
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Straightforward synthesis of aliphatic polydithiocarbonates from commercially available starting materials†
Timo
Sehn
,
Birgit
Huber
,
Julian
Fanelli
 and
Hatice
Mutlu
*
and
Hatice
Mutlu
*
Soft Matter Synthesis Laboratory, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany. E-mail: hatice.mutlu@kit.edu
First published on 11th October 2022
Abstract
Herein, a novel 1,1′-carbonyldiimidazole (CDI) mediated polymerization methodology that complements ROP and unlocks a greater synthetic window to less-recognized polydithiocarbonates is presented.
Sulfur containing polymers1,2 are fascinating macromolecules that have received increasing attention due to their unique properties (e.g., affinity to metals, improved refractive index, divergent redox and thermal characteristics, and enhanced chemical and biological properties). As a matter of fact, sulfur tethered macromolecules [particularly, polythiocarbonates, polydithiocarbonates (depicted in Scheme 1A) and polytrithiocarbonates] show higher susceptibility to light induced degradation (although they are recognized to be non-bio-degradable),3,4 compared to their oxygen counterparts (i.e., polycarbonates). In this way, those polymers could even facilitate new light-induced chemical recycling methods as an answer to the ever-increasing amount of plastic pollution.5 Indeed, several (degradable) polymeric scaffolds decorated with various sulfur-functionalities (e.g., thioethers, thioesters, thiocarbonates, thiourethanes, thioamides, and thioureas among others) are now accessible.1 Still, compared to the more conventional oxygen-rich polymers (such as aliphatic polycarbonates),6 the toolbox of polymerizations to furnish sulfur-rich polymers (particularly, aliphatic polydithiocarbonates in which both of the ethereal oxygen atoms of the carbonate group are replaced by sulfur atoms) remains limited to polycondensation (Scheme 1B)7 and the ring-opening polymerization (ROP) of cyclic sulfur-containing monomers (Scheme 1C).8 The mentioned approaches are usually accompanied by unavoidable drawbacks involving the usage of toxic phosgene (or chloroformates) along with the tedious and low yield multiple synthetic steps of the cyclic monomers. Indeed, the absence of practical synthetic strategies that unlock a greater number of functional aliphatic polydithiocarbonates has most likely inhibited their broader use in application-driven research. Although, aliphatic polydithiocarbonates could offer parallel properties and applications to those of the corresponding polycarbonates.
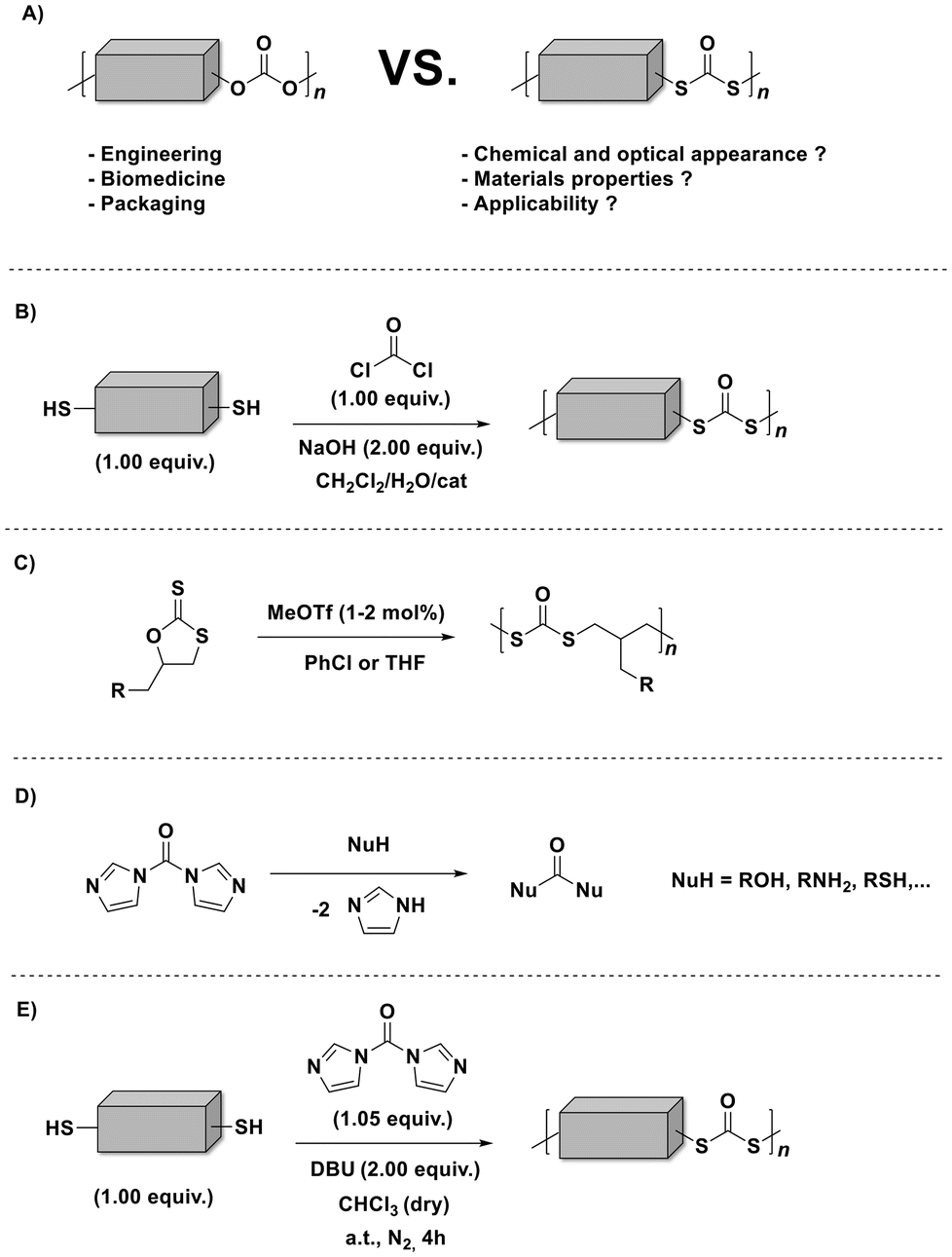 | ||
| Scheme 1 Schematic representation of: (A) the replacement of oxygen with sulfur atoms in polycarbonates, resulting in polydithiocarbonates; previously reported approaches for polydithiocarbonate synthesis: (B) phosgene-based step-growth and (C) ring-opening polymerizations; (D) The reactivity of CDI with diverse nucleophiles; (E) the currently investigated CDI mediated polymerization for polydithiocarbonate synthesis in the presence of DBU. | ||
Aliphatic polydithiocarbonates, as analogues of polycarbonates, are expected to have improved thermal properties. In fact, a literature survey reveals7,9–11 that higher melting temperatures have been reported for aliphatic polydithiocarbonates with respect to the oxygenated analogues.12,13 Thus, in particular, poly(hexamethylene dithiocarbonate) has a melting temperature (Tm) about 60 °C higher than that of poly(hexamethylene carbonate), i.e., 90 °C vs. 30 °C.10 The latter behaviour could be attributed to the higher packing efficiency in the crystalline lattice when sulfur atoms are present in the polymer backbone.14 In other words, poly(hexamethylene dithiocarbonate) is a material with a higher degree of crystallinity compared to poly(hexamethylene carbonate), respectively about 60% vs. 15%, and thus, reflects a faster crystallisation rate. Moreover, polymer chains decorated with –S–CO–S– groups are known to show higher flexibility and mobility compared to the chains containing –O–CO–O– groups.
Besides, the poly(hexamethylene dithiocarbonate) derivative shows a decomposition temperature (Td) of ∼320 °C (as it is discussed below) which is 40 °C higher compared to the oxygen analogue (∼280 °C).15
1,1′-Carbonyldiimidazole (CDI, also known as Staab's reagent)16 has been employed for various applications in organic chemistry,17,18 and quite recently for polymer synthesis by virtue of its reactivity with various nucleophiles (carboxylic acids, alcohols, and amines) as shown in Scheme 1D.19,20
Moreover, CDI is safe, cheap (3 € per gram), and easy to handle, compared to the commonly used toxic phosgene or chloroformates. In fact, Malkoch20 and colleagues reported the detailed phosgene-free synthesis of aliphatic polycarbonates with a broad range of properties. Surprisingly, literature survey has revealed that the synthesis of polydithiocarbonates in the presence of CDI is so far an unexplored research ground (Scheme 1E), despite the obvious advantages including easily available monomers (dithiols, which possess higher nucleophilicity compared to diols) and the relatively low toxicity of the imidazole by-product. Accordingly, the continuously increasing need for advanced materials prompted us to investigate a new synthetic approach with emphasis on operational practicality. In other words, we herein disclose a novel synthetic strategy that expands the window of aliphatic polydithiocarbonates that are synthesized from simple dithiol building blocks in the presence of a slight excess of CDI (i.e., 1.05 equiv.). In fact, the main idea of utilizing the slight excess of CDI is to achieve tailor-made imidazole end-groups, which subsequently could be easily transformed into the corresponding methyl monothiocarbonate end-groups during the precipitation process in ice-cold methanol (the mechanism of the polymerization is depicted in Scheme S1†). In this way we ensure that no free-thiol end groups are present, which could give rise to polydisulfide formation. Given the fact that diazabicyclo[5.4.0]undec-7-ene (DBU) has been recognized as an effective organocatalyst for the CDI-mediated21 nucleophilic reactions as well as in a variety of thiol-based22 organic syntheses, we sought to examine the effect of DBU on the polymerization. Within the broad family of aliphatic polydithiocarbonates, the synthesis of poly(hexamethylene dithiocarbonate) is the most widely explored as this polymer distinguishes itself by virtue of its complex thermal behaviour.10 Hence, to prove our hypothesis as a suitable strategy, an initial screening of the reaction conditions was performed using 1,6-hexane dithiol M1 (shown in Fig. 1A) and CDI (Scheme 1E and Table S1†) in the presence of different equivalents of DBU (Fig. S1†) in 0.8 M chloroform to deliver polymer P1. In particular, by screening 2.00, 0.10 and 0.01 equivalents of DBU, the contribution of the base to the reaction and its impact on the degree of polymerization were investigated. Indeed, the efficiency of the base could be confirmed by using 2.00 equivalents of DBU resulting in polymer P1 with an apparent number average molecular weight (Mn) of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 and dispersity (Đ) of 2.1 in 4 hours at ambient temperature (Fig. 1B). Notably, the reaction was also effective using lower loadings of DBU (0.01 equivalent), however resulting in a lower Mn. The polymerization reaction which was carried out with no base (P1.3 in Table S1†) showed that only a very low molecular weight polymer was formed regardless of the reaction time. These results underpin the non-catalytic nature of the base in this reaction. It is assumed that DBU is removed from the reaction mixture by scavenging the formed by-product imidazole to yield an ionic liquid (compare Scheme S1†),23 thus reducing the concentration of the active base in the reaction solution. Nevertheless, the formation of an ionic liquid (IL) could be beneficial for the synthesis of the targeted polydithiocarbonates, as it has been recently reported that ionic liquids (ILs) can pave the way towards an eco-compatible route for synthesizing polycarbonates from sterically hindered alcohol derivatives (i.e., isosorbide) and dimethyl carbonate.24 The authors clearly emphasized that the cation and anion of ILs should synergistically activate the substrates. Indeed, the formation of the ionic liquid was also clearly observed with the aid of NMR analysis (particularly 13C NMR), which was performed on the crude reaction mixture after 2 hours of the polymerization to deliver P1 (Fig. S2 in the ESI†). The respective NMR data were compared with the 13C NMR spectrum of the freshly prepared DBU–imidazole ionic liquid mixture in addition to the NMR data of pure DBU and pure imidazole (all data were collected in CDCl3). Accordingly, a magnetic resonance at 162.4 ppm (compare Fig. S2†) that can be assigned to the protonated DBU species (DBUH+) of the formed ionic liquid (which is consistent with the literature value, i.e., 162.1 ppm)25 was detected. Although, the literature survey24 has emphasized that the cation and anion of ILs could synergistically activate the polymerization reaction, the stoichiometric ratio of DBU was increased, yielding the highest molecular weights, when 2.00 equiv. were employed. Crucially, the beneficial features of CDI include its low nucleophilicity and distinct neutral character, enabling the effective formation of polydithiocarbonates without the need for the continuous removal of the imidazole by-product (without the need for any additional setup).
500 g mol−1 and dispersity (Đ) of 2.1 in 4 hours at ambient temperature (Fig. 1B). Notably, the reaction was also effective using lower loadings of DBU (0.01 equivalent), however resulting in a lower Mn. The polymerization reaction which was carried out with no base (P1.3 in Table S1†) showed that only a very low molecular weight polymer was formed regardless of the reaction time. These results underpin the non-catalytic nature of the base in this reaction. It is assumed that DBU is removed from the reaction mixture by scavenging the formed by-product imidazole to yield an ionic liquid (compare Scheme S1†),23 thus reducing the concentration of the active base in the reaction solution. Nevertheless, the formation of an ionic liquid (IL) could be beneficial for the synthesis of the targeted polydithiocarbonates, as it has been recently reported that ionic liquids (ILs) can pave the way towards an eco-compatible route for synthesizing polycarbonates from sterically hindered alcohol derivatives (i.e., isosorbide) and dimethyl carbonate.24 The authors clearly emphasized that the cation and anion of ILs should synergistically activate the substrates. Indeed, the formation of the ionic liquid was also clearly observed with the aid of NMR analysis (particularly 13C NMR), which was performed on the crude reaction mixture after 2 hours of the polymerization to deliver P1 (Fig. S2 in the ESI†). The respective NMR data were compared with the 13C NMR spectrum of the freshly prepared DBU–imidazole ionic liquid mixture in addition to the NMR data of pure DBU and pure imidazole (all data were collected in CDCl3). Accordingly, a magnetic resonance at 162.4 ppm (compare Fig. S2†) that can be assigned to the protonated DBU species (DBUH+) of the formed ionic liquid (which is consistent with the literature value, i.e., 162.1 ppm)25 was detected. Although, the literature survey24 has emphasized that the cation and anion of ILs could synergistically activate the polymerization reaction, the stoichiometric ratio of DBU was increased, yielding the highest molecular weights, when 2.00 equiv. were employed. Crucially, the beneficial features of CDI include its low nucleophilicity and distinct neutral character, enabling the effective formation of polydithiocarbonates without the need for the continuous removal of the imidazole by-product (without the need for any additional setup).
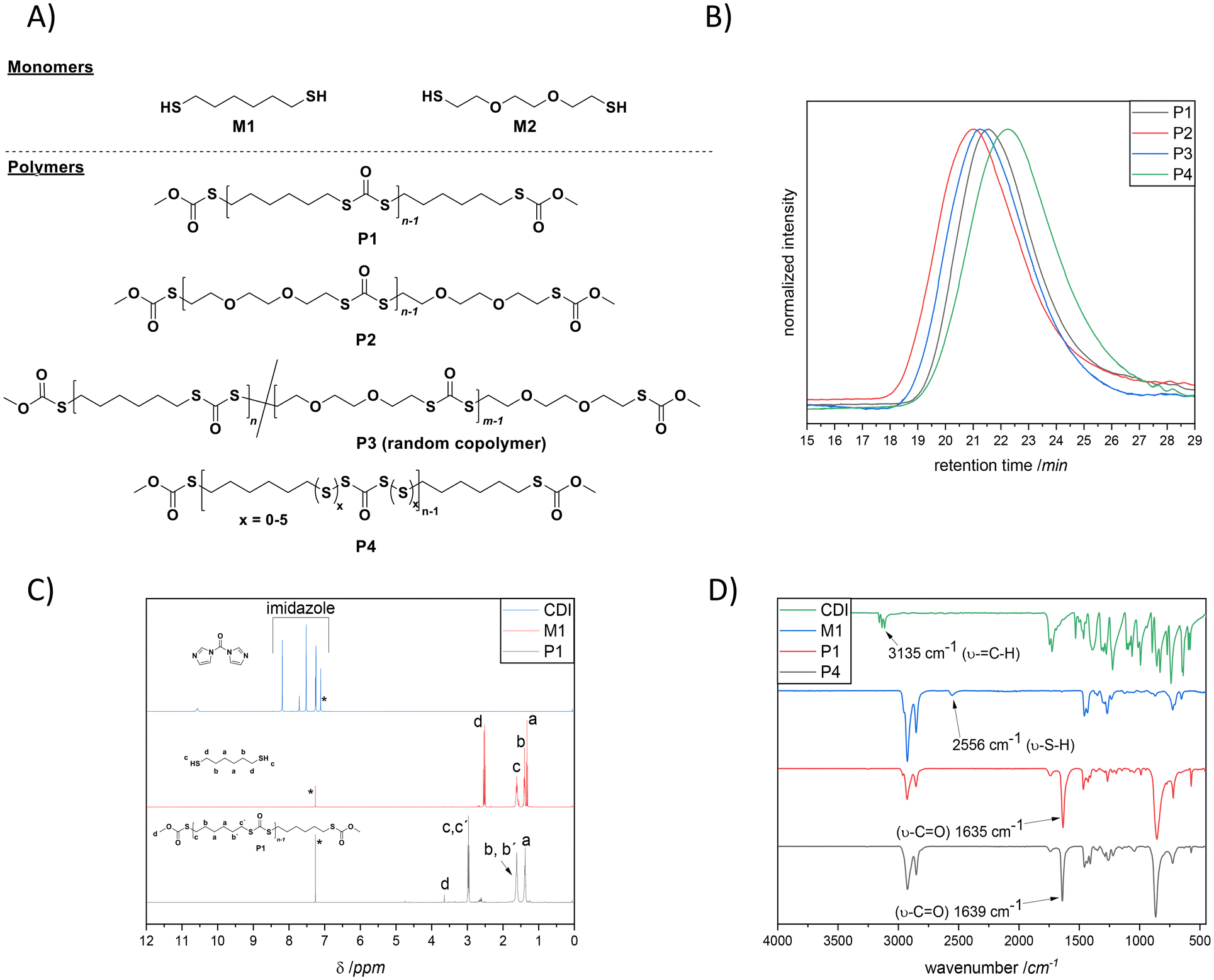 | ||
| Fig. 1 (A) Chemical structures of monomers M1 and M2 in addition to the synthesized (co)polymers P1–P4. (B) SEC traces of step growth polymers P1 (Mn = 11500 g mol−1, Đ = 2.1, black line), P2 (Mn = 14700 g mol−1, Đ = 2.4, red line), P3 (Mn = 14000 g mol−1, Đ = 2.1, blue line) and P4 (Mn = 6500 g mol−1, Đ = 2.5, green line) in THF + 0.2% w/v BHT. (C) 1H NMR spectra (400 MHz, CDCl3, 298 K) of CDI (up, blue line), M1 (middle, red line) and P1 (bottom, black line). (D) ATR-IR spectra of CDI (green line), M1 (blue line), P1 (red line) and P4 (black line). | ||
The polymerization of P1 was monitored by Size Exclusion Chromatography (SEC) of reaction aliquots withdrawn at 30, 90, 120 and 240 min. In fact, the results depicted in Fig. S3† together with the mechanism postulated in Scheme S1† suggest a step-growth polymerization mechanism. Nevertheless, further studies (which are currently performed in our laboratories) are necessary in order to conclude the latter statement.
The successful incorporation of dithiocarbonate units into the polymer backbone was further demonstrated by NMR (1D and 2D), ATR-IR and UV-Vis analyses. The 1H NMR spectrum and peak assignments of P1 are shown in Fig. 1C, where explicitly the appearance of magnetic resonance corresponding to the –CH2– groups at the α-position to the dithiocarbonate, i.e., at 2.97 ppm, was observed. Moreover, the complete disappearance of the magnetic resonances of terminal –SH at 1.33 ppm and imidazole protons (appearing from 7.11 to 8.19 ppm) also confirmed the successful polymerization. In a complementary way, a new magnetic resonance that arose at 190 ppm was detected in 13C NMR (Fig. S4†), attributable to the carbonyl carbon of the dithiocarbonate moiety in P1 (which is also consistent with the literature).26 Furthermore, 2D NMR, i.e.1H–1H correlation spectroscopy (COSY) and heteronuclear single quantum coherence (HSQC) spectroscopy (Fig. S5 and S6 in the ESI†), confirmed in a supplementary manner the formation of the targeted polymer structure. Besides, the attenuated total reflectance infrared (ATR-IR) spectrum of P1 shown in Fig. 1D revealed the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration band of dithiocarbonate moieties at ∼1635 cm−1, while neither the C–H stretch vibration band of imidazole nor the S–H stretch vibration band of thiol groups could be observed (appearing in the ranges of 3000 to 3150 cm−1 and 2550 to 2598 cm−1, respectively). Moreover, a band at 871 cm−1 due to the C–S stretching was also detectable. Importantly, NMR and ATR-IR analyses affirmed that the resulting polymers contain only the expected polydithiocarbonate segments. Neither polydisulfide and polymonothiocarbonate segments nor poly(S-dithiocarbonate) derivatives (polymers containing the oxythiocarbonylthio or xanthate ester) were observed. The latter products are attributed to the side reactions occurring during the ROP of cyclic monomers. Furthermore, the experimentally obtained elemental analysis values for P1 (C: 49.7%, H: 6.81%, S: 37.9% in Table S2†) were in good agreement with theoretical ones, reaffirming the fact that the dithiocarbonate units were installed.
O stretching vibration band of dithiocarbonate moieties at ∼1635 cm−1, while neither the C–H stretch vibration band of imidazole nor the S–H stretch vibration band of thiol groups could be observed (appearing in the ranges of 3000 to 3150 cm−1 and 2550 to 2598 cm−1, respectively). Moreover, a band at 871 cm−1 due to the C–S stretching was also detectable. Importantly, NMR and ATR-IR analyses affirmed that the resulting polymers contain only the expected polydithiocarbonate segments. Neither polydisulfide and polymonothiocarbonate segments nor poly(S-dithiocarbonate) derivatives (polymers containing the oxythiocarbonylthio or xanthate ester) were observed. The latter products are attributed to the side reactions occurring during the ROP of cyclic monomers. Furthermore, the experimentally obtained elemental analysis values for P1 (C: 49.7%, H: 6.81%, S: 37.9% in Table S2†) were in good agreement with theoretical ones, reaffirming the fact that the dithiocarbonate units were installed.
From a commercial point-of-view, a relevant advantage of the proposed strategy is that it allowed the synthesis of copolymers. The latter was manifested by the efficient random copolymerization of M1 with M2 (shown in Fig. 1A). Note to the reader: M2 was the choice of dithiol because such a hydrophilic ethylene glycol segment could be introduced; hence the final copolymer was anticipated to show unique thermal properties. Prior to the copolymerization of M1 with M2, M2 was also homopolymerized with CDI under the optimized conditions (depicted in the ESI†), to ensure that chloroform, which has been utilized for the polymerization of M1, was also a suitable solvent for the polymerization of this slightly hydrophilic dithiol derivative. Indeed, the SEC results shown in Fig. 1B affirmed the successful homopolymerization and copolymerization attempts by delivering polymers with the respective Mn of 14700 g mol−1 (Đ = 2.4, red line, P2), and Mn = 14000 g mol−1 (Đ = 2.1, blue line, P3). The detailed characterization of the homopolymer P2 and the copolymer P3 is additionally provided in the ESI (Fig. S7 to S12†). Nevertheless, it is of crucial importance to emphasize that the incorporation of both dithiol monomers in P3 was confirmed via in-depth spectroscopic analysis, e.g., 1D and 2D-NMR in addition to ATR-IR. Indeed, the magnetic resonances at 2.95 and 3.18 ppm (1H NMR, Fig. 2A) in addition to 30.2 and 30.5 ppm (13C NMR, Fig. S10†) respectively affirmed the postulated chemical structure. On the one hand, the integral ratio of the magnetic resonances corresponding to c and d suggests 50:50 random copolymer formation (Fig. 2A). On the other hand, one could not observe any distinguishable pattern in the comparative IR spectra of P2 compared with P3 (Fig. 2B). Nevertheless, a complementary elemental analysis affirmed the copolymer composition (C: 48.5%, H: 6.77%, S: 35.4%).
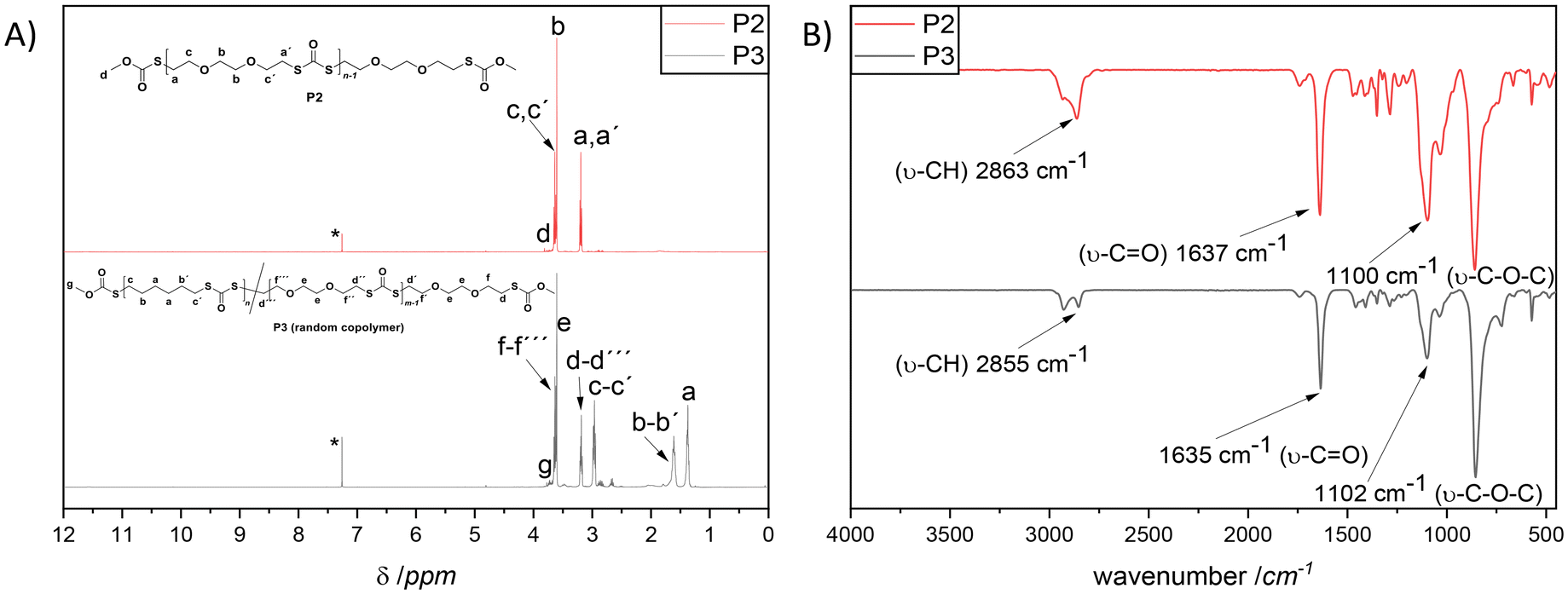 | ||
| Fig. 2 (A) 1H NMR spectra (500 MHz, CDCl3, 298 K) of P2 (up, red line) and P3 (bottom, black line), respectively, homopolymer of M2 in addition to a copolymer of M1 and M2. (B) ATR-IR spectra of P2 (up, red line) and P3 (bottom, black line). | ||
Additionally, the efficiency of the polymerization approach was demonstrated by the synthesis of a polydithiocarbonate possessing a higher sulfur content, i.e., P4 (Fig. 1B and D, and Table S3 in addition to Scheme S2†) which was realized by introducing 0.5 equivalent of elemental sulfur, an abundant side product of the petrochemical industry, into the polymerization media. It is important to note that superbases, such as DBU, are known to catalyse diverse reactions of elemental sulfur at ambient temperature.27 Subsequently, in addition to SEC, NMR and ATR-IR analyses (depicted in Fig. 13 to 17 in the ESI†), the elemental analysis of the isolated polymer, P4, showed a 4.4% higher sulfur content (42.3%, Table S3 in the ESI†) compared to P1 (37.9%, Table S2 in the ESI†), hence underpinning the efficient incorporation of additional sulfur atoms into the polymer backbone. It is important to mention that the sulfur content in the repeating unit of any polymeric structure is thermodynamically limited to a maximum of 9 sulfur atoms before the homopolymerization of elemental sulfur starts to compete with depolymerization. The latter is observed as the average bond dissociation energy of the –S–S– bond is expected to decrease to a certain value, resulting in an accelerated sulfur exchange reaction.28 Hence, at the most, six sulfur atoms can be integrated. Indeed, the multiplet in the region between 2.62 and 2.50 ppm associated with the hydrogen atom at the α-carbon adjacent to the disulfide bond was shifted to the following ranges: 3.03–2.93, 2.93–2.87 and 2.85–2.79 ppm, respectively, in such a way that each series of multiplets is assigned to the respective sulfur rank, hence nicely showing the high sulfur content of the polymer. In fact, this shift in the resonances of the disulfide bond in NMR analyses has been noted for polysulfides whose α-carbon protons exhibit multiplets, as in the current case.28
The synthesized polydithiocarbonate derivatives P1–P4 were investigated regarding their thermal characteristics by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). On the one hand, the decomposition temperature (Td), defined as the temperature at which 5% weight loss takes place, was observed for P1 at 321 °C (Fig. 3A), because of the highly crystalline behaviour. On the other hand, P2–P4 showed lower thermal stability, i.e., 258 °C < Td < 280 °C. Importantly, all (co)polymers displayed a single step thermal degradation with no char residue (aside P1). Complementary to TGA, the DSC analysis confirmed the crystalline behaviour of P1. In other words, the second heating scan (β = 10 °C min−1) of P1 was characterised by the presence of a well distinct endothermic peak located at 58 °C (Fig. 3B, black line). The DSC study of P2 indicated that the polymer was in the amorphous state, being characterized by a glass transition temperature, Tg, value of −40 °C. Whereas, copolymer P3 exhibited a crystalline polymer behaviour and possesses amorphous domains, reflected with a glass transition at −50 °C (resulting from the hydrophilic monomer corporation, M2), followed by crystallization at −8 °C and the respective melting at 29 °C (the latter two underpinning the presence of a crystalline domain and hence the incorporation of monomer M1 (Fig. 3B, blue line)). The high sulfur content counterpart of the crystalline polymer P1, i.e., polymer P4, showed an amorphous state with a Tg of −67 °C (Fig. 3B, green line). Indeed, this is consistent with the results obtained by Fitch and Helgeson, who have shown that the increase in the sulfur content introduced into a polymer results in a lowering of the Tg.29 Moreover, the domination of the amorphous phase and the lack of crystallinity in P4 confirm the random arrangement of the repeating units of the high sulfur macromolecules.
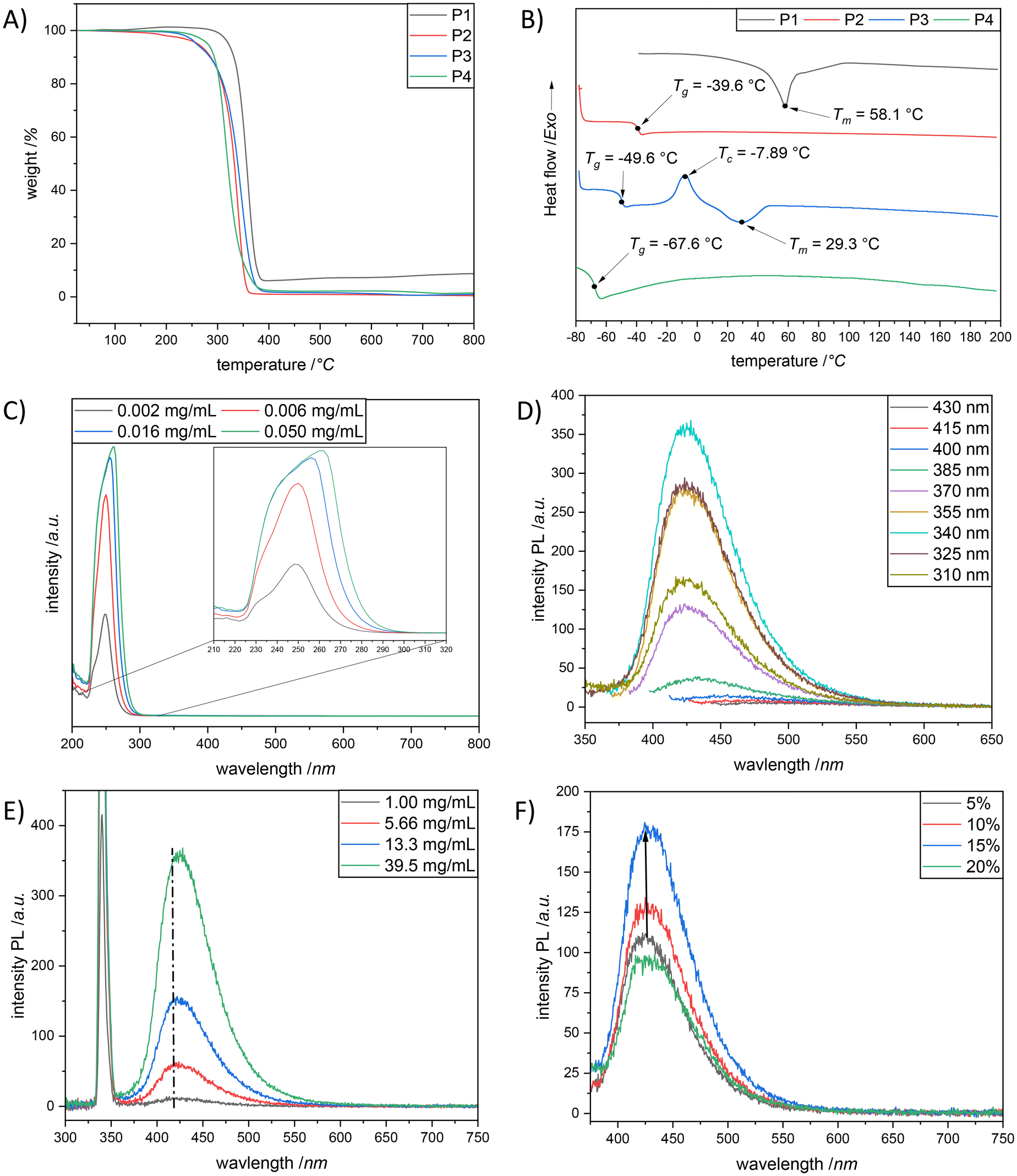 | ||
| Fig. 3 (A) Thermogravimetric analysis (TGA) of P1 (black line), P2 (red line), P3 (blue line) and P4 (green line) from 25 to 800 °C with a heating rate of 10 K min−1 under a nitrogen flow. (B) Differential scanning calorimetry (DSC) studies (second heating run) of P1 (black line), P2 (red line), P3 (blue line) and P4 (green line) from −80 °C to 200 °C with a heating rate of 10 K min−1 under a nitrogen flow (as for P1, measurement was from −40 °C). (C) Absorption traces of P2 at different concentrations (black: 0.002 mg mL−1, red: 0.006 mg mL−1, blue: 0.016 mg mL−1 and green: 0.050 mg mL−1) in DCM (298 K). (D) Emission spectra of P2 at various excitation wavelengths (from 310 nm to 430 nm) in DCM (c = 39.5 mg mL−1). (E) Emission spectra of P2 at different concentrations (black: 1.00 mg mL−1, red: 5.66 mg mL−1, blue: 13.3 mg mL−1 and green: 39.5 mg mL−1) in DCM (298 K) at λexc = 340 nm. (F) Emission spectra of P2 in DCM/water mixtures with an increasing water ratio (black: 5%, red: 10%, blue: 15% and green: 20%) at 298 K; the arrow indicates the slight shift of the maxima of each emission spectrum. | ||
The discovery of intrinsic fluorescence in non-aromatic materials (such as proteins, peptides, and rice amongst others,30 which was later termed a clustering-triggered emission (CTE)31 mechanism) has advanced the current fluorescence paradigm. In particular, because this discovery not only extends the traditional principles and establishes new theories,32–36 but also improves the virtue of biocompatibility and biodegradability, aliphatic fluorescent polymers have shown more privileges in the biomedical and biological fields over the existing fluorescent chromophore containing aromatic moieties.37,38 The so-called non-conventional luminescent compounds generally comprise various isolated chromophores, such as electron-rich heteroatoms (i.e. N, O, and P),39,40 hydroxyl (OH), amino (NH2), carbonyl (CO), cyano (CN),30 carboxyl (COOH),36 amide (CONH),41 and anhydride42 groups and carbon–carbon double bonds (CC).43 Accordingly, the formation of clusters of those isolated chromophores with π and/or n electrons is essential for the emission, which in turn establishes the CTE mechanism. While sulfur atom (S) has been proved to facilitate fluorescence due to larger atomic radius,44 the non-conventional luminophores (small- or macro-molecules) involving the S moiety have rarely been reported.45,46 As a matter of fact, we were interested if the herein synthesized polymers would be emissive under UV irradiation in aggregates, despite the absence of remarkable conjugations. In fact, all (co)polymers appeared as “see-through” transparent solids (or highly viscous liquids) under natural light, whereas a blue emission was observed under 365 nm UV light (as shown in Fig. S18†). It is important to mention that recently non-conjugated (hyperbranched) polycarbonate derivatives were reported to exhibit bright blue photoluminescence without any additional treatment under 365 nm UV light.47–49 The authors attributed the fluorescence emission to the cluster-induced emission (CIE) mechanism which resulted from the clustering of the locked carbonyl groups, although they could not postulate a clear mechanism to these non-aromatic fluorescent materials. Inspired by the above research, the absorbance and photoluminescence properties of P2 in dichloromethane (DCM) were carefully studied, as preliminary screening of all polymers revealed that the strongest emission was observed for P2 (compare Fig. 3D and Fig. S23, S25 and S26 in the ESI†). As depicted in Fig. 3C, two absorption bands, centered at ∼ 232 nm and 248 nm, were detected, respectively, when the measurement was performed in a low concentrated solution, i.e., 0.002 mg mL−1P2. Eventually, the absorption band appearing at 232 nm can be associated with the π–π* electron transition of dithiocarbonate functional groups, whereas the absorption peak arising around 248 nm can be assigned to the n–π* electron transitions of dithiocarbonate moieties (that is common for molecules containing a CO group in S,S-dialkyl-dithiocarbonates). Importantly, this information is in accordance with the literature.50 At this point it is also noteworthy that the intensities of the absorption bands rise gradually while increasing the solution concentration of P2 accompanied by a small bathochromic shift. The latter phenomenon was presumably due to the aggregation of polymer chains at the increased solution concentrations of P2, which respectively resulted in dithiocarbonate (–S–C(O)–S–) cluster formation (Fig. S24 in the ESI†). Indeed, overlapping electron clouds and delocalized electrons in the dithiocarbonate clusters ensure an extensive electron delocalization system, which is responsible for the red shift of the absorption bands (Fig. 3E). To further characterize the photoluminescence properties of P2 in DCM, a series of fluorescence emission spectra at various excitation wavelengths (310 nm to 430 nm) were also recorded (Fig. 3D). As expected, the fluorescence intensity of the polymer solution of P2 in DCM, at the constant concentration of 39.5 mg mL−1, significantly altered with the increase of the excitation wavelength from 310 nm to 430 nm; a maximum emission wavelength of 423 nm was detected at λexc = 340 nm (Fig. 3D). Additionally, a significant increase of the fluorescence emission intensity was observed for P2, while increasing the polymer concentration from 1.00 mg mL−1 to 39.5 mg mL−1 (Fig. 3E). The slight red shift of the emission band located at 416 nm to a higher wavelength of 423 nm (Fig. 3E) was also detected with the increase of the emission intensity as a result of the enhanced formation of rigid dithiocarbonate clusters at elevated polymer concentrations, which in turn underpins a possible cluster induced emission mechanism.48 Because the dithiocarbonate functional groups are structural analogues of their oxygen analogues, i.e., carbonate moieties, it was assumed that the cluster formation of dithiocarbonates is also triggered by space and n–π* electronic interactions as it was previously reported for carbonate moieties.48 To further investigate the fluorescence properties of P2, the influence of incorporating different water fraction ratios into the solution of P2 (dissolved in THF) was examined. Unsurprisingly, keeping the fact in mind that P2 was not soluble in water, the fluorescence emission intensity of the corresponding polymer solution increased with elevated water fractions according to the typical aggregation-enhanced emission (AEE) effect (Fig. 3F).48 The maximum fluorescence emission intensity of the polymer solution of P2 was achieved with a water fraction of 15%. However, when the water fraction reached 20%, the polymer, i.e., P2, precipitated, and the mixture started to become opaque. Consequently, the fluorescence emission intensity of the polymer solution faded at a water content of 20%. Hence, the experiment revealed in a different way that the macromolecular mechanics, i.e., AEE effect, gives rise to the unexpected fluorescence character of the aliphatic polydithiocarbonates, specifically P2 in our case.
Last but not least, in order to validate the abovementioned results and get more insights into the emission mechanism, a model compound, MC, mimicking the repeating unit of P2 (Fig. 4A) was synthesized (Sections B6 and B7, and Fig. S25–S28 in the ESI†). The respective absorbance and emission data of this compound in DCM were recorded and compared with the optical properties of the starting compound 3,6-dioxa-1,8-octanedithiol in addition to the intermediate product 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole (Fig. 4B and C). The spectral patterns of these compounds (particularly, 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole and MC at 10 mg mL−1) were similar to each other with a strong absorption band around 284 nm (with an obvious red-shift of 50 nm in comparison with that of 3,6-dioxa-1,8-octanedithiol) suggesting increased intramolecular cluster triggered interaction plausibly owing to the clusterization of the dithiocarbonyl moiety. Moreover, a shoulder around 370 nm was observed for 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole as a result of the n–π* electron transitions of the imidazole moieties.51 Meanwhile, as a maximum emission wavelength of 423 nm was detected at λexc = 340 nm for P2, 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole and MC were subjected to analogue analysis; a slightly lower maximum emission wavelength of 351 nm was recorded at λexc = 340 nm. The intensity of 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole was slightly higher in comparison with that of MC (185 vs. 140 a.u.), which can be attributed to the presence of the imidazole and thiourethane units. In contrast, no emission was observed for 3,6-dioxa-1,8-octanedithiol. In a similar manner to P2, a significant increase of the fluorescence emission intensity was observed for MC while increasing the compound concentration from 1.00 mg mL−1 to 50 mg mL−1 (Fig. 4D). The slight red shift of the emission band located at 404 (5.00 mg mL−1) nm to a slightly higher wavelength of 412 nm (50 mg mL−1) was also in accordance with the results observed for P2.
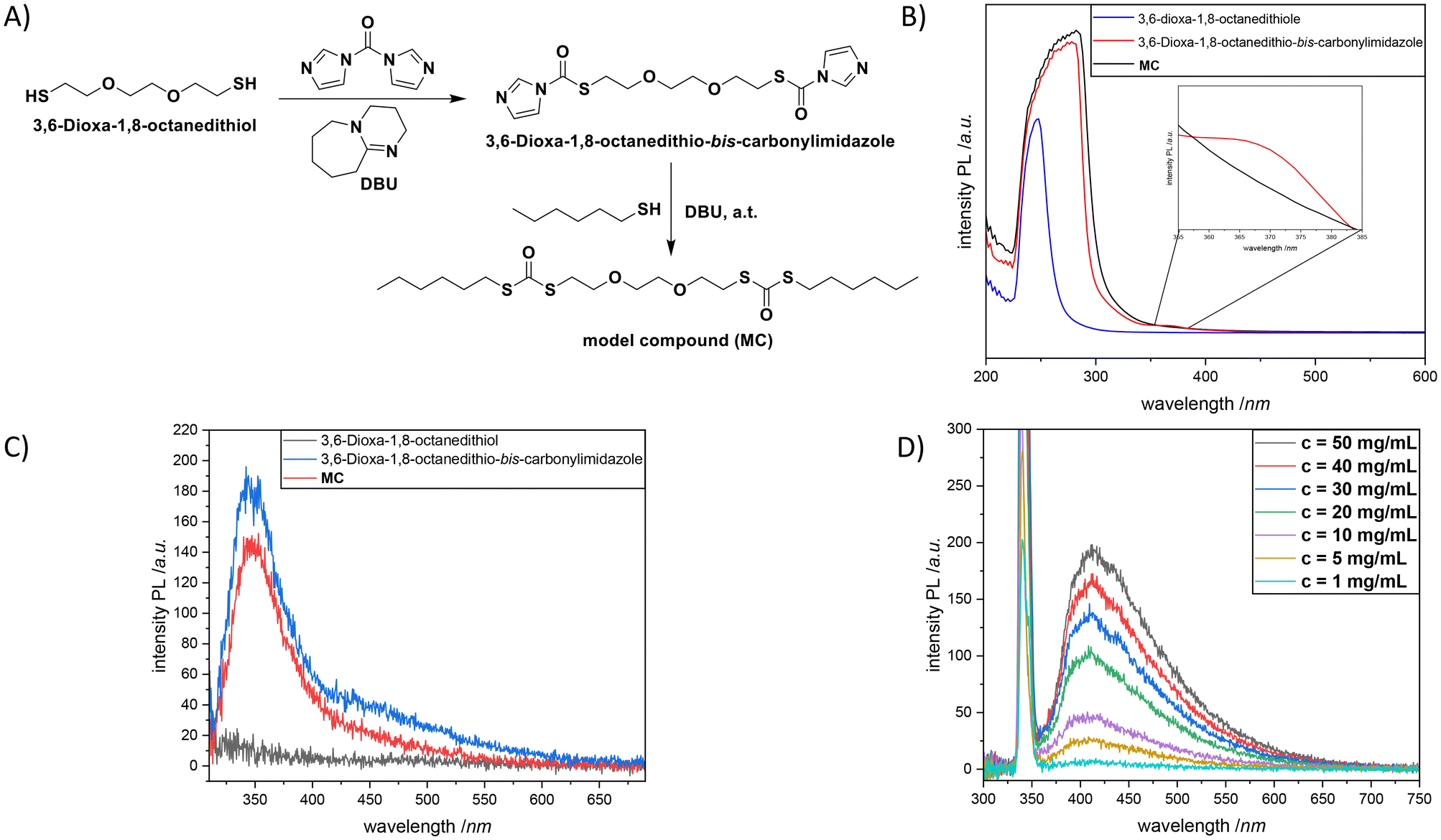 | ||
| Fig. 4 (A) The synthesis of a model compound, MC, mimicking the repeating unit of P2. (B) Absorption traces of MC, 3,6-dioxa-1,8-octanedithiol and the intermediate product 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole in DCM (298 K). (C) Emission spectra of MC, 3,6-dioxa-1,8-octanedithiol and the intermediate product 3,6-dioxa-1,8-octanedithio-bis-carbonylimidazole in DCM (10 mg mL−1) at λexc = 340 nm. (D) Emission spectra of MC at different concentrations (from 1.00 mg mL−1 to 50.0 mg mL−1) in DCM (298 K) at λexc = 340. | ||
On the one hand, these results may show that the dithiocarbonate unit could be both a CTEgen and an AIEgen, demonstrating the important role of clusterization in the manipulation of the photophysical behaviour of a molecular unit either in solution or in an aggregated state. On the other hand, to further understand the fluorescence behaviour of those classes of polymers and the respective small molecules at the molecular level, density functional theory (DFT) calculations are essential.
Conclusions
In conclusion, we have presented a facile synthetic strategy to obtain polydithiocarbonates (particularly poly(hexamethylene dithiocarbonate) and its copolymers) under benign and mild reaction conditions by taking advantage of the versatile reactivity of 1,1′-carbonyldiimidazole, CDI, with dithiols in the presence of diazabicyclo[5.4.0]undec-7-ene, DBU, as an efficient base. Crucially, the beneficial features of CDI include its low nucleophilicity and distinct neutral character, enabling the effective formation of polydithiocarbonates without the need for continuous removal of the imidazole by-product. The polymerization followed a traditional step-growth mechanism which was favored over depolymerization, and it is postulated that this approach should serve as a complement to the existing methodologies by combining the operational benefits of the ROP and scalability of traditional step-growth polymerization protocols. Indeed, the methodology facilitates the fabrication of previously inaccessible functionally and structurally diverse dithiocarbonate-decorated macromolecules, which exhibit unexplored photophysical properties. It is important to mention that studies on the degradation properties of those polymers are currently investigated. Thus, we truly believe that this approach will play a crucial role in the future availability of advanced polydithiocarbonates, and their use as materials in the technical and biomedical engineering fields, particularly on account of their unexplored intrinsic emission properties (which still need to be investigated in detail).Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. M. acknowledges the continuous financial support from the Karlsruhe Institute of Technology, in addition to the Federal Ministry of Education and Research (BMBF) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments. Prof. M. A. R. Meier and Prof. P. Levkin, respectively, are acknowledged for the access of diverse analytical characterization facilities. Prof. P. Theato is acknowledged for his continuous support and access of the SML facilities.References
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- H. Mutlu and P. Theato, Macromol. Rapid Commun., 2020, 41, 2000181 CrossRef CAS PubMed.
- B. A. Fultz, D. Beery, B. M. Coia, K. Hanson and J. G. Kennemur, Polym. Chem., 2020, 11, 5962 RSC.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253 RSC.
- T.-J. Yue, L.-Y. Wang and W.-M. Ren, Polym. Chem., 2021, 12, 6650 RSC.
- E. A. Gilbert, M. L. Polo, J. M. Maffi, J. F. Guastavino, S. E. Vaillard and D. A. Estenoz, J. Polym. Sci., 2022 DOI:10.1002/pol.20220119.
- G. Montaudo, C. Puglisi, C. Berti, E. Marianucci and F. Pilati, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2277 CrossRef CAS.
- S. Krishnamurthy, Y. Yoshida and T. Endo, Polym. Chem., 2022, 13, 267 RSC.
- F. Pilati, E. Marianucci, C. Berti, P. Manaresi and G. D. Fortuna, Polym. Commun., 1990, 31, 431 CrossRef CAS.
- C. Berti, A. Celli, P. Marchese, E. Marianucci, C. Marega, V. Causin and A. Marigo, Polymer, 2007, 48, 174 CrossRef CAS.
- T. P. Zhao, A. Celli, X. K. Ren, J. R. Xu, S. Yang, C. Y. Liu and E. Q. Chen, Polymer, 2017, 113, 267 CrossRef CAS.
- H. R. Kricheldorf and A. Mahler, Polymer, 1996, 37, 4383 CrossRef CAS.
- V. Pokharkar and S. Sivaram, Polymer, 1995, 36, 485 CrossRef.
- E. Riande, J. Guzman and J. S. Roman, J. Chem. Phys., 1980, 72, 5263 CrossRef CAS.
- W. Liu, W. Zhu, C. Li, G. Guan, D. Zhang, Y. Xiao and L. Zheng, Polym. Degrad. Stab., 2015, 112, 70 CrossRef CAS.
- H. A. Staab, Justus Liebigs Ann. Chem., 1957, 609, 75 CrossRef CAS.
- R. Paul and G. W. Anderson, J. Am. Chem. Soc., 1960, 82, 4596 CrossRef CAS.
- S. P. Rannard and N. J. Davis, Org. Lett., 1999, 1, 933 CrossRef CAS.
- J. D. Wolfgang, B. T. White and T. E. Long, Macromol. Rapid Commun., 2021, 42, 2100163 CrossRef CAS PubMed.
- J. V. Olsson, D. Hult, S. García-Gallego and M. Malkoch, Chem. Sci., 2017, 8, 4853 RSC.
- C. Larrivée-Aboussafy, B. P. Jones, K. E. Price, M. A. Hardink, R. W. McLaughlin, B. M. Lillie, J. M. Hawkins and R. Vaidyanathan, Org. Lett., 2010, 12, 324 CrossRef PubMed.
- Y. H. Wang, Z. Y. Cao, Q. H. Li, G. Q. Lin, J. Zhou and P. Tian, Angew. Chem., 2020, 132, 8080 CrossRef.
- Z. Li, DBU-Based Ionic Liquids, in Encyclopedia of Ionic Liquids, ed. S. Zhang, Springer, Singapore, 2022. DOI:10.1007/978-981-10-6739-6_126-1.
- W. Qian, X. Ma, L. Liu, L. Deng, Q. Su, R. Bai, Z. Zhang, H. Gou, L. Dong, W. Cheng and F. Xu, Green Chem., 2020, 22, 5357 RSC.
- F. Gao, Z. Wang, P. Ji and J.-P. Cheng, Front. Chem., 2019, 6, 658 CrossRef PubMed.
- X.-H. Zhang, F. Liu, X.-K. Sun, S. Chen, B.-Y. Du, G.-R. Qi and K. M. Wan, Macromolecules, 2008, 41, 1587 CrossRef CAS.
- Y. Hu, Z. Yin, T. Werner, A. Spannenberg and X.-F. Wu, Eur. J. Org. Chem., 2018, 1274 CrossRef CAS.
- R. Steudel, Angew. Chem., Int. Ed. Engl., 1975, 14, 655 CrossRef.
- R. M. Fitch and D. C. Helgeson, J. Polym. Sci., Polym. Symp., 1969, 22, 1101 CrossRef.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586 CrossRef CAS PubMed.
- P. Liao, J. Huang, Y. Yan and B. Z. Tang, Mater. Chem. Front., 2021, 5, 6693 RSC.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- H. K. Zhang, Z. Zhao, P. R. McGonigal, R. Q. Ye, S. J. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. P. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275 CrossRef CAS.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35 CrossRef CAS.
- W. Z. Yuan and Y. M. Zhang, J. Polym. Sci. Polym. Chem., 2017, 55, 560 CrossRef.
- Y. Wang, X. Bin, X. Chen, S. Zheng, Y. Zhang and W. Z. Yuan, Macromol. Rapid Commun., 2018, 39, 1800528 CrossRef PubMed.
- J. Yang, Y. Zhang, S. Gautam, L. Liu, J. Dey, W. Chen, R. P. Mason, C. A. Serrano, K. A. Schug and L. Tang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10086 CrossRef CAS PubMed.
- R. B. Wang, W. Z. Yuan and X. Y. Zhu, Chin. J. Polym. Sci., 2015, 33, 680 CrossRef CAS.
- J. Yan, B. Zheng, D. Pan, R. Yang, Y. Xu, L. Wang and M. Yang, Polym. Chem., 2015, 6, 6133 RSC.
- R. B. Restani, P. I. Morgado, M. P. Ribeiro, I. J. Correia, A. Aguiar-Ricardo and V. D. B. Bonifácio, Angew. Chem., Int. Ed., 2012, 51, 5162 CrossRef CAS PubMed.
- X. Zhou, W. Luo, H. Nie, L. Xu, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 4775 RSC.
- L. Yang, L. Wang, C. Cui, J. Lei and J. Zhang, Chem. Commun., 2016, 52, 615 Search PubMed.
- B. Liu, H. K. Zhang, S. J. Liu, J. Z. Sun, X. H. Zhang and B. Z. Tang, Mater. Horiz., 2020, 7, 987 RSC.
- Z. Zhang, S. Feng and J. Zhang, Macromol. Rapid Commun., 2016, 37, 318 CrossRef CAS PubMed.
- Z. Zhao, X. Chen, Q. Wang, T. Yang, Y. Zhang and W. Z. Yuan, Polym. Chem., 2019, 10, 3639 RSC.
- W. Huang, H. Yan, S. Niu, Y. Du and L. Yuan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3690 CrossRef CAS.
- Y. Du, Y. Feng, H. Yan, W. Huang, L. Yuan and L. Bai, J. Photochem. Photobiol., A, 2018, 364, 415 CrossRef CAS.
- Y. F. Zhang, W. M. Lai, S. Xie, H. Zhou and X. B. Lu, Polym. Chem., 2022, 13, 201 RSC.
- M. W. Fichtner and N. F. Haley, J. Org. Chem., 1981, 46, 3141 CrossRef CAS.
- X. Lin, M. Huang, T. Lu, W. Zhao, C. Hu, X. Gu and W. Zhang, Atmosphere, 2022, 13, 970 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00990k |
| This journal is © The Royal Society of Chemistry 2022 |