
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignin for energy applications – state of the art, life cycle, technoeconomic analysis and future trends
Anne
Beaucamp
a,
Muhammad
Muddasar
 a,
Ibrahim Saana
Amiinu
b,
Marina
Moraes Leite
b,
Mario
Culebras
c,
Kenneth
Latha
d,
María C.
Gutiérrez
e,
Daily
Rodriguez-Padron
f,
Francisco
del Monte
e,
Tadhg
Kennedy
bg,
Kevin M.
Ryan
bg,
Rafael
Luque
f,
Maria-Magdalena
Titirici
d and
Maurice N.
Collins
*ag
a,
Ibrahim Saana
Amiinu
b,
Marina
Moraes Leite
b,
Mario
Culebras
c,
Kenneth
Latha
d,
María C.
Gutiérrez
e,
Daily
Rodriguez-Padron
f,
Francisco
del Monte
e,
Tadhg
Kennedy
bg,
Kevin M.
Ryan
bg,
Rafael
Luque
f,
Maria-Magdalena
Titirici
d and
Maurice N.
Collins
*ag
aStokes Laboratories, School of Engineering, Bernal Institute, University of Limerick, Limerick, Ireland. E-mail: Maurice.collins@ul.ie
bDepartment of Chemical Sciences, Bernal Institute, University of Limerick, Limerick, Ireland
cInstitute of Material Science, University of Valencia, Valencia, Spain
dDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ UK
eInstituto de Ciencia de Materiales de Madrid (ICMM), Consejo Superior de Investigaciones Científicas (CSIC), Calle Sor Juana Inés de la Cruz, 3, Campus de Cantoblanco, 28033, Madrid, Spain
fDepartamento de Química Orgánica, Universidad de Córdoba, Campus de Rabanales, Edificio Marie Curie (C-3), Ctra Nnal IV-A, Km 396, 14014 Cordoba, Spain
gSFI AMBER Centre, University of Limerick, Ireland
First published on 12th October 2022
Abstract
Lignin is produced in large quantities as a by-product of the papermaking and biofuel industries. Lignin is the most abundant aromatic biopolymer on the planet with its chemical structure rendering it ideal for carbon materials production and finely tailored architectures of these sustainable carbon materials are beginning to find use in high value energy applications. This review focuses on lignin chemistry, various lignin extraction and fractionation techniques, and their impact on lignin structure/property relationships for energy applications are discussed. Chemistries behind important and emerging energy applications from recent research on this increasingly valuable sustainable polymer are described.
1. Introduction
It is now widely accepted that global warming is a threat to our planet, and it needs to be addressed urgently. To replace petroleum-based products with sustainable materials is a major step in the fight against climate change, as highlighted by the United Nations Sustainable Development Goal (SDG) 9 for Industry, Innovation, and Infrastructure and SDG 12 for Responsible Consumption and production.1–3 Therefore, the development of sustainable technologies capable of producing new devices using sustainable resources within the circular use of products approach is of critical importance.Materials used as electrodes greatly influence the performance of supercapacitors, batteries, and thermoelectric materials.4 Carbon-based materials, such as activated carbon, carbon nanotubes, and graphene nanosheets, have great potential as electrodes owing to their lightweight, high conductivity, and adjustable porosity.5,6 However, high-quality carbon compounds are usually synthesized using sophisticated and expensive synthesis methods that involve strict chemical conditions, elevated carbonization temperatures, and non-renewable precursors, limiting their extensive commercial use.7,8 Thus, it is crucial to find easy, efficient, and eco-friendly pathways for producing carbon materials by converting low-cost, sustainable precursors into carbon materials. The utilization of lignocellulosic biomass (cellulose, lignin, hemicellulose) could be a promising alternative route for the synthesis of carbon materials. The scientific community has been attracted to cellulose because of its abundance, adaptability, sustainability, and affordability. The low carbon content in cellulose (44.4 wt%), however, makes cellulose-based carbon materials economically unsuitable for extensive usage.9 On the other hand, lignin, another biopolymer with high carbon content (≈60 wt%), is the main source of aromatic moieties in the natural world and could be a promising carbon precursor source.10,11 Lignin is mainly found in plant cell walls, accounting for 20–25% of plant dry weight, making lignin the second most abundant biopolymer after cellulose. However and crucially, the valorisation of lignin is currently limited with 95% of the worldwide lignin being a underutilised by-product of the paper and pulp production and the remainder is used for low value applications.12,13 The main extraction of lignin is through the Kraft process, which separates efficiently cellulose from biomass. The paper industry burns the black liquor containing the lignin to generate heat, reused in the production plant through energy cogeneration. The combustion also regenerates the inorganic pulping agent for further use. If the lignin is recovered by acidification of the black liquor, the pulping process produces low-quality lignin, with high sulphur, ash and carbohydrate contents. Therefore, kraft lignin is mainly burnt as a biofuel.14 Another reason for the poor valorisation of lignin is the complexity and variation of its chemical structure, which is highly dependent on its source and extraction method. Therefore, more profitable value-streams are necessary to motivate a need for a new generation of biorefineries capable of producing high quality lignin fractions. Recent reviews have focused on the refining of lignin,15 its use for civil engineering,16 for polymer production,17 and its use in nanocomposites systems.18
This review focuses on the role of lignin in energy applications. The first section briefly describes lignin chemistry and the impact of the extraction method on the quality of the isolated lignin. It also discusses the emerging benefits of fractionation of lignin via deep eutectic solvents (DES). Section two details the use of lignin in battery component development with emphasis on anodes, cathodes, binders, electrolytes, separators and redox flow batteries. The following section describes the latest technologies in creating greener and more sustainable supercapacitor electrodes from lignin, starting with simple activation methods and moving towards templated and free-standing carbon electrodes. This leads to a section on an emerging application for lignin in thermoelectric materials where lignin precursor materials are producing nanomaterials with very promising Seebeck coefficients. Section 5 summaries, the production of biofuels from lignin, with particular emphasis on both heterogenous and photo-assisted catalytic conversion of lignin, along with some recent findings on mechanochemical conversion. Finally, we conclude the review with an outlook on the use of lignin in future energy applications.
1.1. Lignin: chemical structure and separation
The word lignin comes from the Latin lignum, meaning wood. It is a polyphenolic component of the vascular plant cell wall that cements the cellulose and hemicellulose rods together, creating mechanical support while protecting the plants from microbial attack.19–21 The amount of lignin depends on the origin of the plant, with softwood (SW) comprising 25–39% of lignin, hardwood (HW) 20–25% and grasses 15–25%.20 Lignin is the most abundant aromatic biopolymer, with a structure containing over 60% carbon making lignin the source of over 30% of the organic carbon on Earth.22It is biosynthesised by enzymatic dehydrogenation of three monolignols monomers, p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol that differ only by the degree of substitution on the phenolic ring. In the lignin structure, monolignols are present in the form of residue units, respectively p-hydroxyphenyl (H), guaiacyl (G) and sinapyl (S). The composition of S, G and H units varies with plant type: SW lignin is mainly composed of S units while HW lignin contains S and G units. Interestingly, grass offers a source of non-methoxylated lignin, containing up to 35% of H units.23 Within a plant type, proportions of G vs. S units would vary depending on the position in the plant and the age of the sample (Fig. 1).24
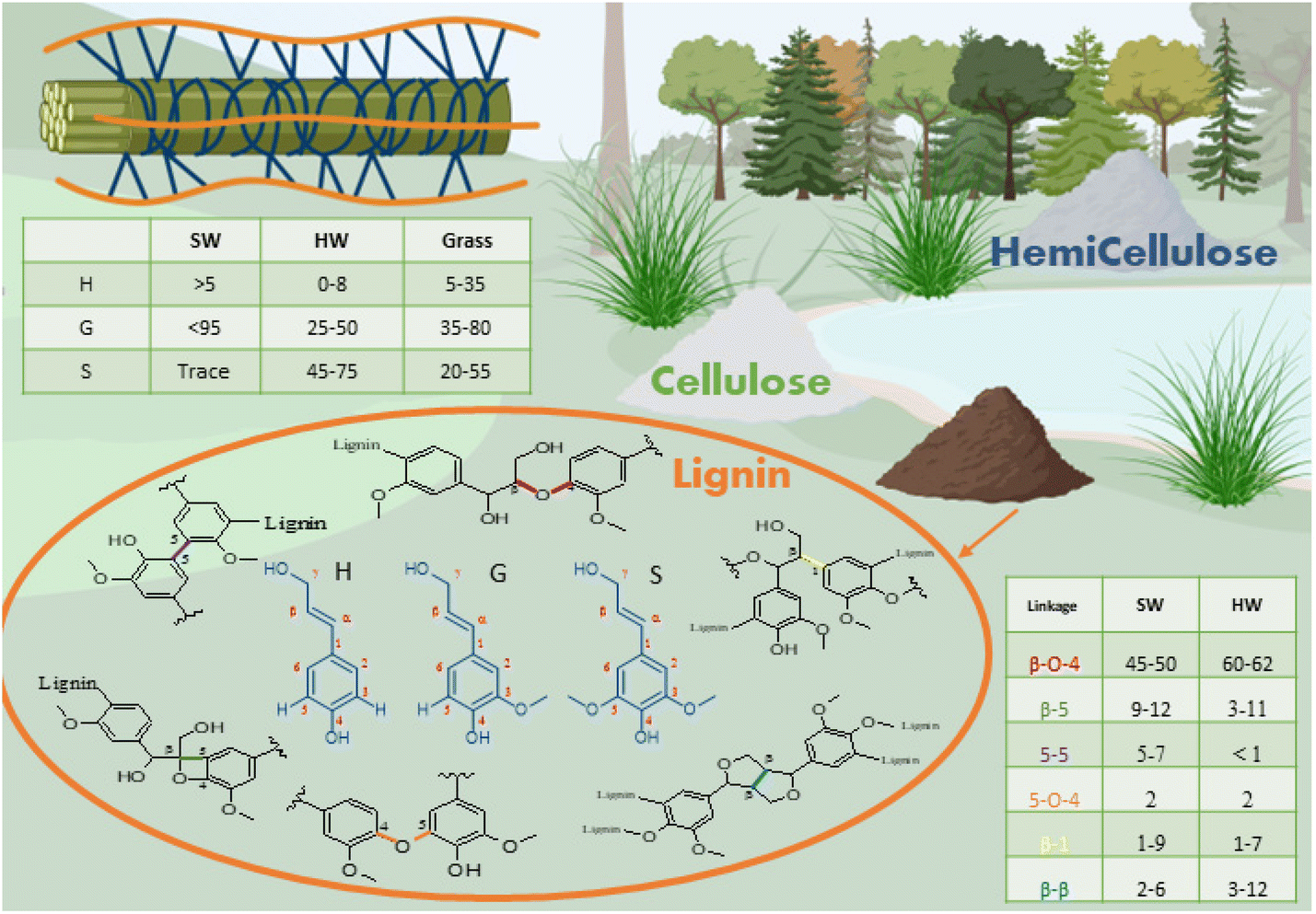 | ||
| Fig. 1 Structure and chemistry of lignin extracted from plants cell walls. | ||
Lignin phenylpropanoid units interlink randomly via crosslinking and branching to form an amorphous three-dimensional network.25 The polymeric structure forms by radical polymerisation, initiated by the action of laccase and peroxidase enzymes.12,26 Various linkages are formed during the polymerisation, with the most common being ether and carbon–carbon linkages. The carbon in Cβ position and the phenoxy oxygen are the most reactive species, making the β-O-4 (β-aryl ether) the most frequent linkage, which can easily be cleaved in an alkaline or acidic medium. The other linkages are more resistant to chemical degradation.12,24 The formation of chemical bonds during lignification is believed to be mediated by kinetically controlled radical coupling and this has recently been computationally modelled by Gani et al.27 They calculated the value of the activation barrier during radical formation and chain growth of various linkages in G and S rich lignins, with results showing that formation of β-O-4 chains is favourable. Empirically, it was found that the presence of methoxy groups on the 5th aromatic carbon prevents the formation of C–C bonds by β-5 and 5-5 links, rendering branched, condensed lignin rare in its native form.21,23
The molecular structure of lignin is dependent on its origin. In particular, hardwood lignins are more rich in phenolic hydroxyl groups (4.3 mmol g−1) displaying a lower molecular weight in comparison with softwood lignins.28
To allow separation from the cellulose and hemicellulose rods within wood, native lignin is subjected to chemical processes, which modify the bonding of the polymer by partial depolymerisation – so called technical lignins.23 The type of technical lignin is as important as the source of lignin. There are three main processes that lead to technical lignins, Kraft, Soda and Organosolv each of which are detailed below.
The kraft process is the largest lignin stream by volume (55 million tons), this high yield process treats wood fibres with a Na2S/NaOH white liquor at a temperature of 155–175 °C for 1 to 2 hours. Lignin and hemicellulose are dissolved in the solution to form a “black liquor” while the cellulose forms a solid residue. The black liquor, containing 70–80% solid, is usually used as a biofuel in the recovery boiler. This combustion, at 760 °C, produces molten salts of Na2S and NaOH. These are precipitated and regenerated for circular use within the mill.36 The kraft lignin can be recovered from the black liquor by precipitation in the presence of acetic acid, which improves the overall mill production.14 The Kraft process modifies the lignin structure via cleavage of the β-ether (C–O) links as well as by demethylation. The formed lignin has a higher degree of condensation through the formation of 5-5 linkages with sulphur added in the form of thiol groups.23,24 The high ash, sulphur and carbohydrate content of kraft lignin restricts its industrial application to fertilizers, pesticides, binders, aromatic chemicals and resins.37
The Soda lignin process is similar to the kraft process, as it uses an alkaline solution (NaOH) at high temperature to separate lignin from cellulose by solubilisation. The lignin is then recovered by precipitation at lower pH. This is a sulphur free process, with a yield of 80%, which produces moderately pure lignin, due to the presence of minerals (Na, K) and carbohydrates. The process cleaves the α- and β-aryl ether bonds and produces free phenolic groups but also generates condensation of the aromatic rings.24,35 The sulphur free soda lignin is used industrially in phenolic resins, animal nutrition, dispersants, and polymer synthesis.
Organosolv lignins are dissolved in organic solvent/water solution at high temperature and pressure. In particular, the Alcell © process uses a mixture of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol:water to dissolve lignin at 180 °C at 13 bars of pressure. The lignin obtained is depolymerised by cleavage of the β-ether links and exhibits a low molecular weight and polydispersity. It also has low ash content and low residual carbohydrate with a general low impurity level.23,24,35
:1 ethanol:water to dissolve lignin at 180 °C at 13 bars of pressure. The lignin obtained is depolymerised by cleavage of the β-ether links and exhibits a low molecular weight and polydispersity. It also has low ash content and low residual carbohydrate with a general low impurity level.23,24,35
Water soluble lignosulfonate or sulphite lignin is formed by reaction between lignin and metal sulphite salts. The reaction can be processed at pH varying between 2 and 12. This process cleaves α and β-ethers and forms a high quantity of sulfonic acid groups on the Cα position.38 Lignosulfonate has the highest molecular weight (up to 50000 g mol−1) due to condensation between the aromatic rings. Sulphite lignin is mostly used as an additive in the construction industry thanks to its solubility in water.37
Other methods such as Milled Wood Lignin (MWL) or enzymatic lignin produce low sulphur content lignin. MWL is obtained by washing finely milled biomass by a neutral solvent such as dioxane/water. The solution is then centrifuged, the solid freeze-dried and washed.39,40 This process extracts lignin that is essentially structurally unchanged from the biomass it originates. This method has a low yield and is usually carried out for native lignin characterisation studies.39,41,42 Steam explosion lignin is produced by rapidly decompressing a high-pressure biomass-steam mixture to separate the biomass. The liquid recovered is washed at low pH and the lignin is isolated. This method produces high quality sulphur-free lignin, with minor structural modifications and at a low cost.43 Enzymatic hydrolysis involves the hydrolysis and dissolution of carbohydrates by cellulase enzyme. This process aims to produce bioethanol and the lignin is recovered as a by-product, with moderate amount of impurities.44
The water solubility of lignin is displayed in Table 1. Most lignins are soluble in alkaline conditions due to the presence of phenolic groups, whose amount can be increased depending on extraction conditions. While lignosulfonates are fully soluble in water due to their extraction conditions.
| Production volume (tonnes per year) | Yield (%) | Impurities (%) | Molecular weight (Mw) and polydispersity | Solubility in water | |
|---|---|---|---|---|---|
| Kraft lignin | 55000000 |
90–95 | Sulphur: 1–3 | 1500–5000 (2.5–3.5) | Fully soluble in pH > 12 |
| Ash: 0.5–3.0 | |||||
| Carbohydrates: 1.0–2.3 | |||||
| Lignosulfonate | 1000000 |
70–95% | Sulphur 3.0–8.0 | 1000–50000 (6.0–8.0) |
Fully soluble |
| Ash: 4.2–7.0 | |||||
| Carbohydrates: N/A | |||||
| Soda lignin | 6000 | >80 | Sulphur: 0 | 1000–10000 (2.5–3.5) |
Fully soluble in pH > 12 |
| Ash: 0.7–3.0 | |||||
| Carbohydrates: 1.5–3.0 | |||||
| Organosolv | 1000 (pilot Scale) | 25–50 | Sulphur: 0 | 500–5000 (1.5–4.4) | Fully soluble in pH > 12 |
| Ash: 1.7 | |||||
| Carbohydrates: 1.0–3.00 | |||||
| Milled Wood Lignin (MWL) | Lab scale | 20 | Sulphur: 0 | 5500–20000 (1.8–2.5) |
Poor |
| Ash: 1.5 | |||||
| Carbohydrates: 0.30 | |||||
| Steam Explosion Lignin (SEL) | Lab scale | >90 | Sulphur: 0–0.5 | 1000–15000 (2.5–7.0) |
Fully soluble in pH > 12 |
| Ash: 5.0–8.0 | |||||
| Carbohydrates: 2.5–4.0 | |||||
| Enzymatic lignin/hydrolysis lignin | Lab scale | 95 | Sulphur: 0–1 | 2000–4500 (1.5–3.2) | Fully soluble in pH > 12 |
| Ash: 1.0–3.0 | |||||
| Carbohydrates: 2.5–4.0 |
Current research uses all lignin streams for the development of high-end applications such as nanostructured materials, fine chemicals, carbon materials and biofuels. Kraft lignin and organosolv lignin are the preferred materials due to the availability of Kraft and the high purity of organosolv. While lignin is heterogeneous in terms of secondary groups, the main characteristic of the macromolecule are its phenolic groups that allow for reactivity, solubility and modification. The thiolation observed on Kraft lignin can be reversed by thermal treatment: the thermal decomposition of sulphur containing lignin releases sulphur dioxide (SO2) and methylated sulphur compounds as the thiol groups are breaking down. Dondi et al. measured the amount of sulphur compounds released during pyrolysis. The release of SO2 was maximum at 253 °C while CH3SH and CH3SCH3 were released at 266 °C. The authors also investigated the release of CO2, and it was found to be one order of magnitude higher for Kraft lignin. It was hypothesised that this increase is due to the oxidative nature of the sulphur compounds.45 A similar study from Han46 showed that below 250 °C, SO2 is the only compound released during fast pyrolysis.
1.2. Deep eutectic solvents and aqueous dilutions thereof for lignin fractionation
Valorisation of lignocellulosic biomass into high-value-added products including biofuels, bio-based chemicals and biomaterials relies on the development of suitable biorefineries. The lignocellulosic-based biorefineries suffer major bottlenecks, such as the design of pre-treatment techniques.47 For pre-treatment the most important requirements include (1) sustainability (with cost-effective and environmentally friendly solvents/reagents), (2) universality (suitable for a wide range of lignocellulosic biomass materials) and (3) delignification efficiency (capable to provide the individual constituents of lignocellulosic biomass with high yield and purity whilst allowing their conversion into value-added biochemicals).In this context, Deep eutectic solvents (DESs) have attracted much attention for lignocellulosic pretreatment.48–51 DESs are formed by hydrogen bond (HB) complexation between (at least) two molecules, one hydrogen-bond donor (HBD) and one hydrogen-bond acceptor (HBA). Examples of HBAs typically used for DESs formation include many different ammonium and phosphonium salts while alcohols, acids, amides, etc. are representative of HBDs.52 The most often cited rationalisation for this phenomenon is that the charge delocalisation occurring through HB between halide anions with hydrogen-donor moieties is responsible for the decrease in the freezing point of the mixture relative to the melting points of the individual components. Recent ab initio molecular dynamic simulations53 and neutron diffraction studies54 have revealed the occurrence of many possible HB interactions of different strengths among DES constituents, forming an extended HB network similar to those found in crystalline structures. In this regard, inelastic neutron scattering studies have indicated that eutectic behaviour emerges when the components mix via HBs, the strength of which is weak enough to prevent them from settling into a co-crystal.55 Moreover, ternary or quaternary DESs can be formed by combination of more than one HBA and/or more than one HBD.56,57
DESs are particularly well-suited to fulfil most of the above-mentioned requirements as they exhibit remarkable green features (i.e., main DESs components are non-toxic, highly biodegradable and biocompatible), they succeed in the treatment of a large number of lignocellulosic biomass materials (e.g., douglas fir, poplar, sorghum, corncob, walnut endocarps cells, peach endocarp cells, wheat straw, corn straw, rice straw, castor seed coat, oil palm empty fruit, bunch, willow, switchgrass, Eucalyptus camaldulensis, Eucalyptus globulus, Cunninghamia lanceolate, Cortex albiziae, Arabidopsis thaliana and Pinus pinaster Ait, among the most relevant) and they provided delignification efficiencies of up to 95% with purities of the obtained lignin in the range of 76–98% with certain abundance of β-O-4 linkages, the presence of which is critical for the subsequent valorisation of lignin (e.g., β-O-4 linkages determine the high-yield production of aromatic monomers). Moreover, DES can be recovered after lignin fractionation and reused in subsequent pre-treatment processes (Fig. 2).58 For further details on this topic can be found in some excellent reviews recently published.48–51
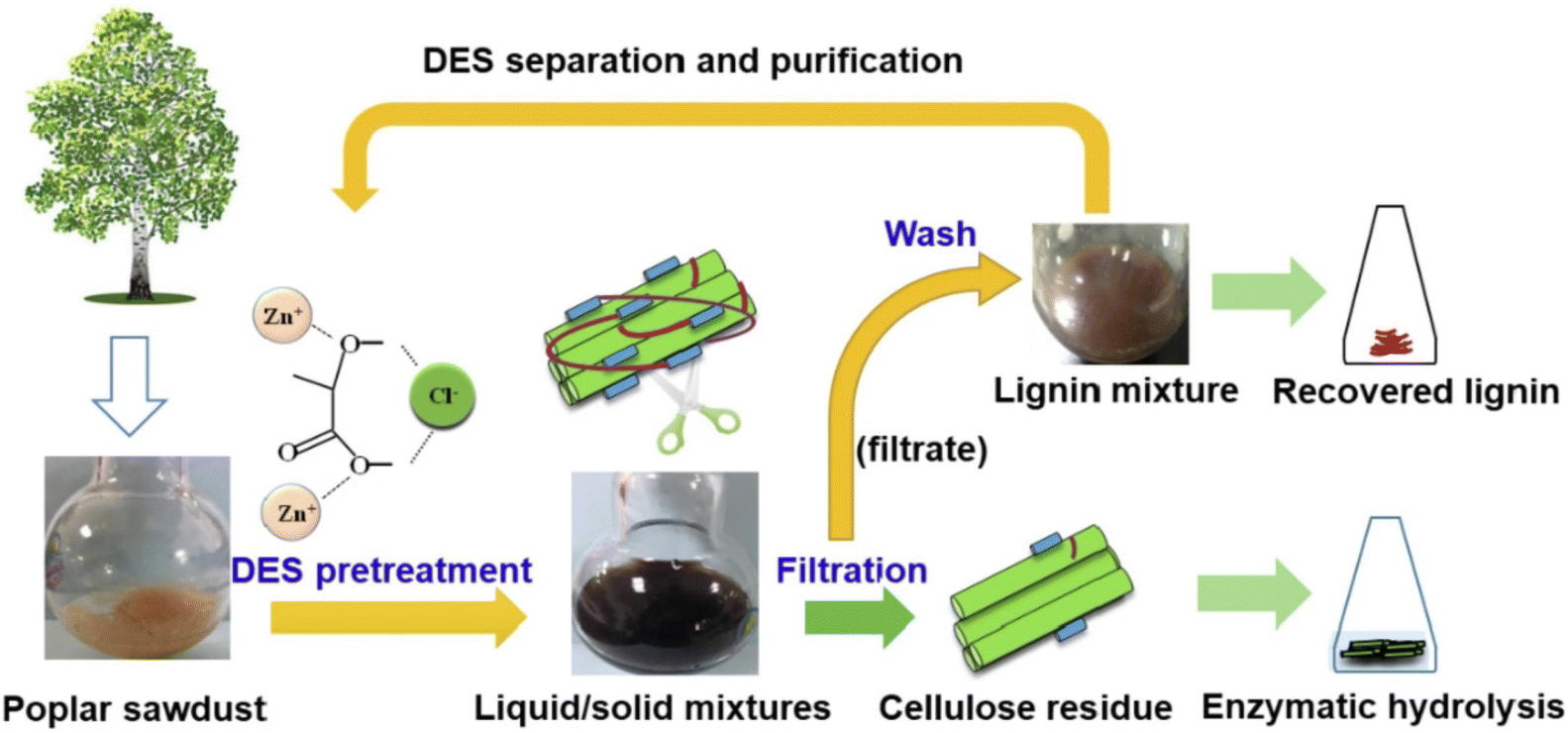 | ||
| Fig. 2 Schematic representation of the DESs pre-treatment process. Reprinted with permission from ref. 58. Copyright 2022, Elsevier. | ||
Nonetheless, it is worth noting that while the fulfilment of requirements (1) and (2) by DESs is remarkable, work must yet be done to obtain well balance results in (3). For instance, lower temperature, shorted time, or higher solid loading give rise to a higher percentage of preserved β-O-4 bonds in DES lignin.59 Moreover, DES constituents, both the HBA and the HBD, also play a role in lignin fractionation performance. For instance, it has been demonstrated that the relevance of the nature and number of functional groups of the HBD id of importance. Thus, most effective HBDs for lignin fractionation are monocarboxylic acids. In this sort of acid-based DESs, the stronger and the higher the molar ratio of the acid, the higher the fractionation yield (e.g., up to 93.1% for a DES composed of lactic acid, LA, and choline chloride, ChCl, mixed in a LA:ChCl molar ratio of 15:1).60 Hydroxyl groups in HBDs have proved less effective than carboxylic acids for lignin fractionation.61 In either case, the increase of the number of functional groups exerted a detrimental effect on delignification. It seems that the eventual HBA participation in a more extended HB network weakens its ability to compete with intra-molecular bonding in the lignin moieties of biomass.62 Particularly interesting among this hydroxyl-based DESs are those formed with phenolic compounds given their capability to form new strong π–π stacking interactions and HBs with lignin moieties.63 It is also worth noting that the ability of DESs with amine/amide-based for lignin fractionation. In this case, it is hypothesized that the presence of amine/amide groups in HBDs endows the DES with strong basicity that facilitates lignin fractionation by loosening various chemical bonds of LCC and contributes to selective dissolution through the deprotonation of phenolic moieties in lignin. Finally, although less studied, HBAs can also play a significant role in the performance of lignin fractionation by the contribution of the halide anions to the breakage of β-O-4′ bonds thus preventing lignin condensation.64
However, it is worth noting that, as mentioned above, stabilisation of β-O-4 linkages are critical for the subsequent valorisation of lignin and this stabilization generally occurs at the expense of lignin fractionation yield and purity. Thus, the challenge for DES-based lignocellulosic pretreatments is the achievement of not only high lignin fractionation yield and purity but also with good preservation of β-O-4 linkages. In this regard, this review focuses on recent and promising research emphasising what we believe should be the future directions to explore. Among others, we think, most intriguing attempts are lately involving the use of aqueous dilutions of DESs.65–68 The nature of aqueous dilutions of DESs (e.g., reline, a DES formed by complexation between choline chloride and urea mixed in a 1:2 molar ratio) was first studied in detail in 2009.69 That work described how, in a highly diluted regime, the original HB complexes of DES were broken and, as most of the DES components are soluble in water, the system became a simple aqueous solution of the individual DES components. Interestingly, the scenario in a less diluted regime is quite the opposite with the HB complexes of DES solvating H2O molecules. Actually, neutron scattering measurements have revealed how H2O molecules are interstitially accommodated within the DES-based HB network.70 This accommodation not only happens for H2O but also for other solvents with HB capabilities (e.g., methanol, benzyl alcohol, DMSO, etc.) as indicated the deviation from ideality observed in excess properties such as molar volume (VE), viscosity (ηE) or compressibility (ΔβS).71–74 Interestingly, all these liquid binary mixtures exhibited viscosities that are below that of the original DES, which is indeed of relevance for practical applications.
This reduction in viscosity is obviously of help for the effectiveness of aqueous dilutions of DESs no matter the mechanism (e.g., hydrotropic or co-solvency, Fig. 2) behind lignin solubilisation. The hydrotropic mechanism is particularly effective for DESs composed of HBDs and HBAs capable to act as hydrotropes.75 More intriguing is the co-solvency mechanism.76 DESs following the co-solvency mechanism are all formed by water-soluble and/or water-miscible compounds (e.g., ethylene glycol, formic acid, propionic acid, lactic acid as HBDs and tetrapropylammonium chloride and choline chloride as HBAs). In these cases, the amount of H2O is by no means trivial, changing its role from co-solvent to anti-solvent when it surpasses a certain content. Whether there is a correlation between amount of H2O providing the best results of lignin dissolution and that in which DES aqueous solutions exhibit the largest deviations from ideality may bring some light to better understand the rationale behind lignin solubilisation in DES aqueous solutions.
Moreover, this co-solvency mechanism for lignin dissolution resembles that described for the lyocell process, in which N-methylmorpholine-N-oxide (NMMO) in its monohydrate form is used for cellulose dissolution. It is widely accepted that NMMO dissolve cellulose by their capability to form HBs (e.g., NMMO-cellulose-HBs) with cellulosic units, replaces the intermolecular and intramolecular HBs between D-glucose units and thus breaks the complex HB network of cellulose.77,78 H2O is a serious competitor for NMMO to form HBs with cellulose (e.g., H2O-cellulose-HBs). Thus, the 1NMMO:1H2O mixture works well because there are still sufficient NMMO-cellulose-HBs (more so than H2O-cellulose-HBs) and the viscosity of the solution is lower than without H2O. Recent works have demonstrated how the addition of co-solvents with HB capabilities can help cellulose dissolution by further decreasing the viscosity while preserving the favoured balance between NMMO-cellulose-HBs and H2O-cellulose-HBs.79,80 Based on this, the use of environmentally friendly co-solvents with HB capabilities that can replace H2O totally or partially which may offer interesting sustainable perspectives for future processes in the valorisation of lignin for high value energy applications.
2. Lignin for energy applications – batteries
Lignin has been identified as an excellent precursor for the synthesis of various active carbon-based materials for energy storage devices. However, to date, their inherently poor electrical conductivity has largely hindered their direct use as electrode materials in energy storage and conversion devices. Therefore, various synthetic methods and structural transformation strategies have been pursued to enhance their conductivity, electrochemical and thermomechanical properties. The most common strategy for converting lignin to battery active carbon materials is by pyrolysis at high temperatures (600–1100 °C) using chemical activation agents (e.g., KOH, K2CO3 and ZnCO3).81–83 The combination of these synthesis steps effectively converts the large polyaromatic macromolecules of lignin into an active carbon that is suitable for battery applications. This introduces several key features that bequeath the carbon derivative with advanced properties such as a hierarchical nano-micro pore system (Fig. 3a), high conductivity and mechanical robustness which benefit fast kinetics, high stability and high charge storage capacity. Like graphite, lignin derived carbons have been widely explored in various energy storage applications, including metal ion (Li+, Na+, K+, etc.), metal oxygen and redox flow batteries.81 However, unlike graphite, the polymeric nature of lignin allows for functionally tailored modifications, including surface functionalisation and molecular grafting prior to carbonisation and heteroatom doping, leading to carbon materials with various morphologies and enhanced functional properties. This enables lignin-derivatives to exhibit beneficial properties spanning the whole chain of the battery component development (i.e., anode, cathode, binder, separator and electrolyte). Therefore, the high structural flexibility of lignin to exhibit various functions makes it a highly attractive precursor material for carbon-based battery materials. | ||
| Fig. 3 (a) Synthesis route to hierarchical porous carbon anode from lignin. Adapted with permission from ref. 96. Copyright 2020, Royal Society of Chemistry (b) SEM image showing hierarchical porosity of lignin-derived carbon anode. Adapted with permission from ref. 85. Copyright 2015, Elsevier (c) TEM image showing hierarchical porosity of lignin-derived carbon nano fibre anode. (d) Anode cycling performance of lignin/PLA derived CNFs at C/2. Adapted with permission from ref. 86. Copyright 2019, Wiley-VCH. | ||
2.1. Lignin derived anode materials
Carbon based materials of various morphologies and structures have been widely used as anode materials in different battery chemistries and have been modulated to meet different requirements. Porous and fibrous carbons are the most researched electrode materials and are typically obtained from organic chemical compounds with high cost and negative environmental impacts. Lignin on the other hand is eco-friendly and a naturally abundant precursor for low-cost production of hierarchically porous carbon and nano fibre-based electrode materials. The porosity enhances the surface area for rapid mass transport of ionic species. For example, Lu's group85 developed a hierarchical porous carbon via the pyrolysis of a lignin/KOH mixture at 700 °C for use as an anode in LIBs (Fig. 3b). The introduction of 3D network of pores within the carbon framework enhances the surface area (907 m2 g−1) for efficient diffusion of ions, leading to a better charge storage capacity (386 mA h g−1) at 200 mA g−1 after 560 cycles compared to the carbon without pores (77.1 m2 g−1, 124 mA h g−1). The significance of pore structures, high surface area and networking was demonstrated by Culebras et al.86 in which a homogenized blend of lignin and polylactic acid (PLA) was electrospun and subsequently carbonized at 900 °C into fine porous carbon nano fibres (CNFs) (Fig. 3c). The lignin/PLA (50:50%) based CNFs exhibited a surface area of 670 m2 g−1 which enhances the ion transport and cycling stability as a LIB anode, delivering a capacity of 611 mA h g−1 after 500 cycles (Fig. 3d), which is almost twice the theoretical capacity of graphite (372 mA h g−1). While the capacity of lignin derived carbons is higher than graphite, they still exhibit low initial coulombic efficiency (CE) and the use of organic polymer blends and chemical activation agents at high temperatures tend to weaken the environmental benefits. Moreover, the demand for high energy density batteries particularly for electric mobility applications cannot be satisfied with only lignin derived carbon materials. However, the capacity of carbon anodes can be improved by incorporating high capacity materials such as Si (theoretical capacity: 3579 mA h g−1) into the carbonized lignin matrix.87–89 It is important to note that while Si exhibits high capacity characteristics, the application of anodes based solely on bulk Si or micron sized particles is limited by the huge volumetric change (>300%) during cycling which can lead to pulverisation of the material and delamination from the current collector with irreversible capacity losses.90–95 Therefore, composite anodes of Si/carbon are critical for the next generation of high performance LIBs as the stability and energy density can be significantly boosted.
Such composite electrode concepts have been demonstrated by Niu and coworkers87 where the lignin-derived carbon networks cross-linked the Si nanoparticles and delivered a stable capacity of ∼2670 mA h g−1 at 300 mA g−1 after 100 cycles with the CE increasing from 62 to >99%. Although the capacity is largely owed to the Si, the lignin derivative played the key role of enhancing mechanical stability, boosting the conductivity while also contributing to the capacity. While lignin anodes exhibit high promise as next-generation electrodes to replace graphite in LIBs, the study is largely limited to half-cells. To advance the development of lignin-based anodes in practical batteries, their characterisation and detailed analysis in full cell configurations should be explored. In a full cell, the actual working voltage range of the anodes can be reduced, which may lead to a decrease in capacity especially when the voltage profile has a sloping region. This will affect the overall battery energy density, but it is hard to distinguish the contribution of each electrode since the signal is a contribution of both cathode and anode. Such studies are lacking to date. This is particularly crucial for composites anodes such as Si/lignin electrodes which hold high promise as high energy density battery components for electric vehicle and robotic applications.
For lignin-derived anodes, in most cases thy display electrochemical cycling stability for over 100 cycles, it is essential to investigate the contributing factors and the mechanism inducing the longer cycling capacity decay that is unique to lignin. It is also essential to investigate the influence of safety issues such as rate of formation and potential impact of delamination effects, and the mechanical and thermal tolerance characteristics on the long-term battery performance and stability.
Dou et al. compared the performance of HC derived from pectin, hemicellulose and lignin in Na-ion batteries.111 The lignin-derived material presented the highest capacity (298 mA h g−1 at 20 mA g−1) and highest initial coulombic efficiency (68%) among the three. Despite having the lowest BET surface area (only 30 cm2 g−1, in comparison to 223 cm2 g−1 for the hemicellulose-derived HC), their lignin-derived hard carbon showed higher mesoporosity. The pore size, combined with the presence of graphitic carbon and the higher purity of the material explain its better performance. A low-temperature pre-oxidation step was found to have an important effect on the produced HC performance by increasing the interlayer distance of the carbon sheets112 and stabilizing the structure to prevent it from rearranging the graphene layers during the high-temperature carbonisation.113 Using a phenolic resin precursor, the pre-oxidation step promoted better cycling stability, but did not improve the capacity of the materials significantly.114 Ghimbeu et al. noticed an abrupt capacity fade after 30 cycles in a lignin–sulfonate-derived HC, caused by the presence of impurities (mainly Na, K and S from the lignin extraction process).115 Including an aqueous washing step in their synthesis promoted a higher capacity and better cycle stability. Other works include washing steps with acids to remove the impurities from lignin precursors.111,116–119 Peuvot et al. tested different temperatures to carbonize stabilized lignin fibres.120 fibre diameter, surface area and the cycling performance of the produced materials varied depending on the carbonisation temperature (from 800 °C and 1700 °C), with 1200 °C found to be the optimum temperature in their study. Many works investigate the effect of carbonisation temperature, with the best results between 1200 °C and 1400 °C (see Table 2).121 Doping HC with anions can improve their performance, inducing the presence of defects and increasing the number of active sites. Several methods can be used to dope HC from lignin. For instance, Fan et al.,122,123 used a hydrothermal method to produce a mixture of lignin and 3-aminophenol. After carbonisation, the N-doped carbons presented more defects, larger interlayer d-spacing, lower charge transfer resistance and higher porosity, leading to a 2.7-time increase in capacity at 50 mA g−1 (115 mA h g−1 to 315 mA h g−1).122 The adsorption of Na+ on defects can take place at a wide range of potentials, thus contributing to the capacity at the sloping region. In this case, the sloping region capacity increased from 62 mA h g−1 (non-doped material) to 104 mA h g−1 (N-doped material) at 100 mA g−1. In this case, high reversibility was achieved, however it should be noted if the binding energy of Na+ to the defects is too high, as in the case of B doping,124 adsorption is not reversible. Other sources of N can be used, such as melamine and urea.125 The methods and modifications that can be applied to lignin precursors are countless and much research is still necessary to reach a full understanding of their impact on the performance of the produced carbons.
| Source | Pre-treatment | Carbonisation | Surface area (m2 g−1) | Electrode | Electrolyte | ICE | Capacity (mA h g−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Oak sawdust | 1. H2SO4 | N2 100 mL min−1 | 208 | 10% AB | 1 M NaClO4 EC/PC/DMC (9:9:2) |
68% | 297 (50 mA g−1) | 118 |
| 2. KOH | 1300 °C (6 h) | 20% PVDF | 116 (2.5 A g−1) | |||||
| 3. HCl | 10 °C min−1 | |||||||
| Peanut shell | H3PO4 (80 wt%) | Ar | 30 | 10% C45 | 1 M NaClO4 | 68% | 298 (20 mA g−1) | 111 |
| 1100 °C (1 h) | 10% CMC (3-electrode) | EC/PC | 77 (2 A g−1) | |||||
| 1 °C min−1 | ||||||||
| Cocoa pod husk | 1 M HCl 60 °C 24 h | N2 100 mL min−1 | 118 (47% mesopores) | 10% AB | 1 M NaClO4 | 87% | 317 (50 mA g−1) | 119 |
| 1300 °C | 20% PVDF | EC/PC/DMC (9:9:2) |
134 (250 mA g−1) | |||||
| 5 °C min−1 | ||||||||
| Phenolic resin + lignin (30:70) |
No pre-treatment | Ar | 2.3 | 5% Na alginate | 1 M NaPF6 | 88% | 373 (30 mA g−1) 250 (300 mA g−1) | 137 |
| 1400 °C (2 h) | EC/DMC (1:1) |
|||||||
| Alkali lignin | Pre-oxidation: air 200 °C (24 h) | Ar | 31 | 5% CB | 1 M NaClO4 | 81.4% | 285 (50 mA g−1) | 112 |
| 5 °C min−1 | 1350 °C (2 h) | 5% Na alginate | PC | 175 (250 mA g−1) | ||||
| 2 °C min−1 | 1 M NaPF6 DEGDME | 86% | 331 (50 mA g−1) | |||||
| 307 (250 mA g−1) | ||||||||
| Pitch | Pre-oxidation: air 300 °C (3 h) | Ar | n/a | 5% Na alginate | 1 M NaPF6 ED/DMC (1:1) |
88.6% | 301 (30 mA g−1) | 113 |
| 1400 °C (2 h) | ∼250 (150 mA g−1) | |||||||
| Kraft lignin | Stabilisation | N2 0.3 L min−1 | 94 | Free standing electrode | 0.6 M NaPF6 DEGDME | 89% | 310 (30 mA g−1) | 120 |
| Air 10 L min−1 250 °C (30 min) | 1200 °C (20 min) | |||||||
| 0.5 °C min−1 | 5 °C min−1 | |||||||
| Lignin + epoxy resin (1:1 wt) |
Air | Ar | n/a | 5% Na alginate | 0.8 M NaPF6 EC/DME (1:1) |
82% | 316 (30 mA g−1) | 138 |
| 150 °C (24 h) | 1400 °C (1 h) | 161 (300 mA g−1) | ||||||
| 2 °C min−1 | ||||||||
| Lignin | Hydrothermal process + 3-aminophenol (11 wt%) 250 °C (12 h) | N2 | 727 (CO2) | 10% CB | 1 M NaClO4 EC/DEC (1:1) |
85% | ∼325 (50 mA g−1) ∼150 (250 mA g−1) | 122 |
| 1100 °C (2 h) | 151 (N2) | 2% CMC | ||||||
| 3% SBR | ||||||||
| Lignin | Hydrothermal treatment with GO and ethylene glycol 180 °C (20 h) | Ar | 94 | 15% CB | 1 M NaClO4 EC/DEC (1:1) |
n/a | ∼210 (200 mA g−1) | 131 |
| 750 °C (2 h) | 10% PVDF | |||||||
| Lignin sulfonate | Spray dried; Ar 500 °C (3 h) | Ar | 12 | 10% AB | 1 M NaClO4 EC/DEC (1:1) |
88.3% | 339 (0.1C) | 139 |
| 1.2 M HCl | 1300 °C (2 h) | 5% CMC | 187 (1C) | |||||
| 5% SBR | ||||||||
| Phenolic resin | Air | Ar | 47 | 5% Super P | 1 M NaPF6 EC/DEC (1:1) + 5% FEC |
76.4% | ∼315 (20 mA g−1) | 114 |
| 300 °C (3 h) | 1300 °C (4 h) | 5% PVDF | ∼75 (200 mA g−1) | |||||
| Alkali lignin + PVA | Electrospinning with KOH (5 wt%) | Ar | 93 | Free-standing | 1 M NaClO4 EC/DMC (1:1) + 5 wt% FEC |
65% | 137 (50 mA g−1) | 140 |
| 600 °C (1 h) | 87 (300 mA g−1) | |||||||
| Corn stalk | Purification (acetone); composite with amphiphilic carbonaceous material | N2 400 °C (1 h) | 5.3 | 10% super P | 1 M NaClO4 EC/DEC (1:1) |
82% | 297 (25 mA g−1) | 141 |
| H2/Ar 1300 °C (3 h) | 10% PVDF | ∼160 (200 mA g−1) | ||||||
| Lignin | Washing with KOH (20 wt%) and HCl (1 M) | N2 100 mL min−1 | 48 | 10% AB | 1 M NaClO4 EC/PC/DMC (9:9:2) |
69% | ∼270 (50 mA g−1) | 117 |
| 1300 °C (6 h) | 20% PVDF | ∼175 (250 mA g−1) | ||||||
| 5 °C min−1 | ||||||||
| Lignin sulphonates | Ar | Ar | 5.6 (N2) | 10% carbon SP | 1 M NaClO4 EC/DMC | 79% | 270 (25 mA g−1) | 115 |
| 600 °C (1 h) | 1200 °C (1 h) | 377 (CO2) | 10% CMC | |||||
| Water washing | 5 °C min−1 | |||||||
| Scrap wood | Washing with 1 M H2SO4 | Ar 20 mL min−1 | 30 | 10% CMC | 1 M NaPF6 EM/DEC (1:1) |
86% | 270 (30 mA g−1) | 116 |
| 1000 °C (6 h) | ||||||||
| 13 °C min−1 | ||||||||
| Pitch + lignin | — | Ar | 1.3 | 5% Na alginate | 0.6 M NaPF6 EC/DMC (1:1) |
82% | 254 (0.1C) | 142 |
| 1400 °C (2 h) | ||||||||
| Lignin | Washing with HCl + formaldehyde | N2 | 16 | 10% Super P | 1 M NaClO4 EC/DEC (1:1) 5% FEC |
74% | 325 (25 mA g−1) 140 (250 mA g−1) | 143 |
| 2 °C min−1 400 °C (1 h) | 10% PVDF | |||||||
| 5 °C min−1 1300 °C (1 h) | ||||||||
| Lignin + PAN (5:5) |
Air | N2 | 26.6 | Free standing | 1 M NaClO4 EC/DEC (1:1) |
70.5% | 296 (20 mA g−1) 80 (1 A g−1) | 144 |
| 400 °C (1 h) | 1300 °C (0.5 h) | |||||||
| Lignin from corn stalks | Extraction with acetone (purification) | N2 | 14 | 10% Super P | 1 M NaClO4 EC/DEC (1:1) |
79% | ∼300 (50) | 145 |
| +20% (NH4)2HPO4 | (1) 400 °C (1 h) | 10% PVDF | ∼160 (50) | |||||
| (2) 1300 °C (2 h) | ||||||||
| Alkali lignin derived AZO polymer | Formation of composite with SiO2, later removed with HF | N2 | 449.7 | 10% CB | 1 M NaClO4 EC/DMC (1:1) |
50% | 190 (50 mA g−1) | 146 |
| 5 °C min−1 | 10% CMC | 161 (200 mA g−1) | ||||||
| 700 °C (4 h) | ||||||||
| Cocklebur fruit | Soaking in NH3·H2O | 1100 °C (3 h) | 64 | 10% super P | 1 M NaOTf DEGDME | 69% | 253 (50) | 121 |
| 10% PVDF | 106 (1 A g−1) | |||||||
| Lignin + 3-aminophenol formaldehyde resin (3:7) |
— | Ar | 18.4 | 10% GVXC-72 | 1 M NaClO4 EC/DEC (1:1) |
81% | 310.4 (25 mA g−1) | 123 |
| 1100 °C (2 h) | 5% CMC | 125 (200 mA g−1) | ||||||
| Alkaline lignin + melamine + urea (1:1:5) |
Formation of composite | N2 | n/a | 10% Ketjen black | 1 M NaPF6 EC/DEC (1:1) |
26% | 247 (30 mA g−1) | 125 |
| 5 °C min−1 | 10% CB | 167.1 (300 mA g−1) | ||||||
| 400 °C (2 h) | 10% PTFE | |||||||
| 800 °C (4 h) | ||||||||
| Enzymatic lignin | Water washing | Ar | 4 | 1 M NaPF6 DEGDME | 74.4% | 303 mA g−1 (100 mA g−1) | 147 | |
| 5 °C min−1 | ∼260 (400 mA g−1) | |||||||
| 1600 °C (2 h) |
Biomass-derived hard carbons have also been applied as potassium-ion battery (KIB) anodes.126–132 The K+ storage mechanism is based on adsorption of ions onto the surface of graphene sheets and the intercalation of ions in graphitic regions133 similar to the Na-ion case. In KIBs, the mechanism originates a galvanostatic charge/discharge profile characterized by two different regions: a high-inclination slope at higher potentials (from 1.2 to 0.4 V vs. K/K+) and a low-inclination slope below <0.4 V. Despite the low first cycle coulombic efficiency (<50%), recent progress led to lignin-derived HC with capacities as high as 355 mA h g−1 at low current rates.131 Wu et al. compared lignin with different molecular weights, reaching 300 mA h g−1 at 50 mA g−1 for their medium weight precursor (9660 g mol−1). Liu et al.132 produced a HC from maple leaves and further treated it with concentrated HNO3 for chemical activation and doping with N and O. Their product exhibited good cyclability, with a capacity of 141.9 mA h g−1 at 1 A g−1 in the 1000th cycle. The development of similar hard carbons from lignin and other bio sources could be a key to boost K-ion technology to a competitive standard.
Despite the numerous examples of high capacity and good cyclability of carbons from lignin as anodes in metal-ion batteries, proper control of carbon structure and porosity is still a challenge. Different bio sources can be used to obtain lignin as a hard carbon precursor.134 The characteristics of the precursor lignin depend both on its source and on the separation/extraction process. Composition, amount of impurities, molecular weight, polydispersity and solubility are some of the characteristics that can change drastically with the extraction method.135 On their turn, they will affect the carbons’ morphology, porous structure, surface composition and hence the final performance in a battery. Unfortunately, a thorough characterization of the precursor lignin is not always shown in the literature, making it hard to draw trends correlating lignin features with the derived carbon properties. Adding to the heterogeneity of lignin precursors is the variety of possible modification steps and the carbonization process itself, rendering a multitude of variables to be considered in the transformation of lignin into metal-ion battery anodes. Working with so many variables, although a challenge, is also encouraging due the great potential to achieve better materials.
SIBs, similar to LIBs, can also work with alloying anodes, where a reversible reaction takes place by alloying and de-alloying Na in the negative electrode instead of adsorption and intercalation of Na+. Despite the high capacities delivered by alloying, it is accompanied by drastic volume changes that can degrade the electrode in just a few cycles. To achieve good cyclability with a Sn anode (91% of capacity retention after 1000 cycles) Wang et al. employed lignin as a precursor to form Sn/C particles.136 Similar approaches could be investigated to enable different alloying metal anodes for high energy density SIBs and KIBs in the future.
2.2. Lignin derived cathode materials
2.3. Lignin based binders
The primary role of binders (typically 2–5 wt% of the electrode) is to ensure an excellent mechanical adhesion and stability of the electrode active materials by maintaining uniformity and intimate contact between active material constituents, conductive additives and the current collector (electrode coating). These features render binders critical to battery safety, cycle life and capacity retention. Traditional binders such as polyvinylidene fluoride (PVDF) require expensive and toxic organic solvents (e.g., N-methyl-2-pyrrolidinone and acetonitrile), necessitating demands for greener, sustainable and less expensive alternatives.81,161,162 In this regard, binders that are dispersible in water (e.g., poly(tetrafluoroethylene, tapioca starch and carboxymethyl cellulose) have been developed recently.81,161,162 Lignin is a good binder material as it satisfies all the above binder requirements in addition to eco-friendliness, abundance and good thermal stability. In particular, the ample functional groups of lignin make it easy for surface modification and functional tailoring of its properties. Moreover, its highly cross-linked structure is suitable for achieving mechanically robust adhesion while the polar functional groups render it hydrophilic, making it processable in aqueous solvents which is suitable for green battery production. Cheng's group163 for example demonstrated that lignin could act as a binder for Si NPs and be subsequently converted at 400–800 °C into a conductive ligament. This allowed for designing a mechanically robust Si/carbon composite electrode free of traditional binders with enhanced electrochemical stability for over 250 cycles compared to a PVDF binder. Luo et al.164 also developed a water-soluble binder consisting of sodium polyacrylate grafted lignin for micro silicon anodes via free radical graft copolymerisation and alkaline hydrolysis. The anode exhibited high performance stability against the high volumetric changes of Si for over 100 cycles compared to that using a CMC binder. Lignin binders with additional capabilities for specific functions have also been explored for LSBs, including a sodium lignosulfonate salts-based binder containing negatively charged sulfonate groups that are beneficial for suppression of lithium polysulfide diffusion, thus, allowing stable cycling performance for 100 cycles at 0.2 C. The rich phenolic groups in lignin have also been exploited as a free radical scavenger in LiNi0.5Mn1.5O4 cathode based high voltage LIB which suppressed the free radical chain reaction, allowing for the generation of a compatible multi-dimensional interphase between the electrolyte and electrode. This enabled the electrode to maintain high stability with high capacity retention (94.1%) after 1000 cycles compared to that of a PVDF (46.2%).165 Although the study of lignin based binders is still at the infancy stage, a critical issue to be resolved is that by thermally converting the lignin binder into an active carbon ligament within the electrode, the robustness may be severely diminished during the charge/discharge due to repeated expansion/contraction and structural changes, however minimal, which can compromise the mechanical functions of the binder in the electrode and lead to capacity decay and poor cell performance. The use of unconverted lignin extracts as binders could also pose the challenge of low conductivity and dead mass in addition to increased parasitic reactions by the surface functional groups or the added structure-improving polymers and lead to rapid electrolyte decomposition and consumption which are undesirable in practical batteries. Therefore, detailed electrochemical studies of the lignin binder redox behaviour and stability mechanism with respect to the electrode structure and cycling characteristics is needed. Furthermore, all conventional binders do not always function adequately with all kinds of electrode materials in different cell types, thus, it will be useful to undertake rigorous electrochemical screening to identify the most suitable combinations and cycling conditions for their application in various battery systems.2.4. Integration of lignin in electrolytes and separators
The leakage, gas evolution, flammability and toxicity of the organic liquid electrolytes are serious safety concerns in the operation of batteries. Such issues are limited to a large extinct in gel polymer electrolytes (GPEs) although hazardous polymers are still used.166 The demand for “green batteries” requires the exploration of natural biodegradable polymers for GPEs. While lignin as a natural material cannot be used in batteries as an electrolyte, its porous derivates (composite, films, membranes, nanofiber mats, etc.) can be utilized for preparing durable, flexible, and resilient GPEs. These lignin-derived porous structures serve as mechanical supports that can absorb a particular activating agent (liquid electrolytes) and serve as electrolytes in rechargeable batteries. Water-soluble lignin has been thoroughly mixed with poly(N-vinylimidazole)-co-poly(poly(ethylene glycol) methyl ether methacrylate) copolymer (weight ratio of 2:1) for three days to develop a lignin-based thin film. An organic liquid electrolyte was used for activating the lignin-based film before its use as a Li-ion battery electrode. This synthesized GPE suppressed Li dendrite formation in a Li metal anode and maintained adequate ionic conductivity (6.3 × 10−4 S cm−1). Moreover, this arrangement resulted in a stable performance (∼150 mA h g−1) for over 450 cycles at 1C in a Li/LiFePO4 cell.166 The acidity of fumed silica in the GPE was reduced by blending with lignin to develop a GPE for rechargeable aqueous Zn/LiMn2O4 batteries, that exhibited improved stability of electrochemical properties.166 The acidity of fumed silica in the GPE was reduced by blending with lignin to develop a GPE for rechargeable aqueous Zn/LiMn2O4 batteries, that exhibited improved stability of electrochemical properties.
Lignin also exhibits potential for use in the development of solid polymer electrolytes (SPEs). Liu et al. successfully developed lignin derivatives for use as a SPE in Li–metal batteries. Softwood kraft lignin was dissolved in organic solvent (dimethylformamide (DMF)) and its chemical structure was modified into abundant alkene group via esterification reactions with N,N′ dicyclohexylcarbodiimide, and 4-(dimethylamino)pyridine. Lignin-based SPE was formed by graft-copolymerisation of poly(ethylene glycol)(PEG) with lignin–alkene via a photoredox thiolene reaction (i.e., grafting and crosslinking reaction procedures). Newly synthesized lignin-graft-PEG combines the mechanical and thermal properties of lignin with the flexibility and superior ionic conductivity of PEG.167 The solid electrolytes served as a binder and an ion conductor with the capability of suppressing dendrite formation. While all these findings show high promise for lignin, the requirement of synthetic polymer blends dilutes the eco-friendliness and performance of the lignin-based battery component. Nonetheless the results in the literature to date show that lignin is a promising material for battery electrolyte development.
Meanwhile, the safety of rechargeable batteries hinges on the separator, which is typically a polymeric membrane that must be chemically and electrochemically stable and compatible with the electrolyte and electrode materials and mechanically robust enough to endure any stress that occurs during battery assembly.168 Similar to binders, the structure and properties of the separator significantly affect the energy and power density, safety and cycle life of the battery. Traditional separators are made of polyolefins (polypropylene, polyethylene, polyisobutylene and polymethylpentene) with semi-crystalline structures.169 The polyethylene backbone is often grafted with micro-porous poly(methyl methacrylate) or siloxane to improve performance. Lignin based separators combine high performance with abundance and environmental benignity. For example, the introduction of lignin in organic polymers such as poly(vinyl alcohol) or polyacrylonitrile (PAN) in separators resulted in improved porosity, high electrolyte wettability, improved thermal stability and enhanced ion transport. Zhao et al. employed electrospinning of lignin/polyacrylonitrile solution to prepare a composite fibre-based nonwoven separator. Various amounts of lignin were dispersed in organic aqueous mixtures (PAN–DMF) under continuous mechanical stirring to prepare lignin/PAN solutions. The synthesized separator resulted a remarkably stable cell performance of a Li/LiFePO4 cell with a capacity of ≈148.9 mA h g−1 after 50 cycles at 0.2 °C for the lignin–PAN (3:7 wt/wt) compared to commercial Celgard-2400 (≈132 mA h g−1) at the same rate.170 While these preliminary studies show high promise for lignin derived GPE and solid-state electrolytes, their performance is still limited and far from meeting practical application requirements. Thus, further improvements are needed to enhance the conductivity and compatibility with other cell components. The literatures surveyed suggest that most studies are conducted at room temperature. This should be expanded to at least the −20 °C to 60 °C range with further analysis of the impact of structural properties on the viscosity, decomposition and conductivity. The separator also needs to be designed with an adequate balance of the required properties such as conductivity, wettability, and porosity, and sufficiently thin with excellent mechanical robustness. Thus, it is recommended to improve the mechanical and electrical properties of both separator and electrolyte to a level equivalent and/or exceeding the state of the art to become practically viable. This will also require advanced characterization tools for operando and in situ analysis in both half and full cell testing and analysis under practical conditions.
2.5. Redox flow batteries
Research to date has shown that lignin, a cheap, abundant and renewable material, can be applied to all of the main components of a redox-flow battery (RFB). Carbon fibres produced by electrospinning of lignin have been employed as electrodes in a vanadium RFB. Unlike in metal-ion batteries where the electrode material is electrochemically active, electrodes in redox-flow systems simply have the purpose of supplying active sites for the redox reaction of electroactive species in the electrolytes. Most electrode materials in these systems are carbon cloths, felts, or papers from non-renewable sources,171 so producing carbon from biomass such as lignin is a positive step towards sustainable redox-flow batteries. Ribadeneyra et al. produced carbon fibres from electrospinning a lignin solution.172 After a two-step carbonisation, the high surface area electrodes showed inferior performance to carbon paper in a full cell, but good activity in a vanadium RFB half-cell. The successful use of the electrospun carbon fibres from lignin could be a matter of optimizing electrode processing and more research is necessary.171The beneficial properties of lignin have also been exploited in the development of RFB ion exchange membranes. To separate the anolyte and catholyte in vanadium RFBs, lignin composite membranes with a sulfonated poly(ether ether ketone)173 or Nafion polymers174 were designed by Ye and co-workers. The latter composite membrane presented better performance with higher coulombic efficiency and capacity retention (97.4% and 52.8%, respectively) than the pure Nafion membranes (90% and 34.8%, respectively). Besides the promising application of lignin in RFB electrodes and membranes, the biopolymer has also been investigated as source material for redox-active organic molecules in RFBs. Commonly used active species include expensive metals such as vanadium and chromium, but organic molecules could be a viable option. Mukhopadhyay et al. used lignosulfonate directly derived from lignin as anolyte,175 showing that lignin is not only a viable material to produce electrode materials and separators, but also electrolytes. Redox flow batteries are a promising technology gaining space in the market, but the use of lignin as a precursor or directly as a component is still incipient.
3. Lignin for energy applications – supercapacitors
Commercial electrical double layer capacitors (EDLCs), also known as supercapacitors, store charge using two symmetrical porous activated carbon electrodes in an organic electrolyte. The electrode makes up 28% of the cost of commercial supercapacitors, with 39% of that attributed to the cost of activated carbon (10–20 EUR kg−1) and 59% to the cost of the current collector (foil).176 Thus, there is significant scope to replace the activated carbon with carbon materials synthesised from lignin, due to lignin's high carbon content (∼60%) and lower cost. The 3D polymeric nature of lignin also provides unique advantages over traditional activated carbon to enhance performance and reduce costs. It can be templated, form hydrogels and aerogels, combined with pseudocapacitive materials and spun into free-standing flexible fibre electrodes.177–182 The most common methods used to produce lignin-based carbon electrodes for supercapacitor applications are shown in Fig. 4. The free-standing electrodes have the potential to remove the need for a current collector, simplifying the cell and further reducing environmental and commercial costs for supercapacitors.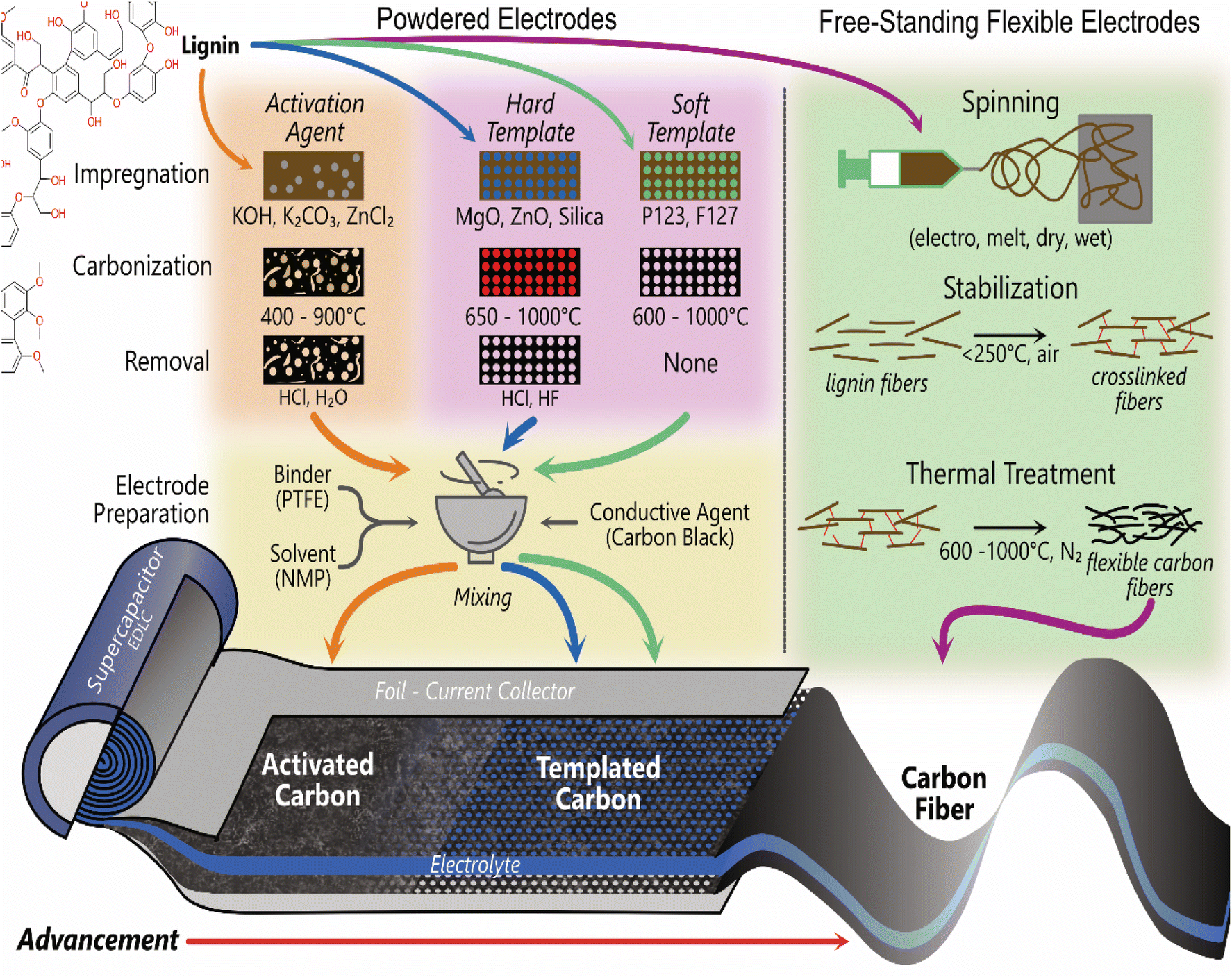 | ||
| Fig. 4 Main methods used to produce carbon electrodes from lignin for supercapacitors. | ||
3.1. Activated carbon lignin electrodes for EDLCs
Activated carbons are highly porous materials with a high specific surface area (SSA, >1500 m2 g−1), reasonable electrical conductivity (0.39 cm−1 for Norit Super 30 and 0.74 S cm−1 for Kansai Maxsorb),183 and low cost compared to other supercapacitor materials (e.g., graphene RuO2). The high SSA of these materials is critical for energy storage in EDLCs, as capacitance is governed by the degree of surface area electrostatically interacting with the electrolyte. Early efforts to generate activated carbon from lignin started in the 1970s for adsorption applications,184 but it was not until the 2010s when lignin based supercapacitors appeared.185 These initial adsorption studies demonstrated that temperature, residence time, activation agent and ratio, as well as the type of lignin all influence the properties of the final activated carbon product.184 Applying this knowledge to supercapacitors, Jianhui et al. demonstrated that the capacitance of KOH activated lignin (900 °C, 1.5 h) could be improved from 196.1 to 267.8 F g−1 (0.05 A g−1, 6 M KOH/LiOH, 1 V window) by adjusting the KOH:lignin ratio from 1:1 to 3:1. This was attributed to the increase in SSA from 718 to 1506 m2 g−1.
The different physicochemical properties of various lignin derivatives resulting from their diverse extraction and growing conditions is a major issue for commercializing lignin. Li et al. examined the EDLC performance of activated carbon from extracted lignin from pine (softwood) and poplar (hardwood). The pine lignin exhibited a specific capacitance of 48.3 F g−1, while poplar lignin was 86.7 F g−1 (0.5 A g−1, 1 M H2SO4, 0.6 V window). The higher performance of poplar lignin was attributed to its higher SSA (621 vs. 314 m2 g−1) after activation resulting from the difference in G and S units between the pine (softwood, only G units) and popular (hardwood, G and S units). It should be noted that the activation temperature in this study (700 °C) is lower than what is commonly used for KOH activation (800–900 °C) and may have resulted in their lower performance. Similarly, Du et al. compared the storage capacity of carbon derivated from hardwood (poplar), softwood (pine) and grass (corn stover). Poplar based carbon had an increased surface area of 1062.5 m2 g−1 and specific capacitance of 349.2 F g−1 (0.1 A g−1, 6 M KOH, 1 V window). Du attributed the increased performances to the increased amount of S units and larger molecular weight.186
Pursuing the highest surface area was the goal of many studies to increase the capacitance of lignin-based supercapacitors. Yu et al. demonstrated that incorporating a pre-pyrolysis step (600 °C, 1 h, N2) before activation (3:1 KOH, 800 °C, 2 h, N2) increased the gravimetric capacitance from 218 F g−1 to 312 F g−1 (0.05 A g−1, 6 M KOH, 1 V window).187 This was attributed to the increase in the Vmicro/Vtotal ratio from 22% to 66%. Hydrothermal carbonisation can also be used as a pre-carbonisation step (Fig. 5a).188,189 Guo et al. hydrothermally carbonized enzymatic hydrolysis lignin at 180 °C in 5% wt H2SO4 for 18 hours before activating the resultant carbon product with KOH at different ratios (800 °C, 3 h, N2).189 They achieved a specific capacitance of 420 F g−1 in 6 M KOH (0.1 A g−1, 1 V window) and 218 F g−1 in neat EMIM TFSI (1 A g−1, 2.5 V window). The high performance of their carbon was attributed to the combination of micro (0.66 cm3 g−1) and mesoporosity (0.10 cm3 g−1) formed in the two-step process and not just the high SSA (1660 m2 g−1). Thus, to achieve the highest surface areas and pore distributions, most studies have adopted a two-step activation method with a pre-carbonisation step.189–193
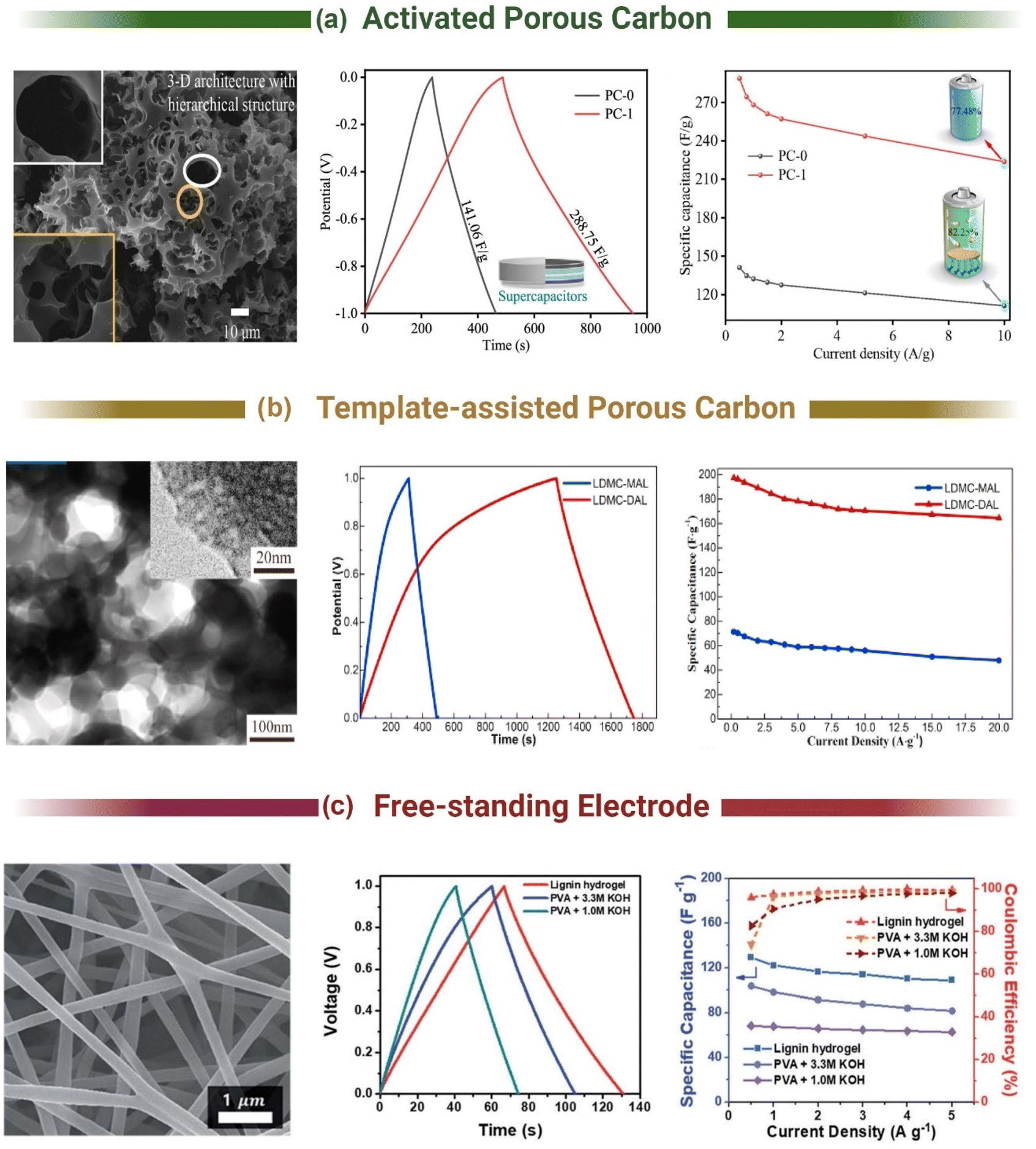 | ||
| Fig. 5 Morphology and electrochemical performance of Lignin-derived carbon materials for supercapacitor applications using (a) activated porous carbon. Adapted with permission from ref. 188. Copyright 2021, American Chemical Society (b) template-assisted porous carbon. Adapted with permission from ref. 213. Copyright 2021, Elsevier and (c) flexible free-standing conductive carbon electrodes. Adapted with permission from ref. 217. Copyright 2019, Royal Society of Chemistry. | ||
One of the issues with activated carbon production is the use of highly corrosive (e.g., KOH,185,187,190–199 NaOH,196,200 H3PO4201) or toxic (ZnCl2202) activation agents for enhancing the surface area and porosity in lignin. Fortunately, efforts are being made to shift towards greener activation agents and methods. For example, Schneidermann et al. demonstrated that ball milling lignin with K2CO3 and urea before thermal treatment at 800 °C achieved a SSA of 3041 m2 g−1. This material had a specific capacitance of 177, 147, 192 F g−1 in 1 M Li2SO4, 1 M TEA–BF4 and EMIM–BF4 at 0.1 A g−1. Alternatively, Jeon et al. applied a different approach examining the self-activation of lignin by thermal treatment without activation agent. A SSA of 1092 m2 g−1 was achieved on a purified Kraft lignin attaining a capacitance of 91 F g−1 (0.5 A g−1, 1 M H2SO4, 1 V window). Liu et al. achieved higher capacitance values by freeze drying alkali lignin before the carbonisation step, negating the need for an activation agent. The resultant carbon had a SSA of 854.7 m2 g−1 and a specific capacitance of 281 F g−1 (0.5 A g−1, 1 M H2SO4, 1 V window). These studies reveal that greener activated carbons can be created with lignin using more sustainable activation agents or agent-free methods.
Increasing the surface area of activated carbons has been the main method of enhancing EDLC performance. Recently, it has become evident that pore size and carbon nanostructure are more crucial in increasing specific capacitance than raw surface area. This can be seen when comparing the results in the previous examples where the relationship between SSA and performance is not linear.185,187,189 As it is difficult to tailor activated carbons towards specific porous structures, researchers have attempted to create tailored carbon materials with specific nanostructures to enhance the energy density of lignin-based supercapacitors.
3.2. Templated porous carbon materials from lignin
Template methods using soft or hard templates (porogens) allow the creation of highly tailorable pore size distributions in carbon materials. Hard templating consists of impregnating inorganic compounds into the lignin structure, such as MgO,203 ZnO,204 silica,205,206 and zeolites,207,208 which are removed after thermal treatment to leave pores in the carbon matrix. For instance, Tain et al. used sodium lignosulfate and tetraethyl orthosilicate as a silica template to create a mesoporous sulphur doped carbon structure. The silica template was removed with 40% HF, resulting in a SSA of 1054 m2 g−1 and an average pore diameter of 6.69 nm. This material achieved a capacitance of 241 F g−1 (1 A g−1, 6 M KOH, 1 V window).209 The use of toxic or corrosive compounds (e.g., HF206,209) is a problem in hard templating and undermines the environmental benefit of using lignin. To address this, Fu et al. combined sodium lignosulfonate, Zn(NO3)2·6H2O, Na2C2O4 and ethanol to form a self-assembled lignin/ZnC2O4 composite. This composite was carbonized at 650 °C creating a lignin/ZnO hybrid with porosity formed by removing the nanosized ZnO template with 1 M HCl. The removal of ZnO created a mesoporous material (12.9% Vmicro/87.1% Vmeso) with a specific capacitance of 274 F g−1 at 0.5 A g−1 and 200 F g−1 at 20 A g−1 in 6 M KOH (1 V window). Although they avoided using toxic or corrosive activation agents to create porosity, the removal of ZnO with HCl is likely to create aqueous ZnCl2, which is toxic to aquatic and plant life.210–212Soft templating methods using polymers and surfactants solve the template removal issue as the template is consumed during the thermal treatment step. For example, Herou et al. demonstrated the green synthesis of mesoporous carbon from organosolv lignin using phloroglucinol, glyoxal and F127 surfactant. The phloroglucinol and the lignin self-assembled around F127 micelles with the glyoxal acting as a cross-linker to form an evaporation-induced self-assembled structure. Subsequent carbonisation at 900 °C resulted in a mesoporous structure with a SSA of 763 m2 g−1 and a specific capacitance of 90 F g−1 (0.1 A g−1, 6 M KOH, 1.2 V window).179 Unfortunately, the low SSA of soft templated carbons tend to hinder their performance in supercapacitors, which has been addressed by combining templating methods with either pre-treatment or post-treatment methods. Sima et al. performed a pre-treatment on lignin using a choline chloride/formic acid based deep eutectic salt before soft templating with F127 (Fig. 5b).213 The deep eutectic salt treatment created an ordered mesoporous carbon, increased the SSA by 56% to 1195 m2 g−1 and more than doubled the pore volume (0.7 cm3 g−1). This improved the specific capacitance from ∼72 F g−1 (0.2 A g−1, 6 M KOH, 1.0 V) in the untreated sample to 197.32 F g−1 (0.2 A g−1, 6 M KOH, 1.0 V) for the pre-treated material. Post treatment of templated carbons typically involves employing activation agents to increase the microporosity in the generally mesoporous structure created from templating. For instance, Saha et al. combined hard wood Kraft lignin with Pluronic F127 and carbonized at 1000 °C to create a mesoporous carbon.214 This material exhibited a SSA of 185 m2 g−1, resulting in a specific capacitance of 77.1 F g−1 (1 mV s−1, 6 M KOH, 0.8 V window). To improve the performance, the templated carbon was activated with CO2 or KOH. This increased the specific capacitance to 91.7 F g−1 for CO2 activation and 102.3 F g−1 for KOH. Finally, a few studies also state a template-free method of creating porous carbon by utilizing a slurry of KOH/lignin before thermal treatment.199,215,216 However, this is more akin to the traditional activation method and still requires a post washing step to remove any residual materials.
Templating methods allow a high level of control over the porosity in carbon materials, allowing the formation of heretical porous carbons matched to the electrolyte. However, these materials tend to be powdered requiring them to be mixed with binders (PTFE), conductive agents (carbon black) and attached to a current collector/foil to form an electrode. These extra components increase the size of the device and are a source of resistive interfaces between the active material and the current collector. Replacing powdered materials with electrically conductive free-standing electrodes provides the opportunity to omit these components, reducing the complexity and increasing the volumetric capacity.
3.3. Flexible free-standing electrically conductive electrodes
Development of lignin based flexible free-standing electrodes is the next step in electrode design for supercapacitors. These electrodes are commonly synthesised from electrospinning,218–221 melt spinning,28,222,223 wet spinning,224,225 gel spinning,226 centrifugal spinning,227 or dry spinning228 the lignin into flexible carbon fibre mats (Fig. 6). In all these techniques, the precursor polymer is initially formed into fibres, followed by thermal treatment at high temperature (>600 °C) under inert atmospheres (N2, Ar). An additional stabilisation step is typically included between spinning and carbonisation to convert the lignin polymer into a thermoset polymer. During this stage, the fibres are heated at a very slow rate (<1 °C min−1) under an oxidative atmosphere until they reach 200 to 300 °C, where they are kept for several hours. Stabilisation is critical as it prevents morphological changes in the fibres when carbonized at high temperatures. Schlee et al. investigated the impact of stabilisation conditions on electrospun softwood and hardwood Kraft lignin.229 Stabilizing at 190 °C created almost no structural changes in the lignin fibres, resulting in the hardwood Kraft lignin melting under carbonisation at 800 °C. At 250 °C, both types of lignin stabilized without fusing with increases in stabilisation temperature leading to higher SSA (918 m2 g−1 at 340 °C). However, this also led to increased mass loss during stabilisation. At 250 °C, 79.1% of the hardwood Kraft lignin remained after stabilisation which dropped to 36.5% after stabilisation at 340 °C. The hardwood Kraft lignin achieved a maximum specific capacitance of 164 F g−1 (0.1 A g−1, 6 M KOH, 1.2 V) when stabilized at 310 °C, whereas the softwood Kraft lignin achieved 150 F g−1 (0.1 A g−1, 6 M KOH, 1.2 V) when stabilized at 340 °C. The differences here were associated with the different side-chain linkages, functional groups and molar mass of the softwood and hardwood Kraft lignins, which in turn influence the stabilisation and final porosity of the carbon fibres.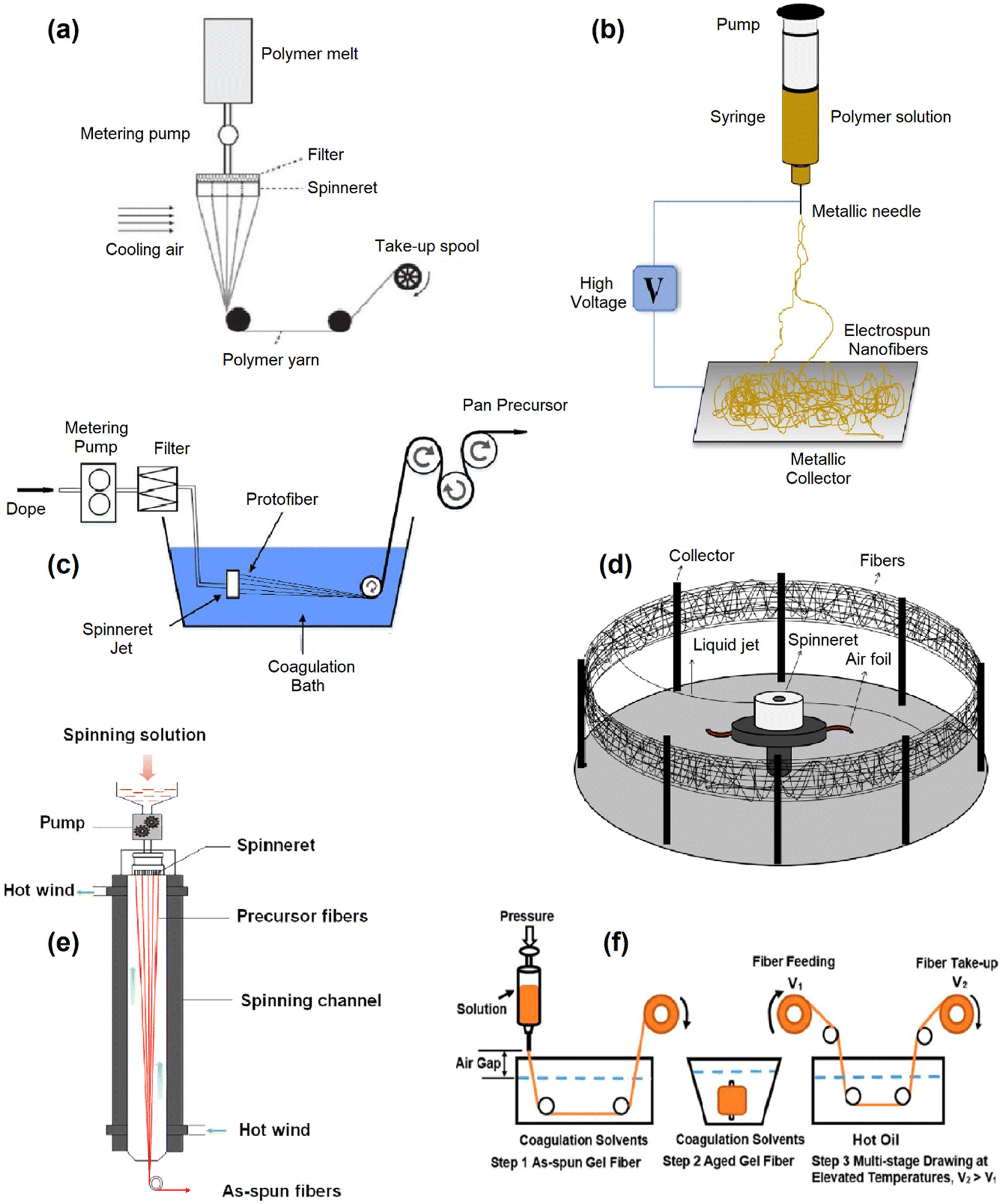 | ||
| Fig. 6 Schematic representation of (a) electrospinning. Adapted with permission from ref. 218. Copyright 2020, American Chemical Society (b) melt spinning. Adapted with permission from ref. 222. Copyright 2021, MDPI (c) centrifugal spinning. Adapted with permission from ref. 227. Copyright 2015, MDPI (d) wet spinning. Adapted with permission from ref. 238. Copyright 2020, Elsevier (e) gel spinning. Adapted with permission from ref. 226. Copyright 2017, American Chemical Society (f) dry spinning. Adapted with permission from ref. 239. Copyright 2020, Wiely. | ||
It is possible to spin pure lignin solutions into carbon fibres,230 but the low molecular weight of lignin makes this difficult. Thus, lignin is typically mixed with polymers, such as polyacrylonitrile (PAN),217,220,231–233 Polyethylene Oxide (PEO),177,178,234 and polyvinyl alcohol (PVA),235–237 to increase the spinnability and enhance the properties of the fibres. Wang et al. created binder-free electrodes by electrospinning PAN and enzymatic hydrolysis lignin at different ratios.220 The precursor mats were stabilized in air at 250 °C (1 °C min−1) and carbonized at 800 °C (10 °C min−1) under N2. They found that a ratio of 40:60 PAN to lignin lead to the highest SSA (675 m2 g−1), which resulted in a specific capacitance of 216.8 F g−1 (1 A g−1, 6 M KOH, 1 V window). However, an issue with lignin spun fibres is beading,235 where small beads of solution are deposited along the fibre length due to viscoelasticity of the lignin solution. Fang et al. addressed this by incorporating surfactants (0.2–1.2%) into the spun solution, which eliminated the beads and enhanced the orientation of the fibre microstructures. This in turn increased the specific capacitance from 66.3 to 80.7 F g−1 (1 A g−1, 6 M KOH, 1 V window). Additionally, the sporadic nature of carbon fibres forming under spinning techniques also leads to large voids between the fibres, reducing their volumetric energy density. Herou et al. addressed this issue by compacting organosolv electrospun lignin fibres with uniaxial compression, reducing the inner-fibre pore size from 1–5 μm to 0.2–0.5 μm. This environmentally friendly and straightforward step, applied prior to carbonisation, improved the volumetric capacitance from 20 to 130 F cm−3 (0.1 A g−1, 6 M KOH, 1.2 V window) outstripping the performance of most commercial and lab-scale porous carbons from bioresources (50–100 F cm−3, 1–3 W h L−1, using 10 mg cm−2). Even higher capacitances have also been achieved with spun lignin fibres (316 F g−1, 1 A g−1, 6 M KOH, 1 V window) although these were not free-standing electrodes.234
Park et al. took the concept of incorporating lignin into supercapacitors one step further by creating the electrolyte and electrode from lignin as shown in Fig. 5c.217 The electrodes were formed from electrospun Alkali lignin/PAN solutions that were stabilized at 250 °C before being carbonized at 900 °C. The electrolyte was a flexible hydrogel, created by adding Alkali lignin (33% w/v) to 3.3 M KOH and 1.2 mmol lignin poly(ethylene glycol) digycidyl ether. This unique combination resulted in a flexible supercapacitor with a specific capacitance of 129.23 F g−1 (0.5 A g−1, 1 V window), which was higher than when using a PVA/KOH gel electrolyte without lignin added (104.09 F g−1). The increase in performance for the lignin hydrogel electrolyte was attributed to its higher conductivity than the PVA/KOH electrolyte.
The use of fossil derived polymers (e.g., PAN, PEO, PVA,235 PMMA, PVP) in lignin carbon fibres remains a large problem for their sustainability. Inspired by trees, Cao et al. investigated the covalent bonding of lignin to cellulose acetate to simulate the linkage between cellulose and lignin in trees to create carbon fibres. The lignin and cellulose acetate were covalently bonded to lignin by introducing epichlorohydrin as a connecting agent. The fibres were thermally stabilized (0.4 °C min−1, 220 °C) and then carbonized at 600 °C under N2. These fibres achieved a maximum SSA of 1061.7 m2 g−1, pore volume of 0.57 cm3 g−1 and specific capacitance of 320.3 F g−1 (1 A g−1, 6 M KOH, 1 V window). When 10% epichlorohydrin was used. Plant proteins can also be used to replace the fossil derived polymers. Yang et al. blended lignin with different hordein/zein ratios to create electrospun carbon fibres.240 A ratio of 50/50 lignin to protein was found to be stable under carbonisation at 900 °C, forming a self-standing flexible carbon fibre mat that were subsequently activated with CO2 at 850 °C. These fibres delivered a specific capacitance of 360 F g−1 (1 A g−1, 6 M KOH, 1 V window). Interestingly, neither study used the carbon fibre mats as is, choosing to mix them with binders (polyvinylidene fluoride or polytetrafluoroethylene) and carbon black, suggesting that the electrode was not conductive enough or not stable as a free-standing mat. Regardless, these studies have demonstrated that it is possible to create carbon fibres without fossil fuel derived polymers and represent the next stage in green, renewable carbon fibres for supercapacitors. Table 3 summarizes the latest studies in the field of lignin-derived carbon materials for supercapacitors.
| Electrolytes | Material | Specific surface area (m2 g−1) | Cycling stability | Specific capacitance (F g−1) | Energy density (W h kg−1) | Ref. |
|---|---|---|---|---|---|---|
| 6 M KOH | LHC-3K | 1660 | 99% after 5000 cycles at 5 A g−1 | 420 at 0.1 A g−1 | 10 at 50 W kg−1 | 189 |
| 284 at 100 A g−1 | ||||||
| 6 M KOH | PLC-650-2 | 1069 | 93.5% after 10000 cycles at 5 A g−1 |
365 at 0.5 A g−1 | 9.75 at 6157.9 W kg−1 | 241 |
| 260 at 20 A g−1 | ||||||
| 6 M KOH | 3D-7-2K | 1504 | 99.7% after 5000 cycles at 5 A g−1 | 324 at 0.5 A g−1 | 17.9 at 458 W kg−1 | 242 |
| 249 at 50 A g−1 | ||||||
| 6 M KOH and EMIM BF4 | L-700 | 1269 | 91.6% after 10000 cycles at 5 A g−1 |
300.5 at 0.5 A g−1 | 8.5 at 100 W kg−1 | 243 |
| 6 M KOH | NSC-700 | 1199 | 95.0% after 3000 cycles at 10 A g−1 | 240.6 at 1 A g−1 | 27.2 at 10 kW kg−1 | 244 |
| 1 M H2SO4 | 3HPC/WO3 | 1305 | 86.6% after 10000 cycles at 10 A g−1 |
432 at 0.5 A g−1 | 34.3 at 237 W kg−1 | 245 |
| 214 at 20 A g−1 | ||||||
| 6 M KOH | AILCFN-3 | 736.14 | 84.7% after 3000 cycles 10 mA cm−2 | 278.9 at 0.14 A g−1 | 30.8 at 800 W kg−1 | 246 |
| 149.6 at 13.6 A g−1 | ||||||
| 6 M KOH | ARS/PGLS-1 | 1727.7 | 99.7% after 2000 cycles at 2 A g−1 | 469.5 at 0.5 A g−1 | 9.45 at 100.06 W kg−1 | 247 |
| 200.2 at 10.0 A g−1 | ||||||
| PVA/H2SO4 gel | sLIG-O/S14 | 181.37 | 81.3% after 8000 cycles at 50 mV s−1 | 53.2 mF cm−2 at 0.08 mA cm−2 | 0.45 mW h cm−3 at 1.6 mW cm−2 | 248 |
| 3.3 M KOH | ECNF | 1176.0 | 99% after 10000 cycles at 5 A g−1 |
129.23 at 0.5 A g−1 | 4.49 at 252 W kg−1 | 217 |
| 6 M KOH | E-CNFs | 2313 | 94.5% after 5000 cycles at 1 A g−1 | 320 at 1 A g−1 | 17.92 at 800 W kg−1 | 249 |
| 200.4 at 20 A g−1 | ||||||
| 1 M Na2SO4 | LCNFs-MSSL-180-3:7 |
1254.5 | 90.6% at 10 A g−1 | 533.7 at 0.5 A g−1 | 69.7 at 780 W kg−1 | 250 |
| 1 M H2SO4 | LCNFs/PPy | 872.60 | 77% after 1000 cycles at 4 A g−1 | 213.7 at 1 A g−1 | — | 251 |
4. Lignin based thermoelectric materials
Generating electricity from low-grade waste heat (from sources below 100 °C) is a key enabler to reduce greenhouse gas emissions. Sources of low-grade heat (<100 °C) are ubiquitous in many industrial processes, electronics data centres and biological processes (metabolism). Significantly, 70% of all energy generated daily is lost as waste heat. Therefore, the conversion of waste heat into useful energy using the thermoelectric effect represents a huge opportunity to decrease carbon footprint of our society. Traditionally, inorganic compounds such as: Bi2Te3, PbTe and SiGe have dominated the manufacturing of energy harvesting devices. However, serious drawbacks such as: toxicity, scarcity of raw materials and high cost have limited their application and pushed research to newer alternative materials which are highly abundant, low cost and non-toxic. Thus, there is no current technology capable to satisfy the efficiency and sustainability requirements for low-grade heat conversion. A thermoelectric generator consists in n-type and p-type semiconductors that are connected electrically in series and thermally in parallel, when heat is apply through the thermoelectric junction a current flow is generated due to the Seebeck effect (voltage generated due to a thermal gradient). Thermoelectric efficiency is measured by the dimensionless figure of merit ZT: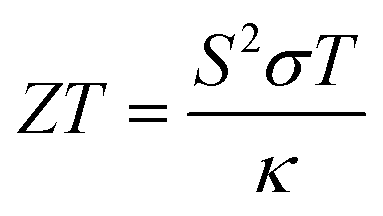 | (1) |
| PF = S2σ | (2) |
Looking at this scenario, lignin has an enormous potential for a promising sustainable source to produce thermoelectric materials due to its ideal molecular structure to produce carbon nanostructures with semiconducting properties. This is an emerging field of application for lignin and there are not many studies published until now. One study published by the University of Limerick259 showed enormous potential of lignin as precursor for carbon nanostructures with TE properties. Fig. 7 shows carbon nano fibres (CNFs) from lignin/PAN blends produced by electrospinning and their TE properties as a function of lignin content and processing conductions. The addition of lignin (up to 70%) reduces the diameter of CNFs from 450 nm to 250 nm, increases sample flexibility and promotes inter-fibre fusion. The results showed the possibility of a conversion of p-type to n-type semiconducting behaviour through doping with hydrazine vapour which allows the production of TEGs utilising both types of semiconductors based on lignin. CNFs depicted a maximum p-type power factor of 9.27 μW cm−1 K−2 for CNFs carbonised at 900 °C with 70% lignin which is a 34.5-fold increase to the CNFs with 0% lignin. For the hydrazine treated samples, the results showed a maximum n-type power factor of 10.2 μW cm−1 K−2 for the CNFs produced in the same way.
 | ||
| Fig. 7 (a) SEM images, (b) electrical conductivity and (c) Seebeck coefficient of lignin derived CNFs. Adapted with permission from ref. 256. Copyright 2019, Elsevier. | ||
Other strategy to use lignin as part of thermoelectric materials is lignin can be used as dopant for carbon-based nanostructures due to its aromatic chemical structure that can tailor thermoelectric properties of multi-walled carbon nanotubes (MWCNTs).260 This study shows how lignin can be valorised as a doping agent for TE devices resulting in outstanding performance levels outperforming fossil equivalents. The addition of lignin to Carbon Nanotube Yarns (CNTYs) improves their TE performance by one order of magnitude, showing for the first time that lignin can influence the transport properties of TE materials such as carbon nanotubes. In this case lignin increases electrical conductivity and Seebeck coefficients simultaneously (Fig. 8), which is considered the “Holy Grail” of TE materials. In addition, these materials show the possibility to manufacture TE generators with an outstanding power of 3.5 μW representing one of the highest values reported in literature for fully organic TE generators.
 | ||
| Fig. 8 SEM images of (a) pristine CNTY and (b) CNTY/lignin nanocomposite doped with 34 wt% lignin; schematic diagrams of (c) densified MWCNT fibre microstructure by incorporating lignin, (e) charge carrier filtering mechanism by introducing lignin; thermoelectric properties of CNTY/lignin nanocomposites with varying dopant levels: (d) electrical conductivity, (f) Seebeck coefficient and power factor. Adapted with permission from ref. 260. Copyright 2020, Wiley. | ||
Another way to use lignin for thermal energy harvesting is the use of lignin as a photothermal material which can be combined with conventional TEGs to produce energy due to the temperature gradient generated due to light absorption of lignin.261 This study demonstrated that lignin nanoparticles (L-NPs) can carry out photothermal conversion, which was attributed to π–π stacking of lignin molecules L-NPs showed a stable photothermal effect (22%). L-NPs were deposited on top of the TEGs as heat source generating around 0.12 V under irradiation of 100 mW cm−2. Other studies have shown this strategy for photoresponsive actuator devices where lignin is blended with castor oil-derived polyamide elastomers to develop the photoactive part.262
This particular valorisation route has not many limitations in terms of lignin structure, morphology and source. In principle, the presence of hydroxyl groups in the vast majority of lignins, facilitates the functionalisation for a targeted doping in organic based thermoelectric materials. In addition, for the case of carbon-based semiconducting nanostructures derived from lignin, these hydroxyls groups can be used for crosslinking points to improve the carbon phase (better electric transport properties) generated by the aromatic ring condensation during the carbonisation process. Therefore, different types of lignin can be easily adapted to meet the requirements in the thermoelectric field.
5. Lignin for energy applications – biofuels
Finding alternative energy sources from renewable feedstocks is also a highly pursued objective. Lignin valorisation in this field is an interesting option due to its high energy density, related to its structure and the presence of aromatic units, outputting numerous patents (70 patents filed with the World Intellectual Property Organization for the last 3 years). Lignin based biofuels could be classified as (i) solid lignin-derived chars (ii) liquid biofuels and (iii) hydrogen. Lignin conversion to fuels is mainly addressed either through depolymerization or gasification approaches. In particular, lignin derived liquid fuels could be accessed via depolymerization, hydrogenation and chemical upgrading steps. Besides, synthetic alcohols and Fischer–Tropsch liquid fuels could be obtained from syngas through gasification strategies.Nowadays, most approaches for lignin utilisation are focused on its combustion, with a low energy efficiency. Hence, integrated schemes, involving depolymerisation towards aromatics and the further gasification of the remaining solids offer promising options which could potentially give rise to liquid, solid and gaseous fuels (Fig. 9). Several strategies have been developed to accomplish the catalytic hydrodeoxygenation of lignin and its derivatives, towards hydrocarbon (cyclohexanes and arenes depending on the catalytic approach) liquid fuels. Such hydrocarbons may also be catalytically transformed into syngas and hydrogen. For example, pure hydrogen may be obtained by conversion of syngas via water-shift reaction and subsequent gas separation.263,264
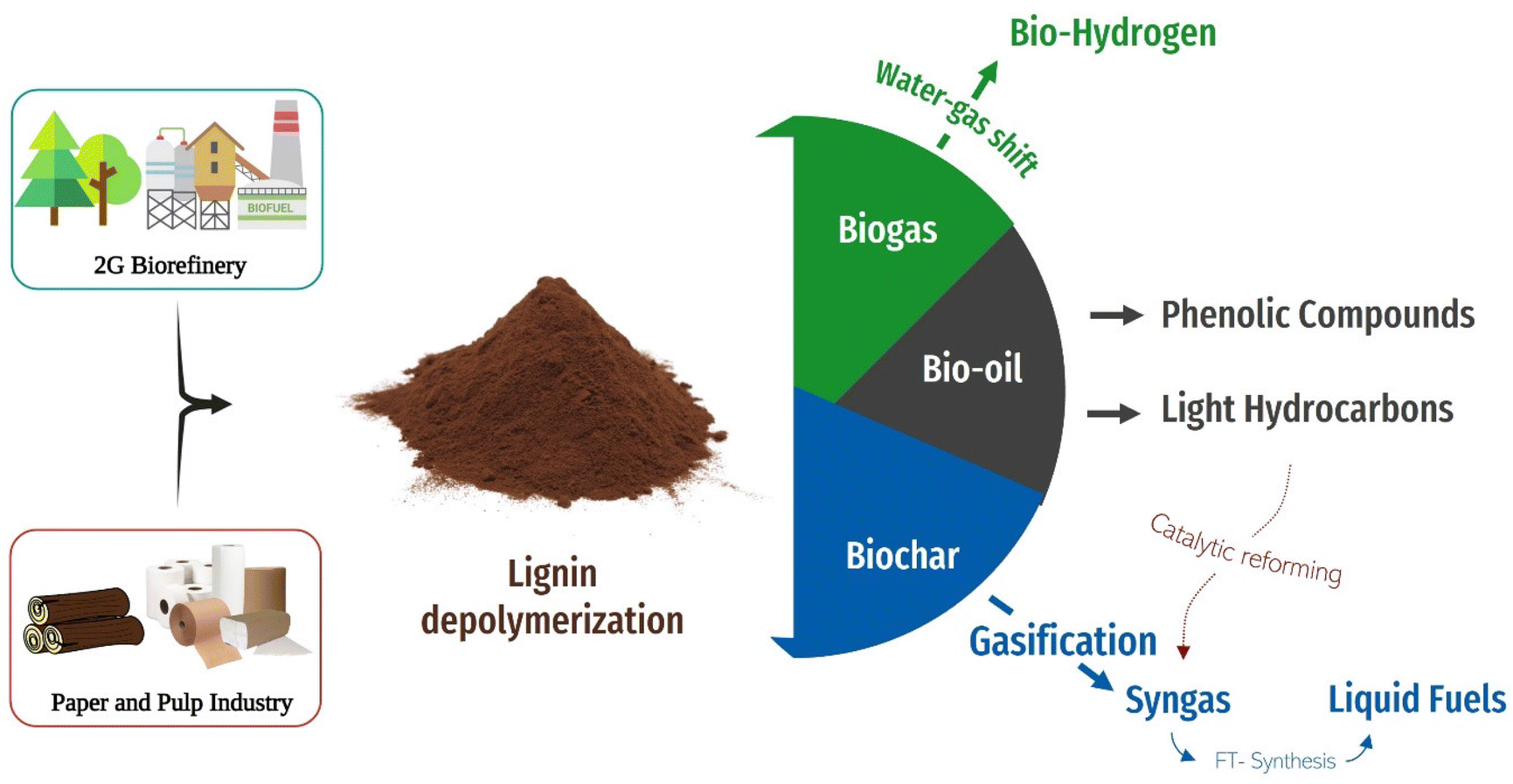 | ||
| Fig. 9 Schematic representation of the processes involved in the upgrading of lignin to fuels. | ||
However, due to (i) the inherent complexity of the lignin structure with the presence of a wide distribution of bond types, including C–C and C–O of different strengths as mentioned earlier in this review, (ii) the heterogeneity of the lignin derived products from depolymerisation and (iii) the trend of the obtained low-weight compounds for recondensation reactions and catalysts poisoning, lignin conversion into fuels still represents a challenging research field.
A myriad of studies have been focused on the depolymerisation of lignin and further hydrodeoxygenation into hydrocarbons, passing by thermo-, photo-, electro and bio-catalytic routes, all of which consider lignin properties, reactivity and catalysts features.263,265–268 Regarding bio-catalytic approaches, inspired by nature, enzymatic systems such as peroxidases and laccases has been investigated for lignin depolymerisation under mild conditions leading to the production of aromatics with low-molecular weight. Nonetheless, the employment of such biomimetic strategies also requires the reduction of the use of enzyme cofactors, whose price could affect the cost-efficiency of the overall process. Also, the natural recalcitrance of lignin hinders the efficiency of biomimetic degradation by providing an hydrophobic surface limiting the biotic and abiotic stresses.269 Thermal strategies, through pyrolysis or its combination with hydrodeoxygenation catalysis towards fuels have also widely explored to overcome these challenges.
Even if not a trivial task, some recent studies have attempted to investigate the influence of lignin properties on the performance and quality of biofuels. For instance, Rodríguez-Soalleiro and co-workers have gotten insights into the influence of the physico-chemical features of lignin from different biomass wastes on the quality of biofuel pellets, noting that higher chlorine and ash contents have a detrimental effect not only on the heating value and energy performance but also on the environmental impact. Besides the relationship biomass composition-pellets quality, some correlations were also performed between the microstructure and distribution of lignin in the pellets and their efficiency.270 The origin of the recalcitrance to enzymatic catalysis are believed to be partially linked to the S/G ratio of the biopolymer through carbon–carbon bonding between the lignin units.269
The design of efficient catalytic systems, considering metal entities and supports, has attained the attention of the scientific community, looking to overcome handicaps related to the material deactivation for coke formation or metal sintering. In this regard, transition metals have been widely investigated, either in one-pot or two-steps approaches involving lignin hydrogenolysis and hydrodeoxygenation reactions. For instance, lignin derived from corn wastes have been treated employing Ru nanoparticles supported over alumina and HY zeolite for the preparation of hydrocarbons, which could be employed as jet fuels. In particular, acid sites of HY zeolite played a crucial role on the depolymerisation of lignin via ether bond cleavage, while the Ru/Al2O3 catalyst promoted the HDO conversion into hydrocarbons. Following the aforementioned strategies, hydrocarbons were obtained with ca. 22% yield, from which alkylcyclohexanes represent a 90%.271 Based on such results, a bifunctional catalyst was designed by supporting ruthenium nanoparticles on HY zeolite. Interestingly, the combination of noble metal nanoparticles and acid zeolite supports exhibited a synergistic effect on the HDO reaction of softwood derived lignin, leading to slightly higher yields of hydrocarbons (26–32 wt%).272 Furthermore, similar catalytic systems, for instance based on Ru/Nb2O5 and Ru/Nb2O5 supported on silica, have been used for the hydrodeoxygenation reaction of birch-derived lignin for the production of hydrocarbons (C7–C9), achieving yields up to ca. 36% with high selectivity (71%) towards arene derivatives.273 Besides the aforementioned ruthenium systems, Ni-based catalytic materials on silica/alumina supports have been also reported for the production of hydrocarbons (C3–C17) from cellulolytic enzyme lignin.263 Moreover, bimetallic materials, based for instance on Ni–Mo or Co–Mo supported on alumina, have been used considering as well the needs to remove heteroatoms, such as sulphur and nitrogen. In this sense, the desired goal is to move toward non-noble metal-based materials to ensure the cost-efficiency of the protocol.
Furthermore, an interesting approach for lignin conversion into fuels has been recently proposed by mechanochemical methods, which could boost the sustainability of the process by decreasing the use of solvents and additional reagents, as well as reducing the required time.274 In this regard, a wet milling oxidative strategy for lignin depolymerisation was proposed by Yao et al. employing KOH and toluene.275 Such mechanochemical process was compared with the results of Baeyer–Villiger oxidation using porphyrin as catalytic specie, displaying improved results. Moreover, a synergistic effect was found by combining mechanochemistry and porphyrin oxidation strategies for lignin depolymersation.276 In addition, solvent-less mechanochemical approaches have been also used for the oxidative cleavage of lignin or lignin β-O-4 model compounds with HO-TEMPO/KBr/oxone, leading to quinones and phenolic derivatives. Outstandingly, such strategy was translated to gram-scale, opening new possibilities for the industrial application of solvent-less mechanochemistry for lignin depolymerisation.277
Furthermore, mimicking photosynthesis process in nature, photocatalytic strategies for lignin conversion into fuels, through C–O and C–C cleavage,278,279 is a promising possibility to move towards more environmentally friendly and economically efficient approaches, especially considering the possibility to use sun-type light irradiation.280–283 Indeed, β-O-4 cleavage in lignin could be attempted through photocatalytic reductive (initiated by electrons or reducing agents), oxidative (initiated by holes or oxidant agents) or redox neutral (initiated by hole or oxidative species, together with electrons) routes, employing metal oxides, metal sulfides, quantum dots (QDs), organometallic complexes and carbon-based materials such as graphitic carbonitride.284,285
In addition, electrocatalytic and photo-assisted electrocatalytic routes have been investigated for the degradation of lignin for instance via lignin-enhanced water electrolysis or for the production of hydrogen. Regarding water electrolysis, the oxidation of lignin, among other molecules or even the direct use of biomass wastes, has been investigated as an economically feasible option to replace anodic oxidation, leading also to the generation of hydrogen at low potentials. As well, electrocatalysis have been employed for the conversion of lignin into valuable chemicals (such as vanillin, phenol and guaiacol) and fuels.286
Environmental and economic viability of lignin transformation into fuels is also an area which requires a lot of attention in order to ensure the competitiveness of such biorefinery scheme. In this direction, some studies have been performed by Life Cycle Assessment (LCA) analysis of the pyrolytic transformation of lignin into two types of biofuels depending on the sulphur content.287
6. Environmental and cost impact of lignin-based materials and products
The environmental and economic impact of the production of lignin can be quantified using tools such as life cycle assessment (LCA) and techno-economic analysis (TEA). The implementation of an LCA is governed by the ISO 14040-2006 and 14044-2006.288 This assessment takes in consideration a product's life cycle in full (cradle-to-grave) or to its intended primary user (cradle-to-gate) to evaluate its environmental impact. Various factors are modelled, such as global warming potential (GWP, in CO2,eq. per kg of product); acidification (in H2SO4,eq. per kg of product), Human Toxicity; ozone depletion etc. An LCA is typically conducted in four phases, the goal and scope definition phase; the inventory analysis phase; the impact assessment phase and the interpretation phase. It is widely used to estimate the environmental impact or benefit of an existing or new technology. Since the interest on lignin as an alternative to fossil-fuels based technologies has gained considerable attention recently, lignin-only LCA studies have emerged, which are either focused on lignin extraction289–299 or lignin-based products (asphalt,300–304 adhesives,305–307 resins,308 fine chemicals,309–311,312–314 carbon fibres315 and fuels.287,316–320 Lignin as a product from biorefineries is a new concept, therefore most of the LCA conducted are based on either well quantified extraction processes such as Kraft or based on experimental lab scale data which has been extrapolated. In the latest case, the LCA allows to evaluate the environmental impact of an upscaled biorefinery.292 Ran in parallel to an LCA, a TEA includes process design, simulation, calculation of capital and operating cost along with installation cost of equipment for commercial scaled biorefinery.321 The output of a TEA is minimal selling price (MSP), revenue and profitability. They are expressed in a similar functional unit as the LCA results. Here we focus on the LCA and TEA performed on lignin processes and materials relevant to energy applications.6.1. Challenges encountered carrying out LCA on lignin
While a very useful tool to quantify the environmental impact of a product life cycle, LCA has its limitations,322 which appear at all level of the process. LCA where lignin is the end product typically uses 1 kg lignin as the functional unit.299,300,323 The country where the product is manufactured affects the outcome of an LCA. For example, the carbon footprint of electricity is significantly lower where renewable or nuclear energy sources are used. The impact on lignin production is illustrated in ref. 292 where the lignin produced in Sweden has a global warming potential GWP three times lower than that of a global lignin (1.68 vs. 5.5 kgCO2,eq. per kg lignin). The lifetime of the product affects how the LCA is conducted. Lignin is a wood-based product. Due to the CO2 sequestration potential of wood, the definition of the system boundaries can be shifted.324,325 It takes between 20 and 80 years for softwood trees to absorb its equivalent in CO2.321 Therefore, most LCA's are conducted over a period of 100 years to assume carbon neutrality.324 While plausible when looking at lignin-based products for buildings, this assumption becomes problematic for carbon materials from lignin. A battery pack has a lifetime of 2–5 years. Incineration at the end of life would provoke a release of CO2 unaccounted for in the LCA. Therefore, a wood-based product with a lifetime less than 100 years would need to account on the amount of CO2,eq. released by the incineration of the wood used in the product manufacture (between 1.95 to 2.08 CO2 eq.,kg per kg of wood, dependant on the plant species and age325).The lignin-only LCA studies are notoriously difficult to conduct, as well outlined by Moretti et al.324 At the inventory phase, the approximation made on the data used (upscale simulated data based on lab scale processes,292,293,299 secondary data from literature301) can impact the results. Most recent LCA conducted on lignin use data provided by biorefineries, leading to more reliable results.300 Due to the nature of a biorefinery, lignin is the product of a multi-output process:323 the allocation of the environmental impact between lignin and its co-products (such as cellulose, electricity, ethanol) from biorefineries is of most importance to quantify the GWP.300,316,326,327 The ISO 14044:2006 guidelines provides a hierarchy to deal with multi-product systems such as biorefineries, with three levels: subdivision, system expansion and allocation.288 This subdivision system, which uses data with higher level of details, is rendered difficult by the integration of the biorefinery systems.323,324 System expansion modifies the system boundaries to include all the production of all functions. For a biorefinery, this means quantifying the impact of the whole process and not lignin extraction alone.288,294,296 The allocation method uses physical parameters (mass, energy, economic values, etc.) to measure the impact of a product. For example, if a biorefinery outputs 100 kg of product, including 41 kg of lignin, lignin will be allocated 41% of the GWP. This method is used by close to a third of the LCA published before 2021.324 However, it doesn't allocate an impact to heat or electricity, which are mass-free.323 Similarly, energy allocation (13% of the studies324) uses energy value to allocate the GWP, which excluded CO2. Economic allocation (16% of the studies324) uses the value of one product to evaluate its GWP. As lignin is typically a by-product, its value is low (0.3 euro per kg), leading to a low GWP by economic allocation. While its value has been low, it could increase due to the overall energy cost increase, therefore increasing lignin environmental impact. Obydenkova et al. allocated the environmental impacts between the three products of a lignocellulosic biorefinery producing ethanol, lignin oligomers and electricity using a subdivision matrix approach. Depending on the allocation method, they quantified the GWP of the lignin oligomers between 6.8 and 31.6 kgCO2-eq. per GJ.328 Hermansen et al. ran different allocation scenarios on the lignin produced from a Kraft mill. The results varied between 0.18 to 0.64 kg CO2 eq. per kg lignin. As mentioned before, the disparity between the functional units renders results difficult to compare. While LCA is a powerful tool, its completion necessitates a well-defined methodology and clearly stated assumptions.324
6.2. LCA of lignin and lignin-based products
The kraft process has been investigated for LCA in several studies.289,299,301,323,329 Bernier focuses on how the extraction of lignin from black liquor positively impacts the overall production output by de-bottlenecking the recovery boiler. He also considers the carbon sequestration from the feedstock, almost cancelling the on-site carbon emissions.299 These assumptions allow for a GWP of 0.57 kg CO2,eq. per kg lignin. However, they assume that the lignin produced has a 100 years lifetime. It is to be noted that the Bernier study is used as secondary data in subsequent articles. Another reference study from Culbertson estimated GWP of a softwood kraft pulp mill between 0.55–0.60 kg CO2,eq. per kg lignin. The de-bottlenecking of the recovery boiler allowed for a 5% increase in the efficiency of the pulp extraction process. As an environmental impact, the acidification was not systematically evaluated. Moretti's studies on Asphalt produced from Kraft mills assigned between 7 to 10% of the whole environmental impact to acidification (vs. 20 to 54% to GWP).300,301 According to Culbertson, between 58%–68% of the acidification is due to the use of natural gas. Bernier evaluated the acidification value to be 5.3 × 10−3 kg SO2,eq. per kg Kraft lignin, due to the H2SO4 wash of the black liquor.299The impact of the organosolv process was estimated in ref. 293, 329 and 330. Arias et al.329 estimated the impact of the organosolv process twice as high as that of the Kraft process during the production of a bioadhesive. However, two pathways are taken in consideration for the lignin chemical modification and bio-adhesive production, rendering the comparison between kraft and Organosolv GWP difficult. Yadav et al.330 compared the organosolv process on a spruce bark using ethanol or bio-ethanol, at lab scale. The bio-ethanol process has a GWP of 1.54 kg CO2,eq. per kg lignin against 2.24 kg CO2,eq. per kg lignin for ethanol. The ethanol and the electricity have the highest impacts on the production (respectively 55% and 30%). The cost of production was estimated between €1.38–2.2 per kg lignin.
Teh et al.295 compared the impact between alkali and organosolv process for the formation of nanoparticles of lignin from birch chip wood. The study uses extrapolated upscale data from lab scale. The alkali process has an impact 4–5 times lower than that of the organosolv treatment. The authors assigned this significant increase to the use of electricity. No TEA was performed.
The lignin-rich oil obtained from reductive catalytic fractionation was accounted for 57% of a biorefinery capital costs, with a MSP of 1.12 USD per kg and a GWP of 0.079 kg CO2,eq. per kg. In this study, the carbon sink of the growing feedstock (hybrid poplar) is accounted for, which offsets the overall GWP of the lignin fraction close to neutral.297 No TEA was performed.
DES pretreatment (choline chloride – oxalic acid dihydrate) to form lignin-containing nanocellulose has an impact that is 2.5 times that of Sulfuric acid hydrolysis. The largest contribution was from the DES chemicals, between 52–63% for GWP.291
Kulas et al.290 recently ran an LCA on kraft lignin as a product of a modified black liquor process. The data used was extrapolated from the lab scale work published and patented by Thies et al.290 In this process, ethanol at 115 °C and 6.2 bar allow for a liquid–liquid solvent recovery from black liquor. The lignin solution is then dried off to recover lignin. The low boiling point of ethanol (78.4 °C) reduces the energy consumption of ethanol. The GWP of the lignin was evaluated at 4.0 kg CO2,eq. per kg lignin, assuming carbon neutrality from lignin. The MSP was estimated at 0.45 USD per kg lignin, cheaper than carbon fibre precursor PAN (2 USD per kg). Furthermore, Kulas performed an LCA on potential production of carbon fibres and activated carbon using an allocation by both mass and economical values, obtained from literature. The GWP of lignin-based carbon fibres was estimated at 20–22 kg CO2,eq. per kg of CF and the activated carbon at 5–12 kg CO2,eq. per kg. The cost of a kg of carbon fibre was estimated at USD 21.78 (depending on grade with higher grades much more expensive), generating a profit of USD 9.76 per kg. For a production based on a production of 6260 tonnes of CF per year for the US, the profit made is of USD 61 million yearly.
The LCA ran for biofuels are different in the terms of product lifetime, usually very short (days-months). The carbon neutrality assumption cannot be made. The studies all refer to the U.S. renewable fuel standard (RFS) which sets a 60% reduction on the greenhouse gas emissions (76% CO2, 16% methane, 6% nitrous oxide, HFC) for cellulosic biofuels.331 The results vary whereas the biorefinery produces its own electricity or not.287 The functional unit is of 1 MJ. Obydenkova uses cornstover as feedstock to an ethanol–lignin biorefinery, from lab scale data. The lignin-derived automotive fuel minimal cost is between 14.4–18.1 USD per GJ, where the cheapest fuel is produced using natural gas as the energy source. The GHG emission targets are only met for lignin-derived automotive fuel for a biorefinery when all the impact is allocated to ethanol production.287 Bartling et al. identified 12 biofuels that reaches the GHG emission targets. However, lignin is considered only as energy cogeneration in this study.320 The LCA on bio-jet fuel from poplar biomass was performed by Budsberg et al.318 This study uses lignin as a source of H2 using hog fuel as the alternative energy source.
The pursuit of LCA for the production and use of lignin shows that lignin is a suitable alternative to fossil sources. It is clear that, when it comes to lignin and its products, LCA data should be analysed with a degree of caution as the methodologies employed have a strong impact on results. Also, current literature lacks assessments carried out on energy applications, outside of the biofuel field. This is mainly due to the emergence of this field and therefore the lack of available data. There is an obvious gap here for future studies.
7. Outlook
Lignin extraction necessitates non-destructive, upscalable extraction processes that preserve the ether bonds and phenolic groups. The emergence of economically viable biorefineries is a welcome step for the use of lignin for energy applications. However many stumbling blocks remain, for example, the depolymerisation processes for the production of biofuels are yet to be fully upscaled. For batteries improvements in performance of lignin-based electrodes in full cell batteries should be the ultimate ambition.For energy storage in general (batteries and supercapacitors), optimisation of electrode architecture is extremely important for maximized electrochemical performance with emerging emphasis on interconnected pore networks, surface area, lignin CNF design features (diameter, length and network rigidity), density/areal loading, and the adoption of cost effective production strategies suitable for scalability such as 3D patterning technologies. These designs need to allow for electrode swelling particularly when combined with high-capacity Li alloying materials like Si which is hugely affected by volumetric expansion (Si > 300%), leading to a long term cycling capacity drop, reduced rate capability, and increased safety concerns. Such issues should be addressed at the capacity enhancing material level (e.g., Si, In, oxides, etc.) by adopting design strategies that will prevent segregation, pulverisation, and cracking. At the electrode level a balanced distribution of voids/pores, network rigidity, and density of active elements must be judiciously controlled to regulate swelling and capacity fading during long term cycling. However, this may require a reasonable trade-off between the electrode features, such as robust structures, energy density/capacity, side reactions, and full cell performance. Consequently, a rational design of composite or more complex electrodes could offer more choices for enhancing the performance of lignin-based electrodes that may become suitable for practical batteries.
To practically deploy lignin electrodes in high-energy density batteries, it is also crucial to investigate the electrochemical behaviour relative to the electrode structure, including the swelling characteristics both in half-cell and full-cell configurations, but this is currently lacking in the literature. The specific impact of non-lignin containing cell components such as conventional electrolytes, binders, and conductive additives on the performance of the lignin-derived electrodes is rarely investigated and should be a focus of future research. We believe that a comprehensive design of lignin-based cells with the specific goal of achieving high energy density, based on high capacity lignin-derived composite anode/cathodes will lay a strong foundation toward the realization of higher-energy batteries than graphite. Despite the significant research progress in developing lignin-based anodes and cathodes, it is still not exactly clear how the specific type of lignin and their functionalized structures or carbonization processes impact the final anode features and performance of various battery types besides the porosity and CNF networking features commonly observed. Therefore, advanced nanoscale characterization techniques for detailed study and in-depth understanding of the structure–property relationship are required to reliably determine the electrochemical mechanisms, particularly for composite structures (e.g., carbon/Si anodes). Operando characterization, in situ studies, and theoretical analysis are also strongly recommended for detailed studies and in-depth understanding of the behaviour of lignin based cell components with respect to ion transport and structural behaviour during the charge/discharge processes. The culmination of these studies could reliably predict and provide clearer understanding of the dynamic electrode kinetics during cycling, thereby providing a better understanding of the redox chemistry, failure dynamics, and the charge storage mechanisms, thus, clearing the path to allow for the efficient design and development of lignin-based materials in practical batteries.
While promising, the valorisation of lignin into a sustainable and economical biofuel necessitates fully developed depolymerisation pathways. The complexity of the lignin structure and its natural recalcitrance to degradation limits the effectiveness of the depolymerisation process. The variety of aromatics outputted from the depolymerisation triggers need for complex separation processes. Thermoelectric future directions will focus on how to develop hybrid carbon-based nanostructures derived from lignin in combination with other inorganic materials to improve their thermoelectric performance. While this field is still very much in its infancy the emerging potential is clear to see.
In terms of LCA and technoeconomic analysis while lignin is typically abundant and a low-cost material (often considered waste), future study on the development and deployment of lignin electrodes in energy storage devices should include practical cost benefits analysis particularly the energy-to-price ratio. While most researchers heavily emphasized its cost and environmental benefits, the combined processing/treatment and transformation steps of pristine lignin into an active carbon electrode material and integration of secondary materials could be relatively costly. Moreover, the use of chemical activation processes and combination with toxic polymeric materials (also requiring further process steps) make the environmental benignity of the electrode materials and “green battery” claims debatable. Also, by blending lignin with such polymer materials in the various cell components (anode, cathode, electrolyte, binder and separator), it is misleading as commonly captured in most literatures to refer to the final composite material as a lignin derivative as the secondary materials do not contain lignin.
8. Concluding remarks
Lignin is a unique polymer within biomass. It presents a one of a kind aromatic structure and a high carbon content. Above all, its production does not compete with food supply chains and its valorisation allows for use of an ever increasing amount of waste. As a result of the active interest of research bodies and industries into candidates to replace oil-based materials, lignin is being extensively investigated for use in functional materials. In particular, the development of lignin-based materials for energy applications has shown enormous promise. Lignin is finding application in a remarkable array of different battery components including electrode materials, separators and electrolytes. Research into lignin-derived carbon materials, especially hard carbons, is rapidly gaining interest in particular for applications beyond lithium ion where larger ions can be accommodated by intercalation between graphitic layers where lignin derived carbons outperform conventional graphite anodes. Clearly, exploitation of lignin as a material for components has enormous potential to lower overall battery cost and contribute to the development of safer and more sustainable energy storage devices. Control of the unique porous carbon architectures derived from lignin also offers advantages in supercapacitors and thermoelectric devices where lignin derived materials have produced devices with beyond state of the art seebeck coefficients and power factors. In particular, for the thermoelectric field, lignin could have an enormous potential not only as carbon precursors for electronic semiconductors but also as platform for ionic thermoelectric materials which have recently emerged as a plausible alternative to classical thermoelectric devices.However, progress in all these applications can only be sustained by the continuous supply of a high-quality lignin, as its properties are strongly dependent on source and extraction method. This calls for the development of versatile processing routes and the provision of industrial scale biorefineries, to possibly produce predefined lignin chemical structures with the aid of artificial intelligence and machine learning. This may help in the elucidation of structure/property/function relationships of lignin precursors and the tailoring of their carbonised products for next generation energy applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Irish Government funding via the DAFM NXTGENWOOD research program 2019PROG704. This work also received support from the Science Foundation Ireland (SFI) (contract no. 11-PI-1148, 16/IA/4629 and SFI 16/M-ERA/3419) and the European Union's Horizon 2020 Research and Innovation Program; grant agreement no. 814464 (Si-DRIVE project). T. K. acknowledges support from the Sustainable Energy Authority of Ireland through the Research Development and Demonstration Funding Programme (grant no. 19/RDD/548) and Enterprise Ireland through the Innovation Partnership Programme (grant no. IP 2019 0910). I. S. A. acknowledges support from the SFI Industry RD&I Fellowship Programme (21/IRDIF/9876) and the EU Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Individual Fellowship Grant (843621). A. B. and M. N. C. acknowleges support from VIBES which has received funding from the Biobased Industries Joint Undertaking (JU) under the European Union’s Horizon 2020 research and innovation programme under grant agreement No 101023190. The JU receives support from the European Union’s Horizon 2020 research and innovation programme and the Bio-based Industries Consortium.References
- T. Kobayashi and L. Nakajima, Curr. Opin. Green Sustain. Chem., 2021, 28, 100439 CrossRef CAS.
- M. Muddasar, R. Liaquat, A. Aslam, M. Z. Ur Rahman, A. Abdullah, A. H. Khoja, K. Latif and A. Bahadar, Int. J. Energy Res., 2022, 5625–5645 CrossRef CAS.
- T. Hák, S. Janoušková and B. Moldan, Ecol. Indic., 2016, 60, 565–573 CrossRef.
- E. E. Miller, Y. Hua and F. H. Tezel, J. Energy Storage, 2018, 20, 30–40 CrossRef.
- S. Iqbal, H. Khatoon, A. Hussain Pandit and S. Ahmad, Mater. Sci. Energy Technol., 2019, 2, 417–428 Search PubMed.
- L. Wang and X. Hu, Chem. – Asian J., 2018, 13, 1518–1529 CrossRef CAS PubMed.
- M. I. Din, S. Ashraf and A. Intisar, Sci. Prog., 2017, 100, 299–312 CrossRef CAS PubMed.
- T. Yoda, K. Shibuya and H. Myoubudani, Measurement, 2018, 125, 572–576 CrossRef.
- S. Li, W. Li and Z. Fan, Biomass Based Energy Storage Mater., 2020, 78, 124–142 CAS.
- W. Zhang, J. Yin, C. Wang, L. Zhao, W. Jian, K. Lu, H. Lin, X. Qiu and H. N. Alshareef, Small Methods, 2021, 5, 2100896 CrossRef CAS PubMed.
- X. Wu, J. Jiang, C. Wang, J. Liu, Y. Pu, A. Ragauskas, S. Li and B. Yang, Biofuels, Bioprod. Biorefin., 2020, 14, 650–672 CrossRef CAS.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- J. C. Carvajal, Á. Gómez and C. A. Cardona, Bioresour. Technol., 2016, 214, 468–476 CrossRef CAS PubMed.
- D. Di Francesco, C. Dahlstrand, J. Löfstedt, A. Orebom, J. Verendel, C. Carrick, Å. Håkansson, S. Eriksson, H. Rådberg, H. Wallmo, M. Wimby, F. Huber, C. Federsel, M. Backmark and J. S. M. Samec, ChemSusChem, 2021, 14, 2414–2425 CrossRef CAS PubMed.
- J. A. Poveda-Giraldo, J. C. Solarte-Toro and C. A. Cardona Alzate, Renewable Sustainable Energy Rev., 2021, 138, 110688 CrossRef CAS.
- P. Jędrzejczak, M. N. Collins, T. Jesionowski and Ł. Klapiszewski, Int. J. Biol. Macromol., 2021, 187, 624–650 CrossRef PubMed.
- J. Sternberg, O. Sequerth and S. Pilla, Prog. Polym. Sci., 2021, 113, 101344 CrossRef CAS.
- E. Lizundia, M. H. Sipponen, L. G. Greca, M. Balakshin, B. L. Tardy, O. J. Rojas and D. Puglia, Green Chem., 2021, 23, 6698–6760 RSC.
- H. Hatakeyama and T. Hatakeyama, in Biopolymers, 2009, ch. 12, pp. 1–63, DOI:10.1007/12_2009_12.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- K. Lundquist and J. Parkås, BioResources, 2011, 6, 920–926 CAS.
- Y. Lu, Y.-C. Lu, H.-Q. Hu, F.-J. Xie, X.-Y. Wei and X. Fan, J. Spectrosc., 2017, 2017, 8951658 Search PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- F. G. Calvo-Flores, J. A. Dobado, J. Isac-García and F. J. Martín-Martínez, Lignin and lignans as renewable raw materials: chemistry, technology and applications, John Wiley & Sons, 2015 Search PubMed.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- Y. Mottiar, R. Vanholme, W. Boerjan, J. Ralph and S. D. Mansfield, Curr. Opin. Biotechnol., 2016, 37, 190–200 CrossRef CAS PubMed.
- T. Z. H. Gani, M. J. Orella, E. M. Anderson, M. L. Stone, F. R. Brushett, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2019, 7, 13270–13277 CrossRef CAS.
- W. Fang, S. Yang, X.-L. Wang, T.-Q. Yuan and R.-C. Sun, Green Chem., 2017, 19, 1794–1827 RSC.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- T. Li and S. Takkellapati, Biofuels, Bioprod. Biorefin., 2018, 12, 756–787 CrossRef CAS PubMed.
- A. G. Vishtal and A. Kraslawski, BioResources, 2011, 6, 3547–3568 Search PubMed.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS PubMed.
- K. Lundquist, R. Simonson and K. Tingsvik, Sven. Papperstidn., 1983, 86, 44–47 Search PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. D. Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- M. A. Karnofski, J. Chem. Educ., 1975, 52, 490 CrossRef CAS.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- S. Y. Lin and I. S. Lin, Ullmann's encyclopedia of industrial chemistry, Verlag Chemie, Hoboken, NJ Search PubMed, 1991.
- A. BjÖRkman, Nature, 1954, 174, 1057–1058 CrossRef.
- J.-L. Wen, B.-L. Xue, F. Xu, R.-C. Sun and A. Pinkert, Ind. Crops Prod., 2013, 42, 332–343 CrossRef CAS.
- J. Rencoret, G. Marques, A. Gutiérrez, L. Nieto, J. I. Santos, J. Jiménez-Barbero, Á. T. Martínez and J. C. del Río, Wood Research and Technology, 2009, 63, 691–698 CAS.
- M.-F. Li, S.-N. Sun, F. Xu and R.-C. Sun, Chem. Eng. J., 2012, 179, 80–89 CrossRef CAS.
- H. Wang, Z. Liu, L. Hui, L. Ma, X. Zheng, J. Li and Y. Zhang, Holzforschung, 2020, 74, 275–285 CrossRef CAS.
- C. Huang, X. Jiang, X. Shen, J. Hu, W. Tang, X. Wu, A. Ragauskas, H. Jameel, X. Meng and Q. Yong, Renewable Sustainable Energy Rev., 2022, 154, 111822 CrossRef CAS.
- D. Dondi, A. Zeffiro, A. Speltini, C. Tomasi, D. Vadivel and A. Buttafava, J. Anal. Appl. Pyrolysis, 2014, 107, 53–58 CrossRef CAS.
- T. Han, N. Sophonrat, P. Evangelopoulos, H. Persson, W. Yang and P. Jönsson, J. Anal. Appl. Pyrolysis, 2018, 133, 162–168 CrossRef CAS.
- V. Ashokkumar, R. Venkatkarthick, S. Jayashree, S. Chuetor, S. Dharmaraj, G. Kumar, W.-H. Chen and C. Ngamcharussrivichai, Bioresour. Technol., 2022, 344, 126195 CrossRef CAS PubMed.
- S. Hong, X.-J. Shen, Z. Xue, Z. Sun and T.-Q. Yuan, Green Chem., 2020, 22, 7219–7232 RSC.
- Y. T. Tan, A. S. M. Chua and G. C. Ngoh, Bioresour. Technol., 2020, 297, 122522 CrossRef CAS PubMed.
- W. Wang and D.-J. Lee, Bioresour. Technol., 2021, 339, 125587 CrossRef CAS PubMed.
- Y. Wang, K. H. Kim, K. Jeong, N.-K. Kim and C. G. Yoo, Curr. Opin. Green Sustain. Chem., 2021, 27, 100396 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71, 10.1039/B210714G.
- C. R. Ashworth, R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2016, 18, 18145–18160 RSC.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- C. F. Araujo, J. A. P. Coutinho, M. M. Nolasco, S. F. Parker, P. J. A. Ribeiro-Claro, S. Rudić, B. I. G. Soares and P. D. Vaz, Phys. Chem. Chem. Phys., 2017, 19, 17998–18009 RSC.
- E. Posada, M. J. Roldán-Ruiz, R. J. Jiménez Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, J. Mol. Liq., 2019, 276, 196–203 CrossRef CAS.
- E. Posada, N. López-Salas, D. Carriazo, M. A. Muñoz-Márquez, C. O. Ania, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. D. Monte, Carbon, 2017, 123, 536–547 CrossRef CAS.
- Y. Bai, X.-F. Zhang, Z. Wang, T. Zheng and J. Yao, Bioresour. Technol., 2022, 347, 126723 CrossRef CAS PubMed.
- V. Provost, S. Dumarcay, I. Ziegler-Devin, M. Boltoeva, D. Trébouet and M. Villain-Gambier, Bioresour. Technol., 2022, 349, 126837 CrossRef CAS PubMed.
- Y. T. Tan, G. C. Ngoh and A. S. M. Chua, Bioresour. Technol., 2019, 281, 359–366 CrossRef CAS PubMed.
- C.-W. Zhang, S.-Q. Xia and P.-S. Ma, Bioresour. Technol., 2016, 219, 1–5 CrossRef CAS PubMed.
- Q. Xia, Y. Liu, J. Meng, W. Cheng, W. Chen, S. Liu, Y. Liu, J. Li and H. Yu, Green Chem., 2018, 20, 2711–2721 RSC.
- Y. Liu, W. Chen, Q. Xia, B. Guo, Q. Wang, S. Liu, Y. Liu, J. Li and H. Yu, ChemSusChem, 2017, 10, 1692–1700 CrossRef CAS PubMed.
- A. M. da Costa Lopes, J. R. B. Gomes, J. A. P. Coutinho and A. J. D. Silvestre, Green Chem., 2020, 22, 2474–2487 RSC.
- Z. Chen, X. Bai and A. Lusi, ACS Sustainable Chem. Eng., 2018, 6, 12205–12216 CrossRef CAS.
- Z. Chen, W. D. Reznicek and C. Wan, Bioresour. Technol., 2018, 263, 40–48 CrossRef CAS PubMed.
- Z. Chen, X. Bai, A. Lusi and C. Wan, ACS Sustainable Chem. Eng., 2020, 8, 9783–9793 CrossRef CAS.
- B. Soares, A. M. da Costa Lopes, A. J. D. Silvestre, P. C. Rodrigues Pinto, C. S. R. Freire and J. A. P. Coutinho, Ind. Crops Prod., 2021, 160, 113128 CrossRef CAS.
- M. C. Gutiérrez, M. L. Ferrer, C. R. Mateo and F. del Monte, Langmuir, 2009, 25, 5509–5515 CrossRef PubMed.
- N. López-Salas, J. M. Vicent-Luna, S. Imberti, E. Posada, M. J. Roldán, J. A. Anta, S. R. G. Balestra, R. M. Madero Castro, S. Calero, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, ACS Sustainable Chem. Eng., 2019, 7, 17565–17573 CrossRef.
- N. López-Salas, J. M. Vicent-Luna, E. Posada, S. Imberti, R. M. Madero-Castro, S. Calero, C. O. Ania, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, ACS Sustainable Chem. Eng., 2020, 8, 12120–12131 CrossRef.
- M. J. Roldán-Ruiz, R. J. Jiménez-Riobóo, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, J. Mol. Liq., 2019, 284, 175–181 Search PubMed.
- H. Zhang, X. Lu, L. González-Aguilera, M. L. Ferrer, F. D. Monte and M. C. Gutiérrez, J. Chem. Phys., 2021, 154, 184501 CrossRef CAS PubMed.
- V. Agieienko and R. Buchner, J. Chem. Eng. Data, 2020, 65, 1900–1910 CrossRef CAS.
- B. Soares, D. J. P. Tavares, J. L. Amaral, A. J. D. Silvestre, C. S. R. Freire and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2017, 5, 4056–4065 CrossRef CAS.
- B. Soares, A. J. D. Silvestre, P. C. Rodrigues Pinto, C. S. R. Freire and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2019, 7, 12485–12493 CAS.
- E. Maia and S. Perez, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 849–852 CrossRef.
- H. Chanzy, S. Nawrot, A. Peguy, P. Smith and J. Chevalier, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 1909–1924 CrossRef CAS.
- B. D. Rabideau and A. E. Ismail, Phys. Chem. Chem. Phys., 2015, 17, 5767–5775 RSC.
- H. Zhang, L. González-Aguilera, D. López, M. Luisa Ferrer, F. del Monte and M. C. Gutiérrez, J. Mol. Liq., 2022, 358, 119113 CrossRef CAS.
- W.-J. Chen, C.-X. Zhao, B.-Q. Li, T.-Q. Yuan and Q. Zhang, Green Chem., 2022, 24, 565–584 RSC.
- X. Wu, J. Jiang, C. Wang, J. Liu, Y. Pu, A. Ragauskas, S. Li and B. Yang, Biofuels, Bioprod. Biorefin., 2020, 14, 650–672 CrossRef CAS.
- J. Zhu, C. Yan, X. Zhang, C. Yang, M. Jiang and X. Zhang, Prog. Energy Combust. Sci., 2020, 76, 100788 CrossRef.
- N. A. Banek, D. T. Abele, K. R. McKenzie and M. J. Wagner, ACS Sustainable Chem. Eng., 2018, 6, 13199–13207 CrossRef CAS.
- W. Zhang, J. Yin, Z. Lin, H. Lin, H. Lu, Y. Wang and W. Huang, Electrochim. Acta, 2015, 176, 1136–1142 CrossRef CAS.
- M. Culebras, H. Geaney, A. Beaucamp, P. Upadhyaya, E. Dalton, K. M. Ryan and M. N. Collins, ChemSusChem, 2019, 12, 4516–4521 CrossRef CAS PubMed.
- X. Niu, J. Zhou, T. Qian, M. Wang and C. Yan, Nanotechnology, 2017, 28, 405401–405401 CrossRef PubMed.
- C.-Y. Chou, J.-R. Kuo and S.-C. Yen, ACS Sustainable Chem. Eng., 2018, 6, 4759–4766 CrossRef CAS.
- L. Du, W. Wu, C. Luo, H. Zhao, D. Xu, R. Wang and Y. Deng, Solid State Ionics, 2018, 319, 77–82 CrossRef CAS.
- H. P. C. K. Chan, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef PubMed.
- I. S. Aminu, H. Geaney, S. Imtiaz, T. E. Adegoke, N. Kapuria, G. A. Collins and K. M. Ryan, Adv. Funct. Mater., 2020, 2003278, DOI:10.1002/adfm.202003278.
- Y. Fan, Q. Zhang, Q. Xiao, X. Wang and K. Huang, Carbon, 2013, 59, 264–269 CrossRef CAS.
- S. Karuppiah, C. Keller, P. Kumar, P.-H. Jouneau, D. Aldakov, J.-B. Ducros, G. R. Lapertot, P. Chenevier and C. D. Haon, ACS Nano, 2020, 14, 12006–12015 CrossRef CAS PubMed.
- T. Kennedy, E. Mullane, H. Geaney, M. Osiak, C. O'Dwyer and K. M. Ryan, Nano Lett., 2014, 14, 716–723 CrossRef CAS PubMed.
- S. Imtiaz, I. S. Amiinu, D. Storan, N. Kapuria, H. Geaney, T. Kennedy and K. M. Ryan, Adv. Mater., 2021, e2105917, DOI:10.1002/adma.202105917.
- Y. Xi, S. Huang, D. Yang, X. Qiu, H. Su, C. Yi and Q. Li, Green Chem., 2020, 22, 4321–4433 RSC.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- W. Zhang, J. Yin, W. Wang, Z. Bayhan and H. N. Alshareef, Nano Energy, 2021, 83, 105792 CrossRef CAS.
- K. Nobuhara, H. Nakayama, M. Nose, S. Nakanishi and H. Iba, J. Power Sources, 2013, 243, 585–587 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- Q. Liu, R. Xu, D. Mu, G. Tan, H. Gao, N. Li, R. Chen and F. Wu, Carbon Energy, 2022, 4(3), 458–479 CrossRef CAS.
- Z. L. Xu, G. Yoon, K. Y. Park, H. Park, O. Tamwattana, S. Joo Kim, W. M. Seong and K. Kang, Nat. Commun., 2019, 10, 2598 CrossRef PubMed.
- I. Hasa, X. Dou, D. Buchholz, Y. Shao-Horn, J. Hassoun, S. Passerini and B. Scrosati, J. Power Sources, 2016, 310, 26–31 CrossRef CAS.
- X.-R. Zhang, J.-Y. Yang, Z.-Y. Ren, K.-Y. Xie, Q. Ye, F. Xu and X.-R. Liu, New Carbon Mater., 2022, 37, 371–379 CrossRef.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- B. Xiao, T. Rojo and X. Li, ChemSusChem, 2019, 12, 133–144 CrossRef CAS PubMed.
- W.-J. Chen, C.-X. Zhao, B.-Q. Li, T.-Q. Yuan and Q. Zhang, Green Chem., 2022, 24, 565–584 RSC.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- D. G. Kizzire, A. M. Richter, D. P. Harper and D. J. Keffer, ACS Omega, 2021, 6, 19883–19892 CrossRef CAS PubMed.
- X. Dou, I. Hasa, M. Hekmatfar, T. Diemant, R. J. Behm, D. Buchholz and S. Passerini, ChemSusChem, 2017, 10, 2668–2676 CrossRef CAS PubMed.
- X. Lin, Y. Liu, H. Tan and B. Zhang, Carbon, 2020, 157, 316–323 CrossRef CAS.
- Y. Lu, C. Zhao, X. Qi, Y. Qi, H. Li, X. Huang, L. Chen and Y.-S. Hu, Adv. Energy Mater., 2018, 8(17), 1703012 CrossRef.
- Z. Wei, H.-X. Zhao, Y.-B. Niu, S.-Y. Zhang, Y.-B. Wu, H.-J. Yan, S. Xin, Y.-X. Yin and Y.-G. Guo, Mater. Chem. Front., 2021, 5, 3911–3917 RSC.
- C. M. Ghimbeu, B. Zhang, A. Martinez de Yuso, B. Réty and J.-M. Tarascon, Carbon, 2019, 153, 634–647 CrossRef.
- C. Marino, J. Cabanero, M. Povia and C. Villevieille, J. Electrochem. Soc., 2018, 165, A1400–A1408 CrossRef CAS.
- S. Alvin, D. Yoon, C. Chandra, H. S. Cahyadi, J.-H. Park, W. Chang, K. Y. Chung and J. Kim, Carbon, 2019, 145, 67–81 CrossRef CAS.
- D. Yoon, J. Hwang, W. Chang and J. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 569–581 CrossRef CAS PubMed.
- R. F. Susanti, S. Alvin and J. Kim, J. Ind. Eng. Chem., 2020, 91, 317–329 CrossRef CAS.
- K. Peuvot, O. Hosseinaei, P. Tomani, D. Zenkert and G. Lindbergh, J. Electrochem. Soc., 2019, 166, A1984–A1990 CrossRef CAS.
- G. Shi, Z. Han, L. Hu, B. Wang and F. Huang, ChemElectroChem, 2022, 9, 304–315 CrossRef.
- L. Fan, Z. Shi, Q. Ren, L. Yan, F. Zhang and L. Fan, Green Energy Environ., 2021, 6, 220–228 CrossRef.
- L. Fan, X. Zhang, L. Fan, L. Yan, Z. Wang, W. Lei, D. Ruan and Z. Shi, ACS Appl. Energy Mater., 2021, 4, 11436–11446 CrossRef CAS.
- Z. Li, C. Bommier, Z. S. Chong, Z. Jian, T. W. Surta, X. Wang, Z. Xing, J. C. Neuefeind, W. F. Stickle, M. Dolgos, P. A. Greaney and X. Ji, Adv. Energy Mater., 2017, 7, 1602894 CrossRef.
- S. Chen, F. Feng and Z.-F. Ma, Compos. Commun., 2020, 22, 106890 Search PubMed.
- X. Yuan, B. Zhu, J. Feng, C. Wang, X. Cai and R. Qin, Chem. Eng. J., 2021, 405, 126897 CrossRef CAS.
- Z. Wu, L. Wang, J. Huang, J. Zou, S. Chen, H. Cheng, C. Jiang, P. Gao and X. Niu, Electrochim. Acta, 2019, 306, 446–453 CrossRef CAS.
- Z. Wu, J. Zou, S. Shabanian, K. Golovin and J. Liu, Chem. Eng. J., 2022, 427, 130972 CrossRef CAS.
- Z. Wu, J. Zou, Y. Zhang, X. Lin, D. Fry, L. Wang and J. Liu, Chem. Eng. J., 2022, 427, 131547 CrossRef CAS.
- L. Tao, L. Liu, R. Chang, H. He, P. Zhao and J. Liu, J. Power Sources, 2020, 463, 228172 CrossRef CAS.
- K. Jiang, X. Tan, S. Zhai, K. Cadien and Z. Li, Nano Res., 2021, 14, 4664–4673 CrossRef CAS.
- M. Liu, D. Jing, Y. Shi and Q. Zhuang, J. Mater. Sci.: Mater. Electron., 2019, 30, 8911–8919 CrossRef CAS.
- J. Xu, C. Fan, M. Ou, S. Sun, Y. Xu, Y. Liu, X. Wang, Q. Li, C. Fang and J. Han, Chem. Mater., 2022, 34, 4202–4211 CrossRef CAS.
- C. del Mar Saavedra Rios, L. Simonin, C. M. Ghimbeu, C. Vaulot, D. da Silva Perez and C. Dupont, Fuel Process. Technol., 2022, 231, 107223 CrossRef CAS.
- J. L. Espinoza-Acosta, P. I. Torres-Chávez, J. L. Olmedo-Martínez, A. Vega-Rios, S. Flores-Gallardo and E. A. Zaragoza-Contreras, J. Energy Chem., 2018, 27, 1422–1438 CrossRef.
- J. Wang, H. Yin, Z. Wang, J. Gao, Q. Jiang, Y. Xu and Z. Chen, Asia–Pac. J. Chem. Eng., 2022, 17, e2768 CAS.
- H. Zhang, W. Zhang and F. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 61180–61188 CrossRef CAS PubMed.
- H. Zhang, W. Zhang, H. Ming, J. Pang, H. Zhang, G. Cao and Y. Yang, Chem. Eng. J., 2018, 341, 280–288 CrossRef CAS.
- C. Li, Y. Sun, Q. Wu, X. Liang, C. Chen and H. Xiang, Chem. Commun., 2020, 56, 6078–6081 RSC.
- E. Stojanovska, E. S. Pampal, A. Kilic, M. Quddus and Z. Candan, Composites, Part B, 2019, 158, 239–248 CrossRef CAS.
- J. Zhang, B. Yu, Y. Zhang and C. Wang, Energy Technol., 2020, 8(3), 1901423 CrossRef CAS.
- Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2016, 4, 96–104 RSC.
- Y. Zhang, Y. Zhu, J. Zhang, S. Sun, C. Wang, M. Chen and J. Zeng, Energy Technol., 2019, 8(1), 1900694 CrossRef.
- J. Jin, B.-J. Yu, Z.-Q. Shi, C.-Y. Wang and C.-B. Chong, J. Power Sources, 2014, 272, 800–807 CrossRef CAS.
- J. Zhang, J. Duan, Y. Zhang, M. Chen, K. Ji and C. Wang, ChemElectroChem, 2021, 8, 3544–3552 CrossRef CAS.
- L. Du, W. Wu, C. Luo, D. Xu, H. Guo, R. Wang, T. Zhang, J. Wang and Y. Deng, J. Electrochem. Soc., 2019, 166, A423–A428 CrossRef CAS.
- M. Chen, F. Luo, Y. Liao, C. Liu, D. Xu, Z. Wang, Q. Liu, D. Wang, Y. Ye, S. Li, D. Wang and Z. Zheng, J. Electroanal. Chem., 2022, 919, 116526 CrossRef CAS.
- M. Ue, K. Sakaushi and K. Uosaki, Mater. Horiz., 2020, 7, 1937–1954 RSC.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- J. B. Robinson, K. Xi, R. V. Kumar, A. C. Ferrari, H. Au, M.-M. Titirici, A. Parra-Puerto, A. Kucernak, S. D. S. Fitch, N. Garcia-Araez, Z. L. Brown, M. Pasta, L. Furness, A. J. Kibler, D. A. Walsh, L. R. Johnson, C. Holc, G. N. Newton, N. R. Champness, F. Markoulidis, C. Crean, R. C. T. Slade, E. I. Andritsos, Q. Cai, S. Babar, T. Zhang, C. Lekakou, N. Kulkarni, A. J. E. Rettie, R. Jervis, M. Cornish, M. Marinescu, G. Offer, Z. Li, L. Bird, C. P. Grey, M. Chhowalla, D. D. Lecce, R. E. Owen, T. S. Miller, D. J. L. Brett, S. Liatard, D. Ainsworth and P. R. Shearing, J. Phys.: Energy, 2021, 3, 031501 CAS.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef CAS PubMed.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- D. Su, D. Zhou, C. Wang and G. Wang, Adv. Funct. Mater., 2018, 28, 1870273 CrossRef.
- F. Yu, Y. Li, M. Jia, T. Nan, H. Zhang, S. Zhao and Q. Shen, J. Alloys Compd., 2017, 709, 677–685 CrossRef CAS.
- Y. Liu, H. Guo, B. Zhang, G. Wen, R. Vajtai, L. Wu, P. M. Ajayan and L. Wang, Batteries Supercaps, 2020, 3, 1201–1208 CrossRef CAS.
- W.-J. Kwak, D. S. Rosy, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- J. Fu, R. Liang, G. Liu, A. Yu, Z. Bai, L. Yang and Z. Chen, Adv. Mater., 2019, 31, e1805230 CrossRef PubMed.
- G. Zhang, Y. Yao, T. Zhao, M. Wang and R. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 16521 CrossRef CAS PubMed.
- A. Arul, H. Pak, K. U. Moon, M. Christy, M. Y. Oh and K. S. Nahm, Appl. Catal., B, 2018, 220, 488–496 CrossRef CAS.
- P. Li, H. Wang, W. Fan, M. Huang, J. Shi, Z. Shi and S. Liu, Chem. Eng. J., 2021, 421, 129704 CrossRef CAS.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 396–3127 RSC.
- A. Cholewinski, P. Si, M. Uceda, M. Pope and B. Zhao, Polymers, 2021, 13, 1–20 CrossRef PubMed.
- T. Chen, J. Hu, L. Zhang, J. Pan, Y. Liu and Y.-T. Cheng, J. Power Sources, 2017, 362, 236–242 CrossRef CAS.
- C. Luo, L. Du, W. Wu, H. Xu, G. Zhang, S. Li, C. Wang, Z. Lu and Y. Deng, ACS Sustainable Chem. Eng., 2018, 6, 12621–12629 CrossRef CAS.
- Y. Ma, K. Chen, J. Ma, G. Xu, S. Dong, B. Chen, J. Li, Z. Chen, X. Zhou and G. Cui, Energy Environ. Sci., 2019, 12, 273–280 RSC.
- S. Wang, L. Zhang, A. Wang, X. Liu, J. Chen, Z. Wang, Q. Zeng, H.-H. Zhou, X. Jiang and L. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 14460–14469 CrossRef CAS.
- H. Liu, L. Mulderrig, D. Hallinan and H. Chung, Macromol. Rapid Commun., 2021, 42, e2000428 CrossRef PubMed.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- W. Luo, S. Cheng, M. Wu, X. Zhang, D. Yang and X. Rui, J. Power Sources, 2021, 509, 230372 CrossRef CAS.
- M. Zhao, J. Wang, C. Chong, X. Yu, L. Wang and Z. Shi, RSC Adv., 2015, 5, 11115–11112 RSC.
- M. O. Bamgbopa, A. Fetyan, M. Vagin and A. A. Adelodun, J. Energy Storage, 2022, 50, 104352 CrossRef.
- M. C. Ribadeneyra, L. Grogan, H. Au, P. Schlee, S. Herou, T. Neville, P. L. Cullen, M. D. R. Kok, O. Hosseinaei, S. Danielsson, P. Tomani, M. M. Titirici, D. J. L. Brett, P. R. Shearing, R. Jervis and A. B. Jorge, Carbon, 2020, 157, 847–856 CrossRef CAS.
- J. Ye, Y. Cheng, L. Sun, M. Ding, C. Wu, D. Yuan, X. Zhao, C. Xiang and C. Jia, J. Membr. Sci., 2019, 572, 110–118 CrossRef CAS.
- J. Ye, D. Yuan, M. Ding, Y. Long, T. Long, L. Sun and C. Jia, J. Power Sources, 2021, 482, 229023 CrossRef CAS.
- A. Mukhopadhyay, J. Hamel, R. Katahira and H. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 5394–5400 CrossRef CAS.
- C. Schütter, S. Pohlmann and A. Balducci, Adv. Energy Mater., 2019, 9, 1900334 CrossRef.
- P. Schlee, S. Herou, R. Jervis, P. R. Shearing, D. J. L. Brett, D. Baker, O. Hosseinaei, P. Tomani, M. M. Murshed, Y. Li, M. J. Mostazo-Lopez, D. Cazorla-Amoros, A. B. Jorge Sobrido and M. M. Titirici, Chem. Sci., 2019, 10, 2980–2988 RSC.
- P. Schlee, O. Hosseinaei, D. Baker, A. Landmér, P. Tomani, M. J. Mostazo-López, D. Cazorla-Amorós, S. Herou and M.-M. Titirici, Carbon, 2019, 145, 470–480 CrossRef CAS.
- S. Herou, M. C. Ribadeneyra, R. Madhu, V. Araullo-Peters, A. Jensen, P. Schlee and M. Titirici, Green Chem., 2019, 21, 550–559 RSC.
- Z. Peng, Y. Zou, S. Xu, W. Zhong and W. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 22190–22200 CrossRef CAS PubMed.
- J. Xu, X. Zhou, M. Chen, S. Shi and Y. Cao, Microporous Mesoporous Mater., 2018, 265, 258–265 CrossRef CAS.
- F. N. Ajjan, N. Casado, T. Rębiś, A. Elfwing, N. Solin, D. Mecerreyes and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1838–1847 RSC.
- E. Raymundo-Piñero, F. Leroux and F. Béguin, Adv. Mater., 2006, 18, 1877–1882 CrossRef.
- P. J. Suhas Carrott and M. M. Ribeiro Carrott, Bioresour. Technol., 2007, 98, 2301–2312 CrossRef PubMed.
- F. Jianhui, Y. Boyuan, N. HaoXiong, H. Xiaojun and W. Ting, 2011 International Conference on Materials for Renewable Energy & Environment, 2011, DOI:10.1109/ICMREE.2011.5930944.
- B. Du, H. Zhu, L. Chai, J. Cheng, X. Wang, X. Chen, J. Zhou and R.-C. Sun, Ind. Crops Prod., 2021, 170, 113745 CrossRef CAS.
- B. Yu, Z. Chang and C. Wang, Mater. Chem. Phys., 2016, 181, 187–193 CrossRef CAS.
- L. Wang, L. Xie, X. Feng, H. Ma, X. Li and J. Zhou, ACS Omega, 2021, 6, 33171–33179 CrossRef CAS PubMed.
- N. Guo, M. Li, X. Sun, F. Wang and R. Yang, Green Chem., 2017, 19, 2595–2602 RSC.
- A. M. Navarro-Suárez, J. Carretero-González, V. Roddatis, E. Goikolea, J. Ségalini, E. Redondo, T. Rojo and R. Mysyk, RSC Adv., 2014, 4, 48336–48343 RSC.
- W. Zhang, M. Zhao, R. Liu, X. Wang and H. Lin, Colloids Surf., A, 2015, 484, 518–527 CrossRef CAS.
- K. Wang, Y. Cao, X. Wang, M. A. Castro, B. Luo, Z. Gu, J. Liu, J. D. Hoefelmeyer and Q. Fan, J. Power Sources, 2016, 307, 462–467 CrossRef CAS.
- K. Wang, M. Xu, Y. Gu, Z. Gu and Q. H. Fan, J. Power Sources, 2016, 332, 180–186 CrossRef CAS.
- M. Klose, R. Reinhold, F. Logsch, F. Wolke, J. Linnemann, U. Stoeck, S. Oswald, M. Uhlemann, J. Balach, J. Markowski, P. Ay and L. Giebeler, ACS Sustainable Chem. Eng., 2017, 5, 4094–4102 CrossRef CAS.
- Z.-Z. Chang, B.-J. Yu and C.-Y. Wang, J. Solid State Electrochem., 2016, 20, 1405–1412 CrossRef CAS.
- S. Hu and Y.-L. Hsieh, RSC Adv., 2017, 7, 30459–30468 RSC.
- H. C. Ho, N. A. Nguyen, K. M. Meek, D. M. Alonso, S. H. Hakim and A. K. Naskar, ChemSusChem, 2018, 11, 2953–2959 CrossRef CAS PubMed.
- W. Li, Y. Zhang, L. Das, Y. Wang, M. Li, N. Wanninayake, Y. Pu, D. Y. Kim, Y.-T. Cheng, A. J. Ragauskas and J. Shi, RSC Adv., 2018, 8, 38721–38732 RSC.
- X. Zhang, W. Jian, L. Zhao, F. Wen, J. Chen, J. Yin, Y. Qin, K. Lu, W. Zhang and X. Qiu, Colloids Surf., A, 2022, 636, 128191 CrossRef CAS.
- Z. Zhao, S. Hao, P. Hao, Y. Sang, A. Manivannan, N. Wu and H. Liu, J. Mater. Chem. A, 2015, 3, 15049–15056 RSC.
- L. Wang, J. Wu, H. Ma, G. Han, D. Yang, Y. Chen and J. Zhou, Energy Fuels, 2021, 35, 8303–8312 CrossRef CAS.
- E. Gonzalez-Serrano, T. Cordero, J. Rodríguez-Mirasol and J. J. Rodríguez, Ind. Eng. Chem. Res., 1997, 36, 4832–4838 CrossRef CAS.
- Y. Song, J. Liu, K. Sun and W. Xu, RSC Adv., 2017, 7, 48324–48332 RSC.
- C. Ma, L. Wu, M. Dirican, H. Cheng, J. Li, Y. Song, J. Shi and X. Zhang, J. Colloid Interface Sci., 2021, 586, 412–422 CrossRef CAS PubMed.
- C. M. Fierro, J. Górka, J. A. Zazo, J. J. Rodriguez, J. Ludwinowicz and M. Jaroniec, Carbon, 2013, 62, 233–239 CrossRef CAS.
- Y. Xi, X. Liu, W. Xiong, H. Wang, X. Ji, F. Kong, G. Yang and J. Xu, Ind. Crops Prod., 2021, 174, 114184 CrossRef CAS.
- M. J. Valero-Romero, E. M. Márquez-Franco, J. Bedia, J. Rodríguez-Mirasol and T. Cordero, Microporous Mesoporous Mater., 2014, 196, 68–78 CrossRef CAS.
- D. Salinas-Torres, R. Ruiz-Rosas, M. J. Valero-Romero, J. Rodríguez-Mirasol, T. Cordero, E. Morallón and D. Cazorla-Amorós, J. Power Sources, 2016, 326, 641–651 CrossRef CAS.
- J. Tian, Z. Liu, Z. Li, W. Wang and H. Zhang, RSC Adv., 2017, 7, 12089–12097 RSC.
- P. L. Kool, M. D. Ortiz and C. A. van Gestel, Environ. Pollut., 2011, 159, 2713–2719 CrossRef CAS PubMed.
- N. M. Franklin, N. J. Rogers, S. C. Apte, G. E. Batley, G. E. Gadd and P. S. Casey, Environ. Sci. Technol., 2007, 41, 8484–8490 CrossRef CAS PubMed.
- A. Perez-Lopez, G. Nunez-Nogueira, C. A. Alvarez-Gonzalez, S. De la Rosa-Garcia, M. Uribe-Lopez, P. Quintana and E. S. Pena-Marin, Environ. Sci. Pollut. Res. Int., 2020, 27, 22441–22450 CrossRef CAS PubMed.
- G. Sima, L. Gan, L. Chang, Y. Cui and R. K. Kankala, Microporous Mesoporous Mater., 2021, 323, 111192 CrossRef CAS.
- D. Saha, Y. Li, Z. Bi, J. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer 3rd, S. Dai, M. P. Paranthaman and A. K. Naskar, Langmuir, 2014, 30, 900–910 CrossRef CAS PubMed.
- W. Zhang, H. Lin, Z. Lin, J. Yin, H. Lu, D. Liu and M. Zhao, ChemSusChem, 2015, 8, 2114–2122 CrossRef CAS PubMed.
- D. Liu, W. Zhang, D. Liu, J. Yang, J. Yin and H. Lin, J. Mater. Sci.: Mater. Electron., 2021, 32, 7009–7018 CrossRef CAS.
- J. H. Park, H. H. Rana, J. Y. Lee and H. S. Park, J. Mater. Chem. A, 2019, 7, 16962–16968 RSC.
- E. Svinterikos, I. Zuburtikudis and M. Al-Marzouqi, ACS Sustainable Chem. Eng., 2020, 8, 13868–13893 CrossRef CAS.
- C. Ma, Z. Li, J. Li, Q. Fan, L. Wu, J. Shi and Y. Song, Appl. Surf. Sci., 2018, 456, 568–576 CrossRef CAS.
- X. Wang, W. Zhang, M. Chen and X. Zhou, Polymers, 2018, 10, 1306 CrossRef PubMed.
- Q. Cao, Y. Zhang, J. Chen, M. Zhu, C. Yang, H. Guo, Y. Song, Y. Li and J. Zhou, Ind. Crops Prod., 2020, 148, 112181 CrossRef CAS.
- A. Bachs-Herrera, O. Yousefzade, L. J. del Valle and J. Puiggali, Appl. Sci., 2021, 11, 1808 CrossRef CAS.
- Y. Aranishi and Y. Nishio, in Blends and Graft Copolymers of Cellulosics: Toward the Design and Development of Advanced Films and Fibers, ed. Y. Nishio, Y. Teramoto, R. Kusumi, K. Sugimura and Y. Aranishi, Springer International Publishing, Cham, 2017, pp. 109–125, DOI:10.1007/978-3-319-55321-4_5.
- S. P. Maradur, C. H. Kim, S. Y. Kim, B.-H. Kim, W. C. Kim and K. S. Yang, Synth. Met., 2012, 162, 453–459 CrossRef CAS.
- X.-Y. Wang, G.-Y. Feng, M.-J. Li and M.-Q. Ge, Polym. Bull., 2019, 76, 2097–2111 CrossRef CAS.
- C. Lu, C. Blackwell, Q. Ren and E. Ford, ACS Sustainable Chem. Eng., 2017, 5, 2949–2959 CrossRef CAS.
- M. Yanilmaz and X. Zhang, Polymers, 2015, 7, 629–643 CrossRef CAS.
- W. Qu, J. Yang, X. Sun, X. Bai, H. Jin and M. Zhang, Int. J. Biol. Macromol., 2021, 189, 768–784 CrossRef CAS PubMed.
- P. Schlee, O. Hosseinaei, C. A. O’ Keefe, M. J. Mostazo-López, D. Cazorla-Amorós, S. Herou, P. Tomani, C. P. Grey and M.-M. Titirici, J. Mater. Chem. A, 2020, 8, 23543–23554 RSC.
- I. Khan, B. Hararak and G. F. Fernando, Sci. Rep., 2021, 11, 16237 CrossRef CAS PubMed.
- X. Xu, J. Zhou, L. Jiang, G. Lubineau, S. A. Payne and D. Gutschmidt, Carbon, 2014, 80, 91–102 CrossRef CAS.
- W. J. Youe, S. J. Kim, S. M. Lee, S. J. Chun, J. Kang and Y. S. Kim, Int. J. Biol. Macromol., 2018, 112, 943–950 CrossRef CAS PubMed.
- D. Lei, X.-D. Li, M.-K. Seo, M.-S. Khil, H.-Y. Kim and B.-S. Kim, Polymer, 2017, 132, 31–40 CrossRef CAS.
- S. Hu, S. Zhang, N. Pan and Y.-L. Hsieh, J. Power Sources, 2014, 270, 106–112 CrossRef CAS.
- W. Fang, S. Yang, T.-Q. Yuan, A. Charlton and R.-C. Sun, Ind. Eng. Chem. Res., 2017, 56, 9551–9559 CrossRef CAS.
- X. Ma, P. Kolla, Y. Zhao, A. L. Smirnova and H. Fong, J. Power Sources, 2016, 325, 541–548 CrossRef CAS.
- C. Lai, Z. Zhou, L. Zhang, X. Wang, Q. Zhou, Y. Zhao, Y. Wang, X.-F. Wu, Z. Zhu and H. Fong, J. Power Sources, 2014, 247, 134–141 CrossRef CAS.
- H. Khayyam, R. N. Jazar, S. Nunna, G. Golkarnarenji, K. Badii, S. M. Fakhrhoseini, S. Kumar and M. Naebe, Prog. Mater. Sci., 2020, 107, 100575 CrossRef CAS.
- Z. Xiong, N. Chen and Q. Wang, J. Appl. Polym. Sci., 2020, 137, 49385 CrossRef CAS.
- J. Yang, Y. Wang, J. Luo and L. Chen, ACS Omega, 2018, 3, 4647–4656 CrossRef CAS PubMed.
- F. Fu, D. Yang, W. Zhang, H. Wang and X. Qiu, Chem. Eng. J., 2020, 392, 123721 CrossRef CAS.
- H. Li, F. Shi, Q. An, S. Zhai, K. Wang and Y. Tong, Int. J. Biol. Macromol., 2021, 166, 923–933 CrossRef CAS PubMed.
- F. Liu, Z. Wang, H. Zhang, L. Jin, X. Chu, B. Gu, H. Huang and W. Yang, Carbon, 2019, 149, 105–116 CrossRef CAS.
- W.-M. Yin, L.-F. Tian, B. Pang, Y.-R. Guo, S.-J. Li and Q.-J. Pan, Int. J. Biol. Macromol., 2020, 156, 988–996 CrossRef CAS PubMed.
- F. Shi, J. Li, J. Xiao, X. Zhao, H. Li, Q. An, S. Zhai, K. Wang, L. Wei and Y. Tong, Int. J. Biol. Macromol., 2021, 190, 11–18 CrossRef CAS PubMed.
- M. Zhou, A. Bahi, Y. Zhao, L. Lin, F. Ko, P. Servati, S. Soltanian, P. Wang, Y. Yu, Q. Wang and Z. Cai, Chem. Eng. J., 2021, 409, 128214 CrossRef CAS.
- T. Wang, S. Hu, D. Wu, W. Zhao, W. Yu, M. Wang, J. Xu and J. Zhang, J. Mater. Chem. A, 2021, 9, 11839–11852 RSC.
- M. Yuan, F. Luo, Y. Rao, Y. Wang, J. Yu, H. Li and X. Chen, J. Power Sources, 2021, 513, 230558 CrossRef CAS.
- Z. Dai, P.-G. Ren, W. He, X. Hou, F. Ren, Q. Zhang and Y.-L. Jin, Renewable Energy, 2020, 162, 613–623 CrossRef CAS.
- B. Du, X. Wang, L. Chai, X. Wang, Z. Pan, X. Chen, J. Zhou and R.-C. Sun, Int. J. Biol. Macromol., 2022, 194, 632–643 CrossRef CAS PubMed.
- Z.-R. Hu, D.-D. Li, T.-H. Kim, M.-S. Kim, T. Xu, M.-G. Ma, S.-E. Choi and C. Si, Front. Chem., 2022, 10, 841956 CrossRef CAS PubMed.
- M. Culebras, C. M. Gómez and A. Cantarero, J. Mater. Chem. A, 2014, 2, 10109 RSC.
- M. Culebras, C. Gómez and A. Cantarero, Materials, 2014, 7, 6701–6732 CrossRef PubMed.
- M. Culebras, B. Uriol, C. M. Gomez and A. Cantarero, Phys. Chem. Chem. Phys., 2015, 17, 15140–15145 RSC.
- M. Culebras, M. M. de Lima, C. Gómez and A. Cantarero, J. Appl. Polym. Sci., 2017, 134(3), 43927 CrossRef.
- J. Gao, C. Liu, L. Miao, X. Wang, C. Li, R. Huang, Y. Chen and S. Tanemura, Synth. Met., 2015, 210, 342–351 CrossRef CAS.
- H. Shi, C. Liu, J. Xu, H. Song, B. Lu, F. Jiang, W. Zhou, G. Zhang and Q. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 12811–12819 CrossRef CAS PubMed.
- M. Culebras, A. M. Igual-Muñoz, C. Rodríguez-Fernández, M. I. Gómez-Gómez, C. Gomez and A. Cantarero, ACS Appl. Mater. Interfaces, 2017, 9, 20826–20832 CrossRef CAS PubMed.
- N. Dalton, R. P. Lynch, M. N. Collins and M. Culebras, Int. J. Biol. Macromol., 2019, 121, 472–479 CrossRef CAS PubMed.
- M. Culebras, G. Ren, S. O'Connell, J. J. Vilatela and M. N. Collins, Adv. Sustainable Syst., 2020, 4, 2000147 CrossRef CAS.
- X. Zhao, C. Huang, D. Xiao, P. Wang, X. Luo, W. Liu, S. Liu, J. Li, S. Li and Z. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 7600–7607 CrossRef CAS PubMed.
- J. Chen, J. Qi, J. He, Y. Yan, F. Jiang, Z. Wang and Y. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 12748–12757 CrossRef CAS PubMed.
- R. Shu, R. Li, B. Lin, C. Wang, Z. Cheng and Y. Chen, Biomass Bioenergy, 2020, 132, 105432 CrossRef CAS.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
- H. Wang, L. Zhang, T. Deng, H. Ruan, X. Hou, J. R. Cort and B. Yang, Green Chem., 2016, 18, 2802–2810 RSC.
- H. Wang, H. Wang, E. Kuhn, M. P. Tucker and B. Yang, ChemSusChem, 2018, 11, 285–291 CrossRef CAS PubMed.
- M. M. Ambursa, J. C. Juan, Y. Yahaya, Y. H. Taufiq-Yap, Y.-C. Lin and H. V. Lee, Renewable Sustainable Energy Rev., 2021, 138, 110667 CrossRef CAS.
- M. Zhao, J. Hu, P. Lu, S. Wu, C. Liu and Y. Sun, Fuel, 2022, 326, 125020 CrossRef CAS.
- C. G. Yoo, A. Dumitrache, W. Muchero, J. Natzke, H. Akinosho, M. Li, R. W. Sykes, S. D. Brown, B. Davison, G. A. Tuskan, Y. Pu and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2018, 6, 2162–2168 CrossRef CAS.
- H. J. Pegoretti, L. de Souza, F. Muñoz, R. T. Mendonça, K. Sáez, R. Olave, C. Segura, D. P. L. de Souza, T. de Paula Protásio and R. Rodríguez-Soalleiro, Renewable Energy, 2021, 163, 1802–1816 CrossRef.
- H. Ohta, K. Yamamoto, M. Hayashi, G. Hamasaka, Y. Uozumi and Y. Watanabe, Chem. Commun., 2015, 51, 17000–17003 RSC.
- H. Wang, H. Ruan, M. Feng, Y. Qin, H. Job, L. Luo, C. Wang, M. H. Engelhard, E. Kuhn, X. Chen, M. P. Tucker and B. Yang, ChemSusChem, 2017, 10, 1846–1856 CrossRef CAS PubMed.
- T. Guo, Q. Xia, Y. Shao, X. Liu and Y. Wang, Appl. Catal., A, 2017, 547, 30–36 CrossRef CAS.
- Y. Cao, S. S. Chen, S. Zhang, Y. S. Ok, B. M. Matsagar, K. C. W. Wu and D. C. W. Tsang, Bioresour. Technol., 2019, 291, 121878 CrossRef CAS PubMed.
- S. G. Yao, Ph.D., University of Kentucky, 2018.
- S. G. Yao, J. K. Mobley, J. Ralph, M. Crocker, S. Parkin, J. P. Selegue and M. S. Meier, ACS Sustainable Chem. Eng., 2018, 6, 5990–5998 CrossRef CAS.
- S. Dabral, H. Wotruba, J. G. Hernández and C. Bolm, ACS Sustainable Chem. Eng., 2018, 6, 3242–3254 CrossRef CAS.
- Y. Wang, J. He and Y. Zhang, CCS Chem., 2020, 2, 107–117 CrossRef CAS.
- Y. Kang, X. Lu, G. Zhang, X. Yao, J. Xin, S. Yang, Y. Yang, J. Xu, M. Feng and S. Zhang, ChemSusChem, 2019, 12, 4005–4013 CrossRef CAS PubMed.
- H. Chen, K. Wan, F. Zheng, Z. Zhang, Y. Zhang and D. Long, Renewable Sustainable Energy Rev., 2021, 147, 111217 CrossRef CAS.
- Z. Hao, S. Li, J. Sun, S. Li and F. Zhang, Appl. Catal., B, 2018, 237, 366–372 CrossRef CAS.
- S. Li, Z. Hao, K. Wang, M. Tong, Y. Yang, H. Jiang, Y. Xiao and F. Zhang, Chem. Commun., 2020, 56, 11243–11246 RSC.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- Z. Xiang, W. Han, J. Deng, W. Zhu, Y. Zhang and H. Wang, ChemSusChem, 2020, 13, 4199–4213 CrossRef CAS PubMed.
- H. Yoo, M.-W. Lee, S. Lee, J. Lee, S. Cho, H. Lee, H. G. Cha and H. S. Kim, ACS Catal., 2020, 10, 8465–8475 CrossRef CAS.
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS PubMed.
- S. V. Obydenkova, P. D. Kouris, E. J. M. Hensen, H. J. Heeres and M. D. Boot, Bioresour. Technol., 2017, 243, 589–599 CrossRef CAS PubMed.
- I. O. f. Standardization, Environmental management: life cycle assessment; requirements and guidelines, ISO Geneva, Switzerland, 2006 Search PubMed.
- C. Culbertson, T. Treasure, R. Venditti, H. Jameel and R. Gonzalez, Nord. Pulp Pap. Res. J., 2016, 31, 30–40 CrossRef CAS.
- D. G. Kulas, M. C. Thies and D. R. Shonnard, ACS Sustainable Chem. Eng., 2021, 9, 5388–5395 CrossRef CAS.
- S. Zargar, J. G. Jiang, F. Jiang and Q. S. Tu, Biofuels, Bioprod. Biorefin., 2022, 16, 68–80 CrossRef CAS.
- D. Koch, M. Paul, S. Beisl, A. Friedl and B. Mihalyi, J. Cleaner Prod., 2020, 245, 118760 CrossRef CAS.
- L. Zeilerbauer, J. Lindorfer, R. Suss and B. Kamm, Biofuels, Bioprod. Biorefin., 2022, 16, 370–388 CrossRef CAS.
- P. N. Shinde, S. A. Mandavgane and V. Karadbhajane, Environ. Sci. Pollut. Res., 2020, 27, 25785–25793 CrossRef CAS PubMed.
- K. C. Teh, C. Y. Tan and I. M. L. Chew, J. Environ. Chem. Eng., 2021, 9, 105381 CrossRef CAS.
- F. Liu, X. Y. Dong, X. B. Zhao and L. Wang, Energy Convers. Manage., 2021, 246, 114653 CrossRef CAS.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. M. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Roman-Leshkov and G. T. Beckham, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- N. M. Kosamia, M. Samavi, K. Piok and S. K. Rakshit, Fuel, 2022, 324, 124532 CrossRef.
- E. Bernier, C. Lavigne and P. Y. Robidoux, Int. J. Life Cycle Assess., 2013, 18, 520–528 CrossRef CAS.
- C. Moretti, R. Hoefnagels, M. van Veen, B. Corona, S. Obydenkova, S. Russell, A. Jongerius, I. Vural-Gürsel and M. Junginger, J. Cleaner Prod., 2022, 343, 131063 CrossRef CAS.
- C. Moretti, B. Corona, R. Hoefnagels, M. van Veen, I. Vural-Gursel, T. Strating, R. Gosselink and M. Junginger, Sci. Total Environ., 2022, 806, 150316 CrossRef CAS PubMed.
- O. O. Tokede, A. Whittaker, R. Mankaa and M. Traverso, Structures, 2020, 25, 190–199 CrossRef.
- Y. C. Yue, M. Abdelsalam, A. Khater and M. Ghazy, Constr. Build. Mater., 2022, 323, 126608 CrossRef CAS.
- A. Khater, D. Luo, M. Abdelsalam, M. Ghazy and Jianxun, Materials, 2021, 14, 6589 CrossRef CAS PubMed.
- J. E. McDevitt and W. J. Grigsby, J. Polym. Environ., 2014, 22, 537–544 CrossRef CAS.
- L. O. Cortat, N. C. Zanini, R. F. S. Barbosa, A. G. de Souza, D. S. Rosa and D. R. Mulinari, J. Polym. Environ., 2021, 29, 3210–3226 CrossRef CAS.
- M. L. Yang and K. A. Rosentrater, Environ. Processes, 2020, 7, 553–561 CrossRef CAS.
- J. Hildebrandt, M. Budzinski, R. Nitzsche, A. Webe, A. Krombholz, D. Thran and A. Bezama, Resour., Conserv. Recycl., 2019, 141, 455–464 CrossRef.
- C. Isola, H. L. Sieverding, A. Numan-Al-Mobin, R. Rajappagowda, E. A. Boakye, D. E. Raynie, A. L. Smirnova and J. J. Stone, Int. J. Life Cycle Assess., 2018, 23, 1761–1772 CrossRef CAS.
- M. Montazeri and M. J. Eckelman, ACS Sustainable Chem. Eng., 2016, 4, 708–718 CrossRef CAS.
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Green Chem., 2018, 20, 3857–3866 RSC.
- R. Akmalina and M. G. Pawitra, Asia–Pac. J. Chem. Eng., 2020, 15, e2436 CAS.
- E. Budsberg, R. Morales-Vera, J. T. Crawford, R. Bura and R. Gustafson, Biotechnol. Biofuels, 2020, 13, 154 CrossRef CAS PubMed.
- S. V. Mankar, M. N. G. Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Z. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- F. Hermansson, M. Janssen and M. Svanström, J. Cleaner Prod., 2019, 223, 946–956 CrossRef CAS.
- M. Secchi, V. Castellani, M. Orlandi and E. Collina, BioResources, 2019, 14, 4832–4865 CAS.
- S. Soam, M. Kapoor, R. Kumar, R. P. Gupta, S. K. Puri and S. S. V. Ramakumar, J. Cleaner Prod., 2018, 197, 732–741 CrossRef CAS.
- E. Budsberg, J. T. Crawford, H. Morgan, W. S. Chin, R. Bura and R. Gustafson, Biotechnol. Biofuels, 2016, 9, 170 CrossRef PubMed.
- J. K. Raman and E. Gnansounou, Bioresour. Technol., 2015, 185, 202–210 CrossRef CAS PubMed.
- A. W. Bartling, P. T. Benavides, S. D. Phillips, T. Hawkins, A. Singh, M. Wiatrowski, E. C. D. Tan, C. Kinchin, L. W. Ou, H. Cai, M. Biddy, L. Tao, A. Young, K. Brown, S. Y. Li, Y. H. Zhu, L. J. Snowden-Swan, C. R. Mevawala and D. J. Gaspar, ACS Sustainable Chem. Eng., 2022, 10(10), 6699–6712 CrossRef CAS.
- N. Mahendrasinh Kosamia, M. Samavi, K. Piok and S. Kumar Rakshit, Fuel, 2022, 324, 124532 CrossRef CAS.
- T. Bicalho, J. Richard and C. Bessou, Sustainability Account. Manage. Policy J., 2012, 3, 218–234 CrossRef.
- F. Hermansson, M. Janssen and M. Svanstrom, Int. J. Life Cycle Assess., 2020, 25, 1620–1632 CrossRef.
- C. Moretti, B. Corona, R. Hoefnagels, I. Vural-Gursel, R. Gosselink and M. Junginger, Sci. Total Environ., 2021, 806(1), 150316 Search PubMed.
- K. Navare, W. Arts, G. Faraca, G. V. D. Bossche, B. Sels and K. V. Acker, Resour., Conserv. Recycl., 2022, 186, 106588 CrossRef CAS.
- J. Wenger, S. Pichler, A. Nayha and T. Stern, Sustainability, 2022, 14, 2619 CrossRef.
- G. Sandin, F. Royne, J. Berlin, G. M. Peters and M. Svanstrom, J. Cleaner Prod., 2015, 93, 213–221 CrossRef.
- S. V. Obydenkova, P. D. Kouris, D. M. J. Smeulders, M. D. Boot and Y. van der Meer, Biofuels, Bioprod. Biorefin., 2021, 15, 1281–1300 CrossRef CAS.
- A. Arias, S. González-García, S. González-Rodríguez, G. Feijoo and M. T. Moreira, Sci. Total Environ., 2020, 738, 140357 CrossRef CAS PubMed.
- P. Yadav, D. Athanassiadis, I. Antonopoulou, U. Rova, P. Christakopoulos, M. Tysklind and L. Matsakas, J. Cleaner Prod., 2021, 279, 123515 CrossRef CAS.
- U. S. E. P. Agency, Overview for Renewable Fuel Standard, https://www.epa.gov/renewable-fuel-standard-program/overview-renewable-fuel-standard, (accessed 22/08/2022, 2022).
| This journal is © The Royal Society of Chemistry 2022 |