
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the capacity of supercapacitive swing adsorption CO2 capture by tuning charging protocols†
Trevor B.
Binford
,
Grace
Mapstone
,
Israel
Temprano
and
Alexander C.
Forse
 *
*
Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, UK. E-mail: acf50@cam.ac.uk
First published on 19th May 2022
Abstract
Supercapacitive swing adsorption (SSA) is a recently discovered electrochemically driven CO2 capture technology that promises significant efficiency improvements over traditional methods. A limitation of this approach is the relatively low CO2 adsorption capacity, and the underlying molecular mechanisms of SSA remain poorly understood, hindering optimization. Here we present a new device architecture for simultaneous electrochemical and gas-adsorption measurements, and use it to investigate the effects of charging protocols on SSA performance. We show that altering the voltage applied to charge the SSA device can significantly improve performance. Charging the gas-exposed electrode positively rather than negatively increases CO2 adsorption capacity and causes CO2 desorption rather than adsorption with charging. We also show that switching the voltage between positive and negative values further increases CO2 capacity. Previously proposed mechanisms of the SSA effect fail to explain these phenomena, so we present a new mechanism based on movement of CO2-derived species into and out of electrode micropores. Overall, this work advances our knowledge of electrochemical CO2 adsorption by supercapacitors, potentially leading to devices with increased uptake capacity and efficiency.
Climate change is one of the greatest challenges the world faces in the 21st century. The Intergovernmental Panel on Climate Change (IPCC) estimates that to limit global temperature increase to 1.5 °C above pre-industrial levels, the world needs to achieve net carbon neutrality by 2050.1 Although expanding renewable energy will be vital to reach this goal,2 certain industries such as cement manufacturing are intrinsically linked to CO2 emissions, while in the short term, fossil fuels continue to be important to the world economy. Carbon dioxide capture at point sources is currently one of the cheapest ways to reduce industrial greenhouse gas emissions,3 and can help to close this gap. The IPCC asserts that all pathways to limit global warming to 1.5 °C will require “carbon dioxide removal (CDR) on the order of 100–1000 GtCO2 over the 21st century”,1 where CDR refers to removal of carbon dioxide from the atmosphere rather than point emissions. Other estimates require annual CO2 capture of 75–175 MtCO2 in the UK alone to meet net-zero targets.4
The best-developed carbon capture technique is solvent scrubbing, where aqueous amine solvents are used to selectively absorb CO2. Heating the solvent, known as a temperature swing, drives CO2 out for storage and returns the system to its original state.5 However, raising the temperature of a large volume of solvent requires substantial energy input, limiting efficiency. Amine solvents can also corrode equipment, be poisoned by impurities in the flue gas, or escape as vapor to cause environmental damage.6 These issues mean that electrochemical swing adsorption, an emerging CO2 capture technology driven by electrochemistry, may have potential advantages.7–12
Supercapacitive swing adsorption (SSA) is a form of electrochemical swing CO2 capture based on charging supercapacitors.13 One electrode of the supercapacitor is exposed to a CO2-containing gas and the other is completely soaked in electrolyte. When the supercapacitor is charged, CO2 is selectively adsorbed from the gas (and released with discharging). The effect has primarily been studied with activated carbon electrodes and aqueous sodium chloride electrolyte,14–16 an affordable and environmentally-friendly model system.
SSA systems, however, have limited CO2 capacities compared to amine scrubbing, typically ∼60 mmol of CO2 per kg of adsorbent,14 while amine scrubbing can reach ∼800 mmol of CO2 per kg of solvent.17 Performance has been improved by adding ion exchange membranes above the electrodes to increase the selectivity for the CO2-derived ions H+, HCO3− in the electric double layer.11,18 Little energy is wasted adsorbing electrolyte ions, so the membrane capacitive deionization approach is significantly more energy efficient. However, the added cost and complexity from the ion-exchange membranes is a drawback compared to the simple SSA approach. Besides low capacities, an additional challenge is the lack of fundamental understanding of electrochemical CO2 capture by supercapacitors. To address these challenges, here we explore new charging protocols and experimental set-ups to obtain new insights into the mechanisms of supercapacitive swing adsorption.
To monitor the electrochemical adsorption of CO2 by supercapacitors we adopted an electrochemical cell equipped with a gas pressure sensor for monitoring gas uptake and release19 (Fig. 1, also see ESI†). Briefly, symmetric activated carbon-based supercapacitors (YP50-F carbon, Kuraray) with 1 M NaCl (aq.) electrolyte are housed in a gas-tight Swagelok cell assembly. The top electrode is directly in contact with a gas reservoir filled with pure CO2. As in previous work on SSA, we chose activated carbon electrodes as they have large capacitances, excellent electronic conductivities, and low cost. Our choice of YP50-F activated carbon, rather than the previously studied BPL activated carbon, was motivated by the higher capacitance of YP50-F in 1 M NaCl (aq.) electrolyte (see Fig. S1†). Gas adsorption measurements showed that YP50-F is a microporous carbon with a BET surface area of 1690 m2 g−1 (see Fig. S2†).
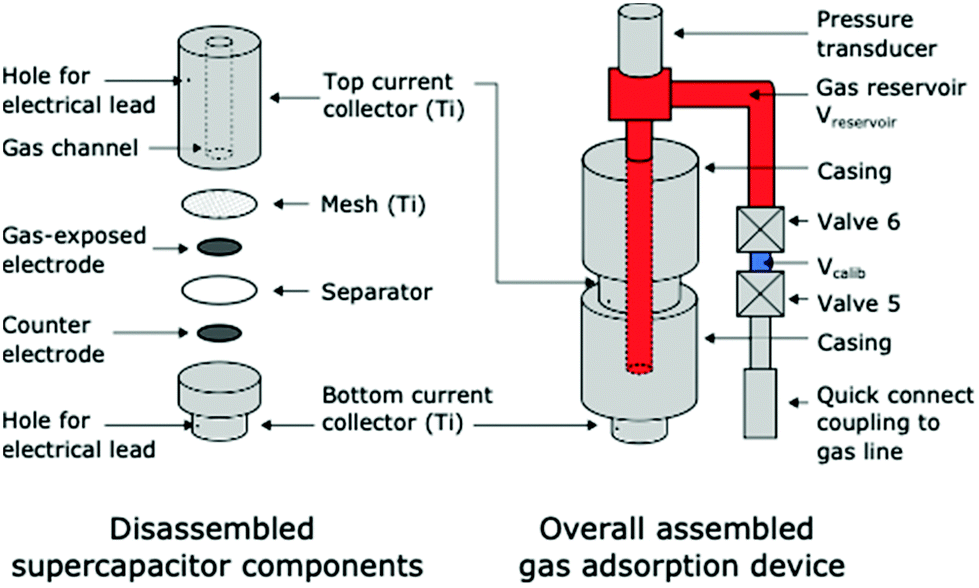 | ||
| Fig. 1 Schematic diagram of the supercapacitive swing adsorption device used to monitor gas pressure during electrochemical measurements. | ||
To date, the bulk of the SSA literature has focused on charging the supercapacitor with the negative electrode exposed to the gas, which we will term negative charging. We are only aware of one published experiment in which the positive electrode is exposed to the gas (positive charging).13 Therefore, we examined the effect of both negative and positive charging (representative data shown in Fig. 2 and the overall experiment in Fig. S3†).
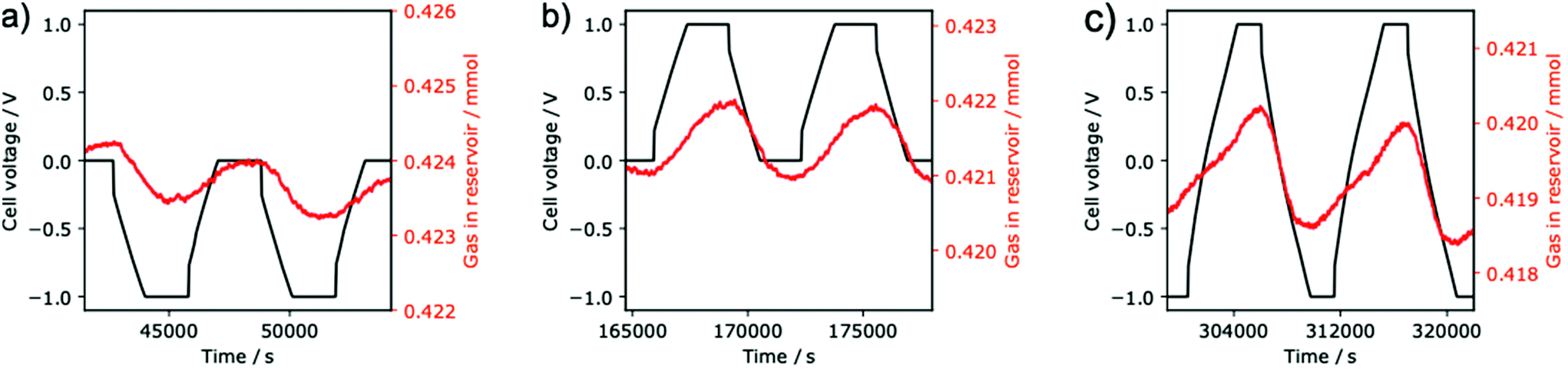 | ||
| Fig. 2 Gas adsorption data from application of a negative (a) or positive (b) voltage between the gas-exposed and counter electrodes, as well as from changing the applied voltage between positive and negative (c). Conducted with 1 M NaCl (aq) electrolyte, 15 mg electrodes, and 30 mA g−1 current density, and at a temperature of 30 °C. | ||
Initial experiments with negative charging revealed reversible electrochemical adsorption (Fig. 2a). As in the literature,13 we observed CO2 adsorption when carrying out negative charging i.e. when the cell voltage is varied between 0 and –1 V, CO2 is adsorbed by the supercapacitor. When discharging back to 0 V, the adsorbed CO2 is released. In contrast to previous work,13–16 here we employed YP50-F activated carbon, demonstrating that electrochemical CO2 adsorption by supercapacitors is not limited to the previously studied BPL activated carbon.
Based on the measured pressure changes and the calibrated gas reservoir volume (see ESI†), for negative charging we obtain a CO2 adsorption capacity of 50 ± 1 mmol kg−1 (calculated per kg of carbon in the gas exposed electrode, error bars represent cycle to cycle variation on a single cell). This value is comparable to the 62 ± 3 mmol kg−1 reported for a similar SSA system by Zhu et al. (Table S1†), though we note their study was under mixed gas conditions and under gas flow.16 We obtained an energy consumption of 628 ± 12 kJ mol−1 of adsorbed CO2, which is larger than the previously reported consumption for SSA (202 ± 14 kJ mol−1)16 or membrane capacitive deionization (27 kJ mol−1)11 systems. The performance is likely hindered by the non-optimized nature of the SSA system in this study, and its relatively high resistance (the equivalent series resistance of this cell was 939 ± 8 Ω).
When then charging the same cell positively (Fig. 2b), the adsorption capacity increases significantly to 75 ± 1 mmol kg−1 and the energy consumption decreases to 356 ± 17 kJ mol−1. When examining the gas adsorption data for insights to this difference (Fig. 2a and b), variations in the adsorption behavior are obvious. When charging negatively to −1.0 V the cell adsorbs CO2 (decreasing reservoir pressure, Fig. 2a), but when charging positively to +1.0 V the cell desorbs CO2 (increasing pressure, Fig. 2b); the different charging polarities have opposite effects on gas adsorption. An equivalent statement is that decreasing the cell voltage always causes CO2 adsorption and increasing the voltage always causes desorption, regardless of the absolute voltage. This implies that the limiting voltages can be chosen arbitrarily, rather than one limit always being 0 V. We therefore hypothesized that we could increase the adsorption capacity by combining the positive and negative charging protocols into a “switching” protocol, with −1 and +1 V as voltage limits.
Excitingly, the switching protocol (Fig. 2c) gives an even higher adsorption capacity of 112 ± 7 mmol kg−1, which is significantly larger than the capacity observed for the conventional negative charging protocol (50 ± 1 mmol kg−1), as well as our positive charging protocol (75 ± 2 mmol kg−1). One limitation of this new approach, however, is the increased energy consumption (751 ± 31 kJ mol−1). We predict that further optimization of (i) the SSA device (to reduce resistance) and (ii) the charging protocol will lead to significantly lower energy consumption values in the future. For example, increasing the mechanical pressure applied between the two electrodes should lead to decreases in resistance, a reduced ohmic drop, and improved energy efficiency. Moreover, reducing the length of the potential hold steps in the charging protocol may further improve energy efficiencies. Interestingly the shape of the adsorption profile for the switching protocol differs to that of negative and positive charging protocols, suggesting a change in the underlying capture mechanism.
The relative performances of these different charging protocols are consistent between independent electrochemical cells, though we observe some variation in the magnitudes of the adsorption capacities from cell to cell. The above results are from a cell that was first charged negatively, then charged positively, and finally charged with the switching protocol (Fig. S3†). When instead applying a positive, then negative, then switching voltage, to an independent cell (Fig. S4†), we obtain adsorption capacities of 66 ± 4, 38 ± 4, and 97 ± 2, respectively. Very similar results were obtained when the experiments were repeated on two further cells by another researcher (Fig. S5 and S6, Tables S2 and S3†). A limitation of our study is that we observed some irreversible pressure decreases over the course of our experiments (Fig. S3–S6†). This may be due to irreversible electrochemical processes such as corrosion, and suggests that further device and material optimization are needed for practical applications. A further limitation arises from our volumetric method of CO2 detection, which prevents the quantification of CO2 uptake under more practical mixed gas conditions. Previous work on SSA has shown selective adsorption of CO2 in the presence of N2, suggesting similar results would be obtained for our new charging protocols here.13,16
The previously proposed mechanisms14,16 for SSA struggle to account for our new observations, and we therefore outline a new mechanism to rationalize the results. The mechanism must account for the key finding that CO2 adsorption is observed for negative charging (Fig. 2a) and CO2 desorption is observed for positive charging (Fig. 2b). Given that a supercapacitor is a symmetric electrochemical cell, one would initially anticipate identical CO2 adsorption whether charging positively or negatively. However, our cell design (Fig. 1) breaks the cell symmetry, placing one electrode in closer contact with the CO2 gas reservoir. In our hypothesized mechanism, we therefore focus on the gas-exposed electrode (Fig. 3), and the movement of CO2 derived species into and out of this electrode when charging. At an electrolyte pH of 7, bicarbonate ions are expected to be the dominant form of dissolved CO2,18 though we do note that pH changes may occur in the device due to (i) CO2 dissolution which causes pH decreases,20,21 (ii) any selective H+ adsorption by the carbon surface (due its basicity) which would increase the pH, and (iii) ionic migration in the supercapacitor during charging which could lead to pH gradients.18
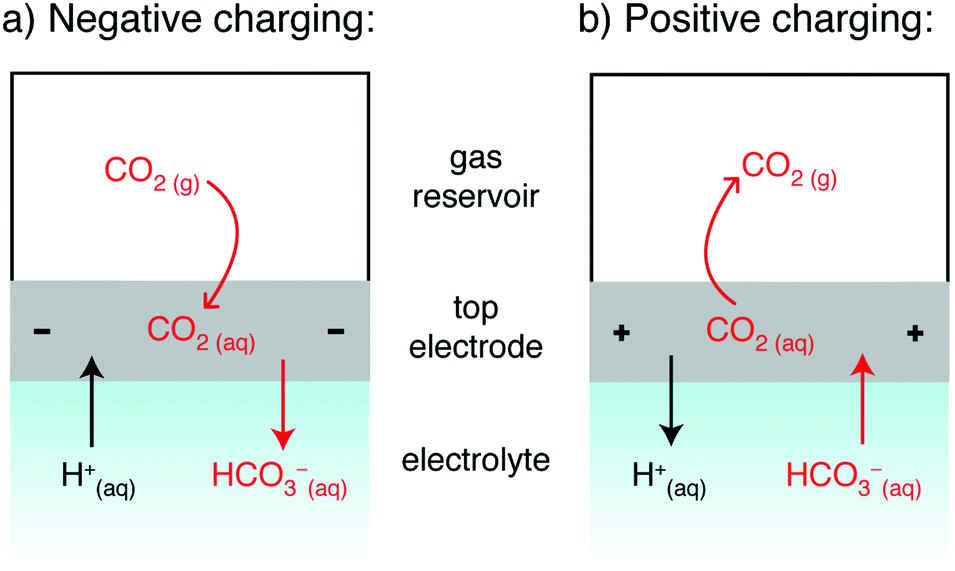 | ||
| Fig. 3 Schematic showing the proposed mechanism for the movement of CO2 and ions with charging. Other cations (e.g. Na+) will behave analogously to H+, and anions (e.g. CO32− and Cl−) to HCO3+. | ||
The direction of movement of CO2 gas observed experimentally is depicted in Fig. 3 (by curved red arrows), along with the expected movement of CO2-derived ions based on charge balancing arguments (straight arrows). For negative charging, we observe that CO2 adsorbs into the cell during charging. Under these conditions, we anticipate HCO3− desorption from the negative electrode.22 This desorption should reduce the concentration of CO2 in the negative electrode (since CO2 and HCO3− are in equilibrium), thereby providing a driving force for CO2 adsorption into the negative electrode. Conversely, for positive charging, the electrosorption of bicarbonate into the positive electrode provides a driving force for CO2 release. We note electrosorption of H+ may also impact CO2 capture.7,10 though the concentration of these species is expected to be very low if the pH remains close to 7.
Finally, the electrolyte ions (Na+ and Cl−) must also play a significant role in electrochemical CO2 adsorption. When deionized water was used as an electrolyte in the literature, excluding any supporting electrolyte ions, a somewhat lower adsorption capacity was obtained.14 We hypothesize that there is either cooperativity or competition between the electrosorption of these ions and the CO2-derived ions. The electrolyte ions may promote CO2 adsorption (e.g. via Lewis acid–base interactions) or desorption (perhaps through competition for adsorption sites). The differences in diffusivities between Na+, Cl−, H+, and HCO3− further complicates this issue. Ultimately, more experimental and theoretical work must be done to fully unravel the mechanisms of electrochemical CO2 capture by supercapacitors, and to test our hypotheses.
Overall, this study has shown that simple changes to charging protocols can significantly increase the adsorption capacities of supercapacitive swing adsorption. Charging with the positive electrode exposed to gas increases adsorption capacity and decreases energy consumption, and a “switching” protocol further increases capacity. This moves SSA further towards the performance required for commercial viability. It also brings new insights into the mechanism of CO2 capture, and we propose a new model to account for supercapacitive swing adsorption.
Author contributions
Conceptualization: A.C.F., T.B.B.; methodology: T.B.B., G.M., I.T., A.C.F.; investigation: T.B.B., G.M.; analysis: T.B.B., G.M., I.T., A.C.F.; writing – original draft: T.B.B.; writing – review and editing: T.B.B., G.M., I.T., A.C.F.; funding acquisition: A.C.F.Data availability
All raw experimental data files are available in the Cambridge Research Repository, Apollo, with the identifier https://doi.org/10.17863/CAM.83307.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by a UKRI Future Leaders Fellowship to A.C.F. (MR/T043024/1), and the Yusuf Hamied Department of Chemistry at Cambridge for the award of a BP Next Generation Fellowship to A.C.F. This work was further supported by the NanoDTC Cambridge EP/S022953/1. We thank Prof. Michael De Volder and Dr Céline Merlet for helpful discussions, and Prof. Clare Grey for support to I.T. We also thank the mechanical workshop in the Dept. of Chemistry for preparation of gas cells, and we thank Jamie Gittins and Dongxun Lyu for carrying out gas adsorption measurements and analysis. A.C.F. thanks Dr Kristian Knudsen and Prof. Bryan McCloskey for helpful discussions and help with feasibility tests at U.C. Berkeley.For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising.
References
- Global Warming of 1.5 C. An IPCC Special Report on the Impacts of Global Warming of 1.5 C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty, ed. V. Masson-Delmotte, P. Zhai, H.-O. Portner, D. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Pean, R. Pidcock, S. Connors, J. B. R. Matthews, Y. Chen, X. Zhou, M. I. Gomis, E. Lonnoy, T. Maycock, M. Tignor and T. Waterfield, Series Eds.; IPCC, 2018 Search PubMed.
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, The Role of Renewable Energy in the Global Energy Transformation, Energy Strategy Rev., 2019, 24, 38–50, DOI:10.1016/j.esr.2019.01.006.
- C. G. F. Bataille, Physical and Policy Pathways to Net-Zero Emissions Industry, Wiley Interdiscip. Rev. Clim. Change, 2020, 11(2), e633, DOI:10.1002/wcc.633.
- Committee on Climate Change. Net Zero: The UK's Contribution to Stopping Global Warming; Technical report; Committee on Climate Change, 2019.
- G. T. Rochelle, Amine Scrubbing for CO2 Capture, Science, 2009, 325(5948), 1652–1654, DOI:10.1126/science.1176731.
- A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Carbon Capture and Utilization Update, Energy Technol., 2017, 5(6), 834–849, DOI:10.1002/ente.201600747.
- M. Rahimi, G. Catalini, M. Puccini and T. A. Hatton, Bench-Scale, Demonstration of CO 2 Capture with an Electrochemically Driven Proton Concentration Process, RSC Adv., 2020, 10(29), 16832–16843, 10.1039/D0RA02450C.
- S. E. Renfrew, D. E. Starr and P. Strasser, Electrochemical Approaches toward CO2 Capture and Concentration, ACS Catal., 2020, 10(21), 13058–13074, DOI:10.1021/acscatal.0c03639.
- J. S. Kang, S. Kim and T. A. Hatton, Redox-Responsive Sorbents and Mediators for Electrochemically Based CO2 Capture, Curr. Opin. Green Sustainable Chem., 2021, 31, 100504, DOI:10.1016/j.cogsc.2021.100504.
- S. Jin, M. Wu, R. G. Gordon, M. J. Aziz and D. G. Kwabi, PH Swing Cycle for CO2 Capture Electrochemically Driven through Proton-Coupled Electron Transfer, Energy Environ. Sci., 2020, 13(10), 3706–3722, 10.1039/D0EE01834A.
- L. Legrand, O. Schaetzle, R. C. F. de Kler and H. V. M. Hamelers, Solvent-Free CO2 Capture Using Membrane Capacitive Deionization, Environ. Sci. Technol., 2018, 52(16), 9478–9485, DOI:10.1021/acs.est.8b00980.
- S. Voskian and T. A. Hatton, Faradaic Electro-Swing Reactive Adsorption for CO2 Capture, Energy Environ. Sci., 2019, 12(12), 3530–3547, 10.1039/C9EE02412C.
- B. Kokoszka, N. K. Jarrah, C. Liu, D. T. Moore and K. Landskron, Supercapacitive Swing Adsorption of Carbon Dioxide, Angew. Chem., Int. Ed., 2014, 53(14), 3698–3701, DOI:10.1002/anie.201310308.
- S. Zhu, J. Li, A. Toth and K. Landskron, Relationships between Electrolyte Concentration and the Supercapacitive Swing Adsorption of CO2, ACS Appl. Mater. Interfaces, 2019, 11(24), 21489–21495 CrossRef CAS PubMed.
- S. Zhu, J. Li, A. Toth and K. Landskron, Relationships between the Elemental Composition of Electrolytes and the Supercapacitive Swing Adsorption of CO 2, ACS Appl. Energy Mater., 2019, 2(10), 7449–7456, DOI:10.1021/acsaem.9b01435.
- S. Zhu, K. Ma and K. Landskron, Relationships between the Charge–Discharge Methods and the Performance of a Supercapacitive Swing Adsorption Module for CO2 Separation, J. Phys. Chem. C, 2018, 122(32), 18476–18483, DOI:10.1021/acs.jpcc.8b03968.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. M. Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Carbon Capture and Storage Update, Energy Environ. Sci., 2014, 7(1), 130–189, 10.1039/C3EE42350F.
- L. Legrand, Q. Shu, M. Tedesco, J. E. Dykstra and H. V. M. Hamelers, Role of Ion Exchange Membranes and Capacitive Electrodes in Membrane Capacitive Deionization (MCDI) for CO2 Capture, J. Colloid Interface Sci., 2020, 564, 478–490, DOI:10.1016/j.jcis.2019.12.039.
- I. Temprano, T. Liu, E. Petrucco, J. H. J. Ellison, G. Kim, E. Jónsson and C. P. Grey, Toward Reversible and Moisture-Tolerant Aprotic Lithium-Air Batteries, Joule, 2020, 4(11), 2501–2520, DOI:10.1016/j.joule.2020.09.021.
- J. W. McBain, The Use of Phenolphalein as an Indicator. The Slow Rate of Neutralisation of Carbonic Acid, J. Chem. Soc. Trans., 1912, 101, 814–820 RSC.
- T. Loerting and J. Bernard, Aqueous Carbonic Acid (H2CO3), ChemPhysChem, 2010, 11(11), 2305–2309, DOI:10.1002/cphc.201000220.
- A. C. Forse, C. Merlet, J. M. Griffin and C. P. Grey, New Perspectives on the Charging Mechanisms of Supercapacitors, J. Am. Chem. Soc., 2016, 138(18), 5731–5744, DOI:10.1021/jacs.6b02115.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, and additional electrochemical gas adsorption data (PDF). See DOI: https://doi.org/10.1039/d2nr00748g |
| This journal is © The Royal Society of Chemistry 2022 |