
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStatus and gaps toward fossil-free sustainable chemical production†
Gabriele
Centi
 * and
Siglinda
Perathoner
*
* and
Siglinda
Perathoner
*
Department ChiBioFarAm – Section of Industrial Chemistry, University of Messina, ERIC aisbl and CASPE–INSTM, V.le F. Stagno d'Alcontres 31, 98166, Messina, Italy. E-mail: centi@unime.it; perathon@unime.it
First published on 24th August 2022
Abstract
Chemical production needs to transform radically toward fossil-free sustainable chemical production to meet the targets for net-zero emissions by the year 2050. The feasibility of this transformation, the motivations, status and gaps, and perspectives are discussed after introducing how this change also implies a change in the model of production. Realizing the defossilization of chemical production involves electrifying the chemical processes, especially crucial elements such as chemical reactors, and the direct use of renewable energy to drive the chemical reaction. With a focus on electrocatalysis, the most relevant cases of (i) light olefin production, (ii) direct synthesis of main intermediates such as formaldehyde and acetic acid, and (iii) the production of aromatics are analyzed. The feasibility of these routes in the short–medium term is shown, while other cases such as the direct synthesis of ammonia from N2 require turning the approach to other directions. On a global scale, defossilization of chemical production is feasible in the medium–long term with a cut of over 800 Mt per year CO2 eq. emissions in line with the expectation to reach the net-zero emission target. A final section introduces a short discussion about some critical questions regarding the sustainability of fossil-free chemical production.
 Gabriele Centi | Gabriele CENTI is Full Professor of Industrial Chemistry at the University of Messina, Italy, and President of the European Research Institute of Catalysis (ERIC). He was the coordinator of the EU Network of Excellence IDECAT and is currently President of the IACS (International Association of Catalysis Societies). He recently initiated and coordinated an ERC Synergy project on plasma-catalysis. He is also a member of the board of SUNERGY, the European initiative on solar fuels. He has received numerous awards, including the International Fellowship Initiative of the President of the Chinese Academy of Sciences, PIFI, as a Distinguished Scientist, and the Humboldt Research Award, and is involved in various publishing activities. He is the author of over 600 scientific publications, 12 books and editor of over 20 special issues of journals. The current h-index is 90 with around 33 |
 Siglinda Perathoner | Siglinda PERATHONER gained her PhD in Chemical Science in 1988 working on the photophysics and photochemistry of supramolecular systems with V. Balzani and Nobel Laureate J. M. Lehn. In 2001 she joined the University of Messina and is presently a full professor of Industrial Chemistry. She has coordinated many EU projects. She was co-chair of Europacat 2017, an important event in the catalysis community, and chaired several other conferences. Recent recognition includes the 2021 G.M. Levi Medal from the Italian Chemical Society (for innovation leading to industrial realization) and a President's International Fellowship Initiative (PIFI) award from CAS (Chinese Academy). The current h-index is 74 with over 23 |
Introduction
Chemical production is facing a major challenge in the next decades, due to a combination of factors:- The pressure to phase out the dependence on fossil fuels (FFs) to reduce greenhouse gas (GHG) emissions and meet socio-political targets such as reaching Net Zero Emissions by the year 2050.
- The change in the nexus between energy and chemistry, which is largely related to the ongoing energy transition, and its impact on the availability and costs of raw materials for chemical production.
- The modification in the economics of the overall value chain, due to increased competitiveness of renewable energy sources, and the loss of profitability windows in several elements of petrochemical production.
- The geopolitical pressure to overcome the limitations related to the large monopoly of FF production and distribution.
Cetinkaya et al.1 of McKinsey & Co. (a major consulting company) some years ago warned petrochemistry that “the old models for value creation are losing traction and that the advantaged-feedstock-opportunity window is closing”, indicating the need to change the model of development.
Drivers to develop a new model of chemical production
We could identify two main driving factors in the current model of petrochemical production:2 the use of FFs and the scale economy. These are two interlinked components of the model of development because over 90% of the raw materials for chemical production are based on FFs, but less than 40% on average is used as carbon sources or to increase the energy value of the product (the ratio of the output energy potential useful – exergy – to the potential exergy input for chemical production is around 30%).3Most of the FFs used in petrochemistry are needed to produce the heat and the energy used in the chemical processes. In addition, most of the chemicals are not produced directly, but involve an often-complex sequence of processes, thus with a progressive loss of energy efficiency (in the global process). Chemical production is strongly linked to the combustion of FFs used to produce the heat and energy necessary to run the chemical transformation and separation. Heat recovery is thus a critical element to achieve acceptable overall energy efficiency, and from here the motivation to have a scale economy, e.g., large plants integrated into complex chemical/refinery sites. Heat recovery is efficient only in large-scale plants.
This model of chemical production is now suffering from significant limitations, from the large environmental impact to the low circularity and integration with local resources. Especially critical now is the low capacity to adapt to a changing world where flexibility in production, time to market and the possibility of accessing a wider range of investors are the keywords for success. Transformation of chemical production is thus motivated not only by the need to reduce the carbon footprint, but also derives from several interlinked social, economic and industrial factors. The result is a push to develop new models of chemical production, generating new windows of opportunity overcoming the strong hierarchical structure of current chemical production: oil refinery, then production of chemical raw materials, then base and intermediate chemicals, etc. Distributed chemical manufacturing, modularity and scale-up of chemical processes by numbering are among the emerging elements of a novel model of chemical production,4–7 requiring new technologies largely based on the new opportunities offered by renewable energy sources (RES) and process intensification.8
Developing FF-free chemical production is thus an opportunity to create new models of growth and economic development, value chains and innovation, and not only a target to reduce GHG emissions.
The new model of fossil-free sustainable chemical production
There are different opinions on whether phasing-out FFs from chemical production is feasible from an economic perspective, or even needed. In addition, quite a spread of opinions exists on timing; for example, whether substituting the use of FFs will be a key element to meet 2050 net-zero emissions targets, or whether instead it will occur in an even longer-term perspective.More detailed discussion of these aspects could defocus this perspective. Some further considerations and supporting data for the discussion were earlier reported.9–11 Here we limit the analysis to the status, gaps and limits of the developments in the area to provide suggestions for this debate from a technical and socio-economic perspective. In addition, the aim is to offer a different, vision-oriented analysis of the opportunities for research in this area, rather than to debate in detail the pros/cons of substituting FF use in chemical production.
We call this novel chemical production alternative to the current petro-chemistry ‘e-chemistry’.12 We assume a high-tech scenario of transformation of chemical production, driven by the forces and motivations indicated before. This high-tech scenario requires an intense R&D effort to develop the new routes necessary to substitute the use of FFs in chemical production, with the energy largely provided by renewable sources, if not directly from solar light, and the use of alternative carbon sources to FFs.
This high-tech scenario considers that a significant part of the chemical products will be transformed in this direction before 2050. From around the years 2030–35, it is expected that new plants based on the old petro-chemical routes will not be introduced. These are assumptions based on the realization of this high-tech scenario, which is difficult to prove, especially regarding the timing. However, even if this scenario is realized later, it is still necessary to consider and prepare the technologies, catalysts, etc. for this transformation. There are already various indications that this transformation has irreversibly started.
Alternatives in transforming chemical production
The chemical and refinery industry is already showing signs of recognition of the change in the value chain and nexus between refinery and chemical production,13 because of the loss of economic value in producing liquid fuels in refineries. One of the signs is the increasing attention given to the so-called crude oil-to-chemicals (COTC) processes.14–16 This term indicates the direct conversion of crude oil to high-value chemical products (olefins, in particular) rather than producing them from refinery side streams. The industrial effort in this direction is a clear sign that the energy–chemistry nexus and the refinery–chemical production value chain are transforming.Several COTC projects (announced or started) plan the reconfiguration of refineries to enhance the production of chemicals (>40% per barrel of oil to chemicals, rather than <10% as in a conventional refinery).15,16 Among the strategies explored in COTC plants are (i) direct processing of crude oil in steam cracking, (ii) integrated hydro-processing/de-asphalting and steam cracking and (iii) processing of middle distillates and residues using hydrocracking technology.
These projects aim to preserve the use of FFs as the backbone of chemical production, refocusing the refinery production on higher added-value products. Thus, the production of raw materials for petrochemistry is directly integrated into the refinery scheme. The impact of COTC in meeting low or zero-carbon targets is negligible, or only indirect (COTC plants will decrease the production of transport fuels). In addition, adapting the refineries to the production of raw materials for petrochemistry will increase further the size of the actual steam crackers (the most used technology to produce olefins, with plants having an average size of 500 kt per year) by a factor of 4–10. The consequence is a further lowering of the capability to adapt to current market dynamics, the long-distance transport of chemicals and the environmental/safety impact, etc; in other words, the opposite of what is required to transform chemical production into a more sustainable model. Thus, the COTC solution, although actively considered by refineries, does not appear to be the necessary strategy to move to fossil-free sustainable chemical production, but rather is a short-term strategy to continue to preserve the dominant role of FFs in chemical production.
Another general objection to moving to FF-free chemical production is that a reduction of the carbon footprint could be achieved by substituting the use of FFs as the source of heat/energy (electrification of furnaces, etc.) while still maintaining hydrocarbons as the preferable carbon source for chemical production. The use of FFs preserves the linear model of the use of carbon, from the extraction of FFs to the production of waste and CO2 as the final products.9 A sustainable model of production must have as a target the closure of the C-cycle (circular carbon use), by increasing the use of waste/CO2 as a carbon source, integrated eventually with the use of low-carbon footprint raw materials (biomass).
Thus, the motivations for substituting the use of FFs as a carbon source in chemical production are to enhance sustainability, realize the integration with local resources, and application of a different model of production with reduced dependence on external resources that have a high monopoly character, etc. This does not necessarily correspond to higher costs but to a different window of opportunities, which is a direct consequence of the change in future societal and industrial models of development.
Looking to FF-free sustainable chemical production is thus not an academic exercise, but an effective, industrially relevant direction to explore for a new model of chemical production.
Scope and limits
This perspective aims to provide a holistic analysis of the transformation to fossil-free chemical production which is not present in the literature. It is thus organized into a first part in which the socio-economic motivations are analysed, because often a comprehensive analysis of these elements is missing, even though the discussion is limited to the main elements and depends on the scenario assumed.The second part analyses the feasibility of implementing a high degree of substitution in the use of FFs for chemical production, and the key technologies, and their status/gaps, to realize this challenge. Transforming chemical production toward a fossil-free model is a deep transition, requiring a simultaneous and synergic convergence of the various technologies necessary. For this reason, the focus here is to provide this general analysis, rather than to discuss each technology in depth, which would defocus the discussion. At the same time, some of the aspects that need to be better understood to implement the technologies under development are highlighted.
The third part then attempts to analyse the impact in terms of reduction of GHG associated with the transformation to fossil-free chemical production, with the fourth and final part dedicated to analysing some of the questions about the sustainability of this transformation.
To focus this manuscript, the discussion here is limited to technologies that have direct use of renewable energy (RE) and alternative carbon sources to FFs. Power-to-X (PtX) technologies, where the only step of direct use of RE is the production of H2, by electrolysis, are not analysed here. These technologies have been developed at already relatively high TRL (technology readiness levels) and are discussed elsewhere.17–21 They suffer, in general, from high costs and ongoing issues in coupling the different steps (electrolysis followed by one or more downstream thermocatalytic steps). Direct technologies to the final products, for example, the direct electrocatalytic conversion of CO2 and H2O to methanol or ethylene, offer potential advantages in terms of energy efficiency, process intensification and cost reduction, although their development is still challenging.22
To demonstrate the need for direct electrocatalytic production technologies, the exergy loss by comparing the direct versus multistep processes in olefin production could be analysed.23 For future low-carbon production, exergy analysis offers a better, integrated evaluation tool to consider the energy and material efficiency of a process.24 We can compare two solutions, both based on the production of light olefins from CO2, H2O and renewable energy. The first route A is based on technology already at an advanced stage of development, based on the production of H2 by electrolysis, then thermocatalytic conversion of CO2 + H2 to methanol, and a further stage of methanol-to-olefin (MTO) conversion. The exergy loss, in this case, is about 50%, i.e., about half of the exergy (the capacity of energy to do physical work) is lost in the transformation. If biomass is used via gasification and methanol synthesis, the exergy loss is even higher (around 70%),25 while slightly below 50% for waste to olefins via methanol synthesis and for the conventional steam cracking process.23 Even for an advanced electrocatalytic process, like the co-electrolysis at high temperature of CO2 and H2O to directly produce syngas followed by methanol/MTO processes step,25 the exergy loss is about 50%. This can be estimated by combining the data on the exergy of methanol production via syngas (by co-electrolysis)26 with the data on the exergy of the MTO. In a direct CO2 to olefin electrocatalytic transformation, an energy efficiency above 70% could be a feasible target. Thus, developing a low-carbon chemical production alternative to current petrochemistry would always require considering direct electrocatalytic processes, when possible, even if challenging.
It should be pointed out that in the MTO process a range of hydrocarbons is produced, requiring complex separation procedures as outlined in Fig. 1a. In the case of the direct electrocatalytic conversion of CO2 and H2O (Fig. 1b), a simplified procedure of separation is possible thanks to the higher selectivity of the process. Following the scheme proposed by Pérez-Ramírez and coworkers,27 a sequence of three PSA (pressure swing absorption) units may allow the separation, with recycling of CO2, H2O and eventually H2 in the case of a pressurized electrocatalytic unit. As visually emerges from the comparison of these two routes, route B of direct electrocatalytic conversion, concerning the multistep process involving electrolytic production of H2 followed by a sequence of thermocatalytic steps, requires fewer steps, with thus a potential reduction of fixed and operative costs, in addition to the potential higher exergy efficiency.
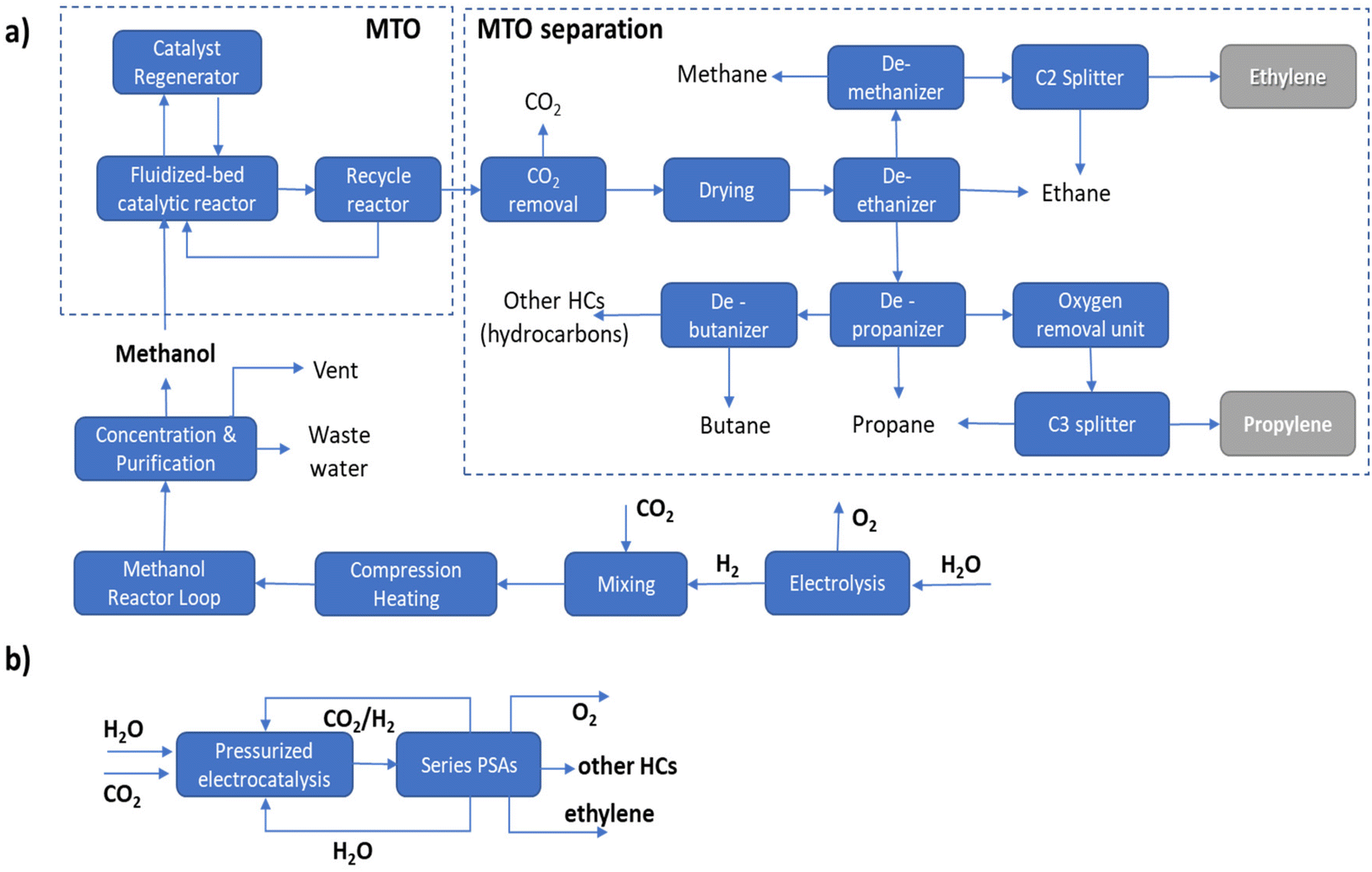 | ||
| Fig. 1 Simplified block scheme of the process to synthesize olefins from CO2: (a) route A via producing H2 by electrolysis followed by thermocatalytic synthesis of methanol then converted to olefins by MTO process; (b) route B of direct production of ethylene by electrocatalytic conversion of CO2 and H2O. | ||
The potential benefits in intensified processes (route B versus route A) are maximized in distributed chemical production models, e.g., when small–medium-scale bio/solar refineries well integrated with the use of local resources will supply the needs of materials and other chemicals for the territory.28,29 The current model of chemical production is based on applying scaling laws in process engineering, pushing to realize larger-scale productions which do not account for factors like environmental impact, transport costs, the capability of adapting to a highly variable demand and cost in raw materials, etc. These factors are currently pushing the need to change the production model toward distributed chemical production. In distributed production models, a requirement is a change of the technology, because intensified and highly adaptable technologies would be required.8 Direct electrification of chemical production offers advantages in this sense over conventional thermocatalytic processes.
Many aspects contribute to determining the industrial feasibility, as reviewed recently by Mbatha et al.30 for the power-to-methanol case. Among them, the availability of the technology at sufficiently high TRL is crucial. However, when considering the future of chemical production from a fossil-free sustainable perspective, the question is not only which technologies will be ready in the short term, but which technologies should be pushed to realize future production. For this reason, we focus discussion here mainly on direct electrification routes.
While other papers have reviewed the state-of-the-art of some of the topics addressed later, an analysis integrated into a framework assessment of this FF-free chemical production is generally missing. Furthermore, rather than focus on reviewing the scientific results with the identification of some design criteria to improve the performances (the scope of most of the reviews in the sector), the aim here is to analyse the technical feasibility, or identify gaps and limits, for the application as a novel sustainable production route to be part of a distributed chemical production model based on the use of local RES and carbon sources.
In addition, for the motivations explained above, we do not consider here technologies to produce CO from CO2, being necessary then the further thermocatalytic upgrading to convert CO with H2, the latter also produced separately by electrolysis or together with CO in co-electrolysis SOEC (solid oxide electrolyzer cell) technology.31–35 Many researchers believe that this is the preferable route, in particular, syngas by co-electrolysis followed by thermocatalytic syngas conversion. We consider instead that investing in the development of this path as a preferential route to produce the raw materials for the future low-carbon chemistry may result in a slow-down of the transformation to a future fossil-free sustainable chemical production. This is a matter for discussion, and clear demonstrations of support for one or the other position are lacking. However, considering the investments which are needed, and the amortization time necessary (typically two decades), investing in intermediate technologies may result in a delay in the whole transformation process, rather than realising a step-by-step renovation. Although challenging, the process intensification possible by direct synthesis using RE should be the target,8,36 even if the multistep routes via syngas production (for example, by co-electrolysis) is a more mature technology. We thus limit the discussion here to this scenario, but with the remark that its realization would require intensifying the research effort, and industrial convergence on it.
Defossilizing chemical production
The recent report of the International Energy Agency (IEA) on the role of chemical production to meet the NZE target37 showed that direct CO2 emissions from primary chemical production were 920 Mt CO2 in 2020, the third largest industry subsector in terms of direct CO2 emissions, behind cement and iron and steel. However, it is expected that it will be difficult for these two industrial sectors to reduce emissions to meet the target of Net Zero Emissions by 2050 scenario. Chemical production should instead achieve the NZE target even before 2050 according to the IEA scenario. Thus an even higher rate of decrease in greenhouse gas emissions is required for the chemical sector than the average for the industrial sector. If the actual trend in the reduction is extrapolated to 2050 (see Fig. 2), only around half of the target reduction necessary to arrive at NZE by 2050 will be achieved. Defossilization of chemical production thus requires (i) to more than triple the average yearly rate of CO2 reduction with reference to what was made in the last decade,38 and (ii) have a rate of decrease for the chemical sector even higher than that of other industrial sectors. Thus, these are challenging objectives.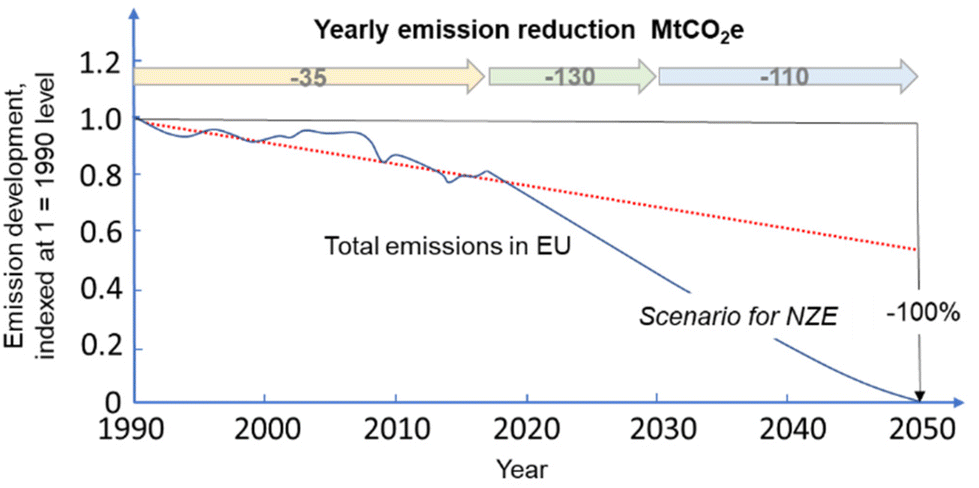 | ||
| Fig. 2 Trend in total emissions in EU (indexed at 1 = 1990) and predicted trend to meet the NZE scenario. Based on indications in ref. 38. | ||
Direct CO2 emissions in the production of chemicals derive both from energy and process emissions. Around half of the chemical energy input is consumed as feedstock, i.e., fuel used as raw material input rather than as a source of energy. If the process emissions are also added, between 30–40% of the inlet carbon as FF is retained in the final chemicals/materials (even less in some cases), while the remainder is used for the transformation and is emitted as CO2.
Even the part of the carbon stored in the chemicals/materials (the final products of the chemical transformation) at the end of the life result in further emissions. The average non-biogenic content of municipal solid wastes (MSW) is typically around 40–50%, thus resulting in additional CO2 emissions, for example when these wastes are sent to incineration. Furthermore, additional chemicals and materials are released into the environment and are not part of MSW.
Thus, together with the electrification of unit operations (especially chemical reactors) in industrial plants to reduce the use of FFs as an energy source for the operation of these units, it is also necessary to substitute FFs as the carbon source for the chemical industry to meet the requirements for NZE targets. The strategies to achieve emissions targets of the chemical industry should thus include:
(i) Phasing out non-renewable energy resources,
(ii) Fostering a transition to a circular economy and the use of green electricity, and
(iii) Electrifying heating and industrial processes.39–41
Primary chemicals include ethylene, propylene, benzene, toluene, mixed xylenes, ammonia and methanol. Their production accounts for two-thirds of energy consumption in the chemical and petrochemical sector. An analysis of the possibilities and scenario toward a fossil-free sustainable chemical production could thus start from the analysis of how to realize the defossilization of these primary chemicals. As these chemicals are also largely the input for many intermediate and final chemicals,2 focusing attention on them implies a cascade effect on industrial chemical production as a whole.
We will start with the analysis of the electrification of the chemical processes, with a focus on reactors.
Electrification of the chemical processes and reactors
Electrification of the chemical processes involves a broad range of options. The objective is to substitute with green electricity operations currently involving FFs, thus mainly those related to furnaces (accounting for about half of the current emissions of CO2 in energy-intensive industries),42 but also those associated with the production of steam, hot water, and space cooling/heating.An estimation of the electrification potential in the EU for the chemical sector is reported in Table 1.43 The electricity demand can be divided into two aspects: that necessary to supply heating or cooling (indicated as electricity thermal), and that used in mechanical power and lighting, indicated as electricity other. The energy from combustible fuels can be separated by considering the temperature range of use. In general, about one-third of the energy input is lost due to inefficiencies and energy losses within the plant.
| Energy service | End-use | Share, % | Electrification technology |
|---|---|---|---|
| Other | - General appliances & machinery | 13 | |
| Thermal | - Space heating | 9 | |
| - Process cooling | |||
| < 100 °C | - Space heating | 6 | - Electric boilers |
| - Compression heat pumps | |||
| - Thermal cooling | - Microwave and infrared dryers | ||
| - Thermal drying | - Compression chillers | ||
| - Thermoelectric cooling | |||
| 100–400 °C | - Steam | 59 | - Electric boilers |
| - Polymers manufacture (e.g. extruders, melters) | - MVR | ||
| - Microwave and infrared dryers | |||
| - Electric reactors | |||
| 400–1000 °C | - Crackers & reformers (fuels only) | 13 | - Electric crackers and reformers |
| - Superheaters | - Electric boilers | ||
| - Limekilns | - Electric calciners |
Power-to-heat is a term often used to indicate these technologies. They include the use of various electromagnetic (e.g., induction, magnetic, microwave, plasma) and heat pump technologies to substitute those actually in use, mainly related to heat exchangers and FF combustion. Electromagnetic heating offers in general a more precise control of the temperature, faster response, and in some cases also the possibility to induce rapid modulations in the temperature, and a broader turndown across a range of operating conditions. However, most of these technologies are still at the laboratory or pilot scale. The deployment will also require more efficient power sources and modular platforms.41
Electricity can also provide the driving force for pressure-driven membrane separation processes or the production of H2 by electrolysis of water.44 Hydrogen can be then used for high-temperature heating. Note that in moving to distributed models of production, the use of power-to-heat technologies offers the additional advantage of process intensification, which is a requirement for small-scale production. In addition, heat integration is typically less efficient in small-scale processes, where high flexibility in production is another requirement. These aspects have to be accounted for when adding further benefits in the introduction of power-to-heat technologies for distributed, small-size chemical production plants.
Heat pumps are an alternative to traditional steam or oil heating. By using novel fluids (liquid CO2, for example), heat pumps can be adapted to lower and higher temperature ranges than the current ones. However, especially for high-temperature operations, costs and stability are still issues. Electromagnetic heating offers novel possibilities for optimizing reactor and furnace design, in addition to a drastic cut in CO2 emissions (possible only when green electricity is available in the amount and with the continuity necessary for chemical processes operations) and process intensification.
Different possibilities for heating reactors using electricity exist. Fig. 3 illustrates schematically the different options possible concerning the case currently most used of (catalytic) chemical reactors where the energy is provided (directly, indirectly) by FF combustion. For temperatures below about 550 °C, an external fluid is typically used to transfer the energy. A temperature gradient is present with respect to the walls of the reactor element where the reaction occurs (in a fixed-bed reactor, a tube of about 3 cm diameter where the catalyst in the form of pellets is contained – see Fig. 3.1b for a case of an endothermic reaction). The fluid external to this tube where the reaction occurs is then externally processed in a furnace (as in Fig. 3.1b, or an, analogous system when cooling could be necessary; eventually a further intermediate exchanging fluid, such as high-pressure steam could be used). When reaction temperatures higher than about 550 °C are necessary, the direct firing of the reaction tubes is necessary, as in crackers or reformers. Thermal transfer is less efficient in these cases and thus more accentuated temperature gradients are present (see Fig. 3.1a). The process is typically limited by thermal transfer. The possible options to electrify the catalytic reactors are summarized below:
1. Electrical furnaces/boilers: when a heat-transferring fluid is present, an electric boiler could be simply used (Fig. 3.2). This is the simple electrically heated reactor, easy to retrofit and already at a high TRL, but essentially not influencing the mechanisms and limits in heat transfer. Thus, this does not offer opportunities for process optimization.
2. Ohmic or Joule effect (inductive heating): electrical resistances are inserted directly in a body (like monolithic support, on which the catalyst is then washcoated) present in the catalytic bed of the reactor. An alternative is that the electric current passes through a resistive conductor, such as metallic support, on which the catalyst is deposited. The catalyst is in direct contact with the electrically heated surface resulting in better heat transfer mechanisms. It is also possible to heat at the temperature necessary for the catalyst layer while maintaining at a lower temperature the fluid in contact with the catalyst. Thus, selectivity or stability could be improved. The catalyst must be designed specifically for operations in this reactor (for example as a thin coating). As for cons, a specific redesign for intimate contact between the electric heat source and the catalyst is necessary. It is also necessary to avoid the possibility that a heated surface enhances side reactions.
3. Magnetic heating: a rapidly alternating magnetic field either generates eddy currents in conducting materials resulting in the Joule heating of those materials, or generates heat in ferromagnetic materials by magnetic hysteresis losses. As reported in the literature from the long term, and used industrially in some cases, it remains a challenge for wider industrial use. The core–shell (or analogous) design has to be realized, with a core of components which is heated magnetically. Often these magnetic nanoparticles can be also catalytically active in side reactions, and their direct contact with reactants should be avoided. It is possible to realize fast changes, and also periodic fast modulations in the temperature. Very localized heating at the catalyst, with the possible creation of thermal gradients with a flowing gas phase (rapid quench), is possible.
4. Microwave/RF (radio frequency) heating: are both volumetric heating methods; the rapidly alternating electric field of the microwave generates heat by moving dipolar molecules or ions in liquids, or by getting absorbed in the so-called “dielectric lossy” solid nonmagnetic materials. There is industrial experience, especially in the area of synthesis of fine chemicals. As for cons, the approach is effective with liquids, but less so with gas reactions. The main challenge with heterogeneous catalysts is to achieve uniform heating, in pilot as well as industrial-size reactors. Furthermore, the proposed solutions are difficult to scale up.
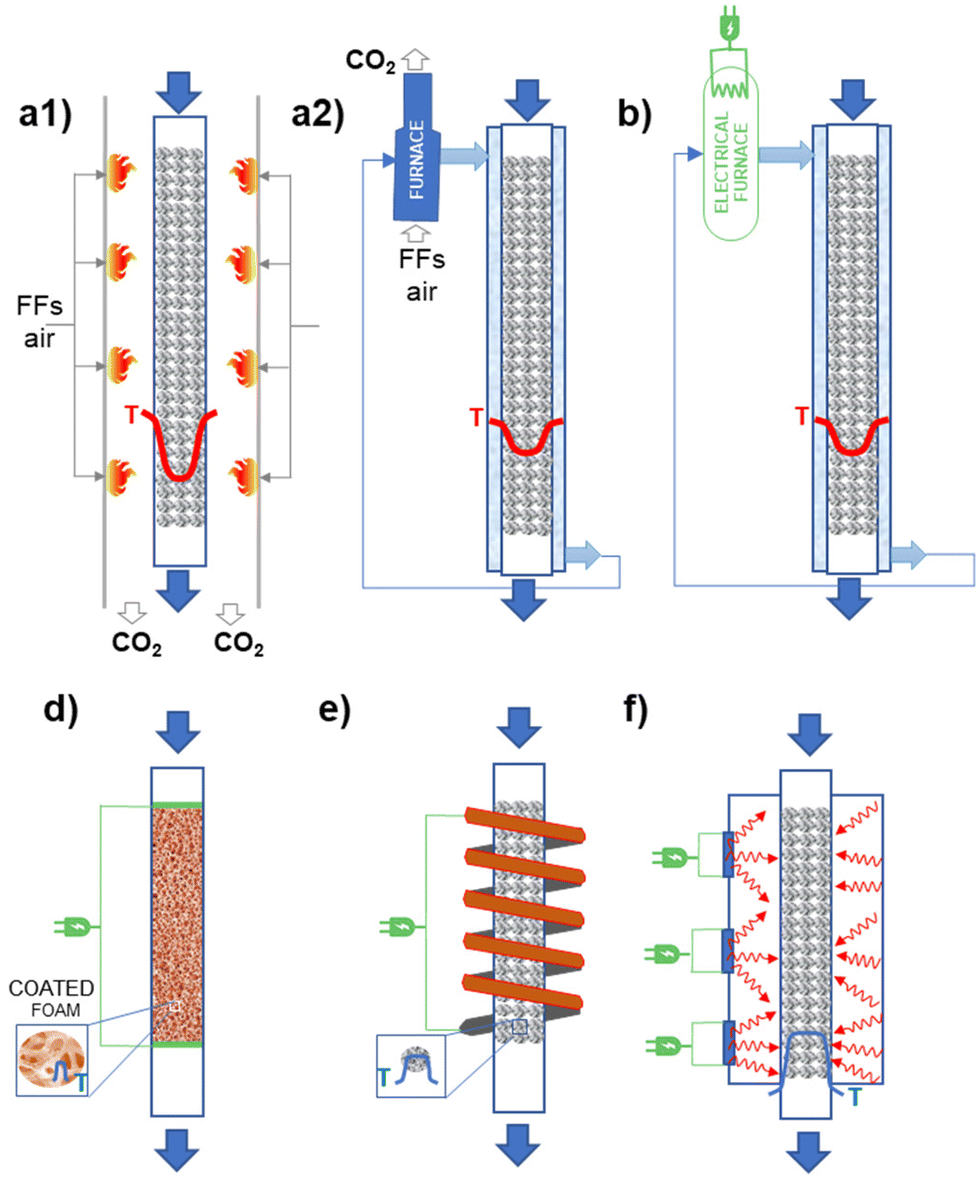 | ||
| Fig. 3 Schematic illustration of catalytic fixed-bed reactors where the energy for the process is provided directly or indirectly by FFs combustion (a1 and a2, respectively) and the alternative possible for electrification: (b) electrical furnaces/boilers, (c) ohmic or Joule effect (a metallic foam), coated with a catalyst layer, directly heated by an electrical current, (d) magnetic inductive heating (for core–shell catalysts pellet, where the core is a magnetic nanoparticle) and (e) microwave/RF heating of a fixed-bed catalyst bed. The temperature profile in the reactors is also schematically presented. | ||
Except for the first case, a general redesign of both the catalysts and reactor geometry and characteristics is necessary to take full advantage of electrification. Thus, substituting the use of FFs with electrical energy as supply also becomes an opportunity for innovation and improvement in performance. On the other hand, scale-up of these solutions, except the first, remains challenging, with a technology maturity which can be indicated at a TRL value between 3 and 5.
Among the important challenges, the possibility of realizing spatial and temporal control of electricity-generated thermal fields also requires the design and fabrication of energy-responsive catalysts.8 Controlling and optimizing the overall energy efficiency is another necessary objective, which involves a better-combined understanding from a multidisciplinary perspective of the underlying phenomena.
Studies in the literature on electrically heated reactors are still limited. One of the first studies was that presented together by Haldor Topsoe and DTU45 (Denmark) for a steam-methane-reforming (SMR) reactor for hydrogen production. By closely integrating the electric heat source and the catalysts it is possible to increase catalyst utilization and limit unwanted byproduct formation. The process intensification is of a factor of around 100. Fig. 4 illustrates the reactor concept and the profile of temperature present in the electrical reactor based on direct resistive (ohmic) heating.
 | ||
| Fig. 4 Conventional fired reactor versus electric resistance-heated reactor with an indication of the radial temperature profiles. Adapted from ref. 45 with permission. Copyright © 2019, The American Association for the Advancement of Science. | ||
The authors tested the concept in a laboratory-scale reactor based on a FeCrAl-alloy tube, which is the resistive heating element, chosen for having the electrical resistance independent of the temperature in the range of interest. This tube (around 6 mm diameter, length about 0.5 m) was inner-coated with a nickel-impregnated washcoat of about 130 microns.45,46 This reactor allows around 90% conversion to syngas with a feed of about 1.7 L min−1. The AC current was applied to copper sockets mounted at opposite ends of the external surface of the reactor tube. The tube was then inserted into high-temperature insulation material. The authors indicate that this reactor is scalable to industrial conditions and capacities. However, for a typical industrial reformer (50.000 m3 h−1), around 500,000 of these (lab-size) reactors should operate in parallel. Even considering the use of this technology for small-scale distributed production of H2, the problem of providing the renewable energy necessary for operating in parallel a large number of reaction tubes still represents a major challenge.
Ghent University (Belgium) has reported a recent assessment, mainly based on ex-ante LCA analysis, of electrified processes for methanol production: (i) plasma-assisted and (ii) electrically heated thermocatalytic dry methane reforming.47 The first path has lower greenhouse gas (GHG) emissions only when the plasma reactor is powered by renewable electricity. However, both routes have a low climate change impact (around 0.6–0.7 kg CO2-eq. per kgMeOH) when all the process units are electrified by renewable electricity. Similar results were obtained for CO2-based methanol production utilizing electrolytic H2. Over 40% CO2 emission reduction is possible by these electrified routes with respect to conventional, state-of-the-art, natural gas steam reforming-based methanol process. However, the availability of fully renewable electricity for continuous operations is required.
Politecnico of Milano's (Italy) group has recently made a numerical assessment of electrically heated (Joule) methane steam reforming over structured catalysts.48 The structured catalysts are used as internal heating elements. They suggest that SiC is the preferred material to be used as catalyst support since the currents and voltages to be used can be safely employed. Differently, metallic materials require higher current densities and applications at very low voltages. The reactor models consider two types of structured catalysts: (i) open-cell foams and (ii) honeycombs, both coated with an Rh-based catalyst. The first, however, was indicated as preferable.
Palma and coworkers49 and Rieks et al.50 also studied the steam reforming and dry reforming of methane, respectively, using a commercial electric heating element in silicon carbide coated with a thin layer of nickel catalyst. However, the geometry of these materials was not optimized for efficient radiative heat transfer. The geometry of these systems must allow a large area for the catalyst and avoid significant bypass.
Catalysts should be also specifically designed to take advantage of the possibilities given by electrified chemical reactors. However, studies are still quite limited. Wismann et al.51,52 showed that by using alumina-supported CoxNi(100−x) nanoparticles with defined alloy compositions, the induction heating for steam methane reforming may be tuned. The approximately 30 nm Co–Ni particles were identified as optimal for operating both as a catalyst and as an induction heating susceptor.
Pham-Huu, Giambastiani and coworkers53 have analysed the opportunities offered by electromagnetic induction heating technology to improve catalytic heterogeneous processes. The possibility to overcome heat transfer limitations and associated effects, such as slow heating/cooling rates, nonuniform heating environments, and low energy efficiency, particularly in strong endo- and exothermic catalytic reactions, was evidenced. The use of microwave (MW)-assisted heterogeneous catalysis was reviewed recently by Muley et al.54 Microwave heating applications ranging from methane conversion to ammonia production, biomass conversion, etc. were discussed. Vlachos and coworkers55 reviewed very recently the advantages and possibilities offered by MW heating for multiphase reactors. The use of MW in combination with heterogeneous catalysts was also recently reviewed by Palma et al.56 They also discussed the mechanistic effects and energy losses, as well as the problems associated with scale-up. A recent paper discussing how to pass from the bench to pilot scale in MW-driven structured reactors for methane dehydroaromatization was published by Santamaria and coworkers.57
An example of process intensification by electro-magnetic induction heating for water catalytic treatment was reported by Munoz et al.58 Yassine et al.59 used the possibility of spatial resolution in temperature at the micro-level offered by induction heating of magnetic nanoparticles for the conversion of resazurin into the fluorescent resorufin, in solution and when confined in small hydrogels, to prove this concept. Chaudret and coworkers60 developed iron-carbide nanoparticles as promoters for magnetic heating of nickel-based catalysts for CO2 hydrogenation. The same goup61 also reported the optimization of the reactor and Ni/CeO2 catalyst (by adding iron nanoparticles) for magnetic CO2 methanation.
Much attention to this topic is also given by industry. For example, BASF, SABIC and Linde have signed a joint agreement to develop and demonstrate solutions for electrically heated steam cracker furnaces. The interest of Haldor Topsoe was indicated previously. Many internal projects are underway by major chemical companies.
Direct electrification of the catalytic reactions
The area often generically indicated as power-to-chemicals lumps together processes where the renewable energy directly provides the energy necessary for the reaction, in the form of either activated reactants (as in plasma-catalysed processes)62 or of species generated on the surface of the catalyst by application of potential, i.e., electrocatalysis.37 Light adsorption on semiconductors also generates a charge separation and thus reactive species, overcoming (in principle) the need to provide externally the energy (heat) to overcome the activation barrier.However, there are some differences in photoactivated processes concerning the electrocatalytic processes: (i) the catalytic surface must be directly irradiated by solar light, and thus process intensifications (such as those possible by using stacked electrocatalytic reactors) are difficult to realize; (ii) the current density generated by light adsorption is typically low, of the order of 10 A cm−2, and thus productivity is low; in electrocatalytic processes, the current density can be up to two orders of magnitude higher; (iii) while different sources of green electricity could be used, direct solar-driven processes are highly dependent on irradiation intensity, thus depend on meteorological aspects, localization, etc.
Although direct photodriven processes have other advantages, the issues to solve before commercialization, except perhaps in a few cases (such as H2 photo- or photoelectro-catalytic production),63 are more severe with respect to electrocatalysis. To restrict the discussion, only electrocatalytic and plasma-catalysis are considered here. For analogous reasons, concentrated solar panel (CSP) technologies are not considered in the power-to-heat section.
In addition to generating an alternative path providing the energy necessary to overcome the activation barriers, direct electrification paths can also generate in situ the other reactants needed to make the catalytic reaction, for example, the H+/e− equivalent of H2 for hydrogenation reactions. Less studied, but also active oxygen species, including peroxo ones, could be generated in situ for direct or mediated oxidation reactions. Thus, electrification also offers the advantage of eliminating the need to separately produce these redox reagents. This could result in a novel process offering potential cost reduction, improved efficiency and reduced GHG emissions.
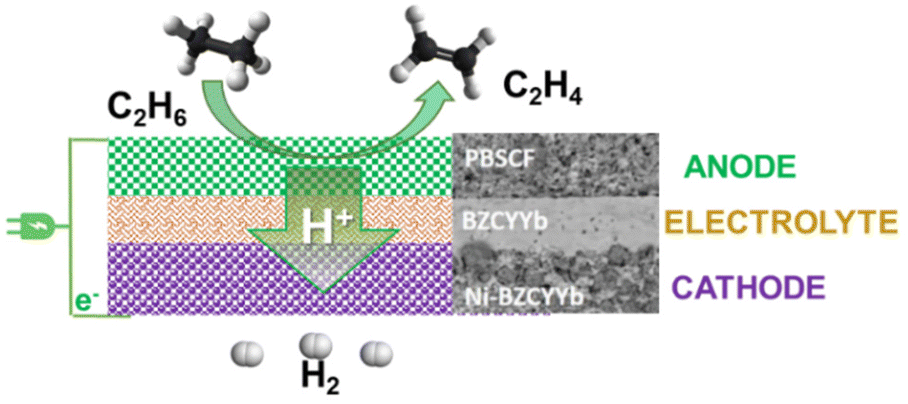 | ||
Fig. 5 Schematic illustration of the ethane to ethylene + H2 coproduction on solid proton-conductive membranes in an electrochemical cell. Adapted from ref. 65 with permission. PBSCF = PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (porous double perovskite, 30 μm). BZVYYb = BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (doped barium zirconate cerate, 10 μm). Ni-BZCYYb = NiO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BZCYYb 6:4 wt. Copyright © 2018, Royal Society of Chemistry. :BZCYYb 6:4 wt. Copyright © 2018, Royal Society of Chemistry. | ||
This consists of a dense 10 mm-thick BZCYYb electrolyte thin film on porous BZCYYb-Ni anode support (300 mm) and a porous double perovskite PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) layer (30 mm) as a cathode. BZCYYb is a barium zirconate cerate (BaZr0.1Ce0.7Y0.1Yb0.1O3−δ), a low-temperature ionic conductive solid electrolyte. At 400 °C (thus about 200–250 °C lower temperatures than for the catalytic ethane dehydrogenation), a current density of 1 A cm−2, corresponding to an H2 production rate of 0.448 mol cm−2 per day (about 0.4 L h−1 cm−2), near 100% ethylene selectivity could be achieved under a low electrochemical overpotential of 140 mV (thus high energy efficiencies). This approach can save about 65% in the process energy and reduce the carbon footprint by over 70% compared with an industrial ethane steam cracker. Scalability and stability still have to be demonstrated. However, the concept of direct electro-assisted dehydrogenation opens new possibilities in chemical processes. By using low-temperature proton-membranes, and suitable electrocatalysts, many valuable alternative chemical processes could be developed.
The concept is not novel. Wang et al.66 about 15 years ago used a similar yttrium-doped barium cerate electrolyte, but for ethane fuel cells. A similar yttrium-doped barium cerate was used by Shi et al.67 also over a decade ago for the co-production of power and ethylene by dehydrogenation of ethane. Power densities, however, were lower than those of Wu et al.65 Various other papers and patents were also published later. Propane could be also efficiently dehydrogenated to propylene, with selectivities up to 80% but decreasing to 50% on increasing current density up to 80 mA cm−2.68 Thus, the approach is not new, but recent results raised the performances to interesting conditions for exploitation and linked specifically to the electrification of the production.
Various recent reviews have specifically discussed this reaction.69–80 The focus of these reviews, however, was mainly on the mechanistic aspects triggering the C–C bond formation and selectivity to ethylene, or C2 oxygenates (such as ethanol). Features analysed included designing the electrocatalysts and the influence of the reaction conditions, with connected aspects such as how the pH at the electrode surface determines the performance and formation of ethylene or other products. Fewer studies were instead centred on the identification of the industrial feasibility of this approach. One of them is by Jaramillo and Sargent's group,81 who undertook a techno-economic and carbon emissions assessment of CO2 to ethylene (or other products) electrocatalytic production from the perspective of practical application. They indicated that to become competitive on this path with FF-derived feedstocks, electrical-to-chemical conversion efficiencies need to reach at least 60–65%, and renewable electricity prices need to fall below 2–4 cents per kilowatt per hour.
To produce 1 kg ethylene (C2H4), 0.428 kg H2eq. generated by water electrolysis are required to form the 12H+ per mole ethylene necessary in CO2 electro-conversion. Around 50 kW h kgH2−1 is typically necessary for water electrolysis by considering the overpotential necessary. Data at the end of 2021 indicate a approximate cost of 0.21 € per kW per h (Eurostat data). Thus, around 4.5 € per kgC2H4 would be necessary, even considering 100% selectivity to ethylene. The commercial value of ethylene depends on various factors but can be considered on average around 1.0–1.2 €/ per kgC2H4. Thus, even considering the cost of CO2 and the process as zero, producing ethylene by electrocatalytic reduction would become attractive only when the cost of renewable electrical energy decreases by a factor over 5, and efficiency in generating the protons necessary for the CO2 reduction is increased. According to Fraunhofer ISE June 2021 estimations,82 the levelized cost of electricity at the best (PV utility-scale) is between 3–6 €cents per kW per h, decreasing to 2–3 in 2040, thus in line and above with the requirements indicated above.
Note that we indicate H2eq., e.g., H+/e−, because the latter are those needed in the electrocatalytic reduction of CO2. This would in part avoid the overpotential necessary to form H2 from this hydrogen equivalent. The largest contribution to overpotential in water electrolysis, however, is related to the oxygen evolution reaction (OER). Substitution of this reaction with anodic reactions alternative to water oxidation and requiring a lower overpotential, eventually providing also a better-added value than O2, is thus a target to decrease the overall costs. Khan et al.83 indicate a 32% decrease in the energy and an 80% in the cost by anodic coproduction of glycolic acid.
In a recent assessment, Sargent's group84 analysed the cost of producing ethylene in a neutral MEA (membrane electrode assembly), an alkaline flow cell electrolyzer, and a high-temperature SOEC (high-temperature solid-oxide electrolyzer cell). They remarked that low eEE (electrical energy efficiency), loss of CO2 to carbonate and crossover were among the critical issues influencing the cost. In a tandem process, the first CO2 to CO followed by CO to C2H4 was indicated as the solution to decrease the cost at an optimistic value of 0.8–0.9 € per kgC2H4. However, this is based on the indication that it would be not possible to prevent carbonate and membrane crossover. The use of gas-diffusion electrodes and the choice of electrolyte and membrane (for example, bipolar membranes) can considerably reduce, if not eliminate, these issues. Thus, a single-cell approach, rather than a tandem one, could be still preferable.
This example illustrates the many difficulties existing in estimating the potentiality of CO2 in the ethylene electro-conversion process. It is necessary to enter into the details of evaluations and analyse whether the assumptions and considerations made are correct or just a challenge to realize. Pappijn et al.85 indicate that the electrochemical production of ethylene from CO2 is not feasible and also has a lower CO2 avoidance potential than the substitution of grey electricity by green electricity. However, the process scheme considered has a conventional CO2 capture and separation step which significantly impacts the overall process (“the thermal energy required in the CO2 capture step is equal to approximately 87% of the total energy need”). In addition, the separation section downstream to the electrocatalytic reactor is also complex, with stages of (i) gas/liquid separation, (ii) gas purification, (iii) compression, (iv) caustic wash and drying, (v) demethanization, (vi) deethanization, (vii) acetylene hydrogenation and (viii) final C2 fractioning. The challenge in all electrocatalytic devices for CO2 conversion is to avoid a preliminary stage of CO2 capture, via the use of tailored gas-diffusion electrodes which include elements for CO2 direct capture from low-concentration streams.86 Separation schemes, as illustrated in Fig. 1b, could be much simpler and less costly, but this depends on the operational conditions (pressurized electrolyzers) and selectivity of the electrodes. The message here is thus that there exists a potential to drastically reduce these negative elements and realize a technologically feasible process.
There are also other critical aspects such as availability and continuity of the supply of renewable electrical energy. Experience in industrial electrolyzers also indicates that water purity for the electrolyzers and the presence of solubilized O2 in it are other factors sensitively influencing the costs, affecting stability. In CO2 electrolyzers the TRL of development is still lower, and thus aspects related to these questions, as well as purity of the CO2 stream, have not yet been analysed.
Note also that most of the studies are focused on the electrocatalytic reduction of CO2 in an aqueous electrolyte, with consequent limits related to the solubility of CO2 in water. De Luna et al.81 remarked on the need to move toward flow-cell and gas diffusion-type architectures that operate at more industrially relevant current densities (>100 mA cm−2). This solution will also in part reduce the purification issues in the process scheme presented by Pappijn et al.85 We already, several years ago, showed that passing to this architecture is a requirement to industrialize electrocatalytic and photoelectro-catalytic reactions.87–89 We also showed that the products, and thus the mechanisms of reaction, are significantly modified in passing from conventional electrocatalytic studies in the presence of a bulk electrolyte to a flow-cell and gas diffusion-type architectures.
Another crucial parameter often not considered is the current density of the specific target product, because this is a measure of the productivity of the electrode and thus is a critical parameter for exploitability. Table 2 reports some selected literature results for copper-based electrocatalysts.69 Copper is the most studied electrocatalyst for CO2 to ethylene electro-conversion. Although it is often cited in literature that copper has unique properties to form ethylene, and this concept is supported by theoretical determinations, other metals can also selectively form C2 products by direct electrocatalytic reduction of CO2 under proper reaction conditions.86,90,91
| Electrode | Electrolyte | Current density, mA cm−2 | Faradaic efficiency to C2H4% | V vs. RHE | Ref. |
|---|---|---|---|---|---|
| Cu nanocubes | 1 M KOH | 300 | 57 | −0.75 | 92 |
| Cu + Al | 1 M KOH | 400 | 80 | −1.50 | 93 |
| Cu modified (N-arylpyridinium) | 1 M KHCO3 | 320 | 72 | −0.83 | 94 |
| Cu modified (poly-N acrylamide) | 1 M KOH | 430 | 72 | −0.97 | 95 |
| Cu + iron porphyrin | 1 M KHCO3 | 300 | 38 | −0.82 | 96 |
| Optimized Cu2O/Nafion | 1 M KHCO3 | 300 | 44 | −0.85 | 97 |
| Cu nano + PFSA ionomer | 7 M KOH | 1550 | 60 | −0.91 | 98 |
| Cu + Ag | 1 M JOH | 330 | 48 | −0.70 | 99 |
| Cu modified F | 0.75 M KOH | 1600 | 65 | −0.89 | 100 |
| Sn-Doped CuO nanosheets | 0.1 M KHCO3 | 16 | 48 | −1.1 | 101 |
| Nanodefective Cu nanosheets | 0.1 M K2SO4 | 60 | 83 | −1.2 | 102 |
Table 2 summarizes several aspects.92–102 The first general consideration is that copper must be modified to obtain good results in CO2 to ethylene conversion. However, a variety of modifiers have been found to significantly enhance the properties, even though these modifiers have different characteristics and mechanisms of promotion. While theoretical studies and many mechanistic hypotheses identify the selective formation in the presence of specific copper active centres,69–80 the fact that quite different types of promoters and mechanisms of promotion equally influence the selectivity in ethylene formation puts a question mark on the mechanistic hypotheses determining the selective formation of ethylene and related electrocatalyst design indications.
The second aspect emerging from the data in Table 2 is that significantly high current densities (J) and faradaic efficiencies (FE) can already be achieved, although not always at the same time. However, quite basic conditions are required, which in general may question the stability of operations in extended continuous tests. When diluted electrolytes are used, current densities are over 1–2 orders of magnitude lower, as exemplified in the last two entries in Table 2 (selected among many others to illustrate the concept). The need for strong basic conditions, up to pH 15, is not convincingly clarified.98 From one side, these strong basic conditions could be necessary for modifying in situ the electrocatalyst characteristics, such as generating a hydroxide-type surface, when the potential is applied, similarly to what we observed with operando EXAFS when a potential is applied to small iron nanoparticles.103
Negative charges generate on the electrocatalyst surface when a potential is applied to the cathode. The electron transfer to CO2 molecules generates a negative charge. The anion-radical molecule after the first electron transfer thus tends to move far from the electrocatalyst surface, preventing further conversion if a sequential electron-transfer mechanism is present (as assumed in almost all mechanisms of CO2 electroreduction, differently from enzymatic mechanisms73). This crucial aspect is typically not considered. Even chemisorption of CO2 through the oxygen atoms104 or in an electrolyte of carbonate ions is inhibited on a negatively charged surface. Thus, a metallic copper surface, that is typically assumed to be active in electrocatalysis for CO2 to ethylene, would be unable to chemisorb CO2 and reduce it, when a negative potential is applied. This crucial aspect suggests that likely an in situ transformation will occur91,103 and this would be the key to the electrocatalytic behaviour, although this aspect is typically not considered even if would significantly influence the reaction mechanism.
As noted before, a very strong basic electrolyte (up to pH 15)98 is sometimes used to inhibit the side reaction of H2 generation and to provide the necessary ionic conductivity to sustain high current densities, but could be critical for stability and industrial exploitability. Understanding how to use reaction conditions and reactor design which are better compatible with industrial scalability while maintaining high performance is still an open question. At the same time, however, another issue to clarify is whether the use of some specific reaction conditions, such as a quite high pH of the electrolyte, could be instead related to an in situ dynamic modification of the electrocatalyst.91
The third consideration emerging from Table 2 is that experimentation with gas-diffusion type electrodes in continuous-flow electrocatalytic reactors (suitable for scale-up and industrialization), and under pressure, is still limited in the literature, although indicated sometimes by the same authors publishing data reported in Table 2. The effect of using diluted streams of CO2, eventually containing some of the typical possible contaminants, is also not well explored. Cross-over effects through the membrane are indicated, but often how this issue could be eliminated is not properly considered.
There are thus many aspects which still need to be developed, but the current state-of-the-art in CO2 electro-conversion to ethylene shows that some of the targets for the process to be considered feasible from an industrial perspective have already been reached, except perhaps stability. The main limiting factor is thus the availability of low-cost renewable electrical energy, below about a few cents per kW h.
As indicated, this renewable energy cost of a few cents per kW per h is feasible, although the possibility of obtaining continuous (24 h) operations with renewable energy remains an issue. This is a requirement to reach the necessary impact on CO2 emission reduction. In a decade from now, the share of renewable energy in the electrical energy mix will be likely not higher than one-third, and by using this share, any of the electrocatalytic processes will meet the targets of strong GHG reduction (>50–70%). The design of electrocatalytic processes integrating a PV unit, a transient energy storage unit (realizing a temporal decoupling of anodic and cathodic reactions through redox storage elements105), and an electrocatalytic component to perform the direct power-to-chemical storage is one of the directions to explore more systematically.
Methanol is used in current petrochemistry for different uses and is one of the chemicals with the largest prospects of growth in the near future.106 There are two main drivers for this forecast: (i) the expected use for fuels (for direct blending or reactants to make other fuel additives) and (ii) the expanding sector of MTO (methanol to olefins) and related processes. The use of liquid fuels will be definitively depressed in the next decades, with the shift to electrical or hydrogen-powered engines. The production of olefins will be substituted by alternative low-carbon routes such as those discussed. Thus, notwithstanding the indications of an expected high compound annual growth rate (CAGR) for the methanol market, it is not reasonable to expect that its market will greatly increase in the future to justify the introduction of a new technology of direct electrocatalytic synthesis of methanol from CO2. Rather than developing this route, it would be better to search for electrocatalytic processes directly producing the chemicals derived from methanol. Excluding fuels, in methanol to olefins the main chemicals produced by using methanol as a reactant or co-fed are, in order of relevance: (i) formaldehyde (∼24 Mt per year), (ii) acetic acid (∼8 Mt per year), and (iii) other chemicals such as methyl methacrylate, methyl mercaptan, methylamines, methyl chloride; altogether about (∼7 Mt per year. Thus, rather than synthesize methanol from CO2 by electrocatalysis (various reviews on this reaction have been published recently107–111), it would be better to synthesize directly the products of further methanol conversion, particularly formaldehyde and acetic acid.
The production of formaldehyde by CO2 electrocatalytic conversion is not the subject of specific reviews and is typically also not discussed, or only marginally, in general reviews on the electrocatalytic reduction of H2. However, as one of the main products synthesized from methane, and requiring fewer electrons with respect to methanol (4e− against 6e− for the methanol case), it should be evident that more attention should be given to this reaction, even with the intrinsic difficulties, such as an easy polymerization. Formaldehyde is the intermediate in going from CO2 to methanol in the enzymatic pathways, summarized below:112
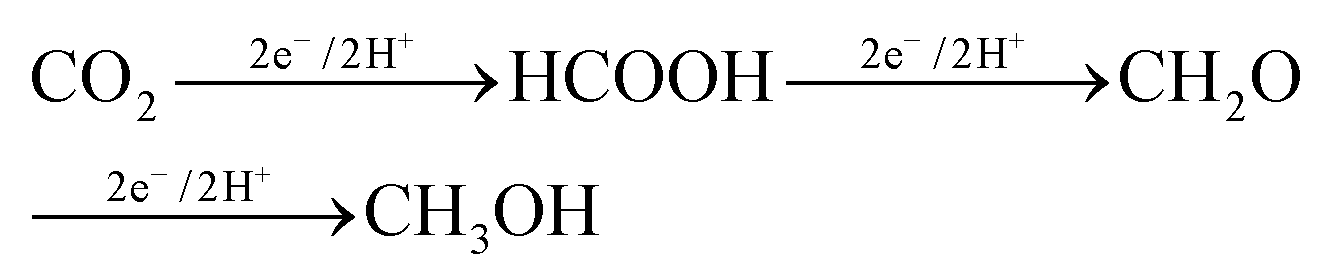 | (1) |
A way to produce formaldehyde (CH2O) from CO2 is thus by microbial electroreduction of CO2. However, also in this case specific studies are limited. Guo et al.113 reported recently that CO2 is converted into formaldehyde by coupled photoenzyme catalysis, although the enzyme reduction of CO2 is predominant compared with the photoreduction. Results, however, are not optimal, with the yield of formaldehyde reaching 3.8% with the addition of 1 mM NAD+, the oxidized form of nicotinamide adenine dinucleotide coenzyme. Liu et al.114 also studied in which experimental conditions it is possible to maximize the formation of formaldehyde, rather than to arrive at the final production (methanol). However, most of the studies in this area are centred on methanol production.
Among the best results in this reaction are those of Nakata et al.,115 who studied the electrocatalytic reduction of CO2 in seawater (to increase CO2 concentration in solution) on boron-doped diamond (BDD) electrodes under ambient conditions to produce formaldehyde. BDD electrodes have a wide potential window and high electrochemical stability, giving high faradaic efficiency (74%). The sp3-bonded carbon of the BDD was indicated as the active centre. More recently, Pawar et al.116 reported the use of a photoelectrocatalytic approach (Ca and Fe co-doped TiO2 photoanode and Cu/rGO/PVP/Nafion multi-layered hybrid composite cathode), but at the conditions of maximum formaldehyde formation (about 480 μM) the energy efficiency is low (around 20%). Thus, data for the electroreduction of CO2 to formaldehyde are still quite limited, and the results still have to be improved.
More studies have been reported on the electrocatalytic reduction of CO2 to acetic acid/acetate. As for the ethylene case, this synthesis requires the formation of a C–C bond. Concerning other C2 products formed in CO2 electroreduction, it is an 8e− reduction (to acetic acid) with respect to a 12e− (to ethanol, or ethylene).
Acetic acid has a market value around three times higher than that of ethanol, and about 30% higher than that of ethylene, although these are average indications, depending highly on regions, time and other aspects. If we consider that the number of electrons necessary for the transformation is over 30% lower, and the higher market value, it is evident that electrochemically producing acetic acid/acetate from CO2 is economically more interesting than the ethylene case, while producing ethanol is likely not a worthwhile area.
Ethanol could be better produced by fermentation.117 Acetic acid could be also produced by fermentation,118,119 and was produced by this route in the past. Although the process is well established, the main issue is the combination of separation costs and productivity. For this reason, almost all industrial production of acetic acid today is via catalytic routes (mainly methanol carbonylation). The fermentative route (oxidative fermentation using acetic acid bacteria) is currently mostly used only for food-grade acetic acid (vinegar). The introduction of advanced separation processes, for example by membranes, could make the fermentation route competitive. In general, the issue is the development of biofactories with an optimized integration between biocatalytic and photo/electrocatalytic routes.29,120–122
Acetic acid/acetate finds application as a large-volume chemical in many applications: manufacturing of coatings, greases, polyesters and sealants which are used in several industries such as electronics, automobiles, textiles, and packaging.
Nguyen et al.123 have recently made an LCA comparative analysis of the production of acetic acid by electrocatalytic reduction of CO2 (direct one-step or step-step via first the electrocatalytic reduction of CO2 to CO), with respect to other products which can be obtained in the reaction (formic acid, CO, CH4, CH3OH, C2H4, C3H7OH). They estimated the carbon intensity (i.e., global warming impact) of one and two-step electrochemical routes with respect to the thermochemical CO2 utilization and FF-based synthesis routes. Due to carbonate formation or CO2 crossover in one-step CO2 electrolysis using a neutral pH membrane electrode assembly, the two-step electrosynthesis pathways (i.e., CO2 to CO in solid oxide electrolysis cell followed by CO electroreduction in an alkaline flow cell) were indicated as preferable, but as noted before the possibility of overcoming this problem (recirculating CO2, avoiding CO2 crossover by suitable membranes, an improvement anyway necessary) was not considered. Thus, the preferred two-step solution over the direct one does not account for possible technological advances.
Furthermore, Nguyen et al.'s123 analysis further indicated that the carbon intensity of electrosynthesis products is due to a significant energy requirement for conversion (77–83% of total energy consumption for gas products) and product separation (30–85% of total energy consumption for liquid products) phases. This is again based on assumptions, such as the need to use liquid electrolytes, and the need for product recovery by distillation. As noted before, technological developments in cell design and integrated separation units could largely change the indications. Thus, the estimated cradle-to-gate carbon intensity (GWI – Global Warming Impact) should be made in the perspective scenario of technology development, which also more precisely defines the objectives. Most of the reviews and perspective papers on CO2 do not make this effort to link needs from a techno-economic feasibility perspective with research targets. Most of these papers link to generic objectives of improving performances, rather than attempting to identify the priorities. Improving the electrocatalysts’ performances such as the faradaic efficiency (FE) is useful, but if this is achieved by using reaction conditions (electrolyte, current densities, etc.) and materials not practicable from an industrial perspective, this could translate to a negative cost advantage. Similarly, FE itself is not an absolute value but is about electrocatalytic operations, including separation, stability, impact on raw materials and electrical energy consumption, etc.
A further assumption by Nguyen et al.123 is that CO2 needs to be captured from emission sources, and then this pure CO2 stream fed to the electrocatalytic reactor. While this is the current approach, this is not a requirement. It is possible to develop electrocatalytic cells directly connected to the CO2-emitting sources, designing more advanced devices which integrate both capture and electrocatalytic functions, operating eventually at the conditions of the emitted CO2-containing stream. In other words, this is an aspect to overcome by proper research rather than a limit that cannot be exceeded. As should emerge from this perspective, and for this reason repeated, it is necessary to have a broad view of the problem, account for the complexity and future developments, identify the technological scenario of development, and link to them the priorities for research. Developing FF-free sustainable chemical production requires a system change and thus an approach going beyond a specific technological evaluation and a too-focused research perspective.
In these terms, another limit of Nguyen et al.'s123 assessment is that not only should the cradle-to-gate carbon intensity be accounted for, but also the different “services” which can be provided by the product. Formic acid, for example, has limited uses itself and should be further converted, while acetic acid is a chemical with direct wide use.
Nguyen et al.123 also compared the GWI (Global Warming Impact) of the electrochemical CO2 conversion with that of the thermocatalytic route (from CO2) and concerning the current (incumbent) production of the same chemical product starting from FFs. They estimated that only in the case of propanol (C3H7OH) are the GWI results inferior to those of the incumbent route, while they do not consider feasible the thermochemical conversion of CO2 to ethanol, acetic acid and propanol. Producing propanol via the fermentation route results in lower GWI than the incumbent route (hydroformylation of ethylene) considered by these authors. In addition, the production of ethanol and acetic acid from the thermocatalytic conversion of CO2 is feasible. With all these limitations, the validity of the conclusions can thus be questioned: preference of two-step versus direct CO2 electrocatalytic conversion, feasibility only of production of CO and propanol, and in part ethylene by CO2RR (CO2 electrocatalytic reduction). We have analysed this more in detail in this assessment123 and note the need from a tutorial perspective to enter into details of estimations, their pros/cons and methodology, etc. rather than using only the conclusions. All LCA, in addition, have intrinsic limits related to the database used for estimations, which become crucial in comparing technologies still at an early stage of development with well-established routes.
Dedicated reviews dealing with acetic acid/acetate from CO2 are not present in the literature, but results on this reaction are part of the general reviews on CO2RR. Feng et al.,124 for example, reviewed results on the generation of oxygenates from the electrochemical reduction of carbon dioxide, but dedicated fewer aspects to this reaction, limited to reporting the results of Sun et al.125 that reported the use of an N-based Cu(I)/C-doped boron nitride (BN-C) composite for electrocatalytic reduction of CO2 to acetic acid. They reported a FE of acetic acid as high as 80.3% but at a low current density (about 14 mA cm−2) when an ionic liquid (IL) containing LiI and water is used as the electrolyte, posing some questions about industrial feasibility. The IL was the commercial 1-ethyl-3-methylimidazolium tetrafluoroborate ([Emim]BF4).
Zheng et al.126 discussed the formation of acetate/acetic acid as part of a critical appraisal of the reduction of CO2 to C2 products focusing on the connection between the fundamentals of reaction and efficient electrocatalysts. Their approach started from an atomistic mechanism of various C2 and C3 products, to analyse the factors influencing the behaviour (local pH, overpotential, presence of surface adsorbates). However, often indications of these aspects contradict the first part of atomistic mechanisms. Also, the design principles of C2 electrocatalysts (chemical states, defective sites, nanostructure) are often not consistent with the whole set of experimental results. For example, the experimental evidence shows that the selectivity of acetate/acetic acid depends drastically on the surface coverage by CO2 and cell design. This remark (the lack of a holistic approach considering all the experimental results) can be applied to several reviews discussing the design criteria for CO2 electrocatalytic reduction. Another example is the review of Wu et al.127 In addition, the need to approach electrocatalysis from a different perspective than just the transposition of approaches used in heterogeneous catalysis has to be highlighted.10,22
Some selected results are summarized in Table 3. Guo et al.128 observed good FE attributed to the unique co-coordination of pyridinic N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O with copper whose chemical state is between +1 and +2 and the porous 3D core that is kinetically favourable for the acetate product. However, as shown in Table 3, the current density is rather low, and thus also the productivity.
O with copper whose chemical state is between +1 and +2 and the porous 3D core that is kinetically favourable for the acetate product. However, as shown in Table 3, the current density is rather low, and thus also the productivity.
| Electrode | Electrolyte | Current density, mA cm−2 | Faradaic efficiency to C2H4% | V vs. RHE | Ref. |
|---|---|---|---|---|---|
| Polymeric Cu–ligand complex core–shell microsphere | 0.5 M KHCO3 | ∼1 | 64 | −0.37 | 128 |
| Mo8 clusters@ Cu nanocubes | Saturated NaHCO3 solution | 110 | 49 | −1.13 | 129 |
| Mn-Corrole on carbon paper | 0.1 M phosphate | ∼0.5 | 63 | −0.67 | 133 |
| N-Doped nanodiamond | 0.5 M NaHCO3 | ∼0.5–1.0 | 78 | −0.8 | 135 |
| 3D 3D dendritic CuO–Cu2O composite | 0.1 M KCl | 11 | 48 | −0.4 | 136 |
Zang et al.129 studied a Mo8@Cu/TNA electrocatalyst, where TNA indicates a TiO2 nanotube array, and Mo8 the polyoxometalate (POM) Cu2Mo8O26·2H2O. The current densities are not very relevant, but in addition to H2, various other organic products were detected (ethanol, acetate, methane, ethylene, ethane). They suggested that the interface of Cu planes and polyoxometalate clusters with abundant Cu–O–Mo active sites promotes the generation of *CH3 and successive coupling with CO2 insertion, highlighting the need to realize a Cu–O–Mo interface for the rational design CO2RR to acetate. However, Giusi et al.130,131 showed that a higher FE to acetic acid (∼62%) could be obtained by using Cu2O deposited over TNA. These tests were realized in a different electrochemical set-up, without a liquid electrolyte and with gaseous CO2 flowing through a TNT nanomembrane. de Brito et al.132 instead used a photoelectrocatalytic device, reporting a FE of about 75% to acetate for Cu2O deposited over a TNA electrode. In both these cases, thus higher FE was reported without the need to realize Cu–O–Mo active sites, and nanocomposites with a POM.
Guo et al.'s128 mechanistic indications were based on theoretical – DFT (density functional theory) – calculations. The question which may be posed is whether a mechanistic interpretation is correct (need to form Cu–O–Mo sites), when higher FE could be obtained in different systems also based on copper, but without molybdenum or POM. In turn, the other question mark is whether the mechanistic approach used (by DFT) can prove the reaction mechanism.
De et al.133 used a molecular manganese corrole complex for the electrocatalytic reduction of CO2 to acetic acid. In a moderately acidic aqueous medium (pH 6), a selectivity of 63% was reported with a turnover frequency (TOF) of 8.25 h−1 (thus quite low, as indicated by the very low current density in Table 3). Genovese et al.134 showed the importance of the interface with the electrolyte and of the subsequent reaction of the negatively charged radical species formed by electron transfer of one electron from the catalyst (a supported copper electrocatalyst) to CO2.
Among the best results are those by Liu et al.135 on an N-doped nanodiamond/Si rod array electrode, giving a FE of about 80% to acetate plus about 10% to formate. However, the current density is rather low (Table 3) and no further results on these electrocatalysts have been reported even by the same group. Genovese et al.103 also showed a high acetic acid FE at low applied potential, deriving from the synergistic effect at the interfaces between FeOOH and nitrogen-doped carbons. Based on both calculations and experimental evidence, Fe species were indicated as responsible for producing acetic acid through a suggested multi e−/H+ transfer. Zhu et al.136 reported dendritic copper-cuprous oxide electrocatalysts for CO2RR, showing a low overpotential (about 0.5 V) with 48% FE to acetic acid (together with 32% FE to ethanol). While there is often the assumption that only copper catalysts can give C–C bond formation (typically assumed via chemisorbed CO coupling, as indicated for example by Zhu et al.136), Zhou and Yeo137 analysed the different mechanisms of formation of C–C bonds during CO2RR (including to form acetate/acetic acid) and the catalysts able to form C2+ products, with a focus on non-copper electrodes. They demonstrate that (i) there is no need to have copper catalysts and (ii) the reaction mechanism do not requires coupling of CO chemisorbed species. Both these indications, widely supported by theoretical DFT studies and generally assumed as valid, have proved to be not correct from an analysis of the experimental data.
In all these tests, the acetate is a product of cathodic reduction of CO2, while Zhang et al.138 attributed its formation to the crossover through the membrane of the products of CO2 reduction (ethanol, in particular) and their oxidation at the anodic side. While crossover could be possible, this effect requires that ethanol diffuses through the membrane, is oxidized and acetate back-diffuses to the cathodic side. In addition, often no ethanol or only traces are detected. Thus, the effective role of this mechanism on selective electrocatalysts to acetate is questionable.
There are no significant relevant results in the literature regarding scaling-up and possible industrialization, as well as aspects regarding stability. The only data are from Zhu et al.139 who analysed the production of acetic acid in the electrocatalytic reduction of CO rather than CO2. They used a Cu nanocube electrocatalyst giving a FE of only about 30% in a flow cell allowing a good current density (150 mA cm−2) and stability (for over 150 h), allowing production of a good concentration of acetic acid (the other liquid product is ethanol, and the other products – H2 and C2H4 – are gas phase).
Note finally that we have discussed as equivalent the formation of acetate or acetic acid. Producing acetate requires a further downstream process of either acid addition (which is not convenient and sustainable from an industrial perspective) or using alternative methods for its conversion. Among them, electrochemical acidification (dialysis) is the most interesting.140 The problem of downstream processing after the CO2RR step has been scarcely recognized in most of the studies on the electrocatalytic conversion of CO2 but is crucial for process economics and thus should be an integral part of the technology assessment and the reactor design. There is often the assumption that electrocatalyst selection is independent of the reactor design, which should be a later stage of development, but this is a wrong assumption.
Ramdin et al.141 have specifically addressed downstream processing in the electrochemical reduction of CO2, although to formic acid/formate rather than acetate/acetic acid. However, in terms of process engineering, the two product classes are analogous. The pH of electrocatalytic synthesis (which depends on the electrolyte solution) determines the choice of downstream operations and economics. They investigated specifically the high-pressure electrochemical synthesis because of solubility issues in realizing the high current densities required to operate at high pressure. To suppress HER, CO2RR in aqueous media is typically performed under alkaline conditions, thus products like formic acid, which has a pKa of 3.75, will almost completely dissociate into the formate form. The selection of separation processes depends on the dissociation state of the acids. Liquid–liquid extraction is suitable for formic acid, while electrodialysis (electrochemical acidification) is preferable for formate separation. The formic acid route is more attractive than the formate one. After these separation steps, a conventional distillation allows production of formic acid with a high concentration (>85%) typically required for use or downstream further processing. While liq.–liq. extraction is more economical, realising CO2 electrocatalysts that can operate at low pH without affecting the selectivity of the desired products is not easy, and thus it could be worth exploring solutions to reduce costs by directly integrating the electro-acidification within the electrocatalytic step.
This was analysed, among others, by Chen et al.142 for the formic acid case. They have developed a robust, scalable cell architecture for the electroreduction of CO2 by integrating bipolar membranes (BPM). An up to 90% faradaic efficiency for the conversion of CO2RR to formate at 500 mA cm−2 was realized at a 25 cm2 GDE with a carbon-supported SnO2 electrocatalyst. A 1.27 mm-thick catholyte film was used between the bipolar membrane and the GDE. While the state of the art for CO2-to-formate electrocatalytic conversion (FE > 90%, current densities >0.4–0.5 A cm−2) already allows consideration of the industrial exploitability of this reaction, current results on CO2-to-acetate conversion are still at much lower current densities (Table 3). The effect should thus be to address, for this reaction, the issues of productivity and cell operations in order to reach levels suitable for exploitability.
In conclusion, differently in the CO2 to ethylene case as well as formate/formic acid production, the production of acetic acid/acetate is still at an early stage of development for industrial exploitability, even if, in general terms, it is a reaction of higher potential interest with respect to others. Intensifying the R&D activities is thus necessary.
The main potential low-carbon processes to produce monocyclic aromatic hydrocarbons for FF-free chemical production are the following:143 (i) production from the lignin component of biomass,144–147 (ii) the production of bioethanol by biomass fermentation and then conversion of ethanol to aromatics on zeolitic catalysts,148 (iii) the production of methanol, by biomass gasification or CO2 conversion with green H2, followed by methanol aromatization (MTA, methanol-to-aromatics),149,150 and (iv) the production of biomethane from anaerobic fermentation followed by methane aromatization.151,152 There are also alternative possibilities to produce FF-free aromatics, as summarized in Fig. 6.
 | ||
| Fig. 6 Overview of different possible routes to produce FFs-free aromatics as raw materials for the chemical industry. Surrounded by a broken line indicates technologies still to be developed. HDO indicates hydrodeoxygenation. | ||
Still, limited studies have attempted a comparative analysis among the different possible routes to determine the preferable path on which it is possible to produce aromatic raw materials for FF-free chemical production. Maneffa et al.153 have analyzed the different possibilities for the synthesis of p-xylene from bio-derived compounds.
The routes considered are:
1. Methanol to aromatics (MTA)
2. The production of ethanol → ethylene → 1-hexene (trimerisation of ethylene) → (2E,4E)-2,4-hexadiene (via disproportionation of 1-hexene) → cis-3,6-dimethyl cyclohexane (by cycloaddition of ethylene on the previous intermediate) → p-xylene (by dehydrogenation of the previous chemical)
3. Acetic acid (by carbonylation of biomethanol or by fermentation) → isobutene → isooctenes (dimerization) → xylenes (dehydrocyclization)
4. Isobutanol (by fermentation) → isobutene → iso-octene (dimerization) → p-xylenes (dehydrocyclization)
5. Dimethylfuran – DMF (glucose isomerization to fructose, dehydration to HMF – hydroxymethylfurfural, then hydrogenolysis) → p-xylene (Diels–Alder cycloaddition with ethylene then dehydration)
6. Diels–Alder cycloaddition of acrolein (from glycerol dehydration) and DMF followed by dehydration and further conversion to p-xylene
7. Fast pyrolysis with catalytic conversion inside a zeolitic material
8. Lignin depolymerisation/hydrotreating route
All these routes thus involve complex transformations with several issues to control the product distribution and separation costs. Maneffa et al.153 concluded that Diels–Alder cycloaddition of DMF with bio-ethylene and the synthesis from isobutanol are the more advanced and attractive routes in the short/medium term. However, precise data on techno-economic and environmental assessment were not given to support these indications. Even if the chemistry is known, many aspects still have to be optimized, starting from the relatively low final yields. For example, the maximum theoretical yield of p-xylene from isobutanol is below 40%. We believe that ethanol to aromatics (on zeolites such as modified H-ZSM-5)154,155 is likely the preferable route, combining the use of a relatively cheap biomass source and the possibility of avoiding too many process steps. Still, too broad a range of products is formed, with a maximum aromatic yield of 23 wt% on post-synthesis modified H-ZSM-5.156
There are more studies on the production of the aromatic via FF-free methanol,149 which can be obtained from many routes, including waste conversion and CO2 hydrogenation. The methanol-to-aromatics (MTA) process is a promising route to produce aromatics commodities, and can also be realized in one direct step from CO2. Among the best results directly from CO2 are those reported by Liu et al.156 using a catalyst composed of spinel structural ZnAlOx oxide and H-ZSM-5 zeolite. This catalyst shows a ∼70% aromatics selectivity (on CO-free bases) with only 0.4% CH4 selectivity in CO2 hydrogenation (∼9% conversion at 320 °C). Stability was proved for ∼100 h. In syngas conversion to aromatics over Zn-doped ZrO2 nanoparticles dispersed on zeolite H-ZSM-5,157 with the syngas produced for example by waste gasification, aromatics with 80% selectivity at CO conversion of 20% could be achieved, without catalyst deactivation for over 1000 h.
In MTA a relatively broad range of aromatics and other hydrocarbons is obtained, while the interest is to maximize the formation of BTX (benzene–toluene–xylenes). Among the best results in this direction are those by Zhang et al.158 reporting a BTX selectivity of up to ∼68%, and by Wang et al.159 with a BTX selectivity of ∼60%. Operations at high pressure have been reported recently to give a highly selective and stable production of aromatics in MTA over ZSM-5,160 but the best results (selectivity to aromatics up to 50%, with 20% BTXs) do not appear to be outperforming. An interesting recent result is the possibility of a computer-aided selection of the zeolite catalyst to perform “ab initio” zeolite design to maximize aromatics formation.161 However, the approach has been used only to maximize DMF conversion to p-xylene (with best yields around 17–18%) on delaminated ITQ-2.
Benzene has been reported recently to be produced from lignin hydrogenolysis on RuW supported on a high-silica HY zeolite.162 Yields of benzene range between 10 and 20% in the in situ refining of lignin, depending on the type of starting woody materials, with pine giving the highest benzene yield. Around 17 g benzene could be produced for each 100 g lignin. In general, producing aromatics from lignin has been indicated by LCA methods to offer better environmental performances than FF-based products, especially with respect to climate change.163 Note finally that not only aromatics could be obtained by lignin conversion, but also aromatic amines.164
Improving the design of zeolites for all these reactions to form aromatics requires a better understanding of weakly acidic sites on zeolites,165 because too strong acidity will lead to acceleration of the rate of carbon deposit in the zeolite pores, with fast deactivation. Quite unexplored is the possibility of using alternative microporous materials analogous to zeolites, such as the zeolite-template carbon (ZTC), which could offer also the possibility of developing electrodes combining a tailored and ordered microporosity, a high surface area and good conductivity.166 Design of bifunctional zeolite not based on noble metals167 also has to be improved for better control of the reaction of aromatic production. These examples highlight that many possibilities still exist for improving the design of catalysts for the selective production of aromatics not based on FF use.
In conclusion, even if the preferable route to produce FF-free aromatics has to be still identified and the performances of the catalysts need to be further improved, it appears to be a feasible possibility for the production of aromatics raw materials for FF-free chemical production. However, more attention also has to be given to developing novel innovative routes to the cost-efficient separation of the different products formed, or to develop truly selective synthesis methods.
The production of ammonia is one of the processes responsible for the largest GHG impact of the chemical industry, around 1.2% of the global anthropogenic CO2 emissions. This is associated with the use of methane (or other FFs) to produce the hydrogen necessary for N2 conversion to ammonia. A modern, optimised and highly efficient methane-fed Haber Bosch (HB) process for ammonia synthesis emits about 1.5–1.6 tCO2-eq per ton of ammonia produced.168 The Sankey diagram showing the breakdown of CO2 emissions in a modern HB plant with respect to an electrified HB process (Fig. 7) provides relevant indications. The electrified HB process considers the production of H2 by electrolysis, and then the use of this H2 for the thermocatalytic synthesis of ammonia. The range of variability is associated with different sources of renewable energy used for electrolysis.
 | ||
| Fig. 7 Sankey diagram comparing CO2 emissions breakdown for conventional HB process and the electrified version, where H2 is produced by electrolysis then used in the HB thermocatalytic process. Based on the data reported in ref. 168. | ||
The electrified process significantly decreases the CO2 emissions with respect to the conventional process, but still, emissions remain significantly and largely associated with H2 generation. To decrease further the impact and reach a decrease in the GHG emissions by over 70%, as would be necessary to meet the NZE target, it is necessary to pass from the two-step electrified process (H2 production by electrolysis, then thermocatalytic ammonia synthesis – high temperature and pressure) to the direct electrocatalytic synthesis of ammonia. In the direct process, rather than produce H2, compress it to over 100 bars and then heat to the ammonia synthesis conditions – over 400 °C; the hydrogen-equivalent (H+/e−) are used in the process of N2 conversion to NH3. Near ambient conditions are used for electrocatalytic synthesis.
In this way, losses by overpotential (due to generation of H2 and its activation) and operations at high temperature/pressure (and associated thermodynamic limitations on equilibriums and thus need to recycle) can be reduced. Thus, meeting the targets requires passing from two-step electrification to direct electrification. The other advantage is that direct electrification is suited for distributed productions, e.g., for small-scale plants, while realizing highly energy-efficient small-scale ammonia synthesis plants using H2 produced by electrolysis is challenging. Furthermore, direct electrified synthesis of ammonia could be directly coupled to a PV (photovoltaic) module, to develop artificial-leaf type devices63 for distributed fertilizer production169 from air and sunlight. Note that a challenging direction, although not yet explored, is to realize on the cathodic site the N2 reduction to NH3170–173 and on the anodic site the N2 oxidation to NOx.174,175 By using gas-diffusion layer (GDL) electrodes and a “gas-phase” electrocatalytic reactor,176,177 continuous operations with the recovery of NH3 and NOx from the reactor gas streams at cathodic and anodic sections, respectively, could be achieved. By adsorbing the two gas outlet fluxes from the (photo)electrocatalytic cell in water, an ammonium nitrate fertilizer solution is produced which can be used directly as fertilizer. This is one example of the new possible directions to radically change the way we consider chemical production, by shifting to a distributed on-site model.
Many reviews have been published recently on N2 reduction (also indicated N2 fixation) to ammonia by a direct electrocatalytic process.178–185 There are alternatives for direct NH3 synthesis using renewable energy sources: photocatalytic and plasma-catalysis.62,186–188 Note also that early studies on electrocatalytic ammonia synthesis were made by feeding H2 together with N2, but now this is no longer used.189 In a sustainable framework, water appears to be the most advisable source of hydrogen atoms.
There is fast progress in the direct synthesis of ammonia, although care should be taken to avoid possible errors.190–192 While often it appears that this is the most critical issue in ammonia direct electrocatalytic synthesis, we would stress that there are other issues. The productivities are still too low, and not enough attention has been paid to this aspect. This means that even minor contaminations could alter significantly the data. Avoiding possible errors in reporting the data is correct, but the question is different: how to make the necessary step-change to obtain more reliable results. We have remarked171 that despite the large variety of proposed reaction mechanisms, all supported by experimental data and theoretical studies, the reported results fail within a quite restricted range of faradaic efficiency versus current density when the results are seen from the perspective of possible industrial exploitability. This suggests that the current studies are still unable to catch the crucial elements to reach the necessary performances for application, notwithstanding the endless reviews discussing the design of electrocatalysts for ammonia direct synthesis.
Table 4 reports a selection of literature results to illustrate further the concepts presented above. Many indications emerge from this table. The first is the very large range of materials used as electrocatalysts (just a minor subset of the large variety of materials tested) which provide relatively similar behaviour if data are analysed with respect to necessary targets (a productivity two-three order of magnitude greater, e.g. a current density above at least 100 mA cm−2). Even if each of these results claims high performances, the results are all still far from applicability. As the very different types of materials give relatively comparable results, identification of the (highly) active sites in the selective electrocatalytic conversion of N2 to NH3 has been not yet successfully made, notwithstanding the claims and mechanistic indications. It may be noted that current densities are over two orders of magnitude lower than those which have been obtained in the other reactions discussed before and indicated as the target for industrial exploitability.
| Electrode | Electrolyte | Current density, mA cm−2 | Yield rate, nmol−1 s−1 cm−2 | FE to NH3% | V vs. RHE | Ref. |
|---|---|---|---|---|---|---|
| Fe/SnO2 | 0.1 M HCl | ∼0.5 | 14 | 20 | −0.3 | 193 |
| CNx | 0.1 M HCl | <0.1 | 0.04 | 62 | −0.1 | 194 |
| Au (faced) | 0.1 M Li2SO4 | ∼0.4 | 0.15 | 73 | −0.3 | 195 |
| Pd3Pb | 0.1 M Na2SO4 | <0.5 | 0.06 | 22 | −0.2 | 196 |
| Bi nano | 0.5 M K2SO4 | ∼0.5 | 14 | 66 | −0.6 | 197 |
| Li–S doped MoS2 | 0.1 M Li2SO4 | ∼1.0 | 7.1 | 10 | −0.2 | 198 |
| B-COF | 0.1 M KOH | ∼0.1 | 0.2 | 45 | −0.2 | 199 |
| FePc/pyrene | 0.01 M H2SO4 | <0.1 | 0.5 | 32 | −0.1 | 200 |
Most of the studies have been focused on FE, with values reported up to over 70% as indicated in Table 4. However, the results are not always reproducible. The issue, however, as emerges from the table, is that these FE results are often obtained under conditions for which the productivity in ammonia formation is quite low, often close to the detection limit. From here comes the difficulty in reproducibility. As indicated, however, this is not the crucial problem, because in any case, the results remain of limited interest. After all, what is needed is to obtain high FE, above 50% at high current densities, over 100 mA cm−2. The REFUEL program of the US Department of Energy set a target of 90% FE at 300 mA cm−2. Systems which can achieve, or at least indicate a possible direction to reach, these targets have been not identified. The conclusion is that it is necessary to rethink the research in this area and use perhaps different approaches.10,171
Note also that FE complement to 100% is due to the side formation of H2. In an industrial device, this H2 could be recycled in the electrocatalytic reactor. There are no studies evaluating the impact of recycling this H2 on FE and performance. In addition, most of the studies use a liquid electrolyte, from which recovery of the ammonia could be costly and may impact stability. What is necessary is to perform the synthesis under conditions (electrolyteless201–204) allowing continuous operations and easy recovery of the ammonia formed from the gas stream of the electrocatalytic cell.
Recent results showing the possibility of obtaining quite high selectivities using the so-called lithium-mediated ammonia synthesis (LiNR) have received much attention.205–207 Particularly when small amounts of O2 are co-fed and high pressure (20 bar) operations are made,207 FE could reach a value around 80% for about 0.6% mol O2 addition. This is presented as a breakthrough result, with a patent application titled “Oxygen enhancement of lithium-mediated electrochemical nitrogen reduction” submitted in February 2021. Current density achieved or productivity are not explicitly reported. Experiments are made in an autoclave by application of a steady current density of −4 mA cm−2 or a cycling method, and the experiment is stopped after 50 C of charge is passed, with part of the current used to reduce the LiClO4 (Li plating). Continuous tests are thus not made, and the stability or effective performances cannot be established. By extrapolating the results given in the paper,207 an average rate of ammonia formation (at 20 bar) of about 9 nmol−1 s−1 cm−2 could be estimated, which is thus within the same range of best productivities reported in Table 4, but obtained at atmospheric pressure, and without applying the large overpotential (about 3–4 V) used in the Li et al.207 results.
In electrochemistry, the rate of electron transfer is proportional to the overpotential. In conventional electrocatalytic tests, the overpotential should be maintained as low as possible to avoid an increase in the side reaction of H2 formation, and thus obtain high HE. In these tests of LiNR, the trick is that the hydrogen does not derive from water electrolysis, but from a sacrificial agent ethanol, which is dehydrogenated likely to acetaldehyde. Aldehydes can easily polymerize, and thus stability could be an issue. The critical issue is the use of a sacrificial agent, the cost of which is higher than the product formed. Thus, this approach cannot be realized from a practical perspective.
An important component of the LiNR system is the solid electrolyte interface (SEI) that forms from the decomposition products of an organic electrolyte during Li deposition on the cathode. The mechanism is still a matter of question, but the first step is likely the electrochemical reduction of Li+ ions in the electrolyte to metallic Li, which is a very reactive material. This freshly plated Li is believed to then dissociate N2, and the N at the surface is finally reduced in a series of electron and proton transfers to form NH3 by using a proton source like ethanol. The possibility of realizing this process under continuous conditions presents a question mark.
Cai et al.206 used a different approach for LiNR. One of the sides of an electrochemical cell is in contact with a stainless steel cloth acting both as electrode and membrane through which N2 diffuses. One side of this membrane is in contact with N2, the other with the electrolyte and Li deposit on it. The performances reported are of slightly better productivity (∼25 nmol−1 s−1 cm−2), but lower FE (∼40%). The overpotential is quite high, at a fixed potential of −6.7 VAg/Ag+ used in these tests. Ethanol is also used as a sacrificial agent (proton donor).
A closer analysis of these results on LiNR thus indicates that they are not a true breakthrough and the issue of developing novel approaches for ammonia direct synthesis remains. It is thus probably necessary to reconsider the whole approach and explore new directions, as well as to analyse from a more critical perspective the whole set of results and possibilities of the approach. Few studies have attempted this effort. Among them is the review by Martín et al.208 which used performance maps to analyse the gap concerning practical implementation and identify sources of inefficiency. Among the novel directions that could be mentioned in the ammonia synthesis is the use of an H2-permeable nickel membrane/electrode that spatially separates the electrolyte and hydrogen reduction side from the dinitrogen activation and hydrogenation sites.209
The impact of FF-free chemical production
To analyse the potential impact of FF-free chemical production, the mapping of the global flow of chemicals prepared by Levi and Cullen may be considered.210 Although the data refer to about one decade ago (the year 2013), this analysis could be still considered valid and no further attempts were later made to update the data. Fig. 8 reports the summary of the input and output data for chemical production reported by Levi and Cullen.210 | ||
| Fig. 8 Summary of input and output data (M ton per year) reported by Levi and Cullen210 in mapping fluxes in chemical production. Output data do not consider secondary products such as CO2, H2O, HCl, CaCl2, etc. | ||
What is evident from Fig. 8 is that when secondary products are not considered, only about one-third of the FFs are incorporated in the final products of petrochemistry. However, part of the use of FFs as an energy source cannot be substituted by RES only. The target of 70% cut of CO2 eq. emissions in the chemical industry thus also requires reduction of the use of FFs as carbon sources, by closing the carbon cycle and using bio-based raw materials. By considering an average factor of 1.3 CO2 eq. ton emitted per ton of fuel combusted, around 470 Mt per year CO2 eq. could be saved. We can also consider a possible saving in the production of N fertilizers of ∼50% using electrified production, saving around 130 Mt per year CO2 eq. By introducing direct electrified processes using alternative carbon sources it may be estimated that an additional 200–230 Mt per year CO2 eq. emissions could be saved. Thus in total, the potential cut is around 830 Mt per year CO2 eq. emissions, which corresponds to a reduction by about 70% of the total emissions by chemical production. It is also evident that it is not possible to meet this target by electrification of process units alone.
Sustainability of the transition to a fossil-free chemical production
After having examined (i) the socio-techno-economic motivations, (ii) the feasibility of this objective and (iii) the potential impact as well, it is necessary to discuss briefly whether the transition to a fossil-free chemical production is sustainable. A sustainable transition happens when economic, environmental, and social improvements are realized at the same time. The question of whether the transition to a fossil-free chemical production is sustainable must be thus addressed in a joint manner from these three perspectives. In addition, a deep transition requires the synchronization of the development of many technologies together with the economic and social elements.211 Thus, the technological and scientific advances must be put in this general perspective (i) to identify whether the directions investigated are sustainable, (ii) what are the gaps, limits and opportunities, and (iii) whether the timing for their implementation is compatible with the necessary synchronization with the other elements of the deep transition. The main motivation for this perspective is to provide the elements for this holistic analysis; the scientific and technological advances alone cannot provide indications of whether this deep transformation in the chemical production, in parallel with that in energy, is sustainable and what the conditions are to improve the sustainability. Thus, scientists must be aware of the general context to contribute effectively to the implementation of the transition. Note that a perspective on future, long-term transformation necessarily contains personal elements of evaluation, which depend on the scenario assumed and related uncertainty and assumptions. For this reason, even if in part the elements discussed here are personal, they are mediated by the larger on-going discussion in the frame of the large initiative SUNERGY (fossil-free fuels and chemicals for a climate-neutral Europe; https://www.sunergy-initiative.eu). As part of the related coordination and support actions (CSA) SUNER-C, a strategic roadmap for the transition toward a fossil-free EU economy, is in preparation.A first general question concerns what is a fossil-free sustainable chemical production. The discussion in this perspective assumes that to meet the NZE targets by the year 2050, it is necessary to achieve by this year a substitution of over 50% in FF use in the chemical industry (at least in Europe). This value is above the 30% target already indicated a decade ago by industrial organizations such as CEFIC (European Chemical Industry Council) as part of large initiatives such as SusChem, later becoming A-SPIRE. Although tight, the 50% substitution, in view of the acceleration in recent years on energy transition, seems an acceptable target also from an industrial perspective. Completely fossil-free chemical production could be realized thus only beyond 2050. It must also be recognized that the transition to fossil-free chemical production will be realized when investment in new plants will be only on technologies not based on FFs as raw or energy materials. However, plants using old technologies based on FFs will still be operative. A typical amortization time of chemical plants is over 20 years.
To produce any chemical, one needs raw materials, energy and reactors that realize the defossilization of the chemical production. Thus, a fossil-free chemical production process needs to defossilize all these elements to be sustainable. It may be estimated that by the year 2050, the green electricity share in the energy mix will be in the 70–90% range, depending on the scenario assumed.213 However, without the introduction of effective energy vectors (solar fuels), the coupling of chemical production (and in general of energy-intensive industries9) and RES will be not effective. Thus, the sustainability of the transition to fossil-free chemical production will depend on the acceleration of the technologies to produce solar fuels (also called e-fuels) on a large scale. A crucial element to realising this objective is to decrease the costs of production while at the same time increasing the energy efficiency of the process. As mentioned in the previous section, avoiding the production of molecular H2 and instead using the hydrogen-equivalent H+/e− is an important factor to increase energy efficiency. By coupling with the process intensification, developing directly the second-generation electrocatalytic routes rather than implementing the first-generation technologies (so-called power-to-X) is one of the critical elements to accelerating the process.22 However, discordant opinions still exist on this question.
In addition, a process using only electricity, but not also defossilizing all the other process and material components, realizes only in part the sustainability objective. Electricity should be generated in a sustainable way, as should the raw materials for the process. Chemical production is a chain process, and for this reason we addressed here especially the building blocks for chemical production, e.g., olefins, aromatics, and syngas, evidencing that not only energy but also the input raw materials should be fossil-free or based on carbon circularity.
At the same time, new process performance indicators such as (i) efficiency in energy and carbon circularity, (ii) overall degree of substitution of FFs, and (iii) sustainable energy transition readiness212 should be introduced to better monitor the transition.
Therefore, is “fossil-free” sustainable? To reply to this final question, it must be noted that this transformation of chemical production is likely to be irreversible, and driven only in part by the need to reduce GHG emissions. The main economic drivers are geopolitical (security) and the change of the windows of opportunity for using FFs. From a social perspective, there is a need to change the modality of production and for better integration with local resources and reduction of the environmental impact of large plants. The sustainability of this transition thus derives from other aspects, rather than only from specific technological aspects. The assessment thus requires the use of new methodological approaches beyond the conventional.214,215 However, a main limiting factor appears to be the rate of technological development for the required new processes. Accelerating this development requires a better identification of the key target objectives beyond the scientific and technological ones.
Conclusions
Chemical production needs to significantly restructure the modalities of production to meet the NZE targets, and in particular, accelerate in moving to FF-free production. This perspective attempted to analyse the status, gaps and perspectives of progress in this direction, noting also that it is an opportunity to develop a novel model of chemical production and create innovation and benefits for society. Defossilization of chemical production is a challenge and requires the combination of electrification of the chemical processes with the direct electrification of the catalytic reactions. Although challenging, accelerating the progress in the direct electrified processes is also a necessary condition to meet the target of NZE in 2050, as shown from the analysis of the potential impact of these technologies in reducing CO2 eq. emissions from chemical production.Electrification of the process units, and in particular of the chemical reactors, requires overcoming the simpler approach of just providing the heat of reaction by electrical heating rather than by burning fossil fuels (electrical versus fired furnaces). More advanced approaches require redesigning the catalyst and the reactor itself to take full advantage of the possibilities of overcoming heat transfer limitations and realize tailored thermal gradients between the catalyst surface and the fluid phase.
Direct electrification of the processes entails developing novel modalities to supply directly the energy for the chemical transformations. We have focused the discussion on electrocatalytic solutions because we commented that this is the technology that likely will be first introduced commercially in a broad mode, while other technologies such as photo- and plasma-catalysis will require a longer time for application or wider use out of specific cases. To focus the discussion, only the most relevant cases for defossilizing the chemical production were analysed: (i) light olefin production, (ii) direct synthesis of main intermediates such as formaldehyde and acetic acid, and (iii) the production of aromatics (BTX) alternative to those starting from FFs. Some considerations on the electrocatalytic production of methanol from CO2 were also given, although we believe that there are better alternatives for this reaction. The analysis of the status of research aimed not to provide a status of the art, but to identify the key elements to assess whether in the short–medium term introducing these technologies will be feasible. Also, indications of areas to foster research activities were given.
The analysis of the direct electrocatalytic synthesis of inorganic chemicals, in particular ammonia to produce fertilizers, instead reveals that current results are still quite far from the possibility of applications, noting that, however, changing the direction of current approaches and exploring new solutions will be likely necessary.
In conclusion, the analyses presented in this perspective show that proceeding towards defossilization of the chemical production may result in an over 70% cut of the current greenhouse gas impact, with a saving of over 800 Mt per year CO2 eq. emissions. Not only is an intensification of the research necessary, but also a holistic system approach. As noted in this paper, often this vision is missing or partial, and the scientific development is focused on specific aspects which may not be the key factors determining the possibility of accelerating the development. This paper aimed to provide at least some elements allowing researchers to have a more comprehensive view of gaps, limits and possibilities in moving to FF-free chemical production, and explained also why this conversion is necessary and the opportunities for innovation and transformation of the modalities of products it offers.
Abbreviations
Acronyms
| BDD | Boron-doped diamond |
| BN-C | Boron nitride |
| BZVYYb | BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (doped barium zirconate) |
| CAGR | Compound annual growth rate |
| CO2RR | CO2 reduction reaction |
| COTC | Crude oil-to-chemicals |
| CSA | Coordination and support actions |
| DFT | Density functional theory |
| DMF | Dimethylfuran |
| eEE | Electrical energy efficiency |
| EXAFS | Extended X-ray absorption fine structure |
| EU | European Union |
| FE | Faradaic efficiency |
| FFs | Fossil fuels |
| GDL | Gas-diffusion layer |
| GHG | Greenhouse gas |
| GWI | Global warming impact |
| HDO | Hydrodeoxygenation |
| HER | Hydrogen evolution reaction |
| HMF | Hydroxymethylfurfural |
| LCA | Life-cycle assessment |
| MEA | Membrane electrode assembly |
| MSW | Municipal solid wastes |
| Mt | Million tons |
| MTA | Methanol-to-aromatics |
| MTO | Methanol-to-olefin |
| MW | Microwave |
| NZE | Net-zero emissions |
| OER | Oxygen evolution reaction |
| PBSCF | PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (porous double perovskite) |
| POM | Polyoxometalate |
| PSA | Pressure swing absorption |
| PtX | Power-to-X |
| PVP | polyvinylpyrrolidone |
| R&D | Research and development |
| rGO | Reduced graphene oxide |
| RE | Renewable energy |
| RES | Renewable energy sources |
| RF | Radio frequency |
| SMR | Steam-methane-reforming |
| SOEC | Solid oxide electrolyzer cell |
| TNA | TiO2 nanotube array |
| TOF | Turnover frequency |
| TRL | Technology readiness level |
| ZTC | Zeolite-template carbon |
Author contributions
All authors equally contributed to preparing and revising this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EU with the ERC Synergy SCOPE project (810182), Italian MUR by PRIN 2017 projects MULTI–e (20179337R7) and CO2 ONLY project (2017WR2LRS). GC also thanks the Alexander von Humboldt-Stiftung/Foundation (Humboldt Research Award).References
- E. Cetinkaya, N. Liu, T. J. Simons and J. Wallach, Petrochemicals 2030: Reinventing the way to win in a changing industry, McKinsey & Co., 2018 Search PubMed.
- F. Cavani, G. Centi, S. Perathoner and F. Trifiró, Sustainable Industrial Chemistry, Wiley-VCH, Weinheim - Germany, 2009 Search PubMed.
- R. U. Ayres, L. T. Peiró and G. Villalba Méndez, Environ. Sci. Technol., 2011, 45, 10634 CrossRef CAS PubMed.
- R. S. Weber and L. J. Snowden-Swan, J. Adv. Manuf. Process., 2019, 1, e10011 Search PubMed.
- H. Uno and Y. Inada, Potential of small-scale, distributed manufacturing processes in energy and chemical industries, Mitsui & Co. Global Strategic Studies Institute Monthly Report May 2018.
- G. S. Patience and D. C. Boffito, J. Adv. Manuf. Process., 2020, 2, e10039 Search PubMed.
- W. J. Grieco, CEP, 2019, 10, 24 Search PubMed.
- A. I. Stankiewicz and H. Nigar, React. Chem. Eng., 2020, 5, 1005 RSC.
- S. Perathoner, K. M. Van Geem, G. B. Marin and G. Centi, Chem. Commun., 2021, 57, 10967 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2022, 387, 216 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2020, 342, 4 CrossRef CAS.
- G. Papanikolaou, G. Centi, S. Perathoner and P. Lanzafame, ACS Catal., 2022, 12, 2861 CrossRef CAS PubMed.
- S. Abate, G. Centi, P. Lanzafame and S. Perathoner, J. Energy Chem., 2015, 24, 535 CrossRef.
- A. Corma, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Al-Bogami, M. S. Al-Ghrami and A. Bourane, Catal.: Sci. Technol., 2017, 7, 12 RSC.
- FutureBridge, Crude Oil-to-Chemicals: Future of Refinery. Accessed on Jan. 17, 2022 on https://www.futurebridge.com/blog/crude-oil-to-chemicals-future-of-refinery/#:~:text=Crude%20oil%2Dto%2Dchemicals%20(,a%20non%2Dintegrated%20refinery%20complex.
- A. H. Tullo, Why the future of oil is in chemicals, not fuels, C&EN, 2019, 97. Accessed on Jan. 17, 2022 on https://cen.acs.org/business/petrochemicals/future-oil-chemicals-fuels/97/i8#:~:text=Taxes%20on%20chemicals%20are%20lower,change%20petrochemical%20markets%2C%20cautions%20R.%20J.
- M. Hermesmann, K. Gruebel, L. Scherotzki and T. E. Mueller, Renewable Sustainable Energy Rev., 2021, 138, 110644 CrossRef CAS.
- D. M. Amaya-Duenas, M. Riegraf, R. Costa and K. A. Friedrich, Chem. Ing. Tech., 2020, 92, 1283 CrossRef CAS.
- Z. Chehade, C. Mansilla, P. Lucchese, S. Hilliard and J. Proost, Int. J. Hydrogen Energy, 2019, 44, 27637 CrossRef CAS.
- S. R. Foit, I. C. Vinke, L. G. J. de Haart and R.-A. Eichel, Angew. Chem., Int. Ed., 2017, 56, 5402 CrossRef CAS PubMed.
- B. Rego de Vasconcelos and J.-M. Lavoie, Front. Chem., 2019, 7, 392 CrossRef CAS PubMed.
- G. Papanikolaou, G. Centi, S. Perathoner and P. Lanzafame, Chin. J. Catal., 2022, 43, 1194–1203 CrossRef CAS.
- M. Fritz and A. Aydemir, in Industrial Efficiency 2020 - Decarbonise Industry!, European Council for an Energy-Efficient Economy (ECEEE), Stockholm, 2020, pp. 49–57. Available online on Feb. 9th, 2022 at: https://www.isi.fraunhofer.de/content/dam/isi/dokumente/cce/2020/2-023-20_Fritz.pdf Search PubMed.
- C. Michalakakis, J. M. Cullen, A. G. Hernandez and B. Hallmark, (2019). Exergy and network analysis of chemical sites, Sustain. Prod. Consum., 2019, 19, 270 CrossRef.
- Y. Li, Y. Li, X. Zhang, C. Wang, X. Li and L. Ma, Int. J. Hydrogen Energy, 2021, 46, 3669 CrossRef CAS.
- Y. D. Chaniago, M. A. Qyyum, R. Andika, W. Ali, K. Qadeer and M. Lee, J. Cleaner Prod., 2019, 239, 118029 CrossRef CAS.
- I. Ioannou, S. C. D'Angelo, A. J. Martín, J. Pérez-Ramírez and G. Guillén-Gosálbez, ChemSusChem, 2020, 13, 6370 CAS.
- P. Lanzafame, S. Abate, C. Ampelli, C. Genovese, R. Passalacqua, G. Centi and S. Perathoner, ChemSusChem, 2017, 10, 4409 CrossRef CAS PubMed.
- S. Abate, P. Lanzafame, S. Perathoner and G. Centi, ChemSusChem, 2015, 8, 2854 CrossRef CAS PubMed.
- S. Mbatha, R. C. Everson, N. M. Musyoka, H. W. Langmi, A. Lanzini and W. Brilmane, Sustainable Energy Fuels, 2021, 5, 3490 RSC.
- R. Kungas, J. Electrochem. Soc., 2020, 167, 044508 CrossRef CAS.
- F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, J. Mater. Chem. A, 2020, 8, 15458 RSC.
- De. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244 CrossRef CAS.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2019, 52, 656–664 CrossRef CAS PubMed.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- S. Perathoner and G. Centi, Catal. Today, 2019, 330, 157 CrossRef CAS.
- T. Vass, P. Levi, A. Gouy and H. Mandová, Chemicals, International Energy Agency (IEA), Paris (France), 2021. Available on Feb. 16th, 2022 at https://www.iea.org/reports/chemicals Search PubMed.
- P. D'Aprile, H. Engel, G. van Gendt, S. Helmcke, S. Hieronimus, T. Nauclér, D. Pinner, D. Walter and M. Witteveen, Net-Zero Europe, McKinsey & Co, Nov. 2020. Available on 18 Feb. 2022 at https://www.mckinsey.com/business-functions/sustainab-ility/our-insights/how-the-european-union-could-achieve-net-zero-emissions-at-net-zero-cost Search PubMed.
- M. Bonheure, L. Vandewalle, G. Marin and K. Van, Geem. Chem. Eng. Prog., 2021, 117, 37 Search PubMed.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10 CrossRef.
- W. Grieco and I. Palou-Rivera, Chem. Eng. Prog., 2022, 1, 17 Search PubMed.
- S. de Bruyn, C. Jongsma, B. Kampman, B. Görlach and J.-E. Thie, Energy-intensive industries - Challenges and opportunities in energy transition, European Union, 2020.
- S. Madeddu, F. Ueckerdt, M. Pehl, J. Peterseim, M. Lord, K. A. Kumar, C. Krüge and G. Luderer, Environ. Res. Lett., 2020, 15, 124004 CrossRef CAS.
- H. Wiertzema, E. Svensson and S. Harvey, Front. Energy Res., 2020, 8, 192 CrossRef.
- S. T. Wismann, C. Frandsen, I. Chorkendorff, J. S. Engbaek, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen and P. M. Mortensen, Science, 2019, 364, 756 CrossRef CAS PubMed.
- S. T. Wismann, J. S. Engbaek, S. B. Vendelbo, W. L. Eriksen, C. Frandsen, P. M. Mortensen and I. Chorkendorff, Ind. Eng. Chem. Res., 2019, 58, 23380 CrossRef CAS.
- E. Delikonstantis, E. Igos, S.-A. Theofanidis, E. Benetto, G. B. Marin, K. Van Geem and G. D. Stefanidis, Green Chem., 2021, 23, 7243 RSC.
- M. Ambrosetti, A. Beretta, G. Groppi and E. Tronconi, Front. Chem. Eng., 2021, 3, 747636 CrossRef.
- S. Renda, M. Cortese, G. Iervolino, M. Martino, E. Meloni and V. Palma, Catal. Today, 2022, 383, 31 CrossRef CAS.
- M. Rieks, R. Bellinghausen, N. Kockmann and L. Mleczko, Int. J. Hydrogen Energy, 2015, 40, 15940 CrossRef CAS.
- M. R. Almind, M. G. Vinum, S. T. Wismann, M. F. Hansen, S. B. Vendelbo, J. S. Engbaek, P. M. Mortensen, I. Chorkendorff and C. Frandsen, ACS Appl. Nano Mater., 2021, 4, 11537 CrossRef CAS.
- M. G. Vinum, M. R. Almind, J. S. Engbæk, S. B. Vendelbo, M. F. Hansen, C. Frandsen, J. Bendix and P. M. Mortensen, Angew. Chem., Int. Ed., 2018, 57, 10569 CrossRef CAS PubMed.
- W. Wang, G. Tuci, C. Duong-Viet, Y. Liu, A. Rossin, L. Luconi, J.-M. Nhut, L. Nguyen-Dinh, C. Pham-Huu and G. Giambastiani, ACS Catal., 2019, 9, 7921 CrossRef CAS.
- P. D. Muley, Y. Wang, J. Huc and D. Shekhawat, in Catalysis: Volume 33, 2021, p. 1 Search PubMed.
- H. Goyal, T.-Y. Chen, W. Chen and D. G. Vlachos, Chem. Eng. J., 2022, 430, 133183 CrossRef CAS.
- V. Palma, D. Barba, M. Cortese, M. Martino, S. Renda and E. Meloni, Catalysts, 2020, 10, 246 CrossRef CAS.
- I. Julian, C. M. Pedersen, A. B. Jensen, A. K. Baden, J. L. Hueso, A. V. Friderichsen, H. Birkedal, R. Mallada and J. Santamaria, Catal. Today, 2022, 383, 2 CrossRef.
- M. Munoz, J. Nieto-Sandoval, E. Serrano, Z. M. de Pedro and J. A. Casas, J. Environ. Chem. Eng., 2020, 8, 104085 CrossRef CAS.
- S. R. Yassine, Z. Fatfat, G. H. Darwish and P. Karam, Catal. Sci. Technol., 2020, 10, 3890 RSC.
- S. S. Kale, J. M. Asensio, M. Estrader, M. Werner, A. Bordet, D. Yi, J. Marbaix, P.-F. Fazzini, K. Soulantica and B. Chaudret, Catal. Sci. Technol., 2019, 9, 2601 RSC.
- S. Faure, S. S. Kale, N. Mille, S. Cayez, T. Ourlin, K. Soulantica, J. Carrey and B. Chaudret, J. Appl. Phys., 2021, 129, 044901 CrossRef CAS.
- A. Bogaerts, X. Tu, J. C. Whitehead, G. Centi, L. Lefferts, O. Guaitella, F. Azzolina-Jury, H.-H. Kim, A. B. Murphy, W. F. Schneider, T. Nozaki, J. C. Hicks, A. Rousseau, F. Thevenet, A. Khacef and M. Carreon, J. Phys. D: Appl. Phys., 2020, 53, 443001 CrossRef CAS.
- V. Romano, G. D'Angelo, S. Perathoner and G. Centi, Energy Environ. Sci., 2021, 14, 5760 RSC.
- W. Wu, H. Hu and D. Ding, Cell Rep. Phys. Sci., 2021, 2, 100405 CrossRef CAS.
- D. Ding, Y. Zhang, W. Wu, D. Chen, M. Liu and T. He, Energy Environ. Sci., 2018, 11, 1710 RSC.
- S. Wang, J.-L. Luo, A. R. Sanger and K. T. Chuang, J. Phys. Chem. C, 2007, 111(13), 5069 CrossRef CAS.
- Z. Shi, J.-L. Luo, S. Wang, A. R. Sanger and K. T. Chuang, J. Power Sources, 2008, 176, 122 CrossRef CAS.
- Y. Feng, J. Luo and K. T. Chuang, J. Phys. Chem. C, 2008, 112(26), 9943 CrossRef CAS.
- L. Kuo and C.-T. Dinh, Curr. Opin. Electrochem., 2021, 30, 100807 CrossRef CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897 RSC.
- J. Chen, T. Wang, Z. Li, B. Yang, Q. Zhang, L. Lei, P. Feng and Y. Hou, Nano Res., 2021, 14, 3188 CrossRef CAS.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975 CrossRef CAS PubMed.
- D. Mallamace, G. Papanikolaou, S. Perathoner, G. Centi and P. Lanzafame, Int. J. Mol. Sci., 2021, 22, 139 CrossRef CAS.
- W. Ye, X. Guo and T. Ma, Chem. Eng. J., 2021, 414, 128825 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610 CrossRef CAS PubMed.
- D. Gao, R. M. Aran-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198 CrossRef CAS.
- D. Gao, I. Sinev, F. Scholten, R. M. Aran-Ais, N. J. Divins, J. Timoshenko and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2019, 5, 17047 CrossRef.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646 CrossRef CAS PubMed.
- K. Ogura, J. CO2 Util., 2013, 1, 43 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS.
- Fraunhofer ISE, Levelized Cost of Electricity Renewable Energy Technologies, June 2021. Available on 24 Feb. 2022 at https://www.ise.fraunhofer.de/content/dam/ise/en/documents/publications/studies/EN2021_Fraunhofer-ISE_LCOE_Renewable_Energy_Technologies.pdf.
- M. A. Khan, S. Nabil, T. A. Al-Attas, S. Roy, M. M. Rahman, S. Larter, P. M. Ajayan, Ji. Hu and M. Kibria, ChemRxiv. Cambridge: Cambridge Open Engage, 2020, DOI:10.26434/chemrxiv.13331204.v1.
- J. Sisler, S. Khan, A. H. Ip, M. W. Schreiber, S. A. Jaffer, E. R. Bobicki, C.-T. Dinh and E. H. Sargent, ACS Energy Lett., 2021, 6, 997 CrossRef CAS.
- C. A. R. Pappijn, M. Ruitenbeek, M.-F. Reyniers and K. M. Van Geem, Front. Energy Res., 2020, 8, 557466 Search PubMed.
- B. C. Marepally, C. Ampelli, C. Genovese, T. Saboo, S. Perathoner, F. M. Wisser, L. Veyre, J. Canivet, E. A. Quadrelli and G. Centi, ChemSusChem, 2017, 10, 4442 CrossRef CAS PubMed.
- C. Ampelli, G. Centi, R. Passalacqua and S. Perathoner, Catal. Today, 2016, 259, 246 CrossRef CAS.
- C. Ampelli, C. Genovese, B. C. Marepally, G. Papanikolaou, S. Perathoner and G. Centi, Faraday Discuss., 2015, 183, 125 RSC.
- S. Perathoner, G. Centi and D. Su, ChemSusChem, 2016, 9, 345 CrossRef CAS PubMed.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS.
- R. Arrigo, R. Blume, V. Streibel, C. Genovese, A. Roldan, M. E. Schuster, C. Ampelli, S. Perathoner, J. J. Velasco Vélez, M. Hävecker, A. Knop-Gericke, R. Schlögl and G. Centi, ACS Catal., 2021, 12, 411 CrossRef.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178 CrossRef CAS PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509 CrossRef CAS PubMed.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104 CrossRef CAS.
- F. P. García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661 CrossRef PubMed.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478 CrossRef CAS.
- Y. Jiang, C. Choi, S. Hong, S. Chu, T.-S. Wu, Y.-L. Soo, L. Hao, Y. Jung and Z. Sun, Cell Rep. Phys. Sci., 2021, 2, 100356 CrossRef CAS.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606 CrossRef CAS PubMed.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, Vl. Solokha, S. K. Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- S. Ralser, A. Kaiser, M. Probst, J. Postler, M. Renzler, D. K. Bohme and P. Scheier, Phys. Chem. Chem. Phys., 2016, 18, 3048 RSC.
- G. Centi, ChemSusChem, 2022, 15, e202200007 CrossRef CAS.
- IRENA (International Renewable Energy Agency) and Methanol Institute, Innovation Outlook: Renewable Methanol, IRENA, Abu Dhabi, 2021. Available on 24 Feb. 2022 at https://www.irena.org/-/media/Files/IRENA/Agency/Publication/2021/Jan/IRENA_Innovation_Renewable_Methanol_2021.pdf.
- Z. Li, N. H. Attanayake, J. L. Blackburn and E. M. Miller, Energy Environ. Sci., 2021, 14, 624 Search PubMed.
- S. Zhang, X. Jing, Y. Wang and F. Li, ChemNanoMat, 2021, 7, 728 CrossRef CAS.
- H. Guzman, N. Russo and S. Hernandez, Green Chem., 2021, 23, 1896 RSC.
- S. Sarp, S. Gonzalez Hernandez, C. Chen and S. W. Sheehan, Joule, 2021, 5, 59 CrossRef CAS.
- A. Roy, H. S. Jadhav, S. J. Park and J. G. Seo, J. Alloys Compd., 2021, 887, 161449 CrossRef CAS.
- S. Schlager, A. Dibenedetto, M. Aresta, D. H. Apaydin, L. M. Dumitru, H. Neugebauer and N. S. Sariciftci, Energy Technol., 2017, 5, 812 CrossRef CAS.
- M. Guo, F. Gu, L. Meng, Q. Liao, Z. Meng and W. Liu, Sep. Purif. Technol., 2022, 286, 120480 CrossRef CAS.
- W. Liu, Y. Hou, B. Hou and Z. Zhao, Chin. J. Chem. Eng., 2014, 22, 1328 CrossRef CAS.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871 CrossRef CAS PubMed.
- A. U. Pawar, U. Pal, J. Y. Zheng, C. W. Kim and Y. S. Kang, Appl. Catal., B, 2022, 303, 120921 CrossRef CAS.
- P. Karagoz, R. M. Bill and M. Ozkan, Renewable Energy, 2019, 143, 741 CrossRef CAS.
- L. Gao, X. Wu, C. Zhu, Z. Jin, W. Wang and X. Xia, Crit. Rev. Biotechnol., 2020, 40, 522 CrossRef CAS PubMed.
- N. Saichana, K. Matsushita, O. Adachi, I. Frebort and J. Frebortova, Biotechnol. Adv., 2015, 33, 1260 CrossRef CAS.
- P. Lanzafame, G. Centi and S. Perathoner, Catal. Today, 2014, 234, 2 CrossRef CAS.
- S. Perathoner, S. Gross, E. J. M. Hensen, H. Wessel, H. Chraye and G. Centi, ChemCatChem, 2017, 9, 904–909 CrossRef CAS.
- Z. Chen, X. Wang and L. Liu, Chem. Rec., 2019, 19, 1272–1282 CrossRef CAS PubMed.
- T. N. Nguyen, J. Guo, A. Sachindran, F. Li, A. Seifitokaldani and C.-T. Dinh, J. Mater. Chem. A, 2021, 9, 12474 RSC.
- G. Feng, W. Chen, B. Wang, Y. Song, G. Li, J. Fang, W. Wei and Y. Sun, Chem. – Asian J., 2018, 13, 1992 CrossRef CAS.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang and B. Han, Green Chem., 2017, 19, 2086 RSC.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646 CrossRef CAS PubMed.
- J. Wu, T. Sharifi, Y. Gao, T. Zhang and P. M. Ajayan, Adv. Mater., 2019, 31, 1804257 CrossRef PubMed.
- F. Guo, B. Liu, M. Liu, Y. Xia, T. Wang, W. Hu, P. Fyffe, L. Tian and X. Chen, Green Chem., 2021, 23, 5129 RSC.
- D. Zang, Q. Li, G. Dai, M. Zeng, Y. Huang and Y. Wei, Appl. Catal., B, 2021, 281, 119426 CrossRef CAS.
- D. Giusi, F. Tavella, M. Miceli, C. Ampelli, G. Centi, D. Cosio, C. Genovese and S. Perathoner, Chem. Eng. Trans., 2021, 86, 1405 Search PubMed.
- D. Giusi, C. Ampelli, C. Genovese, S. Perathoner and G. Centi, Chem. Eng. J., 2021, 408, 127250 CrossRef CAS.
- J. F. De Brito, C. Genovese, F. Tavella, C. Ampelli, M. V. Boldrin Zanoni, G. Centi and S. Perathoner, ChemSusChem, 2019, 12, 4274 CrossRef PubMed.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U.-P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schöfberger, Angew. Chem., Int. Ed., 2020, 59, 10527 CrossRef CAS PubMed.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, Green Chem., 2017, 19, 2406 RSC.
- Y. M. Liu, S. Chen, X. Quan and H. T. Yu, J. Am. Chem. Soc., 2015, 137, 11631 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef.
- Y. Zhou and B. S. Yeo, J. Mater. Chem. A, 2020, 8, 23162 RSC.
- J. Zhang, W. Luo and A. Zuttel, J. Catal., 2020, 385, 140 CrossRef CAS.
- P. Zhu, C. Xia, C.-Y. Liu, K. Jiang, G. Gao, X. Zhang, Y. Xia, Y. Lei, H. N. Alshareef, T. P. Senftle and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2010868118 CrossRef CAS.
- L. Handojo, A. K. Wardani, D. Regina, C. Bella, M. T. A. P. Kresnowati and I. G. Wenten, RSC Adv., 2019, 9, 7854 RSC.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, E. Irtem, A. T. Laitinen, L. J. P. van den Broeke, T. Breugelmans, J. P. Martin Trusler, W. de Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 22718 CrossRef CAS.
- Y. Chen, A. Vise, W. E. Klein, F. C. Cetinbas, D. J. Myers, W. A. Smith, T. G. Deutsch and K. C. Neyerlin, ACS Energy Lett., 2020, 5, 1825 CrossRef CAS.
- G. Busca, Energies, 2021, 14, 4061 CrossRef CAS.
- Q. Meng, J. Yan, R. Wu, H. Liu, Y. Sun, N. N. Wu, J. Xiang, L. Zheng, J. Zhang and B. Han, Nat. Commun., 2021, 12, 4534 CrossRef CAS.
- H. Li, A. Bunrit, N. Li and F. Wang, Chem. Soc. Rev., 2020, 49, 3748 RSC.
- A. Kumar, M. Jindal, S. Maharana and B. Thallada, Energy Fuels, 2021, 35, 16965 CrossRef CAS.
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470 CrossRef CAS PubMed.
- M. Seifert, M. S. Marschall, T. Gille, C. Jonscher, O. Busse, S. Paasch, E. Brunner, W. Reschetilowski and J. J. Weigand, ChemCatChem, 2020, 12, 6301 CrossRef CAS.
- T. Li, T. Shoinkhorova, J. Gascon and J. Ruiz-Martinez, ACS Catal., 2021, 11, 7780 CrossRef CAS.
- D. Wang, Z. Xie, M. D. Porosoff and J. Chen, Chem, 2021, 7, 2277 CAS.
- T. Zhang, Chem. Sci., 2021, 12, 12529 RSC.
- D. Kiani, S. Sourav, Y. Tang, J. Baltrusaitis and I. E. Wachs, Chem. Soc. Rev., 2021, 50, 1251 RSC.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736 CrossRef CAS PubMed.
- S. M. Seifert, M. S. Marschall, T. Gille, C. Jonscher, O. Busse, S. Paasch, E. Brunner, W. Reschetilowski and J. J. Weigand, ChemCatChem, 2020, 12, 6301–6310 CrossRef.
- M. Seifert, M. S. Marschall, T. Gille, C. Jonscher, P. Royla, O. Busse, W. Reschetilowski and J. J. Weigand, Chem. – Asian J., 2020, 15, 3878–3885 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- G. Q. Zhang, T. Bai, T. F. Chen, W. T. Fan and X. Zhang, Ind. Eng. Chem. Res., 2014, 53, 14932–14940 CrossRef CAS.
- F. Wang, W. Xiao, L. Gao and G. Xiao, Catal. Sci. Technol., 2016, 6, 3074–3086 RSC.
- T. Shoinkhorova, T. Cordero-Lanzac, A. Ramirez, S.-h. Chung, A. Dokania, J. Ruiz-Martinez and J. Gascon, ACS Catal., 2021, 11, 3602–3613 CrossRef CAS.
- V. J. Margarit, E. M. Gallego, C. Paris, M. Boronat, M. Moliner and A. Corma, Green Chem., 2020, 22, 5123–5131 RSC.
- Q. Meng, J. Yan, R. Wu, H. Liu, Y. Sun, N. N. Wu, J. Xiang, L. Zheng, J. Zhang and B. Han, Nat. Commun., 2021, 12, 4534 CrossRef CAS PubMed.
- C. Moretti, B. Corona, R. Hoefnagels, I. Vural-Gürsel, R. Gosselink and M. Junginger, Sci. Total Environ., 2021, 770, 144656 CrossRef CAS PubMed.
- E. Blondiaux, J. Bomon, M. Smolen, N. Kaval, Fi. Lemiere, S. Sergeyev, L. Diels, B. Sels and U. W. Bert, ACS Sustainable Chem. Eng., 2019, 7, 6906–6916 CrossRef CAS.
- P. Lanzafame, G. Papanikolaou, S. Perathoner, G. Centi, G. Giordano and M. Migliori, Microporous Mesoporous Mater., 2020, 300, 110157 CrossRef CAS.
- G. Papanikolaou, P. Lanzafame, S. Perathoner, G. Centi, D. Cozza, G. Giorgianni, M. Migliori and G. Giordano, Catal. Commun., 2021, 149, 106234 CrossRef CAS.
- G. Papanikolaou, P. Lanzafame, G. Giorgianni, S. Abate, S. Perathoner and G. Centi, Catal. Today, 2020, 345, 14–21 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- M. Jewess and R. H. Crabtree, ACS Sustainable Chem. Eng., 2016, 4, 5855–5858 CrossRef CAS.
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- G. Centi and S. Perathoner, Small, 2021, 17, 2007055 CrossRef CAS PubMed.
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- Y. Guo, S. Zhang, R. Zhang, D. Wang, D. Zhu, X. Wang, D. Xiao, N. Li, Y. Zhao, Z. Huang, W. Xu, S. Chen, L. Song, J. Fan, Q. Chen and C. Zhi, ACS Nano, 2022, 16, 655–663 CrossRef CAS PubMed.
- T. Li, S. Han, C. Wang, Y. Huang, Y. Wang, Y. Yu and B. Zhang, ACS Catal., 2021, 11, 14032–14037 CrossRef CAS.
- C. Genovese, C. Ampelli, B. C. Marepally, G. Papanikolaou, S. Perathoner and G. Centi, Chem. Eng. Trans., 2015, 43, 2281–2286 Search PubMed.
- S. Perathoner, G. Centi and D. S. Su, ChemSusChem, 2016, 9, 345–357 CrossRef CAS PubMed.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2021, 31, 2009070 CrossRef CAS.
- A. Sreedhar, I. N. Reddy and J. S. Noh, J. Cleaner Prod., 2021, 328, 129647 CrossRef CAS.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- Q. Qin and M. Oschatz, ChemElectroChem, 2020, 7, 878–889 CrossRef CAS.
- Y. Tanabe and Y. Nishibayashi, Chem. Soc. Rev., 2021, 50, 5201–5242 RSC.
- N. Morlanes, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. A. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef CAS.
- X. Han, Q. Gao, Z. Yan, M. Ji, C. Long and H. Zhu, Nanoscale, 2021, 13, 1515–1528 RSC.
- Y. Fu, Y. Liao, P. Li, H. Li, S. Jiang, H. Huang, W. Sun, T. Li, H. Yu, K. Li, H. Li, B. Jia and T. Ma, Coord. Chem. Rev., 2022, 460, 214468 CrossRef CAS.
- P. Mehta, P. M. Barboun, Y. Engelmann, D. B. Go, A. Bogaerts, W. F. Schneider and J. C. Hicks, ACS Catal., 2020, 10, 6726–6734 CrossRef CAS.
- S. Chen, D. Liu and T. Peng, Sol. RRL, 2021, 5, 2000487 CrossRef CAS.
- D. L. T. Nguyen, M. A. Tekalgne, T. H. C. Nguyen, M. T. N. Dinh, S. S. Sana, A. N. Grace, M. Shokouhimehr, D.-V. N. Vo, C. K. Cheng, C. C. Nguyen, S. Y. Kim and Q. Van Le, J. Environ. Chem. Eng., 2021, 9, 104997 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli and G. Centi, Stud. Surf. Sci. Catal., 2019, 178, 31–46 CrossRef CAS.
- J. Choi, B. H. R. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. Ferrero Vallana, R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 5546 CrossRef CAS PubMed.
- S. Z. Andersen, V. Colic, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- J. Kibsgaard, J. K. Nørskov and I. Chorkendorff, ACS Energy Lett., 2019, 2986–2988 CrossRef CAS.
- L. Zhang, M. Cong, X. Ding, Y. Jin, F. Xu, Y. Wang, L. Chen and L. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10888 CrossRef CAS PubMed.
- G. Peng, J. Wu, M. Wang, J. Niklas, H. Zhou and C. Liu, Nano Lett., 2020, 20, 2879–2885 CrossRef CAS PubMed.
- L. Tan, N. Yang, X. Huang, L. Peng, C. Tong, M. Deng, X. Tang, L. Li, Q. Liao and Zi. Wei, Chem. Commun., 2019, 55, 14482–14485 RSC.
- J. Guo, H. Wang, F. Xue, D. Yu, L. Zhang, S. Jiao, Y. Liu, Y. Lu, d. M. Liu, S. Ruan, Y.-J. Zeng, C. Ma and H. Huang, J. Mater. Chem. A, 2019, 7, 20247–20253 RSC.
- Y. C. Hao, Y. Guo, L. W. Chen, M. Shu, X.-Y. Wang, T.-A. Bu, W.-Y. Gao, N. Zhang, X. Su, X. Feng, J.-W. Zhou, B. Wang, C.-W. Hu, A.-X. Yin, R. Si, Y.-W. Zhang and C.-H. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- Y. Liu, M. Han, Q. Xiong, S. Zhang, C. Zhao, W. Gong, G. Wang, H. Zhang and H. Zhao, Adv. Energy Mater., 2019, 9, 1803935 CrossRef.
- S. Liu, M. Wang, T. Qian, H. Ji, J. Liu and C. Yan, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- H. Zhong, M. Wang, M. Ghorbani-Asl, J. Zhang, K. Hoang Ly, Z. Liao, G. Chen, Y. Wei, B. P. Biswal, E. Zschech, I. M. Weidinger, A. V. Krasheninnikov, R. Dong and X. Feng, J. Am. Chem. Soc., 2021, 143, 19992–20000 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, H. Wei, S. Abate, B. Zhang and G. Centi, J. Energy Chem., 2020, 49, 22–32 CrossRef.
- S. Chen, S. Perathoner, C. Ampelli, H. Wei, S. Abate, B. Zhang and G. Centi, ChemElectroChem, 2020, 7, 3028–3037 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, ACS Sustainable Chem. Eng., 2017, 5, 7393–7400 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., 2017, 129, 2743–2747 CrossRef.
- J. a M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- X. Cai, C. Fu, H. Iriawan, F. Yang, A. Wu, L. Luo, S. Shen, G. Wei, Y. Shao-Horn and J. Zhang, iScience, 2021, 24, 103105 CrossRef CAS PubMed.
- K. Li, S. Z. Andersen, M. J. Statt, M. Saccoccio, V. J. Bukas, K. Krempl, R. Sažinas, J. B. Pedersen, V. Shadravan, Y. Zhou, D. Chakraborty, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2021, 374, 1593–1597 CrossRef CAS PubMed.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- D. Ripepi, R. Zaffaroni, H. Schreuders, B. Boshuizen and F. M. Mulder, ACS Energy Lett., 2021, 6, 3817–3823 CrossRef CAS.
- P. G. Levi and J. M. Cullen, Environ. Sci. Technol., 2018, 52, 1725–1734 CrossRef CAS PubMed.
- J. Schot and L. Kanger, Resour. Policy, 2018, 47, 1045–1059 CrossRef.
- H. Neofytou, A. Nikas and H. Doukas, Renewable Sustainable Energy Rev., 2020, 31, 109988 CrossRef.
- International Renewable Energy Agency (IRENA), Global Renewables Outlook: Energy transformation 2050, IRENA, Abu Dhabi, 2020. Available on 5 Aug. 2022 at https://www.irena.org/publications/2020/Apr/Global-Renewables-Outlook-2020.
- G. Centi, G. Iaquaniello and S. Perathoner, BMC Chem. Eng., 2019, 1, 1–16 CrossRef.
- G. Centi, S. Perathoner, A. Salladini and G. Iaquaniello, Front. Energy Res., 2020, 8, 567986 CrossRef.
Footnote |
| † This manuscript summarizes the concepts and discussion that emerged from the large EU initiative SUNERGY (fossil-free fuels and chemicals for a climate-neutral Europe; https://www.sunergy-initiative.eu). |
| This journal is © The Royal Society of Chemistry 2022 |