 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputationally driven design of an artificial metalloenzyme using supramolecular anchoring strategies of iridium complexes to alcohol dehydrogenase†
Floriane L.
Martins
,
Anca
Pordea
 * and
Christof M.
Jäger
*
* and
Christof M.
Jäger
*
Sustainable Process Technologies, Faculty of Engineering, University of Nottingham, Nottingham, UK. E-mail: christof.jaeger@nottingham.ac.uk; anca.pordea@nottingham.ac.uk
First published on 29th October 2021
Abstract
Artificial metalloenzymes (ArMs) confer non-biological reactivities to biomolecules, whilst taking advantage of the biomolecular architecture in terms of their selectivity and renewable origin. In particular, the design of ArMs by the supramolecular anchoring of metal catalysts to protein hosts provides flexible and easy to optimise systems. The use of cofactor dependent enzymes as hosts gives the advantage of both a (hydrophobic) binding site for the substrate and a cofactor pocket to accommodate the catalyst. Here, we present a computationally driven design approach of ArMs for the transfer hydrogenation reaction of cyclic imines, starting from the NADP+-dependent alcohol dehydrogenase from Thermoanaerobacter brockii (TbADH). We tested and developed a molecular docking workflow to define and optimize iridium catalysts with high affinity for the cofactor binding site of TbADH. The workflow uses high throughput docking of compound libraries to identify key structural motifs for high affinity, followed by higher accuracy docking methods on smaller, focused ligand and catalyst libraries. Iridium sulfonamide catalysts were selected and synthesised, containing either a triol, a furane, or a carboxylic acid to provide the interaction with the cofactor binding pocket. IC50 values of the resulting complexes during TbADH-catalysed alcohol oxidation were determined by competition experiments and were between 4.410 mM and 0.052 mM, demonstrating the affinity of the iridium complexes for either the substrate or the cofactor binding pocket of TbADH. The catalytic activity of the free iridium complexes in solution showed a maximal turnover number (TON) of 90 for the reduction of salsolidine by the triol-functionalised iridium catalyst, whilst in the presence of TbADH, only the iridium catalyst with the triol anchoring functionality showed activity for the same reaction (TON of 36 after 24 h). The observation that the artificial metalloenzymes developed here lacked stereoselectivity demonstrates the need for the further investigation and optimisation of the ArM. Our results serve as a starting point for the design of robust artificial metalloenzymes, exploiting supramolecular anchoring to natural NAD(P)H binding pockets.
1. Introduction
Enzyme catalysis can provide sustainable synthetic alternatives to high-value chemicals.1 The application of biocatalysis can be expanded by engineering enzymes to incorporate non-natural activities. To achieve this, a promising strategy is the development of artificial metalloenzymes (ArMs).2 These advanced chemo-enzymatic hybrids are created by the incorporation of a non-native metal catalytic functionality into a protein scaffold imparting reaction selectivity (substrate, regio-, or stereo-selectivity). ArMs have the potential to address key challenges in green chemistry: tunable reaction control, renewability, enantioselectivity and environmentally friendly process conditions.3 The incorporation of the metal catalyst inside a protein can be achieved using dative, covalent, or supramolecular anchoring approaches.2 Though dative anchoring strategies of a metal ion have been widely used in ArM design,4 they are a challenging option with the need to precisely design an appropriate binding site inside the enzyme. The supramolecular anchoring of a metal complex of an organic ligand, designed to have affinity to the protein, is the most successful strategy to date, exemplified by the versatile biotin–(strep)avidin5 and the LmrR multidrug resistance regulator systems.6The efficiency of ArMs has constantly improved over the past decades, due to advancements in both experimental protein engineering technologies and in computational aided approaches.7–9 Amongst the latter, computational tools for the de novo design and redesign of binding sites for metal cofactors have greatly contributed to the rapid design of ArMs with novel or improved functionality.9 Such tools are focused either on the design of non-native metal binding or on the design of catalytic functionality within the newly created metalloproteins, using software such as the Rosetta suit of programmes,10,11 which combines computational tools applicable for methods like molecular docking, structure prediction, and de novo protein design.
Computationally driven de novo (re-)design strategies of metal containing active sites were demonstrated successfully for a number of examples (see also ref. 9). Rosetta Match12 was applied to optimise the design of a novel abiological hetero-Diels–Alderase, based on density functional theory (DFT) informed transition state (TS) model structures;13 a novel organophosphate hydrolase was also designed de novo by first defining a suitable TS and subsequently redesigning the active site of a natural deaminase.14
With a focus on supramolecular catalyst anchoring, software such as RosettaDesign15 has been used to fine tune the binding of the ligand, as demonstrated in the tailoring of the amino acid sequence of human carbonic anhydrase II to bind an iridium metal complex catalyst with high affinity, thus improving the catalytic performance of the resulting ArM.16 Molecular docking is another useful method of choice for evaluating and designing protein–ligand interactions between the guest catalyst and the protein environment, particularly when combined with methods with higher accuracy (e.g. QM/MM, MD). Docking has commonly been employed as a second step in the design to gain insight into the experimental results from first-generation ArMs and to define key residues influencing optimal and directed substrate binding and thus enzymatic activity and substrate selectivity.13,17–19 Computational analysis of the defined complexes can also guide the selection of bioconjugation sites in the case of the covalent binding of the metal complex to the protein.20,21 In other approaches, molecular docking in combination with QM and MD simulations was used to control the activity and selectivity of ArMs by understanding substrate binding affinity, flexibility, and orientation.22,23
Despite the above examples and the similarity of the approach to computational drug design, where chemical structures are non-covalently incorporated within proteins as strongly binding inhibitors,24 docking is not routinely used in ArM design. This is because binding affinity alone is not sufficient to generate a working ArM. The correct orientations of the incorporated metal catalyst and of the binding substrate(s) to form the Michaelis complex are equally important and thus must be evaluated by computational approaches. Since docking is only an approximate method, more accurate (but more costly) molecular dynamics coupled free energy perturbation (FEP) simulations and QM and QM/MM evaluations should also be considered.
Whilst most successful, the design of ArMs based on supramolecular anchoring relies on non-enzymatic protein structures with a large hydrophobic pocket which is able to accommodate both the metal catalyst and the substrate. An alternative is the use of cofactor-dependent enzymes, where the native cofactor is replaced with a non-native catalytic moiety.25 For example, the heme-binding pocket in P450 was used to accommodate Ir-porphyrins to catalyse C–C and C–N bond formations.26 In our group, we developed ArMs for the reduction of nicotinamide cofactors, starting from the wild type alcohol dehydrogenase from Thermoanaerobacter brockii (TbADH WT), by covalently binding rhodium piano stool complexes at the place of the catalytic zinc site.21,27 In this work, we aim to simplify the ArMs by designing catalysts for supramolecular binding to avoid the need for the covalent modification of the protein. So far, ArMs based on supramolecular anchoring rely on systems where a natural affinity between the protein and ligand is a pre-requisite, thereby limiting the protein and/or ligand choice; whereas systematic rational strategies to specifically design the metal catalysts for supramolecular anchoring to a selected protein scaffold are yet to be developed.
To address this, we present a critically evaluated and refined docking strategy for the design of anchoring functionalities to provide strong interactions between d6 piano stool metal catalysts and TbADH. In this design, the NADP+ cofactor binding site was used to accommodate the catalyst, as schematically shown in Fig. 1. This site has a relatively high binding affinity for the native cofactor (e.g.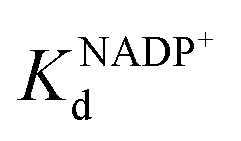 = 9 μM)28 due to a network of hydrogen bond interactions, in addition to being in proximity to the hydrophobic substrate binding pocket. We use the mutant TbADH 5M (H59A–D150A–C203S–C283A–C295A) as a starting point, where catalytic Zn2+ was removed via mutations of two coordinating residues to alanine.27 We selected sulfonamide-based iridium d6 piano stool catalysts for the study, because they are highly active in imine reduction and were previously successfully used in the transfer hydrogenation of imine by ArMs (Fig. 1).29–31 Following computational design, the most promising catalyst structure was synthesised and evaluated for the formation of a functional ArM.
= 9 μM)28 due to a network of hydrogen bond interactions, in addition to being in proximity to the hydrophobic substrate binding pocket. We use the mutant TbADH 5M (H59A–D150A–C203S–C283A–C295A) as a starting point, where catalytic Zn2+ was removed via mutations of two coordinating residues to alanine.27 We selected sulfonamide-based iridium d6 piano stool catalysts for the study, because they are highly active in imine reduction and were previously successfully used in the transfer hydrogenation of imine by ArMs (Fig. 1).29–31 Following computational design, the most promising catalyst structure was synthesised and evaluated for the formation of a functional ArM.
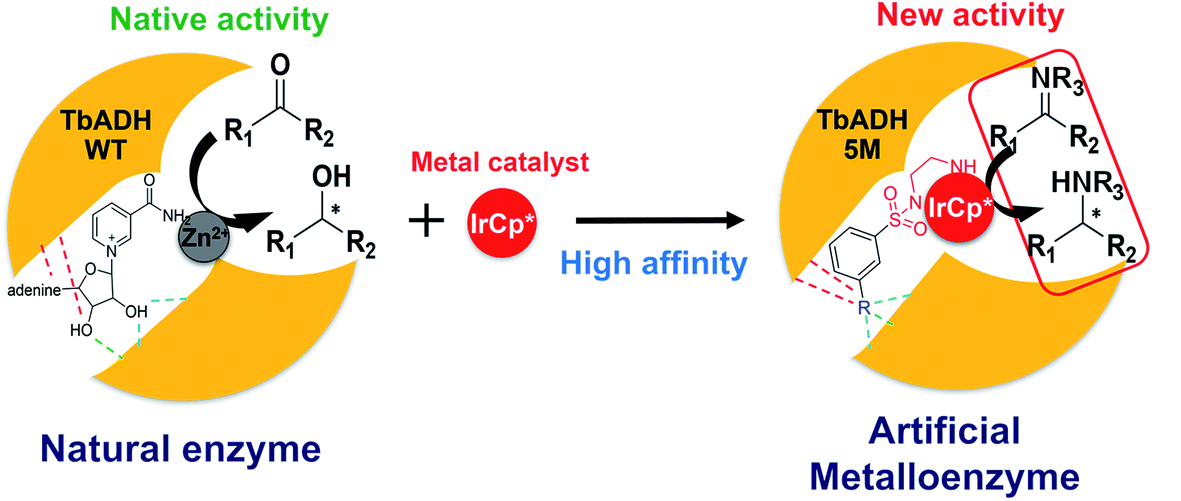 | ||
| Fig. 1 Schematic representation of the design of ArMs: left, the natural catalysis in TbADH WT involving NADP+ and a catalytic Zn2+; right, the supramolecular insertion of an iridium piano stool complex (red) inside the NADP+ cofactor pocket and Zn2+ binding region of TbADH 5M with high affinity induced by the anchor substructure (R). | ||
2. Results and discussion
2.1 Design and evaluation of the docking protocol
Before refining a docking protocol applicable to the design of suitable anchor structures for catalysts to ADH-based ArMs, it was necessary to evaluate whether an approximate method, such as docking, could result in sufficiently accurate predictions of anchor structure binding affinities and orientations. Therefore, we evaluated different docking protocols for their ability to predict the crystallographically known NADP+ cofactor binding to TbADH. This evaluation was additionally used to analyse key binding interactions informing the subsequent anchor design.Different docking accuracy levels were analysed. The standard precision (SP) docking, as implemented in Glide32,33, was compared against the more costly extra precision (XP) docking protocols and the most sophisticated MM-GBSA (molecular mechanics generalized Born and surface area) docking refinement to estimate the relative binding free energies. The crystal structure of TbADH WT co-crystallised with NADP+ was selected from the Protein Data Bank (PDB) for docking studies (PDB 1YKF34). For the subsequent docking approaches in the search of high affinity anchors for transition metal catalysts, TbADH 5M was additionally prepared without the catalytic zinc ion, to make space in the active site for the inserted transition metal, by the creation of five mutations to remove Zn2+.
The co-crystallised natural cofactor NADP+ was re-docked into the cofactor binding site of both TbADH variants. The calculated binding affinities at the different docking accuracy levels are presented in Table 1. The results demonstrate high affinity binding in all cases. The scores were similar for both enzymes, with the MM-GBSA evaluation indicating slightly better binding to TbADH WT. This approach led to very good agreement with the two structures of TbADH WT and TbADH 5M, with an all-atom RMSD of < 2 Å for the bound cofactor (see Fig. S1 in the ESI†). Between the two enzymes, TbADH 5M docking positioned the nicotinamide cofactor slightly closer to the substrate binding site than that for TbADH WT.
| Enzyme | Glide XP | MM-GBSA | RMSD |
|---|---|---|---|
| TbADH WT | −10.1 | −76.4 | 1.7 |
| TbADH 5M | −10.7 | −59.5 | 1.8 |
Following validation of the docking procedure, an iterative docking workflow was developed to design iridium complexes that could be accommodated via supramolecular interactions within the cofactor binding site of TbADH. Initially, an efficient and computationally affordable screening method of a diverse library of chemical structures was implemented to find anchor structures providing high affinity binding to TbADH. The highest affinity anchor structures were subsequently incorporated within whole catalyst structures with a piano stool Ir(Cp*)–sulfonamide functionality, which were computationally evaluated both for their binding affinity and catalytically relevant positioning.
The general docking protocol followed three major steps, as shown in Fig. 2: (1) a large compound library was screened to identify patterns of functional groups responsible for high affinity, leading to a template ligand structure; (2) anchor structures identified by the initial screening were appended to the sulfonamide functionality to create a ligand library of 26 compounds, which were evaluated with higher precision docking methods; (3) 19 of the highest affinity ligands were selected and analysed for their ease of synthesis and a final library of metal catalyst structures was then generated and docked using induced fit docking. Additional filters were applied to evaluate not only the binding affinity, but also the optimal positioning and orientation for catalysis. From this final docking step, six structures were identified as potentially suitable, and one was finally selected for chemical synthesis and testing.
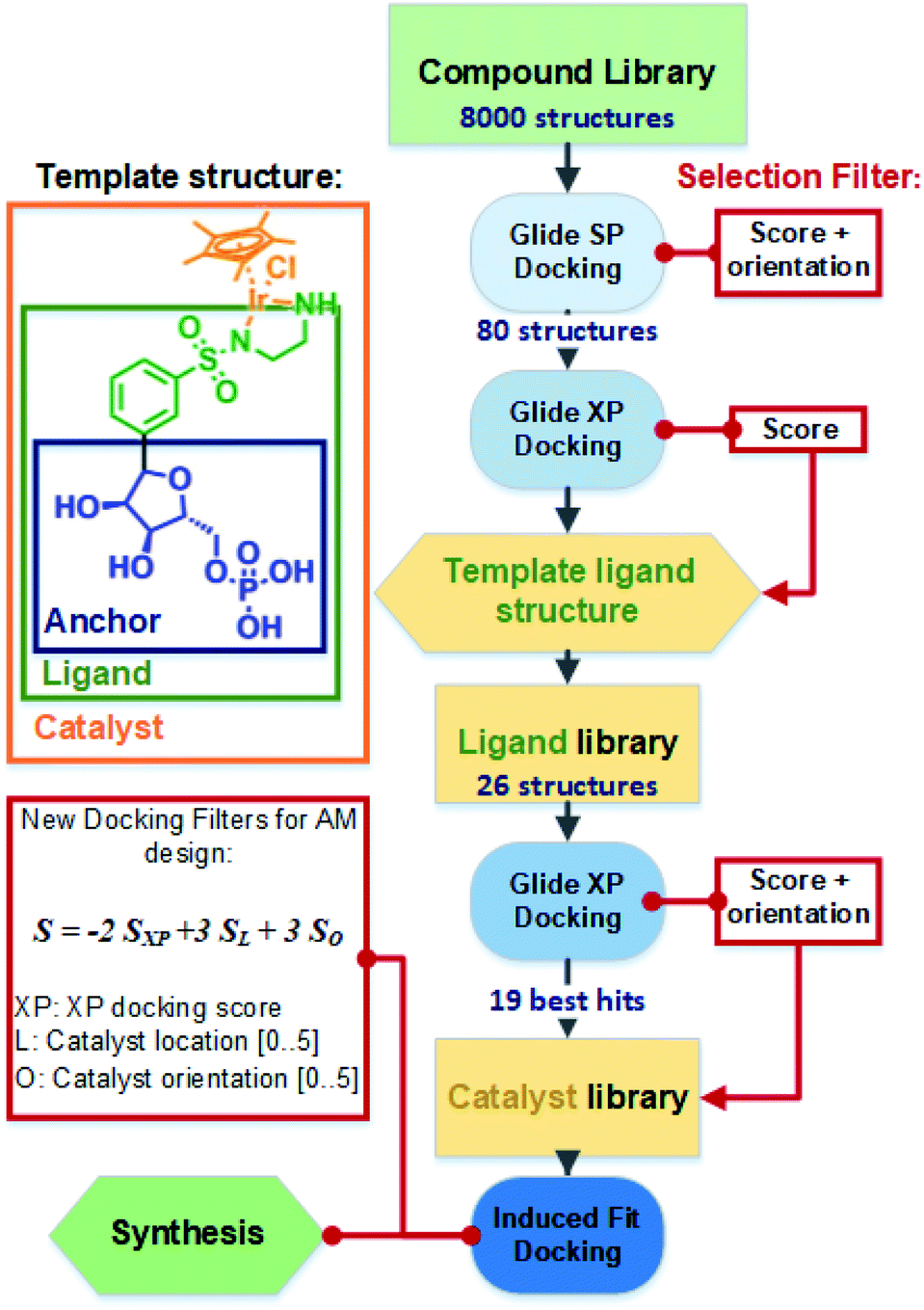 | ||
| Fig. 2 Overall docking and library design protocol used to filter and optimise the anchor, ligand, and catalyst structures. Template structures are shown with definitions of the anchor, ligand, and catalyst structure, as presented in the text. | ||
Both strategies led to similar observations. The best ranking structures from the high throughput docking and the fragment-based approach both demonstrated the importance of hydrophilic functional group anchoring being present in the ribose and phosphate binding regions of NADP+ (see Fig. 3 for the best ranked docking structures). The best ranked structures all shared a hydrophilic centre composed of amino, carboxyl or hydroxyl groups, forming several hydrogen bonding interactions with residues such as Val178, Ser39, Asn266 or Tyr267, which correspond to the NADP+ interactions found in the TbADH crystal structure34 (Fig. S3 in the ESI†).
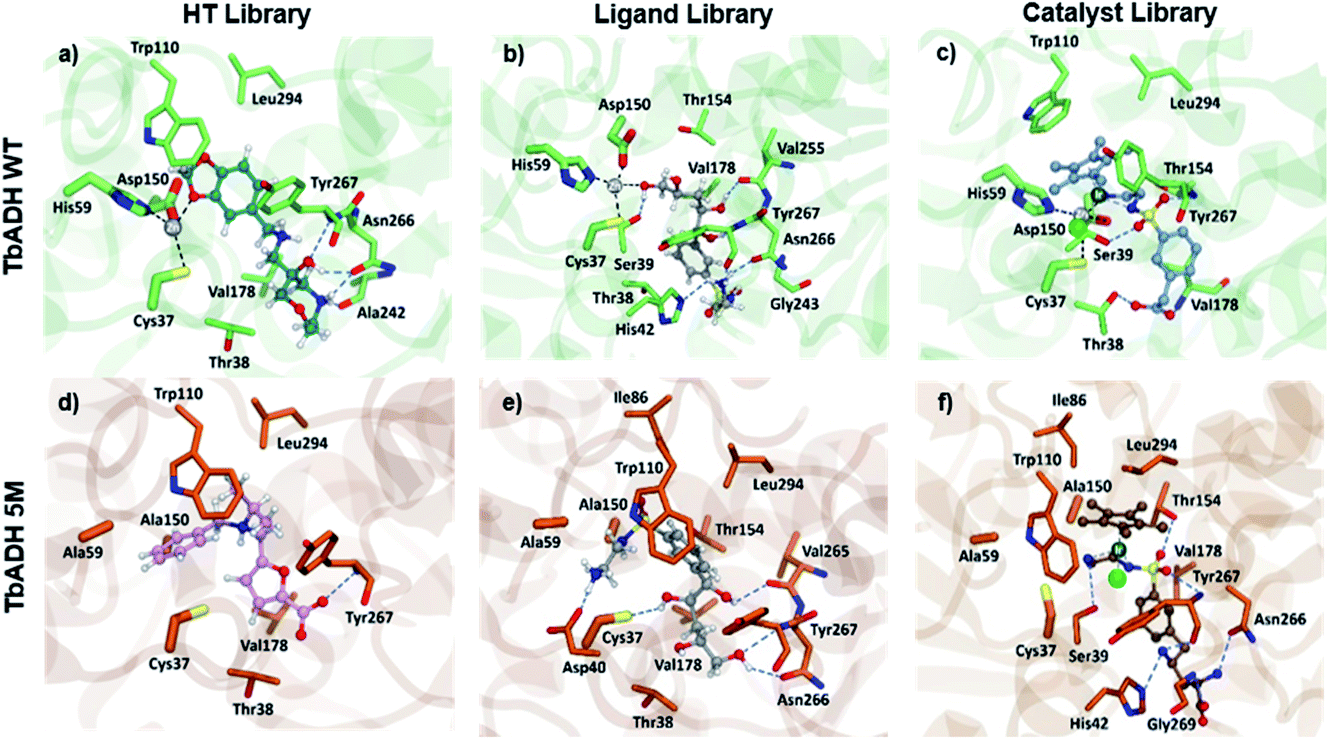 | ||
| Fig. 3 Pictures of the best ranked compounds from ligand libraries screened through XP and IFD docking in TbADH WT and TbADH 5M, respectively: (a) and (d) best ranked ZINC library compounds; (b) and (e) best ligands docked through XP docking ((b) 1, e: 1) best metal complexes ranked by IFD score ((c) Ir2, (f) Ir15). | ||
On the other hand, the fragment-based docking indicated little affinity of the adenosine for the respective binding locations, suggesting a low relevance of high affinity binding. Also, the nicotinamide ring showed no interaction in all the different fragment-based docking experiments. This strongly aligned with the library docking, which ranked hydrophobic structures incorporating aromatic rings lowest. Additionally, larger hydrophobic entities tended to be positioned in the hydrophobic substrate pocket, particularly in TbADH 5M. This would likely also prevent substrate binding and needed to be addressed in the subsequent steps.
As the pyridinium ring demonstrated little effect on the binding affinity, it was adapted to a phenyl ring, with an eye on the subsequent simpler chemical synthesis. As the last feature of the lead structures, the sulfonamide group was attached to the phenyl ring, which served to bind the transition metal. An initial screening of the library additionally revealed that the meta position of substituents on the phenyl delivered the best affinity scores and this was thus chosen for the subsequent library design.
The designed library of 26 structures is presented in the ESI (Fig. S5 in the ESI†). For all of the structures, all possible conformations and configurations were generated and docked inside both TbADH WT and TbADH 5M, using a combined SP and XP workflow (Table S2 in ESI†). The best binding structures all had hydroxyl and carboxyl groups in their anchoring part. This matched the aim to substitute ribose (by hydroxyl) and phosphate (by carboxylic acid) for easier chemical synthesis. Conversely, compounds with only one hydrophilic group and hydrophobic groups (e.g.12 and 23, Fig. S5 in the ESI†) were ranked lowest.
We observed that most configurations of the same anchored structures delivered similar XP scores and only the strongest binding configurations were selected for the next iteration of the docking protocol (see the example for ligand 4 in Table S3 in the ESI†). A comparison between TbADH WT and TbADH 5M showed overall slightly lower XP scores for the 5M mutant, indicating a higher binding affinity. Despite this difference, many of the first ranked structures were common for both enzymes, with only one different structure within the first five highest ranked structures. Analysis of the positions remarkably showed inverted orientations and unfavourable positioning of the majority of ligand structures in the case of TbADH WT, whilst all TbADH 5M bound ligands displayed an orientation favourable for catalysis, similar to the natural cofactor NADP+.
| Iridium catalyst | IFD(S) score | Iridium catalyst | IFD(XP) score |
|---|---|---|---|
| Ir15 | 54.0 | Ir2 | −12.8 |
| Ir3 | 53.6 | Ir1 | −12.2 |
| Ir1 | 53.4 | Ir22 | −11.2 |
| Ir4 | 52.6 | Ir13 | −11.6 |
| Ir14 | 50.4 | Ir15 | −9.5 |
| Ir9 | 46.6 | Ir6 | −9.5 |
| Ir10 | 43.8 | Ir3 | −9.3 |
| Ir13 | 43.6 | Ir14 | −9.2 |
| Ir8 | 42.8 | Ir4 | −8.8 |
| Ir2 | 42.6 | Ir9 | −8.8 |
| Ir6 | 41.0 | Ir8 | −8.4 |
| Ir22 | 39.4 | Ir10 | −7.4 |
From the 19 docked metal complexes fed through the IFD docking, 7 catalysts demonstrated significantly repulsive interactions induced by incorporating the metalation and were subsequently removed from the analysis. The ranking of the remaining 12 catalysts was highly influenced by the nature of the anchoring substituent, which directed the metal complex [IrClCp*] into a catalytic position. This was also demonstrated by a distinctly different ranking order when taking only the XP score from the IFD docking compared to the additionally applied new scoring scheme (Table 2). Thus, our best ranking catalyst Ir15 only scored fifth in pure IFD docking (IFD(XP)) while the best IFD docking score (Ir2) was ranked down by applying the additional score correction due to the unfavourable orientation in the binding site, most likely not leading to catalytic activity.
Similar to the previous ligand docking, TbADH 5M presented higher affinity XP and IFD scores than those of TbADH WT. The low scoring results from the TbADH WT results were obtained because of the mainly reversed position of the metal complexes inside the cofactor binding pocket. Most likely this was due to the Zn coordination site occupying the space needed to fit the catalyst and preventing ideal binding.
Analysis of the structure poses in TbADH 5M demonstrated a general orientation of the metal toward the substrate pocket, except for few inverted poses. Fig. 4 shows an example of Ir4 displaying a high IFD(S) score with its crucial binding interactions in the active site of TbADH 5M, whilst there is an inverted orientation in TbADH WT. All of the high affinity complexes demonstrated the key interactions with residues like Thr38, Ser39, Asn266 or Tyr247.
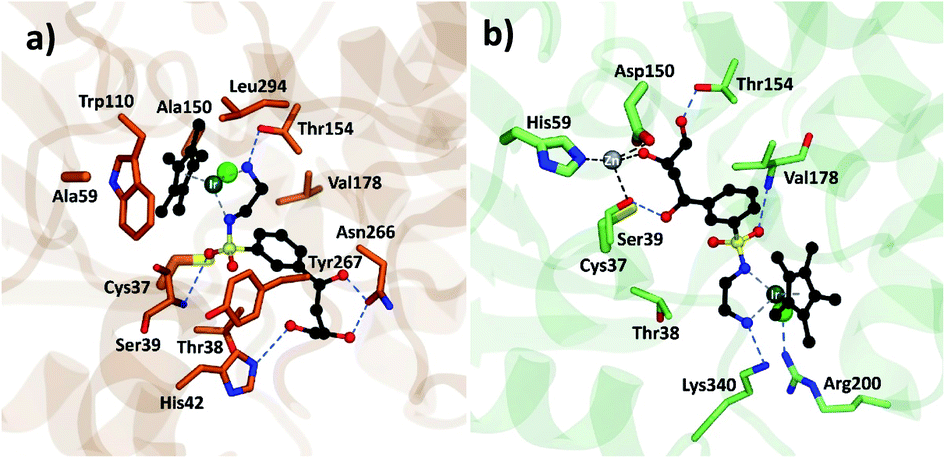 | ||
| Fig. 4 Metal complex Ir4 docked into (a) TbADH 5M with a catalytic orientation and (b) into TbADH WT with an inverted position. | ||
Fig. 5 shows the structures of the six best metal complexes based on the IFD(S) results for TbADH 5M. These structures were expected to bind with high affinity to TbADH 5M and to additionally accommodate a hydrophobic substrate in the catalytic position, therefore representing attractive options for constructing an ADH-based ArM using the supramolecular binding approach. From these structures, we proceeded to select and synthesise one example for experimental validation, based on the ease of synthesis.
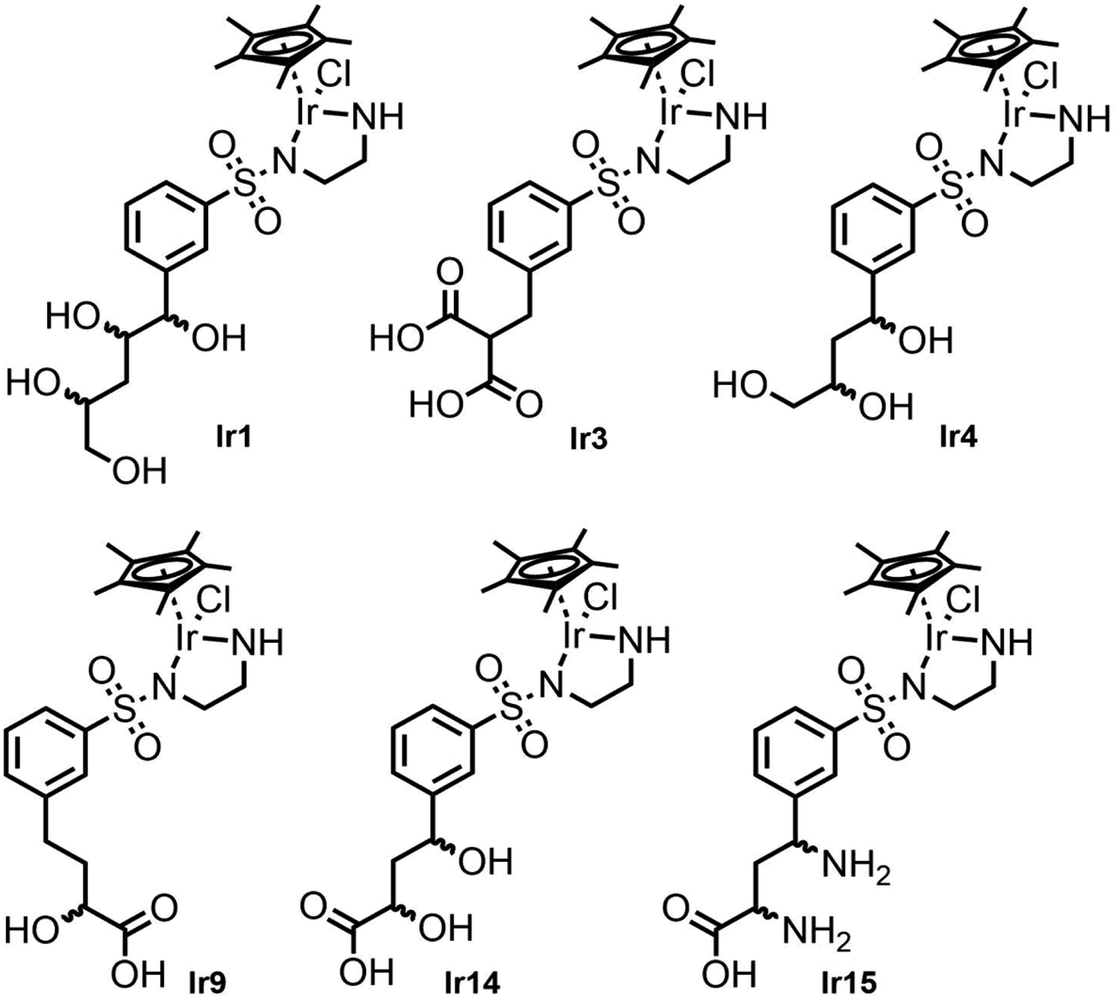 | ||
| Fig. 5 Selected metal complex hit structures from the docking study, with expected high affinities for TbADH 5M. | ||
2.2 Synthesis of iridium complexes
From the preceding docking analysis, the iridium complex Ir14, bearing a 2,4-dihydroxy carboxylic acid anchor, was initially selected for experimental validation, given its high IFD(S) score and its short retro-synthesis path. The synthesis of Ir14 was attempted from either the Boc or the Cbz-protected N-sulfonyl-ethylenediamine-2,4-diketoester structure (compounds 28 and 32 in Fig. 6). This diketoester was obtained as a E/Z stereoisomer mixture of the mono-enol form via the Claisen condensation of the acetophenone derivatives 27/31 and diethyl oxalate under basic conditions (Fig. 6). However, given the lack of the stability of the 2,4-diketoester functionality under both acidic Boc deprotection and palladium catalysed Cbz deprotection conditions (leading to self-Claisen, retro-Claisen or hydrogenolysis by-products), a refinement of the target ligand structure was necessary. The tri-hydroxy ligand Ir4, which was also found to bind with high scores in the preceding docking approach, was synthesised by Pd-catalysed reduction and the concomitant deprotection of the Cbz-protected 2,4-diketoester 28 with H2 (Fig. 6A). Interestingly, when the same synthesis was attempted from the Boc-protected precursor 32, the deprotection of intermediate 33 with trifluoroacetic acid led to cyclisation of the anchor with the formation of a furan ring to give compound 12 (Fig. 6B). This behaviour was similar to the Paal–Knorr reaction, involving the acid catalysed cyclisation of a 1,4-diketone in its mono-enol form, followed by dehydration to form a furan.36 The synthesis of furan derivatives from a 1,4-keto-alcohol intermediate was previously demonstrated in acidic conditions,37 as was the TFA-catalysed deprotection followed by the cyclisation of tert-butyl acetoacetate derivatives.38 Compound 12 and its corresponding iridium complex Ir12 were also prepared to be further tested in artificial metalloenzyme formation, as they had a conveniently available hydrophobic and aromatic anchor substructure to be compared with the hydrophilic triol anchor of 4 for its binding and catalytic potential. Given the similarity of the XP scores for the different configurations of ligand 4 we did not consider stereoconfiguration in the synthesis. Furthermore, intermediate 23 and its corresponding iridium complex Ir23 were prepared, bearing a single hydrophilic (carboxylate) functionality as an anchor. As expected, docking analysis showed a very low ranking in all screenings and a reverted position for the metal complexes (see Table S5 and Fig. S7 in the ESI†).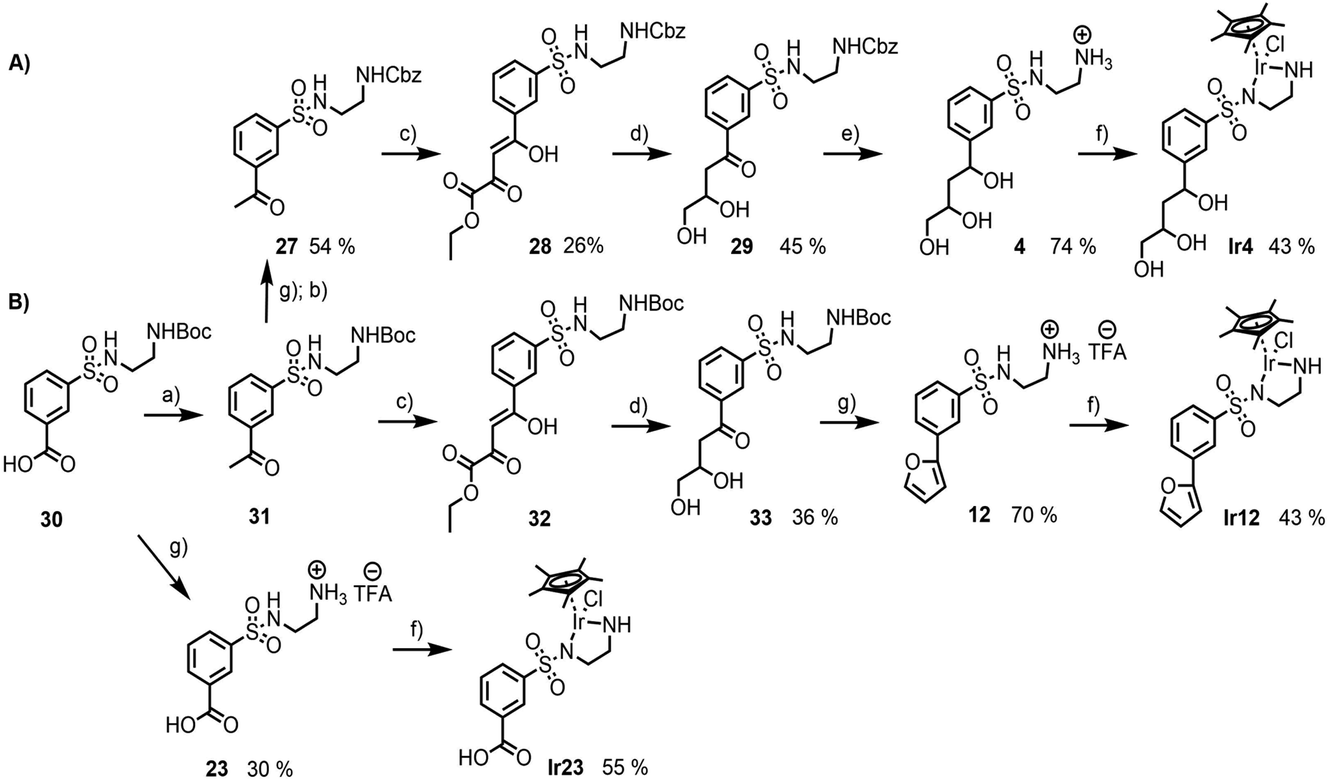 | ||
| Fig. 6 Synthesis of the ligands and iridium complexes used in this study: (A) compounds 4 and Ir4, starting from the Cbz-protected N-sulfonyl-ethylenediamine precursor; (B) compounds 12, 23, Ir12 and Ir23, starting from the Boc-protected N-sulfonyl-ethylenediamine precursor (a) (i) SOCl2, CH3NHOCH3, Et(i-NPr)2, CH2Cl2; (ii) CH3MgBr, THF; (b) Na2CO3, Cbz-Cl, H2O/Et2O; (c) t-BuOLi, (CO2CH2CH3)2, THF; (d) LiAlH4, THF; (e) Pd/C 10%, H2, MeOH; (f) [IrCl2Cp*]2, Et3N, MeOH; (g) TFA, CH2Cl2. | ||
2.3 Artificial metalloenzymes by the supramolecular anchoring of iridium complexes to TbADH
| Ligand | IC50 (mM) |
|---|---|
| 4 | >10 |
| Ir4 | 0.248 ± 0.003 |
| 12 | 16.050 ± 0.390 |
| Ir12 | 0.052 ± 0.012 |
| 23 | 5.459 ± 0.430 |
| Ir23 | 4.410 ± 0.035 |
All of the sulfonamide ligands showed IC50 values higher than 5 mM, indicating lower affinities for TbADH in comparison with the native NADP+ cofactor, as expected from the docking prediction. However, in contrast to docking, the inhibition assays suggested a lower affinity of TbADH for the tri-hydroxy ligand 4 compared to that of ligand 23, which bears a mono-carboxyl anchor. One potential explanation for these results is that the assumption of competitive inhibition of the cofactor binding site was not appropriate in this case. In addition, ligand 4 also presented a mixture of stereoisomers of which some might bind with different binding affinities or in other locations, not adding to competitive inhibition, although docking suggests a similar affinity of the isomers for the cofactor binding pocket. The binding of smaller or hydrophobic anchors at positions where they hinder substrate approach and/or binding, but outside the cofactor binding pocket, may also explain the higher inhibition potential of these compounds.
All of the iridium complexes showed better inhibitory potential than their corresponding sulfonamide ligands alone, with IC50 values between 0.052 (Ir12) and 4.410 mM (Ir23). The iridium functionality was not expected to significantly add to the binding affinity, as its bulky nature is more of a challenge to accommodate in the pocket. This was also demonstrated by the observation from docking that in the presence of the Zn2+ ion in TbADH WT, the structures were inverted, positioning the metal toward the pocket entrance, in order to fit the pocket. Therefore, it can be speculated that the iridium piano stool functionality has a higher impact on enzyme inhibition than the binding conferred by the anchor. One explanation of this could be that the coordination of the iridium piano stool moiety to the surface exposed coordinating (cysteine or histidine) residues, leading to enzyme deactivation, as previously shown with rhodium complexes.39 Furthermore, the slightly better inhibition demonstrated by Ir12 over that of the tri-hydroxy functionalised Ir4 could be attributed again to alternative binding modes with different orientations not predicted by docking that could add to the inhibition.
A detailed comparison of the binding within TbADH WT and the 5M mutant is difficult. This is because only binding to TbADH WT, which contains the Zn-binding site, could be assessed by inhibition studies. Moreover, inversion of binding was observed when docking the iridium structures to TbADH WT, compared to the mutant 5M. In conclusion, whilst inhibition by the designed iridium complexes was observed and indicated an interaction between the designed complexes and TbADH WT, certainty on the exact location and orientation of the bound structures and on the affinity of TbADH 5M for the iridium complexes could not be defined because of the indirect analysis.
|
|
||||
|---|---|---|---|---|
| Entry | TbADH variant | Ir catalyst | TON | TOF (min−1) |
| 1 | — | Ir4 | 90 ± 1.04 | 0.062 ± 0.001 |
| 2 | — | Ir12 | 66 ± 1.45 | 0.046 ± 0.001 |
| 3 | — | Ir23 | 59 ± 8.39 | 0.041 ± 0.006 |
| 4 | 5M | Ir4 | 36 ± 3.21 | 0.025 ± 0.002 |
| 5 | WT | Ir4 | 1.6 ± 0.59 | 0.001 ± 0.000 |
| 6 | 5M | Ir12 | 0 | — |
| 7 | WT | Ir12 | 1.7 ± 0.55 | 0.001 ± 0.000 |
| 8 | 5M | Ir23 | 0 | — |
| 9 | WT | Ir23 | 0 | — |
| 10 | 5M (100 μM) | Ir4 | 34 ± 1.34 | 0.023 ± 0.001 |
| 11 | 5M | Ir4 (200 μM) | 79 ± 3.18 | 0.055 ± 0.002 |
| 12 | WT | Ir4 (200 μM) | 81 ± 0.80 | 0.056 ± 0.001 |
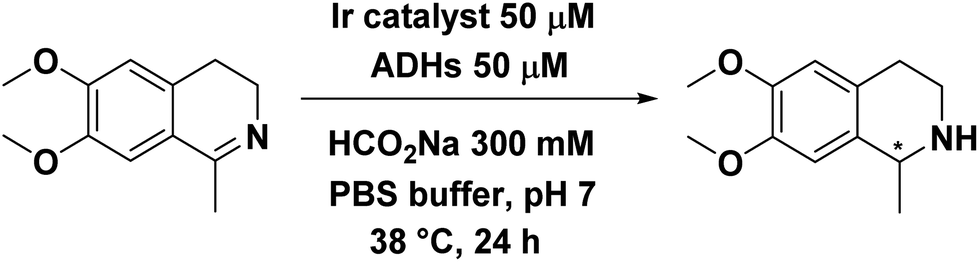
Only complex Ir4 showed activity in the presence of TbADH 5M and no stereoselectivity was observed (Table 4, entries 4, 6 and 8). Furthermore, when the zinc-containing enzyme (TbADH WT) was used, the activity of Ir4 was drastically reduced and this was the case with the other catalysts as well (Table 4, entries 5, 7 and 9).
Two different scenarios could explain the lack of activity in the presence of the WT and the mutual inactivation of the enzyme and catalyst: binding to the surface exposed coordinating residues and binding in the cofactor site, but without space for the imine substrate, or in the wrong orientation, as suggested by docking. Both of these scenarios would lead to the iridium complexes not being available as active free catalysts in solution, implying that any activity in the presence of the protein was likely to come from a TbADH-bioconjugated catalyst.
In addition, there was no difference in the surface exposed coordinating residues between the TbADH WT and 5M variants, which means that the difference in activity observed with the two variants cannot come from bioconjugation to surface exposed residues. These observations, alongside the docking results, suggest that the iridium catalyst Ir4 binds within the cofactor site in the orientation suggested by docking, with the catalytic part towards the hydrophobic substrate pocket of the enzyme. When bound to 5M, there was enough space for the imine substrate to react, whilst within the zinc-occupied WT enzyme, the imine substrate could not access the iridium catalyst. Iridium complexes Ir12 and Ir23 may bind to the cofactor binding site, but not in active poses or orientations, which would explain their inhibitory potential for TbADH WT and their lack of activity within TbADH 5M. This theory was supported by the previous docking studies, which predicted the insertion of hydrophobic structures inside the substrate pocket (Fig. 3d).
To further confirm the theory of the supramolecular binding of the iridium catalyst to TbADH, the concentrations of both TbADH 5M and of the catalyst Ir4 were varied, in order to perturb the equilibrium towards the bound state of the catalyst. When the concentration of TbADH 5M was doubled from the initial stoichiometric ratio, the turnover number remained similar (Table 4, entry 10 compared to 5), suggesting that most of the available binding sites were occupied with the iridium catalyst. This could mean either that the active catalyst binds to TbADH 5M with very high affinity, or that not all binding sites were occupied, therefore providing an excess of binding sites even at stoichiometric monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ir conditions. On the other hand, when the concentration of the metal catalyst was increased four times, an increased TON was observed with both TbADH WT and 5M (Table 4, entries 11 and 12), with the increase in activity likely coming from the free catalyst in solution. Taken together, these results further suggest the formation of a functional artificial metalloenzyme for imine reduction by the supramolecular incorporation of the iridium complex Ir4 within the NADP+ binding site of the zinc devoid TbADH variant 5M.
:Ir conditions. On the other hand, when the concentration of the metal catalyst was increased four times, an increased TON was observed with both TbADH WT and 5M (Table 4, entries 11 and 12), with the increase in activity likely coming from the free catalyst in solution. Taken together, these results further suggest the formation of a functional artificial metalloenzyme for imine reduction by the supramolecular incorporation of the iridium complex Ir4 within the NADP+ binding site of the zinc devoid TbADH variant 5M.
3. Conclusions
This work demonstrates how docking methodologies can be applied and optimised to design ArMs based on the supramolecular anchoring of imine reduction catalysts into the NADP+-binding site of alcohol dehydrogenase scaffolds. The computational workflow started from high throughput virtual screening and was refined to the docking of a selected catalyst library. The refined protocol focused on more accurate calculations to predict a transition metal catalyst with catalytic positioning.The creation of an active ArM was demonstrated for the transfer hydrogenation of salsolidine, however the lack of stereoselectivity represents a drawback. Nonetheless, from the combination of competition experiments (indicating binding affinity) and catalytic activity, there is clear evidence of a functional ArM, strongly suggesting that the catalyst is bound to a defined site that can also accommodate the substrate. Although docking suggested that the iridium piano stool moiety would be oriented towards the substrate binding pocket, it cannot be ruled out that catalyst 4 binds towards the surface of the enzyme or is involved in multiple binding modes involving all its different stereoisomers.
The results presented here show that the first step to create this type of ADH-based ArMs needs further experimental and computational assessment and optimisation. While this study focused on improving the potential and workflow for docking applications in connection to ArM design, future computational work needs to target more advanced and accurate computational methods to test and challenge the original predictions.
Molecular dynamics (MD) simulations will give insights into the flexibility and integrity of the protein catalyst complexes. Catalyst and substrate binding and unbinding simulations will be able to give further into the binding strength, flexibility and alternative binding modes. Finally, the definition of transition state structures for the simulated binding arrangements will make it possible to optimise and re-design the surrounding binding pocket for better TS stabilisation and introduction of stereo selectivity.
4. Experimental
4.1 Docking calculation procedures
The Schrödinger suite41 of programs and the Maestro graphical interphase were used for the design of the molecular docking protocols with the default parameters defined in the Schrödinger program. All compounds included in this work were prepared using Ligprep from the Schrödinger suite,42 with the OPLS3 force field.43 The generation of all possible protonation and ionisation state combinations was performed for each ligand using Epik23 in aqueous solution at pH values of 7.0 ± 2.0. Whenever the ligand contained a ribose, the same stereochemistry as that of the natural cofactor was kept. Structures containing iridium metal were created using the Maestro interface to build a pyramidal metal centre, substituted with chloride and a pentamethylcyclopentadienyl (Cp*) moiety, applying fixed bonds of zeroth bond order. The complex structures were then minimized within the Maestro graphical interphase.The crystal structure of TbADH was obtained from the Protein Data Bank (PDB 1YKF2 (ref. 34)). The monomer of the protein was additionally prepared without the natural zinc ion in the active site. From the crystal structure of TbADH, several mutations were created, Cys283Ala, His59Ala, Asp150Ala, Cys295Ala and Cys203Ser, to yield the structure TbADH 5M. Protein structures were prepared using the Maestro Protein Preparation Wizard in the Schrödinger suite.44 Missing hydrogen atoms and residue side chains were added to the structures by Prime refinement.41 During the refinement, water molecules with less than three hydrogen bonds to other atoms were removed, which resulted in no more water in the binding site. The selection of the position of hydroxyl and thiol hydrogens, the protonation/tautomer states and the “flip” assignment of aspartic acid, glutamic acid, arginine, lysine and histidine were adjusted at pH = 7.0 using PROPKA.45 Finally, the structures were minimized using the OPLS3 force field with a RMSD = 0.3 Å maximal displacement of non-hydrogen atoms as the convergence criterion.
For non-covalent docking using Glide software, the grid box was defined from the optimized protein structure at the centroid of the active site (20 Å radius around the co-crystallized ligand). In the section involving transition metal complex docking, the enclosure box was defined from the optimized protein structure without zinc by a selection of amino acid residues (TbADH 37, 59, 60, 42, 151, 178, 295, 266 and 150). No constraints were added. The standard settings of a van der Waals scaling factor of 1.0 for nonpolar atoms was kept. Nonpolar atoms were defined with an absolute value of partial atomic charges of ≤0.25.
The prepared ligand conformers without the metal were flexibly docked into the receptor grid using Glide SP and Glide XP procedures, with the receptor kept rigid.32,33,46 The default parameters as defined in the Schrodinger program were kept. OPLS3 (ref. 43) was used as the force field and the variation of conformation was applied. The estimations of the free energy binding of each ligand for the enzymes were ranked. The structures with the best Glide XP score were used in the MM-GBSA calculations. The VSGB solvation model (surface generalized Born model and variable dielectric) was selected to approximate the solvation free energy and correct the interactions (H-bond, hydrophobic, π–π).47 Flexibility was added to residues at 5.0 Å from the ligand position in the binding pocket. Prime minimisation was used on the residues selected in the flexible region, without any constraints. For the IFD docking of different metal complexes, the protein residues within 5.0 Å of the catalyst were kept flexible using Prime. The best scores were selected for analysis.
4.2 Synthesis of the ligands and metal complexes
For the detailed and quantified synthesis protocol, see Section 3.1 of the ESI.†The crude extract was purified by flash column chromatography on silica gel (ethyl acetate 60/40 hexane) and directly used in the next step. To a cooled solution of the previously purified compound (1 eq., 0.098 mmol, 38 mg) in anhydrous THF (2 mL), a solution of methylmagnesium bromide was added dropwise under nitrogen (3 eq., 0.294 mmol, 0.15 mL). The solution was then stirred at room temperature for 3 h. The reaction was monitored by TLC (ethyl acetate 60/40 hexane). The solution was treated with saturated aqueous NaHCO3 (pH = 7) and extracted twice with ethyl acetate. The organic layers were washed with brine and water, then dried over Na2SO4, filtered, and evaporated under vacuum. The crude extract was purified by flash column chromatography on silica gel (ethyl acetate 60/40 hexane) to afford product 31 as a yellow oil.
4.3 Artificial metalloenzyme characterisation and catalytic assays
Author contributions
CMJ and AP developed and conceptualised the research idea and supervised the project, contributed to the planning and design of the experimental methodology, aided in the data interpretation and wrote the manuscript. FLM performed the molecular modelling, designed the methodology with input from CMJ and AP, performed all experimental work and data analysis and contributed to manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CMJ gratefully acknowledges the financial support from the Nottingham Advanced Research Fellowship (NRF) scheme for initiating this joint collaboration. The authors further want to thank Prof. Rachel Gomes for her helpful insights in project discussions and Dr Marko Hanževački for support in finalising the manuscript and input into graphical representations. We also gratefully acknowledge access to the University of Nottingham’s Augusta HPC service. The authors would also like to thank Dr Gemma Harris for providing access and support for the ITC experiment at the Harwell research complex.Notes and references
- S. C. Hammer, A. M. Knight and F. H. Arnold, Curr. Opin. Green Sustain. Chem., 2017, 7, 23–30 CrossRef.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- B. Large, N. G. Baranska, R. L. Booth, K. S. Wilson and A. K. Duhme-Klair, Curr. Opin. Green Sustain. Chem., 2021, 28, 1–7 Search PubMed.
- A. S. Klein and C. Zeymer, Protein Eng., Des. Sel., 2021, 34, gzab003 CrossRef PubMed.
- T. Heinisch and T. R. Ward, Acc. Chem. Res., 2016, 49, 1711–1721 CrossRef CAS PubMed.
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS PubMed.
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585–595 CrossRef CAS PubMed.
- L. Alonso-Cotchico, J. Rodríguez-Guerra, A. Lledós and J.-D. Maréchal, Acc. Chem. Res., 2020, 53, 896–905 CrossRef CAS PubMed.
- G. B. Akcapinar and O. U. Sezerman, Biosci. Rep., 2017, 37, BSR20160179 CrossRef CAS PubMed.
- F. Richter, A. Leaver-Fay, S. D. Khare, S. Bjelic and D. Baker, PLoS One, 2011, 6, 1–12 Search PubMed.
- J. K. Leman, B. D. Weitzner, S. M. Lewis, J. Adolf-Bryfogle, N. Alam, R. F. Alford, M. Aprahamian, D. Baker, K. A. Barlow, P. Barth, B. Basanta, B. J. Bender, K. Blacklock, J. Bonet, S. E. Boyken, P. Bradley, C. Bystroff, P. Conway, S. Cooper, B. E. Correia, B. Coventry, R. Das, R. M. De Jong, F. DiMaio, L. Dsilva, R. Dunbrack, A. S. Ford, B. Frenz, D. Y. Fu, C. Geniesse, L. Goldschmidt, R. Gowthaman, J. J. Gray, D. Gront, S. Guffy, S. Horowitz, P. S. Huang, T. Huber, T. M. Jacobs, J. R. Jeliazkov, D. K. Johnson, K. Kappel, J. Karanicolas, H. Khakzad, K. R. Khar, S. D. Khare, F. Khatib, A. Khramushin, I. C. King, R. Kleffner, B. Koepnick, T. Kortemme, G. Kuenze, B. Kuhlman, D. Kuroda, J. W. Labonte, J. K. Lai, G. Lapidoth, A. Leaver-Fay, S. Lindert, T. Linsky, N. London, J. H. Lubin, S. Lyskov, J. Maguire, L. Malmström, E. Marcos, O. Marcu, N. A. Marze, J. Meiler, R. Moretti, V. K. Mulligan, S. Nerli, C. Norn, S. Ó’Conchúir, N. Ollikainen, S. Ovchinnikov, M. S. Pacella, X. Pan, H. Park, R. E. Pavlovicz, M. Pethe, B. G. Pierce, K. B. Pilla, B. Raveh, P. D. Renfrew, S. S. R. Burman, A. Rubenstein, M. F. Sauer, A. Scheck, W. Schief, O. Schueler-Furman, Y. Sedan, A. M. Sevy, N. G. Sgourakis, L. Shi, J. B. Siegel, D. A. Silva, S. Smith, Y. Song, A. Stein, M. Szegedy, F. D. Teets, S. B. Thyme, R. Y. R. Wang, A. Watkins, L. Zimmerman and R. Bonneau, Nat. Methods, 2020, 17, 665–680 CrossRef CAS PubMed.
- A. Zanghellini, L. Jiang, A. M. Wollacott, G. Cheng, J. Meiler, E. A. Althoff, D. Röthlisberger and D. Baker, Protein Sci., 2006, 15, 2785–2794 CrossRef CAS PubMed.
- S. Basler, S. Studer, Y. Zou, T. Mori, Y. Ota, A. Camus, H. A. Bunzel, R. C. Helgeson, K. N. Houk, G. Jiménez-Osés and D. Hilvert, Nat. Chem., 2021, 13, 231–235 CrossRef CAS PubMed.
- S. D. Khare, Y. Kipnis, P. J. Greisen, R. Takeuchi, Y. Ashani, M. Goldsmith, Y. Song, J. L. Gallaher, I. Silman, H. Leader, J. L. Sussman, B. L. Stoddard, D. S. Tawfik and D. Baker, Nat. Chem. Biol., 2012, 8, 294–300 CrossRef CAS PubMed.
- C. E. Tinberg, S. D. Khare, J. Dou, L. Doyle, J. W. Nelson, A. Schena, W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard and D. Baker, Nature, 2013, 501, 212–216 CrossRef CAS PubMed.
- T. Heinisch, M. Pellizzoni, M. Dürrenberger, C. E. Tinberg, V. Köhler, J. Klehr, D. Häussinger, D. Baker and T. R. Ward, J. Am. Chem. Soc., 2015, 137, 10414–10419 CrossRef CAS PubMed.
- V. M. Robles, M. Dürrenberger, T. Heinisch, A. Lledós, T. Schirmer, T. R. Ward and J. D. Maréchal, J. Am. Chem. Soc., 2014, 136, 15676–15683 CrossRef CAS PubMed.
- J. Bos, A. García-Herraiz and G. Roelfes, Chem. Sci., 2013, 4, 3578–3582 RSC.
- L. Alonso-Cotchico, J. R. G. Pedregal, A. Lledós and J. D. Maréchal, Front. Chem., 2019, 7, 211 CrossRef CAS PubMed.
- I. Drienovská, L. Alonso-Cotchico, P. Vidossich, A. Lledós, J. D. Maréchal and G. Roelfes, Chem. Sci., 2017, 8, 7228–7235 RSC.
- M. Basle, H. A. W. Padley, F. L. Martins, G. S. Winkler, C. M. Jäger and A. Pordea, J. Inorg. Biochem., 2021, 220, 111446 CrossRef CAS PubMed.
- C. Esmieu, M. V. Cherrier, P. Amara, E. Girgenti, C. Marchi-Delapierre, F. Oddon, M. Iannello, A. Jorge-Robin, C. Cavazza and S. Ménage, Angew. Chem., Int. Ed., 2013, 52, 3922–3925 CrossRef CAS PubMed.
- L. Villarino, K. E. Splan, E. Reddem, L. Alonso-Cotchico, C. Gutiérrez de Souza, A. Lledós, J. D. Maréchal, A. M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2018, 57, 7785–7789 CrossRef PubMed.
- I. A. Guedes, C. S. de Magalhães and L. E. Dardenne, Biophys. Rev., 2014, 6, 75–87 CrossRef CAS PubMed.
- C. K. Prier and F. H. Arnold, J. Am. Chem. Soc., 2015, 137, 13992–14006 CrossRef CAS PubMed.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102–106 CrossRef CAS PubMed.
- S. Morra and A. Pordea, Chem. Sci., 2018, 9, 7447–7454 RSC.
- I. Kleifeld, O. Shi, S. P. Zarivach, R. Eisenstein and M. Sagi, Protein Sci., 2003, 12, 468–479 CrossRef PubMed.
- D. J. Raines, J. E. Clarke, E. V. Blagova, E. J. Dodson, K. S. Wilson and A. K. Duhme-Klair, Nat. Catal., 2018, 1, 680–688 CrossRef CAS.
- M. Hestericová, T. Heinisch, L. Alonso-Cotchico, J. D. Maréchal, P. Vidossich and T. R. Ward, Angew. Chem., Int. Ed., 2018, 57, 1863–1868 CrossRef PubMed.
- F. W. Monnard, E. S. Nogueira, T. Heinisch, T. Schirmer and T. R. Ward, Chem. Sci., 2013, 4, 3269–3274 RSC.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- Y. Korkhin, A. J. Kalb, M. Peretz, O. Bogin, Y. Burstein and F. Frolow, J. Mol. Biol., 1998, 278, 967–981 CrossRef CAS PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- V. Amarnath and K. Amarnath, J. Org. Chem., 1995, 60, 301–307 CrossRef CAS.
- L. D. Krasnoslobodskaya and Y. L. Gol’dfarb, Russ. Chem. Rev., 1969, 38, 389–406 CrossRef.
- F. Stauffer and R. Neier, Org. Lett., 2000, 2, 3535–3537 CrossRef CAS PubMed.
- M. Poizat, I. W. C. E. Arends and F. Hollmann, J. Mol. Catal. B: Enzym., 2010, 63, 149–156 CrossRef CAS.
- Y. Okamoto, V. Köhler and T. R. Ward, J. Am. Chem. Soc., 2016, 138, 5781–5784 CrossRef CAS PubMed.
- Prime, LLC, New York, NY, 2016, 2002, vol. 320, pp. 597–608 Search PubMed.
- LigPrep, LLC, New York, NY, 2016 Search PubMed.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. SØndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2011, 79, 2794–2812 CrossRef CAS PubMed.
- K. Krohn, A. Vidal, J. Vitz, B. Westermann, M. Abbas and I. Green, Tetrahedron: Asymmetry, 2006, 17, 3051–3057 CrossRef CAS.
- Y. L. Song, M. L. Peach, P. P. Roller, S. Qiu, S. Wang and Y. Q. Long, J. Med. Chem., 2006, 49, 1585–1596 CrossRef CAS PubMed.
- J. G. Kim and D. O. Jang, Bull. Korean Chem. Soc., 2010, 31, 171–173 CrossRef CAS.
- Y. K. Tahara, M. Ito, K. S. Kanyiva and T. Shibata, Chem.–Eur. J., 2015, 21, 11340–11343 CrossRef CAS PubMed.
- J. A. Jiang, W. Bin Huang, J. J. Zhai, H. W. Liu, Q. Cai, L. X. Xu, W. Wang and Y. F. Ji, Tetrahedron, 2013, 69, 627–635 CrossRef CAS.
- K. Sivagurunathan, S. Raja Mohamed Kamil, S. Syed Shafi, F. Liakth Ali Khan and R. V. Ragavan, Tetrahedron Lett., 2011, 52, 1205–1207 CrossRef CAS.
- R. A. Croft, M. A. J. Dubois, A. J. Boddy, C. Denis, A. Lazaridou, A. S. Voisin-chiret, R. Bureau, C. Choi, J. J. Mousseau and J. A. Bull, Eur. J. Org. Chem., 2019, 2019, 5385–5395 CrossRef CAS.
- C. Letondor, A. Pordea, N. Humbert, A. Ivanova, S. Mazurek, M. Novic and T. R. Ward, J. Am. Chem. Soc., 2006, 128, 8320–8328 CrossRef CAS PubMed.
- O. Kleifeld, A. Frenkel, O. Bogin, M. Eisenstein, V. Brumfeld, Y. Burstein and I. Sagi, Biochemistry, 2000, 39, 7702–7711 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fd00070e |
| This journal is © The Royal Society of Chemistry 2022 |