
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic polymers of intrinsic microporosity for hydrogen production from water†
Yang
Bai
 ad,
Liam
Wilbraham‡
b,
Hui
Gao
a,
Rob
Clowes
a,
Haofan
Yang
a,
Martijn A.
Zwijnenburg
*b,
Andrew I.
Cooper
*a and
Reiner Sebastian
Sprick
*ac
ad,
Liam
Wilbraham‡
b,
Hui
Gao
a,
Rob
Clowes
a,
Haofan
Yang
a,
Martijn A.
Zwijnenburg
*b,
Andrew I.
Cooper
*a and
Reiner Sebastian
Sprick
*ac
aDepartment of Chemistry and Material Innovation Factory, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: aicooper@liverpool.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
cDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: sebastian.sprick@strath.ac.uk
dInstitute of Materials Research and Engineering, Agency for Science, Technology and Research, #08-03, 2 Fusionopolis Way, Innovis, Singapore, 138634, Singapore
First published on 13th July 2021
Abstract
The most common strategy for introducing porosity into organic polymer photocatalysts has been the synthesis of cross-linked conjugated networks or frameworks. Here, we study the photocatalytic performance of a series of linear conjugated polymers of intrinsic microporosity (PIMs) as photocatalysts for hydrogen production from water in the presence of a hole scavenger. The best performing materials are porous and wettable, which allows for the penetration of water into the material. One of these polymers of intrinsic microporosity, P38, showed the highest sacrificial hydrogen evolution rate of 5226 μmol h−1 g−1 under visible irradiation (λ > 420 nm), with an external quantum efficiency of 18.1% at 420 nm, placing it among the highest performing polymer photocatalysts reported to date for this reaction.
Introduction
Hydrogen has the potential to be a green energy carrier when generated renewably from water using solar energy. It combines a high gravimetric energy density with the absence of CO2 production at the point of use.1–3 Most photocatalysts studied for water splitting have been inorganic semiconductors, but the demonstration that carbon nitride can act as a hydrogen evolution photocatalyst4 has inspired a large number of subsequent studies5–9 on sacrificial proton reduction by organic materials. Various organic materials that can be obtained via low temperature condensation reactions have been studied as photocatalysts,3,10–12 including conjugated microporous polymers (CMPs),13–19 covalent triazine-based frameworks (CTFs),20–24 covalent organic frameworks (COFs),25–33 linear conjugated polymers34–46 and linear conjugated oligomers.47–49 Carbon nitride loaded with Pt/Co was shown to facilitate overall water splitting.50 Also, hydrogen evolution by various donor–acceptor covalent organic frameworks for high efficiency sunlight-driven has been demonstrated, which might inspire the future development of sunlight-driven porous photocatalysts.33,51The photocatalytic activity is influenced by a range of different factors:3,52 Light absorption43 matching the wavelength and sufficiently high extinction coefficients are a necessary condition for a photocatalyst to be active. However, the polymers' potentials also have to be aligned in such a way that they straddle the water reduction and oxidation/scavenger oxidation potentials with sufficient driving-force for both half reactions to occur.44,53 Excitons have to be separated into individual charge-carriers with fairly high efficiency, though this often occurs at the interface through reaction with the hole scavenger.44 Creating large interfaces with water is another important factor allowing for excitons to migrate from the inside of the material to reactive sites, which can be either be achieved through large internal surface areas15,27,54 or extrinsic surfaces on small particles.45,55 Crystallinity can also play a role here in facilitating the transport of excitons or individual charges and some examples have shown that order within the material can be beneficial.49,56 Another important factor for polymer photocatalysts is their wettability,44,46 which compared to inorganic semiconductors it seems to be a much more important property. This is most likely because of the inherent apolar nature of these materials making it difficult for water to reach the surface and/or making dispersion in aqueous mixtures more difficult reducing activity. However, it is very important to consider the interconnectivity of different factors, which can result in factors being optimized at the expense of one or multiple different factors and hence lowering overall activity.57 For example, increasing hydrophilicity (generally a positive factor) could also decrease the absorption of visible light (a negative factor), if the chromophore is changed adversely.
Previously, we compared non-porous linear conjugated polymers and their CMP analogues as photocatalysts for sacrificial hydrogen evolution from water.15 The internal pore structure of these CMP photocatalysts was studied by gas sorption and this pore structure was shown to be wettable, as evidenced by water uptake measurements. This wettable porosity seemed to enhance the activity towards photocatalytic hydrogen production from water in the presence of a sacrificial hole scavenger, providing that the introduction of porosity did not compromise the activity in some other way.15 Here we explore conjugated polymers of intrinsic microporosity (PIMs)58–60 analogues that contain the same dibenzo[b,d]thiophene sulfone unit as porous alternative to CMPs, where the porosity arises from the inherent inability of the spiro-unit containing polymers to pack densely in the solid-state. These PIMs combine extended conjugation along the aromatic backbone with porosity, without introducing connectivity defects and twisting of the structure that are commonly observed in CMPs, and which might affect the photocatalytic activity for hydrogen production.15,61
Experimental
We studied spirobifluorene-based and spiro[4H-cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-[9H]fluorene]-based polymers and compared them to their (previously reported) 9H-dimethylfluorene polymers analogues, along with a previously reported CMP of dibenzo[b,d]thiophene sulfone and spirofluorene (S-CMP3).15 This allowed us to explore the influence of porosity and water penetration into the material. We were also able to assess the importance of effective conjugation length by comparing the linear polymers with the polymer network, S-CMP3. To do this, we synthesized four new photocatalysts of spirobifluorene and spiro[4H-cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-[9H]fluorene] as co-polymers with phenylene and dibenzo[b,d]thiophene sulfone (Fig. 1).15,34 The spiro[4H-cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-[9H]fluorene] unit was synthesized using a previously reported approach.62 These polymers were synthesized using Suzuki–Miyaura polycondensation conditions, as reported previously39 (see ESI† for synthetic details). All materials were purified using Soxhlet extraction to give solids that are insoluble in common organic solvents. Thermogravimetric analysis indicated that all polymers were thermally stable in air up to 300 °C, and powder X-ray diffraction patterns showed that the materials do not possess significant long-range order with only P36 and P37 showing defined features that possibly relate to oligomers that were not removed during work-up. | ||
| Fig. 1 Structures of the polymer photocatalysts studied here. | ||
Results and discussion
UV/visible absorption spectra were measured for all materials in the solid-state (Fig. 2a). As expected, the co-polymer containing no heteroatoms, P2 and P36, have the most blue-shifted absorption on-sets with optical-gaps estimated to be 2.80 and 2.65 eV, while the dibenzo[b,d]thiophene sulfone co-polymers have narrower band gaps (Table 1). The spiro[4H-cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-[9H]fluorene] co-polymer with phenylene (P37) seemed to have a much smaller band gap (2.28 eV) than its P2 and P36 analogues. This could be an artefact of the Tauc analysis (see Fig. S-7†) and TD-DFT calculations (B3LYP/DZP,63–66 ESI†) suggest that P37 should have a similar gap as P2 and P36. However, repeat experiments and different batches of P37 gave consistent results. The lifetime of the excited state was estimated using time-correlated single-photon counting, with weighted averaged lifetimes ranging from 0.36 to 1.01 ns for P36 and P39 (ref. 39) (Fig. S-12 and S-13†). The lifetimes of all materials are pH 11.5 shortened upon addition of triethylamine (Fig. S-39†) in line with previous observations.44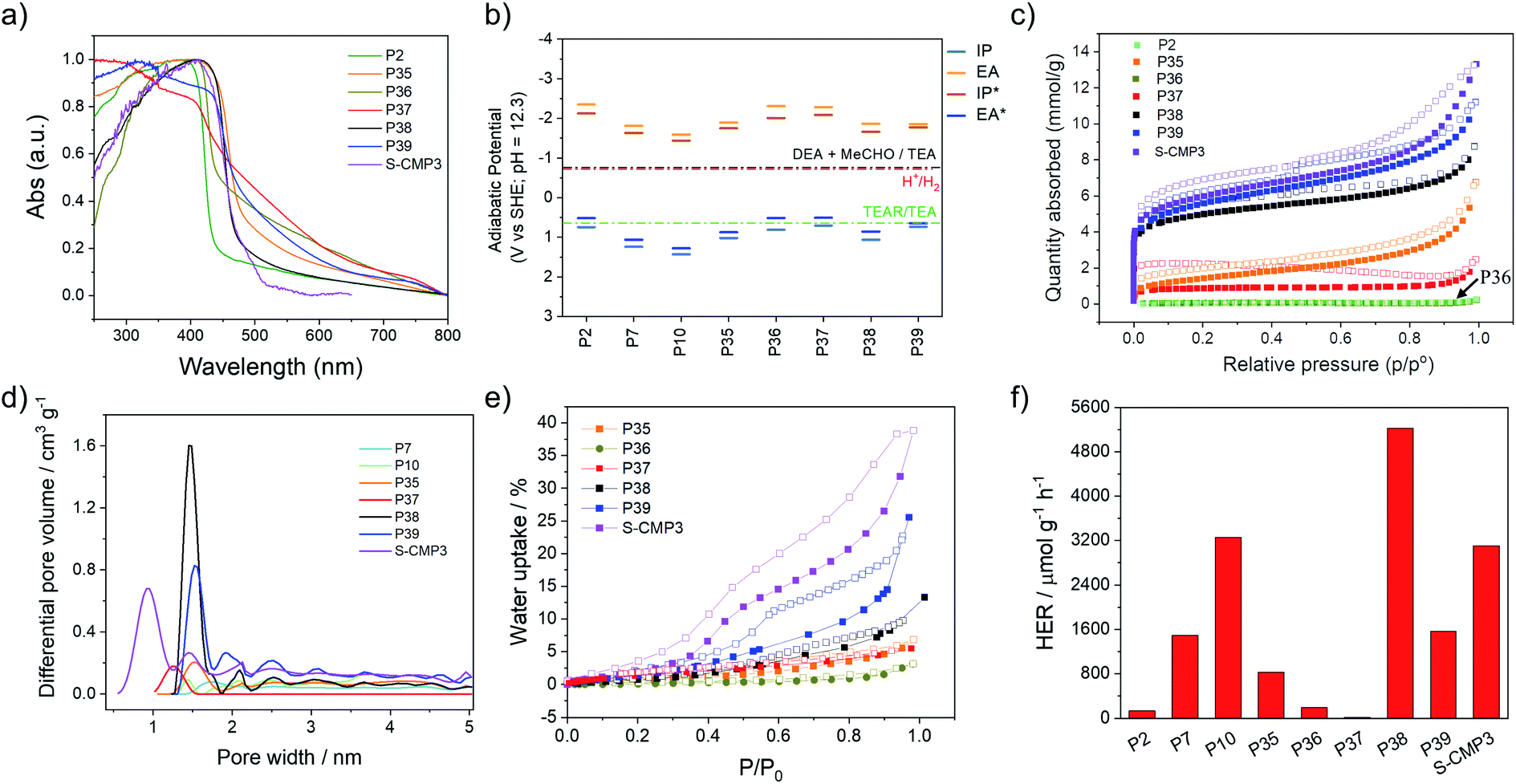 | ||
| Fig. 2 (a) Absorption spectra of polymers measured in the solid-state; (b) predicted charge carrier (IP, EA) and excitons (IP*, EA*) potentials of polymers considered as calculated by DFT for oligomer models. For guidance, reduction potential for hydrogen and oxidation potential for the sacrificial hole scavenger triethylamine (TEA) at pH 12.3. (c) Nitrogen sorption isotherms measured at 77 K; (d) pore size distribution of the polymers. The values were calculated from nitrogen isotherms at 77 K using nonlocal density functional theory; (e) water uptake measurements at 25.0 °C and up to 31.7 mbar; (f) H2 evolution rates of polymers, each measurement was performed with 25 mg catalyst in water/MeOH/triethylamine mixture under visible light irradiation (λ > 420 nm, 300 W Xe light source). | ||
| Photo-catalyst | Optical gapa/eV | t avg /ns | Particle sizec/μm | SABETd/m2 g−1 | Transmittancee/% | IP vs. SHEf/V | EA vs. SHEf/V | IP* vs. SHEf/V | EA* vs. SHEf/V | HER λ > 420 nmg/μmol h−1 g−1 |
|---|---|---|---|---|---|---|---|---|---|---|
a Optical gap calculated from the absorption on-set using Tauc plots.
b Estimated weighted average life-time of the excited state determined by time-correlated single-photon counting.
c Surface area mean diameter (Sauter mean diameter).
d Apparent BET surface area calculated from the nitrogen adsorption isotherm.
e Average transmittance of a polymer suspension in water/methanol/trimethylamine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1).
f Potentials of the charge-carriers (IP, EA) and excitons (IP*, EA*) as calculated by DFT for oligomer models. Values for S-CMP3 were not calculated due to the topological complexity of the network structure but will likely be similar to that of P38.
g Reaction conditions: 25 mg polymer was suspended in 25 mL water/methanol/triethylamine (1:1:1) solution, irradiated by 300 W Xe light source using λ > 420 nm cut-off filter.
h Values taken from previous work (ref. 34 & 44).
i Value found to be in disagreement with prediction.
j Previously reported (ref. 15); n.d. = not determined. :1:1).
f Potentials of the charge-carriers (IP, EA) and excitons (IP*, EA*) as calculated by DFT for oligomer models. Values for S-CMP3 were not calculated due to the topological complexity of the network structure but will likely be similar to that of P38.
g Reaction conditions: 25 mg polymer was suspended in 25 mL water/methanol/triethylamine (1:1:1) solution, irradiated by 300 W Xe light source using λ > 420 nm cut-off filter.
h Values taken from previous work (ref. 34 & 44).
i Value found to be in disagreement with prediction.
j Previously reported (ref. 15); n.d. = not determined.
|
||||||||||
| P2 | 2.81 | 0.59 | 2.94 | 4 | 22.5 | 0.74 | −2.34 | −2.12 | 0.52 | 136.0 |
| P7 | 2.73 | 0.87 | 1.12 | 69 | 3.5 | 1.24h | −1.81h | −1.63h | 1.06h | 1492.0 |
| P10 | 2.62 | 2.06 | 3.32 | 56 | 0.4 | 1.43h | −1.59h | −1.44h | 1.28h | 3260.0 |
| P35 | 2.56 | 0.46 | 8.28 | 114 | 0.2 | 1.02 | −1.89 | −1.75 | 0.87 | 826.0 |
| P36 | 2.65 | 0.36 | 7.63 | 5 | 51.6 | 0.81 | −2.30 | −2.01 | 0.51 | 191.7 |
| P37 | 2.28i | 0.56 | 5.40 | 60 | 2.1 | 0.70 | −2.28 | −2.09 | 0.51 | 17.8 |
| P38 | 2.63 | 0.58 | 4.61 | 352 | 1.5 | 1.06 | −1.87 | −1.66 | 0.86 | 5226.0 |
| P39 | 2.44 | 1.01 | 3.45 | 412 | 0.4 | 0.73 | −1.85 | −1.77 | 0.65 | 1566.3 |
| S-CMP3 | 2.56j | 0.72j | 11.5j | 431j | 2.7j | n.d. | n.d. | n.d. | n.d. | 3106.0j |
Nitrogen sorption isotherms were measured at 77 K for all materials and apparent Brunauer–Emmett–Teller (SABET) surface areas were calculated based on the adsorption isotherms (Fig. 2c). The phenylene derivatives P2, P36 and P37 have low apparent surface areas (4, 5 and 60 m2 g−1) compared to their dibenzo[b,d]thiophene sulfone analogs, P35, P38 and P39 (ranging from 114 to 412 m2 g−1, Table 1). It is possible that the internal voids within the materials that give rise to the microporosity of the materials are due to the bulky spirobifluorene and spiro[4H-cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-[9H]fluorene] units create empty space which is stabilized by stacking of the dibenzo[b,d]thiophene sulfone units in P35, P38 and P39 resulting in microporosity as evident by a micropore step at low relative pressures in their adsorption isotherms. Pore size distributions show that P38, P39, and S-CMP3 have significant microporous character, with the highest volume of pores occurring in the 0–2 nm diameter region (Fig. 2d). To study water penetration into the pores, we measured the water uptake of the polymers (Fig. 2e). Non-porous polymer P36 shows very little water uptake (<5 wt%) compared to P35, P38, P39 and S-CMP3, and this can be ascribed to surface absorption on the polymer particles. The highest water uptake for the linear PIMs (25 wt% at a P/P0 = 1) was measured for 4H-cyclopenta[2,1-b:3,4-b′]dithiophene containing polymer P39, which also has the highest BET surface area in the dry state (412 m2 g−1).
The ionization potential (IP) and electron affinity (EA), and their excited state equivalents (EA* and IP*), for these polymers determine the driving force for reactions where the polymer accepts and donates electrons, respectively. The values of IP, EA, EA* and IP* were predicted using our standard approach67,68 based on a combination of ΔDFT and TD-DFT (B3LYP/DZP63–66) in combination with the COSMO69 dielectric screening model (εr 80.1, water) to describe the screening of charges in the polymers near the polymer–solution interface. Previously, we showed that this approach yields results comparable to those measured using photoelectron spectroscopy for organic polymers.40,68 As in previous work,39,44 the dibenzo[b,d]thiophene sulfone-containing polymers are predicted to have more positive IP/EA* values versus the standard hydrogen electrode than those lacking dibenzo[b,d]thiophene sulfone units, as well as less negative EA/IP* values (Fig. 2b). All polymers are predicted to have ample thermodynamic driving force for proton reduction at pH 12.3, the measured pH of the solution, as well as the ability to drive the overall 2-electron oxidation of TEA. The intermediate 1-electron oxidation of TEA, however, is predicted to be endergonic for P2, P36, P37 and P39. For these materials, this intermediate 1-electron oxidation step might act as a thermodynamic barrier along the path towards the overall oxidation of TEA, slowing down TEA oxidation for these materials relative to polymers for which the 1-electron step is predicted to be exergonic.
Static light scattering was used to study the particle size of dispersions of the materials in the TEA/MeOH/H2O mixtures used for photocatalytic hydrogen production. These measurements showed that the polymer dispersions all display similar particle size volume distributions and Sauter mean diameters of around 5 μm (Table S-3†). Scanning electron microscopy (SEM) was conducted for the P36, P37, P38, and P39 (Fig. S-22 and S-23†), with the SEM images of other polymers in this study reported previously.15,34,44 This showed that most materials consist of small particles of similar sizes that are fused together into micron-sized particles, agreeing well with the static light scattering measurements. However, P2 and P36 are poorly dispersible in TEA/MeOH/H2O mixtures. This evident from the relatively high transmission values of the P2 and P36 dispersions in light obscuration measurements with values of 22.5% and 51.6%, respectively (Table 1), indicating that particles of these polymers rapidly settle from their suspension, which can also be seen in photographs of the suspensions (Fig. S-42†).
All other materials have transmission values ranging from 0.2% for P35 to 2.7% for S-CMP3 and 3.5% for P7, indicating good dispersibility in the reaction medium.
The ability of the materials to act as a photocatalyst for hydrogen production from water was studied for the as-made materials without the addition of any co-catalysts. Previous studies have shown that residual Pd within these materials acts as the co-catalyst and all materials were found to have residual amounts of Pd from the synthesis (ranging from 3000 to 8600 ppm).15,54
For the hydrogen evolution measurements, the materials were dispersed in a mixture of water/methanol/triethylamine. Here, triethylamine (TEA) acts as the hole-scavenger, while methanol enhances the miscibility of TEA with water and improves the wettability of the polymers.34
The phenylene co-polymers P2 (136 μmol h−1 g−1) and P36 (192 μmol h−1 g−1) show limited activity, and P37 (17.8 μmol h−1 g−1) evolves almost no hydrogen under visible illumination (λ > 420 nm, 300 W Xe light source). Similar to previous reports, we found that high activities were observed when dibenzo[b,d]thiophene sulfone was introduced into these polymers.34,39 The co-polymer of dibenzo[b,d]thiophene sulfone and spirofluorene P38 shows the highest activity under visible light, with a rate of 5226 μmol h−1 g−1; that is, higher than the non-porous dibenzo[b,d]thiophene sulfone homopolymer (P10, 3260 μmol h−1 g−1)44 under the same conditions (λ > 420 nm, 300 W Xe light source). The non-porous dibenzo[b,d]thiophene sulfone co-fluorene polymer P35 displays a lower activity (826 μmol h−1 g−1). The activity of P38 was twice as high as its conjugated microporous polymer analog, S-CMP3 (3106 μmol h−1 g−1),15 under the same conditions (λ > 420 nm, 300 W Xe light source). In comparison to P38, P39 had reduced photocatalytic activity under visible light illumination (1566 μmol h−1 g−1).
The absence of hydrogen production in the dark for P38 (Fig. S-25†) and an isotope labelling experiment resulting in predominant production of D2 from D2O/triethylamine mixture under visible light irradiation for P38 (Fig. S-26†) confirms that this is a photocatalytic process. Additional palladium photodeposited onto P38 using [Pd(NH4)2Cl4] resulted in no further improvement of the materials activity (Fig. S-27†). The photostability of P38 in TEA/MeOH/H2O mixture was measured under visible light irradiation (λ > 420 nm, 300 W Xe light source) over the course of 24 hours (Fig. 3a). The hydrogen evolution activity decreased slightly over the course of the run but could be recovered when the TEA/MeOH/H2O mixture was replaced after 14 hours. Characterisation of P38 after this extended run showed no noticeable differences in the FT-IR, UV-vis, and photoluminescence spectra compared to the as-made material (Fig. S-28 and S-29†).
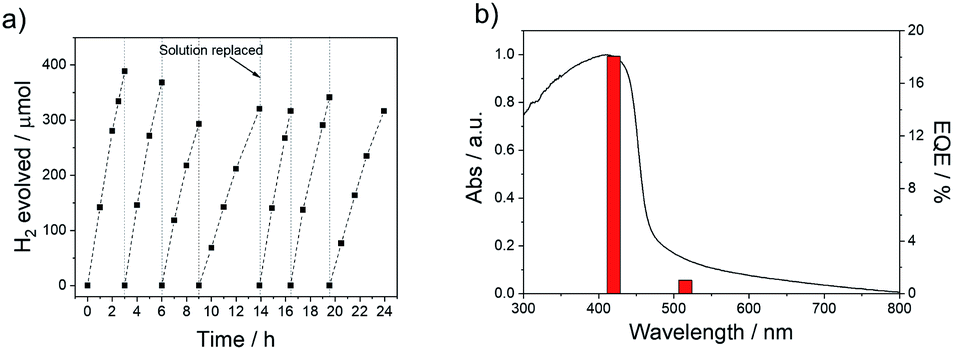 | ||
| Fig. 3 (a) Plot showing sacrificial photocatalytic hydrogen evolution versus time for P38 under visible light irradiation (λ > 420 nm, 300 W Xe light source). The vertical dashed lines indicate degassing; (b) absorption spectrum and external quantum efficiency (EQE) measured with monochromatic light in a water/MeOH/TEA mixture for P38. | ||
The external quantum efficiency (EQE) of P38 at 420 nm was determined to be 18.1%, which is higher than previously reported values for poly(p-phenylene) (P1, EQE420nm = 0.4%), the dibenzo[b,d]thiophene sulfone homopolymer (P10, EQE420nm = 11.6%),44 and the porous dibenzo[b,d]thiophene sulfone-co-9,9′-spirobi[9H-fluorene] S-CMP3 (EQE420nm = 13.2%),15 but slightly lower than a dibenzo[b,d]thiophene sulfone-dibenzo[b,d]-thiophene co-polymer P64 (EQE420nm = 20.7%),3,39 all measured under the same conditions in water/methanol/TEA mixtures. It is also lower than CTF ter-CTF-0.7 measured using water/TEOA (EQE420nm = 22.8%)24 and structurally optimised carbon nitride in water/TEOA (EQE420nm = 57.0%),7 though we note that the experimental conditions for these other measurements were not all identical. The hydrogen evolution rate of P38 (5226 μmol h−1 g−1) was much higher under visible light irradiation (λ > 420 nm, 300 W Xe light source) than commercial carbon nitride in 10 vol% triethanolamine in water loaded with 3 wt% Pt (108 μmol h−1 g−1) and commercial TiO2 P25 in water/methanol/TEA solution with photodeposition of 1 wt% Pt (4 μmol h−1 g−1).70
We found previously20,39,41 that the performance of non-porous polymer photocatalysts could be explained by the interplay of several material properties: IP, EA, optical gap and the dispersibility of the polymers in solution. We used these same properties to analyze the activity of the materials studied here, considering first the linear polymers. The purely hydrocarbon materials, P2 and P36, were the least dispersible in the water/MeOH/TEA mixture, as evidenced by the high transmittance values of their dispersions. They were also predicted to lack thermodynamic driving force for the 1-electron oxidation of TEA, and hence likely will display slow overall TEA oxidation kinetics. Coupled with the most blue-shifted optical gaps this explains why these materials are among the least active studied here. The higher activity of the dibenzo[b,d]thiophene sulfone containing polymers (P7, P10, P35, P38, P39) can be explained because these polymers are much more dispersible in water, are predicted to drive the 1-electron oxidation of TEA (except for P39, see below), and have generally lower optical gaps. The poor performance of P37 is harder to explain, even if we assume that the optical gap extracted from the Tauc analysis is an artefact and that the true optical gap is more in line with the TD-DFT results.
Focussing on P38 and P39, these polymers perform much better than their direct fluorene analogue, P35. Also, P38 outperforms P10. This is superficially surprising as P38 and P39 are slightly less or equivalently dispersible in the reaction medium than P35 and P10, and they are predicted to have a lower driving force for the intermediate 1-electron oxidation of TEA. For P39, indeed, the 1-electron oxidation of TEA is predicted to be endergonic. P39 has a slightly smaller optical gap than P35 and P10, but the optical gap of P38 is similar to that of P10. The main difference between P38/P39 and P10/P35, therefore, lies in the surface area of the materials. All polymers studied here have similar particle sizes, hence this surface area is internal to the particles in the form of micro and macropores (Fig. 2c), which are accessible to water based on water adsorption isotherms (Fig. 2e). We therefore suggest that the water-wettable pores and internal surface area of P38 & P39 compensates for their slightly worse dispersibility and thermodynamic driving forces. This makes sense from a theoretical perspective because porosity allows water and sacrificial electron donor (TEA) to enter the particle interior, and hence excitons (excited electron–hole pairs) must travel less far to dissociate at the polymer–solution interface in comparison with solid particles. We suggest that this lowers electron–hole recombination rates and maximises the volume fraction of the polymer particles that contribute to hydrogen evolution. For solid particles, excitons generated by absorbing light in the centre of the particle will most likely never reach the interface because the particles (Sauter mean diameter ∼ 5 μm) are very large relative to the probable exciton diffusion length (<10 nm).40,71
S-CMP3 is a more complex material than the PIMs as it is a CMP which is typically a network and/or branched with a large number of structural defects and end-groups. It could be that the lower catalytic activity of S-CMP3 relative to P38, despite its higher surface area and water adsorption, is somehow the result of this more complex topology with structural defects and end-groups acting as trapping sites for charge-carriers. We have no direct experimental evidence for this, however. The Sauter mean diameter of the S-CMP3 particles is two times lower than for P38, which could also partly explain the reduction in hydrogen evolution rate. Also, it is conceivable that S-CMP3 might be too porous, and that this lowers exciton diffusion rates to an extent that outweighs the benefits of increased surface area.
Overall, it seems therefore that P38 performs better than all other materials in this study due to a combination of factors. It has small highly dispersible particles with a large accessible surface area, has sufficient driving force for the relevant solution reactions and ability to absorb light. All other materials also fulfil some of these requirements, but not at the same time therefore limiting their performance relative to P38.
Conclusions
In conclusion, we studied two sets of polymer photocatalysts for their activity in proton reduction. As in previous studies, materials containing dibenzo[b,d]thiophene sulfone had a much higher catalytic activity, especially when combined with porosity and wettability. One PIM material, P38, has a high photocatalytic activity that surpasses our ‘benchmark’ linear polymer P10, and we suggest that the reason is the intrinsic microporosity. This is perhaps the first clear example that demonstrates that the introduction of porosity can dramatically increase the activity of these organic proton reduction photocatalysts. In the future it may also be able to introduce the key defining feature of PIMs—solution processability60—for example, by introducing suitable solubilizing groups.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1. Y. B. and H. G. thank the China Scholarship Council for a PhD studentship. Y. B. thanks the Accelerated Materials Development for Manufacturing Program at A*STAR via the AME Pro-grammatic Fund by the Agency for Science, Technology and Research under Grant no. A1898b0043. R. S. S. thanks the University of Strathclyde for financial support through The Strathclyde Chancellor's Fellowship Scheme.Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- W. Ren, J. Cheng, H. Ou, C. Huang, M. M. Titirici and X. Wang, ChemSusChem, 2019, 12, 3257–3262 CrossRef CAS PubMed.
- L. Yin, S. Wang, C. Yang, S. Lyu and X. Wang, ChemSusChem, 2019, 12, 3320–3325 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z.-A. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 129, 13445–13449 CrossRef PubMed.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1–52 Search PubMed.
- C. Zhao, C. Ding, C. Han, X. Yang and J. Xu, Sol. RRL, 2021, 5, 1–7 Search PubMed.
- J. Jayakumar and H. Chou, ChemCatChem, 2020, 12, 689–704 CrossRef CAS.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- M. Liras, M. Barawi and V. A. de la Peña O'Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- Y. Xu, N. Mao, S. Feng, C. Zhang, F. Wang, Y. Chen, J. Zeng and J.-X. Jiang, Macromol. Chem. Phys., 2017, 218, 1700049 CrossRef.
- W. J. Xiao, Y. Wang, W. R. Wang, J. Li, J. Wang, Z. W. Xu, J. Li, J. Yao and W. S. Li, Macromolecules, 2020, 53, 2454–2463 CrossRef CAS.
- C. G. López-Calixto, M. Barawi, M. Gomez-Mendoza, F. E. Oropeza, F. Fresno, M. Liras and V. A. de la Peña O'Shea, ACS Catal., 2020, 10, 9804–9812 CrossRef.
- H. Qiang, T. Chen, Z. Wang, W. Li, Y. Guo, J. Yang, X. Jia, H. Yang, W. Hu and K. Wen, Chinese Chem. Lett., 2020, 31, 3225–3229 CrossRef CAS.
- C. B. Meier, R. Clowes, E. Berardo, K. E. Jelfs, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Chem. Mater., 2019, 31, 8830–8838 CrossRef CAS PubMed.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- L. Guo, Y. Niu, H. Xu, Q. Li, S. Razzaque, Q. Huang, S. Jin and B. Tan, J. Mater. Chem. A, 2018, 6, 19775–19781 RSC.
- K. Wang, L.-M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed.
- L. Guo, Y. Niu, S. Razzaque, B. Tan and S. Jin, ACS Catal., 2019, 9, 9438–9445 CrossRef CAS.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- K. Gottschling, G. Savasci, H. Vignolo-González, S. Schmidt, P. Mauker, T. Banerjee, P. Rovó, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2020, 142, 12146–12156 CrossRef CAS PubMed.
- S. Ghosh, A. Nakada, M. A. Springer, T. Kawaguchi, K. Suzuki, H. Kaji, I. Baburin, A. Kuc, T. Heine, H. Suzuki, R. Abe and S. Seki, J. Am. Chem. Soc., 2020, 142, 9752–9762 CAS.
- W. Huang, W. Luo and Y. Li, Mater. Today, 2020, 40, 160–172 CrossRef CAS.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- Z. Lan, G. Zhang, X. Chen, Y. Zhang, K. A. I. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 10236–10240 CrossRef CAS PubMed.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- W.-H. Wang, L.-Y. Ting, J. Jayakumar, C.-L. Chang, W.-C. Lin, C.-C. Chung, M. H. Elsayed, C.-Y. Lu, A. M. Elewa and H.-H. Chou, Sustainable Energy Fuels, 2020, 4, 5264–5270 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- Y. Bai, D. J. Woods, L. Wilbraham, C. M. Aitchison, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 8700–8705 RSC.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- R. S. Sprick, K. J. Cheetham, Y. Bai, J. Alves Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
- S. Yanagida, K. Murakoshi, M. Kusabac and N. Nakashima, J. Chem. Soc., Perkin Trans. 2, 1996, 1963–1969 RSC.
- S. Matsuoka, H. Fujii, C. Pac and S. Yanagida, Chem. Lett., 1990, 1501–1502 CrossRef CAS.
- C. M. Aitchison, M. Sachs, M. A. Little, L. Wilbraham, N. J. Brownbill, C. M. Kane, F. Blanc, M. A. Zwijnenburg, J. R. Durrant, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 8744–8756 RSC.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
- B. P. Biswal, H. A. Vignolo-González, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- C. M. Aitchison and R. S. Sprick, Nanoscale, 2021, 13, 634–646 RSC.
- S. Bi, Z. Lan, S. Paasch, W. Zhang, Y. He, C. Zhang, F. Liu, D. Wu, X. Zhuang, E. Brunner, X. Wang and F. Zhang, Adv. Funct. Mater., 2017, 27, 1703146 CrossRef.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 RSC.
- C. M. Aitchison, C. M. Kane, D. P. McMahon, P. R. Spackman, A. Pulido, X. Wang, L. Wilbraham, L. Chen, R. Clowes, M. A. Zwijnenburg, R. S. Sprick, M. A. Little, G. M. Day and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7158–7170 RSC.
- Y. Bai, L. Wilbraham, B. Slater, M. Zwijnenburg, R. Sprick and A. Cooper, J. Am. Chem. Soc., 141, 9063–9071 CrossRef CAS PubMed.
- N. B. Mc Keown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- P. Klein, H. J. Jötten, C. M. Aitchison, R. Clowes, E. Preis, A. I. Cooper, R. S. Sprick and U. Scherf, Polym. Chem., 2019, 10, 5200–5205 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- C. Weng, W. Wang, J. Liang, G. Wang, S. Tan and P. Shen, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2330–2343 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- P. Guiglion, A. Monti and M. A. Zwijnenburg, J. Phys. Chem. C, 2017, 121, 1498–1506 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
- S. Dimitrov, B. Schroeder, C. Nielsen, H. Bronstein, Z. Fei, I. McCulloch, M. Heeney and J. Durrant, Polymers, 2016, 8, 14 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta03098a |
| ‡ School of Chemistry, The University of Glasgow, University Avenue, Glasgow G12 8QQ, UK. |
| This journal is © The Royal Society of Chemistry 2021 |