
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustained release of drug-loaded nanoparticles from injectable hydrogels enables long-term control of macrophage phenotype†
Shreya S.
Soni
 ,
Arielle M.
D'Elia
,
Abdulrahman
Alsasa
,
Sylvia
Cho
,
Tina
Tylek
,
Erin M.
O'Brien
,
Ricardo
Whitaker
,
Kara L.
Spiller
and
Christopher B.
Rodell
*
,
Arielle M.
D'Elia
,
Abdulrahman
Alsasa
,
Sylvia
Cho
,
Tina
Tylek
,
Erin M.
O'Brien
,
Ricardo
Whitaker
,
Kara L.
Spiller
and
Christopher B.
Rodell
*
School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA. E-mail: christopher.b.rodell@drexel.edu
First published on 21st October 2022
Abstract
Injectable hydrogels may be pre-formed through dynamic crosslinks, allowing for injection and subsequent retention in the tissue by shear-thinning and self-healing processes, respectively. These properties enable the site-specific delivery of encapsulated therapeutics; yet, the sustained release of small-molecule drugs and their cell-targeted delivery remains challenging due to their rapid diffusive release and non-specific cellular biodistribution. Herein, we develop an injectable hydrogel system composed of a macrophage-targeted nanoparticle (cyclodextrin nanoparticles, CDNPs) crosslinked by adamantane-modified hyaluronic acid (Ad-HA). The polymer-nanoparticle hydrogel uniquely leverages cyclodextrin's interaction with small molecule drugs to create a spatially discrete drug reservoir and with adamantane to yield dynamic, injectable hydrogels. Through an innovative two-step drug screening approach and examination of 45 immunomodulatory drugs with subsequent in-depth transcriptional profiling of both murine and human macrophages, we identify celastrol as a potent inhibitor of pro-inflammatory (M1-like) behavior that furthermore promotes a reparatory (M2-like) phenotype. Celastrol encapsulation within the polymer-nanoparticle hydrogels permitted shear-thinning injection and sustained release of drug-laden nanoparticles that targeted macrophages to modulate cell behavior for greater than two weeks in vitro. The modular hydrogel system is a promising approach to locally modulate cell-specific phenotype in a range of applications for immunoregenerative medicine.
Introduction
Systemic drug administration is often hampered by biodistribution challenges, including rapid renal clearance, an inability to target specific tissue, and poor aqueous solubility of many small molecule drugs.1 Local delivery strategies mitigate these issues by minimizing drug exposure in off-target tissues and extending the window of therapeutically active drug concentrations at the site of action. Hydrogels therefore continue to evolve as tailorable platforms for therapeutic delivery that can support tissue regeneration, alter cell or tissue responses, or present appropriate therapeutic cargo. Such cargo may include pharmacologic drugs, exosomes, or cells intended to support processes of immune modulation and tissue repair.2 In particular, small molecule drugs are attractive payloads due to their ease of synthesis and suitability for optimization; however, their local delivery from hydrogels is often challenging due to their rapid diffusive release and non-specific cellular biodistribution.Many hydrogels are conventionally formed ex vivo via covalent crosslinking, necessitating invasive implantation of the resulting solid material. In contrast, injectable hydrogels may be formed through varying methods to enable minimally invasive delivery, including for the local presentation of encapsulated therapeutic cargo.3,4 One such approach is in situ hydrogel formation after the injection of liquid precursors, accomplished by mechanisms such as radical polymerization, addition crosslinking, or environmental stimuli.5,6 However, the kinetics of hydrogel formation often complicate the delivery process. Rapid gelation risks clogging the delivery device, whereas slow gelation results in material dispersion throughout the tissue after injection.6,7 As an alternative to in situ hydrogel formation, some approaches capitalize on the use of dynamic crosslinks, such as dynamic covalent chemistries, engineered biomolecular interactions, or guest–host chemistries.8–10 Dynamic crosslinking allows hydrogels to be formed ex vivo and then extruded during injection by shear-thinning processes, as the dynamic bonds temporarily break in response to shear stress and rapidly bind again.11 These dynamic material behaviors ultimately allow for injectable delivery that mitigates the risks for invasive implantation, while also increasing material retention at the target site – including precious therapeutic cargo.12–15
As an extension of these self-assembling hydrogel systems, hydrogels may also be formed from discrete micro- or nano-structural units instead of polymeric building blocks alone. Such non-homogenous structures uniquely allow for the structural subunits to take on discrete functions and the unique behavior of the bulk hydrogel formed. For example, granular hydrogels have emerged as a unique class of injectable materials, assembled through inter-particle crosslinking reactions or particle jamming.16–18 Discrete microgel components within these systems may have tunable behavior to control drug delivery.17,19 Other systems have leveraged polymer-nanoparticle interactions to aid in the self-assembly of injectable hydrogels. The inclusion of discrete nanoparticles within these structures has been leveraged to endow polymer-nanoparticle hydrogels with distinct therapeutic, diagnostic, and physical properties.15,20,21 While such polymer-nanoparticle hydrogels open new avenues for local therapeutic delivery, their use as a means to provide controlled release of nanoparticles to naturally target highly phagocytic innate immune cells remains unexplored.
Such local and cell-specific delivery is particularly important for immune modulation. Specifically, the systemic administration of immunosuppressive drugs places patients at an increased risk of infection, which has hampered their clinical acceptance.22,23 Despite these challenges, there remains a growing interest in the use of immunomodulatory drugs to combat inflammatory disease and promote tissue healing.24 Macrophages (MF) are crucial regulators of the tissue-immune microenvironment, and unsolicited MF-derived inflammation commonly underlies failed tissue healing processes and an array of chronic diseases,25,26 making them an attractive therapeutic target.27 While these cells exist across an array of phenotypes, they are often broadly characterized as pro-inflammatory (‘M1-like’) or pro-healing (‘M2-like’), particularly in the context of tissue repair.28 MF phenotypes are highly plastic, subject to modulation by external stimuli such as pharmaceuticals that arrest inflammatory behavior and promote a reparative M2-like phenotype for injury resolution. While immunosuppressants are widely available and used in clinical practice, few M2-polarizing drugs have been reported and necessitate high dosing.29 Immunoregenerative medicine therefore remains limited by a lack of knowledge regarding which drugs or drug classes can potently promote reparatory MF phenotypes.
Small molecule drugs may be of use towards the goal of modulating MF phenotype and are amenable to delivery by guest–host interactions. Guest–host interactions are a subset of supramolecular associations, characterized by the transient complexation of a macrocyclic host with a small molecule guest. In the case of many macrocycles, and β-cyclodextrin (CD) in particular, complexation is driven by hydrophobic interactions that enable the inclusion of a wide variety of guest molecules.30,31 It is therefore a versatile and common excipient in pharmaceutical formulations on market,32 used to enhance drug solubility and bioavailability.33–35 CD is also widely used in biomaterial applications, including in molecular imaging probes, surface coatings, and polymeric drug carriers for affinity-based delivery.36–40 We have previously reported on the development of cyclodextrin nanoparticles (CDNPs) that possess a high drug loading capacity and inherent capacity for MF-targeted therapy.41 The CDNPs are formed only from CD crosslinked by L-lysine, creating a dense network of the host macrocycle that perpetuates a high drug loading capacity. Due to the saccharide-based structure of CD, it is readily internalized by MF, likely via recognition by cell surface receptors that include scavenger receptor A1 (SR-A1) and mannose receptor (MRC1).42,43 The drug loading capacity and MF avidity have been leveraged for systemically administered MF-targeted cancer immunotherapy,41 and similar MF-targeting strategies are highly effective in a range of applications.44–46 However, the delivery of these and other therapeutic nanocarriers is typically accomplished via systemic administration; their local and sustained delivery has not previously been reported.
Herein, we developed an injectable hydrogel platform for the local administration of small molecule drugs that uniquely leverages guest–host interactions for injectable hydrogel assembly and cell-targeted nanoparticle therapy to directly address the need for location-specific modulation of MF behavior. Through supramolecular assembly by guest–host interactions, CDNPs were crosslinked with adamantane-modified hyaluronic acid (Ad-HA) to yield an injectable hydrogel. The same guest–host interactions also serve as a mechanism to retain immunomodulatory therapeutics. The drug of choice was selected by scrutiny of a targeted library of 45 small molecules drugs that spanned a variety of drug classes with reported immunomodulatory capacity. Through a two-step drug screening process and subsequent in-depth transcriptional analysis of both human and murine MF, we identified celastrol as a potent modulator of MF phenotype that suppresses M1-like and promotes M2-like behavior. Celastrol was included within the nanoparticle core by guest–host interaction prior to hydrogel formation. The resulting therapeutic hydrogels degraded by surface erosion over the course of greater than one month, continually releasing drug-loaded nanoparticles that were uptaken by MF and arrested their pro-inflammatory response. The approach represents a promising strategy to achieve functional re-orientation of the local immune microenvironment by the continual release of drug-loaded nanoparticles from the hierarchical hydrogels formed.
Experimental
Materials
Unless otherwise indicated, solvents and general reagents were purchased from Sigma-Aldrich or TCI America and used without additional purification. Pharmaceutical drugs were obtained from Selleckchem, MedChemExpress, or Cayman Chemical Company and prepared at stock concentrations of 100 mM in dimethyl sulfoxide (DMSO). Cell culture reagents were purchased from VWR, unless otherwise stated.Nanoparticle synthesis and characterization
Cyclodextrin nanoparticles (CDNPs) were prepared by methods as previously described.41 Briefly, succinyl-β-cyclodextrin (1.0–3%w/v, 1.0 eq. succinylate groups), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 5–12.5 eq. to succinylate), and N-hydroxysuccinimide (NHS, 0.5 eq. to EDC) were dissolved in MES buffer (50 mM, pH 6.0). The reaction was stirred (30 min, RT) prior to the dropwise addition of L-lysine (0.5–4 eq. to succinylate) and overnight crosslinking. The product was recovered by addition of 100 μL brine and precipitation from a tenfold excess of anhydrous ethanol on ice. Following immediate re-dissolution in water, the product was purified by size-exclusion chromatography (SEC; PD-10, Fisher). Saccharide positive fractions were identified by spotting on a silica gel TLC plate, developing with 5% H2SO4 in ethanol, and then heating the plate. Positive fractions were concentrated by centrifugal filtration (10 kDa MWCO, Amicon), washed repeatedly with water, and lyophilized until dry. The final CDNP products were re-dissolved at 20%w/v in MilliQ water and stored at −20 °C until later use. Particle size was determined by dynamic light scattering (DLS; Zetasizer, Malvern) in triplicate at a concentration of 5 mg mL−1 in phosphate buffered saline (PBS). For scanning electron microscopy (SEM), samples were prepared at 200 μg mL−1 in DI water, lyophilized in a thin layer on conductive scaffolding, attached to SEM stubs using double sided carbon tape, and sputter coated prior to imaging (Zeiss, Supra 50VP).Polymer synthesis and characterization
Hyaluronic acid (HA; MW = 82 kDa or 337 kDa; Lifecore Biomedical) was modified by pendant addition of 1-adamantane acetic acid (Ad), similar to previous reports.47 HA (5 g) was dissolved in DI water at 2%w/v, exchanged against Dowex 50 W resin (15 g), neutralized by tetrabutylammonium hydroxide (TBA) to a final pH of 7.02–7.05, and lyophilized to yield HA-TBA (Fig. S1†). Coupling of 1-adamantane acetic acid to HA-TBA proceeded via esterification. A round bottom flask was charged with a stir bar, HA-TBA (1.5 g, 1 mol equiv. HA repeat units), 4-dimethylaminopyridine (DMAP, 0.38 g, 1.5 mol equiv.), Ad (1.2 g, 3 mol equiv.), and blanketed under dry nitrogen. Anhydrous dimethyl sulfoxide (5 mL per 0.1 g of HA-TBA) was added via cannulation and reactants dissolved. Di-tert-butyl decarbonate (BOC2O) was added via syringe (0.23 mL for 82 kDa Ad-HA (Low), 0.20 mL for 337 kDa Ad-HA (Low), and 0.56 mL for 82 kDa Ad-HA (High)) and the reaction allowed to proceed for 20 h at 45 °C. The product was dialyzed (8–10 kDa MWCO) against DI water, which was changed twice daily for 14 days. After lyophilization, the degree of substitution of the HA backbone by Ad was determined by 1H-NMR (Fig. S2†).Hydrogel formation
Hydrogels were prepared from stock solutions of CDNP (20%w/v) and the denoted Ad-HA polymer (82 kDa Ad-HA (Low), 337 kDa Ad-HA (Low), and 82 kDa Ad-HA (High)) prepared in PBS. Hydrogels were formed by pipette mixing of the two separate solutions, followed by vortexing, manual stirring, and sonication to ensure homogenization with brief centrifugation to remove entrapped air bubbles. Hydrogel formulations were varied by adjusting the initial Ad-HA polymer concentration (2.5–10%w/v), the volumetric ratio of Ad-HA to CDNP (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:4), and anneal time (0–14 days) following mixing.
:1–1:4), and anneal time (0–14 days) following mixing.
Rheological characterization
Characterization was performed on a TA Instruments Discovery HR20 rheometer fitted with a cone and plate geometry (20 mm diameter, 1° cone angle, 27 μm gap). Temperature was maintained at 25 °C through use of a Peltier plate stage. Properties of hydrogel samples were examined by oscillatory time sweeps at varying frequencies (0.1, 1, and 10 Hz; 1% strain), oscillatory frequency sweeps (0.01 Hz to 100 Hz; 1% strain), oscillatory strain sweeps (1% to 500% strain; 10 Hz), and continuous flow experiments with the shear rate linearly ramped from 0.005 to 50 s−1. Recovery experiments were performed using oscillatory time sweeps at 500% strain with recovery at 1% strain.Cell culture
Cells were maintained under standard culture conditions (37 °C, 5% CO2) in the indicated medium which was replenished every two days. The murine MF cell lines, RAW 264.7 (ATCC) and RAW-Blue™ (InvivoGen), were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with heat-inactivated fetal bovine serum (FBS), and 1% penicillin–streptomycin (Pen-Strep). RAW-Blue™ media was additionally supplemented by 100 μg ml−1 Normocin with the addition of 100 μg ml−1 of Zeocin every other passage to maintain selection pressure, as recommended by the manufacturer. Cells were passaged at 70% confluency. Primary MF were derived from murine and human tissues. Bone marrow derived macrophages (BMDMs) were isolated and derived according to standard protocols.48 All animal procedures were performed in compliance with Drexel University's guidelines for the care and use of laboratory animals, with procedures approved by the University's Institutional Animal Care and Use Committee. Marrow was extracted from the surgically resected femur and tibia of male C57BL/6 mice, dissociated, and filtered using a 40 μm strainer. Red blood cells were lysed with ammonium chloride, and recovered cells were plated at 2 × 106 cells per well in 24 well plates and maintained in Iscove's Modification of DMEM (IMDM) supplemented with 10% heat-inactivated FBS, 1% Pen-Strep, and 10 ng mL−1 recombinant mouse macrophage colony-stimulating factor (M-CSF; PeproTech). Primary human monocytes were isolated from peripheral blood (New York Blood Center) from a single healthy human donor via density centrifugation as previously described.49,50 Harvested monocytes were cultured for 5 days on non-tissue culture-treated well plates in RPMI-1640 media supplemented with 10% heat-inactivated human serum, 1% Pen-Strep, and 20 ng mL−1 recombinant human M-CSF (PeproTech).Reporter assays and transcriptional analysis
Our process of drug selection leveraged a unique two-step drug screen that first identified potent M1-inhibitors using a high-throughput reporter cell assay followed by secondary identification of M2-promoting drugs in cell lines. Furthermore, a rigorous approach for validating drug activity was pursued, including in depth transcriptional analysis of primary murine and human cells to ensure cross-species drug activity. For initial drug screens, RAW-Blue™ cells were plated at 1 × 105 cells per well in 384 well plates. To induce polarization towards an M1-like phenotype, media was replaced after 24 h including zymosan (100 μg mL−1). Drug dosing was performed concurrently, spanning concentrations from 100 μM to 0.03 μM in half-log dilutions; n = 4 per group. At 24 h after drug treatment, secreted embryonic alkaline phosphatase (SEAP) reporter activity was quantified using QUANTI-Blue™ Solution following the manufacturer's protocol. Absorbance was read at 620 nm (BioTek Instruments, Synergy H1) and is presented following normalization to zymosan-treated controls.For secondary identification of M2-promoting drugs, transcriptional analysis using qPCR was performed. RAW264.7 cells were plated at 1 × 106 cells per well in 24 well plates. After 24 h, media was replaced, and IL-4 (10 ng mL−1, PeproTech) or zymosan (100 μg mL−1) were included to serve as internal controls for M2-like and M1-like phenotypes, respectively. Treatment groups were concurrently activated via zymosan and treated with 1 μM drug concentrations, n = 3 per group. After 24 h, cells were lysed by freezing, RNA was extracted (RNEasy Mini Kit; Qiagen), and cDNA synthesis was performed (High-Capacity cDNA Reverse Transcription Kit; Fisher). Samples were subject to qPCR using Taqman Fast Advanced Master Mix and probes (Fisher) for analysis of hprt (Mm01545399_m1), nos2 (Mm00440502_m1), il12b (Mm01288989_m1), il6 (Mm00446190_m1), mrc1 (Mm00485148_m1), il10 (Mm01288386_m1), and arg1 (Mm00475988_m1). Data is expressed as a fold change in gene expression using the ΔΔCt method, relative to the hprt and zymosan-treated controls.
For in depth transcriptional profiling of best drug candidates (piclamilast and celastrol), primary murine and human cells were subject to nanoString analysis. BMDMs were differentiated in 24 well plates as described. Zymosan (100 μg mL−1) and IL-4 (10 ng mL−1) treatments were again included as internal references for M1-like and M2-like MF phenotypes, respectively. Cells were treated with 1 μM of the prescribed compounds for 24 h, n = 3 per group. Following RNA extraction, nanoString multiplex gene expression analysis was performed using 100 ng of extracted RNA and a custom-designed panel of 91 genes (Table S1†), which relate primarily to murine MF phenotypes, as well as angiogenesis and fibrosis. Transcriptional analysis was similarly performed for human MF. Following differentiation from peripheral blood monocytes as described, cells were stimulated with lipopolysaccharide (LPS, 100 ng mL−1). After 24 h, celastrol was added (1–10 μM, 24 h). RNA was isolated (RNAqueous-Micro Total RNA Isolation Kit, Fisher) and nanoString was performed with a custom-designed panel of 70 genes related primarily to MF phenotypes, angiogenesis, and fibrosis (Table S2†). For both murine and human datasets, data was normalized to internal positive and negative controls using the nSolver 4.0 software and subsequently normalized to housekeeping genes (geometric mean of hprt and tbp for mouse, gapdh and tbp for human), as recommended by the manufacturer. Data is presented for all genes expressed above background as raw gene counts or as the Z-score of log-transformed data relative to M1-like controls or row means as indicated.
Cell viability
To examine potential cytotoxicity of drug and hydrogel components, RAW-Blue™ cells were plated at 5 × 103 cells per well in 96 well plates. Media was replaced at 24 h containing either drug (100 μM to 0.03 μM in half-log dilutions) or polymeric components (Ad-HA or CDNP, 5 to 0.04%w/v in five-fold dilutions) of interest. After 24 h, metabolic activity was assessed by PrestoBlue™ (Fisher) following the manufacturer's protocol, n = 3 per treatment group. Absorbance was recorded at 570 nm, background subtracted from cell-free control wells, and normalized to untreated controls.Surface plasmon resonance of drug binding to CD
Surface plasmon resonance (Nicoya, OpenSPR) was used to quantify binding affinity between CD and celastrol. The instrument was primed with running buffer (0.5%v/v DMSO in PBS), a high sensitivity carboxyl sensor was installed, and both channels were cleaned with 10 mM HCl at 150 μL min−1. The surface of the sensor was activated via injection of 200 μL of 0.1 M EDC/NHS in DI water in both channels at 20 μL min−1. The ligand, aminated β-cyclodextrin, prepared as previously described,14 was dissolved in sodium acetate buffer (10 mM, pH 6, 1.2 mg mL−1) and immobilized on the surface in channel 2 at 20 μL min−1. Residual succinyl esters were deactivated by ethanolamine. The analyte, celastrol, was dissolved in running buffer and injected at concentrations of 62.5 μM, 125 μM, and 250 μM. Between tests, the injection port was rinsed with 1 mL of running buffer. Curves were analyzed in GraphPad Prism 8 via the association kinetics model.Hydrogel erosion and drug release
To determine the rate at which hydrogels were eroded, established methods of one-dimensional hydrogel degradation were used. Assays were performed in custom made acrylic erosion wells, having a hydrogel chamber (4.3 mm diameter, 7 mm depth) overlaid with a supernatant chamber (1.6 cm diameter, 10 mm depth).14 Hydrogels (n = 4 per group, 30 μL each) were deposited in the hydrogel chamber, wells were centrifuged to provide an even hydrogel surface, and the hydrogel was covered with 1 mL of PBS. Samples were incubated at 37 °C, and the supernatant was collected at regular intervals with replacement by fresh buffer. At the endpoint, hydrogels were degraded in 2 mg mL−1 hyaluronidase (Sigma) for complete sample recovery. Quantification of polymeric content in release buffer was performed via uronic acid assay. For each supernatant sample, 50 μL of the sample was combined with ice-cold sulfuric acid containing sodium tetraborate decahydrate (1 mL, 19 mg mL−1). Samples were incubated at 100 °C for 10 min and cooled on ice prior to addition of carbazole (30 μL, 1.25 mg mL−1 in ethanol). Samples were briefly vortexed, incubated at 100 °C (15 min), and cooled on ice prior to recording absorbance at 525 nm (Thermo, Spectronic BioMate 3). Data is presented as the cumulative erosion over time, normalized to total sample recovery after enzymatic degradation.For release studies, hydrogels were prepared including celastrol (5 mM). Celastrol was loaded into CDNPs by overnight mixing in PBS prior to the addition of Ad-HA. Hydrogels (n = 3 per group) were prepared and loaded into erosion wells as previously described, with 1 mL of RAW-Blue™ cell media used as the supernatant which was subsequently collected with replacement at set time points over 14 days. Samples were stored at −20 °C until analysis of bioactivity in RAW-Blue™ cells, which were plated at 25 × 103 cells per well in 96 well plates. After 24 h, media was replaced with unconditioned media (control) or conditioned media from the release studies, supplemented with zymosan (100 μg mL−1) as an inflammatory stimulus. Inflammatory activity was detected at 24 h after treatment using QUANTI-Blue™ Solution, following the manufacturer's protocol as described above.
In vitro nanoparticle uptake
For in vitro imaging of nanoparticle uptake, CDNPs were dissolved at 50 mg mL−1 in carbonate buffer (0.1 M, pH 8.5) and fluorescently labelled with Alexa Flour 555 NHS (10 μg mL−1, Fisher). The reaction proceeded for 2 h at RT in the dark prior to product recovery by centrifugal filtration (10 kDa MWCO, Amicon). The product was repeatedly washed with water to remove unbound dye and lyophilized.To determine if CDNPs released from hydrogels were uptaken by MF, hydrogels were prepared from the fluorescently labeled nanoparticle (CDNP-AF555) with or without celastrol inclusion (5 mM) and loaded into erosion wells as previously described (n = 3 per group), with 1 mL of RAW-Blue™ cell media used as the supernatant which was collected after 48 hours. Conditioned media was transferred to RAW-Blue™ cells, seeded 24 hours prior at 15 × 103 cells per well in a 96 well glass bottom plate. After 24 h, cells were washed by PBS, fixed with 4% paraformaldehyde (15 min, 37 °C), and stained for cell membrane (5 μg mL−1 AlexaFluor 488 wheat germ agglutinin, Fisher) and nuclei (NucBlue™, Fisher) for 15 min at room temperature. Plates were washed by PBS prior to imaging (Leica, DMI 6000B). CDNP-AF555 uptake was assessed in ImageJ, quantified as the integrated fluorescence density.
Data presentation and statistical analysis
Data presented are mean ± standard deviation (SD), unless otherwise indicated. Statistical analysis was performed using GraphPad Prism v9.3.1 and determined by analysis of variance (ANOVA), using repeated measurements where appropriate in conjunction with post-hoc Tukey's honest significant difference test. Normality was assessed by Shapiro–Wilk test. For two-way ANOVA, the Geisser-Greenhouse's epsilon correction was applied. Significance was determined at p < 0.05.Results and discussion
Development and characterization of CDNPs
Cyclodextrins are versatile macrocyclic hosts, used in the pharmaceutical industry as excipients to improve drug solubility and bioavailability – a use which has been meaningfully extended through the development of nano and bulk materials that leverage this host capacity for drug retention, including for macrophage (MF)-targeted therapies. In biomaterial applications, the transient guest–host complexes formed between CD and hydrophobic guest molecules is also a common means of forming dynamically crosslinked hydrogels. For this purpose, adamantane (Ad) is frequently used, as its size readily complements that of CD's hydrophobic cavity, contributing to formation of a one-to-one inclusion complex with relatively high affinity (Keq = 105 M).51 Here, we look to leverage CD's versatile host capacity to develop a nanotherapeutic drug delivery vehicle which is dynamically crosslinked by a host-modified polymer.To prepare cyclodextrin nanoparticles (CDNPs) of varying size, particles were formed through EDC/NHS mediated amidation between succinyl-β-cyclodextrin and L-lysine (Fig. 1A), similar to reported protocols. While nanoparticles previously formed by these methods were approximately 30 nm in diameter, we rationalized that an increased particle size would afford both a greater surface area for polymer-NP crosslinking as well as an improved volume to surface area ratio essential to forming a discrete drug reservoir. Synthesis conditions were therefore explored as a means to control CDNP size, where related variables include the concentration of CD during crosslinking (Fig. 1B), the ratio of lysine to succinyl groups (i.e., lysine feed ratio, Fig. 1C), and the catalyst concentration relative to succinyl groups (i.e., EDC feed ratio, Fig. 1D). Nanoparticle size was positively correlated with the substrate and catalyst concentrations. Furthermore, a 1:1 ratio of amine to succinate at a lysine feed ratio of 0.5:1 resulted in the largest observed diameter. Further increase in the particle size was not possible, as increased substrate or catalyst concentrations resulted in formation of a solid hydrogel during the reaction. We therefore pursued the largest available CDNPs for hydrogel development; nanoparticles formed with 3.3%w/v CD, a 0.5:1 lysine feed ratio, and a 12.5:1 EDC feed ratio reliably produced CDNPs with a diameter of 80 nm, as confirmed by dynamic light scattering (DLS, Fig. 1E) and scanning electron microscopy (SEM, Fig. 1F). Notably, nanoparticles on the order of 50–100 nm are likewise preferential for phagocytic uptake necessary for subsequent MF-targeted delivery applications.52
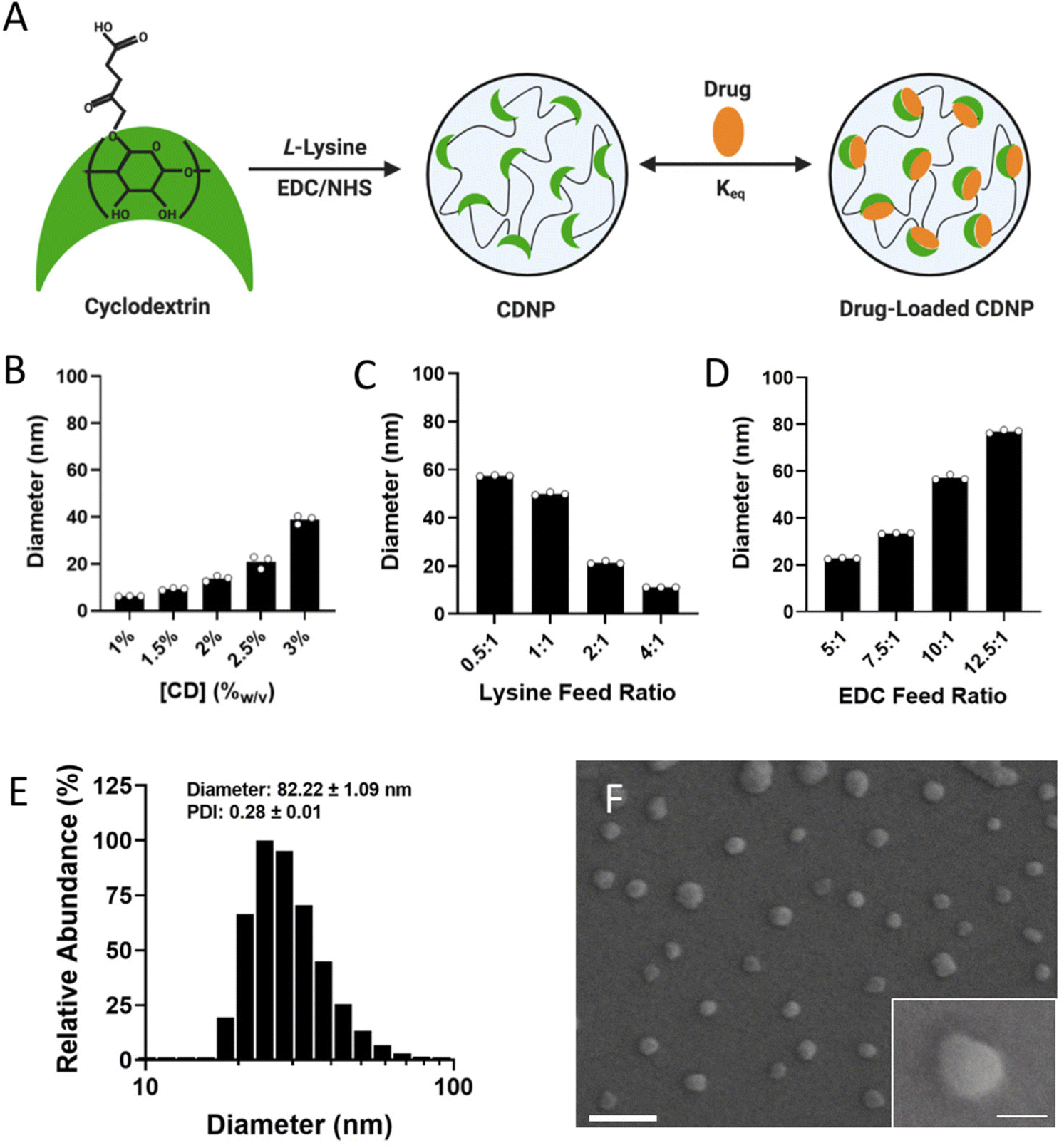 | ||
| Fig. 1 Development and characterization of CDNPs. (A) Schematic of cyclodextrin nanoparticle (CDNP) preparation through EDC/NHS-mediated crosslinking of succinyl-β-cyclodextrin by L-lysine with subsequent drug loading by guest–host interaction. CDNP diameter dependence on CD concentration (B; 10:1 EDC, 0.5:1 lysine), the molar ratio of lysine to succinyl groups (C; 3.3%w/v CD, 10:1 EDC), and the molar ratio of EDC to succinyl groups (D; 3.3%w/v CD, 0.5:1 lysine). (E and F) CDNP characterization (3.3%w/v CD, 0.5:1 lysine, and 12.5:1 EDC). (E) Number average histogram of particle size. Inset values: z-average diameter and polydispersity index (PDI); mean ± SD. (F) Corresponding representative scanning electron microscopy images. Scale bar: 200 nm. Inset: higher resolution image of a single CDNP. Scale bar: 50 nm. | ||
Polymer-nanoparticle interactions form a shear-thinning and self-healing hydrogel
We sought to develop a shear-thinning and self-healing hydrogel by leveraging the high affinity supramolecular interaction of CD at the nanoparticle surface with Ad. Here, hyaluronic acid (HA, MW = 82 kDa or 337 kDa) was exchanged against Dowex-100 resin and neutralized by tetrabutylammonium hydroxide to yield HA-TBA (Fig. S1†), required for subsequent anhydrous reaction. HA was pendantly modified by 1-adamantane acetic acid, where the degree of substitution was controlled by altering the molar ratio of BOC2O catalyst to HA repeat units. The guest-modified polymers were prepared having a range of functionalization determined by 1H-NMR (Fig. S2†): 10% (337 kDa Ad-HA (Low)), 18% (82 kDa Ad-HA (Low)), and 43% (82 kDa Ad-HA (High)). These degrees of substitution were selected to allow for variation in guest–host crosslink density between 82 kDa low and high modifications. Additionally, varying polymer molecular weight affords a similar number of potential crosslinking sites between high and low molecular weight polymers, where 337 kDa Ad-HA (Low) and 82 kDa Ad-HA (High) have an average of 85 and 88 Ad per HA macromer, respectively. The higher molecular weight polymer, however, may better span inter-particle distance to facilitate subsequent hydrogel formation. The relative importance of guest-modification density and polymer molecular weight were subsequently investigated.The guest-polymers prepared serve as a crosslinker between host (CDNP) particles to yield polymer-nanoparticle hydrogels that self-assemble through guest–host crosslink formation (Fig. 2A). Separate components, Ad-HA and CDNP, were initially viscous solutions. After their mixing, rapid formation of a viscoelastic solid was qualitatively observed, as was an increase in the storage (G′) and loss (G′′) moduli by several orders of magnitude (Fig. 2B). Interestingly, we observed that hydrogel properties were time dependent, exhibiting increases in moduli after room temperature incubation over several days. While individual guest–host complex formation is rapid, it is likely that polymer relaxation is required for thermodynamic equilibrium to be achieved. To quantitatively investigate this annealing behavior, rheological properties were assessed over a span of 14 days (Fig. S3†). A moderate increase in moduli was observed up to day 3, with subsequent stabilization thereafter. For following studies, hydrogels were therefore annealed for 3 days prior to evaluation.
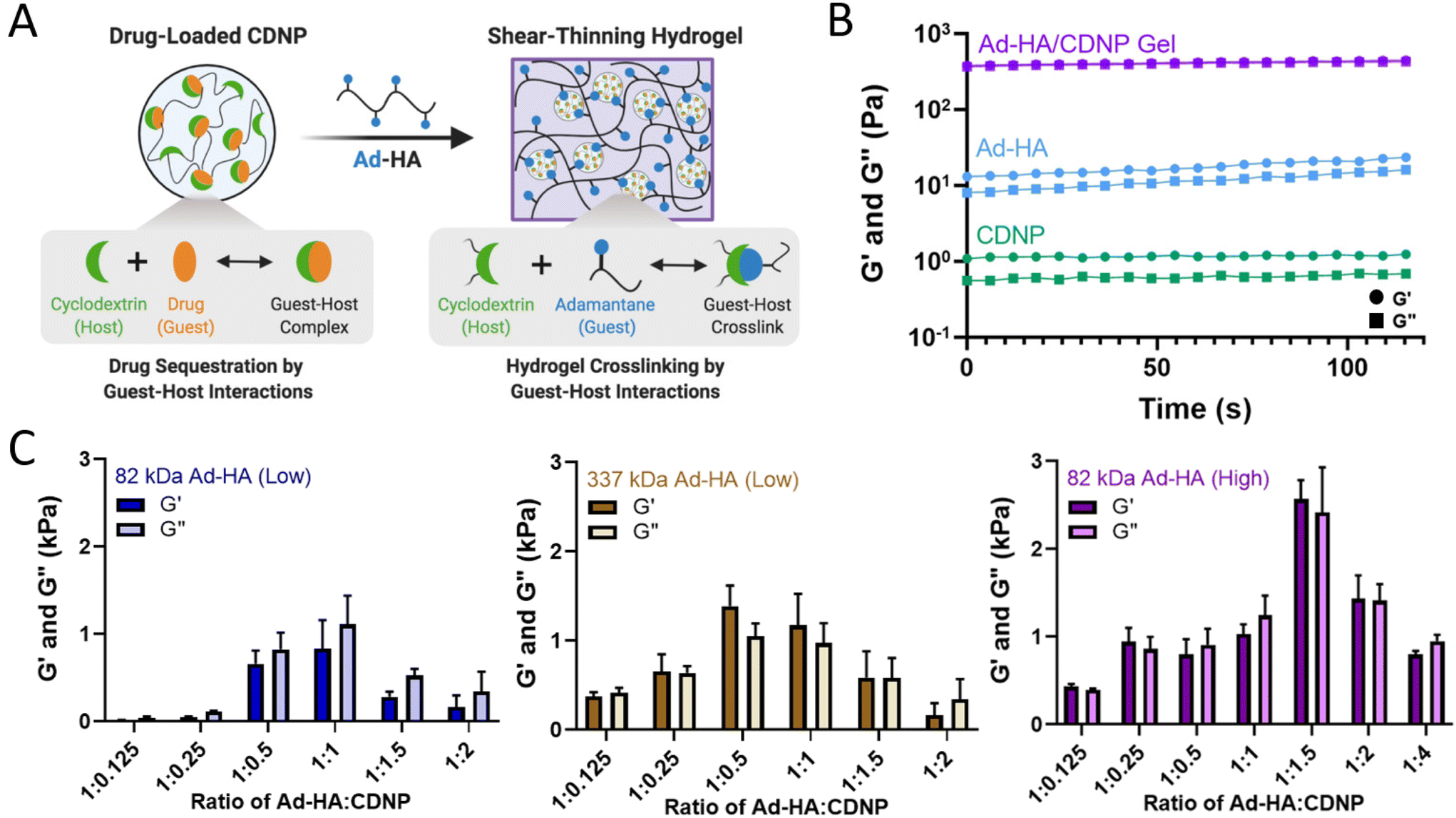 | ||
| Fig. 2 Self-assembly of polymer-nanoparticle hydrogels is composition dependent. (A) Schematic of drug-loaded hydrogel assembly by guest–host interaction. (B) Oscillatory time sweeps of individual components (20%w/v CDNP, green and 7.5%w/v Ad-HA, blue) and formed polymer-CDNP hydrogel (1:1.5 ratio of 7.5%w/v Ad-HA: 20%w/v CDNP, purple); storage (G′, circles), and loss (G′′, squares) moduli at 1.0 Hz, 1.0% strain. (C) Storage and loss moduli of hydrogels formed from 82 kDa Ad-HA (low, left), 337 kDa Ad-HA (low, middle), and 82 kDa Ad-HA (high, right) at varying volumetric ratios of Ad-HA:CDNP. Time sweeps were performed at 10.0 Hz, 1.0% strain; mean ± SD, n = 2. | ||
We further investigated the viscoelastic properties of the hydrogels via oscillatory shear rheology. As the number of host sites accessible for guest-polymer binding at the nanoparticle surface is indeterminate, the ratio of guest to host components was empirically investigated (Fig. 2C). At high ratios of Ad-HA:CDNP, low moduli were observed – potentially due to polymeric coating of the CDNPs as opposed to desirable interparticle crosslinking. Conversely, high ratios of Ad-HA:CDNP likewise formed hydrogels with low moduli, presumably due to a low density of guest–host interactions required for stable gel formation. Greatest moduli were observed for an intermediate polymer to nanoparticle ratio, which were dependent on polymer modification and molecular weight. Across these datasets, it is apparent that crosslink density is critical to stable gel formation as increased polymer modification yielded increased hydrogel moduli when molecular weight remained constant. Consistent with these observations, moduli were likewise dependent on component concentration (Fig. S4†). Moreover, increasing polymer molecular weight afforded hydrogels with moderately increased moduli, relative to 82 kDa Ad-HA (Low). However, most robust hydrogel formation was observed at a relatively high ratio of Ad-HA:CDNP which is undesirable for later drug-loading applications. Hydrogels composed of 82 kDa Ad-HA (High) exhibited the most robust moduli (G′ = 2.72 ± 0.018 kPa) at a 1:1.5 ratio of Ad-HA:CDNP and provide a relatively high CDNP content essential for later therapeutic delivery. In sum, these studies demonstrate a method of tuning the mechanical properties of associative polymer-nanoparticle hydrogels and highlight the dominant role of dynamic bond density as a critical factor in stable hydrogel formation. More specifically, these studies revealed that in our particular hydrogel system, 82 kDa Ad-HA (High) affords the most robust hydrogel formation and does so at polymer-nanoparticle ratios that are beneficial for subsequent drug loading.
Hydrogels formed through dynamic bonds assume varying structural integrity under strained conditions. As the bonds are continually broken by the application of sufficient external force, bond disassembly enables fluid-like flow and shear-thinning hydrogel injection. Continuous flow experiments (Fig. 3A) were used to examine viscosity and stress over an increasing shear rate. As expected for shear-thinning materials, the hydrogels displayed reduced viscosity at high shear-rates and a resulting plateau in the observed shear stress. To examine the potential for rapid self-healing as a result of dynamic bond self-healing, hydrogels were subject to repeated cycles of high (500%) and low (1%) strain, representing processes of disassembly when injected through a syringe needle (shear-thinning) and subsequent re-assembling (self-healing). Under high strain conditions, fluid-like behavior (G′′ > G′) was observed, with recovery of solid properties observed within seconds after onset of low strain conditions (Fig. 3B). Results are consistent with oscillatory strain sweeps (0.01–1000% strain; 10 Hz) wherein a yield stress of approximately 10% was observed, above which the hydrogels underwent forceful disassembly and fluid-like behavior (Fig. 3C). In sum, the hydrogels exhibit fluid-like flow under high strain for ease of injection (shear-thinning). When the strain is removed, the hydrogels rapidly recover such as to allow for retention within the tissue.14 As a result, hydrogels could be pre-formed within a syringe and easily injected as liquids that rapidly re-solidify (Fig. 3D and Fig. S5†).
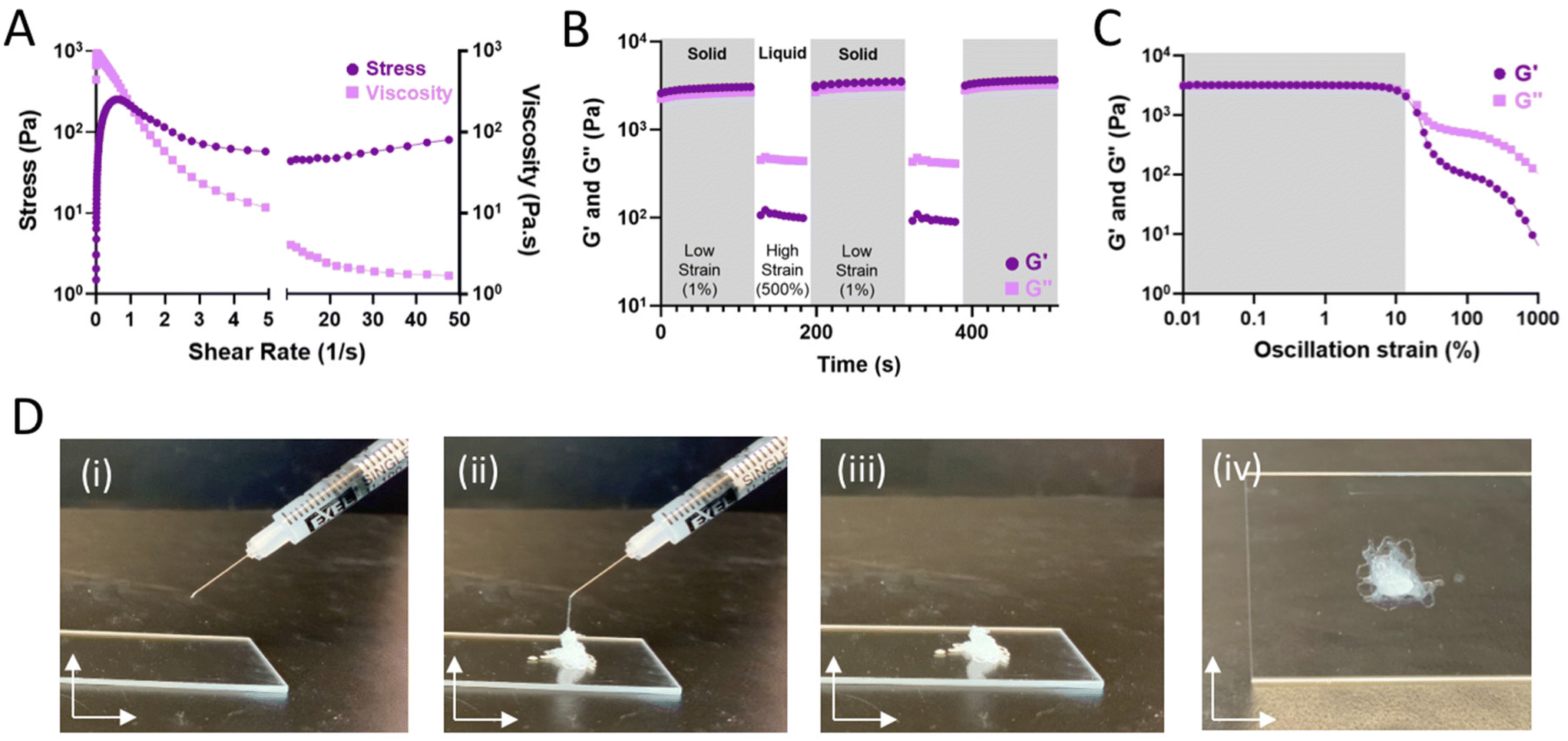 | ||
| Fig. 3 Guest–host assembled polymer-nanoparticle hydrogels are shear-thinning and self-healing for ease of injection. (A) Stress (dark purple, circle) and viscosity (light purple, square) of 82 kDa Ad-HA (high) as a function of shear rate. (B) Shear-thinning and self-healing characterization of optimally formulated 82 kDa Ad-HA (high) hydrogel (7.5%w/v) under repeated deformation at 1.0% (low, shaded) and 500% (high) strain at 10.0 Hz; G′ (dark purple, circle), G′′ (light purple, square). (C) Strain amplitude sweep of 82 kDa Ad-HA (high); G′ (dark purple, circle), G′′ (light purple, square). (D) Representative images of hydrogel injection; 28G needle, 1 mL syringe. | ||
Drug screening to identify immunomodulatory small molecule drugs
We sought to identify small molecule drugs capable of modulating MF phenotype for application in the context of tissue injury and inflammatory disease. Specifically, we looked to identify a candidate that could suppress the damaging M1-like MF phenotype following injury and promote the reparatory M2-like MF phenotype for injury resolution. Therefore, we developed a two-step screening process that first identified pharmacological inhibitors of M1-like transcription using RAW-Blue™ cells, which are a readily available and inexpensive reporter cell line that incorporates a SEAP reporter construct downstream of both AP-1 and NF-κB promoter regions, and thus reports pro-inflammatory activation. The cell line is frequently used to evaluate both agonists and antagonists of M1-like polarization.53–55 While some approaches have been successful in identifying compounds for MF re-polarization such as through morphological analysis,41,56 applications have focused on redirection towards an M1 and not an M2 state. Currently, few approaches exist to directly assay for M2-like polarization. In the second step of the screening process, we therefore used transcriptional analysis via qPCR and multiplex analysis (nanoString) to identify compounds that promote canonical M2-like MF gene expression.We sought to approach initial drug screens through an unbiased evaluation that compared dozens of drugs across multiple drug classes as such direct comparison is currently lacking from literature. Suppression of M1-like activation was therefore examined for a targeted library of 45 small molecule drug candidates that span a diverse set of specific drugs and drug classes that have established immunosuppressive or immunomodulatory capacity, including glucocorticoids, statins, and PPAR inhibitors, among others. For primary evaluation in RAW-Blue™ cells, cells were stimulated by zymosan, a toll-like receptor 2 (TLR2) agonist that mimics sterile inflammation,57 and concurrently treated with the drugs of interest. HDAC inhibitors examined had no effect and were excluded from the data presented and subsequent studies. Other drug classes had varying degrees of efficacy (Fig. 4), with select drugs exhibiting sub-micromolar inhibition of M1-like activation. Viability of RAW264.7 cells was further examined following drug treatments (Fig. S6†). Notable losses of viability were not observed; initial results are therefore a direct result of desired pathway inhibition and not an artifact of drug toxicity. Within each class, drugs were rank-ordered based on the largest cumulative suppression of M1-like activation across doses, and the most potent drug from each class was selected for further in vitro phenotyping studies.
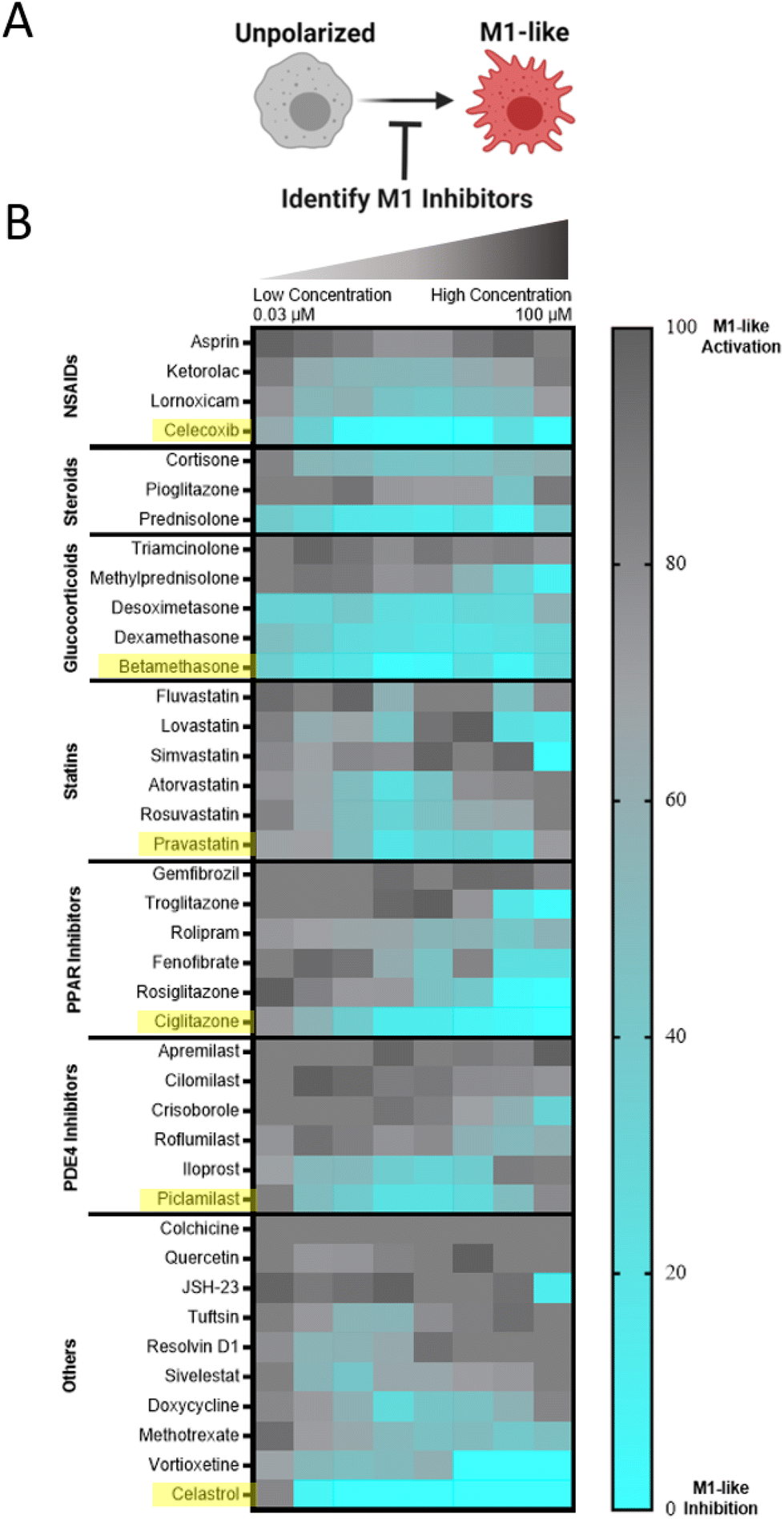 | ||
| Fig. 4 Initial drug screening identifies pharmacological inhibitors of M1-like MF activation. (A) RAW-Blue™ cells were concurrently treated by zymosan (100 μg mL−1) and the indicated drug, spanning a dose response from 100 μM to 31.6 nM in half-log titration to identify M1-like inhibitors. (B) Heatmap of compound bioactivities in reporter cells. Results represent the mean of n = 4 independent samples, subsequent to subtraction of untreated controls and normalization to zymosan-treated cells such that a lighter blue color indicates desired inhibition of M1-like response. Within each drug class, compound bioactivity is rank ordered according to cumulative suppression of M1-supression across all doses. Compounds selected for subsequent evaluation are indicated in yellow. | ||
After initial inhibitory screening, the effects of six selected drugs (celecoxib, betamethasone, pravastatin, ciglitazone, piclamilast, and celastrol) on MF phenotype were further scrutinized by transcriptional analysis in zymosan-activated RAW264.7 MF in order to determine if these drug candidates further promote M2-activation. Canonical markers of both M1-like (nos2, il6, il12b) and M2-like (mrc1, arg1, il10) phenotypes were included (Fig. 5A and B). Two drugs, piclamilast and celastrol, significantly downregulated M1-like genes with concurrent upregulation of M2-like genes. Notably, these drugs showed the greatest decrease in il12b, a pro-inflammatory cytokine critical to adaptive immune activation,58 as well as the greatest increase in mrc1 and il10. MRC1 (CD206) is an established marker of M2-like activation, both in humans and mice that is correlated with decreases in MF migration and serum levels of inflammatory proteins,59 whereas IL10 is a potent anti-inflammatory cytokine critical towards wound healing and tissue repair.60 The relatively simple two-step screening approach therefore identified two drug candidates that exhibit potent anti-inflammatory action via high-throughput reporter screens, and a subset of the best-in-class drug candidates (piclamilast and celastrol) biased cells towards an M2-like activation state even during opposing pressure by TLR agonization.
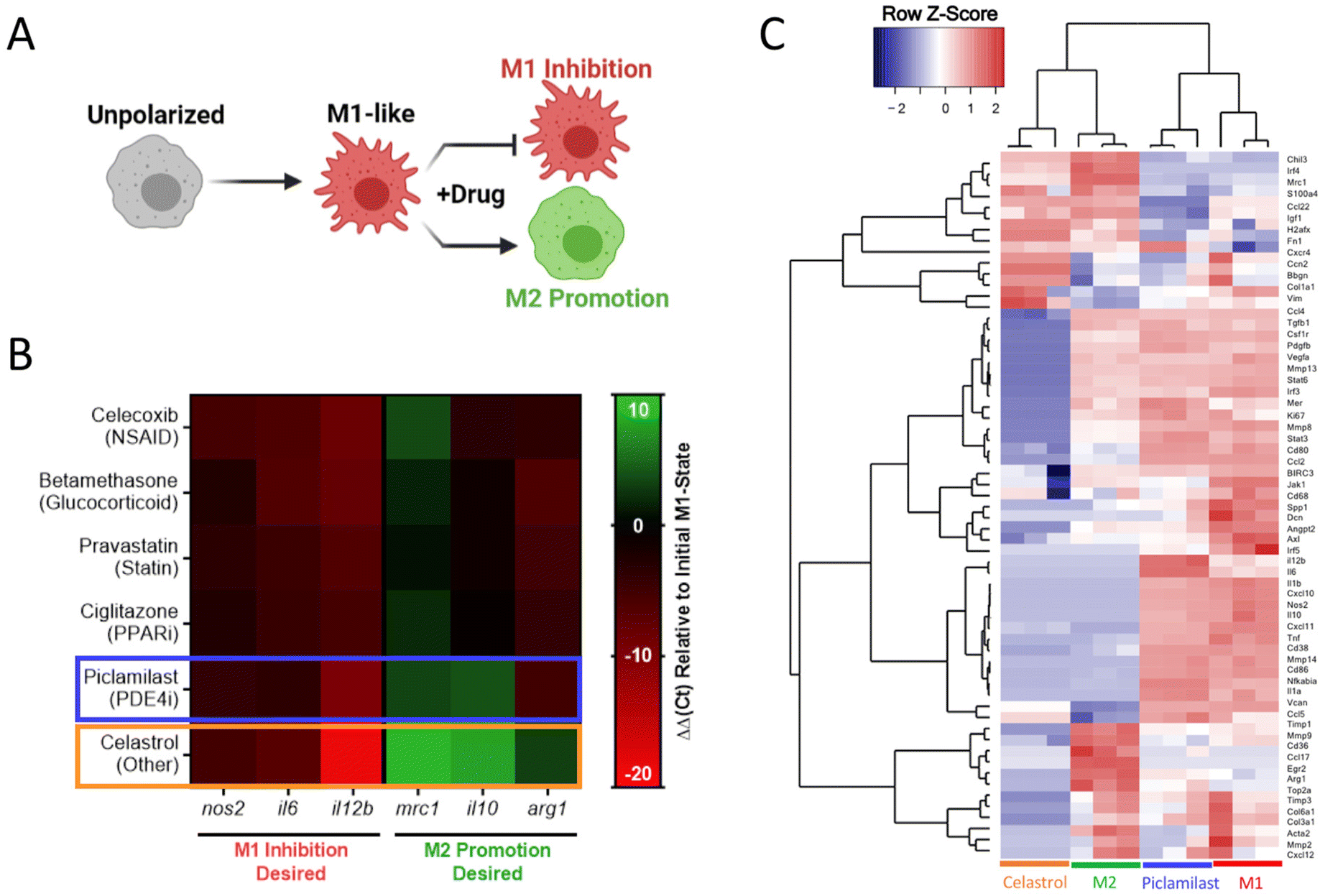 | ||
| Fig. 5 Transcriptional analysis of murine MF phenotype. (A) RAW264.7 cells were concurrently treated by zymosan (100 μg mL−1) and the indicated drug (1 μM) to identify M1-like inhibition and M2-like promotion for all datasets presented. (B) Heat map of gene expression following 24 h treatment, expressed as ΔΔCt relative to hprt and zymosan-treated controls. Compounds selected for further evaluation are boxed in blue (piclamilast) and orange (celastrol). Results represent the mean of n = 3, and are normalized to a housekeeping hprt gene, as well as an internal M1-like control. (C) Heatmap of nanoString data represented as the row Z-score of log-transformed normalized data. Genes that were not expressed above background are excluded from presentation. Bone marrow derived macrophages (BMDMs) were subject to celastrol and piclamilast treatment (1 μM) with concurrent stimulation by zymosan (100 μg mL−1). M1 and M2 controls are included for reference. Accompanying cluster analysis and dendogram most closely associate celastrol treatment with M2 controls, while piclamilast treatments are intermediate to M1 and M2 control phenotypes. | ||
Further in-depth transcriptional analysis of the two drugs candidates was performed by multiplexed gene expression analysis (nanoString) in zymosan-activated bone marrow derived MF (BMDMs) treated with piclamilast and celastrol. The analysis measured gene expression across 91-genes that represent multiple M1-like and M2-like markers, as well as genes associated with ECM regulation, fibroblast activity, angiogenesis, and immune signaling pathways (Table S1†). Cluster analysis and corresponding dendograms (Fig. 5C) most closely associate celastrol treatment with M2 controls, while piclamilast treatment was intermediate to M1 and M2 control phenotypes. Relative to M1 controls, downregulation of M1-associated genes was typically greater for celastrol than for piclamilast, with expression levels approaching or in some cases exceeding that of M2-like (IL-4 treated) controls (Fig. S7A†). A number of these genes are implicated in chronic inflammation and impaired tissue healing. For example, CXCL10 (IP-10) is a MF-derived chemokine critical for the recruitment of inflammatory T-cell subsets.61 Celastrol likewise downregulated pro-inflammatory cytokines, including tnf, il1b, and il6 that perpetuate continued tissue injury and are common biomarkers of organ failure.62–64 Celastrol also preferentially modulated the expression of M2-associated genes (Fig. S7B†).65 For example, celastrol upregulated irf4 and ccl22 to levels consistent with M2-like controls; IRF-4 suppresses innate immune signaling through inhibition of the TLR/MyD88 pathway.66 Interestingly, celastrol treatment also drastically downregulated tgfb1, consistent with prior indications of its use as an antifibrotic agent and further motivating a potential use in tissue injury applications.67,68 For these reasons, celastrol was chosen as the final drug of interest for continuing studies.
While results in cell lines and primary murine cells are promising, there exist nuanced differences between the murine and human inflammatory programs that may hinder translation and warrant early investigation. To ensure drug activity across species, transcriptional analysis was further performed in LPS-activated MF derived from peripheral blood mononuclear cells (PBMCs) using a panel of human genes (Table S2†). The choice of LPS instead of zymosan as a pro-inflammatory stimulus was made to allow comparison to other studies of human MF, which more commonly use LPS. Subsequent to LPS activation, cells were treated with varying concentrations of celastrol (1–10 μM). Both cluster analysis (Fig. S8†) and expression of specific genes (Fig. 6) indicated dose response across celastrol concentrations, with desired upregulation of M2-associated genes and downregulation of M1-associated genes when compared to LPS-treated controls. These include, for example, decreased expression of the T-cell costimulatory signal cd80 that is critical for TH1 activation. The expression of inflammatory chemokines (ccl8) and cytokines (il1b, il6), and immunoregulatory enzymes (ido1) were likewise suppressed in a dose-dependent fashion. In contrast, the expression of M2-associated genes was typically upregulated, including for cell surface markers (mrc1) as well as chemokines and their receptors (ccl18, ccl22, cxcr4). Overall, these findings are consistent with our observations in murine cell lines and primary cells, indicating the immunomodulatory capacity of celastrol is not species specific.
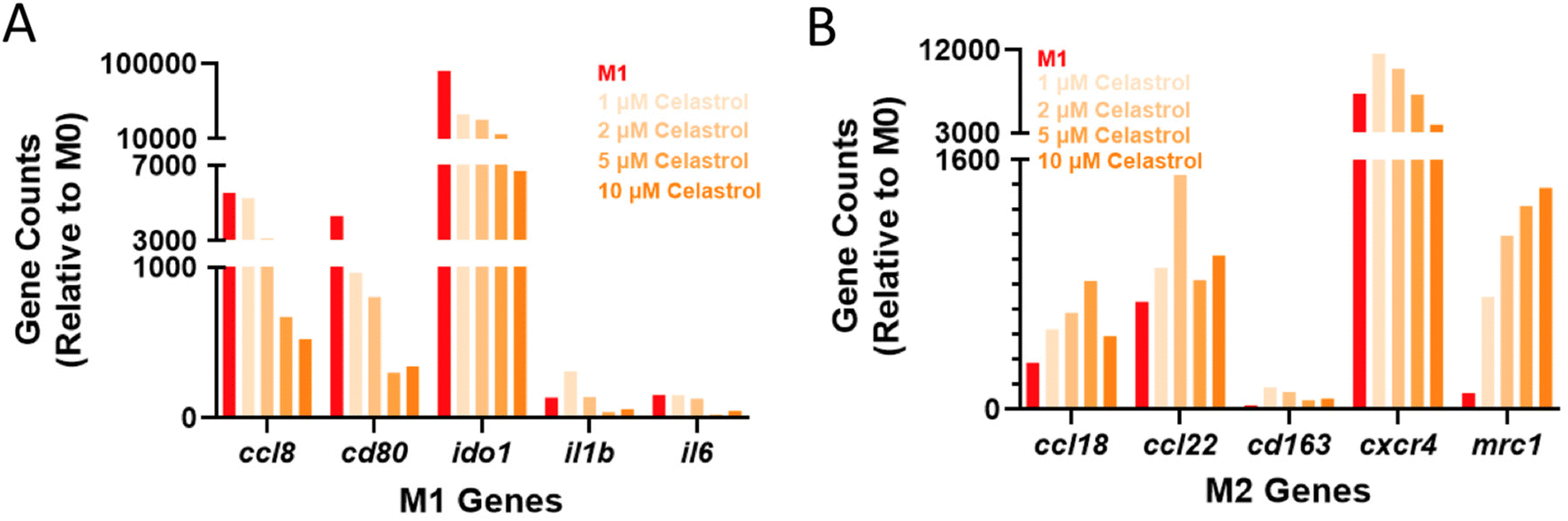 | ||
| Fig. 6 Response of human MF to celastrol treatment. Human monocyte-derived MF were subject to LPS activation (100 ng mL−1) and celastrol treatment at varying doses (1–10 μM). The expression of both M1-associated (A) and M2-associated (B) genes is presented as gene counts relative to untreated (M0) controls; M1 controls (red) are included for reference. | ||
Drug encapsulation and release
While celastrol has moderate oral bioavailability which can be enhanced by formulation,69–71 it displays significant sex-dependences in systemic bioavailability69–71 and such administration is associated with hepatotoxicity,72,73 hematopoietic system toxicity,74,75 nephrotoxicity,76 and undesirable biodistribution.77–79 Moreover, local biomaterial-delivery strategies afford an opportunity to reduce clearance rates, prolong release, concentrate drug dose at the site of action, and enhance cell-specific delivery.80,81 For these reasons, we examine here its potential as a suitable cargo for CDNP encapsulation and delivery from the polymer-nanoparticle hydrogels (Fig. 7A). CD is amenable to guest–host complex formation with a variety of hydrophobic small molecules, particularly polycyclic and aromatic structures with suitable size for host inclusion. In guest–host delivery systems, the diffusive release of guest drug molecules is prolonged by complex dissociation and subsequent re-association events that can sustain release for a period of weeks to months.82–85 We first sought to establish that celastrol is a suitable molecular guest for complexation with CD, necessary for inclusion within the CDNP drug carrier. The equilibrium binding constant (Keq) was determined to be 0.474 mM by equilibrium analysis using surface plasmon resonance (Fig. 7B). These results are consistent with other polyaromatic guests, including tryptophan and doxorubicin, which are typically reported to have single micromolar Keq that is sufficient for drug delivery applications.30,86–89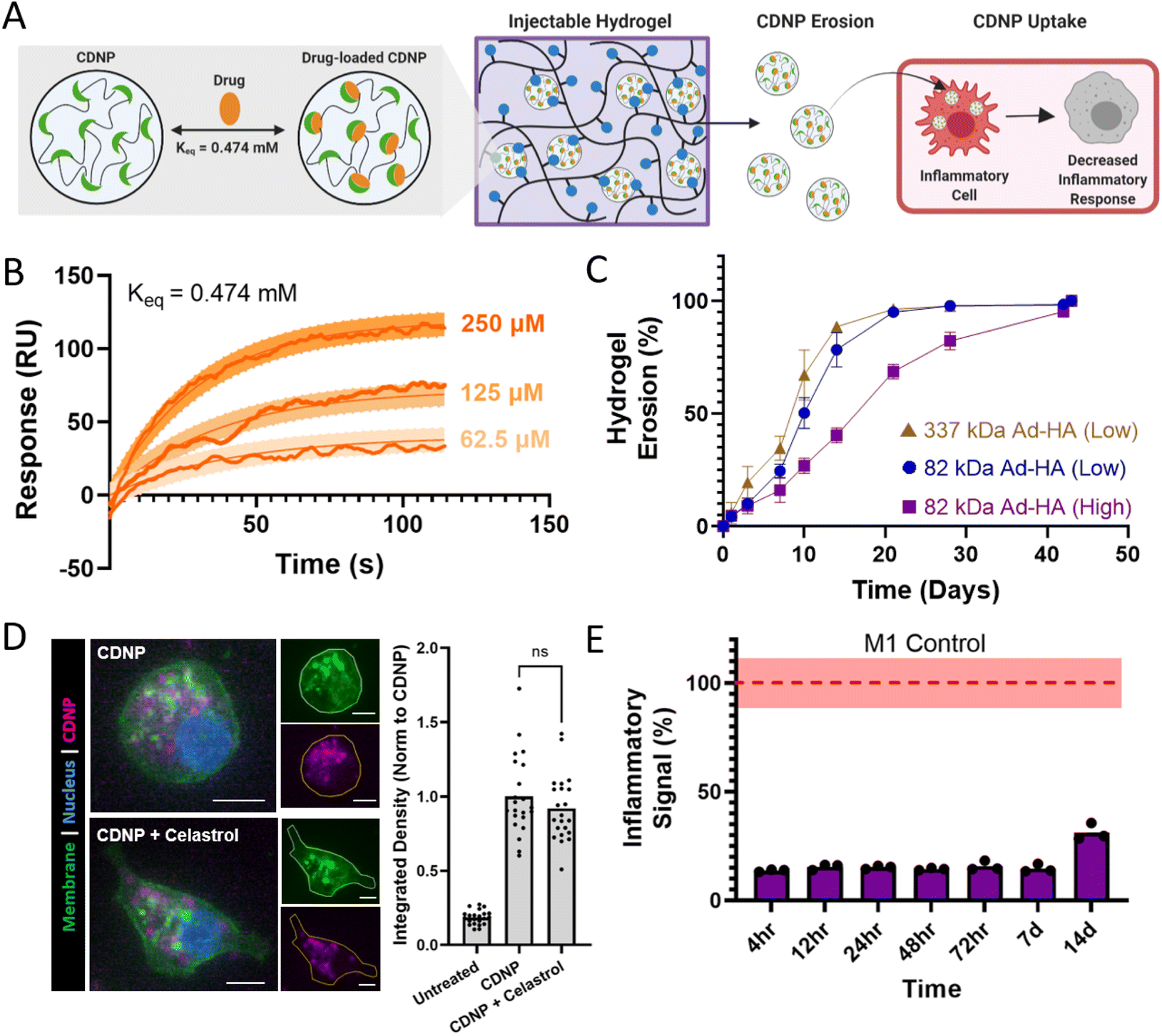 | ||
| Fig. 7 Therapeutic nanoparticle erosion enables long-term modulation of MF phenotype. (A) Schematic of drug loaded CDNP release from shear-thinning hydrogels and uptake by MF for desired decrease in inflammatory response. (B) Binding sensograms between celastrol and CD, assessed at increasing concentrations of celastrol. (C) Cumulative erosion for hydrogels formed from 337 kDa Ad-HA (low, brown), 82 kDa Ad-HA (low, blue), and 82 kDa Ad-HA (high, purple); mean ± SD, n = 4. (D) RAW264.7 cell uptake of unloaded (CDNP) and drug-loaded (CDNP-Cel) nanoparticles from media conditioned by 82 kDa Ad-HA (high) hydrogel erosion. Representative images (left) show punctate accumulation of CDNP-AF555. Scale bars, 10 μm. Quantification of fluorescence per cell (right), normalized to unloaded CDNP. (E) Anti-inflammatory activity of drug release samples, performed in RAW Blue™ cells by concurrent zymosan stimulation (100 μg mL−1) and treatment with conditioned media from 82 kDa Ad-HA (high) hydrogels; p value < 0.0001 for all samples relative to zymosan-treated controls (dashed line, red). Data represent the mean ± SD, n = 3. | ||
Supramolecularly assembled hydrogels typically degrade via surface erosion, with dependence on network structure and composition.15,47 We therefore anticipated that nanoparticle release from the hydrogels would be both a means of hydrogel degradation and a potential method of cell-targeted delivery, owing to the rapid uptake of CDNPs by phagocytic immune cells that occurs both in vitro and in vivo.41,90 Hydrogel erosion was assessed over a six week period at 37 °C, spanning a range of hydrogel compositions. Across these formulations, hydrogels typically exhibited slow erosion over the course of greater than one month, with 82 kDa Ad-HA (High) degrading slower than less effectively crosslinked network architectures (Fig. 7C). Degradation profiles were relatively independent of polymer concentration, but more highly dependent on ratio of guest to host components (Fig. S9†). Notably, reduced relative CDNP concentrations led to rapid erosion, attributable to insufficient hydrogel crosslinking. The release of soluble polymer by either degradation or erosion mechanisms warrants investigation of polymer cytocompatibility. Cells were therefore treated with media containing the separate hydrogel components, Ad-HA and CDNP (Fig. S10†). Cell viability was unaffected by soluble CDNP. While relatively high concentrations of soluble Ad-HA (>1.0%w/v) significantly reduced cell viability, effects were moderate and observed only at concentrations in excess of what would result from hydrogel erosion. These results are in agreement with the literature concerning the use of guest–host hydrogels composed of Ad-HA for both in vitro and in vivo studies without detriment to exposed cells or tissues.14
Celastrol-loaded hydrogel formulations were next prepared by simple mixing of celastrol (Cel) and CDNP under aqueous conditions and the resulting drug-loaded nanoparticle (CDNP-Cel) were used directly in hydrogel formation. Importantly, drug incorporation did not alter the rheological properties (Fig. S11†), including across all hydrogel formulations examined. Reductions in moduli might otherwise indicate disruption of the guest–host mediated crosslinking mechanism, such as by competitive binding. The absence of perturbation in the crosslinking mechanism is attributable to the disparity in binding affinity between Ad and celastrol.
Next, we assessed the ability of CDNP-Cel to be released from the hydrogels by erosion and subsequently uptaken by MF. For this purpose, CDNPs were fluorescently labeled by Alexa Fluor 555 (CDNP-AF555). Both drug loaded and unloaded CDNP-AF555 nanoparticles were formulated in 82 kDa Ad-HA (High) hydrogels that were subsequently allowed to erode in media. RAW264.7 cells were treated with the conditioned media prior to imaging and quantification (Fig. 7D). Cells exhibited punctate accumulation of CDNP-AF555, consistent with prior observations of rapid CDNP uptake and endolysosomal accumulation in phagocytic immune cells.90 While MF polarization has been noted to impact nanoparticle uptake,91,92 CDNP-AF555 uptake was unhindered by celastrol loading. The release of CDNP-Cel by hydrogel erosion is therefore an efficient route of cell-targeted drug delivery from the injectable scaffold.
Finally, we investigated the ability of the drug-laden hydrogels to provide bioactive concentrations of CDNP-Cel release, capable of modulating cell phenotype over time in vitro (Fig. 7E). Media was again conditioned by the degradation of celastrol-loaded hydrogels over a period of two weeks. RAW-Blue™ cells were concurrently stimulated with zymosan and treated with the conditioned media. Inflammatory pathway transduction was attenuated by >80% for one week and more moderately out to day 14. Similar results were observed for drug release from 82 kDa Ad-HA (Low) and 337 kDa Ad-HA (Low) hydrogels (Fig. S12†). Both diffusive and erosion processes likely contribute to celastrol release, as has been observed in other polymer-nanoparticle composites with encapsulated protein cargo.15 For hydrogel formulations with a reduced crosslinking density, such as those formed at reduced polymer or CDNP concentration, inhibitory drug affects waned earlier with moderate recovery of inflammatory signaling beginning at days 3–7 (Fig. S13†). This attributed to exhaustion of CDNP-Cel supply within the hydrogel depot, consistent with the observed rapid erosion of these formulations, as well as an inability to retain the drug within the hydrogel in the case of reduced CDNP concentrations. Overall, these findings demonstrate the release of CDNP-Cel from the hydrogels is a promising strategy for MF-targeted drug delivery that can bias cell phenotype over the course of weeks. This timespan coincides well with the established timeline of exuberant inflammation that frequently underlies failed tissue repair programs, particularly in organ injury,93,94 and is therefore a promising immunoregenerative strategy towards such applications.
Conclusions
Here, we have developed a shear-thinning and self-healing hydrogel formed through the simple mixing of host nanoparticles (CDNPs) and a guest-modified polymer (Ad-HA) in aqueous conditions. The supramolecular association of the guest and host components drives rapid hydrogel formation, while retaining host sites in the nanoparticle interior for drug retention. Physical properties of the hydrogels formed were dependent on composition, including crosslink density, Ad-HA molecular weight, polymer concentration, and the ratio of guest to host components. Across formulations, hydrogels demonstrated shear-thinning and injectable behavior with subsequent rapid self-healing. Rheological properties were independent of drug-loading. After identifying an optimal hydrogel formulation with robust mechanical properties and resistance to erosion, we demonstrated the system's ability to sustain release of a model immunomodulatory drug, celastrol, which was identified through a multi-step screening process and retained via specific inclusion complex formation within the host nanoparticle. The release of celastrol-loaded nanoparticles from the hydrogel arrested pro-inflammatory signaling in MF reporter assays for two weeks, indicating a great potential for long-term immune modulation. In sum, the material platform developed harnesses specific guest–host interactions for both hydrogel assembly and drug sequestration within the host nanoparticle. The polymer-nanoparticle hydrogel presents an injectable controlled release system that can modulate cell function by the continual release of drug-laden nanoparticles – a function that can be readily tuned for use in a number of applications.Author contributions
Shreya S. Soni: conceptualization, methodology, data collection and analysis, writing – original draft, writing – review and editing. Arielle M. D'Elia: data collection and analysis, writing – original draft. Abdulrahman Alsasa: data collection and analysis. Sylvia Cho: data collection and analysis. Tina Tylek: data collection and analysis, writing – original draft, writing – review and editing. Erin M. O'Brien: resources and methodology, writing – review and editing. Ricardo C. Whitaker: resources and methodology, writing – review and editing. Kara L. Spiller: guidance, resources, and writing – review and editing. Christopher B. Rodell: conceptualization, resources, methodology, data analysis, writing – original draft, writing – review and editing, supervision, and funding acquisition.Conflicts of interest
C. B. R. is an inventor on a patent filed by Partners Healthcare pertaining to nanoparticle development. The remaining authors declare no competing interests.Acknowledgements
We gratefully acknowledge the assistance of Kate Vanderburgh, PhD of the Materials Characterization Core at Drexel University for assistance with SEM imaging, as well as Azad Ahmed, MD of the Genomics Core Facility at Drexel University's College of Medicine for assistance with nanoString data collection. This work was supported by Startup Funds from the School of Biomedical Engineering, Science and Health Systems at Drexel University (C. B. R.), NIH R35GM147184 (C. B. R.), and NIH R01HL130037 (K. L. S).References
- W. J. Geldenhuys, M. T. Khayat, J. Yun and M. A. Nayeem, Drug Delivery and Nanoformulations for the Cardiovascular System, Res. Rev. Drug Deliv., 2017, 1(1), 32–40 CAS.
- Y. Li, J. Rodrigues and H. Tomás, Injectable and biodegradable hydrogels: gelation, biodegradation and biomedical applications, Chem. Soc. Rev., 2012, 41(6), 2193–2221 RSC.
- S. J. Buwalda, T. Vermonden and W. E. Hennink, Hydrogels for Therapeutic Delivery: Current Developments and Future Directions, Biomacromolecules, 2017, 18(2), 316–330 CrossRef CAS PubMed.
- H. Zhou, C. Liang, Z. Wei, Y. Bai, S. B. Bhaduri, T. J. Webster, L. Bian and L. Yang, Injectable biomaterials for translational medicine, Mater. Today, 2019, 28, 81–97 CrossRef CAS.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Shear-thinning hydrogels for biomedical applications, Soft Matter, 2012, 8(2), 260–272 RSC.
- D. J. Overstreet, D. Dutta, S. E. Stabenfeldt and B. L. Vernon, Injectable hydrogels, J. Polym. Sci., Part B: Polym. Phys., 2012, 50(13), 881–903 CrossRef CAS.
- C. B. Rodell, J. E. Mealy and J. A. Burdick, Supramolecular Guest–Host Interactions for the Preparation of Biomedical Materials, Bioconjugate Chem., 2015, 26(12), 2279–2289 CrossRef CAS PubMed.
- F. Picchioni and H. Muljana, Hydrogels Based on Dynamic Covalent and Non Covalent Bonds: A Chemistry Perspective, Gels, 2018, 4(1), 21 CrossRef PubMed.
- M. Ehrbar, S. C. Rizzi, R. G. Schoenmakers, B. San Miguel, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomolecular Hydrogels Formed and Degraded via Site-Specific Enzymatic Reactions, Biomacromolecules, 2007, 8(10), 3000–3007 CrossRef CAS PubMed.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Biofunctional hydrogels based on host–guest interactions, Polym. J., 2020, 52(8), 839–859 CrossRef CAS.
- S. Correa, A. K. Grosskopf, H. Lopez Hernandez, D. Chan, A. C. Yu, L. M. Stapleton and E. A. Appel, Translational Applications of Hydrogels, Chem. Rev., 2021, 121(18), 11385–11457 CrossRef CAS PubMed.
- O. S. Fenton, M. W. Tibbitt, E. A. Appel, S. Jhunjhunwala, M. J. Webber and R. Langer, Injectable Polymer–Nanoparticle Hydrogels for Local Immune Cell Recruitment, Biomacromolecules, 2019, 20(12), 4430–4436 CrossRef CAS PubMed.
- F. Vashahi, M. R. Martinez, E. Dashtimoghadam, F. Fahimipour, A. N. Keith, E. A. Bersenev, D. A. Ivanov, E. B. Zhulina, P. Popryadukhin, K. Matyjaszewski, M. Vatankhah-Varnosfaderani and S. S. Sheiko, Injectable bottlebrush hydrogels with tissue-mimetic mechanical properties, Sci. Adv., 2022, 8(3), eabm2469 CrossRef CAS PubMed.
- C. Loebel, C. B. Rodell, M. H. Chen and J. A. Burdick, Shear-thinning and self-healing hydrogels as injectable therapeutics and for 3D-printing, Nat. Protoc., 2017, 12(8), 1521–1541 CrossRef CAS PubMed.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Self-assembled hydrogels utilizing polymer–nanoparticle interactions, Nat. Commun., 2015, 6(1), 6295 CrossRef CAS PubMed.
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks, Nat. Mater., 2015, 14(7), 737–744 CrossRef CAS PubMed.
- A. E. Widener, M. Bhatta, T. E. Angelini and E. A. Phelps, Guest–host interlinked PEG-MAL granular hydrogels as an engineered cellular microenvironment, Biomater. Sci., 2021, 9(7), 2480–2493 RSC.
- L. Riley, L. Schirmer and T. Segura, Granular hydrogels: emergent properties of jammed hydrogel microparticles and their applications in tissue repair and regeneration, Curr. Opin. Biotechnol, 2019, 60, 1–8 CrossRef CAS PubMed.
- J. E. Mealy, J. J. Chung, H. H. Jeong, D. Issadore, D. Lee, P. Atluri and J. A. Burdick, Injectable Granular Hydrogels with Multifunctional Properties for Biomedical Applications, Adv. Mater., 2018, 30(20), 1705912 CrossRef PubMed.
- G. Bovone, E. A. Guzzi, S. Bernhard, T. Weber, D. Dranseikiene and M. W. Tibbitt, Supramolecular Reinforcement of Polymer–Nanoparticle Hydrogels for Modular Materials Design, Adv. Mater., 2022, 34(9), 2106941 CrossRef CAS PubMed.
- G. A. Roth, E. C. Gale, M. Alcántara-Hernández, W. Luo, E. Axpe, R. Verma, Q. Yin, A. C. Yu, H. Lopez Hernandez, C. L. Maikawa, A. A. A. Smith, M. M. Davis, B. Pulendran, J. Idoyaga and E. A. Appel, Injectable Hydrogels for Sustained Codelivery of Subunit Vaccines Enhance Humoral Immunity, ACS Cent. Sci., 2020, 6(10), 1800–1812 CrossRef CAS PubMed.
- P. M. Ridker, B. M. Everett, T. Thuren, J. G. Macfadyen, W. H. Chang, C. Ballantyne, F. Fonseca, J. Nicolau, W. Koenig, S. D. Anker, J. J. P. Kastelein, J. H. Cornel, P. Pais, D. Pella, J. Genest, R. Cifkova, A. Lorenzatti, T. Forster, Z. Kobalava, L. Vida-Simiti, M. Flather, H. Shimokawa, H. Ogawa, M. Dellborg, P. R. F. Rossi, R. P. T. Troquay, P. Libby and R. J. Glynn, Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease, N. Engl. J. Med., 2017, 377(12), 1119–1131 CrossRef CAS PubMed.
- J.-C. Tardif, S. Kouz, D. D. Waters, O. F. Bertrand, R. Diaz, A. P. Maggioni, F. J. Pinto, R. Ibrahim, H. Gamra, G. S. Kiwan, C. Berry, J. López-Sendón, P. Ostadal, W. Koenig, D. Angoulvant, J. C. Grégoire, M.-A. Lavoie, M.-P. Dubé, D. Rhainds, M. Provencher, L. Blondeau, A. Orfanos, P. L. L'Allier, M.-C. Guertin and F. Roubille, Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction, N. Engl. J. Med., 2019, 381(26), 2497–2505 CrossRef CAS PubMed.
- Y.-C. Chen, S. F. Gad, D. Chobisa, Y. Li and Y. Yeo, Local drug delivery systems for inflammatory diseases: Status quo, challenges, and opportunities, J. Controlled Release, 2021, 330, 438–460 CrossRef CAS PubMed.
- C. Nathan and A. Ding, Nonresolving inflammation, Cell, 2010, 140(6), 871–882 CrossRef CAS PubMed.
- K. E. Martin and A. J. García, Macrophage phenotypes in tissue repair and the foreign body response: Implications for biomaterial-based regenerative medicine strategies, Acta Biomater., 2021, 133, 4–16 CrossRef CAS PubMed.
- M. Ponzoni, F. Pastorino, D. Di Paolo, P. Perri and C. Brignole, Targeting Macrophages as a Potential Therapeutic Intervention: Impact on Inflammatory Diseases and Cancer, Int. J. Mol. Sci., 2018, 19(7), 1953 CrossRef PubMed.
- F. O. Martinez and S. Gordon, The M1 and M2 paradigm of macrophage activation: time for reassessment, F1000Prime Rep., 2014, 6(13) DOI:10.12703/P6-13.
- J. C. Gensel, T. J. Kopper, B. Zhang, M. B. Orr and W. M. Bailey, Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment, Sci. Rep., 2017, 7(1), 40144 CrossRef CAS PubMed.
- S. S. Soni, A. Alsasa and C. B. Rodell, Applications of Macrocyclic Host Molecules in Immune Modulation and Therapeutic Delivery, Front. Chem., 2021, 9, 658548 CrossRef CAS PubMed.
- S. Y. Raut, A. S. N. Manne, G. Kalthur, S. Jain and S. Mutalik, Cyclodextrins as Carriers in Targeted Delivery of Therapeutic Agents: Focused Review on Traditional and Inimitable Applications, Curr. Pharm. Des., 2019, 25(4), 444–454 CrossRef CAS PubMed.
- J. Conceicao, O. Adeoye, H. M. Cabral-Marques and J. M. S. Lobo, Cyclodextrins as Drug Carriers in Pharmaceutical Technology: The State of the Art, Curr. Pharm. Des., 2018, 24(13), 1405–1433 CrossRef CAS PubMed.
- M. E. Brewster and T. Loftsson, Cyclodextrins as pharmaceutical solubilizers, Adv. Drug Delivery Rev., 2007, 59(7), 645–666 CrossRef CAS PubMed.
- R. L. Carrier, L. A. Miller and I. Ahmed, The utility of cyclodextrins for enhancing oral bioavailability, J. Controlled Release, 2007, 123(2), 78–99 CrossRef CAS PubMed.
- D. D. Gadade and S. S. Pekamwar, Cyclodextrin Based Nanoparticles for Drug Delivery and Theranostics, Adv. Pharm. Bull., 2020, 10(2), 166–183 CrossRef CAS PubMed.
- T. R. Thatiparti, A. J. Shoffstall and H. A. von Recum, Cyclodextrin-based device coatings for affinity-based release of antibiotics, Biomaterials, 2010, 31(8), 2335–2347 CrossRef CAS PubMed.
- H. Wei and C.-Y. Yu, Cyclodextrin-functionalized polymers as drug carriers for cancer therapy, Biomater. Sci., 2015, 3(7), 1050–1060 RSC.
- W. Xu, X. Li, L. Wang, S. Li, S. Chu, J. Wang, Y. Li, J. Hou, Q. Luo and J. Liu, Design of Cyclodextrin-Based Functional Systems for Biomedical Applications, Front. Chem., 2021, 9, 635507 CrossRef CAS PubMed.
- P. L. Thi, T. Y. N. Tran, H. C. Luu, D. L. Tran, T. T. H. Thi and D. H. Nguyen, In situ forming gelatin: Cyclodextrin hydrogels prepared by “click chemistry” to improve the sustained release of hydrophobic drugs, J. Bioact. Compat. Polym., 2022, 088391152210980 Search PubMed.
- G. Fang, X. Yang, S. Chen, Q. Wang, A. Zhang and B. Tang, Cyclodextrin-based host–guest supramolecular hydrogels for local drug delivery, Coord. Chem. Rev., 2022, 454, 214352 CrossRef CAS.
- C. B. Rodell, S. P. Arlauckas, M. F. Cuccarese, C. S. Garris, R. Li, M. S. Ahmed, R. H. Kohler, M. J. Pittet and R. Weissleder, TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy, Nat. Biomed. Eng., 2018, 2(8), 578–588 CrossRef CAS PubMed.
- Y. Chao, P. P. Karmali and D. Simberg, Role of carbohydrate receptors in the macrophage uptake of dextran-coated iron oxide nanoparticles, Adv. Exp. Med. Biol., 2012, 733, 115–123 CrossRef CAS PubMed.
- S. Pustylnikov, D. Sagar, P. Jain and Z. K. Khan, Targeting the C-type lectins-mediated host-pathogen interactions with dextran, J. Pharm. Pharm. Sci., 2014, 17(3), 371–392 Search PubMed.
- G. Hu, M. Guo, J. Xu, F. Wu, J. Fan, Q. Huang, G. Yang, Z. Lv, X. Wang and Y. Jin, Nanoparticles Targeting Macrophages as Potential Clinical Therapeutic Agents Against Cancer and Inflammation, Front. Immunol., 2019, 10, 1998 CrossRef CAS PubMed.
- Y. Song, Y. Huang, F. Zhou, J. Ding and W. Zhou, Macrophage-targeted nanomedicine for chronic diseases immunotherapy, Chin. Chem. Lett., 2022, 33(2), 597–612 CrossRef CAS.
- M. M. T. van Leent, B. Priem, D. P. Schrijver, A. de Dreu, S. R. J. Hofstraat, R. Zwolsman, T. J. Beldman, M. G. Netea and W. J. M. Mulder, Regulating trained immunity with nanomedicine, Nat. Rev. Mater., 2022, 7(6), 465–481 CrossRef.
- C. B. Rodell, A. L. Kaminski and J. A. Burdick, Rational Design of Network Properties in Guest–Host Assembled and Shear-Thinning Hyaluronic Acid Hydrogels, Biomacromolecules, 2013, 14(11), 4125–4134 CrossRef CAS PubMed.
- W. Ying, P. S. Cheruku, F. W. Bazer, S. H. Safe and B. Zhou, Investigation of macrophage polarization using bone marrow derived macrophages, J. Visualized Exp., 2013,(76), e50323 Search PubMed.
- J. Seager Danciger, M. Lutz, S. Hama, D. Cruz, A. Castrillo, J. Lazaro, R. Phillips, B. Premack and J. Berliner, Method for large scale isolation, culture and cryopreservation of human monocytes suitable for chemotaxis, cellular adhesion assays, macrophage and dendritic cell differentiation, J. Immunol. Methods, 2004, 288(1), 123–134 CrossRef CAS PubMed.
- K. L. Spiller, S. Nassiri, C. E. Witherel, R. R. Anfang, J. Ng, K. R. Nakazawa, T. Yu and G. Vunjak-Novakovic, Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds, Biomaterials, 2015, 37, 194–207 CrossRef CAS PubMed.
- D. Granadero, J. Bordello, M. J. Pérez-Alvite, M. Novo and W. Al-Soufi, Host-Guest Complexation Studied by Fluorescence Correlation Spectroscopy: Adamantane–Cyclodextrin Inclusion, Int. J. Mol. Sci., 2010, 11(1), 173–188 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Cellular uptake of nanoparticles: journey inside the cell, Chem. Soc. Rev., 2017, 46(14), 4218–4244 RSC.
- R. Xiao, J. Zeng, E. M. Bressler, W. Lu and M. W. Grinstaff, Synthesis of bioactive (1→6)-β-glucose branched poly-amido-saccharides that stimulate and induce M1 polarization in macrophages, Nat. Commun., 2022, 13, 1–13 Search PubMed.
- Y. Zhou, T. Takano, X. Li, Y. Wang, R. Wang, Z. Zhu, M. Tanokura, T. Miyakawa and S. Hachimura, β-elemene regulates M1-M2 macrophage balance through the ERK/JNK/P38 MAPK signaling pathway, Commun. Biol., 2022, 5(1), 1–15 CrossRef PubMed.
- P. Deak, H. R. Knight and A. Esser-Kahn, Robust tolerogenic dendritic cells via push/pull pairing of toll-like-receptor agonists and immunomodulators reduces EAE, Biomaterials, 2022, 286, 121571 CrossRef CAS PubMed.
- G. Hu, Y. Su, B. H. Kang, Z. Fan, T. Dong, D. R. Brown, J. Cheah, K. D. Wittrup and J. Chen, High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming, Nat. Commun., 2021, 12(1), 773 CrossRef CAS PubMed.
- R. J. Vagnozzi, M. Maillet, M. A. Sargent, H. Khalil, A. K. Z. Johansen, J. A. Schwanekamp, A. J. York, V. Huang, M. Nahrendorf, S. Sadayappan and J. D. Molkentin, An acute immune response underlies the benefit of cardiac stem cell therapy, Nature, 2020, 577(7790), 405–409 CrossRef CAS PubMed.
- K. Gee, C. Guzzo, N. F. Che Mat, W. Ma and A. Kumar, The IL-12 family of cytokines in infection, inflammation and autoimmune disorders, Inflammation Allergy: Drug Targets, 2009, 8(1), 40–52 CrossRef CAS PubMed.
- T. Rőszer, Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms, Mediators Inflammation, 2015, 2015, 1–16 CrossRef PubMed.
- A. King, S. Balaji, L. D. Le, T. M. Crombleholme and S. G. Keswani, Regenerative Wound Healing: The Role of Interleukin-10, Adv. Wound Care, 2014, 3(4), 315–323 CrossRef PubMed.
- E. Y. Lee, Z. H. Lee and Y. W. Song, CXCL10 and autoimmune diseases, Autoimmun. Rev., 2009, 8(5), 379–383 CrossRef CAS PubMed.
- H. Zelová and J. Hošek, TNF-α signalling and inflammation: interactions between old acquaintances, Inflammation Res., 2013, 62(7), 641–651 CrossRef PubMed.
- B. Bozkurt, D. L. Mann and A. Deswal, Biomarkers of inflammation in heart failure, Heart Failure Rev., 2010, 15(4), 331–341 CrossRef CAS PubMed.
- N. I. Skrypnyk, K. M. Gist, K. Okamura, J. R. Montford, Z. You, H. Yang, R. Moldovan, E. Bodoni, J. T. Blaine, C. L. Edelstein, D. E. Soranno, L. A. Kirkbride-Romeo, B. R. Griffin, C. Altmann and S. Faubel, IL-6-mediated hepatocyte production is the primary source of plasma and urine neutrophil gelatinase-associated lipocalin during acute kidney injury, Kidney Int., 2020, 97(5), 966–979 CrossRef CAS PubMed.
- S. D. Allen, Y.-G. Liu, T. Kim, S. Bobbala, S. Yi, X. Zhang, J. Choi and E. A. Scott, Celastrol-loaded PEG-b-PPS nanocarriers as an anti-inflammatory treatment for atherosclerosis, Biomater. Sci., 2019, 7(2), 657–668 RSC.
- G. Lorenz, F. Moschovaki-Filippidou, V. Würf, P. Metzger, S. Steiger, F. Batz, J. Carbajo-Lozoya, J. Koziel, M. Schnurr, C. D. Cohen, C. Schmaderer, H.-J. Anders, M. Lindenmeyer and M. Lech, IFN Regulatory Factor 4 Controls Post-ischemic Inflammation and Prevents Chronic Kidney Disease, Front. Immunol., 2019, 10, 2162 CrossRef CAS PubMed.
- M. Tang, X. Cao, K. Zhang, Y. Li, Q.-Y. Zheng, G.-Q. Li, Q.-H. He, S.-J. Li, G.-L. Xu and K.-Q. Zhang, Celastrol alleviates renal fibrosis by upregulating cannabinoid receptor 2 expression, Cell Death Dis., 2018, 9(6), 1–12 Search PubMed.
- T. Divya, V. Dineshbabu, S. Soumyakrishnan, A. Sureshkumar and G. Sudhandiran, Celastrol enhances Nrf2 mediated antioxidant enzymes and exhibits anti-fibrotic effect through regulation of collagen production against bleomycin-induced pulmonary fibrosis, Chem.-Biol. Interact., 2016, 246, 52–62 CrossRef CAS PubMed.
- J. Zhang, C. Y. Li, M. J. Xu, T. Wu, J. H. Chu, S. J. Liu and W. Z. Ju, Oral bioavailability and gender-related pharmacokinetics of celastrol following administration of pure celastrol and its related tablets in rats, J. Ethnopharmacol., 2012, 144(1), 195–200 CrossRef CAS PubMed.
- M. S. Freag, W. M. Saleh and O. Y. Abdallah, Self-assembled phospholipid-based phytosomal nanocarriers as promising platforms for improving oral bioavailability of the anticancer celastrol, Int. J. Pharm., 2018, 535(1–2), 18–26 CrossRef CAS PubMed.
- X. Qi, J. Qin, N. Ma, X. Chou and Z. Wu, Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation, Int. J. Pharm., 2014, 472(1–2), 40–47 CrossRef CAS PubMed.
- M. Sun, Y. Tang, T. Ding, M. Liu and X. Wang, Inhibitory effects of celastrol on rat liver cytochrome P450 1A2, 2C11, 2D6, 2E1 and 3A2 activity, Fitoterapia, 2014, 92, 1–8 CrossRef CAS PubMed.
- C. Jin, Z. Wu, L. Wang, Y. Kanai and X. He, CYP450s-Activity Relations of Celastrol to Interact with Triptolide Reveal the Reasons of Hepatotoxicity of Tripterygium wilfordii, Molecules, 2019, 24(11), 2162 CrossRef CAS PubMed.
- P. E. Lipsky and X. L. Tao, A potential new treatment for rheumatoid arthritis: thunder god vine, Semin Arthritis Rheum., 1997, 26(5), 713–723 CrossRef CAS PubMed.
- S. Kusy, E. E. Ghosn, L. A. Herzenberg and C. H. Contag, Development of B cells and erythrocytes is specifically impaired by the drug celastrol in mice, PLoS One, 2012, 7(4), e35733 CrossRef CAS PubMed.
- M. Wu, W. Chen, X. Yu, D. Ding, W. Zhang, H. Hua, M. Xu, X. Meng, X. Zhang, Y. Zhang, A. Zhang, Z. Jia and S. Huang, Celastrol aggravates LPS-induced inflammation and injuries of liver and kidney in mice, Am. J. Transl. Res., 2018, 10(7), 2078–2086 CAS.
- Z. Wang, C. Dabrosin, X. Yin, M. M. Fuster, A. Arreola, W. K. Rathmell, D. Generali, G. P. Nagaraju, B. El-Rayes, D. Ribatti, Y. C. Chen, K. Honoki, H. Fujii, A. G. Georgakilas, S. Nowsheen, A. Amedei, E. Niccolai, A. Amin, S. S. Ashraf, B. Helferich, X. Yang, G. Guha, D. Bhakta, M. R. Ciriolo, K. Aquilano, S. Chen, D. Halicka, S. I. Mohammed, A. S. Azmi, A. Bilsland, W. N. Keith and L. D. Jensen, Broad targeting of angiogenesis for cancer prevention and therapy, Semin. Cancer Biol., 2015, 35(Suppl), S224–S243 CrossRef PubMed.
- Z. Li, Z. Guo, D. Chu, H. Feng, J. Zhang, L. Zhu and J. Li, Effectively suppressed angiogenesis-mediated retinoblastoma growth using celastrol nanomicelles, Drug Delivery, 2020, 27(1), 358–366 CrossRef CAS PubMed.
- J. Shi, J. Li, Z. Xu, L. Chen, R. Luo, C. Zhang, F. Gao, J. Zhang and C. Fu, Celastrol: A Review of Useful Strategies Overcoming its Limitation in Anticancer Application, Front. Pharmacol., 2020, 11, 558741–558741 CrossRef CAS PubMed.
- O. S. Fenton, K. N. Olafson, P. S. Pillai, M. J. Mitchell and R. Langer, Advances in biomaterials for drug delivery, Adv. Mater., 2018, 30(29), 1705328 CrossRef PubMed.
- E. R. Bentley and S. R. Little, Local delivery strategies to restore immune homeostasis in the context of inflammation, Adv. Drug Delivery Rev., 2021, 178, 113971 CrossRef CAS PubMed.
- J. E. Mealy, C. B. Rodell and J. A. Burdick, Sustained Small Molecule Delivery from Injectable Hyaluronic Acid Hydrogels through Host-Guest Mediated Retention, J. Mater. Chem. B, 2015, 3(40), 8010–8019 RSC.
- A. Dogan and H. von Recum, Engineering selective molecular tethers to enhance suboptimal drug properties, Acta Biomater., 2020, 115, 383–392 CrossRef CAS PubMed.
- A. B. Dogan, K. Dabkowski and H. von Recum, Leveraging affinity interactions to prolong drug delivery of protein therapeutics, bioRxiv, 2020.
- C. B. Rodell, M. S. Ahmed, C. S. Garris, M. J. Pittet and R. Weissleder, Development of adamantane-conjugated TLR7/8 agonists for supramolecular delivery and cancer immunotherapy, Theranostics, 2019, 9(26), 8426 CrossRef CAS PubMed.
- J. E. Mealy, C. B. Rodell and J. A. Burdick, Sustained small molecule delivery from injectable hyaluronic acid hydrogels through host–guest mediated retention, J. Mater. Chem. B, 2015, 3(40), 8010–8019 RSC.
- N. Husain, T. T. Ndou, A. M. De La Peña and I. M. Warner, Complexation of Doxorubicin with β and γ-Cyclodextrins, Appl. Spectrosc., 1992, 46(4), 652–658 CrossRef CAS.
- D. F. Suárez, J. Consuegra, V. C. Trajano, S. M. Gontijo, P. P. Guimarães, M. E. Cortés, Â. L Denadai and R. D. Sinisterra, Structural and thermodynamic characterization of doxycycline/β-cyclodextrin supramolecular complex and its bacterial membrane interactions, Colloids Surf., B, 2014, 118, 194–201 CrossRef PubMed.
- M. J. Webber and R. Langer, Drug delivery by supramolecular design, Chem. Soc. Rev., 2017, 46(21), 6600–6620 RSC.
- M. S. Ahmed, C. B. Rodell, M. Hulsmans, R. H. Kohler, A. D. Aguirre, M. Nahrendorf and R. Weissleder, A Supramolecular Nanocarrier for Delivery of Amiodarone Anti-Arrhythmic Therapy to the Heart, Bioconjugate Chem., 2019, 30(3), 733–740 CrossRef CAS PubMed.
- S. A. MacParland, K. M. Tsoi, B. Ouyang, X. Z. Ma, J. Manuel, A. Fawaz, M. A. Ostrowski, B. A. Alman, A. Zilman, W. C. Chan and I. D. McGilvray, Phenotype Determines Nanoparticle Uptake by Human Macrophages from Liver and Blood, ACS Nano, 2017, 11(3), 2428–2443 CrossRef CAS PubMed.
- N. Jain, J. Moeller and V. Vogel, Mechanobiology of Macrophages: How Physical Factors Coregulate Macrophage Plasticity and Phagocytosis, Annu. Rev. Biomed. Eng., 2019, 21, 267–297 CrossRef CAS PubMed.
- Y. Ma, A. J. Mouton and M. L. Lindsey, Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction, Transl. Res., 2018, 191, 15–28 CrossRef PubMed.
- N. A. Hukriede, D. E. Soranno, V. Sander, T. Perreau, M. C. Starr, P. S. T. Yuen, L. J. Siskind, M. P. Hutchens, A. J. Davidson, D. M. Burmeister, S. Faubel and M. P. de Caestecker, Experimental models of acute kidney injury for translational research, Nat. Rev. Nephrol., 2022, 18(5), 277–293 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01113a |
| This journal is © The Royal Society of Chemistry 2022 |