
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface chemistry of metal–organic polyhedra
Jorge
Albalad
 *a,
Laura
Hernández-López
b,
Arnau
Carné-Sánchez
*b and
Daniel
Maspoch
*bc
*a,
Laura
Hernández-López
b,
Arnau
Carné-Sánchez
*b and
Daniel
Maspoch
*bc
aCentre for Advanced Nanomaterials and Department of Chemistry, The University of Adelaide, North Terrace, Adelaide, SA 5000, Australia. E-mail: jorge.albalad@adelaide.edu.au
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC, Barcelona Institute of Science and Technology, Bellaterra 08193, Barcelona, Spain. E-mail: arnau.carne@icn2.cat; daniel.maspoch@icn2.cat
cICREA, 08010 Barcelona, Spain
First published on 25th January 2022
Abstract
Metal–organic polyhedra (MOPs) are discrete, intrinsically-porous architectures that operate at the molecular regime and, owing to peripheral reactive sites, exhibit rich surface chemistry. Researchers have recently exploited this reactivity through post-synthetic modification (PSM) to generate specialised molecular platforms that may overcome certain limitations of extended porous materials. Indeed, the combination of modular solubility, orthogonal reactive sites, and accessible cavities yields a highly versatile molecular platform for solution to solid-state applications. In this feature article, we discuss representative examples of the PSM chemistry of MOPs, from proof-of-concept studies to practical applications, and highlight future directions for the MOP field.
 Jorge Albalad | Jorge Albalad is an ARC-funded Postdoctoral Researcher under the supervision of Prof. Christian J. Doonan and Christopher J. Sumby at the University of Adelaide (Australia). He received his BSc degree in Chemistry at the Autonomous University of Barcelona (UAB) in 2014. Then, he completed his MSc in Industrial Chemistry and Introduction to Chemical Research (2015) and his PhD in Chemistry (2019) at the same university, conducting his research at the ICN2. His research focuses on the post-synthetic modification of metal–organic entities, with particular emphasis on the influence of molecular-level transformations to the macroscopic properties. |
 Laura Hernández-López | Laura Hernández López is currently pursuing a PhD in Chemistry at the ICN2 under the supervision of Prof. Daniel Maspoch and Dr. Arnau Carné. She received her BSc degree in Nanoscience and Nanotechnology at Autonomous University of Barcelona (UAB) in 2017. In 2019, she got her MSc in Applied Materials Chemistry from University of Barcelona (UB). Her research interest lies in understanding the reactivity and assembly of discrete metal–organic polyhedra in solution. |
 Arnau Carné-Sánchez | Arnau Carné-Sánchez is a “La Caixa” Junior Leader fellow at ICN2. He received his BSc degree in Chemistry from the Universitat de Barcelona (UAB) in 2008. In 2014 he got his PhD in Chemistry from the UAB under the supervision of Prof. Daniel Maspoch and Dr. Inhar Imaz. In 2014 he joined Prof. Susumu Kitagawa and Prof. Shuhei Furukawa's group in iCeMS at Kyoto University (Japan) under a JSPS postdoctoral fellowship. He is currently researching the synthesis and reactivity of nano-sized molecular porous materials. |
 Daniel Maspoch | Daniel Maspoch is an ICREA Research Professor at the Institut Català de Nanociència i Nanotecnologia (ICN2). He received his BS degree at the Universitat de Girona and his PhD degree at the Universitat Autònoma de Barcelona & Institut de Ciència de Materials de Barcelona. He worked as a postdoctoral fellow at Northwestern University. His research interests include reticular materials (MOFs, COFs and MOPs) and delivery systems. |
1. Introduction
Research on porous materials dates back centuries but has only bloomed over the past few decades. This is down to two reasons: growing interest in their physical properties, and the ability to rationally design them via bottom-up methods.1–3 A new generation of porous materials such as Metal–Organic Frameworks (MOFs)4,5 and more recently, Covalent–Organic Frameworks (COFs)6,7 has garnered interest from both academia and industry, owing to their modular structures with well-defined microporous environments.8 These materials can be designed reticularly with bespoke structural features (e.g., pore sizes, windows, and inner functionalisation) that, together with strong bonding among constituent components,9 define the mechanical, chemical, and topological attributes of a given framework.10–12 Additionally, the structured building units retain a chemical reactivity similar to that of their molecular counterparts; therefore, the pore domains of framework can be further modified with specific functionalities by post-synthetic modification (PSM).13–15 By applying PSM to porous materials, researchers have been able to blend specialised materials with previously unobtainable properties. Indeed, by bypassing the solvothermal synthetic conditions typically used for porous materials such as MOFs, scientists can now incorporate highly-reactive and sensitive moieties into these materials.16–18Recently, investigators have extended PSM to Metal–Organic Polyhedra (MOPs), which are discrete, intrinsically-porous architectures assembled through metal–linker coordination bonds, similarly to MOFs.19,20 MOPs are an underrepresented family of porous materials whose unique attributes are not found in extended frameworks. Unlike their extended analogues, MOPs are generally bound in the solid state by weak cage-to-cage interactions that can easily be disrupted to dissolve them.21 Thus, while these materials generally lack the long-range order effects that confer MOFs with robustness and high surface areas, researchers can exploit their molecular nature to access a wide library of solution-based PSM chemistry to precisely control their solubility, processability, and solid-state packing.22–24 This in turn can generate unique porous materials with well-defined surfaces poised for specific applications in homogeneous and interfacial regimes, where long-range order and diffusion kinetics have proven detrimental.25,26
In this feature article, we explain the influence of PSM on MOPs and their molecular chemistry, highlighting the interesting properties conferred to MOPs via PSM that are not found in other common porous materials. Whilst MOPs have attracted some interest as potential building blocks for the synthesis of extended materials,27–29 that is beyond the scope of this article. In the first section of the present article, we cover the fundamental structural features of MOPs, aiming to facilitate visualisation of their surface chemistry for the subsequent sections. Next, we discuss the principal challenges that researchers face for PSM of MOPs at their metal nodes and at their organic linkers, and cite significant milestones in the chemistry at each site. In these sections, we mention examples of conceptual and practical value. Finally, we look towards the future, identifying what we believe to be the most important paths to follow for the next decade, in terms of translating the wealth of chemical knowledge that has been amassed to date into the design of porous soluble platforms with unique functionalities.
2. Morphology and structural features of prototypical metal–organic polyhedra
Structurally, MOPs are similar to the well-known coordination cages devised by the groups of Fujita,30,31 Stang,32,33 and others. Thus, MOPs can be considered a subclass of such cages, distinguished by showing permanent porosity in the solid state and for their strong, typically metal–carboxylate, coordination bonds. Just like their extended MOF analogues, MOPs exhibit rich and versatile structural chemistry. However, the polyhedral nature of MOPs provides structural attributes that are not found in MOFs: (i) a single internal cavity, accessible through the cage's windows; (ii) two well-defined surfaces (internal and external); (iii) finite, directional, orthogonal reactive sites throughout both surfaces (i.e. axial metal sites and organic functionalities); and (iv) modular solubility.34 Together, these features confer MOPs with exquisite reactivity, matching that of MOFs, albeit within the molecular solution regime. Thus, MOPs can operate as highly-processable soluble hollow nanoparticles,35 where the finite number of reactive sites enables stoichiometric control of their surface reactivity as well as reversible transition from solution phase to packed phase.36,37 Consequently, the functionalisation of MOPs can be characterised both in solution (e.g. by NMR, mass-spectrometry, DLS) and the solid-state (e.g. by X-ray diffraction/crystallography, infrared spectroscopy, gas sorption).We recognise that ensemble of structural details that we have provided on MOPs so far may be difficult to visualise for some readers, especially for those new to the field. Thus, in this section, we provide a graphic representation of all the features that we described above. Fig. 1 depicts the structure of a MOP from the prototypical M24L24 family, arguably the most studied architecture to date.38 It comprises 12 divalent M–M paddlewheel clusters and 24 angular aromatic linkers perfectly spaced throughout the material, converging into a cuboctahedral shape with an intrinsic inner cavity (diameter: 2.5 nm). The rigidity and directionality imposed from both subunits render a distinct geometry in which all vertices, edges, and facets can be chemically elucidated with atomic-level precision by adapting the principles of reticular chemistry.39 Within the cage shown in Fig. 1, one can clearly depict both an internal (blue) and external (red) MOP surface. These surfaces can be targeted separately during the design, self-assembly, and PSM of the cage. The inner surface is tagged with 12 spaced coordination centres that stem from the axial sites of each paddlewheel cluster. Likewise, 24 distinct organic functionalities point inwards from position 2 in the aromatic ligand (bottom, left) and heavily influence both the hydrophobicity and sterics of the inner cavity. The inner metal sites and the organic functionalities can only be accessed by substrates small enough to fit through the two windows (diameters: 4.5 Å and 6.6 Å) in the MOP structure (right), so that these sites/functionalities are mainly targeted towards adsorption/separation of small substrates or gas-phase molecules.40 Alternatively, the external surface contains 12 independent axial metal sites, and the organic linkers confer this surface with up to 72 different functional sites that stem from positions 4, 5 and 6 of the aromatic linkers (top, left), which dictate the solubility of the cage. Consequently, if for simplicity, one limits the total permutation to a single reaction per position, then this one cage could contain up to 36 moieties on its internal surface and up to 84 moieties at its external one. Importantly, each moiety is perfectly defined, both directionally and structurally, within the cage's backbone.
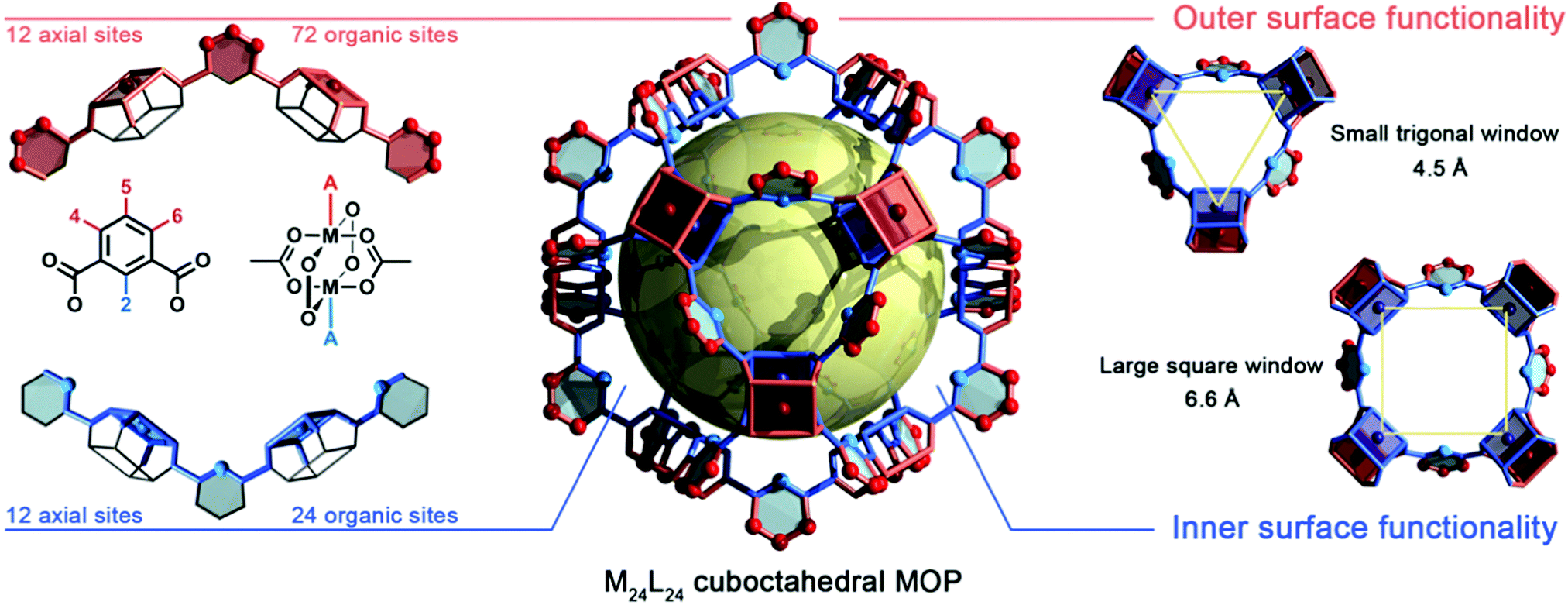 | ||
| Fig. 1 Schematic representation of the internal and external orthogonal reactive sites within the archetypical M24L24 metal–organic polyhedra. Left: The external surface (red) contains twelve axial open-metal sites that stem from the dimetallic M–M paddlewheel SBUs, as well as up to 72 organic functional groups from positions 4, 5, and 6 of the linker. The internal surface (blue) contains 12 axial open-metal sites and 24 organic functionalities that stem from position 2 of the linker. Right: The internal cavity of the cage (2.5 nm; yellow sphere) is accessible through two distinct windows: a trigonal one (diameter: 4.5 Å) and a square-shaped one (diameter: 6.6 Å). | ||
The aforementioned structural analysis can be extended to the entire repertoire of (over fifteen) reported MOP structures, each of which contains distinct metal nodes, linkers, functional groups, and cavities. Representative MOPs include the lantern-type (M2L4),41,42 tetrahedral (M4L4X+),43,44 and octahedral (M6L12)45,46 families, among other, more complex variants with multiple components.26 Thus, researchers now have access to numerous possible molecular platforms with very distinct attributes that offer potential for development of smart soluble materials with molecular-level precision. Importantly, each reactive site can be modified via PSM to exponentially increase the number of variables that scientists can tune to generate a massive library of functionally specialised MOPs for countless applications in solution or solid phase.
3. Post-synthetic modification of metal–organic polyhedra
Reflecting on the impressive features of MOPs, we are shocked that their PSM has remained rather underrepresented in the literature, and that MOPs have been overshadowed by other competing porous materials. Nevertheless, we recognise that this may be partially explained by certain drawbacks inherent to the earliest MOPs. Foremost among these is the structural fragility exhibited by the first generation of Cu(II)-based MOPs, which made them prone to degradation by moisture, heat, nucleophiles, or acids. Accordingly, for years, researchers focused on shielding MOPs within confined environments,47–49 which meant that they could learn very little about the surface chemistry of these materials.In the following sections, we review the most remarkable examples of surface functionalisation of MOPs at both the internal and external surfaces, grouping the examples by the subunit targeted and the type of chemistry employed in each case (Fig. 2). We underscore how the development of alternative modification protocols, and the assembly of more-robust cages—including those of MOPs based on Rh(II), Cr(II), Mo(II), or Zr(IV)—have together enabled researchers to use more-attractive chemistries to develop functional MOP platforms for diverse applications.
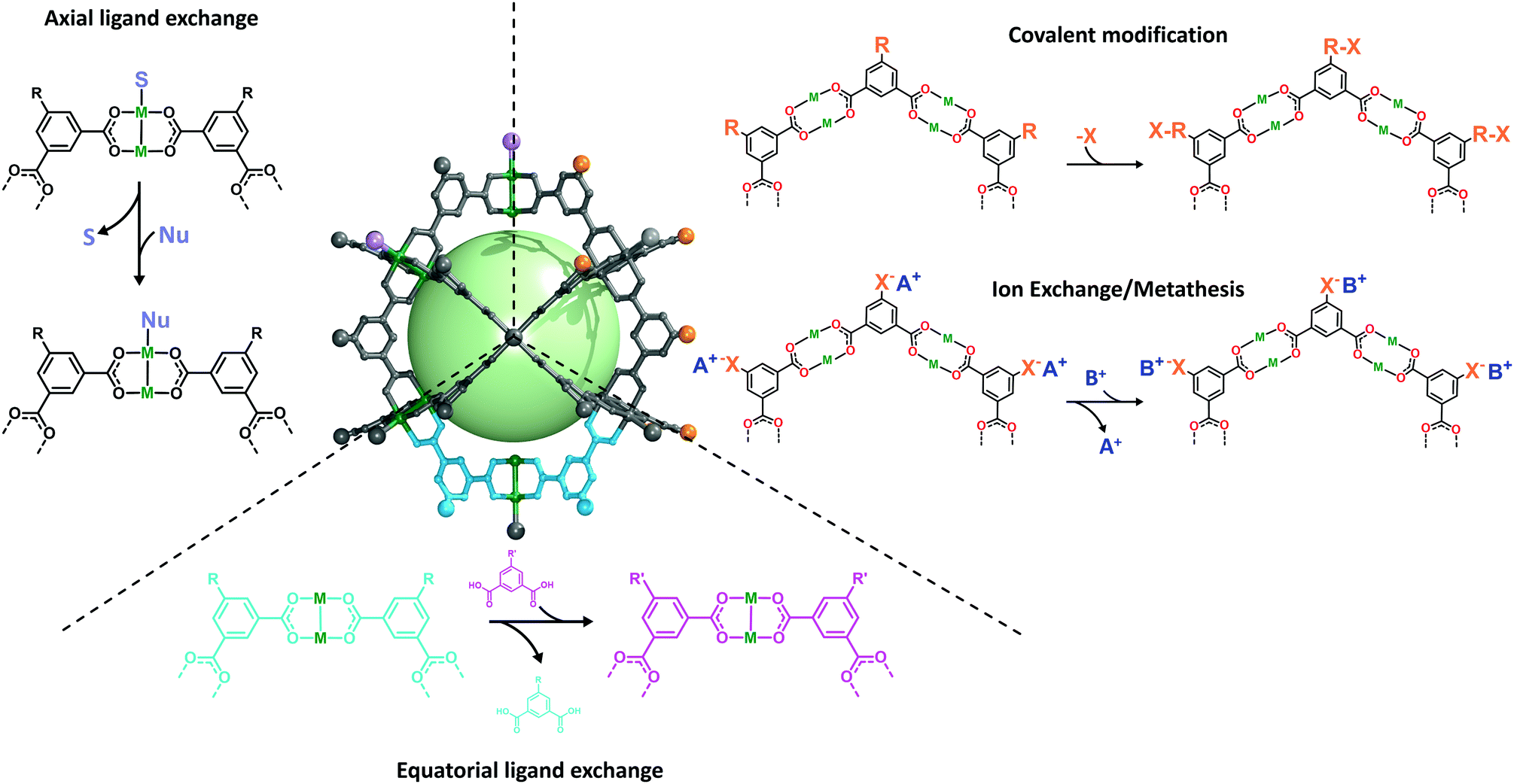 | ||
| Fig. 2 Schematic representation of the surface reactivity of the archetypical M24L24 metal–organic polyhedra. S refers to solvent and Nu refers to a nucleophile. | ||
3.1. Post-synthetic modification of MOPs at their metal centres
Unlike most molecular platforms, MOPs present two defined domains at their periphery: one comprising organic groups from the linkers; and one comprising inorganic groups from the coordination clusters that extend throughout the lattice. In the latter, one can distinguish two distinct types of positions around the metal nodes: one type, which helps hold the cage together; and another type (open-metal site), which does not, but is available and is prone to coordinate extraneous nucleophiles. For example, in a four-connected, dimetallic paddlewheel node, there are four equatorial coordination positions (M-COO) that are directly involved in the MOP formation, and two axial labile sites that generally accommodate weakly-bound solvent molecules. Both types of positions are amenable to PSM. Importantly, they can be targeted independently under distinct reaction conditions.50Zhou and co-workers were the first to demonstrate the feasibility of reacting the equatorial positions in Cu(II)-based paddlewheel MOPs through a ligand-exchange approach.51 These positions are directly implicated in the coordination-driven self-assembly of MOPs under solvothermal conditions. Such assembly requires a certain degree of bond lability, as it facilitates the formation of thermodynamically-favoured, defect-free assemblies.52,53 Certainly, the equatorial metal–linker bonds are not “blocked” after formation of the MOP, and their dynamic reactivity can be further exploited through PSM (Fig. 3a). The authors harnessed the reversible nature of their MOPs, successfully exchanging the initial MOP linkers with structurally-diverse dicarboxylate variants to obtain a set of functionalised isostructural MOPs. The successful exchange was based on two major driving forces that include the equilibrium displacement with saturated linker solutions and the precipitation of the products from solution. Moreover, they were able to change the cage topology, by selecting a substituting ligand with an appropriate bending angle for ligand-exchange. Thus, they first converted their initial lantern-type cages to cuboctahedral cages, which they subsequently converted to octahedral cages (Fig. 3b).
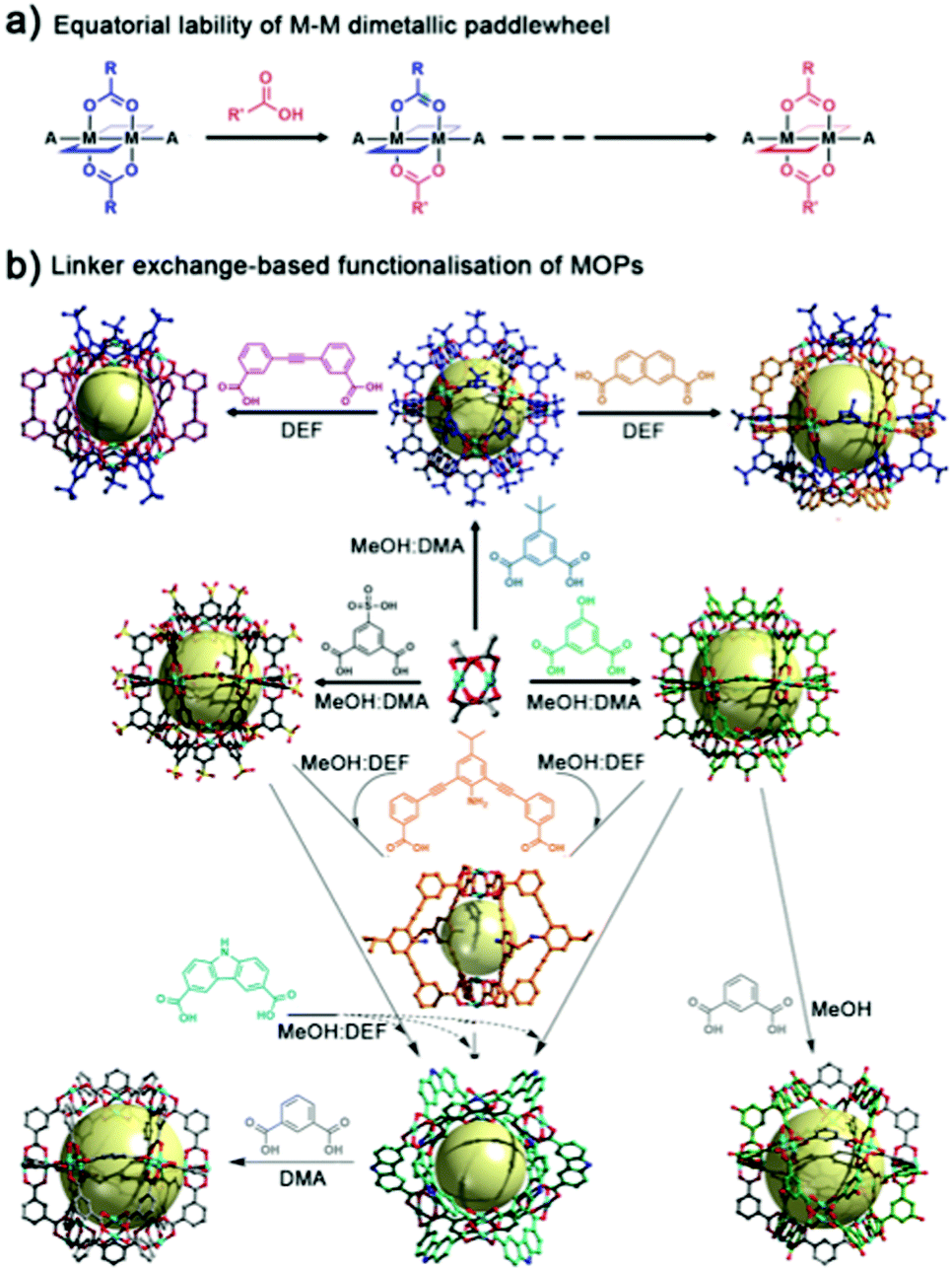 | ||
| Fig. 3 (a) Schematic of the equatorial lability of dimetallic M–M paddlewheel nodes. (b) The combination of Cu(Ac)2 and the corresponding linker in mixed solvents at room temperature affords multiple Cu-based MOPs with labile equatorial positions. To modify the external surface of the resultant MOPs, the labile bonds can be subjected to linker-exchange chemistry. This entails mixing an excess of the new ligand(s) with the (soluble) parent MOP in a suitable solvent at room temperature. Adapted with permission from ref. 51. | ||
Unfortunately, performing PSM on the equatorial positions in paddlewheel MOPs threatens the chemical stability of the structure, as uncontrolled ligand substitutions, whether partial or complete, can induce hydrolytic or defect-promoted degradation.54,55 Accordingly, for PSM of MOPs at their inorganic sites, researchers have prioritised targeting the accessible axial sites under conditions that do not interfere with the cage's integrity. Even the simplest cage containing paddlewheel nodes features accessible open-metal sites that exhibit a strong affinity towards electron-donating nucleophiles and, consequently, can accommodate those nucleophiles within its structure. These PSMs entail ligand-exchange in solution, as said axial positions are inherently occupied by solvent molecules. When electron-donating nucleophiles with higher affinities compete for these coordination sites, the solvent molecules of poor nucleophilicity can be replaced. Therefore, the exploitation of open-metal sites in MOPs is strongly influenced by the solvent in which the MOPs have been dissolved, which can drastically affect the properties and consequently, the reactivity of solvated cages both in solution and in the solid state. Recent work highlights the strong influence of coordinating solvent molecules on the sorption properties42,56 and crystal packing57 of MOPs, and illustrates how the same cage can exhibit drastically different attributes owing to its intrinsic solvatomorphism. Likewise, the coordination-based reactivity of MOPs with accessible open-metal sites can be either enhanced or completely masked by carefully selecting the reaction medium. Strongly coordinating solvents (e.g. DMSO or DMF) generally occlude the unoccupied nodes in the cage, which might restrain their reactivity. Fortunately, this occlusion is not permanent: coordinated solvent molecules can be replaced by a strategically-chosen coordinating moiety of interest that will promote the formation of stronger coordination bonds. Non-coordinating solvents (e.g. alkanes or alcohols) will augment the reactivity of the cage due to their weak interaction with the open-metal sites, but might induce the self-aggregation of the cages in solution. Overall, researchers seeking to maximise the potential of MOPs as molecular platforms should first anticipate the influence of the solvent(s) to be used before studying the reactivity of the MOP(s) in question.
The examples that we cited above laid the foundation for the open-metal site chemistry and reactivity of MOPs. Unfortunately, the lability of Cu(II)-, Zn(II)-, and Co(II)-based nodes in solution has precluded further work on the potential of their coordinative binding-sites. Interestingly, researchers have extensively demonstrated that the presence of mild N-donor nucleophiles (e.g. primary amines, pyridine derivatives, or imidazole derivatives) induces the decomposition of certain MOPs into discrete clusters, via ligand-exchange between the N-donor and MOP bridging-ligands.54 Accordingly, investigators have recently begun to focus on assembling more-robust MOP architectures built from non-labile, equatorial, metal–linker bonds. Among these, we would like to highlight here the rich surface-functionalisation chemistry of Rh(II)-based MOPs that are isostructural to the archetypical Cu(II) paddlewheel analogues. Rh(II)-based MOPs were first presented by the groups of Su (in 2015) and Furukawa (in 2016), who reported unprecedented thermal and chemical robustness in these materials.58,59 This enhanced stability relies on a combination of chemically-inert equatorial positions in the paddlewheel, and strong intermetallic Rh–Rh bonds. Remarkably, although the equatorial position of the dirhodium paddlewheel is “chemically locked”, its axial metal sites remain highly reactive, albeit without compromising the integrity of the nodes. Thus, these sites can readily accommodate a plethora of coordinative nucleophiles, the most-widely studied of which are N-donors, although nucleophiles with other donor atoms (e.g. S-donors, P-donors, O-donors) can establish similar coordination bonds. The pioneering work of the Su and Furukawa groups thus paved the way for researchers to explore molecular functionalisation of the open-metal sites of MOPs.
Our (Maspoch) group and the Furukawa group were the first to interrogate the axial sites in Rh(II)-based MOPs and established the basic chemistry of these understudied platforms. Our research confirmed that up to twelve pyridine-based linkers could be selectively attached to the external axial Rh–Rh sites of the cage. Thus, we demonstrated that these porous molecular platforms could be functionalised at their axial sites with diverse coordinating linkers of different polarity, charge, and chirality. Consequently, certain intrinsic physicochemical properties of these materials (i.e. solubility, hydrophilicity, and chirality) could be selectively modulated in a controlled and reversible way, without compromising their inner porosity. Through this approach, it was possible to modulate the solubility of multiple Rh(II)-based MOPs in a broad spectrum of solvents, encompassing non-polar (diethyl ether), halogenated (DCM & chloroform), polar aprotic (DMF & DMSO), and protic (H2O & alcohols) solvents, by tagging the open-metal sites with suitable pyridine derivatives. Interestingly, the hybrid nature of MOP surfaces confers these materials with well-defined orthogonal positions that can be targeted independently and reversibly without compromising the other active sites in the cage, whether internal or external (Fig. 4).24 This approach allows for further chemistry at the MOP surface, which would otherwise be precluded by the insolubility of the parent MOP in the desired solvent. As proof of concept, we covalently functionalised an OH-functionalised cuboctahedral Rh(II)-based MOP (named OH-RhMOP, of formula [Rh2(5-OH-bdc)2]12, where 5-OH-bdc = 5-hydroxy-1,3-benzenedicarboxylate) in a solvent in which the cage was insoluble and thus, unreactive. By selectively coordinating twelve suitable pyridine derivatives to the axial Rh–Rh sites, we were able to dissolve the cage in the optimal reaction solvent for subsequent etherification of the hydroxyl groups with allyl bromide. Next, we selectively removed the coordination-based solubilising groups from the axial sites by a mild acid wash to obtain the functionalised material as a precipitate.
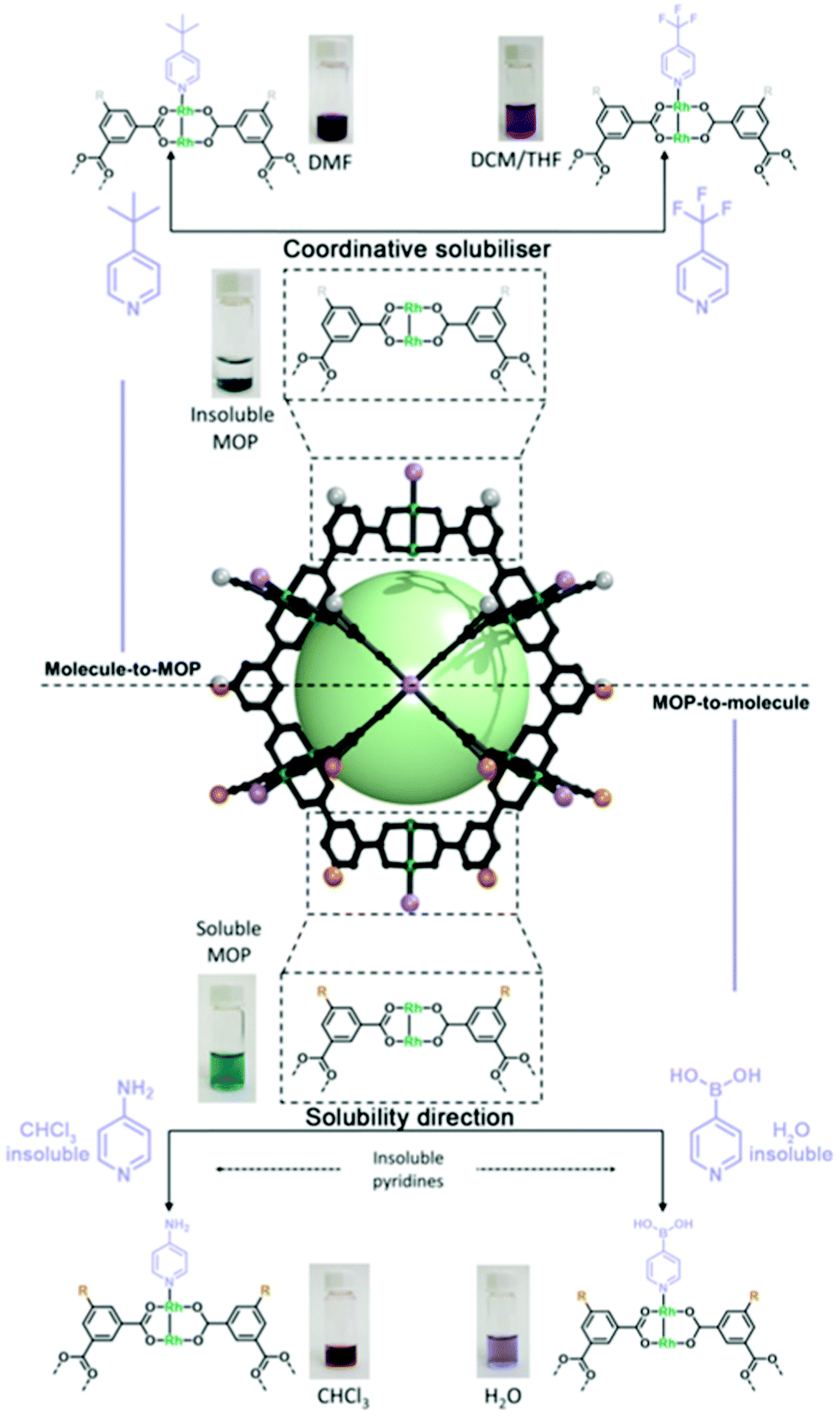 | ||
| Fig. 4 Schematic of the molecule-to-MOP and MOP-to-molecule transfer of properties via coordination of organic molecules to the external axial sites of Rh(II)-based MOPs. Top: The solubility of insoluble MOP platforms can be modulated via a coordinative solubiliser method, whereby the anchoring of (up to twelve) pyridinyl nucleophiles modifies the surface properties. Bottom: Conversely, the solubility of organic substrates can be modified by anchoring them to the surface of suitable MOP platforms that have well-defined solubility profiles. | ||
Just as the properties of a MOP cage can be tuned by attached coordinated moieties, as explained above, the opposite is also true: certain MOP properties can be transferred to surface-coordinated molecules. Accordingly, MOP surfaces can be coordinatively functionalised for reasons other than modulation of physicochemical properties of the MOP. For example, MOP-to-molecule transfer of properties can be exploited to direct the solubility of important organic substrates by coordinating them to a suitable MOP platform, such that the entire MOP-bound substrate molecules dissolve in a media where the substrate molecules alone would normally be insoluble. This approach is best suited for MOPs that contain pendant organic groups that can heavily influence their solubility profile, such as long aliphatic chains or charged polar groups, despite the coordination of small molecules to the axial sites. In 2021, this technique was implemented for the solubilisation of strongly hydrophobic or hydrophilic pyridines into aqueous or non-polar phases, respectively. Specifically, we used fully-deprotonated OH-RhMOP (named ONa-RhMOP; vide infra), a water-soluble species, to dissolve the strongly hydrophobic substrate 4-pyridinylboronic acid in water. Conversely, we used a hydrophobic MOP with 24 aliphatic chains (named C12-RhMOP) to dissolve the strongly hydrophilic substrate 4-aminopyridine into CHCl3 (Fig. 4).60 This strategy can be implemented into separation systems, as the MOP-bound substrate molecules can be easily separated from the corresponding unbound substrate molecules, thanks to its differentiated solubility. Therefore, molecules can be separated according to their capacity to coordinate to Rh(II)-based MOPs, which is influenced by the basicity and steric hindrance of their constituent coordinating heteroatoms.61 This approach could prove highly valuable for diverse applications, including currently low-yielding catalytic reactions that are limited by solubility restraints of the reagents and/or catalysts. Thus, we believe that further refinement of this coordinative solubilising methodology will enable researchers to surpass the solubility constraints in homogeneous organic chemistry.
3.2. Post-synthetic modification of MOPs at their organic linkers
Having overviewed the PSM of MOPs at their metal centres, we now turn to their organic linkers. From a strictly chemical perspective, soluble coordination cages should not behave any differently than the simplest aromatic molecule: in both cases, the organic reactivity is shaped by the presence of functional groups. Thus, the richer the reactivity of those appendices, the broader the scope of possible bond formation and the wider the range of accessible surface-modification techniques. Unfortunately, the chemistry of MOPs is not always as straightforward as that of organic molecules: consequently, PSM of MOPs at their organic sites faces two major hindrances that arise from the supramolecular nature of the MOP backbone. Firstly, the functional groups incorporated within the linkers must be able to tolerate the conditions to synthesise the cage, which can range from room temperature crystallisation to solvothermal couplings in basic media.62 Secondly, the reaction conditions available for covalent functionalisation are restricted to those that do not affect other elements in the structure, particularly the labile equatorial M–COO bonds.54 Fortunately, organic chemistry is replete with reactions to reliably assemble, cleave, or rearrange covalent bonds under mild conditions that can be safely applied to fragile MOP platforms. Most of the reported covalent PSMs of MOPs have revolved around condensation reactions (e.g. formation of ester, imine, or amide bonds),57,63,64 click chemistry (azide-alkyne cycloadditions),65,66 or alkene polymerisation.22,67,68One of the first examples of the covalent PSM of a MOP was reported in 2010 by the group of Zhou, who performed a Huisgen cycloaddition reaction between an alkyne-functionalised cuboctahedral Cu(II)-based MOP (named C2H2-CuMOP, of formula [Cu2(5-C2H2-bdc)2]12, where 5-C2H2-bdc = 5-ethynyl-1,3-benzenedicarboxylic acid) and azide-terminated polyethylene glycol (PEG) chains. Despite the fragility of the mother cage in aqueous media, the combination of reagents in situ led to formation of water-stable, PEG-grafted cages with up to four hydrophilic, polymeric, PEG side-chains at the periphery.65 Intriguingly, the authors later employed these PEG side-chains to encapsulate and release anti-cancer drugs in physiological media. In 2016, Kitagawa et al. reported the first-ever quantitative surface-functionalisation (24 positions) of another Cu(II)-based cuboctahedral MOP, using dithiobenzoate or trithioester moieties (of formula [Cu2(5-Z-SCS-bdc)2]12, where 5-Z-SCS-bdc = 5-dithiobenzoate-1,3-benzenedicarboxylic acid, or 5-trithiobutane-1,3-benzenedicarboxylic acid, respectively). They grafted up to 24 polymeric arms around the MOP core by direct coupling with monomeric building blocks via soft Reversible-Addition-Fragmentation Chain Transfer (RAFT) polymerisation, without compromising the integrity of the cage. These results offered a rational strategy for the synthesis of highly-processable MOP-based star polymer materials with controlled grafting of multiple side-chains.69
Although researchers can access new and exciting MOP reactions through mild covalent chemistry, if they truly wish to exploit MOPs as functional platforms, then they must address the lability of M–COO equatorial bonds. Indeed, although a diverse array of MOPs derivatised with broadly reactive moieties (e.g. hydroxyl, alkyl, or alkynyl groups) is now available, only a mere fraction of their potential reactivity can be harnessed for surface chemistry due to stability concerns. In particular, the chemistry of hydroxyl-functionalised MOPs is limited by their instability under basic conditions, which are the most common reaction conditions for quantitative formation of ether or ester bonds. In fact, the cuboctahedral OH-CuMOP suffers spontaneous phase-transition upon exposure to N-based bases or even simply to water;70 consequently, it has not been subjected to deprotonation or PSM by this type of chemistry at its surface. Interestingly, this limitation was circumvented by reacting a hydroxyl-functionalized lantern-type Cu-MOP (named OH-CuMOP(L), of formula [Cu2(OH-L)2]2, where OH-L= 3,3′-(5-hydroxy-1,3-phenylene)-bis(ethyne-2,1-diyl)dibenzoic acid) with hydrophobic alkyl anhydrides in the presence of an esterification catalyst at room temperature in DMF. Using these very mild conditions, the authors were able to quantitatively transform the pendant hydroxyl groups of OH-CuMOP(L) into alkyl-ester side-chains, which drastically enhanced the solubility and stability of the cage in organic solvents.71
Adamant to overcome the aforementioned limitations of OH-functionalised MOPs, our group, in close collaboration with the group of Furukawa, investigated the chemistry of the Rh(II) analogue, OH-RhMOP.59 Specifically, we explored the possibility of functionalising OH-RhMOP under standard interfacial conditions (biphasic system made of aqueous and non-polar organic solvents) for esterification and etherification (Fig. 5a).24 Remarkably, OH-RhMOP endured the quantitative deprotonation of its hydroxyl groups with excess NaOH, which generated a water-soluble platform with 24 negative charges at its periphery. We then coupled this charged platform with various organic moieties (acyl chlorides, anhydrides, alkyl halides, etc.) in high yields, under conditions that would completely disintegrate any Cu(II)-based analogue. More recently, the group of Bloch applied similar chemistry to Cr(II) and Mo(II)-based MOPs to obtain functionalised platforms that would be inaccessible via direct synthesis.63
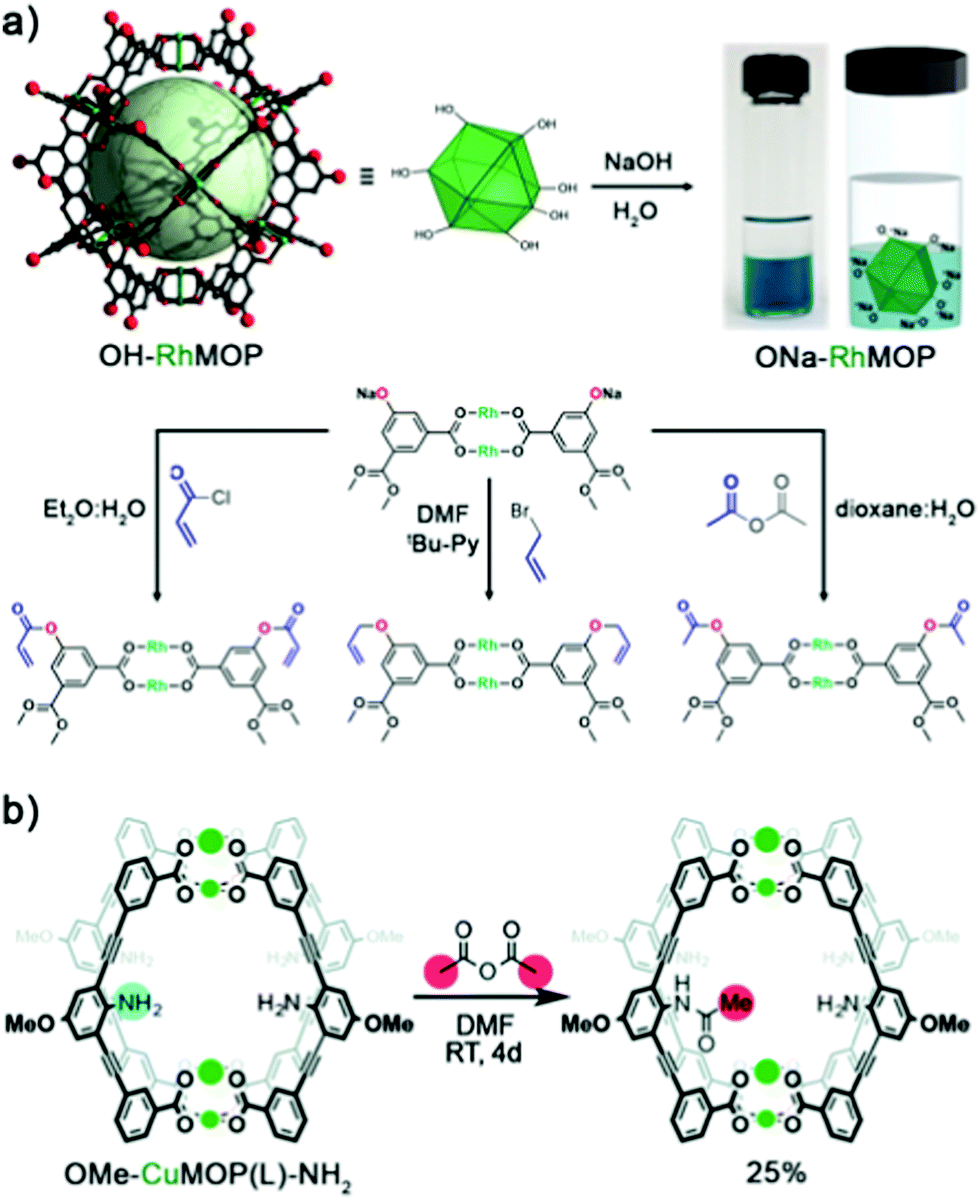 | ||
| Fig. 5 Covalent PSM of OH-RhMOP under interfacial conditions. Reacting OH-RhMOP with at least 24 mol eq. of NaOH in H2O affords the quantitative deprotonation of the terminal hydroxyl groups within the cage, forming a 24-charged, water-soluble species. ONa-RhMOP reacts under interfacial conditions (aqueous/non-polar organic solvents), or under pyridine-assisted solution, with multiple substrates to afford PSM of the cage in high yields. (b) Synthesis and PSM of internal NH2 moieties in the lantern-type OMe-CuMOP(L)-NH2. Despite the nucleophilicity of the amine groups, the cage can be assembled without having to mask the reactive sites, thanks to steric constraints. After cage assembly, the free NH2 groups retain their reactivity, and can be subsequently reacted with acetic anhydride to afford the mono-substituted amide product; further amide formation is prevented by sterics. Reproduced (adapted) with permission of ref. 24 (Copyright 2019 American Chemical Society) and 72 (Copyright Royal Society of Chemistry). | ||
To further enhance the versatility of MOPs, researchers must develop alternative pathways to expand the catalogue of surface functional groups, especially those that are normally incompatible with the conditions for cage synthesis. For example, linkers with free coordinating moieties such as primary amine (–NH2) or carboxylic acid (–COOH) groups have been challenging to incorporate into MOP structures due to their nucleophilicity and their affinity for metal sources, both of which can disrupt the assembly of the MOP. One way to circumvent these issues is to use ligands with sterically-hindered amine groups, which essentially renders them those non-coordinative. For instance, Klosterman and co-workers functionalised the inner cavity of a lantern-type MOP named OMe-CuMOP(L)-NH2, of formula [Cu2(L)2]2, where L = 3,3′-((2-amino-5-methoxy-1,3-phenylene)-bis(ethyne-2,1-diyl)-dibenzoic acid) via direct assembly of Cu(II) precursors and a linker with free NH2 groups. The position of the amine groups on the linker made them too sterically hindered to interfere with the Cu(II) ions during the cage assembly, yet sufficiently reactive with small molecules that had entered the lantern-type cavity. The authors demonstrated this concept by reacting the lantern cage with acetic anhydride in DMF to yield the mono-functionalised amide product. Molecular modelling corroborated the experimental results, indicating that the inclusion of a second acetate is disfavoured due to sterics (Fig. 5b).72
Unfortunately, to synthesise MOPs that contain free amines at their external surface, researchers cannot employ the steric hindrance approach described above, because of the high degree of exposure of these coordinating groups within the bent-linker structure. To date, two opposing methodologies have been developed to circumvent the nucleophilicity of free amino groups during MOP synthesis, both of which entail the use of protecting groups to temporarily mask undesired reactivity. The group of Yaghi was the first to introduce free amino groups onto the surface of MOPs, by developing a protocol that blocked the axial reactivity of Cu(II) precursors. They exploited the affinity of copper(II) acetate towards nitrogen-donors to protect the axials sites with bulky monodentate auxiliary ligands, rendering the first-ever M24L24 NH2-functionalised MOP (NH2-CuMOP, of formula [Cu2(5-NH2-BDC)2]12, where 5-NH2-bdc = 5-amino-1,3-benzenedicarboxylate) (Fig. 6a).73 More recently, our group reported the use of covalent protecting groups to incorporate sensitive moieties at the external surface of MOPs. Through a two-step synthesis involving temporary masking of the linker functional groups prior to the assembly of the cage, followed by chemoselective deprotection, we obtained the first-ever cuboctahedral Rh(II)-based MOPs with 24 appended NH2 or COOH moieties (named NH2-RhMOP and COOH-RhMOP, of formulae [Rh2(5-NH2-bdc)2]12 and [Rh2(5-COOH-bdc)2]12, respectively, where 5-COOH-bdc = 1,3,5-benzenetricarboxylate) (Fig. 6b).74 These two examples established the basis for incorporating reactive groups at the surface of MOPs independently of their stability, as the protecting group and the deprotection step can each be adapted to address concerns over chemical compatibility or stability.75,76 Indeed, this strategy was later employed by Bloch et al. to control the self-condensation of a new family of lantern Cu-MOPs with external NH2 groups, via controlled deprotection of Fmoc groups, which afforded products ranging from isolated cages to crystalline self-condensation crystalline networks.77
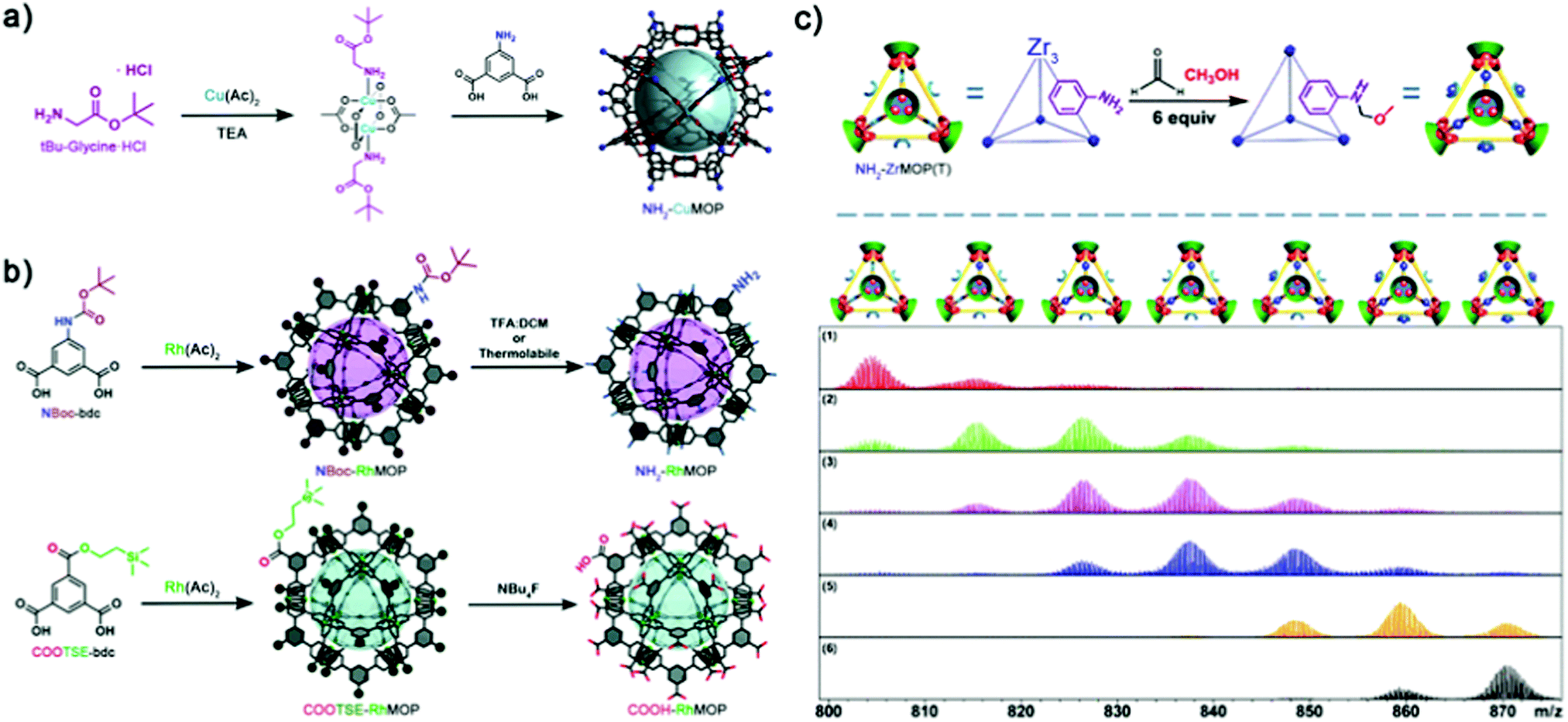 | ||
| Fig. 6 (a) Nucleophile-based masking of the axial sites of copper(II) acetate with tert-butyl glycine ester affords the synthesis of the cuboctahedric NH2-CuMOP instead of evolving to extended products. (b) Protecting group-based strategy for the synthesis of NH2- and COOH-tagged Rh(II)-based MOPs, unobtainable via direct synthesis. A selective deprotection pathway affords the quantitative deprotection of the cages without compromising their integrity, and revealing new functionality poised for further PSM steps. (c) Top: Scheme for the PSM of NH2-ZrMOP(T) with up to 6 sites per cage. Bottom: ESI-TOF-MS-based process-tracing of the PSM products of a Mannich reaction with formaldehyde and methanol after (1) 1 min; (2) 20 min; (3) 50 min; (4) 120 min; (5) 600 min; (6) 1400 min. Reproduced (adapted) with permissions of ref. 73, 74 and 78. Copyright 2008/2018 American Chemical Society – 2019 Royal Society of Chemistry. | ||
An alternative way to develop NH2-tagged MOPs is based on using hard-metal sources with no affinity for such reactive groups (e.g. Zr(IV)-based MOPs). In this case, exploitation of surface NH2 groups enables the use of PSM pathways with higher degrees of stoichiometric control, since the risk of self-condensation is precluded. In 2018, Yuan, Zhao and colleagues published the first process-tracing study on the stepwise PSM of a NH2-tagged Zr(IV)-based MOP (NH2-ZrMOP(T), of formula [Zr3(CP)3(2-NH2-bdc)]44+, where CP = cyclopentadienyl and 2-NH2-bdc = 2-amino-1,4-benzenedicarboxylate). The authors employed ESI-TOF Mass Spectrometry to unambiguously determine the molecular formula of their cages at different stages of PSM, which comprised Mannich reaction of the free NH2 groups with formaldehyde and methanol. The mass spectra confirmed the sequential stoichiometric formation of amino–methyl methoxy side-chain products throughout the PSM, from a mono-functionalised cage to a quantitatively tagged (six side-chains) one (Fig. 6c).78 Importantly, the authors not only contributed to the field by elucidating stoichiometric control over MOP surface chemistry, but also achieved a milestone towards the processability and manipulation of these materials by controlled switching from the solid-state to solution via PSM.
Finally, we would like to highlight that covalent PSM is not the only post-synthetic strategy that can be applied to modify the chemistry of MOP linkers. As in MOFs, the introduction of stimuli-sensitive moieties in the cage backbone in MOPs can yield platforms whose structure responds stimuli such as changes in temperature, light, or solvent.79 In 2014, Zhou et al. synthesised the first example of stimuli-responsive MOP (srMOPs), which they functionalised with azobenzene groups. The cage exhibited UV-irradiation-induced isomerisation from the insoluble trans-conformer to the soluble cis-conformer, whereas irradiation with blue light reversed this process to precipitate out the trans-conformer. Impressively, the authors were able to trap guest molecules inside the srMOPs, and then selectively release the guest molecules upon cis-to-trans and trans-to-cis isomerisation, laying the groundwork for a new class of optically-responsive soluble platforms.80 More recently, Bloch et al. exploited this concept to engineer a series of self-sorting lantern-type cages composed of rotationally isomeric ligands with different porosity profiles. The authors were able to selectively prepare the most favourable cage isomers from among the 34 configurationally-distinct MOPs possible, by applying different crystallisation steps based on solubility and crystal-packing effects. They thus obtained crystallographic evidence of three distinct C2h-symmetric MOPs, with two of these comprising different ligand conformations within the same cage assembly. In the solid-state, the respective cage configurations were locked, allowing the authors to probe the respective solid-state transformations and porosity profiles of each conformer.81
We believe that the above examples reflect how covalent chemistry and other chemistries of organic subunits are the most powerful post-synthetic techniques with which to modify the surface of MOPs and convert them into a specialised platform. Recent advances regarding the overall stability of MOPs, in combination with techniques that introduce functionality by eschewing aggressive synthetic conditions, have enabled researchers to functionalise MOPs with unprecedented diversity. The recent development of Zr(IV)- or Rh(II)-based MOPs, among other, less-represented sources of strong metals, has enabled researchers to revisit post-synthetic chemistries that were once incompatible with the platforms. Only time can tell what exciting possibilities will arise from exploring surface functionalisation of robust MOPs with alkyne, NH2, or other equally reactive groups now that the full repertoire of chemistry is becoming accessible, beyond the mere use of mild condensation pathways.
3.3. Post-synthetic ion-exchange/metathesis
MOPs with a charged backbone can undergo PSM by an additional type of reactivity, which is based on salt metathesis. Akin to the behaviour of charged inorganic nanoparticles,82 the type of counter ions on the surface of nano-sized molecules influence their surface chemistry and consequently, their behaviour in solution (i.e. solubility and reactivity). For example, Nitschke et al., among other groups, have elegantly employed ion-exchange reactions to alter the solubility of coordination cages.83–85In addition to modifying charged MOPs, researchers can now confer neutral MOPs with charge. For example, although the backbone of paddlewheel-based MOPs is neutral, these MOPs can be conferred with charge through the strategic use of pendant pH-responsive groups that, upon protonation (or deprotonation), become positively (or negatively) charged. For instance, strong acids such as sulfonic acid can be easily deprotonated to yield the corresponding negatively-charged sulfonate groups. Through this approach, Zhou et al. synthesised the first negatively-charged MOP with formula (named SO3X-CuMOP, of formula X24[Cu2(5-SO3-bdc)2]12, where X = Li(I) or Na(I)).51 Later, Bloch et al. used SO3X-CuMOP as a charged monomer to synthesise porous salts.86,87 However, the amenability of SO3X-CuMOP in to solution-phase metathesis has not yet been studied, probably due to its structural fragility in solution, especially in water.
Alternatively, our group has shown that the 24 hydroxyl groups at the periphery of OH-RhMOP could be quantitatively deprotonated with NaOH to afford the negatively-charged ONa-RhMOP, of formula Na24[Rh2(5-O-bdc)2]12. The resultant negatively-charged species with 24 hydrophilic Na+ cations on its surface proved to be highly soluble and stable in an aqueous alkaline solution, whereas the parent OH-RhMOP was only soluble in aliphatic alcohols. The deprotonation of the surface hydroxyl groups was both fast and quantitative, enabling us to perform it in situ to transfer the Rh-MOPs from an organic phase (1-butanol) into an aqueous phase. Importantly, we were also able to do the reverse transfer. Additionally, ONa-RhMOP was not only responsive towards a change in pH but also to cation-exchange reactions. Thus, it was possible to quantitatively exchange the 24 Na(I) ions with organic hydrophobic cations, such as cetyltrimethylammonium (CTA), to yield a MOP with formula CTA24[Rh2(5-O-BDC)2]12 that was highly soluble in polar organic solvents (Fig. 7a). Furthermore, this cation exchange reaction could also be triggered in situ to transfer the Rh-MOPs from an aqueous phase to an organic phase (i.e. chloroform or 1-butanol). Remarkably, the pH- and cation-triggered phase transfer steps could be coupled in a triphasic system to induce the autonomous transport of OH-RhMOP from the 1-butanol phase into the aqueous phase and finally, into the chloroform phase (Fig. 7b).60
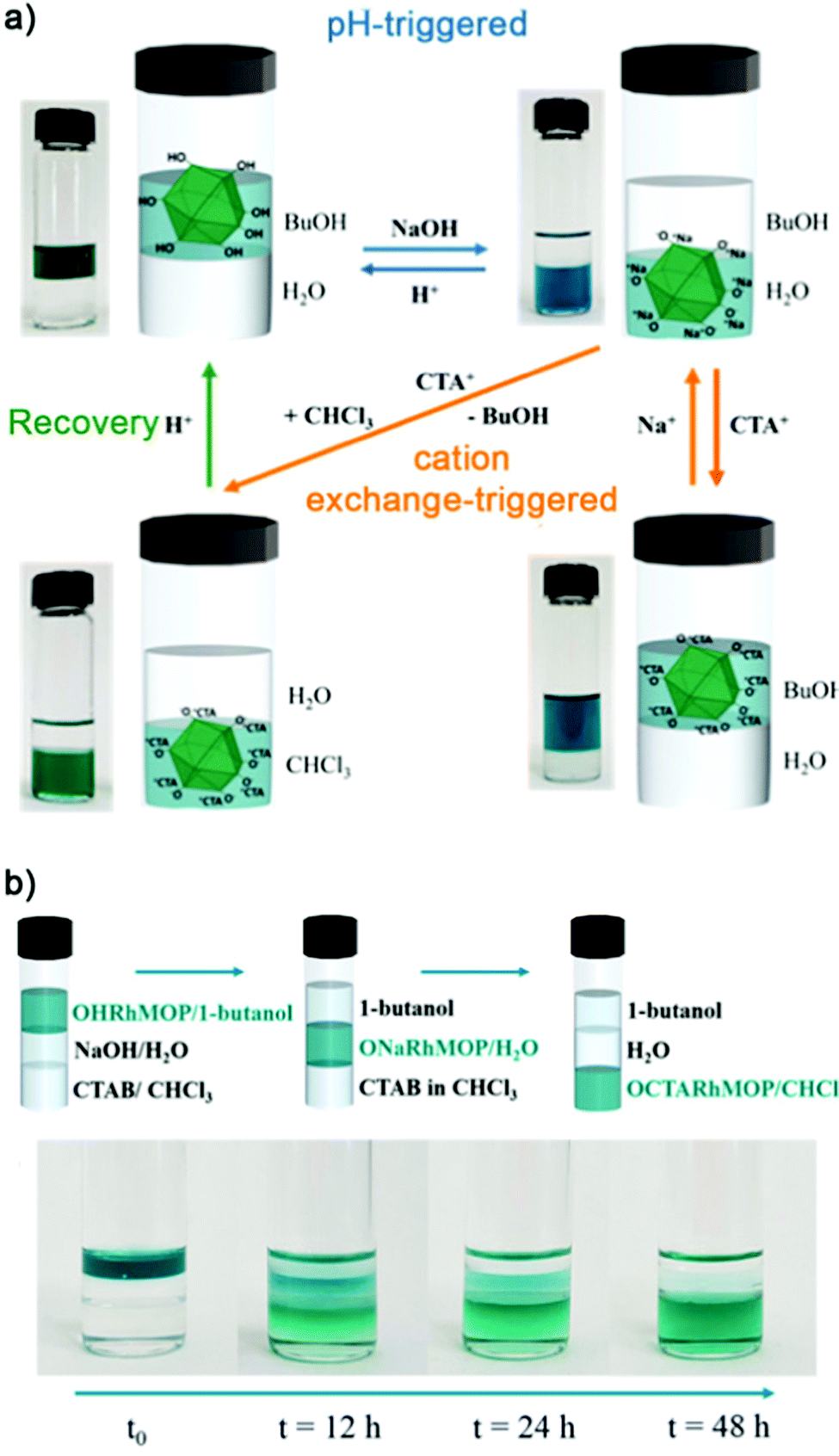 | ||
| Fig. 7 (a) Reversible pH-triggered (blue arrows) and cation exchange-triggered (orange arrows) phase transfer of OH-RhMOP after quantitative deprotonation of surface hydroxyl groups in water. (b) The pH- and cation-triggered phase transfer steps were coupled in a triphasic system, to trigger autonomous transport of OH-RhMOP through three immiscible solvent media, from a 1-butanol phase into water and finally, into chloroform. Reproduced with permission of ref. 60. Copyright 2019 American Chemical Society. | ||
We believe that the synthesis of charged MOPs through protonation/deprotonation reactions could be extended to other MOPs containing pH-sensitive pendant groups such as amines, sulphonic acids, or carboxylic acids. However, these functionalised MOPs would have to be sufficiently robust to withstand the pH required for the protonation/deprotonation. Alternatively, one could use pH-independent charged pendant groups such as imidazolium rings or tetrasubstituted amines to develop positively charged MOPs. Importantly, such charged groups can also be incorporated within the axial sites of paddlewheel-based MOPs via coordination chemistry, using charged N-donor or S-donor nucleophiles.34
Unlike paddlewheel-based MOPs, Zr-MOPs are intrinsically charged, owing to their trinuclear zirconocene clusters. Zr-MOPs are generally balanced by Cl− ions,44 which makes them soluble in polar solvents, including water.78 In a pioneering example, the Cl− ions in a Zr-MOP of formula Cl4[Cp3Zr3μ3-O(μ2-OH)3(Me2-bdc)6] (where Me2-bdc is 2,5-dimethyl-1,4-benzenedicarboxylic acid) could be quantitatively exchanged by larger anions such as triflate (OTf−), and that by doing so, the solubility of the resulting cage in methanol is significantly greater than that of the parent MOP.86 This example nicely illustrates the possibility of leveraging the zirconocene-cluster counterion as an additional source of reactivity to selectively tune the properties of Zr-MOPs in solution.
Overall, salt metatheses are rapid and quantitative reactions that can dramatically change the surface chemistry of MOPs, and consequently, enable in situ tuning of their solubility and reactivity. We believe that the synergistic combination of ionic exchange reactions, the inherent porosity of MOPs, and their coordination and/or covalent reactivity, is especially attractive for the development of novel vehicles for molecular transport, separation, and phase-transfer catalysts.
4. Outlook and perspectives
Researchers were initially slow to advance the field of MOPs, given their widespread preference for other materials with more-robust bulk properties, such as MOFs. Nevertheless, the chemistry of MOPs has gradually matured, proving to be as attractive and precise as that of other porous materials. In fact, MOPs are poised to take the spotlight for applications in which the long-range structure or insolubility of their extended counterparts can prove detrimental.88,89 For example, MOPs hold great potential for the development of new porous liquids. The surface reactivity of MOPs can be leveraged for versatile functionalisation to confer MOPs with unusual properties, such as melting (e.g. polymer melts) or solubility in bulky solvents (e.g. ionic liquids), to generate new type 1 or 2 porous liquids, respectively.90 The molecular nature of MOPs, their defined cavities, and their open-metal sites, amenable to rich and selective coordination chemistry, together allow for the synthesis of new metal–organic porous liquids with potential as working fluids for tasks such as separation, catalysis, or storage.We believe that the stability and solubility of certain robust MOPs in physiological pH should inspire researchers to explore using MOPs for biomedical applications. Given the surface chemistry of MOPs, we envision conjugation of them to biomolecules (e.g. peptides, proteins, or even DNA) by conventional click chemistry or condensation chemistry. Similarly, we reason that MOPs could be used for Drug Delivery, by incorporating drug molecules onto the MOP surface and/or entrapping them within the MOP cavities. In this regard, the proven capacity of MOPs to solubilise hydrophobic molecules in water could prove invaluable for engineering delivery vehicles for drugs with poor water-solubility. This use of MOPs would merge the benefits of inorganic or MOF nanoparticles, with those of molecular systems, by combining rich surface chemistry, defined cavities, and stoichiometric reactivity. These combined features would enable highly versatile delivery vehicles that could be characterised at the molecular level, thus enabling facile study of their structure-property relationships.
We are confident that the surface of MOPs is an excellent platform at which to localise and arrange active molecules while keeping them in solution. The possibility of localising chromophores on the surface of a MOP, either through covalent or coordinative linkages, would enable the synthesis of solutions with optical properties in which the distance and symmetry between the dyes would be controlled to regulate or suppress their energy transfer processes. This approach would circumvent the use of solid scaffolds to regulate energy transfer processes between dyes, thus enhancing the liquid processability of optical materials.
However, the full potential of MOPs for practical applications such as those that we mentioned above cannot be reached unless researchers take full advantage of their latent reactivity. Fig. 1 (Section 2) clearly illustrates that the potential reactivity of MOPs has scarcely begun to be explored. To date, most literature on the PSM of MOPs concerns position 5 of the standard cuboctahedral M24L24 linkers. Indeed, although positions 4 and 6 cover a much higher proportion of the external surface and could provide a new type of directionality, there have not yet been any reports on their chemistry. Likewise, the internal axial sites and position 2 in M24L24 MOPs have never been functionalised or targeted. To access these sites, reagents must pass through minuscule windows; even if some substrates could penetrate through the framework, the internal pore is too small to accommodate more than two or three moieties at its accessible sites. Importantly, access to the internal cavity in less-connected cages such as tetrahedral, octahedral or lantern-type MOPs is much easier. Indeed, researchers have reported some success in functionalising these positions despite steric concerns,91 albeit with the trade-off of a less furnished external surface. Thus, we see a clear demand for larger MOPs with larger windows and pore sizes, which could be targeted via the same type of isoreticular expansion that has enabled the synthesis of systematically larger MOFs.92 Regardless of these challenges, we remain convinced that unlocking the internal reactivity of MOPs, and fully exploiting their surface reactivity, will afford porous nanomaterials of unprecedented designs and massive potential for applications such as catalysis, delivery, molecular transport/separation, and storage, both in solution and in the solid-state.
Author contributions
J. A., L. H.-L. and A. C.-S. contributed to the bibliographic research and manuscript redaction. J. A. and L. H.-L. designed and created the manuscript figures. D. M. supervised the work and redacted the final draft. All the authors collectively discussed the manuscript and provided insightful inputs prior to submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish MINECO (project RTI2018-095622-B-I00) and the Catalan AGAUR (project 2017 SGR 238). It was also funded by the CERCA Programme/Generalitat de Catalunya and through a fellowship (LCF/BQ/PR20/11770011) from the “la Caixa” Foundation (ID 100010434). ICN2 is supported by the Severo Ochoa programme from the Spanish MINECO (Grant No. SEV-2017-0706).References
- K. Otsubo, Y. Wakabayashi, J. Ohara, S. Yamamoto, H. Matsuzaki, H. Okamoto, K. Nitta, T. Uruga and H. Kitagawa, Nat. Mater., 2011, 10, 291–295 CrossRef CAS PubMed.
- F. Dai, J. Zai, R. Yi, M. L. Gordin, H. Sohn, S. Chen and D. Wang, Nat. Commun., 2014, 51, 1–11 Search PubMed , 2014, 5.
- X. Jia, W. Khan, Z. Wu, J. Choi and A. C. K. Yip, Adv. Powder Technol., 2019, 30, 467–484 CrossRef CAS.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 110, 1–19 Search PubMed , 2016, 1.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- T. He, X. J. Kong and J. R. Li, Acc. Chem. Res., 2021, 54, 3083–3094 CrossRef CAS PubMed.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1295–1300 CrossRef CAS PubMed.
- H. Jiang, D. Alezi and M. Eddaoudi, Nat. Rev. Mater., 2021, 66, 466–487 CrossRef , 2021, 6.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- S. M. Cohen, J. Am. Chem. Soc., 2017, 139, 2855–2863 CrossRef CAS PubMed.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef CAS PubMed.
- M. Kalaj and S. M. Cohen, ACS Cent. Sci., 2020, 6, 1046–1057 CrossRef CAS PubMed.
- R. A. Peralta, M. T. Huxley, J. Albalad, C. J. Sumby and C. J. Doonan, Inorg. Chem., 2021, 60, 11775–11783 CrossRef CAS PubMed.
- S. P. Argent, I. Da Silva, A. Greenaway, M. Savage, J. Humby, A. J. Davies, H. Nowell, W. Lewis, P. Manuel, C. C. Tang, A. J. Blake, M. W. George, A. V. Markevich, E. Besley, S. Yang, N. R. Champness and M. Schröder, Inorg. Chem., 2020, 59, 15646–15658 Search PubMed.
- E. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef PubMed.
- S. Mollick, S. Fajal, S. Mukherjee and S. K. Ghosh, Chem. – Asian J., 2019, 14, 3096–3108 CrossRef CAS PubMed.
- X. Y. Xie, F. Wu, X. Q. Liu and L. B. Sun, Dalton Trans., 2019, 48, 17153–17157 RSC.
- K. Omoto, N. Hosono, M. Gochomori and S. Kitagawa, Chem. Commun., 2018, 54, 7290–7293 RSC.
- A. Carné-Sánchez, J. Albalad, T. Grancha, I. Imaz, J. Juanhuix, P. Larpent, S. Furukawa and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 4094–4102 CrossRef PubMed.
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- A. W. Markwell-Heys, M. Roemelt, A. D. Slattery, O. M. Linder-Patton and W. M. Bloch, Chem. Sci., 2022, 13, 68–73 RSC.
- H.-N. Wang, X. Meng, G.-S. Yang, X.-L. Wang, K.-Z. Shao, Z.-M. Su and C.-G. Wang, Chem. Commun., 2011, 47, 7128–7130 RSC.
- H. Takezawa, K. Shitozawa and M. Fujita, 126, Nat. Chem., 2020, 574–578 CrossRef CAS PubMed , 2020, 12.
- H. Takezawa, R. Tabuchi, H. Sunohara and M. Fujita, J. Am. Chem. Soc., 2020, 142, 17919–17922 CrossRef CAS PubMed.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- Y. Li, S. S. Rajasree, G. Y. Lee, J. Yu, J. H. Tang, R. Ni, G. Li, K. N. Houk, P. Deria and P. J. Stang, J. Am. Chem. Soc., 2021, 143, 2908–2919 CrossRef CAS PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, V. Guillerm, K. Urayama, D. Maspoch and S. Furukawa, Angew. Chem., Int. Ed., 2019, 58, 6347–6350 CrossRef PubMed.
- M. A. Andrés, A. Carné-Sánchez, J. Sánchez-Laínez, O. Roubeau, J. Coronas, D. Maspoch and I. Gascón, Chem. – Eur. J., 2019, 26, 143–147 CrossRef PubMed.
- M. Kitchin, J. Teo, K. Konstas, C. H. Lau, C. J. Sumby, A. W. Thornton, C. J. Doonan and M. R. Hill, J. Mater. Chem. A, 2015, 3, 15241–15247 RSC.
- C. R. P. Fulong, J. Liu, V. J. Pastore, H. Lin and T. R. Cook, Dalton Trans., 2018, 47, 7905–7915 RSC.
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed.
- D. J. Tranchemontagne, Z. Ni, M. O’Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- C. Serre and J. Pelta, Chem, 2017, 2, 459–460 CAS.
- M. Jaya Prakash, M. Oh, X. Liu, K. N. Han, G. H. Seong and M. S. Lah, Chem. Commun., 2010, 46, 2049–2051 RSC.
- G. A. Craig, P. Larpent, S. Kusaka, R. Matsuda, S. Kitagawa and S. Furukawa, Chem. Sci., 2018, 9, 6463–6469 RSC.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110–7118 CrossRef CAS PubMed.
- G. Liu, Z. Ju, D. Yuan and M. Hong, Inorg. Chem., 2013, 52, 13815–13817 CrossRef CAS PubMed.
- J. R. Li, A. A. Yakovenko, W. Lu, D. J. Timmons, W. Zhuang, D. Yuan and H. C. Zhou, J. Am. Chem. Soc., 2010, 132, 17599–17610 CrossRef CAS PubMed.
- Y. T. Zhang, X. L. Wang, E. L. Zhou, X. S. Wu, B. Q. Song, K. Z. Shao and Z. M. Su, Dalton Trans., 2016, 45, 3698–3701 RSC.
- L. B. Sun, J. R. Li, W. Lu, Z. Y. Gu, Z. Luo and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 15923–15928 CrossRef CAS PubMed.
- Y. H. Kang, X. D. Liu, N. Yan, Y. Jiang, X. Q. Liu, L. B. Sun and J. R. Li, J. Am. Chem. Soc., 2016, 138, 6099–6102 CrossRef CAS PubMed.
- S. Mollick, S. Mukherjee, D. Kim, Z. Qiao, A. V. Desai, R. Saha, Y. D. More, J. Jiang, M. S. Lah and S. K. Ghosh, Angew. Chem., 2019, 131, 1053–1057 CrossRef.
- D. T. Richens, Chem. Rev., 2005, 105, 1961–2002 CrossRef CAS PubMed.
- J. R. Li and H. C. Zhou, Nat. Chem., 2010, 210, 893–898 CrossRef PubMed , 2010, 2.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- P. J. Stang, J. Am. Chem. Soc., 2012, 134, 11829–11830 CrossRef CAS PubMed.
- C. M. Vetromile, A. Lozano, S. Feola and R. W. Larsen, Inorg. Chim. Acta, 2011, 378, 36–41 CrossRef CAS.
- M. Tonigold and D. Volkmer, Inorg. Chim. Acta, 2010, 363, 4220–4229 CrossRef CAS.
- G. A. Craig, P. Larpent, H. Urabe, A. Legrand, M. Bonneau, S. Kusaka and S. Furukawa, Chem. Commun., 2020, 56, 3689–3692 RSC.
- W. M. Bloch, R. Babarao and M. L. Schneider, Chem. Sci., 2020, 11, 3664–3671 RSC.
- L. Chen, T. Yang, H. Cui, T. Cai, L. Zhang and C. Y. Su, J. Mater. Chem. A, 2015, 3, 20201–20209 RSC.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carné-Sánchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Inorg. Chem., 2016, 55, 10843–10846 CrossRef CAS PubMed.
- T. Grancha, A. Carné-Sánchez, L. Hernández-López, J. Albalad, I. Imaz, J. Juanhuix and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 18349–18355 CrossRef CAS PubMed.
- L. Hernández-López, J. Martínez-Esaín, A. Carné-Sánchez, T. Grancha, J. Faraudo and D. Maspoch, Angew. Chem. Int. Ed., 2021, 133, 11507–11514 CrossRef.
- Z. Lu, C. B. Knobler, H. Furukawa, B. Wang, G. Liu and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 12532–12533 CrossRef CAS PubMed.
- G. A. Taggart, A. M. Antonio, G. R. Lorzing, G. R. Lorzing, G. P. A. Yap, E. D. Bloch and E. D. Bloch, ACS Appl. Mater. Interfaces, 2020, 12, 24913–24919 CrossRef CAS PubMed.
- G. Liu, Z. Yang, M. Zhou, Y. Wang, D. Yuan and D. Zhao, Chem. Commun., 2021, 57, 6276–6279 RSC.
- D. Zhao, S. Tan, D. Yuan, W. Lu, Y. H. Rezenom, H. Jiang, L.-Q. Wang, H.-C. Zhou, D. Zhao, D. Yuan, W. Lu, Y. H. Rezenom, H. Zhou, S. Tan, H. Jiang and L. Wang, Adv. Mater., 2011, 23, 90–93 CrossRef CAS PubMed.
- S. K. Samanta, D. Moncelet, B. Vinciguerra, V. Briken and L. Isaacs, Helv. Chim. Acta, 2018, 101, e1800057 CrossRef PubMed.
- D. Nam, J. Huh, J. Lee, J. H. Kwak, H. Y. Jeong, K. Choi and W. Choe, Chem. Sci., 2017, 8, 7765–7771 RSC.
- X. Y. Xie, F. Wu, X. Liu, W. Q. Tao, Y. Jiang, X. Q. Liu and L. B. Sun, Chem. Commun., 2019, 55, 6177–6180 RSC.
- N. Hosono, M. Gochomori, R. Matsuda, H. Sato and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 6525–6531 CrossRef CAS PubMed.
- G. Lal, B. S. Gelfand, J. Bin Lin, A. Banerjee, S. Trudel and G. K. H. Shimizu, Inorg. Chem., 2019, 58, 9874–9881 CrossRef CAS PubMed.
- W. Lu, D. Yuan, A. Yakovenko and H. C. Zhou, Chem. Commun., 2011, 47, 4968–4970 RSC.
- V. Brega, M. Zeller, Y. He, H. Peter Lu and J. K. Klosterman, Chem. Commun., 2015, 51, 5077–5080 RSC.
- H. Furukawa, J. Kim, N. W. Ockwig, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650–11661 CrossRef CAS PubMed.
- J. Albalad, A. Carné-Sánchez, T. Grancha, L. Hernández-López and D. Maspoch, Chem. Commun., 2019, 55, 12785–12788 RSC.
- M. Schelhaas and H. Waldmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2056–2083 CrossRef CAS.
- K. Jarowicki and P. Kocienski, J. Chem. Soc., Perkin Trans. 1, 2001, 2109–2135 RSC.
- M. L. Schneider, O. M. Linder-Patton, W. M. Bloch and R. Li, Chem. Commun., 2020, 56, 12969–12972 RSC.
- G. Liu, Y. Di Yuan, J. Wang, Y. Cheng, S. B. Peh, Y. Wang, Y. Qian, J. Dong, D. Yuan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 6231–6234 CrossRef CAS PubMed.
- Y. Jiang, P. Tan, S. C. Qi, C. Gu, S. S. Peng, F. Wu, X. Q. Liu and L. B. Sun, CCS Chem., 2021, 3, 1659–1668 CrossRef CAS.
- J. Park, L. B. Sun, Y. P. Chen, Z. Perry and H. C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 5842–5846 CrossRef CAS PubMed.
- A. W. Markwell-Heys, M. L. Schneider, J. M. L. Madridejos, G. F. Metha and W. M. Bloch, Chem. Commun., 2021, 57, 2915–2918 RSC.
- T. Bian, A. Gardin, J. Gemen, L. Houben, C. Perego, B. Lee, N. Elad, Z. Chu, G. M. Pavan and R. Klajn, Nat. Chem., 2021, 1310, 940–949 CrossRef PubMed , 2021, 13.
- A. B. Grommet, J. B. Hoffman, E. G. Percástegui, J. Mosquera, D. J. Howe, J. L. Bolliger and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 14770–14776 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 18949–18953 CrossRef CAS PubMed.
- N. Mihara, T. K. Ronson and J. R. Nitschke, Angew. Chem. Int. Ed., 2019, 131, 12627–12631 CrossRef.
- A. J. Gosselin, G. E. Decker, A. M. Antonio, G. R. Lorzing, G. P. A. Yap and E. D. Bloch, J. Am. Chem. Soc., 2020, 142, 9594–9598 CrossRef CAS PubMed.
- A. J. Gosselin, A. M. Antonio, K. J. Korman, M. M. Deegan, G. P. A. Yap and E. D. Bloch, J. Am. Chem. Soc., 2021, 143, 14956–14961 CrossRef CAS PubMed.
- E. Haldoupis, S. Nair and D. S. Sholl, J. Am. Chem. Soc., 2010, 132, 7528–7539 CrossRef CAS PubMed.
- K. A. S. Usman, J. W. Maina, S. Seyedin, M. T. Conato, L. M. Payawan, L. F. Dumée and J. M. Razal, NPG Asia Mater., 2020, 121, 1–18 Search PubMed , 2020, 12.
- S. S. Nagarkar, M. Tsujimoto, S. Kitagawa, N. Hosono and S. Horike, Chem. Mater., 2018, 30, 8555–8561 CrossRef CAS.
- J. Guo, Q. Chang, Z. Liu, Y. Wang, C. Liu, M. Wang, D. Huang, G. Chen, H. Zhao, W. Wang and X. Fang, Chem. Sci., 2021, 12, 7361–7368 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |