63 for the 1S3, 2S3, 3S3, 4S3, and 5S3 respectively. The unmodified flavonoids had lower drug scores, save for luteolin (5S0) which had a score of 0.84. The data in Table 3 presents the flavonoid acetamide derivatives (S3 series) as better drug candidates than the unmodified flavonoids (S0 series).
AMES toxicity test was used to identify whether a compound is mutagenic or not. The tested compound displayed a negative AMES toxicity test. The carcinogenic profile showed that the flavonoids (S0 series) and the flavonoid acetamide derivatives (S3 series) were non-carcinogenic. The toxicity prediction assessment predicted that the S3 series compounds were nontoxic, with the exception of 2S3 and 5S3, which are predicted to be carcinogenic (in mouse). The toxicity prediction data for the S0 and the S3 compounds is presented in Table 4. Our previous experimental studies indicated that 2S3 did not show any mutagenic activity for the strain TA 98. Some of its metabolites could be active at high doses.2 The toxicity data in Table 4 correlates with the experimental data in our previous studies.2 More experimental studies will follow to fully understand the possible ADMET toxicity of these compounds.
| Compounds | Toxicity prediction | Test Name | Value |
|---|---|---|---|
| 1S3 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Negative | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Negative | ||
| 2S3 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Negative | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Positive | |
| Carcinogenicity (rat) | Negative | ||
| 3S3 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Negative | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Negative | ||
| 4S3 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Negative | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Negative | ||
| 5S3 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Negative | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Positive | |
| Carcinogenicity (rat) | Negative | ||
| 2S0 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Positive | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Positive | ||
| 3S0 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Positive | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Positive | ||
| 4S0 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Positive | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Positive | ||
| 5S0 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Negative | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Positive | ||
| 1S0 | Ames | Ames TA100 (+S9) | Negative |
| Ames TA100 (−S9) | Positive | ||
| Ames TA1535 (+S9) | Negative | ||
| Ames TA1535 (−S9) | Negative | ||
| Carcinogenicity | Carcinogenicity (mouse) | Negative | |
| Carcinogenicity (rat) | Positive |
4. Conclusion
We successfully synthesized three new flavonoid acetamide derivatives namely: (3S3); 2,2′,2′′-((2-(4-(2-amino-2-oxoethoxy)phenyl)-4-oxo-4H-chromene-3,5,7-triyl)tris(oxy))triacetamide (4S3) and 2,2′-((4-(5,7-bis(2-amino-2-oxoethoxy)-4-oxo-4H-chromen-2-yl)-1,2-phenylene)bis(oxy))diacetamide (5S3). To our knowledge, this is the first time the synthesis of these derivatives is being reported. The flavonoid acetamide derivatives were tested for bioavailability, antioxidant, and ADMET drug properties. The flavonoid acetamide derivatives showed enhanced bioavailabilities and reduced antioxidant capacities relative to the unmodified flavonoids. The ADMET drug properties present these flavonoids as favorable drug candidates. This knowledge could be utilized to enhance the drug delivery of drug candidates. Structure–activity relationship (SAR) could be probed to understand the effect of the number and the position of different functional groups in flavonoids on their biological properties.Author contributions
(1) Daniel K. Isika: methodology, wet-lab experiment studies, first draft preparation, writing–review & editing. (2) Fatma Nur Özkömeç: ADMET and computational studies, writing–review & editing. (3) Mustafa Çeşme: computational studies, visualization, investigation, writing–review & editing. (4) Omowunmi A. Sadik: conceptualization and supervision, project administration, methodology, writing–review & editing and proofreading.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the New Jersey Institute of Technology (NJIT) Star-up funds, the National Science Foundation Grant # IOS-1543944, and Bill & Melinda Gates Foundation for funding.References
- A. Massi, O. Bortolini, D. Ragno, T. Bernardi, G. Sacchetti, M. Tacchini and C. De Risi, Molecules, 2017, 22, 1270 CrossRef PubMed.
- D. Isika, M. Çeşme, F. J. Osonga and O. A. Sadik, RSC Adv., 2020, 10, 25046–25058 RSC.
- B. Havsteen, Biochem. Pharmacol., 1983, 32(7), 1141–1148 CrossRef CAS PubMed.
- S. Das and J. P. N. Rosazza, J. Nat. Prod., 2006, 69, 499–508 CrossRef CAS PubMed.
- A. Faria, M. Meireles, I. Fernandes, C. Santos-Buelga, S. Gonzalez-Manzano, M. Dueñas, V. De Freitas, N. Mateus and C. Calhau, Food Chem., 2014, 149, 190–196 CrossRef CAS PubMed.
- N. Koirala, N. H. Thuan, G. P. Ghimire, D. Van Thang and J. K. Sohng, Enzyme Microb. Technol., 2016, 86, 103–116 CrossRef CAS PubMed.
- H. Halbwirth and K. Stich, Phytochemistry, 2006, 67, 1080–1087 CrossRef CAS PubMed.
- B. Hofer, Appl. Microbiol. Biotechnol., 2016, 100, 4269–4281 CrossRef CAS PubMed.
- H. M. Awad, M. G. Boersma, S. Boeren, P. J. Van Bladeren, J. Vervoort and I. M. C. M. Rietjens, Chem. Res. Toxicol., 2001, 14, 398–408 Search PubMed.
- J. Zhao, J. Yang and Y. Xie, Int. J. Pharm., 2019, 570, 118642 CrossRef CAS PubMed.
- M. M. Vuolo, V. S. Lima and M. R. Maróstica Junior, in Bioactive Compounds: Health Benefits and Potential Applications, Woodhead Publishing, 2018, pp. 33–50 Search PubMed.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT—Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- F. Grande, O. I. Parisi, R. A. Mordocco, C. Rocca, F. Puoci, L. Scrivano, A. M. Quintieri, P. Cantafio, S. Ferla, A. Brancale, C. Saturnino, M. C. Cerra, M. S. Sinicropi and T. Angelone, Eur. J. Pharm. Sci., 2016, 82, 161–170 CrossRef CAS PubMed.
- A. Chimento, M. Sala, I. M. Gomez-Monterrey, S. Musella, A. Bertamino, A. Caruso, M. S. Sinicropi, R. Sirianni, F. Puoci, O. I. Parisi, C. Campana, E. Martire, E. Novellino, C. Saturnino, P. Campiglia and V. Pezzi, Bioorg. Med. Chem. Lett., 2013, 23, 6401–6405 CrossRef CAS PubMed.
- M. Camilleri, L. J. Colemont, S. F. Phillips, M. L. Brown, G. M. Thomforde, N. Chapman and A. R. Zinsmeister, Am. J. Physiol., 1989, 257(2), 284–290 Search PubMed.
- A. A. T. Bioquestand Inc, Quest GraphTM IC50 Calculator, https://www.aatbio.com/tools/ic50-calculator, accessed 8 February 2022.
- Very Simple IC50 Tool Kit – calculate, measure, determine IC50 online, http://ic50.tk/, accessed 23 February 2022 Search PubMed.
- M. Rashid, Bioorg. Chem., 2020, 96, 103576 CrossRef CAS PubMed.
- R. Dang, M. Saks, S. Upreti and S. V. Rajendra, Global Journal of Pharmacy and Pharmacology, 2017, 1(5), 133–138 Search PubMed.
- S. B. Bukhari, S. Memon, M. Mahroof-Tahir and M. I. Bhanger, Spectrochim. Acta, Part A, 2009, 71, 1901–1906 CrossRef PubMed.
- S. V. Jovanovic, S. Steenken, M. Tosic, B. Marjanovic and M. G. Simic, J. Am. Chem. Soc., 1994, 116, 4846–4851 CrossRef CAS.
- C. Tian, X. Liu, Y. Chang, R. Wang, T. Lv, C. Cui and M. Liu, S. Afr. J. Bot., 2021, 137, 257–264 CrossRef CAS.
- W. Zheng and S. Y. Wang, J. Agric. Food Chem., 2001, 49, 5165–5170 CrossRef CAS PubMed.
- R. Hirano, W. Sasamoto, A. Matsumoto, H. Itakura, O. Igarashi and K. Kondo, J. Nutr. Sci. Vitaminol., 2001, 47, 357–362 CrossRef CAS PubMed.
- M. S. Blois, Nature, 1958, 181, 1199–1200 CrossRef CAS.
- S. B. Kedare and R. P. Singh, J. Food Sci. Technol., 2011, 48, 412–422 CrossRef CAS PubMed.
- S. K. Chidambaram, D. Ali, S. Alarifi, S. Radhakrishnan and I. Akbar, Journal of Infection and Public Health, 2020, 13, 1671–1677 CrossRef PubMed.
- S. V. Stoddard, S. D. Stoddard, B. K. Oelkers, K. Fitts, K. Whalum, K. Whalum, A. D. Hemphill, J. Manikonda, L. M. Martinez, E. G. Riley, C. M. Roof, N. Sarwar, D. M. Thomas, E. Ulmer, F. E. Wallace, P. Pandey and S. Roy, Viruses, 2020, 12, 942 CrossRef CAS PubMed.
- M. U. Mirza, S. Ahmad, I. Abdullah and M. Froeyen, Comput. Biol. Chem., 2020, 89, 107376 CrossRef CAS PubMed.
- İ. Şahin, M. Çeşme, F. B. Özgeriş, Ö. Güngör and F. Tümer, J. Mol. Struct., 2022, 1247, 131344 CrossRef.
- İ. Şahin, M. Çeşme, N. Yüce and F. Tümer, J. Biomol. Struct. Dyn., 2022 DOI:10.1080/07391102.2022.2025905.
- A. Ubani, F. Agwom, O. RuthMorenikeji, S. Nathan, P. Luka, A. Umera, U. Umar, S. Omale, N. E. Nnadi and J. C. Aguiyi, bioRxiv, 2020 DOI:10.1101/2020.03.31.017657.
- V. V. Kouznetsov, Eur. J. Med. Chem., 2020, 203, 112647 CrossRef CAS PubMed.
- K. F. Azim, S. R. Ahmed, A. Banik, M. M. R. Khan, A. Deb and S. R. Somana, Inform. Med. Unlocked, 2020, 20, 100367 CrossRef PubMed.
- M. Venkateshan, M. Muthu, J. Suresh and R. Ranjith Kumar, J. Mol. Struct., 2020, 1220, 128741 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01375d |
| This journal is © The Royal Society of Chemistry 2022 |