
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of titanium ‘POCOP’ pincer complexes†
Leah
Webster
a,
Tobias
Krämer
 b and
F. Mark
Chadwick
*a
b and
F. Mark
Chadwick
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, 82 Wood Lane, W12 0BZ, UK. E-mail: m.chadwick@imperial.ac.uk
bDepartment of Chemistry, Maynooth University, Maynooth, Co. Kildare, Ireland
First published on 26th October 2022
Abstract
The ‘POCOP’ pincer ligand, [2,6-(OPR2)2C6H3], has been attached to titanium in both Ti(III) and Ti(IV) complexes for the first time. Using a lithium-halogen exchange route [2,6-(OPR2)2C6H3]Li ([RPOCOP]Li) can be synthesised. Both the iso-propyl and tert-butyl derivatives can be made, but only the latter isolated. These can be reacted with the Ti(III) and Ti(IV) synthons to make a range of [POCOP]TiClx species. In the presence of Ti(IV), THF and [RPOCOP]Li, an unprecedented ligand rearrangement occurs. (tBuPOCOP)TiCl2, 1, can be derivatised with alkylating agents to make bis-methyl, phenyl and neopentyl complexes. The last of these can activate H2 to make a rare example of a titanium chlorohydride, with the metal pincer fragment staying attached. EPR has been used to characterise the paramagnetic complexes and locate their electron spins, which is further validated with DFT calculations. This opens the door for this archetypical pincer ligand to be used with early transition metals.
Introduction
Since their initial discovery by Shaw in 1976 pincer complexes have held a privileged position in organometallic chemistry.1–3 Pincer complexes are widely applied in catalysis for example in hydrogenations using first-row transition metals,4 iridium and ruthenium catalysed alkane dehydrogenation,5 and cross-coupling reactivity.6 Pincer ligands also support interesting reactivity, for example acting as a two-electron sink to augment reactivity.7 Of the many different pincer ligands developed some of the most recognisable are those of the RPCP and RPOCOP design, where a central arene is flanked by two phosphines connected via a methylene group or an oxygen atom respectively (RPCP = 2,6-(CH2PR2)2C6H3; RPOCOP = 2,6-(OPR2)2C6H3)).The RPCP ligand is the progenitor of pincer ligands. It, and its close cousin RPOCOP have been extensively used with late transition metals. The ease of changing the phosphine and arene substituents has resulted in these ligands being adopted in a multitude of challenging reactivity.8 For example Ir RPOCOP complexes have been used in tandem with olefin metathesis catalysts to perform alkane metathesis,9 and have been able to support single-crystal to single-crystal ligand exchange.10 However to date there are only two reports of the RPOCOP motif being used with early transition metals.11,12 The origin of this scarcity is that the traditional route to these complexes relies on the oxidative addition of a strong C–H bond, which is disfavoured for early transition metals (both because of the relatively weak bonds formed, and the lack of suitable synthons in an N-2 oxidation state).
However early transition metal pincer complexes are enjoying a renaissance.13 Pioneered by Fryzuk's work on aliphatic ‘PNP’ pincer complexes,14 more recently others have taken up the baton making early transition metal pincer complexes capable of activating small molecules (e.g. N2, CH4) in interesting reactivity.15–24 An issue with these systems is that they can be challenging to derivatise in order to tune the steric and electronic profile of the pincer ligand. Of particular interest would be the isolation of early transition metal hydrides. These have been invoked as intermediates in the activation of N2 and isolable titanium monohydrides are able to C–H activate arenes.25–27 Arguably even rarer than isolable titanium hydrides are titanium chlorohydrides, which are ‘often invoked but never (sic.) observed’.28 Though this quote refers to titanocene chlorohydride there are still only two structurally characterised titanium chlorohydrides (excluding those where the hydride is supported by another element, e.g. boron).29,30
Herein we report the selective synthesis of RPOCOP Titanium complexes. We demonstrate ligand platforms with different steric profiles and binding to the metal in multiple oxidation states (with divergent chemistries). We have taken one of these synthons further onwards, derivatising it to make a number or alkyl and aryl Ti(RPOCOP) complexes. One of these is reacted with dihydrogen to form a rare example of an isolable Ti chlorohydride, displaying the RPOCOP ligands’ ability to support interesting reactivity from the metal centre.
Results and discussion
Synthesis of titanium RPOCOP complexes was achieved via the salt metathesis reaction of the lithium salt of the RPOCOP ligand and either TiCl3(THF)3 or TiCl4. A similar method from the iodo-RPOCOP compound has been described, however the lithiated material was not isolated nor characterised.31,32[tBuPOCOP]Li (RPOCOP = 2,6-(OPR2)C6H3) was isolated via the lithium halogen exchange reaction of (tBuPOCOP)Br with a slight excess of nBuLi (Scheme 1, yield 99%).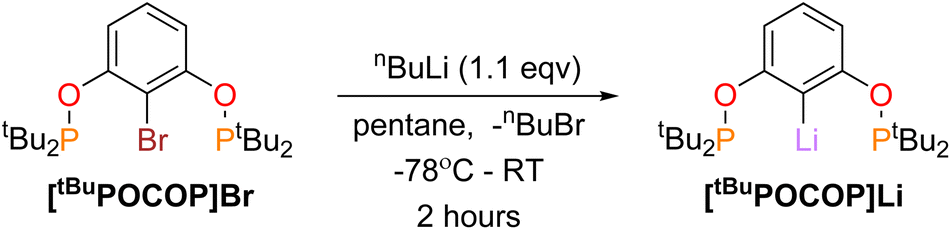 | ||
| Scheme 1 Synthesis of [tBuPOCOP]Li. | ||
[tBuPOCOP]Li can be stored at −40 °C in an inert atmosphere as a colourless solid for over two months with no degradation. [tBuPOCOP]Li is extremely moisture-sensitive and exposure to water leads to intractable contamination by (tBuPOCOP)H. [tBuPOCOP]Li is extremely soluble in hydrocarbon solvents, including aliphatic solvents. Crystals of [tBuPOCOP]Li suitable for X-ray diffraction were grown from slow evaporation of a pentane solution (Fig. 1).
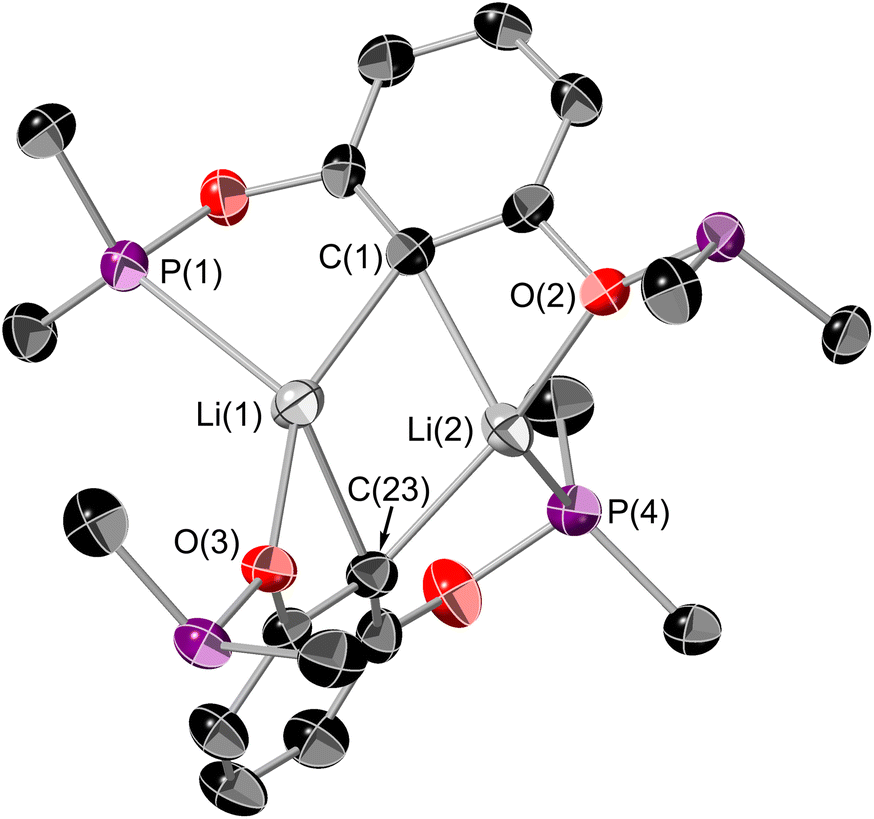 | ||
| Fig. 1 ORTEP plot of [tBuPOCOP]Li. Thermal ellipsoids at 50% probability, hydrogens omitted for clarity. tBu groups on phosphines only have the central carbon shown for clarity. Colour key: black (carbon); purple (phosphorus), red (oxygen), grey (lithium). Selected bond lengths (Å): Li(1)–C(1) 2.197(11), Li(1)–C(23) 2.264(11), Li(1)–O(3) 2.164(10), Li(1)–P(1) 2.614(10), Li(2)–C(1) 2.234(11), Li(2)–C(23) 2.192(11), Li(2)–O(2) 2.116(9), Li(2)–P(4) 2.544(10). | ||
Organolithium compounds aggregate in the solution phase (and indeed in the solid and gaseous phase) to varying degrees.33 There are only a couple of other examples of a lithium ‘PCP’ complexes.34,35[tBuPOCOP]Li has a similar structure to the related [MePCP]Li complex.35 The dimeric structure of [tBuPOCOP]Li is retained in non-donor solvents at room temperature. This is demonstrated by a quintet resonance in the 7Li{1H} NMR spectrum, though some fluxionality must be present to allow the Li centres to couple equally to all four phosphorus atoms in the dimer. Such fluxionality in pincer complexes has precedence.35,36
Synthesis of Titanium(III) POCOP Chloride, (tBuPOCOP)TiCl2, (1)
The synthesis of (tBuPOCOP)TiCl2, (1) in pentane (Scheme 2) yielded an electric blue solution from which 1 was isolated as a light blue solid. A similar synthesis has recently been reported, however with minimal characterising data.12 An improved crystalline yield can be achieved by recrystallising 1 from cyclopentane (59%).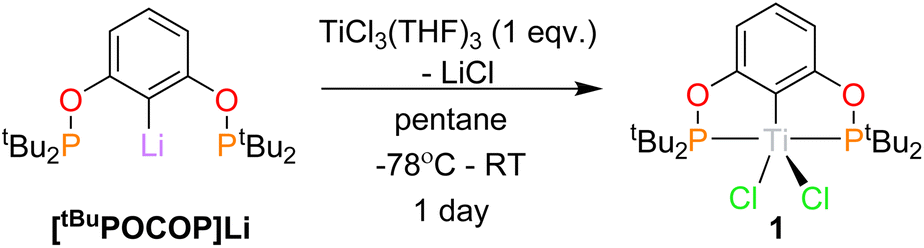 | ||
| Scheme 2 Synthesis of 1. | ||
Evan's NMR of 1 determined an μeff value of 1.86 μB, corresponding to one unpaired electron on the titanium centre, which was confirmed by the DFT-calculated spin density (see ESI†). Unfortunately, due to the paramagnetic nature of the complex, only broad resonances were observed in the 1H NMR spectrum (see ESI†) and no resonances were observed in the 31P{1H} NMR spectrum.
The presence of a titanium(III) metal centre was further confirmed by X-Band CW EPR of 1 (Fig. 2). The room temperature EPR signal is a triplet with a giso value of 1.9665 and isotropic hyperfine coupling value, a0 of 2.31 mT, This is representative of a single unpaired electron centred on the titanium atom coupled to two equivalent phosphorus (I = 1/2) nuclei.15
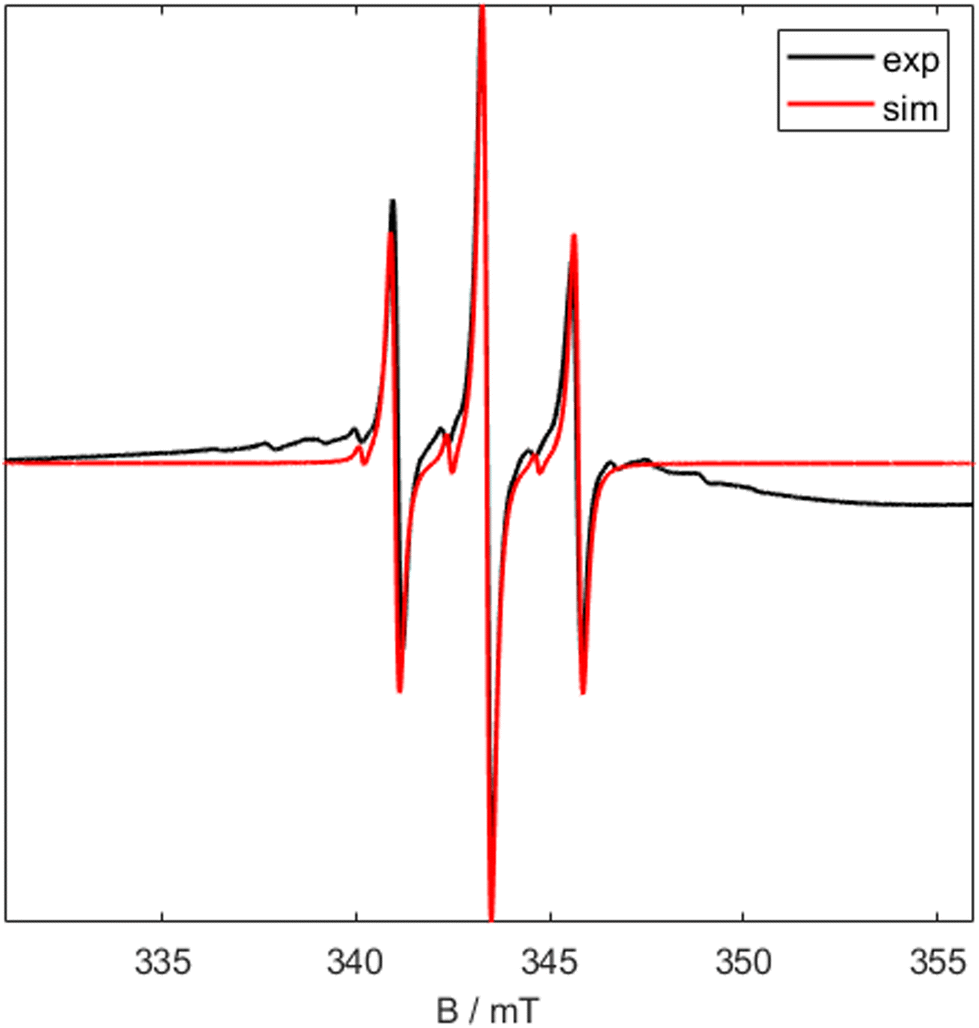 | ||
| Fig. 2 X Band CW EPR spectrum of 1 at room temperature. Black shows experimental data and red shows simulated data (giso = 1.9665, a0 = 2.31 mT). | ||
Synthesis of titanium(IV) POCOP chlorides, (RPOCOP)TiCl3, (2)
The reaction of [tBuPOCOP]Li and TiCl4 in pentane (Scheme 3) yields (tBuPOCOP)TiCl3, 2, as a red solid in 57% yield. Single crystals suitable for X-ray diffraction studies were grown from a saturated hexane solution cooled to −40 °C. 2 is the Ti(IV) analogous complex of 1.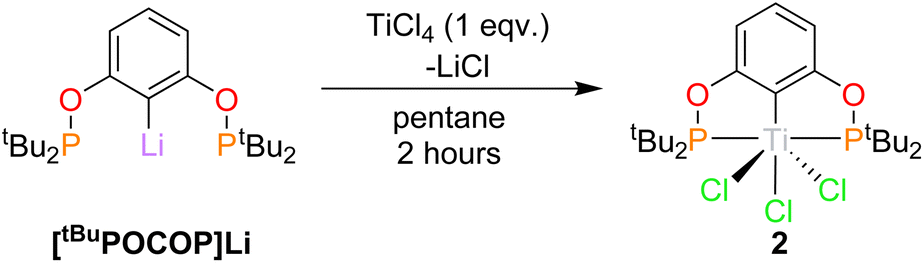 | ||
| Scheme 3 Synthesis of 2. | ||
The solid-state structure of 2 (Fig. 3) shows titanium in an extremely distorted octahedral geometry, with the sum of the angles around the equatorial plane (featuring the POCOP ligand and Cl(2)) being 364.46(4)° but the axial Cl(1)–Ti(1)–Cl(3) angle being only 143.16(4)°. The origin of this distortion appears to be due to a second-order Jahn Teller effect, but a more comprehensive theoretical analysis is currently beyond the scope of this paper. Generally the Ti(1)–X bond lengths show a small increase when compared to 1, presumably due to the increased steric encumbrance in 2.
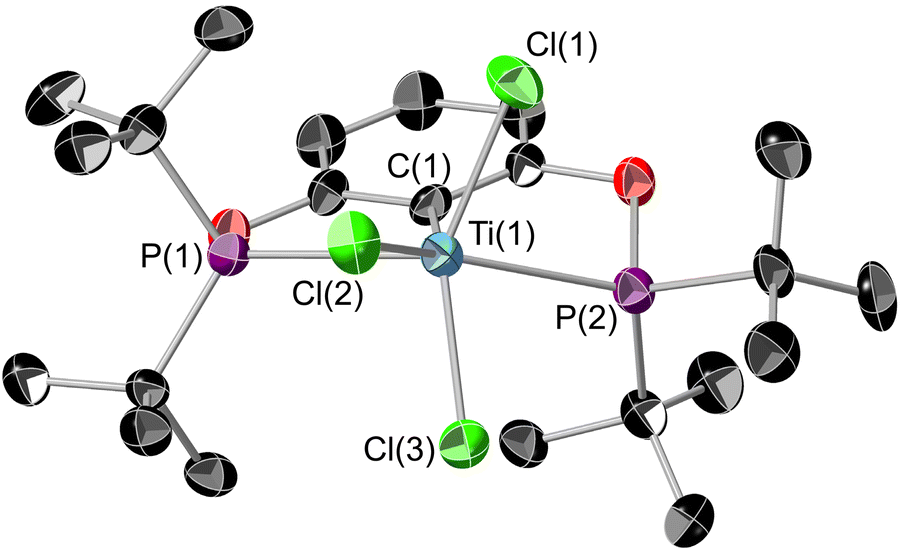 | ||
| Fig. 3 ORTEP plot of 2. Thermal ellipsoids at 50% probability, hydrogens omitted for clarity. Colour key: sky blue (titanium), black (carbon); purple (phosphorus), red (oxygen), green (chlorine). Selected bond lengths (Å) and bond angles (°): Ti(1)–C(1) 2.2081(11), Ti(1)–P(1) 2.6133(11), Ti(1)–P(2) 2.6330(11), Ti(1)–Cl(1) 2.2643(11), Ti(1)–Cl(2) 2.3831(10), Ti(1)–Cl(3) 2.3006(10); Σ equatorial angles: 364.46(4) [C(1)–Ti(1)–P(1) 71.80(4), C(1)–Ti(1)–P(2) 68.64(4), P(1)–Ti(1)–Cl(2) 79.55(3), P(2)–Ti(1)–Cl(2) 144.47(4)], Cl(1)–Ti(1)–Cl(3) 143.16(4). | ||
The 1H NMR spectrum shows the characteristic doublet resonance for the PtBu2 groups (1.41 ppm, 2J(H–P) 13 Hz) and the 31P{1H} NMR spectrum shows a single resonance at 187.6 ppm, roughly 30 ppm downfield from the free protonated ligand, (tBuPOCOP)H at 153.1 ppm.37
Unexpectedly, targeting 2 using [tBuPOCOP]Li and a different titanium synthon, TiCl4(THF)2, instead promoted ligand rearrangement, yielding (κ2-O,P-(1-O-2-PtBu2-3-(OPtBu2)-C6H3)2TiCl2, 3 (Scheme 4). This structure sees one of the O–P bonds cleaved and the phosphine group migrating to the original ipso carbon. Two rearranged ligands bind to the single titanium metal centre. This is the first example of such a rearrangement of the POCOP ligand.
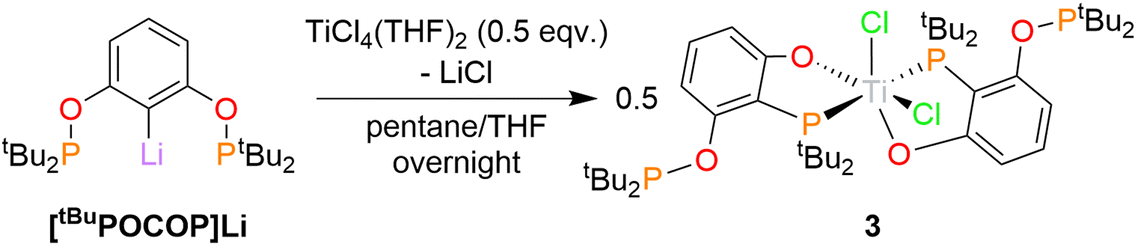 | ||
| Scheme 4 Synthesis of 3. | ||
THF is required for this rearrangement to occur – conducting the reaction in toluene or diethyl ether does not produce either 2 or 3. 2 cannot be converted to 3 despite heating in THF and excess [tBuPOCOP]Li. Heating [tBuPOCOP]Li in THF in the absence of stoichiometric titanium does not induce ligand rearrangement. 3 is still exclusively the only observable product even with a strict 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalence of TiCl4(THF)2 and [tBuPOCOP]Li. This rearrangement is curiously not seen with TiCl3(THF)3. This appears to be an unique rearrangement in the presence of both Ti(IV) and THF.
:1 equivalence of TiCl4(THF)2 and [tBuPOCOP]Li. This rearrangement is curiously not seen with TiCl3(THF)3. This appears to be an unique rearrangement in the presence of both Ti(IV) and THF.
The rearrangement is clearly seen in the 1H and 31P{1H} NMR spectra with two resonances observed corresponding to the tert-butyl protons; one tBu 1H resonance still observed as a doublet at 1.10 ppm (2JH–P = 12 Hz) whilst the other observed as a broad singlet (1.76 ppm). The 31P{1H} NMR spectrum also displays two resonances which integrate 1:1, one at 159.0 ppm, which is a similar chemical shift to the free protonated ligand,37 whilst the other is upfield at 78.2 ppm. These are assigned to the free and ligating phosphine respectively.
Single crystals of 3, suitable for X-ray diffraction, could be grown by cooling a saturated hexane solution to −40 °C (Fig. S1†). The poor quality of the X-ray data means detailed structural analysis is not appropriate, however the titanium clearly adopts an approximately octahedral geometry, as demonstrated by the P(1)–Ti(1)–P(3) and Cl(1)–Ti(1)–Cl(2) angles (178.4(2)° and 95.3(2)° respectively). The deviation from ideal octahedral geometry arises due to the constraint of the P and O being 1,2-substituted on the arene ring, thus pulling the oxygen away (P(1)–Ti(1)–O(1) 71.3(4)°). This is also presumably the cause for the slight lengthening of the Ti–P bonds as compared with 2.
Only a single stereoisomer of this product is observed, with the two phosphine groups trans to each other and the chloride and oxo groups in cis geometries. This is demonstrated by only one ligating 31P resonance observed in the 31P{1H} NMR spectrum and no P–P coupling as the P centres are equivalent. The isomer shown in Scheme 4 has been determined to be the lowest energy isomer by DFT, with all possible isomers and their relative energies shown in Fig. 4.
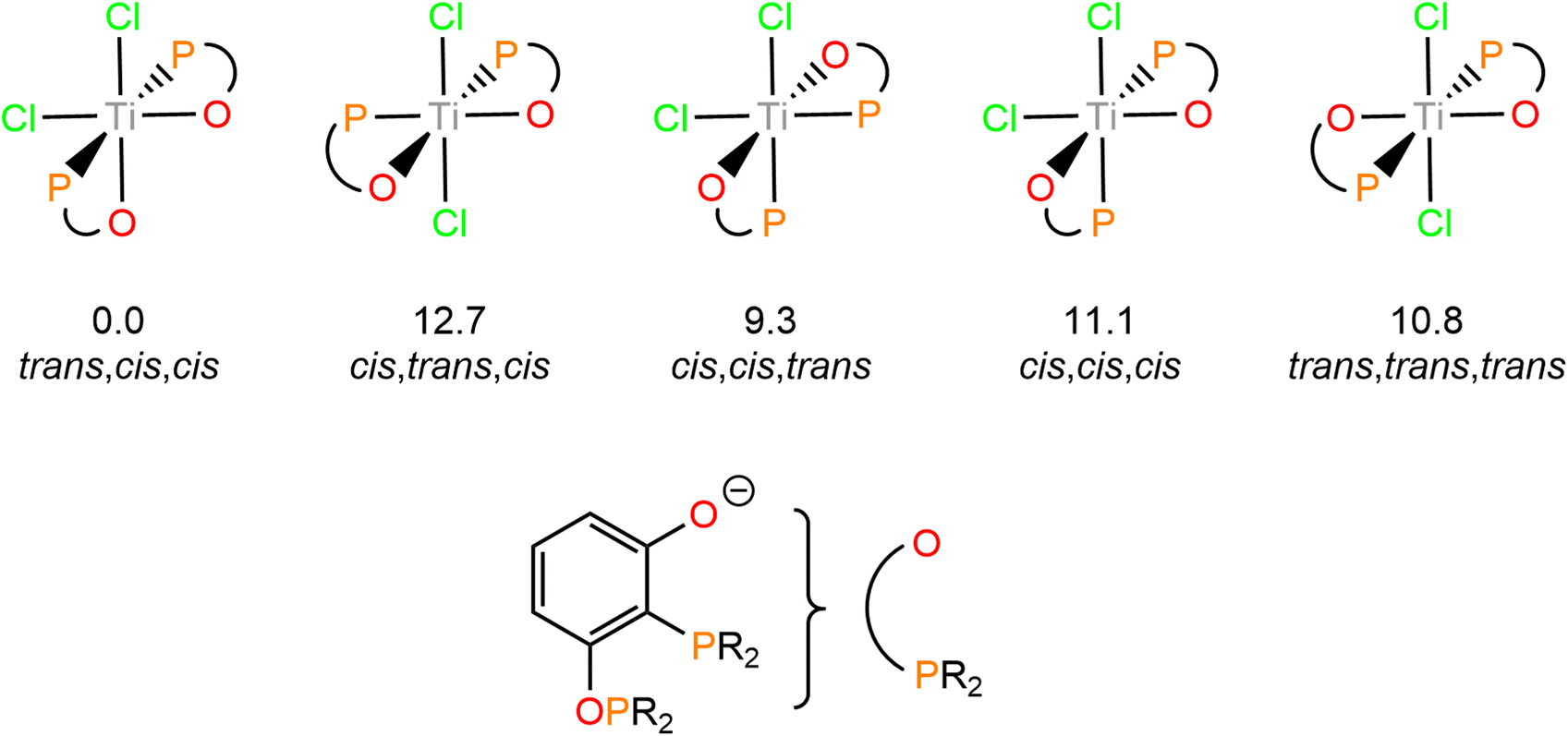 | ||
| Fig. 4 Isomers of 3 and their relative Gibbs Free Energies (in kcal mol−1) calculated at the BP86-D3(BJ)/def2-TZVP/def2-SVP level of theory. | ||
To explore the influence of sterics within the ligand, and prove the broader applicability of this route to tuneable ligands, the iso-propyl derivative was also targeted. It was found tBuLi was needed to affect the lithium-halogen exchange – [iPrPOCOP]Li is an off-white oil, and could not be isolated. However it could be made in situ and used directly onwards in reaction with TiCl4 in pentane. This yielded dimeric {(iPrPOCOP)TiCl2(μ-Cl)}2 (4, Scheme 5).
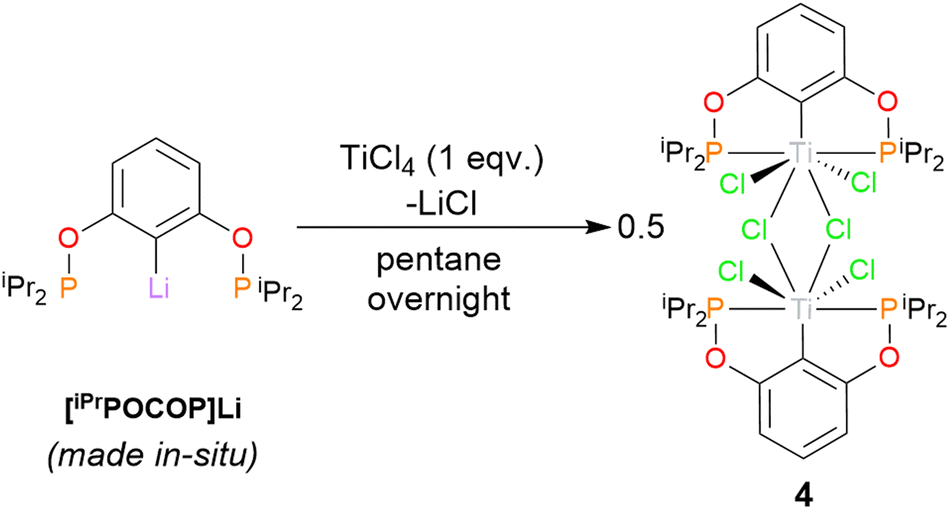 | ||
| Scheme 5 Synthesis of 4. | ||
Single crystals of 4 suitable for X-ray diffraction can be grown from a saturated hexane solution cooled to −40 °C (Fig. 5). The geometry around the titanium is a near-perfect pentagonal bipyramid (Σ equatorial angles 360.89(5)°). The structural parameters are in line with similar motifs,38 for example the Ti–Cl distance for the terminal Cl is nearly identical to that found in 2.
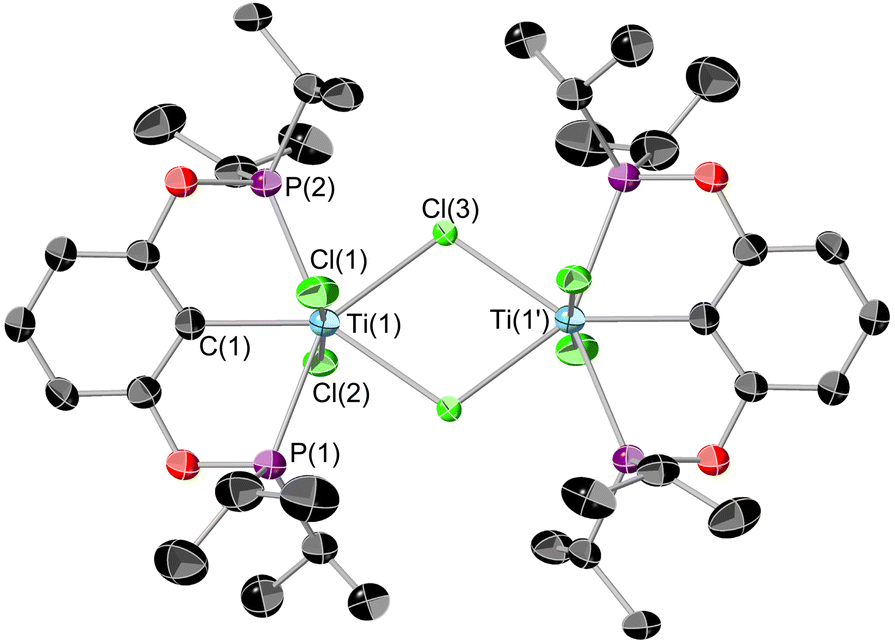 | ||
| Fig. 5 ORTEP plot of 4. Thermal ellipsoids at 50% probability, hydrogens omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ti(1)–Cl(1) 2.293(1), Ti(1)–Cl(2) 2.336(1), Ti(1)–Cl(3) 2.587(1), Ti(1)–P(1) 2.578(1), Ti(1)–P(2) 2.587(1), Ti(1)–C(1) 2.252(4), Ti(1)–Ti(1′) 4.116(1), Σ equatorial angles 360.89(5), Cl(1)–Ti(1)–Cl(2) 174.05(5). | ||
The origin of the structural difference between 2 and 4 arises due to the reduction of steric encumbrance due to the ligands. The dimeric structure is 15 kcal mol−1 more favoured than the monomeric structure for 4. However making the substituents tBu rather than iPr reverses this, making the monomeric form 1 kcal mol−1 more stable than the dimer. There is still a small steric clash in the iso-propyl derivative when compared to a simple methylated model (see ESI†)
Derivatisations of (tBuPOCOP)TiCl2
With a high-yielding synthesis of 1 devised alkylation and arylation experiments were undertaken. Attempts at alkylating 2 with Grignard and alkyl lithium reagents resulted in reduction.Methyl
1 can be reacted with two equivalents of MeMgCl resulting in an immediate colour change from blue to teal, forming (tBuPOCOP)TiMe2 (5, Scheme 6). To date, there has been no observation of a monomethylated complex (tBuPOCOP)TiMeCl. Teal crystals suitable for X-ray diffraction studies can be grown from a concentrated pentane solution cooled to −40 °C (Fig. 6). The geometry around the titanium centre remained unchanged from the starting material as a square based pyramid, with a τ5 parameter of 0.017 and C(1) determined to be the axial ligand.39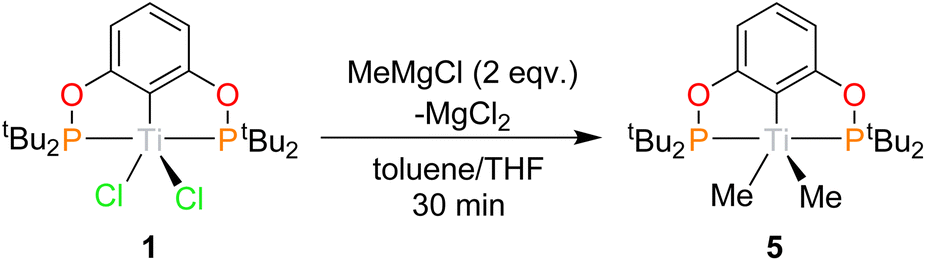 | ||
| Scheme 6 Synthesis of 5. | ||
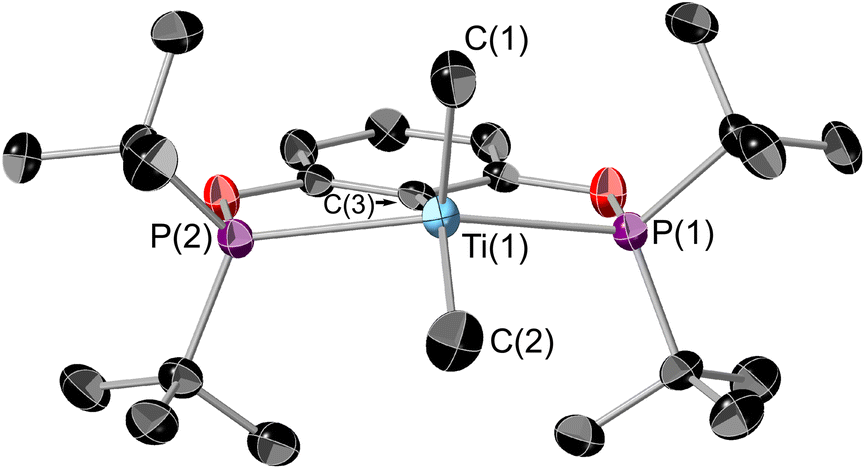 | ||
| Fig. 6 ORTEP plot of 5. Thermal ellipsoids at 50% probability, hydrogens omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ti(1)–P(1) 2.6243(9), Ti(1)–P(2) 2.6180(9), Ti(1)–C(1) 2.138(4), Ti(1)–C(2) 2.143(4), Ti(1)–C(3) 2.241(3), P(1)–Ti(1)–P(2) 141.44(3), C(2)–Ti(1)–C(3) 140.53(14). | ||
The Ti–P bond lengths are slightly elongated in 5 when compared to 1. The Ti–Me distances are comparable to other Ti pincer dimethyl complexes.19,405 is paramagnetic with a μeff = 2.03μB (Evan's method). Fig. 7 shows the X band CW EPR of 5 measured at 298 K.
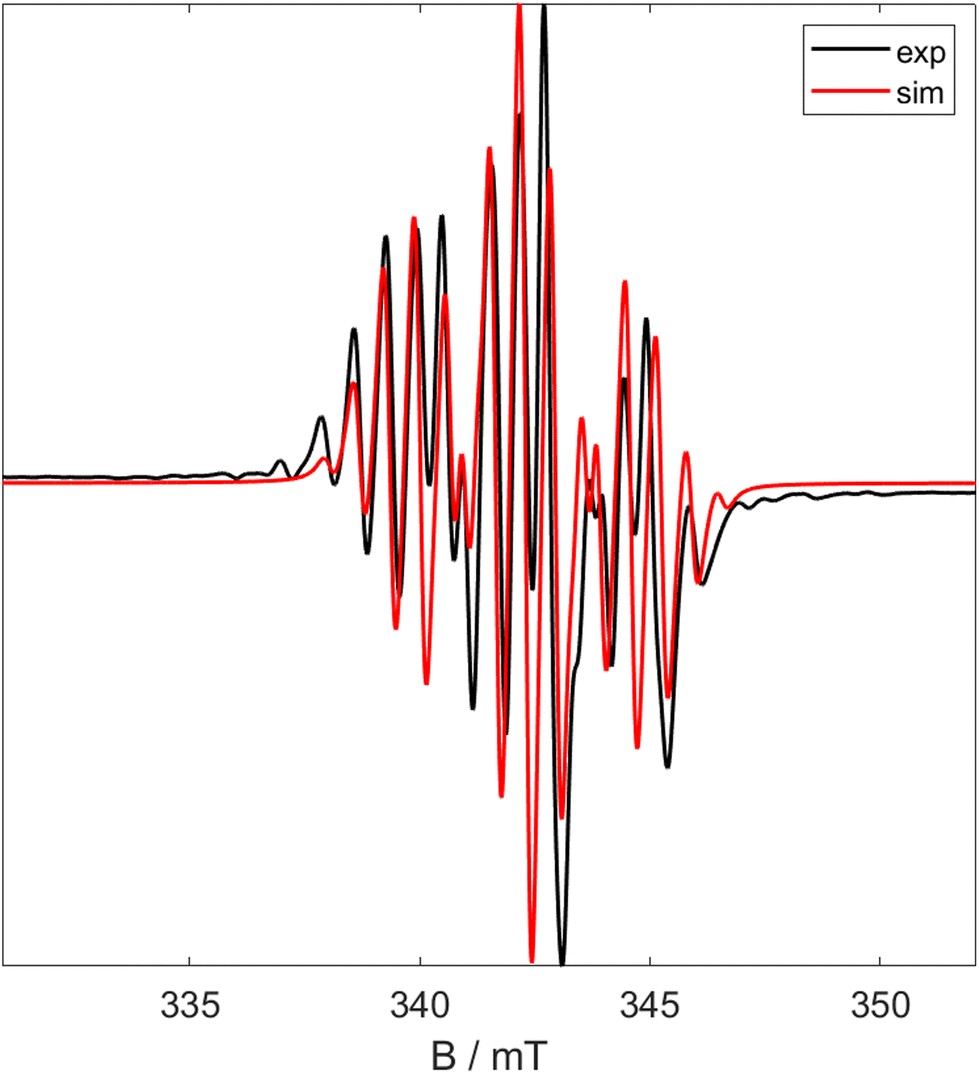 | ||
| Fig. 7 X Band CW EPR spectra of 5 at room temperature. Black shows experimental data and red shows simulated data (giso = 1.970, a0 = 2.25 mT, 0.64 mT). | ||
The experimental spectrum in Fig. 7 can be simulated as a triplet of septets (giso = 1.970; a0 = 2.251 mT, 0.642 mT). This arises due to coupling of the electron to the two 31P nuclei and slightly weaker coupling to the six 1H attached to the methyl groups. This implies that the SOMO is primarily positioned on the titanium centre (implied by the g value) but has significant character on both the phosphorus and methyl ligands.
Phenyl
When 1 was reacted with PhMgCl, only the monosubstituted product was isolated, (tBuPOCOP)TiPhCl (6), even if excess PhMgCl is used or the reaction is heated overnight (Scheme 7). Use of PhMgBr on one occasion resulted in halogen exchange resulting in (tBuPOCOP)TiPhBr (see ESI†). Single crystals of 6 suitable for X-ray diffraction studies could be grown from cooling a concentrated pentane solution of 6 to −40 °C (Fig. 8). | ||
| Scheme 7 Synthetic route to (tBuPOCOP)TiPhCl. | ||
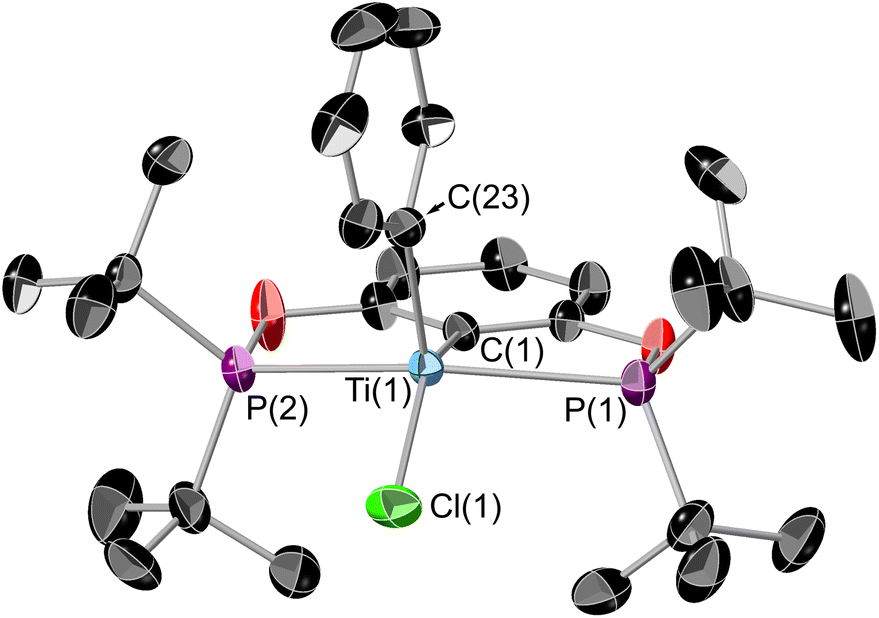 | ||
| Fig. 8 ORTEP plot of 6. Thermal ellipsoids at 50% probability, hydrogens omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ti(1)–P(1) 2.6109(6), Ti(1)–P(2) 2.6109(6), Ti(1)–C(1) 2.2129(19), Ti(1)–C(23) 2.100(2), Ti(1)–Cl(1) 2.3157(6), P(1)–Ti(1)–P(2) 143.72(2), C(1)–Ti(1)–Cl(1) 144.36(5). | ||
The geometry around the titanium centre remained unchanged from the starting material as square based pyramid, with a τ5 parameter of 0.011 and the phenyl ring in the axial position. The plane of the phenyl ring is perpendicular to the POCOP aryl ring. Interestingly this is only the second example of a crystallographically characterised ‘TiPhCl’ fragment with an unsubstituted phenyl ring.40
The room temperature X band CW EPR spectrum of 6 (see ESI, Fig. S4†) consistently appeared to have two signals, one major (60%, giso = 1.9665, a0 = 2.26 mT) and one minor (40%, giso = 1.9720, a0 = 2.24 mT). These apparent two signals occur even if excess PhMgCl is used in the synthesis and are both due to 6 (see later Q-band EPR studies of (tBuPOCOP)TiNpCl, 7). The magnetic moment was measured by Evan's method (μeff = 2.08μB). These data imply there is a single unpaired electron that is almost entirely located on the titanium centre with some coupling to the phosphorus atoms.
Neopentyl
1 can also be derivatised using organolithiums, and the addition of 1 equivalence of neopentyl lithium yields (tBuPOCOP)TiClNp (7) as dark green crystals in 63% yield (Scheme 8). Similarly to 6, substitution of two chloride ligands for neopentyl fragments does not occur, even when using excess neopentyl lithium and forcing conditions. Single crystals suitable for X-ray crystallography can be grown from a saturated pentane solution cooled to −40 °C (Fig. 9). | ||
| Scheme 8 Synthesis of 7. | ||
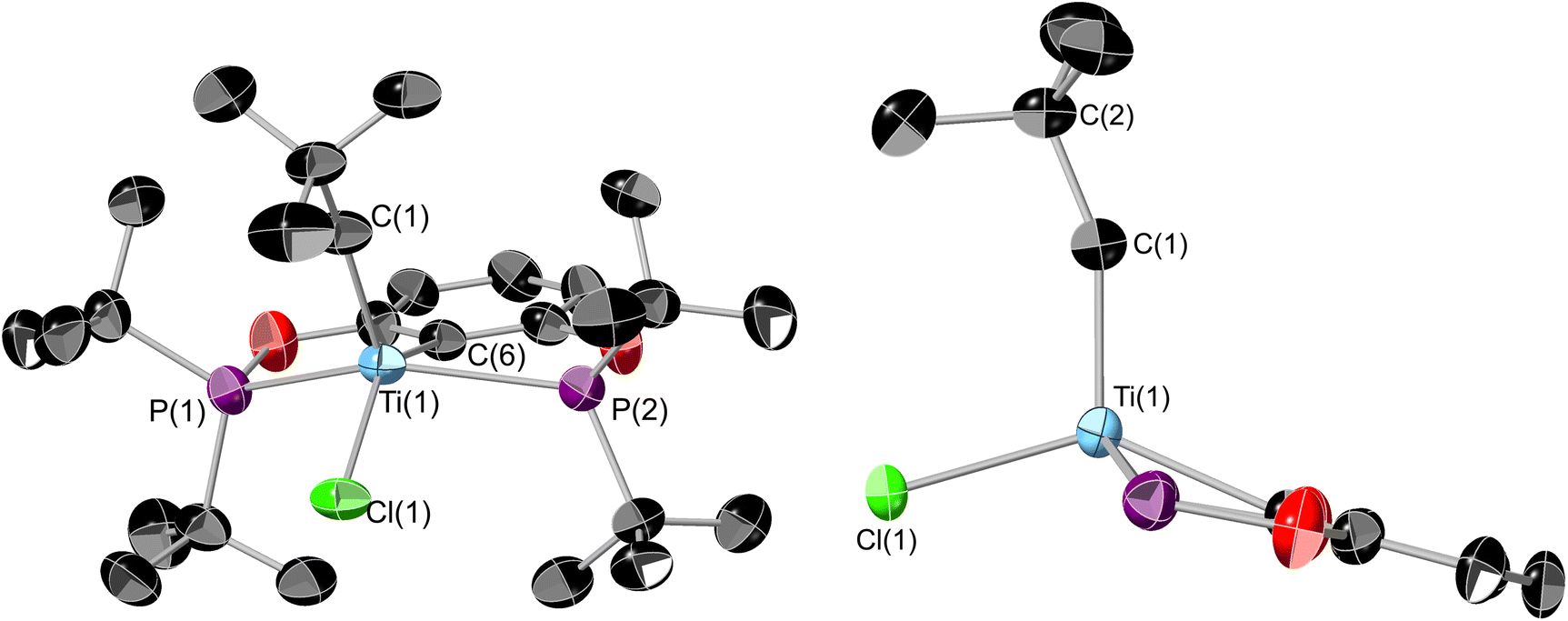 | ||
| Fig. 9 Two ORTEP plots of 7. Thermal ellipsoids at 50% probability, hydrogens omitted for clarity. The phosphine tert-butyl groups are omitted from the side-on view. Selected bond lengths (Å) and bond angles (°): Ti(1)–P(1) 2.6223(17), Ti(1)–P(2) 2.6232(17), Ti(1)–C(1) 2.033(6), Ti(1)–C(6) 2.222(5), Ti(1)–Cl(1) 2.3799(14), P(1)–Ti(1)–P(2) 139.03(6), C(6)–Ti(1)–Cl(1) 140.40(15), Ti(1)–C(1)–C(2) 156.3(5). | ||
The geometry around the titanium centre is still a square based pyramid, however significantly more distorted than 1 (τ5 = 0.265 for 7). The neopentyl fragment is remarkably distorted towards linerarity around C1 (Ti(1)–C(1)–C(2) = 156.3(5)°), such a distortion could arise from some form of agostic interactions with the C–H bonds around C(1). This interaction must be somewhat fluxional however since no coupling to these protons is observed in the room-temperature solution EPR (see ESI, Fig. S5†). The single crystal X-ray data was of insufficient quality to refine the H-atom positions. The optimised geometry of 7 employing the BP86-D3 functional allows for a more detailed analysis of the interaction between the neopentyl fragment and titanium. The bond metrics associated with the crystallographic structure are reproduced very well. The computed Ti⋯H(1) distance of 2.04 Å is significantly shorter than Ti⋯H(2) with 2.44 Å. Additionally, both C(1)–H bonds are somewhat elongated with respect to the remaining C–H bonds (1.11 Å), although distances of 1.14 Å and 1.12 Å for C(1)–H(1) and C(1)–H(2), respectively, imply that this effect is more pronounced for the former (Fig. S45†). The weakening of these bonds is also reflected in their stretching frequencies which are red-shifted to νC(1)H(1) = 2689 cm−1 and νC(1)H(2) = 2888 cm−1, while the other C–H bonds show symmetric and antisymmetric modes at around 3087 cm−1 and 2959 cm−1, respectively. These data are diagnostic of α-agostic interactions between Ti and the C(1)H2 group, where exchange of the symmetry-equivalent hydrogen atoms would imply a degree of fluxionality. This is further validated by NBO and QTAIM analyses of the computed electron density (see Fig. S46 and S47†). For the former, several contributions to σCH → dTi donor/acceptor interactions can be identified, with the leading interactions stabilising the structure by ∼7–13 kcal mol−1, considering both α-and β-spin manifolds. The topology analysis reveals reduced QTAIM parameters associated with the bond critical points (BCPs) of C(1)–H(1) (ρ(r) = 0.245 au; ∇2ρ(r) = –0.734 au) and C(1)–H(2) (ρ(r) = 0.257 au; ∇ρ2(r) = –0.835 au), diagnostic of weakened bonds due to their interaction with the Ti centre. No BCP for the Ti⋯H interaction could be located, however, the complete absence of BCPs and associated bond paths in these types of systems is not uncommon and is not considered a requirement for agostic interactions.41,42
Similarly to 1 and 6, 7 only displays a triplet in the EPR, indicating the unpaired electron is centred on the metal centre (major component g = 1.9665) but couples to the two 31P nuclei (a0 = 2.20 mT). Also similarly to 6 the X band spectrum shows a minor component. In order to deduce whether this is due to an impurity measurements were carried out using Q band radiation. The resultant spectrum is shown in Fig. 10 and shows the signal has collapsed to a simple triplet. This shows that the minor component in the X band EPR is due to coupling and not due to an impurity. The effective magnetic moment (μeff = 1.75μB) was measured using Evan's method and corresponds to a single unpaired electron.
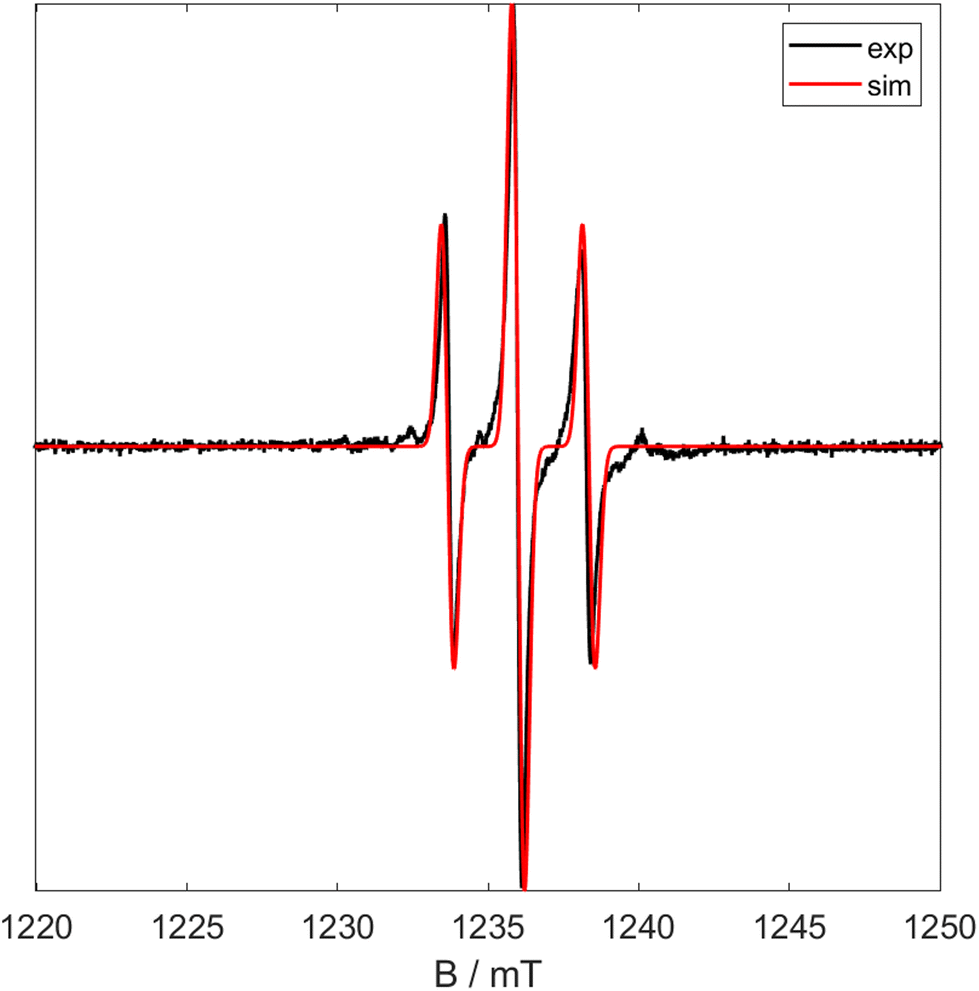 | ||
| Fig. 10 Q Band CW EPR spectra of 5 at room temperature. Black shows experimental data and red shows simulated data (giso = 1.9665, a0 = 2.30 mT). | ||
Hydrogen activation studies
Having demonstrated the ability of the ‘(tBuPOCOP)Ti’ platform to allow for derivatisation the possibility that these complexes could be used to activate small molecules was explored. For the ‘(RPOCOP)Ti’ to be useful in reactivity it is imperative that the pincer-metal fragment remains intact in reactive conditions. One of the most fundamental reactions is the activation of hydrogen. When a pentane solution of 7 was charged with 1 bar of dihydrogen the solution quickly changed from green to royal purple. Single crystals X-ray diffraction studies showed this product to be [(tBuPOCOP)TiCl(μ-H)]2 (8, Scheme 9). The reaction can also proceed in the solid state, using crystalline 7 and charged with 1 bar of H2 at room temperature. The green solid changes to purple over 10 minutes, with a loss of crystallinity.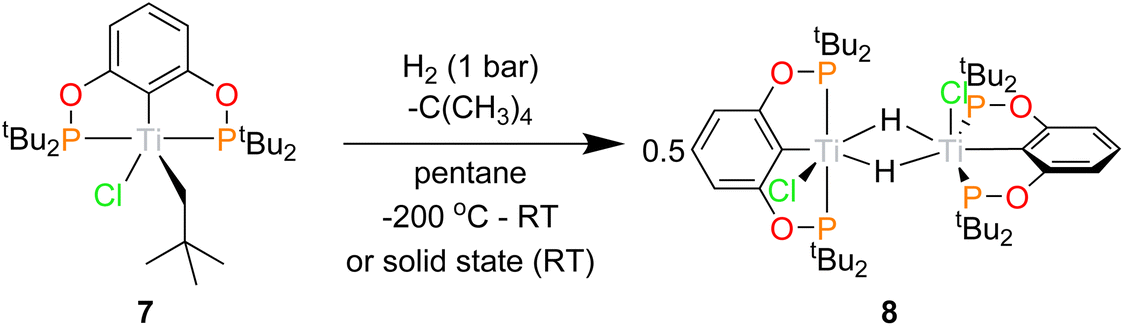 | ||
| Scheme 9 Hydrogen activation by 7 to form 8. | ||
This is a rare example of a titanium chlorohydride, with only two others being isolated and structurally characterised (excluding those that are due to another element-hydride binding to the metal centre e.g. BH4−).29,30 Titanium chlorohydride are invoked as intermediates in a range of catalytic processes but are extremely challenging to observe.27,28 The mechanism of the above reaction was probed by DFT calculations (see Fig. S48†). It traverses a σ-bond metathesis pathway that involves a typical four-membered kite-shaped transition state with an overall Free Energy activation barrier of 22.5 kcal mol−1. Formation of monomeric (tBuPOCOP)TiCl(H) after release of neopentane is exergonic (−9.9 kcal mol−1). Subsequent dimerization of two units (tBuPOCOP)TiCl(H) then forms 8 with an overall exergonicity of −20 kcal mol−1.
Single crystals suitable for X-ray diffraction could be grown from a saturated SiMe4 solution cooled to −40 °C. The resultant structure is shown in Fig. 11. The bridging hydrides could be located on the electron density map but the data was not of sufficient quality to be able to refine their positions reliably. Interestingly these crystals were grown in a N2 atmosphere, showing 8 to be stable in the presence of N2. This is in contrast to previous titanium pincer hydrides.26
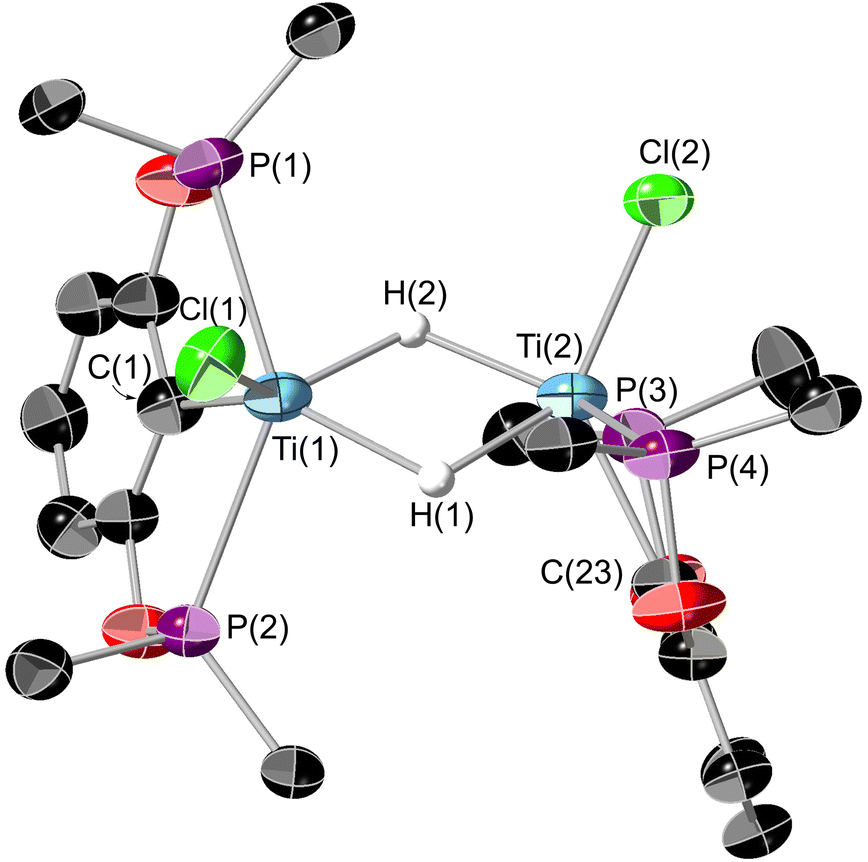 | ||
| Fig. 11 ORTEP plot of 8. Thermal ellipsoids at 50% probability, hydrogens (except for H(1) and H(2)) and tert-butyl methyl groups omitted for clarity. White sphere represent hydrogen atoms. Selected bond lengths (Å) and bond angles (°): Ti(1)–Ti(2) 3.1708(12), Ti(1)–P(1) 2.6858(13), Ti(1)–P(2) 2.6898(13), Ti(1)–C(1) 2.212(4), Ti(2)–P(3) 2.6906(13), Ti(2)–P(4) 2.6799(13), Ti(2)–C(23) 2.221(4), Ti(1)–Cl(1) 2.3157(14), Ti(2)–Cl(2) 2.3113(14), P(1)–Ti(1)–P(2) 138.03(5), C(1)–Ti(1)–Cl(1) 129.99(14), P(3)–Ti(2)–P(4) 137.77(5), C(23)–Ti(1)–Cl(2) 129.79(13). Angle between Cl(1)–Ti(1)–Ti(2) plane and Cl(2)–Ti(2)–Ti(1) plane: 72.25°. | ||
The geometry of 8 is quite distorted away from the optimal octahedron. The Ti(1)–Ti(2) distance is in the region where there could be a Ti–Ti bond,43 however the retention of the unpaired electrons (as evidenced by the EPR spectrum, see ESI Fig. S6†), implies there is no bond present. Looking down the Ti–Ti axis the bonding around the metal centres are eclipsed, however with the POCOP ligands at approximately 90° to one another, presumably this results in minimising the steric clash between the tert-butyl groups. The DFT-optimised geometry of 8 is in good agreement with its experimental counterpart, although the BP86 functional somewhat underestimates the Ti–Ti separation. A survey of different functionals reveals that this trend is general for meta-GGA and GGA functionals, whilst hybrid functionals tend to give Ti–Ti distances closer to the experimental value. Nonetheless, the calculations support the assignment of two bridging hydrides in the structure of 8, with other isomers being energetically disfavoured (see Fig. S49 and S50†). In the computed triplet electronic ground state of 8, stabilised relative to the antiferromagnetically coupled singlet diradical by ∼1 kcal mol−1, one unpaired electron is centred on each Ti ion.
1H NMR studies of the reaction mixture at 5 °C showed formation of neopentane and a broad resonance at a chemical shift of −0.52 ppm which is assigned to the Ti–(μ-H)–Ti environment, as upon reaction with deuterium gas this resonance is absent. 8, like 1, 6 and 7, gives a triplet in its X band CW EPR spectrum at room temperature (see ESI, Fig. S6†). The principle resonance comes at a coincident giso value as the other examples measured, with coupling to two phosphorus resonances. This implies the electron is localised onto the titanium centre and there is no bonding interaction between the metal centres. The effective magnetic moment (measured by Evan's method) is 3.37μB. This is somewhat higher than the ideal spin-only value for two electrons.
Conclusion
The RPOCOP ligand framework has been successfully deployed onto titanium. Both (RPOCOP)Ti(III) and (RPOCOP)Ti(IV) complexes can be made, and different substituents on the POCOP ligand can be used. For Ti(IV), using the bulky tBuPOCOP ligand results in monomeric species 2 to be formed whereas changing the ligand for the slightly smaller iPrPOCOP allows for dimerization (4). Interestingly in the presence of THF, Ti(IV) and [tBuPOCOP]Li reacts in an unprecedented rearrangement of the POCOP ligand. Such a rearrangement does not occur when Ti(III) is used. (tBuPOCOP)TiCl2, 1, can be made on the gram-scale and can be easily derivatised with alkylating and arylating reagents. All of these species have been well-characterised including using X-ray crystallography and EPR.The neopentyl compound (tBuPOCOP)TiNpCl, 7, was reacted with H2, demonstrating the ability of the POCOP ligand to stay attached even in reactive condition. This resulted in the formation of a rare titanium chlorohydride complex, which is remarkably stable under an inert atmosphere. Overall this opens the door to using this valuable and easily customisable ligand platform, the fundamental pincer ligand, with early transition metals.
Conflicts of interest
The authors declare there are no conflicts of interest.Acknowledgements
The EPR measurements were performed at the Centre for Pulse EPR at Imperial College London (PEPR), supported by EPSRC Grant EP/T031425/1. The authors thank Dr Alberto Collauto for useful discussions. FMC thanks Imperial College London for provision of a research fellowship (Imperial College Research Fellowship). The authors also wish to acknowledge the Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support.References
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- G. van Koten and R. A. Gossage, The Privileged pincer-metal platform: Coordination chemistry & applications, Springer International Publishing, 2015 Search PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed.
- R. Shi, Z. Zhang and X. Hu, Acc. Chem. Res., 2019, 52, 1471–1483 CrossRef CAS PubMed.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS.
- D. M. Roddick, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer-Verlag, Berlin, 1st edn, 2013, pp. 49–88 Search PubMed.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257–261 CrossRef CAS PubMed.
- Z. Huang, P. S. White and M. Brookhart, Nature, 2010, 465, 598–601 CrossRef CAS PubMed.
- D. Himmelbauer, B. Stöger, L. F. Veiros, M. Pignitter and K. Kirchner, Organometallics, 2019, 38, 4669–4678 CrossRef CAS.
- J. Zhang, W. Huang, K. Han, G. Song and S. Hu, Dalton Trans., 2022, 51, 12250–12257 RSC.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- R. J. Burford, A. Yeo and M. D. Fryzuk, Coord. Chem. Rev., 2017, 334, 84–99 CrossRef CAS.
- B. C. Bailey, J. C. Huffman, D. J. Mindiola, W. Weng and O. V. Ozerov, Organometallics, 2005, 24, 1390–1393 CrossRef CAS.
- C. M. Brammell, E. J. Pelton, C. H. Chen, A. A. Yakovenko, W. Weng, B. M. Foxman and O. V. Ozerov, J. Organomet. Chem., 2011, 696, 4132–4137 CrossRef CAS.
- S. S. Nadif, M. E. O'Reilly, I. Ghiviriga, K. A. Abboud and A. S. Veige, Angew. Chem., Int. Ed., 2015, 54, 15138–15142 CrossRef CAS PubMed.
- Y. Sekiguchi, F. Meng, H. Tanaka, A. Eizawa, K. Arashiba, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Dalton Trans., 2018, 47, 11322–11326 RSC.
- Z. Mo, T. Shima and Z. Hou, Angew. Chem., Int. Ed., 2020, 59, 8635–8644 CrossRef CAS PubMed.
- S. Dey and T. K. Hollis, Inorganics, 2021, 9, 15 CrossRef CAS.
- P. Zatsepin, E. Lee, J. Gu, M. R. Gau, P. J. Carroll, M. H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2020, 142, 10143–10152 CrossRef CAS PubMed.
- T. Kurogi, J. Won, B. Park, O. S. Trofymchuk, P. J. Carroll, M. H. Baik and D. J. Mindiola, Chem. Sci., 2018, 9, 3376–3385 RSC.
- D. P. Solowey, M. V. Mane, T. Kurogi, P. J. Carroll, B. C. Manor, M. H. Baik and D. J. Mindiola, Nat. Chem., 2017, 9, 1126–1132 CrossRef CAS PubMed.
- T. Kurogi, B. Pinter and D. J. Mindiola, Organometallics, 2018, 37, 3385–3388 CrossRef CAS.
- T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed.
- B. Wang, G. Luo, M. Nishiura, S. Hu, T. Shima, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2017, 139, 1818–1821 CrossRef CAS PubMed.
- K. Ma, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2006, 128, 3303–3312 CrossRef CAS PubMed.
- J. Gordon, S. Hildebrandt, K. R. Dewese, S. Klare, A. Gansäuer, T. V. RajanBabu and W. A. Nugent, Organometallics, 2018, 37, 4801–4809 CrossRef CAS PubMed.
- N. Tsoureas, J. C. Green and F. G. N. Cloke, Dalton Trans., 2018, 47, 14531–14539 RSC.
- E. G. Perevalova, I. F. Urazowski, D. A. Lemenovskii, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1985, 289, 319–329 CrossRef CAS.
- T. J. Hebden, R. R. Schrock, M. K. Takase and P. Müller, Chem. Commun., 2012, 48, 1851–1853 RSC.
- T. J. Hebden, A. J. St. John, D. G. Gusev, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, Angew. Chem., Int. Ed., 2011, 50, 1873–1876 CrossRef CAS PubMed.
- H. J. Reich, Chem. Rev., 2013, 113, 7130–7178 CrossRef CAS PubMed.
- J. S. Ritch, D. Julienne, S. R. Rybchinski, K. S. Brockman, K. R. D. Johnson and P. G. Hayes, Dalton Trans., 2014, 43, 267–276 RSC.
- A. Pape, M. Lutz and G. Müller, Angew. Chem., Int. Ed. Engl., 1994, 33, 2281–2284 CrossRef.
- C. Idelson, L. Webster, T. Krämer and F. M. Chadwick, Dalton Trans., 2020, 49, 16653–16656 RSC.
- I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- N. Serpone, P. H. Bird, D. G. Bickley and D. W. Thompson, J. Chem. Soc., Chem. Commun., 1972, 217–218 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- N. Rahimi, B. de Bruin and P. H. M. Budzelaar, Organometallics, 2017, 36, 3189–3198 CrossRef CAS.
- P. L. A. Popelier and G. Logothetis, J. Organomet. Chem., 1998, 555, 101–111 CrossRef CAS.
- W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782–1806 CrossRef CAS PubMed.
- R. H. Duncan Lyngdoh, H. F. Schaefer and R. B. King, Chem. Rev., 2018, 118, 11626–11706 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2204753–2204761. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03291k |
| This journal is © The Royal Society of Chemistry 2022 |