
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible polycondensations outside the Jacobson–Stockmayer theory and a new concept of reversible polycondensations†
Hans R.
Kricheldorf
*a,
Steffen. M.
Weidner
 b and
Jana
Falkenhagen
b
b and
Jana
Falkenhagen
b
aUniversität Hamburg, Institut für Technische und Makromolekulare Chemie, Bundesstrasse 45, 20146 Hamburg, Germany. E-mail: kricheld@chemie.uni-hamburg.de
bBundesanstalt für Materialforschung und -prüfung, Richard-Willstätter-Strasse 11, 12489 Berlin, Germany
First published on 29th July 2021
Abstract
L-Lactide was polymerized with tin(II)acetate, tin(II)2-ethyl hexanoate, diphenyltin dichloride and dibutyltin bis(pentafluorophenoxide) at 130 °C in bulk. When an alcohol was added as initiator, linear chains free of cycles were formed having a degree of polymerization (DP) according to the lactide/initiator (LA/In) ratio. Analogous polymerizations in the absence of an initiator yielded high molar mass cyclic polylactides. Quite similar results were obtained when ε-caprolactone was polymerized with or without initiator. Several transesterification experiments were conducted at 130 °C, either with polylactide or poly(ε-caprolactone) indicating that several transesterification mechanisms are operating under conditions that do not include formation of cycles by back-biting. Furthermore, reversible polycondensations (revPOCs) with low or moderate conversions were found that did not involve any kind of cyclization. Therefore, these results demonstrate the existence of revPOCs, which do neither obey the theory of irreversible polycondensation as defined by Flory nor the hypothesis of revPOCs as defined by Jacobson and Stockmayer. A new concept encompassing any kind of revPOCs is formulated in the form of a “polycondensation triangle”.
Introduction
When Flory elaborated the first theory of polycondensations he formulated a theory of irreversible polycondensations, although his experimental experience was based on syntheses of polyesters and polyamides under conditions typical for a reversible polycondensation (revPOC).1,2 The differentiation between irreversible and reversible step-growth polymerizations was defined and established in 1950 by two papers of Jacobson and Stockmayer (J + S).3,4 An irreversible polymerization is defined by a kinetically controlled course, where every growing and cyclization step is irreversible. In the reversible case every growing and cyclization reaction is reversible and thus part of an equilibrium, so that the reaction mixture presents sooner or later the thermodynamically most favourable scenario. The experimental work of J + S was based on proton-catalyzed polycondensations of 1,10-decane diol with adipic acid, and the permanent equilibration of the reaction mixture was attributed to the reversible formation of cyclic oligomers via “back-biting” (Scheme 1). The reversibility of ring formation includes that any revPOC is to a large extent also a ring-opening polymerization and thus, comparable with the curse of a ring-opening polymerization involving simultaneous polycondensation (ROPPOC) as discussed below. As already formulated by J + S, the ring-chain equilibration also involves ring–ring and chain–chain equilibration, so that total equilibration of all reaction products occurs.4 Other equilibration reactions, such as the attack of an activated end group on bonds in the polymer backbone (so-called random transesterification – RT), were not taken into account. At the time, when J + S performed their work, analytical methods allowing for the detection of such reactions (e.g. transesterification or transamidation) did not yet exist.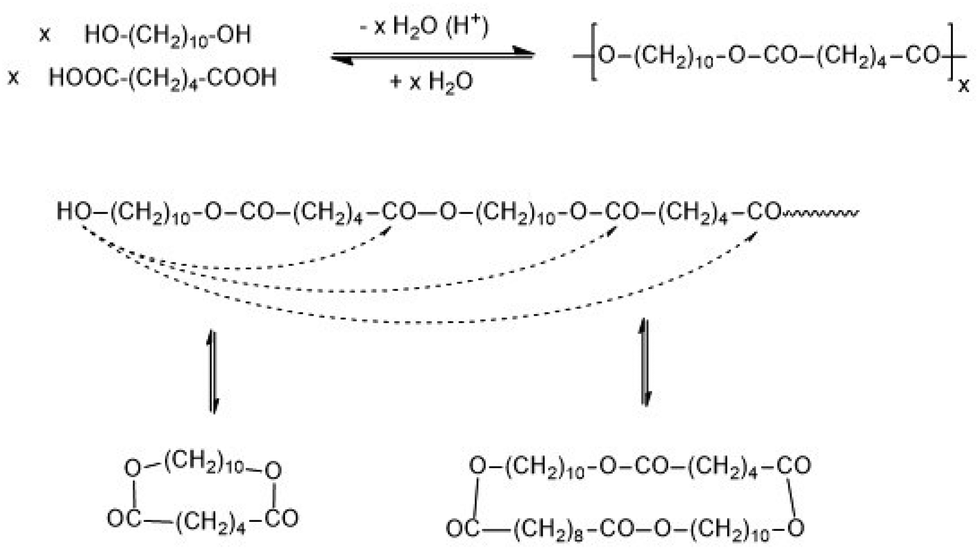 | ||
| Scheme 1 Synthesis of poly(decamethylene adipiate) and reversible formation of cyclic polymers. | ||
Critique of part of the JS-theory was recently published by the authors5,6 who demonstrated that formation of cyclic oligomers and polymers does not only result from “back-biting” but also from end-to-end (ete) cyclization, which is in contrast to the assumption of J + S. However, previous results and the results presented below demonstrate that ete-cyclization is a very efficient process. Moreover, it is unproven that the cycles indirectly detected by J + S (mass spectrometry and NMR spectroscopy did not exist at that time) mainly or exclusively result from back-biting. A consequence of ete-cyclization is that the JS theory does not provide a correct description of all revPOCs. Furthermore, it is necessary to emphasize that J + S did never study a stoichiometric polycondensation up to high conversions (>99.5%). Nonetheless, the widely accepted view and understanding of revPOCs is still such that the reversibility and thermodynamic control is mainly or exclusively the result of ring chain equilibration via “back-biting”.
Recently the authors have detected and defined a special kind of polymerization which combines ring-opening polymerization (ROP) with simultaneous polycondensation (ROPPOC).7,8 Such ROPPOC polymerizations arise from ROPs initiated with a compound that introduces a reactive end group, so that linear chains having two reactive chains will be formed. ROPPOC polymerization may be equivalent to a normal revPOC of a non-cyclic monomer as illustrated by syntheses of nylons from ω-amino acids alone or in combination with the corresponding lactams (Scheme 2).9
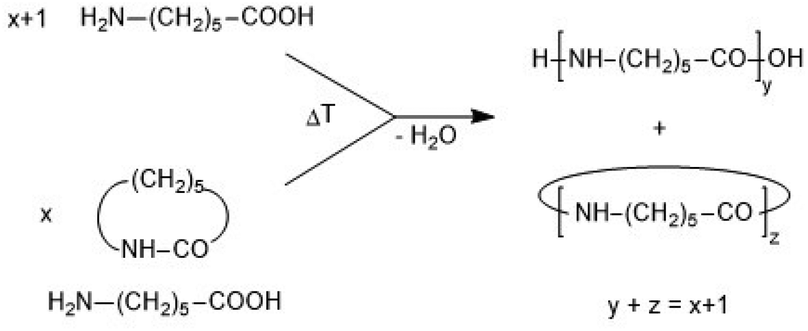 | ||
| Scheme 2 Synthesis of nylon-6 by polycondensation of aminocapronic acid or equilibration of caprolactam with aminocapronic acid. | ||
From a preparative and theoretical point of view, ROPPOCs have the advantage that they allow for simulation of polycondensations with conversions >99.9%. Consider a monomer/Cat ratio of 1000/1 as used in this work, the ROP of the monomer corresponds to a polycondensation with 99.9% conversion. Any inter or intramolecular condensation following or accompanying the ROP will then raise the conversion above 99.9% and conversions of 99.999% or higher may be achieved, which can hardly be realized by a normal polycondensation starting out from linear monomers. Such extreme conversions are interesting, because the modern theories of reversible and irreversible polycondensations predict that the fraction of cycles increases with the conversion until all reaction products have a cyclic topology at 100% conversion. For ROPPOC syntheses of cyclic polyesters two classes of catalysts have been found, namely strong nucleophiles, such as pyridines10–14 heterocyclic carbenes15–22 on the one hand, and covalent tin(II) or tin(IV) compounds23–26 on the other. A simplified polymerization mechanism involving covalent end groups catalyzed by dibutyltin bis(pentafluorophenoxide) BuSnOPF is outlined in Scheme 3.24 These ROPPOCs of lactide or CL play an important role in the present work.
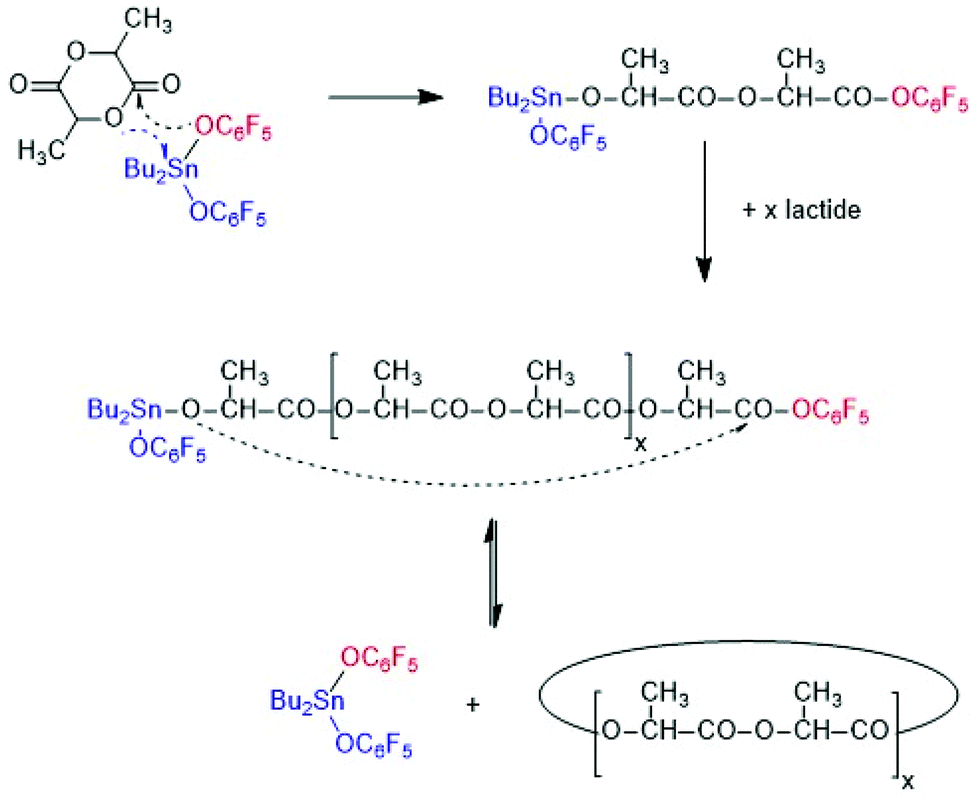 | ||
| Scheme 3 Simplified ROPPOC mechanism catalysed by BuSnOPF. | ||
The present study was stimulated by an observation mentioned by several research goups,27–30 which reported that alcohol-initiated ROPs of lactide catalyzed by tin(II)2-ethylhexanoate (SnOct2) proceed without back-biting at temperatures around 130 °C or below. Yet, despite absence of back-biting a smaller or larger fraction of odd-numbered chains was detectable (depending on temperature) indicating occurrence of a transesterification reaction. In the absence of transesterification reactions, a clean polymerization of lactide must yield even-numbered species regardless if linear or cyclic (if the initiator does not incorporate an additional unit). Hereinafter, this type of transesterification will be called “single unit exchange mechanism” (SUE).
As a typical example, Fig. 1A presents a MALDI mass spectrum of an ethyl L-lactate-initiated polymerization of L-lactide catalyzed by SnOct2 at 130 °C with a lactide/initiator ratio of 40/1.
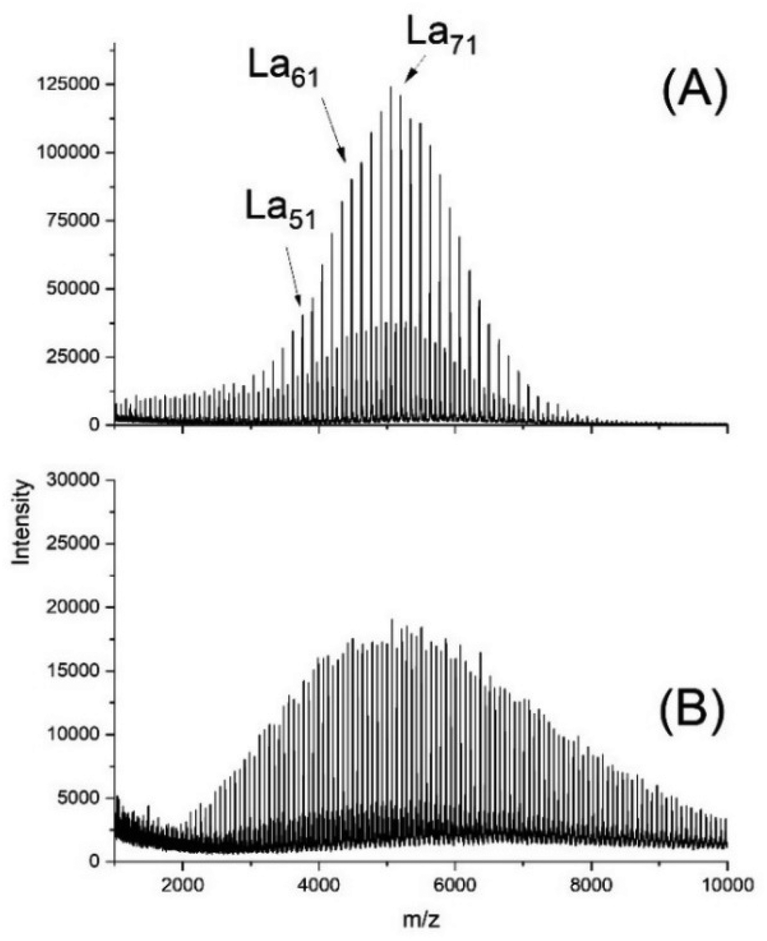 | ||
| Fig. 1 MALDI TOF mass spectra of polyLA polymerized at 130 °C with SnOct2 as catalyst: (A) initiated with ELA (No.1A, Table 1), (B) initiated with pentaerythritol (No. 6, Table 1). | ||
This spectrum shows a predominance of odd-numbered chains (due to incorporation of an ethyl lactate end group) along with a considerable fraction of even-numbered species. These findings raise the question, if it is possible to perform ROPPOC syntheses of cyclic polyesters in the absence of back-biting, but in the presence of other transesterification/equilibration reactions. Since in this case the formation of cyclic polyesters will exclusively result from ete-cyclization, such a ROPPOC synthesis of cyclic polyesters will be outside the JS-hypothesis of revPOCs. To avoid misunderstanding, it should be clarified that the authors differentiate between two aspects of the JS-theory. On the one hand, the work of J + S is a theory of ring-chain equilibration, which allows for the calculation of equilibrium constants and their correlation with the conformational properties of the polyester chains under investigation. On the other hand, J + S have developed a hypothesis of revPOCs on the basis of ring chain equilibration with a mathematical approach predicting the composition of the reaction mixture with increasing conversion. Both aspects of the JS theory overlap, of course, but are not identical. The critique of the authors in their previous publications5,6 and in the present work is exclusively directed against the JS-hypothesis of revPOCs.
In this context, the present work was aimed at finding catalysts and reaction conditions allowing for the realization of revPOCs proceeding without equilibration via back-biting, which is the chemical basis of the JS concept.
Experimental
Materials
L-Lactide (Lac), a product of Corbion Purac, was kindly supplied by Thyssen-Uhde AG (Berlin) and purified by crystallization from “Toluene 99.89% extra dry” (ACROS Organics, Geel, Belgium). ε-Caprolactone (CL) was purchased from Alfa Aesar (Karlsruhe, Germany) and distilled over P4O10 in a vacuum <1 mbar. For this purpose, CL was shaken with a small amount of P4O10 for approx. 1 min, and decanted. Again, a small amount of P4O10 was added, followed immediately by distillation at a bath temperature of 95–97 °C, which was raised to 107 °C in the course of 1 h. Yield 60–70%. Tin(II)2-ethylhexanoate (SnOct2), diphenyltin dichloride (Ph2SnCl2), dibutyltin oxide, pentafluorophenol and ethyl L-lactate were all purchased from Alfa Aesar and used as received. Tin(II) acetate (SnAc2), 2-hydroxymethyl 3,4 benzo-1,4-dioxane (HMBD) and pentaerythritol (PENT) were also purchased from Alfa Aesar and dried in vacuo in the presence of solid NaOH. Dibutyltin bis(pentafluorophenoxide), (BuSnOPF) was prepared as described previously.24Polymerizations
Synthesis of Ac-PLA-Et
Ethyl lactate (4 mmol) was weighed into a flame-dried 100 mL Erlenmeyer flask; a magnetic bar and L-lactide (160 mmol) were added under blanket of argon. SnOct2 (0.4mLof a 0.4 M solution in toluene) was injected. The reaction vessel was immersed into an oil bath thermostated at 130 °C. After 1.5 h the crystallized product was dissolved in 100 mL of dichloromethane and acetic anhydride (3 mL) and pyridine (0.5 mL) were added. After 3d at 22 °C the reaction mixture was refluxed for 1 h and precipitated into ligroin (1 L). The crystalline precipitate was isolated by filtration and dried at 60 °C in vacuo. In the 1H NMR spectrum the triplet signal of the ethyl ester end group at 1.28 ppm and the singlet signal of the acetyl end group at 2.15 ppm had equal intensities.Synthesis of Ac-PCL-Et
This synthesis was performed with ε-caprolactone (160 mmol) as described above. The crystalline polyester was dried at 40 °C in vacuo. In the 1H NMR spectrum the CH3 triplet signal of the ethyl ester end group at 1.28 ppm and the singlet signal of the acetyl end group at 2.04 ppm had equal intensities.Transesterification experiments
When SnOct2 was used as catalyst, 0.1 mL of a 0.4 M solution in toluene was added to the lactide by means of a syringe.
An analogous experiment was performed with 0.1 mmol of SnOct2 and a duration of 24 h.
Measurements
The 400 MHz 1H NMR spectra were recorded with a Bruker Avance 400 in 5 mm sample tubes. CDCl3 containing TMS served as solvent and shift reference. MALDI TOF mass spectrometry was performed using an AutoflexMax mass spectrometer (Bruker Daltonik GmbH, Bremen). Trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB) dissolved in tetrahydrofuran (10 mg mL−1) was doped with potassium trifluoroacetate and served as matrix. 1 μL of premixed solutions of matrix and analyte (chloroform, 4 mg mL−1) in a ratio of 5/1 (v/v) were prepared and deposited on the sample target. 8000 single spectra recorded at four different positions within the spots were accumulated in the linear positive mode. The instrument was previously calibrated with PEO standards.For the GPC experiments a modular system kept at 40 °C (isocratic pump, 1 mL min−1, refractive index detector, RI-501 -Shodex) was applied. Samples were manually injected (100 μL, 2–4 mg mL−1). For instrument control and data calculation Clarity software (GPC extension, DataApex) was used. The calibration was performed using polystyrene standard sets (Polymer Standards Service – PSS, Mainz, Germany). For the determination of the Mark–Houwink–Sakurada (MHS) relationship a viscometer (Viscostar, Wyatt, Germany) and a multiangle laser light scattering (MALS) detector (Dawn EOS, Wyatt, Germany) were used. Astra 6.1 software (Wyatt) served for calculating the MHS curves.
Results and discussion
General considerations
The polymerization experiments conducted in this work are subdivided into two groups. The first group is characterized by the use of an alcohol as initiator (mainly ethyl L-lactate, ELA), whereas neat catalysts were used for the second group of polymerizations under identical or similar conditions as the alcohol-initiated ROPs. The alcohol-initiated experiments had the purpose to find out, if cycles are formed by back-biting (intramolecular transesterification). The second series had the purpose to find out, if intermolecular transesterification occurs and if cycles are formed by end-to-end (ete) cyclization via the ROPPOC mechanism (e.g.Scheme 3). Most polymerizations were performed with L-lactide because the dimeric character of this monomer includes an important analytical advantage. Any clean polycondensation and polymerization initiated by an alcohol will yield even-numbered polylactides and the appearance of odd-numbered species evidences the occurrence of transesterification reactions. ELA as initiator will, of course, result in the inverse pattern. Since MALDI TOF mass spectrometry enables a sensitive and unambiguous differentiation between odd- and even-numbered chains or cycles in the mass range up to m/z 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, this type of mass spectrometry was used for the characterization of all reaction products. The MALDI-TOF mass spectra of the alcohol-initiated polylactides typically displayed a pattern of four signals: two strong signals of even and odd-numbered linear chains doped with K ions ad two accompanying weak signals resulting from unintended Na doping.
000, this type of mass spectrometry was used for the characterization of all reaction products. The MALDI-TOF mass spectra of the alcohol-initiated polylactides typically displayed a pattern of four signals: two strong signals of even and odd-numbered linear chains doped with K ions ad two accompanying weak signals resulting from unintended Na doping.
The LA/In ratio was fixed at the low value of 40/1 for all experiments of this work, for the following reason. It was found in a previous study of SnOct2-catalyzed ROPs that a high initiator/catalyst ratio (typically >10/1) is beneficial for the suppression of back-biting. Presumably, a high catalyst concentration has the consequence that the ROPPOC mechanism can compete with the alcohol-initiated ROP, so that cycles are formed by ete cyclization and appear to result from back-biting. Therefore, a LA/Cat ratio of 1000/1 was preferentially used, when SnOct2 served as catalyst (Table 1). However, for other catalysts, such as Ph2SnCl2 or BuSnOPF it was found that higher catalyst concentrations (LA/Cat = 100/1 up to 400/1) are beneficial to avoid side reactions including cyclization. Finally, it should be mentioned that most experiments were performed at 130 °C, because it was learned from ROPs catalyzed with SnOct2 that at this temperature and short reaction times (<3 h) formation of cycles by back-biting is avoidable. The following description of results is subdivided into sections according to the catalysts used for the polymerizations.
| Exp. no. | Initiator | LA/Cat | Temp. (°C) | Time (h) | M n | M w | Đ | Cycles |
|---|---|---|---|---|---|---|---|---|
| 1 | ELA | 400/1 | 130 | 1 | 11100 |
12400 |
1.1 | — |
| 1B | ELA | 400/1 | 130 | 3 | 11000 |
12300 |
1.1 | — |
| 2A | ELA | 1000/1 | 130 | 1 | 10600 |
11700 |
1.1 | — |
| 2B | ELA | 1000/1 | 130 | 3 | 10800 |
11900 |
1.1 | — |
| 3 | ELA | 1000/1 | 160 | 1 | 10900 |
15000 |
1.4 | Traces |
| 4A | HMBD | 1000/1 | 130 | 1 | 10700 |
12300 |
1.2 | — |
| 4B | HMBD | 1000/1 | 130 | 3 | 11000 |
12300 |
1.1 | — |
| 5 | PENT | 1000/1 | 130 | 1 | 8 900 | 10400 |
1.2 | — |
| 6A | — | 400/1 | 130 | 1 | 44000 |
76000 |
1.7 | + |
| 6B | — | 400/1 | 130 | 3 | 48000 |
84000 |
1.7 | + |
| 7A | — | 1000/1 | 130 | 1 | 118500 |
188000 |
1.6 | + |
| 7B | — | 1000/1 | 130 | 3 | 121500 |
217000 |
1.8 | + |
| 8 | — | 1000/1 | 160 | 1 | 91000 |
240000 |
2.6 | ++ |
ROPs and ROPPOCs of L-lactide catalyzed by SnOct2
A first series of polymerizations was performed with SnOct2 as catalyst for three reasons. First, this tin(II) salt is the standard catalyst for the technical production of polylactide, via an alcohol-initiated polymerization process outlined in Scheme 4. Second, it is the most widely studied catalyst for polymerizations of lactide and third, the authors have recently found that neat SnOct2 catalyses a ROPPOC mechanism yielding high molecular weight cyclic poly(L-lactide)s (Scheme 5). Most ROPs summarized in Tables 1, were conducted in bulk at 130 °C and a few experiments were performed at 160 °C. These reaction conditions were selected for the following reasons. A temperature of 130 °C was the highest temperature reported in literature where alcohol-initiated ROPs (catalyzed by SnOct2) were shown to proceed without formation of cyclics, meaning without back-biting. Previous studies of the authors have confirmed that at 120 °C and polymerization times of several hours may result in formation of cyclics. The authors have also shown for ROPs at 160 °C, that formation of cyclics is favoured by lower initiator/catalyst ratios. Alcohol/SnOct2 ratios above 5, better above 10, are needed to avoid formation of cycles. The ROPs initiated by ELA yielded polyLAs having a low dispersities (1.1–1.2) and a kind of broadened Poisson distribution of peak intensities in the MALDI TOF mass spectra (Fig. 1A and 2A). The even/odd equilibration was above 50% for the ELA experiments (No. 1–5 Table 1). It increased with the catalyst concentration (No. 1 versus No. 2A), with time (No. 2A versus No. 2B) and with temperature (No. 2A versus No. 3 and Fig. 1Aversus S2A†).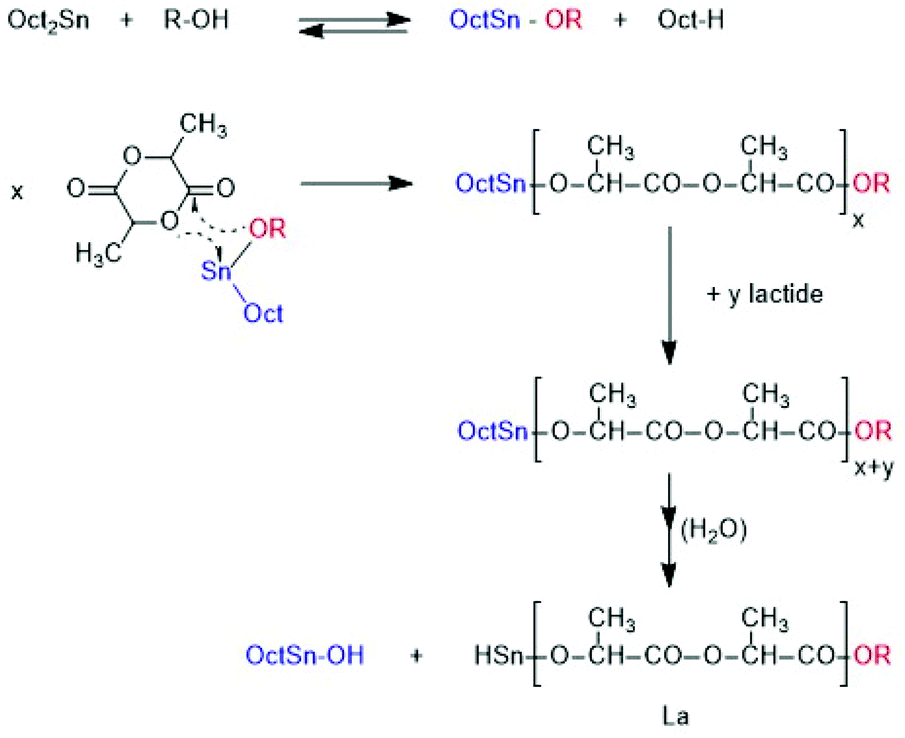 | ||
| Scheme 4 Alcohol-initiated ROP of L-lactide catalyzed by SnOct2 at low temperature and high In/lac ratio. | ||
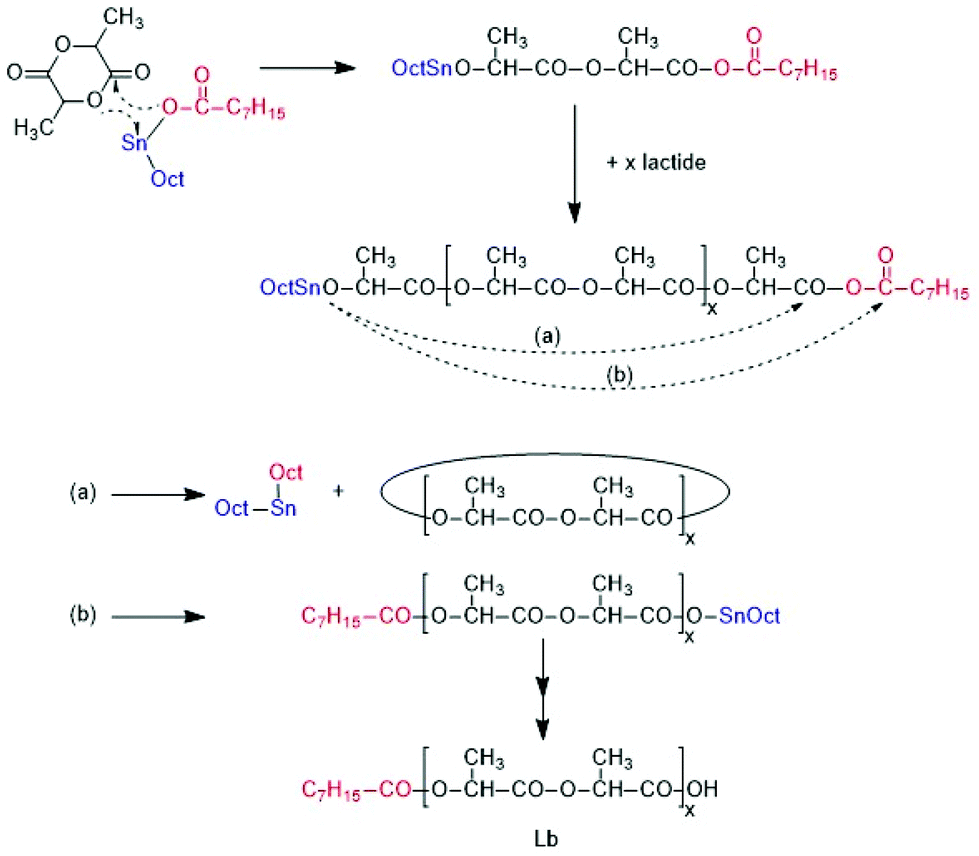 | ||
| Scheme 5 Formation of cyclic polylactides via the SnOct2 catalysed ROPPOC mechanism. | ||
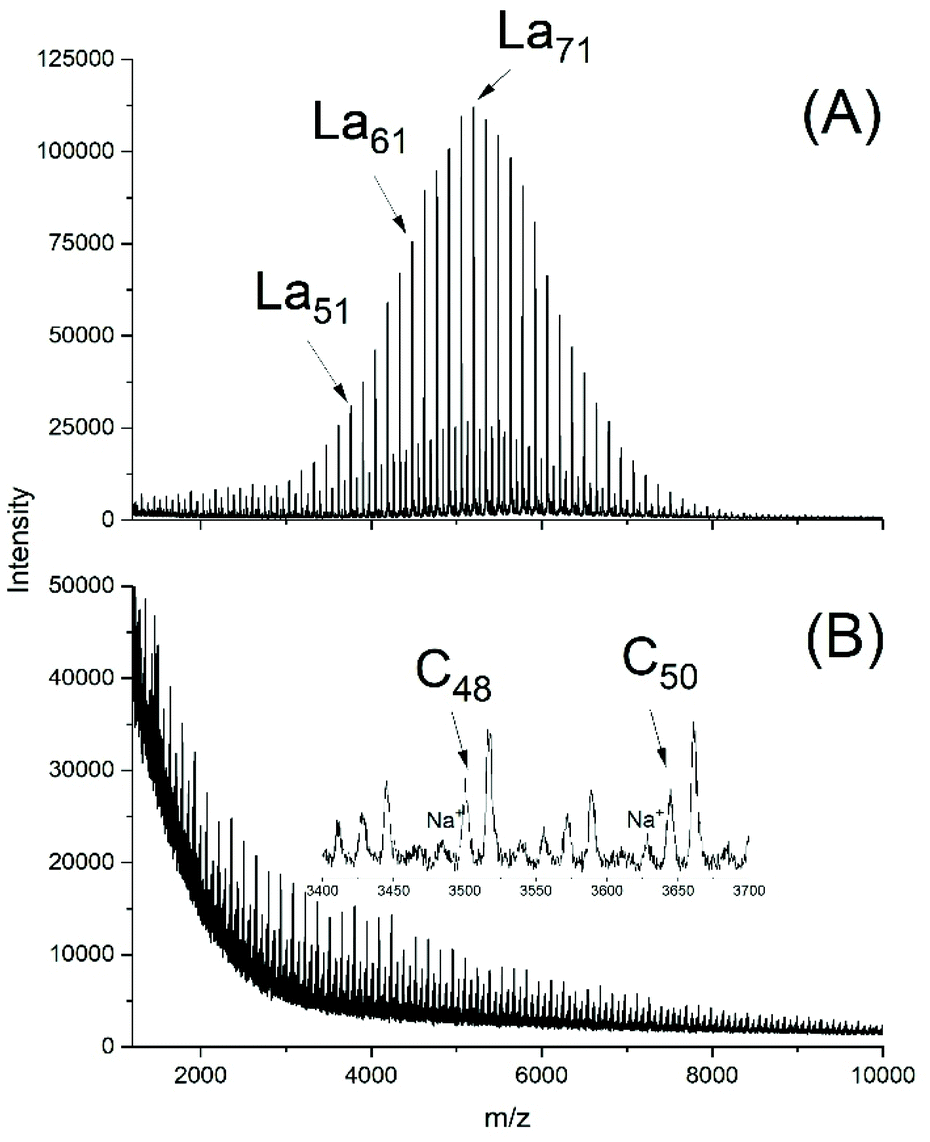 | ||
| Fig. 2 MALDI TOF mass spectra of polyLA polymerized at 130 °C/3 h with SnOct2 as catalyst (LA/Cat 1000/1): (A) initiated with ELA (No. 2B, Table 1), (B) without initiator (No. 7B, Table 1). | ||
These three trends were also valid for all alcohol initiated ROPs based on other catalysts (Tables 2–6). An even/odd equilibration around 50% (Fig. S1†), which increased with time, was also found for the ROPs initiated with the primary alcohol HMBD (No. 4A and 4B).
| Exp. no. | Init. | Lac/Cat | Temp. (°C) | Time (h) | M n (meas.) | M w | Đ | Cycles |
|---|---|---|---|---|---|---|---|---|
| 1 | ELA | 200/1 | 130 | 1 | 9200 | 10600 |
1.2 | — |
| 2 | ELA | 400/1 | 130 | 1 | 9100 | 10200 |
1.2 | — |
| 3 | ELA | 400/1 | 160 | 1 | 9100 | 14000 |
1.5 | — |
| 4 | ELA | 400/1 | 160 | 3 | 10000 |
15100 |
1.5 | — |
| 5 | — | 200/1 | 130 | 1 | 47000 |
105000 |
2.2 | + |
| 6 | — | 400/1 | 130 | 1 | 63000 |
153000 |
2.4 | + |
| 7 | — | 400/1 | 160 | 1 | 79500 |
205000 |
2.6 | ++ |
| Exp. no. | Init. | Lac/Cat | Temp. (°C) | Time (h) | M n (meas.) | M w (meas.) | Đ | Cycles |
|---|---|---|---|---|---|---|---|---|
| 1A | ELA | 200/1 | 130 | 1.0 | 9000 | 10700 |
1.2 | — |
| 1B | ELA | 200/1 | 130 | 2.0 | 10100 |
11200 |
1.1 | — |
| 1C | ELA | 200/1 | 130 | 4.0 | 10200 |
11300 |
1.1 | — |
| 2 | ELA | 400/1 | 130 | 1.0 | 8600 | 10800 |
1.2 | — |
| 3A | ELA | 400/1 | 160 | 1.0 | 8700 | 11000 |
1.3 | — |
| 3B | ELA | 400/1 | 160 | 3.0 | 9000 | 13800 |
1.4 | Traces |
| 4A | — | 200/1 | 130 | 2.0 | 44000 |
69500 |
1.6 | — |
| 4B | — | 200/1 | 130 | 4.0 | 43500 |
70500 |
1.6 | + |
| 5A | — | 400/1 | 130 | 2.0 | 41000 |
64000 |
1.6 | + |
| 5B | — | 400/1 | 130 | 4.0 | 37000 |
58000 |
1.6 | — |
| 6 | — | 100/1 | 160 | 1.0 | 42000 |
107000 |
2.5 | ++ |
| 7 | — | 200/1 | 160 | 1.0 | 54000 |
124000 |
2.3 | + |
| 8 | — | 400/1 | 160 | 1.0 | 55000 |
132000 |
2.4 | + |
| Exp. no. | Init. | Lac/Cat | Temp. (°C) | Time (h) | M n (meas.) | M w (meas.) | Đ | Cycles |
|---|---|---|---|---|---|---|---|---|
| 1 | ELA | 200/1 | 130 | 1 | 8500 | 12000 |
1.4 | — |
| 2 | ELA | 400/1 | 130 | 1 | 8300 | 11500 |
1.4 | — |
| 3 | ELA | 400/1 | 160 | 1 | 8800 | 15000 |
1.7 | Traces |
| 4 | ELA | 400/1 | 160 | 3 | 8500 | 14700 |
1.7 | Traces |
| 3A | — | 200/1 | 130 | 0.5 | 53000 |
111000 |
2.1 | ++ |
| 3B | — | 200/1 | 130 | 1 | 50000 |
103000 |
2.1 | ++ |
| 4 | — | 400/1 | 130 | 1 | 55000 |
139000 |
2.5 | ++ |
| 5A | — | 600/1 | 130 | 1 | 75000 |
181000 |
2.4 | ++ |
| 5B | — | 600/1 | 130 | 2 | 68000 |
157000 |
2.3 | ++ |
| Exp. no. | Init. | CL/Cat | Time (h) | M n | M w | Đ | Cycles |
|---|---|---|---|---|---|---|---|
| 1 | EHH | 200/1 | 1.0 | 8100 | 16000 |
2.0 | — |
| 2 | EHH | 400/1 | 1.0 | 8300 | 17000 |
2.0 | — |
| 3 | EHH | 1000/1 | 1.0 | 9000 | 18500 |
2.1 | — |
| 4A | HMBD | 400/1 | 1.0 | 6500 | 15700 |
2.5 | — |
| 4B | HMBD | 400/1 | 2.0 | 4100 | 15300 |
3.7 | — |
| 5A | HMBD | 600/1 | 1.0 | 6100 | 15300 |
2. | — |
| 5B | HMBD | 600/1 | 2.0 | 5400 | 13400 |
2. | — |
| 6 | — | 200/1 | 1.0 | 46000 |
92000 |
2.0 | ++ |
| 7 | — | 400/1 | 1.0 | 61000 |
127000 |
2.1 | ++ |
| 8 | — | 600/1 | 1.0 | 70500 |
143000 |
2.1 | ++ |
| 9 | — | 1000/1 | 1.5 | 85000 |
168000 |
2.0 | ++ |
Their mass spectra were quite similar to those of Fig. 1A. For a project dealing with biodegradable coatings, a ROP initiated with pentaerythritol was performed at 130 °C and in this case the odd-/even equilibrium was even complete as displayed in Fig. 1B. All these mass spectra have in common, that no cycles were detectable (s. Fig. S1B†). In the case of experiments 2B and 3, the low molar mass fraction was also subject to ESI† mass spectroscopy and again no cycles were found for No. 2B (130 °C) and only a trace for No. 3 (160 °C). The 160 °C experiment also revealed almost complete odd-/even equilibration along with broadening of the molecular weight distribution indicating random intermolecular transesterification. (Fig. S2A†). Hence these experiments demonstrate that under the given reaction conditions ROPs including intermolecular transesterification exist without contribution of back-biting.
With neat SnOct2 completely different results were obtained. The polymerizations were slower without addition of alcohol (a well-known phenomenon), and with a LA/Cat ratio of 1000/1 only a conversion of 91% was reached after 1 h, but after 3 h the conversion had reached the equilibrium level of 97%. With a LA/Cat ratio of 400/1, a conversion of 97% was reached within 1 h.
Regardless of the LA/Cat ratio, high molecular weights were achieved, and quite similar mass spectra were obtained which displayed strong peaks of cycles (Fig. 2B) along with a peak of an unidentified linear species.
Complete odd/even equilibration was not achieved, what indicates that the cycles were formed by ete-cyclization and not by back-biting. At 160 °C complete conversion, complete equilibration and a high molecular weight was already obtained after 1 h. MALDI TOF mass spectra exclusively displaying peaks of cycles (Fig. S2B†) and intrinsic viscosity measurements confirming a cyclic topology also for the high molar mass fraction were found, results that were recently published and thus, do not need extensive discussion at this point.26 All these properties were in agreement with the ROPPOC mechanism outlined in Scheme 3 as discussed in a recent publication.26
ROPs and ROPPOCs of L-lactide catalyzed by SnAc2 or Ph2SnAc2
The mass spectra of polyLAs prepared with SnOct2 have the shortcoming that they cannot differentiate between cycles and linear chains terminated by a 2-ethyl hexanoate end group, because 2-ethyl hexanoic acid has the same mass as lactide.As outlined in Scheme 3, formation of linear chains having a COOH chain end may compete with the formation of cycles although to a low extent. This analytical problem does not exist for tin acetates. As published recently,26 the reactivity of SnAc2 is quite similar to that of SnOct2. At LA/In ratios <100/1 linear acetate terminated chains are predominantly formed, but at higher ratios formation of cycles is prevalent and at LA/In ratios >400 linear chains are not detectable in the mass spectra anymore. Furthermore, SnAc2 can catalyse alcohol-initiated ROPs, so that the average degree of polymerization (DP) parallels the LA/In ratio and low dispersities are obtained. The results listed in Table 2 are in line with previous results.26
The MALDI TOF mass spectra of the ELA-initiated poyLAs (No. 1–4, Table 2) were quite similar to those obtained with SnOct2, and Fig. 3 also illustrates the dependence of the even/odd equilibration on the catalyst concentration. Particular important is again the absence of cycles. The results obtained with neat SnAc2 were also analogous to those found with SnOct2, because a considerable fraction of cycles was formed after 1 h at 130 °C (Fig. 4A). At 160 °C the predominance of cycles was more complete (Fig. 4B). Regardless of temperature, again high molecular weights and high dispersities were obtained along with full odd/even equilibration. Therefore, the SnAc2 experiments were in full agreement with the SnOct2-catalyzed polymerizations and their interpretation.
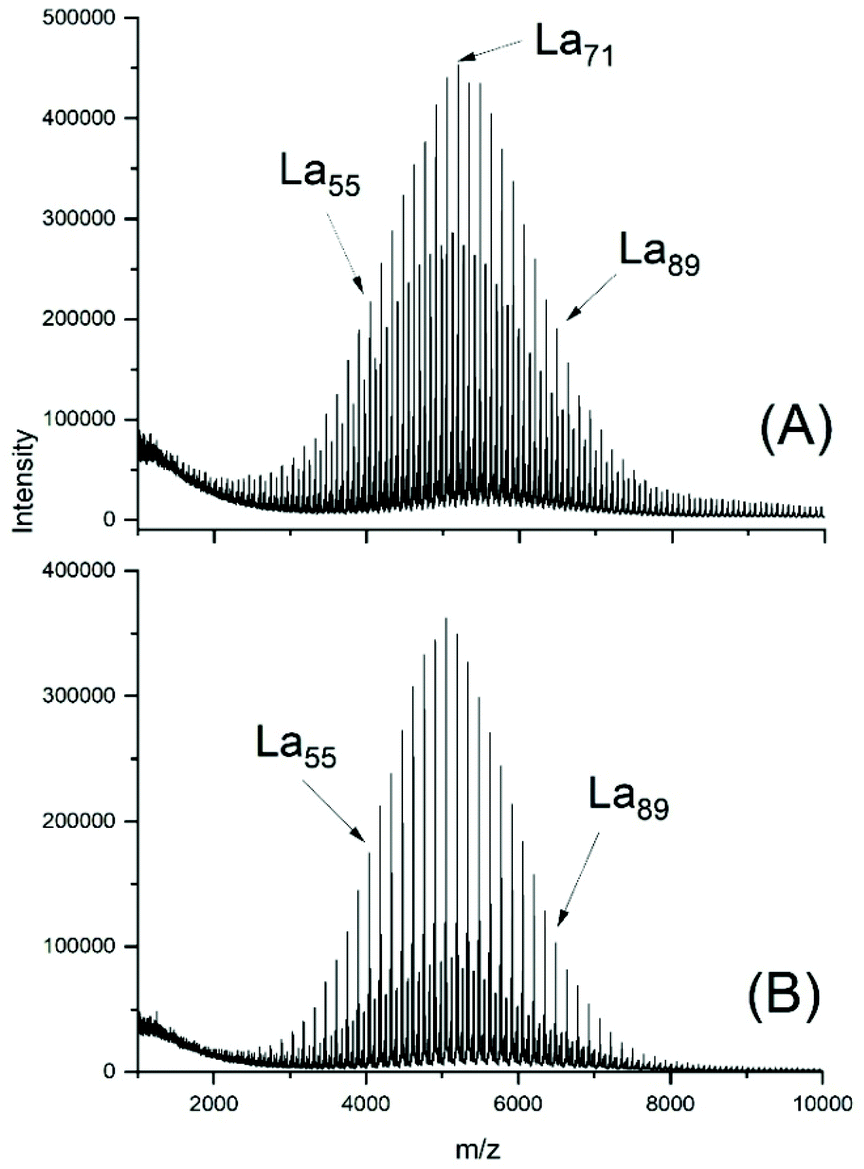 | ||
| Fig. 3 MALDI TOF mass spectra of polyLA polymerized at 130 °C with SnAc2 as catalyst and ELA as initiator (LA/In = 40/1): (A) LA/Cat = 200/1 (No. 1, Table 2), (B) LA/Cat = 400/1 (No. 2, Table 2). | ||
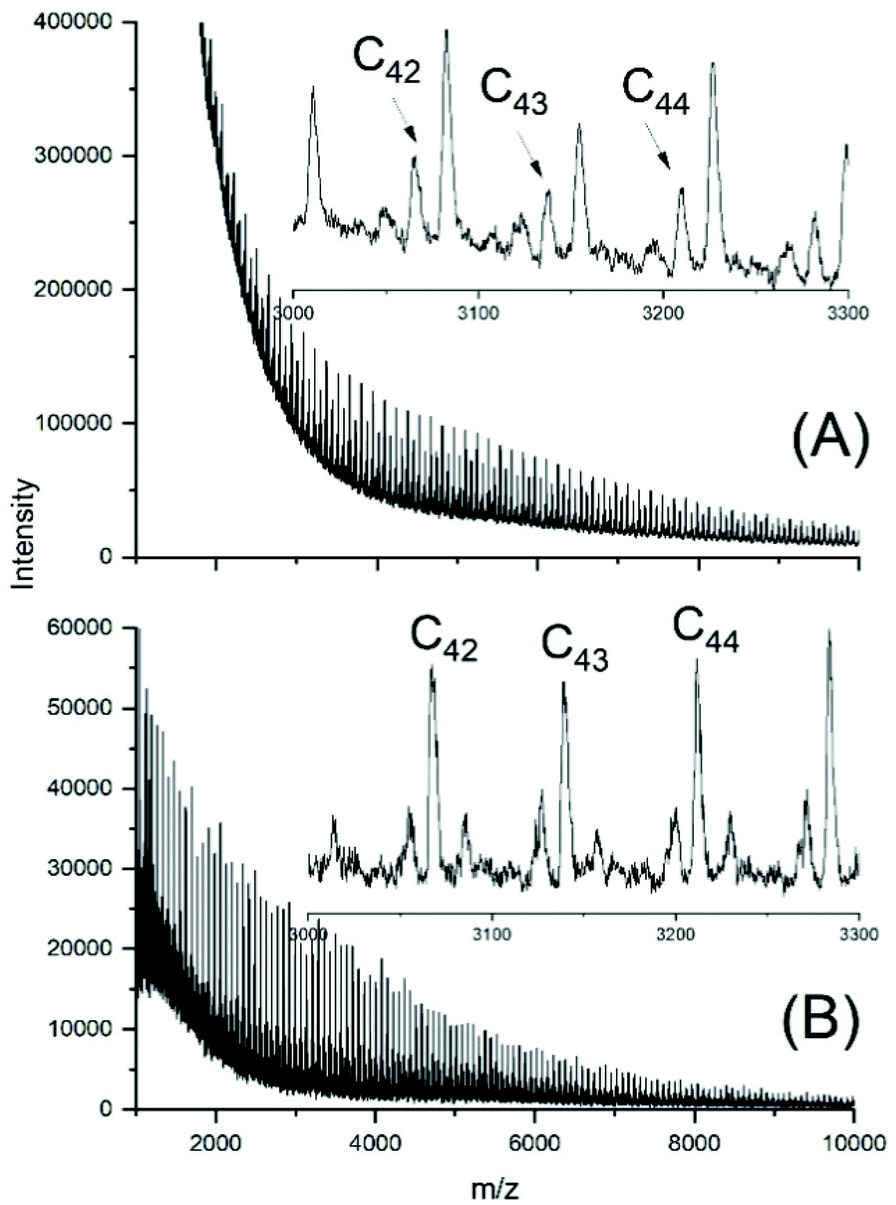 | ||
| Fig. 4 MALDI TOF mass spectra of polyLA polymerized with SnAc2 as catalyst (LA/Cat = 400/1): (A) at 130 °C/1 h (No. 6, Table 2), (B) 160°/1 h (No. 7, Table 2). | ||
ROPs and ROPPOCs of L-lactide catalyzed by Ph2SnCl2
Again, two series of polymerizations were performed with Ph2SnCl2 as catalyst and compiled in Table 3. Relatively low LA/Cat ratios were used and varied, because it was found in a previous study where neat Ph2SnCl2 served as catalyst at 160 °C that the highest molecular weights and the most uniform topology (i.e. cyclic polylactide)s were obtained when LA/Cat ratios <400/1 were used.25The (hitherto unpublished) ROPs initiated with ELA yielded polylactides of low dispersity and the molecular weights were almost independent on the LA/Cat ratio but corresponded to the LA/ELA ratio. At this point, it should be mentioned that all tin catalyst mentioned above yielded at 160 °C higher Mw values, whereas the increase of Mn was small. In agreement with the mass spectra this effect may be attributed to more efficient transesterification reactions resulting in higher dispersities. Condensation steps of the ethyl ester end groups (see discussion below) may be responsible for a slight increase of Mn. The MALDI TOF spectrum presented in Fig. 5 not only illustrates the relatively narrow MWDs, but also the absence of cycles.
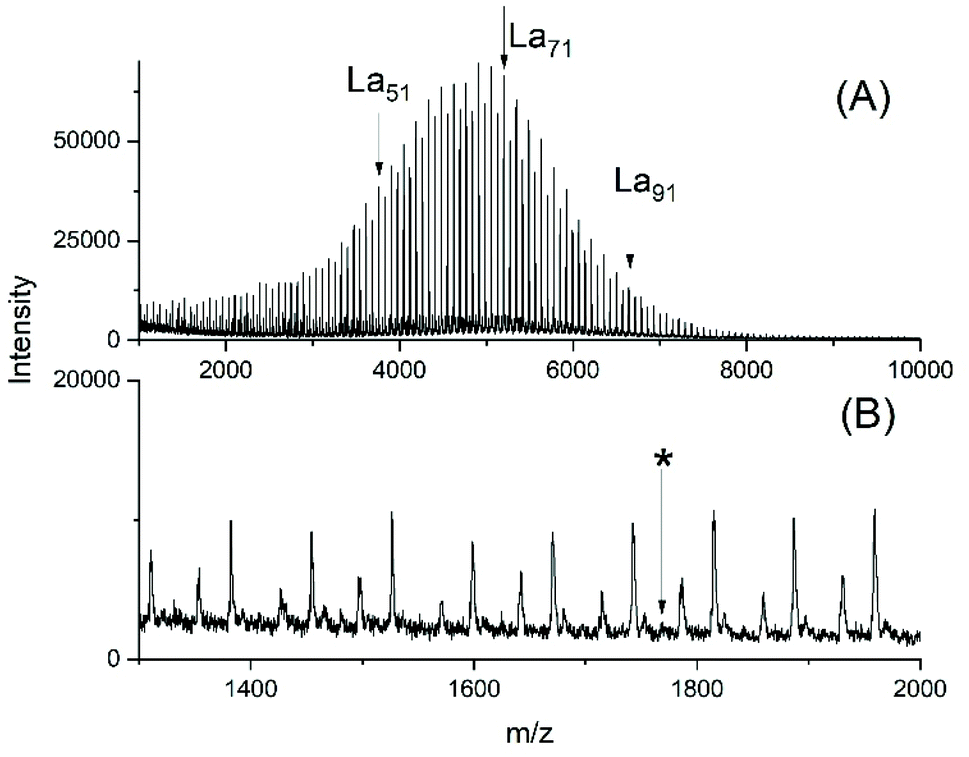 | ||
| Fig. 5 MALDI TOF mass spectra of polyLA polymerized at 130 °C/2 h with Ph2SnCl2 as catalyst (LA/Cat 200/1) and initiated with ELA: (A) full spectrum, (B) expanded segment (No. 1B, Table 4), the asterisk indicates the position of a hypothetical cycle. | ||
In contrast, all polymerizations conducted with neat Ph2SnCl2 yielded considerably higher molecular weights and higher dispersities. Furthermore, the mass spectra evidenced that mainly cyclic polylactides were formed as demonstrated in Fig. 6. In summary, the results obtained with Ph2SnCl2 were similar to those obtained from SnOct2 and SnAc2, and the predominant formation of high molar mass cyclic polylactides is also in good agreement with the previously published experiments. The results of the neat catalyst are also in perfect agreement with the hypothesis of a ROPPOC mechanism involving the intermediate formation of CO-Cl end groups as discussed previously.25
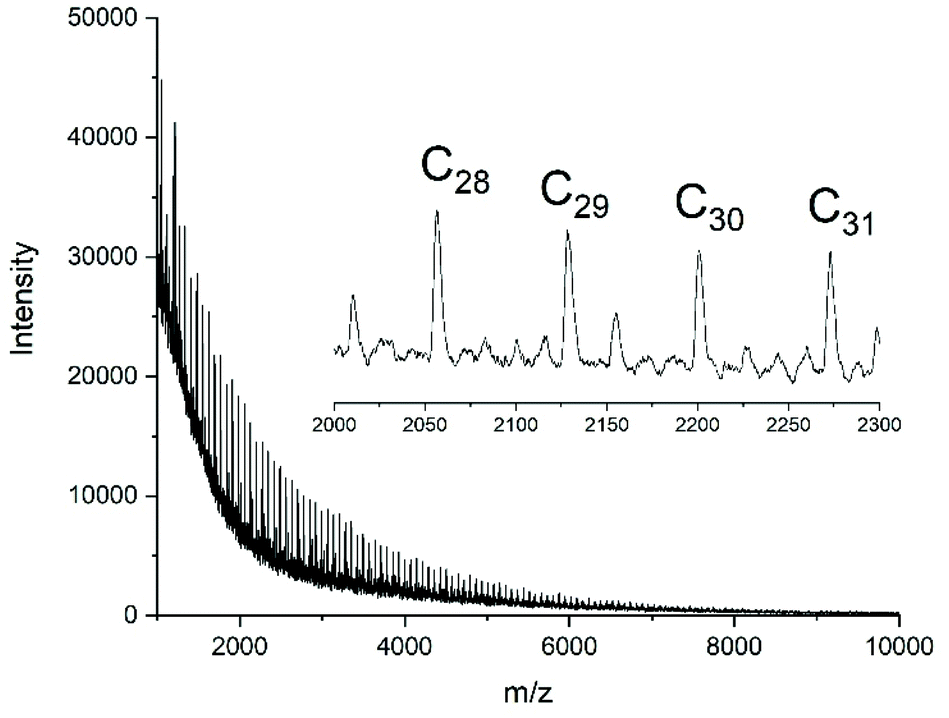 | ||
| Fig. 6 MALDI TOF mass spectrum of polyLA polymerized with neat Ph2SnCl2 in bulk at 130 °C/2 h (No. 4A, Table 2). | ||
ROPs and ROPPOCs of L-lactide catalyzed by Bu2Sn(OC6F5)2
From a previous study ROPs catalyzed with neat BuSnOPF it was learned that the best results in terms of high molecular weights and homogeneous topology were obtained at relatively low LA/Cat ratios such as 100/to 400/1, quite analogous to Ph2SnCl2.24 Therefore, the experiments of this work were performed with such low LA/Cat ratios. Two ROPS initiated with ELA were conducted at 130 °C and two ROPs at 160 °C (Table 4). The results were, in principle analogous to those described above for Ph2SnAc2 and Ph2SnCl2, but with two noteworthy differences. The odd/even equilibration was almost complete after 1 h at 130 °C favoured by the high catalyst concentration (Fig. 7A). A comparison of Fig. 7A and S3A† illustrates the role of the catalyst concentration. The absence of cycles underlines that here again transesterification reactions occur without contribution of back-biting. The second difference concerns the relatively high dispersities of 1.4 at 130 °C and 1.7 at 160 °C. The Mn values agree with the theoretical value (6000) calculated from the LA/In ratio taking a correction factor of 0.68 (±0.01) into account.31 However, the Mw values were somewhat higher than in previous experiments (Tables 1–3), possibly due to more intensive transesterification reactions.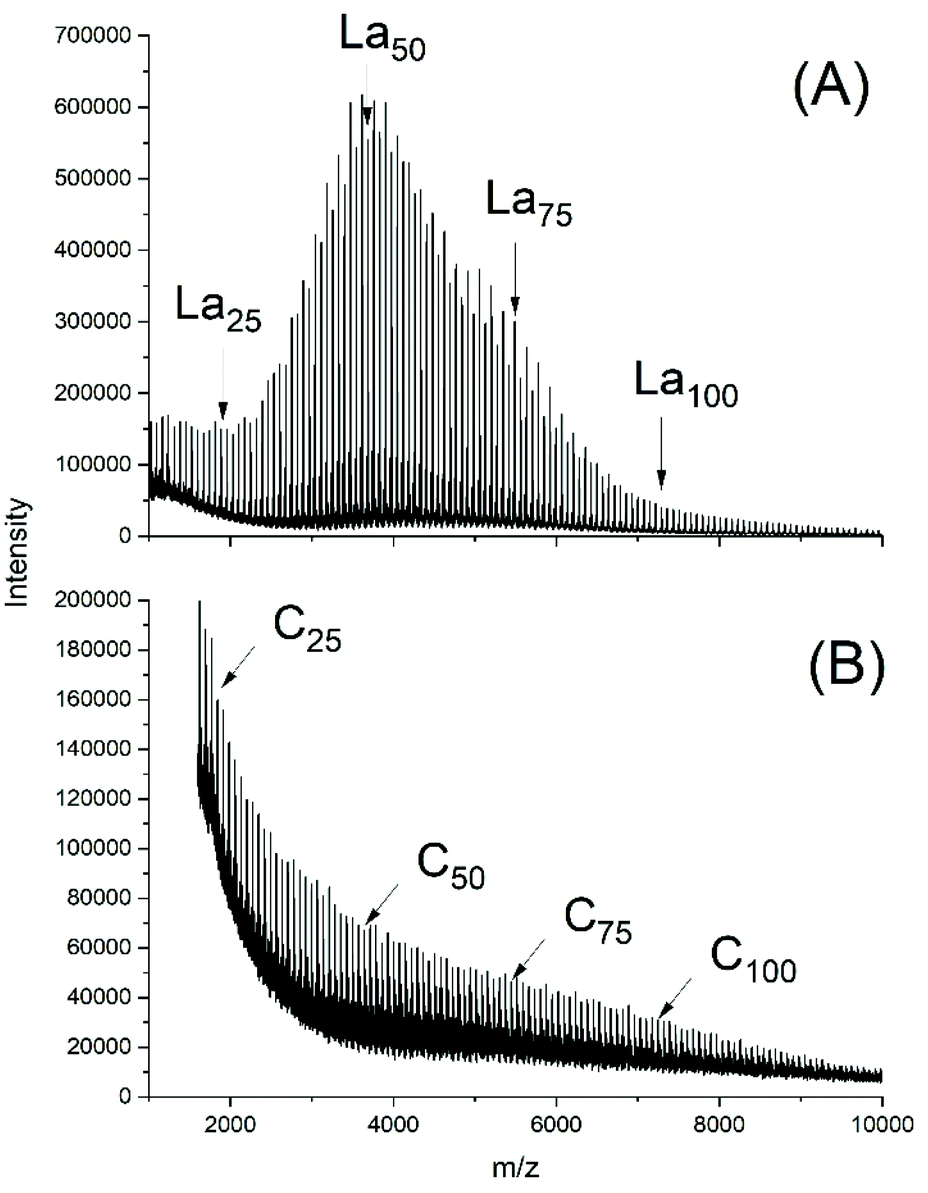 | ||
| Fig. 7 MALDI TOF mass spectra of polyLA polymerized with BuSnOPF as catalyst (LA/Cat = 200/1) at 130 °C/1 h: (A) initiated with ELA (No.1, Table 4), (B) without initiator, (No. 3B, Table 4). | ||
Noteworthy is the absence of cycles in the 130 °C samples, whereas traces of cycles were detected in the 160 °C samples. The polymerizations catalyzed with neat BuSnOPF yielded polylactides with the four characteristics typical for a ROPPC process (see Scheme 4). First, the molecular weights were considerably higher, second the dispersities were higher, the odd/even equilibration was complete, and cycles were the predominant reaction products as demonstrated in Fig. 7B and S3B.†
ROP and ROPPOC of CL with and without initiator catalyzed by BuSnOPF
To demonstrate that the results described above were not a peculiarity of the lactide chemistry, two series of experiments was performed with CL. In the first series (No. 1–5B, Table 5) two primary alcohols were added as initiators at a LA/In ratio of 40/1. In agreement with analogous ROPs of lactide linear chains were the main polymerization products, but a small amount of a linear by-product of unknown structure was also formed below m/z 2 000. However, cycles were not detectable (Fig. S6†).A remarkable difference relative to analogous ROPs of lactide is the higher dispersity which is immediately evident from the MALDI TOF mass spectra (Fig. 8A and S4A†). In absence of an initiator higher molecular weights were achieved and the mass spectra exclusively displayed peaks of cycles (Fig. 8B).
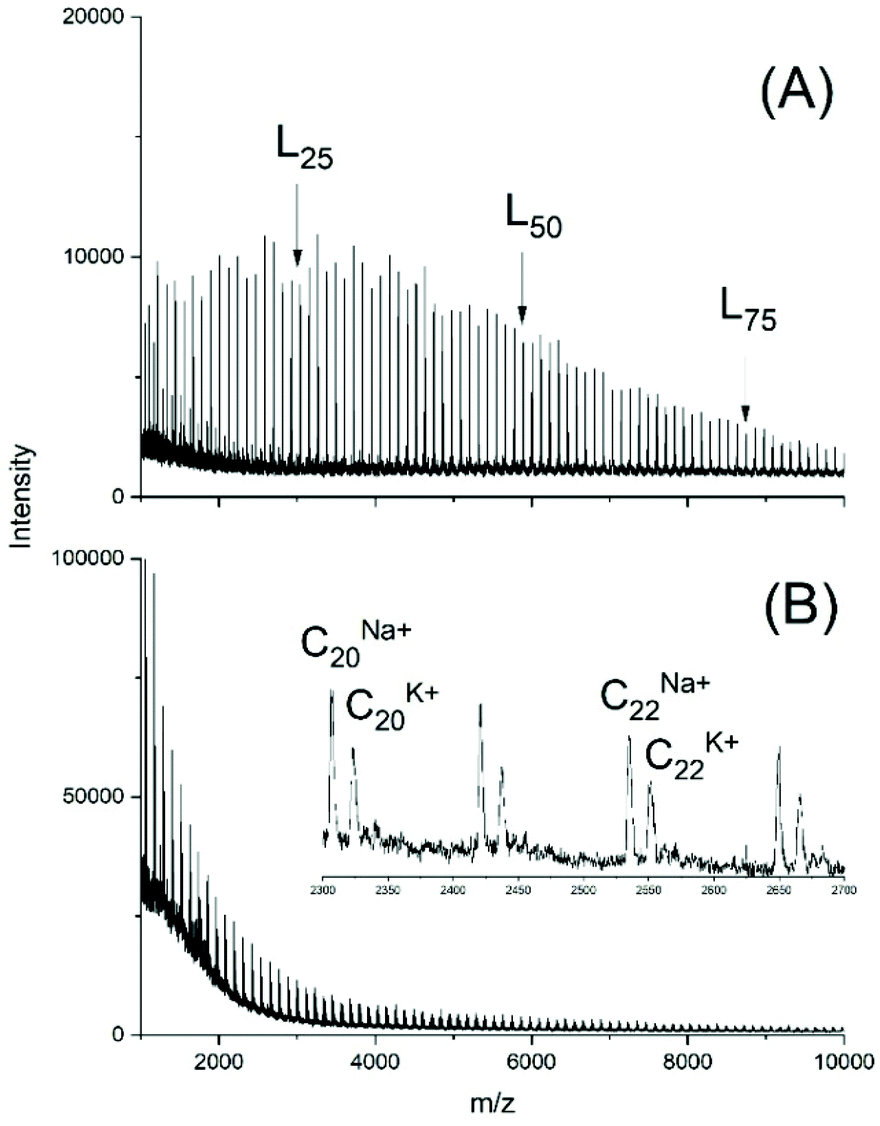 | ||
| Fig. 8 MALDI TOF mass spectra of polyCL polymerized with BuSnOPF as catalyst at 130 °C (LA/Cat = 200/1): (A) initiated with ethyl 6-hydroxyhexanoate (No. 1, Table 6), (B) without initiator (No. 6, Table 6). L – linear chains having an ethyl ester end group. | ||
Although information about transesterification yielding odd/even equilibration is, of course, lacking in the mass spectra of polylactones, the results obtained from the BuSnOPF catalyzed polymerizations listed in Table 6 clearly support the conclusions extracted from the polymerizations od L-lactide.
Model reactions of intermolecular transesterifications
The results described above demonstrate that ROPs conducted with addition and without addition of an alcohol take a quite different course. Without alcohol low and high molar mass cycles are the main reaction product as expected from a polycondensation process with high conversion (>99.9%). However, formation of cycles via ete-cyclization also occurs in irreversible polycondensations as predicted by Gordon et al.32,33 and Stepto et al.34 and experimentally confirmed by the first author.35Therefore, the results presented in this work would not be new, if equilibration reactions were absent. As mentioned in the Introduction reversible back-biting was considered by J + S to be the only source of ring-chain, ring–ring and chain–chain equilibration. Therefore, it was a particular important aspect of the present study to demonstrate that equilibration reactions occur in the absence of back-biting. As discussed above, the SUE mechanism, which is perhaps confined to the chemistry of lactide is a first example of a transesterification/equilibration mechanism which operates at temperature below which back-biting sets out. However, intermolecular transesterification reactions that broaden the MWD occur more frequently and at lower temperatures than back-biting, which was already reported previously.36 That model reaction was performed at 120 °C in such a way that L-lactide was polymerized by means of SnOct2 + benzyl alcohol in the presence of preformed polylactide having blocked end groups (Ac-PLA-Et). Three series of additional experiments was performed in the present work. In the first series L-lactide was polymerized at 160 °C with neat SnOct2, SnAc2 or BuSnOPF, so that cyclic polylactides were formed. After 1 h the reaction was stopped by cooling. The polyLA was dissolved in dry dichloromethane and combined with diethyl succinate.
After homogenization the dichloromethane was evaporated at 80 °C and the remaining reaction mixture was thermostated at 130 °C for 1 or 2 h (see Table 6). Diethylsuccinate was selected as reaction partner of polyLA for three reasons. First its boiling point is high enough to avoid premature vaporization. Second it seemed to be partially miscible with molten poly(L-lactide) and the masses of reaction products expected from transesterification (Lc and Ld chains, Scheme 6) allowed for easy identification by mass spectrometry (Lc and Ld are isomers and give identical mass peaks). The mass spectra proved that Lc and Ld chains were formed in the experiment with BuSnOPF after 2 h (Fig. S6†).
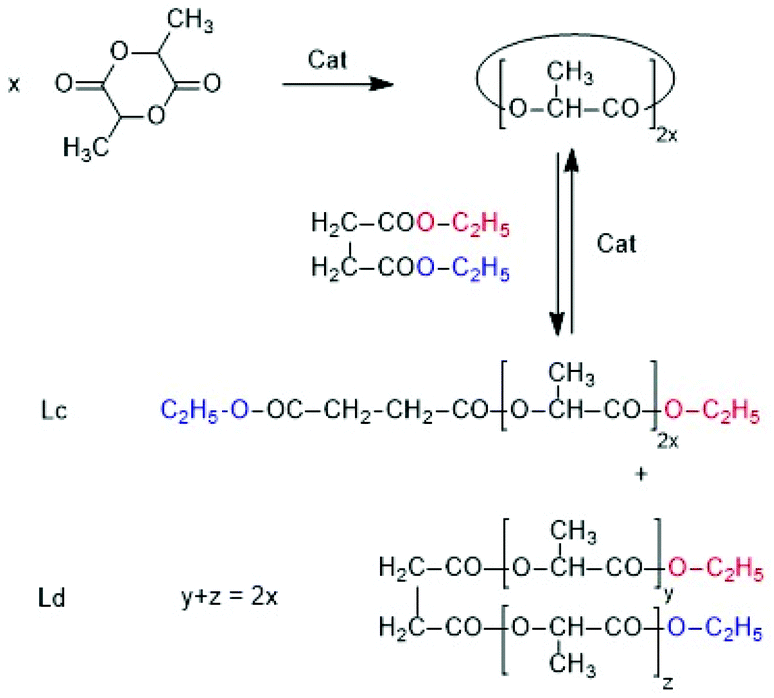 | ||
| Scheme 6 Synthesis and transesterification of poly L-lactide with diethyl succinate. | ||
A second series of experiments was performed in such a way that the potential polycondensation of ethyl L-lactate was studied at 130 °C with addition of SnOct2 or BuSnOPF as catalysts (Table 7).
| Exp. no. | Catalyst | LA/Cat | Temp (°C) | Time (h) | La Chains | Cyclics |
|---|---|---|---|---|---|---|
| 1 | SnOct2 | 1000/1 | 130 | 4 | + | — |
| 2 | SnOct2 | 400/1 | 130 | 24 | ++ | — |
| 3 | SnOct2 | 400/1 | 160 | 4 | ++ | — |
| 4A | BuSnOPF | 400/1 | 130 | 4 | + | — |
| 4B | BuSnOPF | 400/1 | 130 | 24 | ++ | Traces |
| 5 | BuSnOPF | 400/1 | 130 | 4 | ++ | — |
Due to the low reactivity of the ethyl ester group a first experiment was performed with the relatively low LA/SnOct2 ratio of 400/1 and La chains were indeed detectable after 4 h (Fig. 9A). Their concentration was higher after 4 h at 160 °C (Fig. 9B). Even with a LA/SnOct2 ratio of 1000/1 oligoLAs were formed, when the time was prolonged to 24 h and the La chains became detectable in the MALDI mass spectrum up to a DP of 25. In all three experiments peaks of cycles were absent.
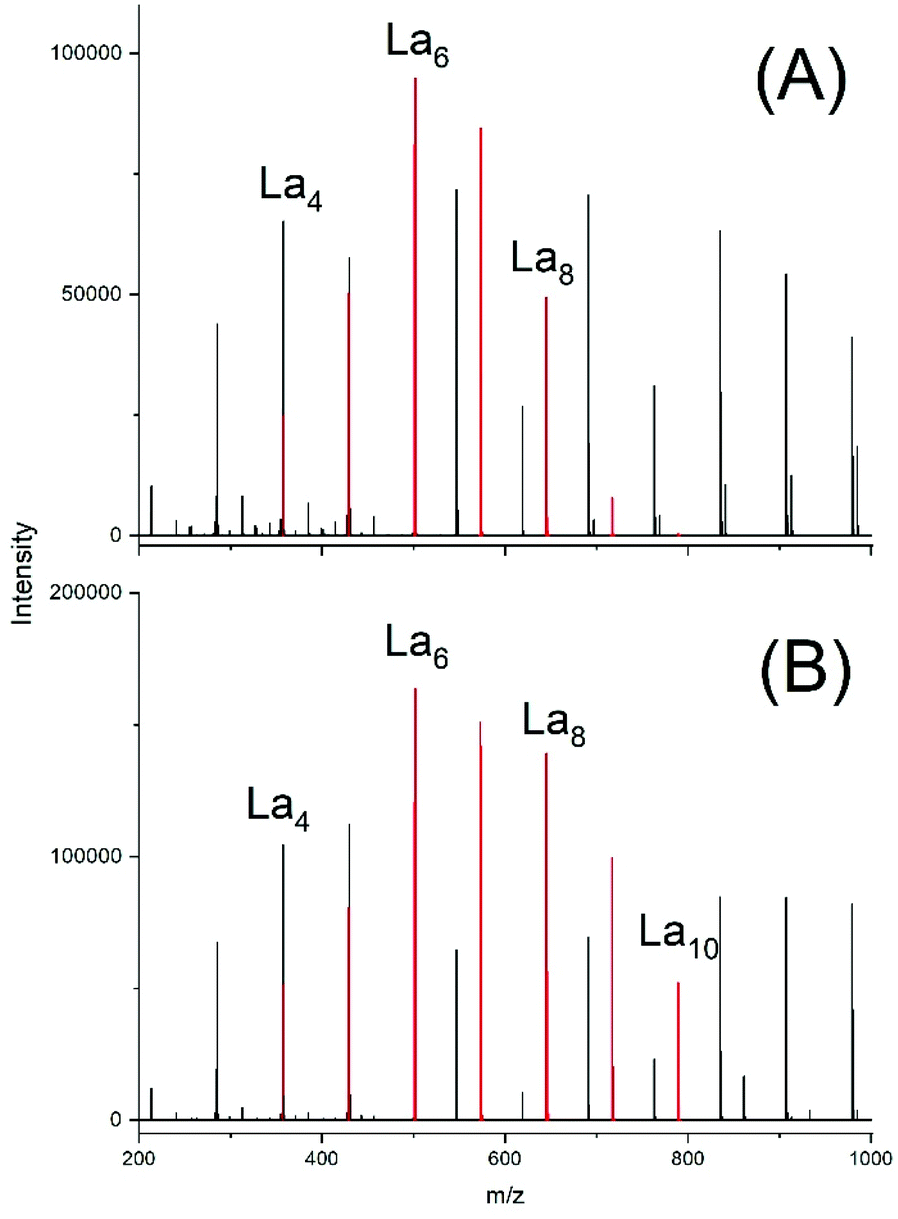 | ||
| Fig. 9 ESI† mass spectra of oligoLA prepared by polycondensation of ELA with SnOct2 (A) at 130 °C/4 h in, (B) 160 °C/4h. | ||
Similar results were obtained with BuSnOPF, which proved to be slightly more reactive than SnOct2 Cycles were again absent in the 4 h experiments (No. 4A and 5, Table 7 and Fig. S7†), but traces of cycles were observable in the ESI spectrum after 24 h at 130 °C (Fig. S8†). In the MALDI mass spectra La chains were detectable up to DPs around 40 (No. 5 (Fig. S9A†), and up to DPs around 140 (No. 4B, Fig. S9B†), whereas peaks of cycles were absent. These results include three remarkable aspects. First, they demonstrated the existence of an intermolecular transesterification of the ethyl ester end groups, which is reversible as long as the liberated alcohol is present in the reaction mixture. Second, they indirectly indicate that the active chain ends (Sn–O–CH) can also react with the lactyl-lactyl bonds in the polymer backbone, because those ester bonds are more reactive than the ethyl ester end groups. This conclusion is confirmed by previously published transesterification experiments.36 In other words, the polycondensations of ELA involve two types of intermolecular transesterification reactions. Third, the experiments of Table 7 present in this work the first examples of reversible polycondensations without back-biting outside the ROPPOC chemistry discussed before.
A third series of transesterification experiments was performed with Ac-PCL-Et (Scheme 7 and Table 8) as reaction partner of dimethyl succinate. Ac-PCL-Et having a DP of 40 was prepared by SnOct2-catalyzed and ethyl 6-hexanoate-initiated polymerization of CL at 130 °C followed by acylation of the OH–CH end group with acetic anhydride and pyridine.
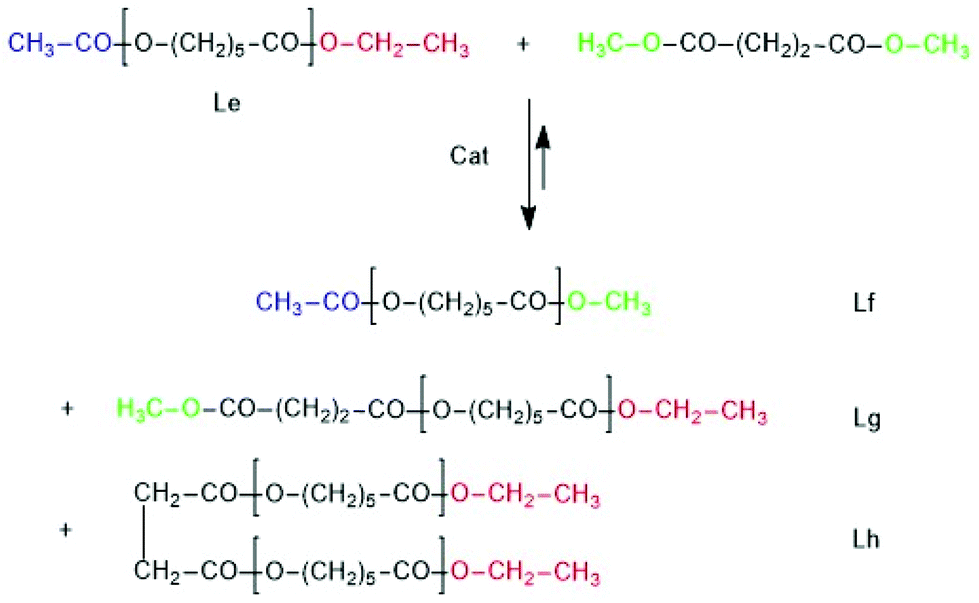 | ||
| Scheme 7 Simplified course of the transesterification of Ac-PCL-Et with dimethylsuccinate. | ||
| Exp. no. | Catalyst | CL/Cat | Time (h) | Lf, Lg, Lh-chains |
|---|---|---|---|---|
| 1A | SnOPF | 200/1 | 1 | + |
| 1B | SnOPF | 400/1 | 2 | ++ |
| 1C | SnOPF | 400/1 | 4 | ++ |
| 2A | SnOct2 | 200/1 | 1 | + |
| 2B | SnOct2 | 200/1 | 2 | ++ |
The MALDI TOF mass spectrum presented in Fig. 10A proved the formation of the expected structure. This model polymer was dissolved in dimethyl succinate and thermostated at 130 °C in the presence of BuSnOPF. The results listed in Table 8 and Fig. 10 demonstrate that the Lf, Lg and Lh chains indicating transesterification were detectable after 1 h, when a LA/Cat ratio of 200/1 was used or after 2 h with a LA/Cat ratio of 400/1. Analogous results, although with different intensity ratios of the Lf, Lg and Lh peaks were obtained with SnOct2 as catalyst.
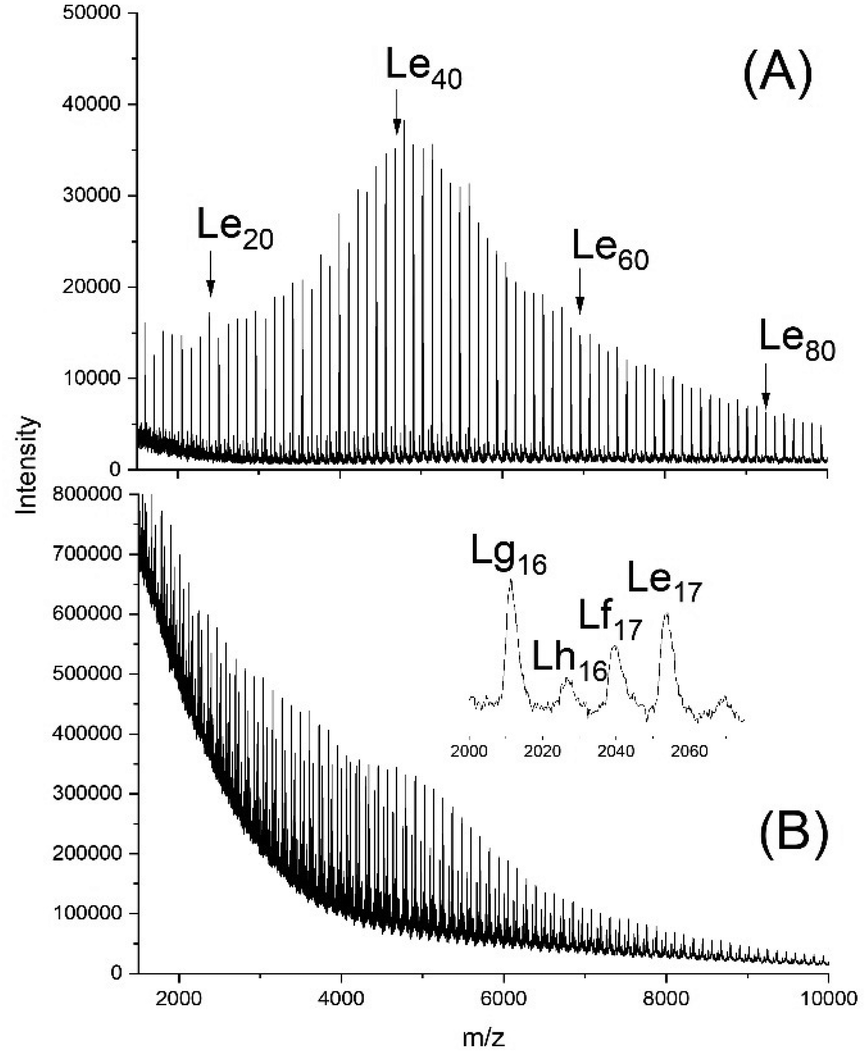 | ||
| Fig. 10 MALDI TOF mass spectra of (A) Ac-PCL-Et (average DP = 40), (B) reaction product of Ac-PCL-Et and dimethyl succinate catalyzed by BuSnOPF (LA/Cat = 200/1) after 1 h at 130 °C (No. 1A, Table 8) measured with Na doping. | ||
In summary, including previous results four different kinds of intermolecular transesterifications were performed at 120 or 130 °C which all proceeded under conditions where back-biting was absent.
Therefore, it is justified to conclude that the ROPPOCs presented in Tables 1–6 may be called reversible polymerizations and polycondensations despite absence of back-biting.
Discussion
Identification of the SUE-type transesterification in the case of lactide, the broadening of the MWD observed in the mass spectra and the results of the transesterification experiments summarized in Tables 7 and 8 or reported previously36 clearly demonstrate that intermolecular transesterification reactions exist under conditions, where back-biting is either totally absent or does at least not play a significant role. Nonetheless, the ROPPOC experiments include formation of large amounts of low and high molar mass cycles which are exclusively formed by ete-cyclization. In most cases, the odd/even equilibration is complete indicating a nearly complete ring–ring equilibration. These ring–ring equilibria are, of course, the same as those resulting from the ring chain equilibration, when back-biting occurs. In a recently published comment on the JS-theory Szymanski37 has calculated that polymerizations of lactide may yield a nearly quantitative fractions of cycles even when conducted in bulk, a result which was not immediately evident from the papers of J + S. From this viewpoint, the ROPPPOC experiments of this work may be understood to be part of the JS-theory. Yet, the authors of this work do not share this view.First: As already mentioned in the Introduction, the work of J + S has two aspects. On the one hand, it is a theory of ring-chain equilibria, on the other hand, it has been presented as a general theory of revPOCs. It is this second aspect, which is criticized by the authors. Characteristic for the JS-hypothesis of revPOCs is the assumption that equilibration exclusively results from back-biting. In contrast, the experiments presented above prove that revPOCs may proceed without back-biting. Any evidence is lacking that polycondensations exist, where according to J + S the reversibility is exclusively based on back-biting.
Second: J + S excluded that ete-cyclization plays a noteworthy role in the course of revPOCs. The ROPPOC experiments presented in this work (and previously5) demonstrate that ete-cyclization plays an important role in revPOCs, even when back-biting takes place. The results presented above demonstrate, that efficient formation of cycles may also occur in absence of back-biting. Any experimental evidence is lacking that polycondensations exist, which involve formation of cycles exclusively on the basis of backbiting. Therefore, the results described above are in total contradiction to the J + S hypothesis of revPOCs and thus, are considered to be outside the JS concept of revPOC, even when a ring-chain equilibration takes place.
Third: Even when back-biting is the main or only source of equilibration, the JS hypothesis does not provide a correct description of the course of revPOCs up to 100% conversion. For polycondensations at high conversions the JS theory predicts that a group of monomers yields 100% cycles at 100% conversion, whereas, for other groups of monomers only a few percent of cycles is predicted. This calculation is a total failure of chemical logic, because 100% conversion yields 100% of cycles for any kind of monomers and all reaction conditions.
Furthermore, it should be considered that all the experiments presented above have in common that they are based on cyclic esters and thus, indirectly on polycondensations of hydroxy acids. However, the technical production of most polyesters and the Jacobson–Stockmayer–Beckmann experiments3 are based on polycondensations of α,ω-diols and dicarboxylic acids or their dimethyl esters. Hence, such polycondensations need to be considered (and (re)investigated) to find out, to what extent the results and conclusions achieved in this work agree with a2 + b2 polycondensations. However, first pertinent experiments that shed light on this aspect have already been published by the first author in another context.38–40 For example, telechelic polyesters were prepared from dimethyl adipate or dimethyl sebacate and excess of ethane diol.38 With bismuth catalysts no cycles were formed up to temperatures of 240 °C. A typical MALDI TOF mass spectrum is displayed in Fig. S10.† Furthermore, telechelic polyesters were prepared from dimethyl terephthalate and excess 1,4-butane diol and with bismuth catalysts and no cycles were formed up to 240 °C. Moreover, 1,6-hexane diol or 1,10-decane diol were polymerized with dimethyl isophthalate in bulk catalyzed by Ti(OnBu)4 with variation of time and temperature. At temperatures up to 210 °C low molar mass polyesters free of cycles were obtained. All these syntheses of telechelic polyesters have in common, that at higher temperatures and longer times formation of small amounts cycles was observed. If these cycles were formed by backbiting, by ete-cyclization or by both reaction pathways remains an open question. Anyway, when those polycondensations are stopped because a sufficiently high molecular weight is achieved, they are certainly still far from complete equilibration, because perfect equilibration either requires extremely fast trans esterification reaction or long reaction times, which are usually not applied in preparative experiments.
In all those polycondensations, chain growth steps are reversible, because the liberated methanol in combination with catalyst can cleave any ester bond in the polymer chains. Hence, those experiments indicate that also in the field of a2 + b2 polycondensations reaction conditions exist that allow for polyester syntheses involving intermolecular transesterification in the absence of back-biting or with a negligible contribution of back-biting. Hence, also those polycondensations are outside the JS-hypothesis. All these facts and arguments together clearly demonstrate that the JS concept cannot serve as general theory of revPOCs.
As a consequence of this conclusion, the authors propose a new concept of revPOCs based on the definition of three classes of polycondensations:
Class I: Irreversible polycondensations that may also be labelled kinetically controlled polycondensations. This definition, in principle, agrees with Florýs polycondensation theory with the exception that ete-cyclization occur, which was denied by Flory.
Class II: RevPOCs exclusively involving back-biting as the only source of reversibility and including such a high rate of equilibration that thermodynamic control is given at any state of the polycondensation process.
Class III: RevPOCs involving rapid intermolecular trans-reactions in the absence of backbiting. Again, equilibration is so efficient that thermodynamic control is given at any stage of the polycondensation.
To avoid misunderstanding the term “thermodynamically controlled polycondensation” needs to be clarified. At long reaction times (i.e. weeks or months), all revPOC will end up with complete equilibration of all reaction products and thus, justify being called “thermodynamically controlled” polycondensations. Yet, as demonstrated by the ROPPOC experiments above and by the experiments in refs.38–40 in many real polycondensations the equilibration reactions will not be efficient enough to reach thermodynamic control at any stage of the process.
The three classes of polycondensations defined above should be considered as extreme cases which form the corner of a triangle. The vast majority of revPOCs will, in fact, be located somewhere inside this triangle. Consider, a series of class III-type polycondensations with decreasing rate constants of trans-reactions. When the rate constants approach zero the definition of Class I is reached and thus, this series of polycondensations is located on the line connecting Class III and Class I. An analogous thought experiment can be formulated for Class II polycondensations. The line connecting class I and II is formed by polycondensations that involve fast equilibration reactions, so that thermodynamic control is established at any time, but the contributions of back-biting and intermolecular equilibration reactions varies. If all the equilibration reactions are not fast enough to establish full thermodynamic control at any stage, this polycondensation will be positioned somewhere inside the triangle. In other words, revPOCs cover a wide area of kinetic and thermodynamic properties and exact knowledge of the effectiveness of all involved trans-reactions is required for a detailed description of an individual polycondensation process.
Finally, a publication of Montaudo et al.41 should be cited which, as early as 1997, described MALDI TOF mass spectrometric analyses of polylactides prepared by means of an Al complex containing one Al-OCH3 group. This methoxide group played the role of initiator and yielded methyl ester terminated polylactides. Despite the low temperature of 70 °C and short times complete odd/even equilibration was observed although cyclic oligomers were absent and only appeared after long polymerization times. Hence, these results demonstrate that a significant predominance of intermolecular transesterification reactions (entailing broadening of the Mw) over back-biting is not limited to the chemistry of tin catalysts. This conclusion and the results presented in ref. 39–41 also suggests that catalyst of other elements (e.g. Zn, Ti and Bi) may be found, that enable ROPPOC of cyclic esters obeying to the principles of Class III polycondensations.
Conclusions
The numerous ROP and ROPPOC experiments performed in this work, based on two different monomers, three initiators and four different catalysts carry one message: revPOCs free of back-biting but involving intermolecular transesterification exist under appropriate reaction conditions. The low and high molar mass cycles formed in absence of initiators exclusively result from ete-cyclization. Furthermore, revPOCs with low or moderate conversions were found which do not involve formation of cycles. Hence, it may be concluded that the JS theory is not useful as a general theory of revPOCs. These results suggest formulation of a new concept of revPOCs based on the definition of three classes of polycondensations (Class I–III). All three classes have in common that when the conversion approaches 100% of functional groups, the final polymerization product almost exclusively consists of cycles with a broad molecular weight distribution. Class I and class III have in common that all cycles small and large ones, are formed by ete-cyclization, whereas in the case of class II both, back-biting” and ete-cyclization contribute to the formation of cycles. The three classes of polycondensations defined above should not be considered as three isolated regimes of polycondensation, but as three corners of triangle (see TOC graphic) which encompasses almost real experiments.Author contributions
HRK – project administration, conceptualization, synthesis, writing original draft, SMW – investigation, methodology, validation, visualization, writing – review & editing, JF – investigation, methodology, visualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Prof. R. Szymanski (Center of Molecular and Macromolecular Studies, Lodz, Poland) for useful discussions and we thank Dipl. Ing. Anett Myxa for the GPC measurements.Notes and references
- P. J. Flory, Chem. Rev., 1946, 39, 137–197 CrossRef CAS
.
-
P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed
- H. Jacobson, C. O. Beckmann and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1607–1612 CrossRef CAS
- H. Jacobson and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1600–1606 CrossRef CAS
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2020, 11, 2595–2604 RSC
- H. R. Kricheldorf and S. M. Weidner, Polym. Chem., 2020, 11, 6226–6228 RSC
- H. R. Kricheldorf, Macromol. Rapid Commun., 2009, 30, 1371–1381 CrossRef CAS
- H. R. Kricheldorf and S. M. Weidner, Macromol. Rapid Commun., 2020, 41, e2000152 CrossRef PubMed
- H. R. Kricheldorf, M. Al Masri and G. Schwarz, Macromolecules, 2003, 36, 8648–8651 CrossRef CAS
- H. R. Kricheldorf, Die Makromol. Chem., 1974, 175, 3325–3342 CrossRef CAS
- H. R. Kricheldorf and K. Bosinger, Makromol. Chem., 1976, 177, 1243–1258 CrossRef CAS
- H. R. Kricheldorf, C. Von Lossow and G. Schwarz, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4680–4695 CrossRef CAS
- H. R. Kricheldorf, N. Lomadze and G. Schwarz, Macromolecules, 2007, 40, 4859–4864 CrossRef CAS
- H. R. Kricheldorf, N. Lomadze and G. Schwarz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6229–6237 CrossRef CAS
- W. Jeong, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2007, 129, 8414–8415 CrossRef CAS
- E. J. Shin, W. Jeong, H. A. Brown, B. J. Koo, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2011, 44, 2773–2779 CrossRef CAS
- E. J. Shin, H. A. Brown, S. Gonzalez, W. Jeong, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2011, 50, 6388–6391 CrossRef CAS PubMed
- H. A. Brown, S. L. Xiong, G. A. Medvedev, Y. A. Chang, M. M. Abu-Omar, J. M. Caruthers and R. M. Waymouth, Macromolecules, 2014, 47, 2955–2963 CrossRef CAS
- L. Guo, S. H. Lahasky, K. Ghale and D. H. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS PubMed
- S. Praban, S. Yimthachote, J. Kiriratnikom, S. Chotchatchawankul, J. Tantirungrotechai and K. Phomphrai, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2104–2112 CrossRef CAS
- A. V. Prasad, L. P. Stubbs, M. Zhun and Y. H. Zhu, J. Appl. Polym. Sci., 2012, 123, 1568–1575 CrossRef CAS
- G. Si, S. Zhang, W. Pang, F. Wang and C. Tan, Polymer, 2018, 154, 148–152 CrossRef CAS
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2017, 8, 1589–1596 RSC
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 952–960 CrossRef CAS
- H. R. Kricheldorf and S. M. Weidner, Polym. Chem., 2020, 11, 5249–5260 RSC
- S. Penczek, R. Szymanski, A. Duda and J. Baran, Macromol. Symp., 2003, 201, 261–269 CrossRef CAS
- M. Jalabert, C. Fraschini and R. E. Prud'Homme, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1944–1955 CrossRef CAS
- Y. C. Yu, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2011, 50, 7927–7940 CrossRef CAS
- S. Sosnowski and P. Lewinski, Polym. Chem., 2015, 6, 6292–6296 RSC
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS
- M. Gordon and W. B. Temple, Makromol. Chem., 1972, 160, 263–276 CrossRef CAS
- M. Gordon and W. B. Temple, Makromol. Chem., 1972, 152, 277–289 CrossRef CAS
- J. L. Stanford, R. F. T. Stepto and D. R. Waywell, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 1308–1326 RSC
- H. R. Kricheldorf and G. Schwarz, Macromol. Rapid Commun., 2003, 24, 359–381 CrossRef CAS
- S. M. Weidner and H. R. Kricheldorf, Macromol. Chem. Phys., 2018, 219, 1800445 CrossRef
- R. Szymanski, Polym. Chem., 2020, 11, 6221–6225 RSC
- H. R. Kricheldorf, G. Behnken and G. Schwarz, Polymer, 2005, 46, 11219–11224 CrossRef CAS
- H. R. Kricheldorf, M. Al Masri, N. Lomadze and G. Schwarz, Macromolecules, 2005, 38, 9085–9090 CrossRef CAS
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 197–208 CrossRef
- G. Montaudo, M. S. Montaudo, C. Puglisi, F. Samperi, N. Spassky, A. LeBorgne and M. Wisniewski, Macromolecules, 1996, 29, 6461–6465 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00704a |
| This journal is © The Royal Society of Chemistry 2021 |