
1,2,3-Triazole-based sequence-defined oligomers and polymers
Xiaojun
Wang
,
Xueyan
Zhang
and
Shengtao
Ding
 *
*
State Key Laboratory of Organic-Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: stding@mail.buct.edu.cn
First published on 12th April 2021
Abstract
Mimicking nature to create polymers with well-defined sequences and precise chain lengths for multitudinous applications is an increasingly attractive but challenging field. In the absence of biological machineries, robust and efficient chemical reactions are important for the construction of artificial sequence-defined polymers. Due to the specific characteristics of the 1,2,3-triazole skeleton and its facile accessibility from the “click” reaction of an azide with an alkyne, research on the synthesis and application of sequence-defined polymers involving 1,2,3-triazole substructures has attracted much attention in the recent two decades. This review summarizes the developments in this field from two parts: (i) creation of biomimetic oligomers in which 1,2,3-triazoles were used as amide bioisosteres or as linkers between natural monomers and (ii) establishment of diverse efficient synthetic strategies on the basis of azide–alkyne cycloaddition reactions and their utilization in the construction of non-biomimetic sequence-defined polytriazoles. Application of these 1,2,3-triazole-linked sequence-defined polymers in different areas is also briefly discussed.
 Xiaojun Wang | Xiaojun Wang obtained her Bachelor's degree from Beijing Forestry University in 2018. She is now pursuing her M.Sc. studies in Prof. Ding's group at Beijing University of Chemical Technology. Her major research interest is the application of click chemistry in sequence-defined polymer synthesis. |
 Xueyan Zhang | Xueyan Zhang received her Bachelor's degree from Beijing University of Chemical Technology in 2017. In the same year she joined Prof. Ding's group to start her Ph.D. study. She is now working on the construction of novel sequence-defined structures and their applications in functional materials. |
 Shengtao Ding | Shengtao Ding received his B.Sc. (2009) and M.Sc. (2011) from Peking University under the supervision of Prof. Ning Jiao. He then joined Prof. Jianwei Sun's group at the Hong Kong University of Science and Technology and received his Ph.D. in 2014. After his postdoctoral work with Prof. Tobias Ritter at Harvard University and Prof. Xiaodong Shi at the University of South Florida, he began his independent research at Beijing University of Chemical Technology in 2016. His current research is focused on the development of novel synthetic methods and their applications in medicinal and polymer chemistry. |
1. Introduction
Biopolymers, such as polynucleotides, polypeptides and polysaccharides, are essential components of creatures. One distinct feature of these natural polymers is that they are well defined with precise monomer sequences and exact chain lengths, which has been disclosed to be vital for their sophisticated structures and functions. Researchers have long sought to imitate nature to construct sequence-defined polymers due to their potential applications in diverse realms.1 Besides artificial creation of oligonucleotides and oligopeptides in a certain alignment, various synthetic sequence-defined polymers, including peptide nucleic acids (PNA),2 peptoids3 and β-peptides,4 were likewise developed as nucleotidomimetics or peptidomimetics (Scheme 1). Principal strategies to achieve these molecularly defined oligomers include iterative sequential growth (ISG)5 and iterative exponential growth (IEG)6 approaches. In the recent two decades, research on non-natural sequence-defined polymers has experienced rapid growth.7 One reason contributing to this is the development of click chemistry, which allows the design of more simple and efficient iterative synthetic methods.8 One emblematic example of “click” reactions is the copper-catalyzed azide–alkyne cycloaddition (CuAAC), which was initially demonstrated by Sharpless9 and Meldal,10 respectively, in 2002 and it shows high efficiency and simplicity in accessing the 1,2,3-triazole skeleton – a stable structure towards enzymatic degradation and environmental variables that has been widely used in various realms.11 In the area of sequence-controlled polymers, CuAAC has evolved as a powerful method for the creation of sequence-defined structures via diverse synthetic protocols in the liquid- or solid-phase. For instance, CuAAC has been successfully employed to build biopolymer analogues with unnatural triazole-linked backbones via iterative monomer additions. It also proves to be an ideal basis for the establishment of IEG strategies, enabling the efficient and rapid extension of the polymer chain-length. While 1,4-disubstituted 1,2,3-triazoles are usually obtained from CuAAC and used as linkers to connect different components, metal-catalyzed regioselective cycloaddition reactions of azides with internal alkynes, termed MAIAC,12 bring opportunities for the construction of sequence-defined polymers bearing programmable side chains at the fully substituted 1,2,3-triazole rings, which is still an under-developed area.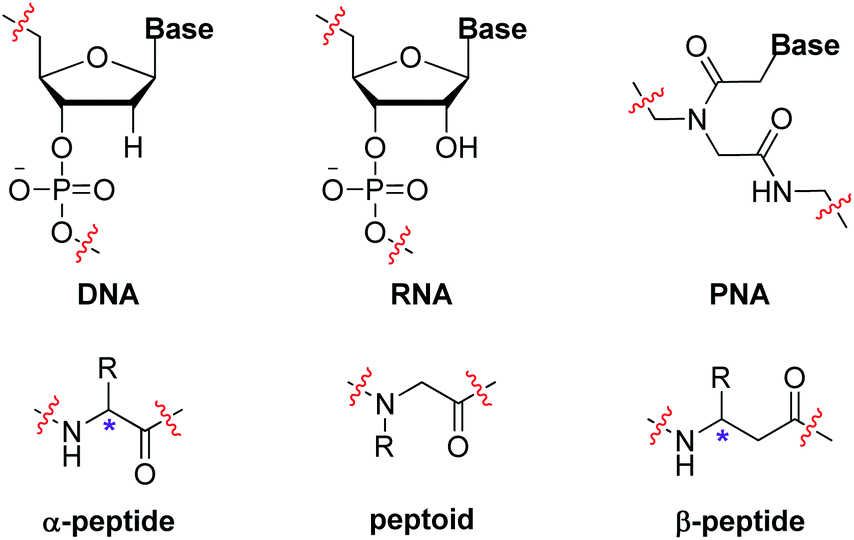 | ||
| Scheme 1 Biopolymers and biomimetic polymers. | ||
In consideration of the huge amount of oligomers and polymers involving triazoles, this review offers a particular summary on linear sequence-defined oligomers and polymers in which all the monomeric units are tethered by triazole linkages, or termed “1,2,3-triazole-based sequence-defined oligomers and polymers”, which will be discussed in two parts. The first one is about the fabrication of biomimetic sequence-defined oligotriazoles, in which 1,2,3-triazoles were used as amide bioisosteres, or as linkers between natural or biomimetic monomers. The construction of non-biomimetic sequence-defined polytriazoles, as well as diverse efficient synthetic strategies established on the basis of MAAC reactions, will be summarized in the second section. A brief discussion on the application of these 1,2,3-triazole-based sequence-defined polymers in different areas will be included as well. As the syntheses of triazolophanes and their applications in anion binding have been well documented in numerous reviews,13 related discussions will not be included in this review. For clarity of the results, 1,2,3-triazole will be simplified to triazole.
2. Biomimetic sequence-defined oligotriazoles
Though there are various applications of biopolymers, their instability towards enzymatic degradation, hydrolysis and other environmental variations limits their potential utilization in diverse areas. One protocol to circumvent this is replacing the degradable chemical bonds, such as the amide bond in polypeptides and the phosphodiester bonds in polynucleotides, with more robust structures. The triazole ring is well-known for its marked stability under diverse conditions, which makes it an attractive unit to connect biological monomers to afford stable biomimetic polymers.14 Besides, the structural features of triazole, including its polarity, rigidity, and ability to act as both hydrogen bond donors and acceptors, make it an ideal surrogate for peptide and phosphodiester bonds.15 Since the first solid-phase synthesis of peptidotriazoles under CuAAC conditions achieved by the Meldal group,10 the triazole unit has evolved as a remarkable linkage in the design and synthesis of biomimetic oligomers.2.1. Triazole-linked analogues of peptides
Due to its physicochemical resemblance to the amide bond, the triazole ring is widely used as an amide bond bioisostere in drug discovery. By replacing the peptide bonds with triazole rings, the Arora group first reported the construction of nonpeptidic triazole-based oligomers, named triazolamers, in 2005 (Scheme 2a).16 As shown in Scheme 2b, both of the initial amine and alkyne substrates were derived from amino acids. Monomer 1 with an amino group was obtained through a three-step iterative reaction sequence involving the conversion of the amine to azide, CuAAC of the azide with alkyne, and deprotection of the Boc group.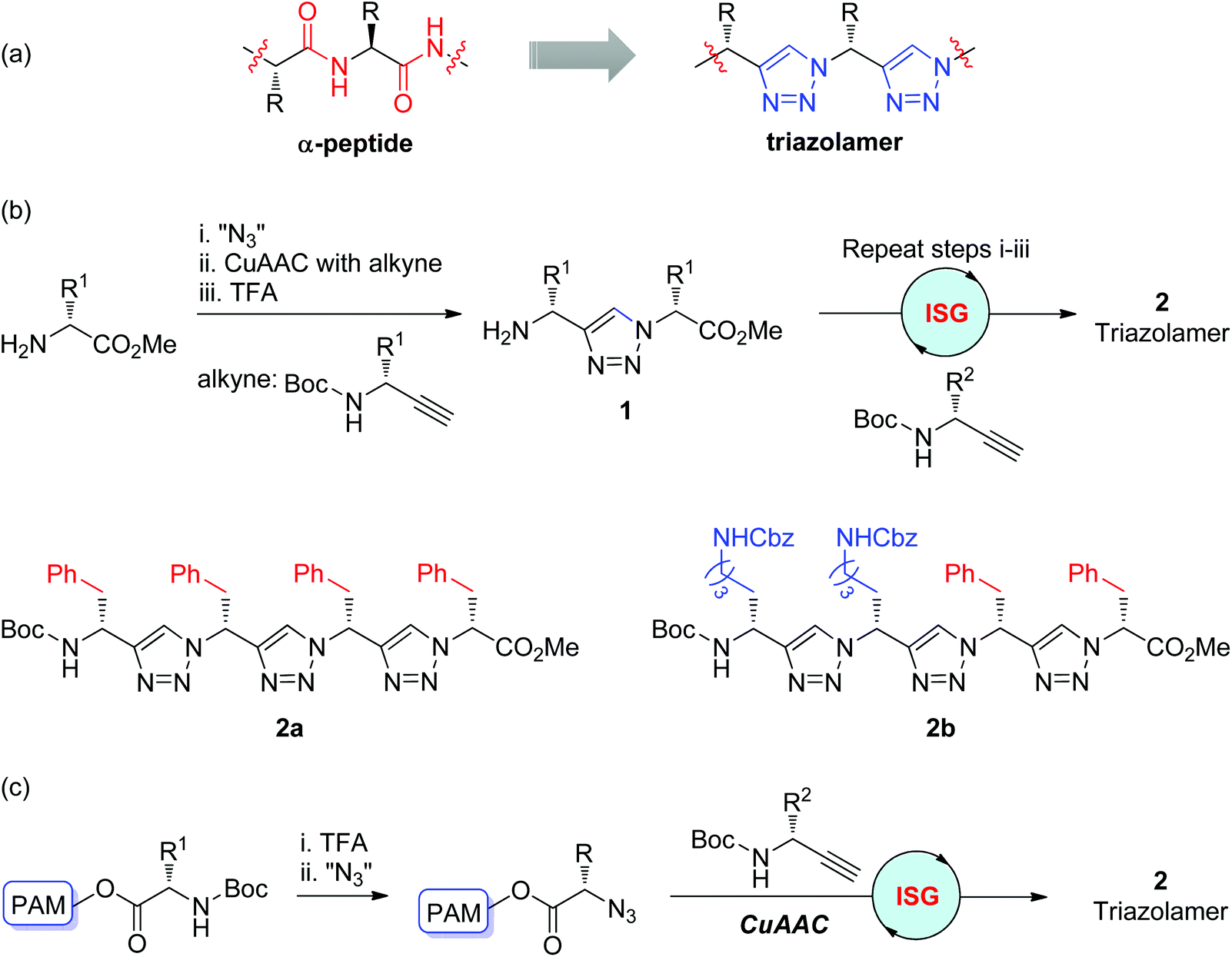 | ||
| Scheme 2 (a) Triazolamers as peptidomimetics; (b) Synthesis of triazolamers in solution phase; (c) Synthesis of triazolamers in solid phase.16,17 TFA: trifluoroacetic acid. Boc: tert-butyloxycarbonyl. Cbz: benzyloxycarbonyl. PAM: polyacrylamide. | ||
Through this CuAAC-based iterative sequential growth strategy, monomer 1 could be further extended to triazolamers 2 with up to three monomer units in poor overall yields (6–12%), which was supposed to be due to the instability of the amino acid-derived azide and low conversion in the azidation step. One more efficient protocol involving the utilization of ZnCl2 as a diazotransfer catalyst and microwave irradiation was released by the same group, which allowed the execution of the iterative sequential growth cycle in one-pot and the synthesis of triazolamer 2b in an overall yield of 62%.17 This improved strategy was also suitable for solid-phase synthesis, through which diverse triazolamers were efficiently fabricated in up to 78% total yields (Scheme 2c). The chiral main-chain and amino acid side chains were preserved in all of these triazole-based oligomers. Structural studies of these oligomers disclosed their zigzag conformations, which might be stabilized by the dipole–dipole interactions between adjacent triazole rings. The bioactivity evaluations of these nonpeptidic β-strand mimetics by Arora and co-workers showed their potential as protease inhibitors.18
Instead of replacing amide bonds, triazole rings were introduced into the peptide backbone by the Zhang group in 2006, affording peptidotriazolamers with alternating triazole and amide connectors (Scheme 3a).19 In this solid-phase supported protocol, the reaction of pentynoic acid with Rink amine was first carried out to generate the Rink amide resin 3. Under one set of optimized conditions involving CuI (5 equiv.), ascorbic acid (5 equiv.), and DMF/piperidine (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), the CuAAC reaction of N-Fmoc-protected amino azides with the Rink amide resin 3 and the deprotection of the Fmoc group were realized simultaneously, providing product 4 in quantitative yield. The high efficiency of this protocol and its good tolerance towards diverse functional groups enabled the iterative sequential synthesis of 4-mer peptidotriazolamer 5 in an overall 56% yield after purification by high performance liquid chromatography (HPLC). In 2012, the Sze group reported the solution-phase synthesis of peptidotriazolamers bearing alternating triazole and amide linkages in a similar manner (Scheme 3b).20 This synthetic route was started from the coupling of N-Boc-α-amino acid 6 with propargylamine to afford the corresponding amide 7, which could then react with α-azido acid 8 under CuAAC conditions to generate triazole-linked peptidomimetic 9. Elongation of 9 to trimer 10 was realized by repeating these amide/click coupling steps. The self-dimerization of these oligomers through hydrogen bonds was revealed by structural analysis.
:2), the CuAAC reaction of N-Fmoc-protected amino azides with the Rink amide resin 3 and the deprotection of the Fmoc group were realized simultaneously, providing product 4 in quantitative yield. The high efficiency of this protocol and its good tolerance towards diverse functional groups enabled the iterative sequential synthesis of 4-mer peptidotriazolamer 5 in an overall 56% yield after purification by high performance liquid chromatography (HPLC). In 2012, the Sze group reported the solution-phase synthesis of peptidotriazolamers bearing alternating triazole and amide linkages in a similar manner (Scheme 3b).20 This synthetic route was started from the coupling of N-Boc-α-amino acid 6 with propargylamine to afford the corresponding amide 7, which could then react with α-azido acid 8 under CuAAC conditions to generate triazole-linked peptidomimetic 9. Elongation of 9 to trimer 10 was realized by repeating these amide/click coupling steps. The self-dimerization of these oligomers through hydrogen bonds was revealed by structural analysis.
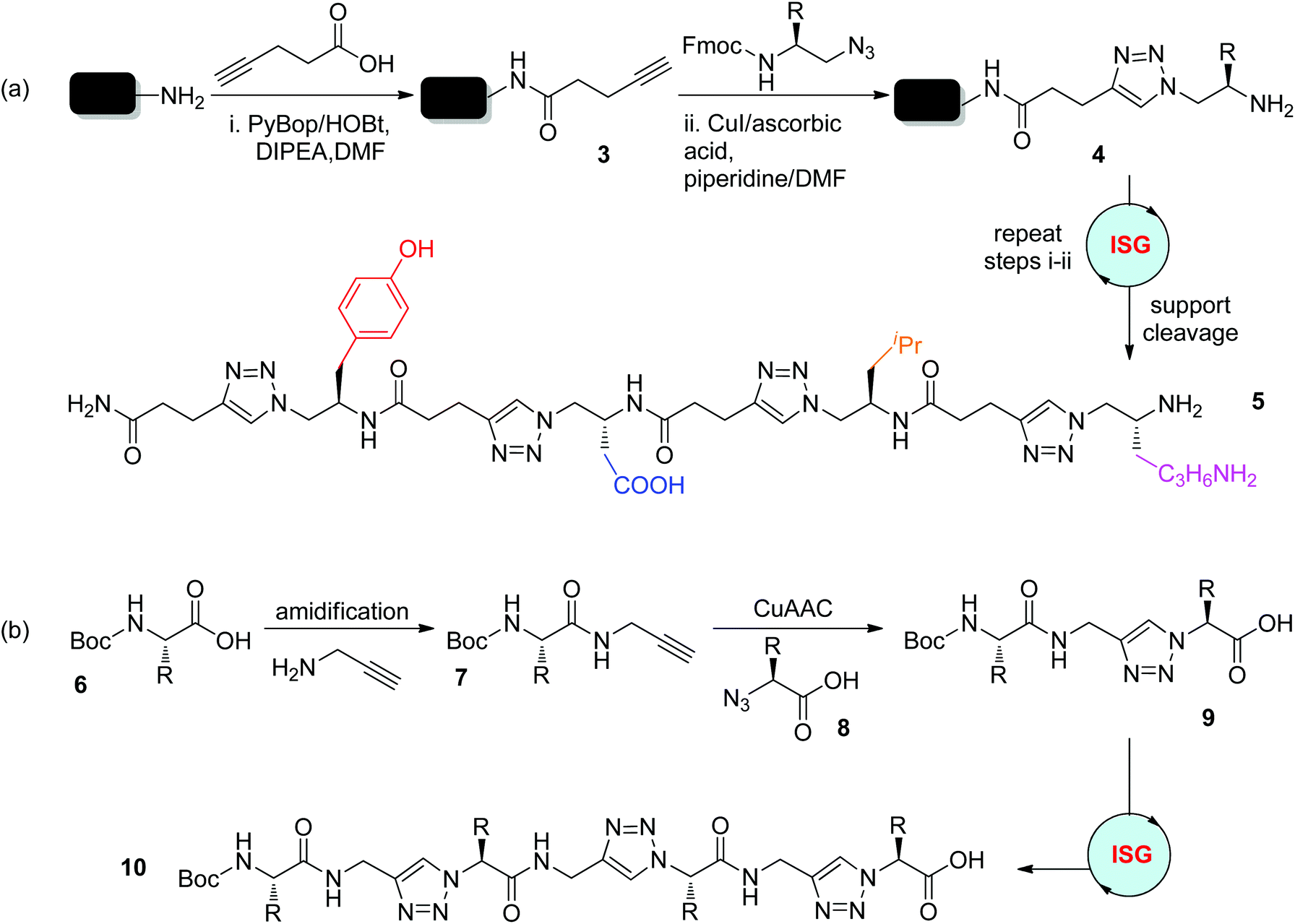 | ||
| Scheme 3 Synthesis of peptidotriazolamers with alternating triazole and amide linkages in solid (a) and solution (b) phase.19,20 PyBop: benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate. HOBt: hydroxybenzotriazole. DIPEA: N,N-diisopropylethylamine. DMF: dimethylformamide. Fmoc: fluorenylmethoxycarbonyl. | ||
The thermal Huisgen 1,3-dipolar cycloaddition of the azide with the terminal alkyne usually affords two regioisomers: 1,4- and 1,5-disubstituted triazoles, which are structurally similar to trans- and cis-amide bonds, respectively. While the CuAAC reaction of the azide with the terminal alkyne selectively generates the 1,4-disubstituted regioisomer of triazole, their ruthenium-catalyzed annulation (RuAAC) developed by Fokin and Jia paved an efficient way for the selective construction of 1,5-disubstituted triazoles.21 One RuAAC-based iterative sequential growth strategy was reported by the Kubik group in 2011 for the synthesis of peptidotriazolamers bearing 1,5-disubstituted triazole rings as connectors (Scheme 4).22 Compound 13 was first constructed from the RuAAC reaction of compounds 11 and 12, both of which were derived from the same precursor. Peptidotriazolamer 14 was obtained after one more ISG cycle. Due to the specific conformation of the 1,5-disubstituted triazole motif, the macrocyclic triazolamer 15 was successfully constructed from compound 14 through further deprotection, azidation and intramolecular RuAAC reaction. X-ray crystallographic analysis of macrocycle 15 disclosed its C3 symmetrical structure, which is structurally closely related to cyclic peptides. Qualitative binding studies showed the potential of this kind of cyclic pseudopeptide in absorbing different anions.
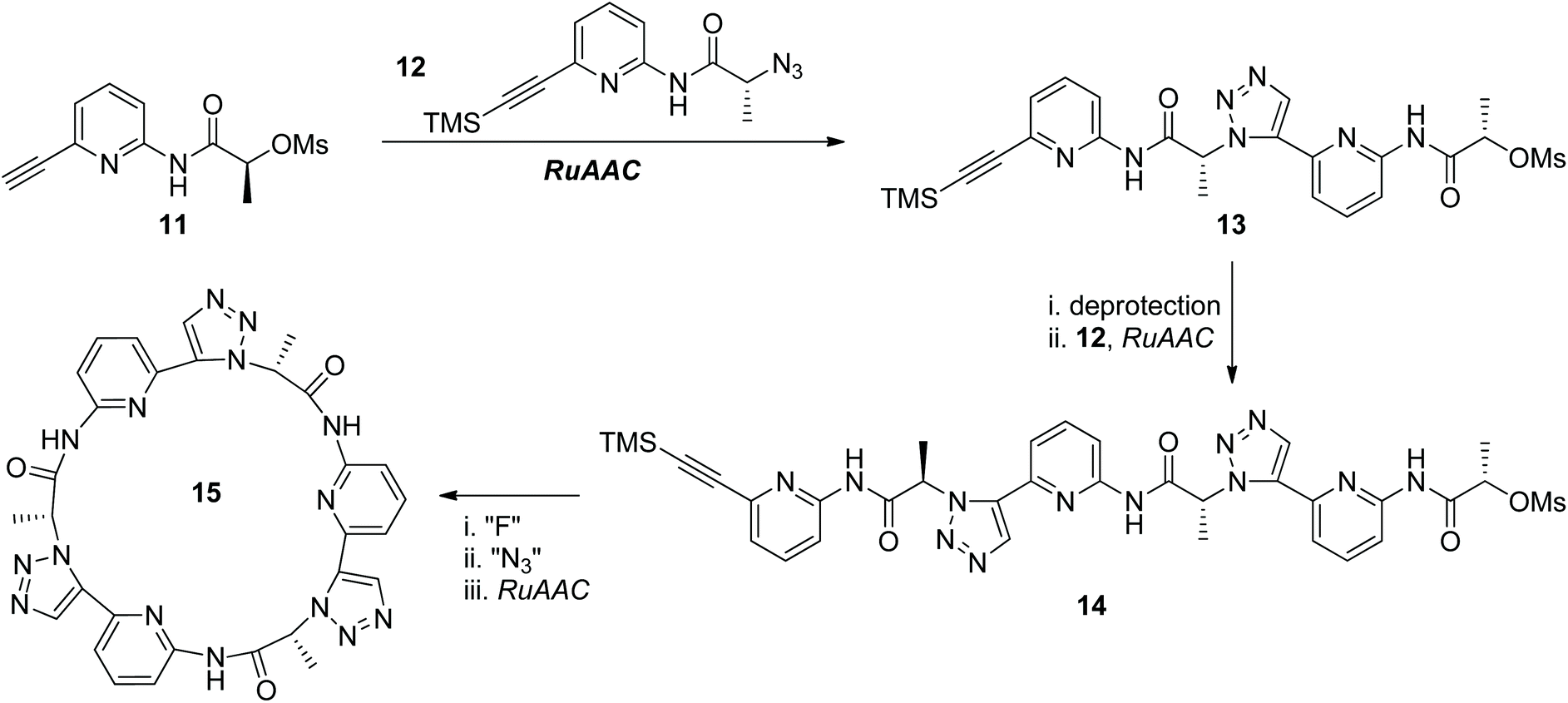 | ||
| Scheme 4 Synthesis of peptidotriazoles through the RuAAC-based ISG strategy.22 | ||
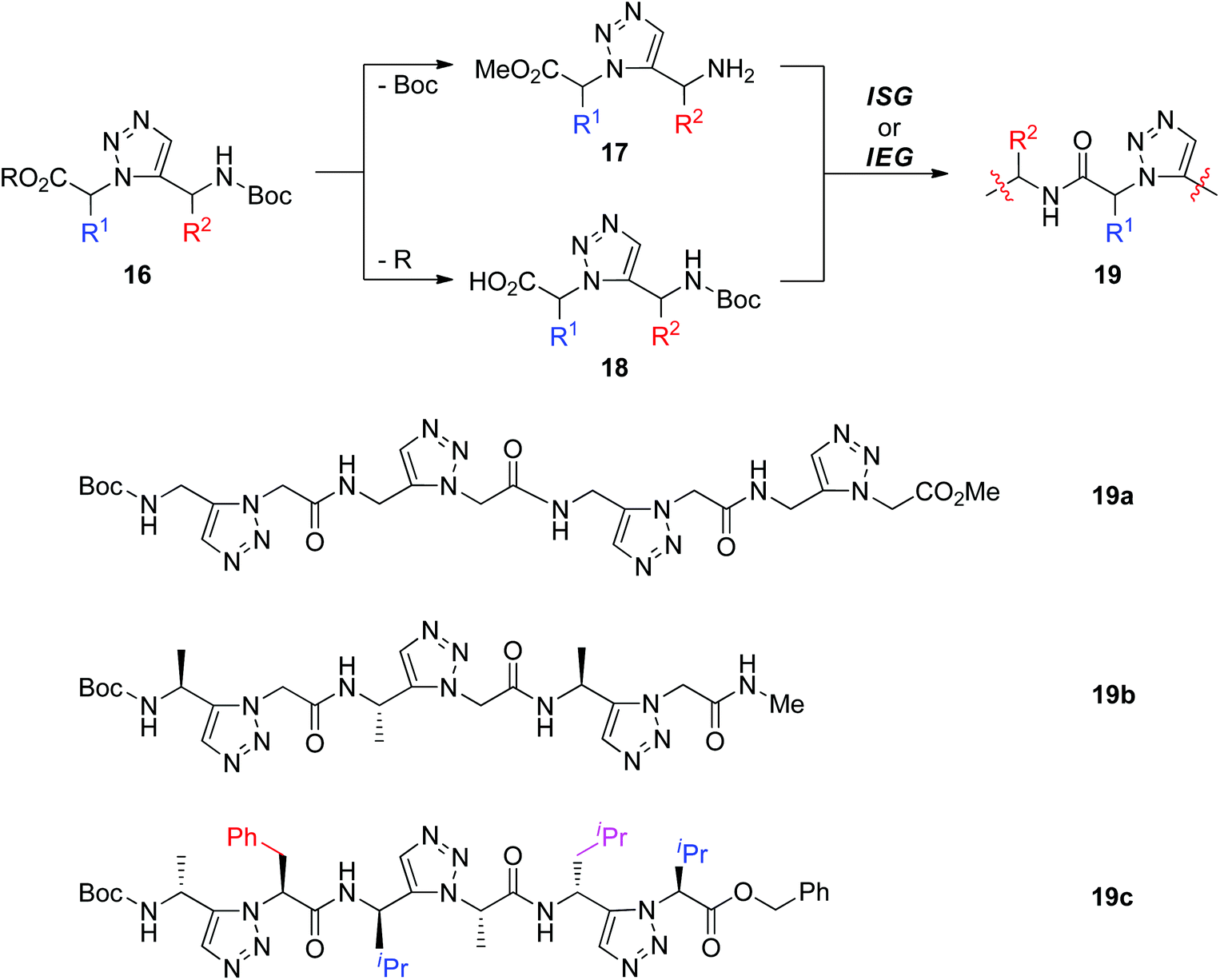
Another method used to fabricate peptidotriazolamers involving 1,5-disubstituted triazole is assembling triazole building blocks through ISG or IEG strategies. As shown in Scheme 5, the 1,5-disubstituted triazole unit 16 could be synthesized from amino acid-derived azides and terminal alkynes under RuAAC conditions. Chemoselective deprotection of 16 generated amine 17 and acid 18, allowing further construction of peptidotriazolamers 19 with alternating 1,5-disubstituted triazole and amide bond connectors. By using this protocol, the Beke-Somfai group reported the synthesis of tetramer 19a from one simple 1,5-disubstituted triazole monomer (R = Me, R1 = R2 = H) that was prepared by Ru-catalyzed cycloaddition of methyl 2-azidoacetate with N-Boc-propargylamine.23 Employment of a chiral alkyne and azide would offer chiral 1,5-disubstituted triazoles, which could act as building blocks for the construction of chiral peptidotriazolamers. As a continuation of their research, Beke-Somfai and Kann recently released the fabrication of the chiral trimer 19b through a similar synthetic approach.24 Through one microwave-assisted RuAAC reaction, six different homo- and heterochiral 1,5-disubstituted triazoles were synthesized by Sewald and co-workers from chiral amino acid-derived azide and alkyne substrates.25 Diverse homo- or heterochiral peptidotriazolamers (such as 19c) were further built by connecting these monomers via an ISG route. Conformational analysis by means of NMR measurements and molecular dynamics simulations revealed the polyproline l-like structure of heterochiral peptidotriazolamer 19c, while homochiral peptidotriazolamers prefer to form a compact β-turn-like conformation.
 | ||
| Scheme 5 Synthesis of peptidotriazoles from 1,5-disubstituted triazole building blocks.23–25 | ||
2.2. Triazole-linked analogues of peptoids
Peptoids have been widely studied and applied in various areas since their first introduction as analogues of peptides.3 In 2015, Schepers and coworkers explored the combination of this intriguing structure with triazole rings to afford triazole-incorporated peptoids.26 As outlined in Scheme 6, this solid-phase synthesis protocol relied on the amidification reaction of azido acid 21 with an amine precursor and subsequent CuAAC reaction with functionalized propargylamine 23. The obtained trimeric triazole-linked peptoids 25 bearing different functional groups were further labelled with rhodamine B as a fluorophore to generate 26 for the investigation of their capability in cell penetration. Related results disclosed the important influence of the side chains on their ability in molecular transport and particular cell tissue targeting.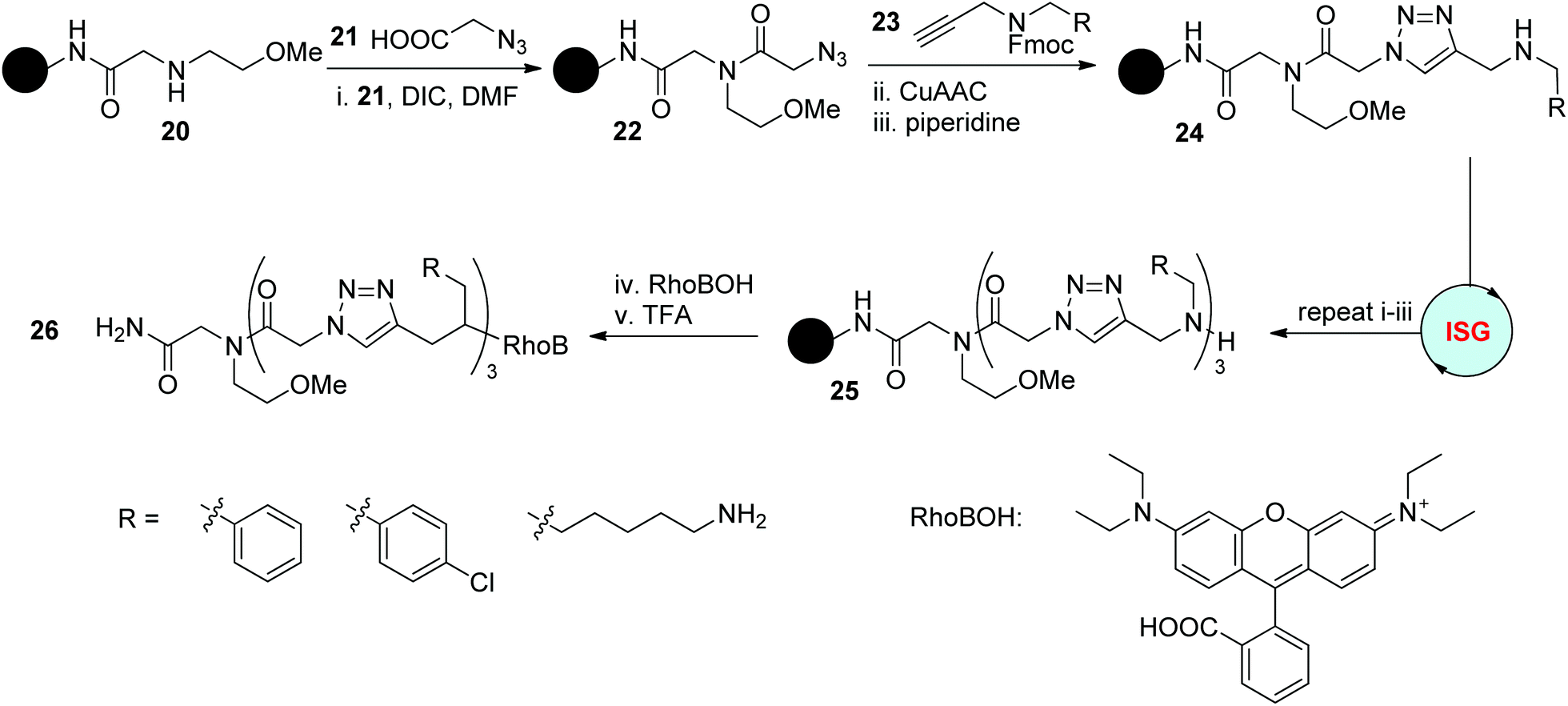 | ||
| Scheme 6 Synthesis of triazole-linked analogues of peptoids.26 DIC: N,N′-diisopropylcarbodiimide. | ||
2.3. Triazole-linked analogues of nucleic acids
Stimulated by the beautiful structures of nucleic acids and their wide applications in various areas, replacing the native phosphodiester moieties in them with different artificial linkages to prepare analogues of nucleic acids has evolved as one important realm. In 2007, Dondoni and Massi demonstrated the synthesis of oligothymidine analogues in which the phosphodiester linkages were replaced by triazole rings through one CuAAC-based iterative ligation methodology (Scheme 7).27 Both of the synthetic precursors 27 and 28 were prepared from thymidine. Coupling them under CuAAC conditions generated dinucleotide 29, which could further react with monomer 30 to form trinucleotide 31via ethynylation and subsequent CuAAC reaction. This explorative work disclosed the feasibility and fidelity of CuAAC in connecting nucleosides. However, some reaction conditions and operations need further optimization in this synthetic route if higher oligonucleotides are intended to be obtained. For instance, the ethynylation step involved in each iterative cycle was realized through two subsequent harsh operations, leading to the formation of the desired products only in moderate yields. Anchoring the ethynyl group to the building blocks in advance is one efficient way to avoid these manipulations, which has been extensively applied in later reports.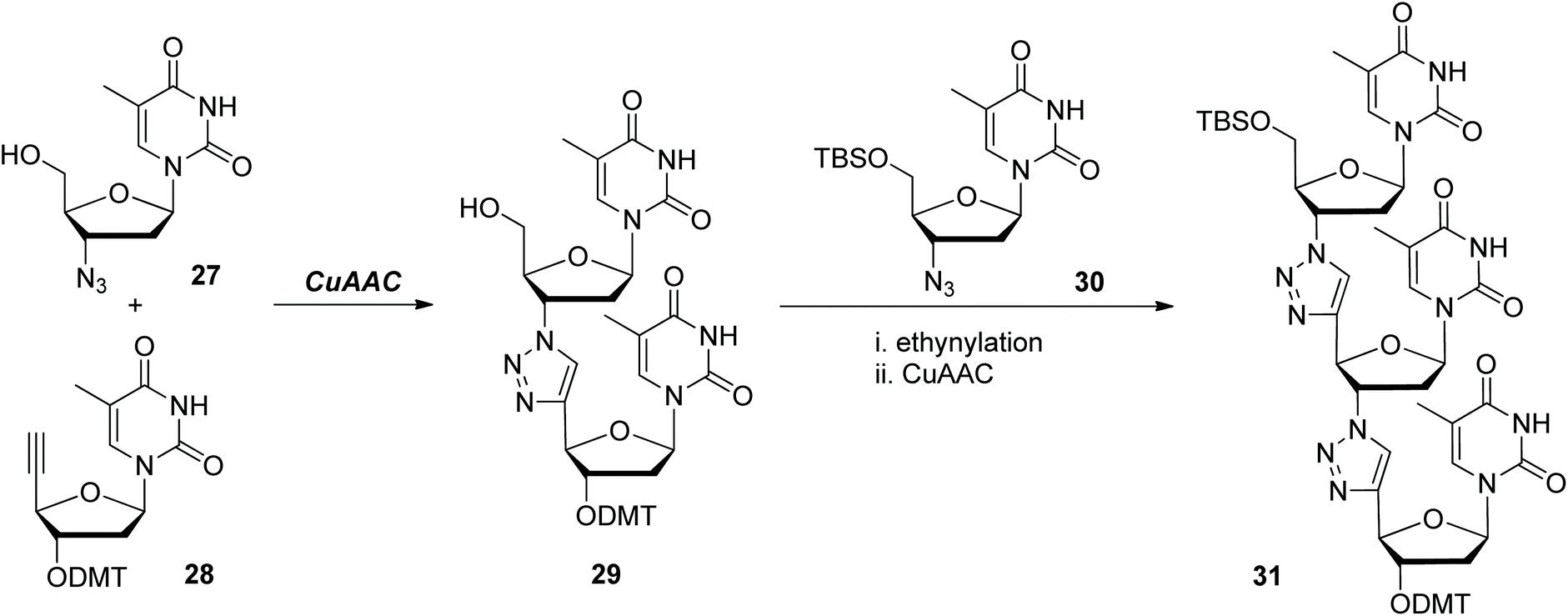 | ||
| Scheme 7 Synthesis of thymidine oligonucleotides with triazole linkages.27 DMT: 4,4′-dimethoxytrityl. TBS: tert-butyldimethylsilyl. | ||
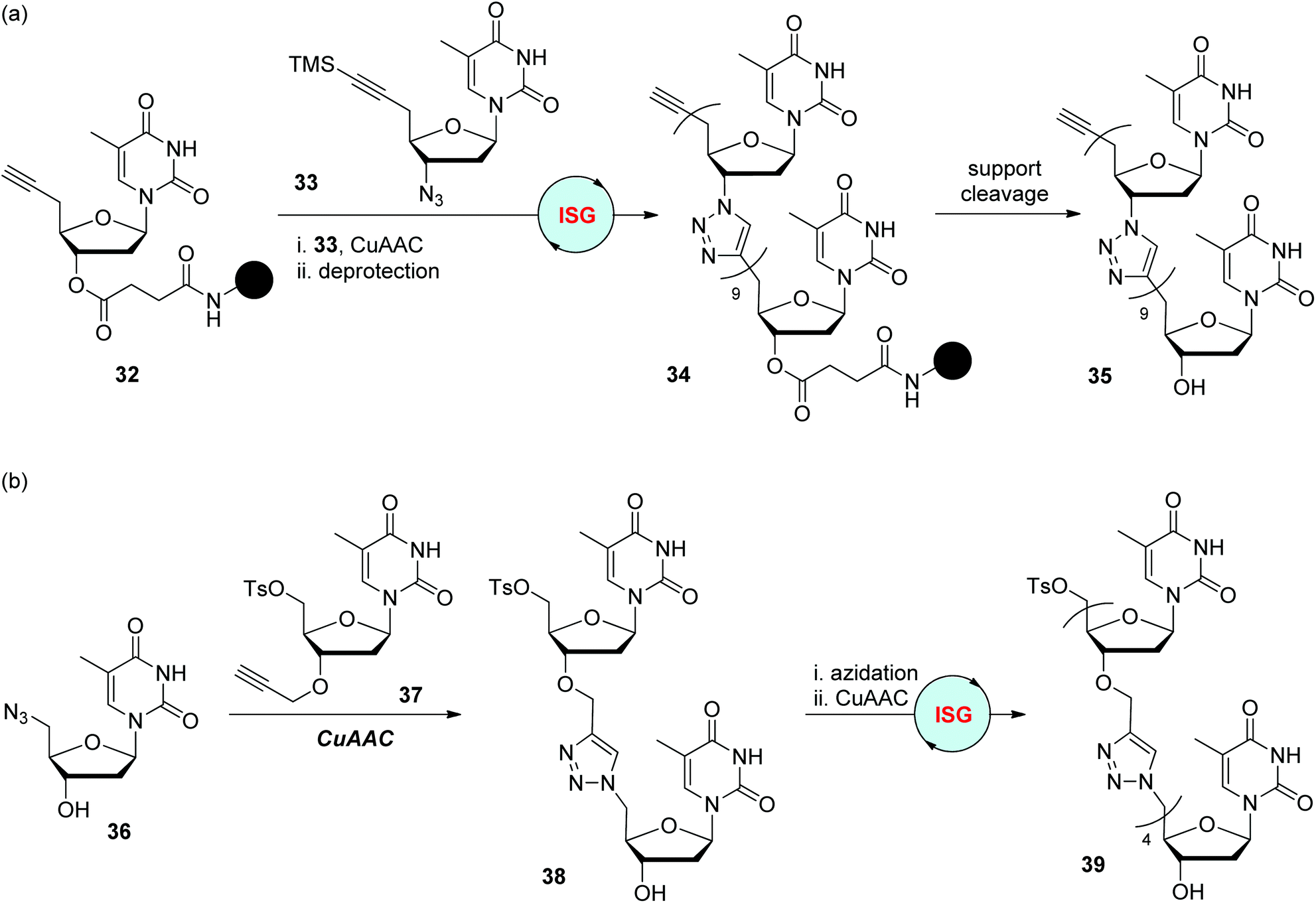
In 2008, the Isobe group designed one type of modified nucleoside monomer bearing a TMS-protected ethynyl group at the 5′-position of 2′-deoxyribose for the convenient fabrication of triazole-linked DNA analogues (Scheme 8a).28 It could be easily converted to monomers featuring a terminal alkynyl group at the 5′-position by TMS group deprotection (such as 32), or an azide group at the 3′-position by 3′-OH mesylation and subsequent azidation (such as 33), for CuAAC reactions in the liquid- or solid-phase. The dimer was obtained by iterative CuAAC reaction of 32 with 33 and deprotection of the TMS group in nearly quantitative yield, which then could be elongated to 10-mer 34 by repeating these two simple steps. Through further cleavage and HPLC purification, water-soluble triazole-linked oligothymine 35 was obtained in a total yield of 0.61%, which was confirmed to be able to form a stable double strand with a natural complementary DNA strand. An improved synthetic protocol was introduced by the same group in 2012, in which TIPS was used as the protecting group instead of TMS (as shown in compound 41), as well as a prolonged chain linked to the solid support (as shown in compound 40).29 Under adjusted reaction conditions for CuAAC and desilylation, a 12-mer oligothymine analogue was afforded in an overall yield of 17% after 12 cycles of iterative synthesis. The feature of this nucleoside monomer in selective transformation was also utilized to build oligomers through a convergent synthetic route in the solution phase, allowing the assembly of triazole-linked oligonucleotides bearing different monomers in an efficient and high-yield manner.30
 | ||
| Scheme 8 Preparation of triazole-linked analogues 35 (a) and 39 (b) of DNA through different strategies.28,31 TMS: trimethylsilyl. Ts: 4-toluenesulfonyl. | ||
The Zerrouki group reported a similar route for the construction of triazole-linked oligothymine in 2008 (Scheme 8b).31 Somewhat differently, the azide group in precursor 36 is anchored at the 5′-position of thymidine, which could be achieved by selective tosylation of the primary hydroxyl group of thymidine and successive azidation. Its CuAAC partner 37 was obtained through regioselective 3′-O-alkylation of the 5′-tosylthymidine. With these two monomers in hand, 5-mer 39 was constructed via an ISG strategy involving CuAAC/azidation in 46% total yield.
On the basis of their success in triazole-linked DNA analogues, Isobe and coworkers reported the synthesis of triazole-linked analogues of RNA in 2012, which were less developed (Scheme 9).32 Unlike the preparation of deoxyribonucleoside analogues from nucleosides, ribonucleoside analogues were accessed from D-xylose, allowing the synthesis of monomer 41 bearing different nucleobases and further solid-phase construction of diverse trinucleotide analogues 42. These trimers could easily connect with natural RNA oligonucleotides through phosphoramidite protocols to form elongated chimeric RNA oligonucleotides, which were found to be applicable in the cell-free translation reaction as mRNA substrates.33 Under improved CuAAC reaction conditions, these trinucleotide analogues were successfully elongated to up to 11-mer oligonucleotide 44 with a nucleobase sequence of 5′-UCUCUCUUUCT-3′, which was able to form a stable duplex with a complementary natural RNA oligonucleotide.34
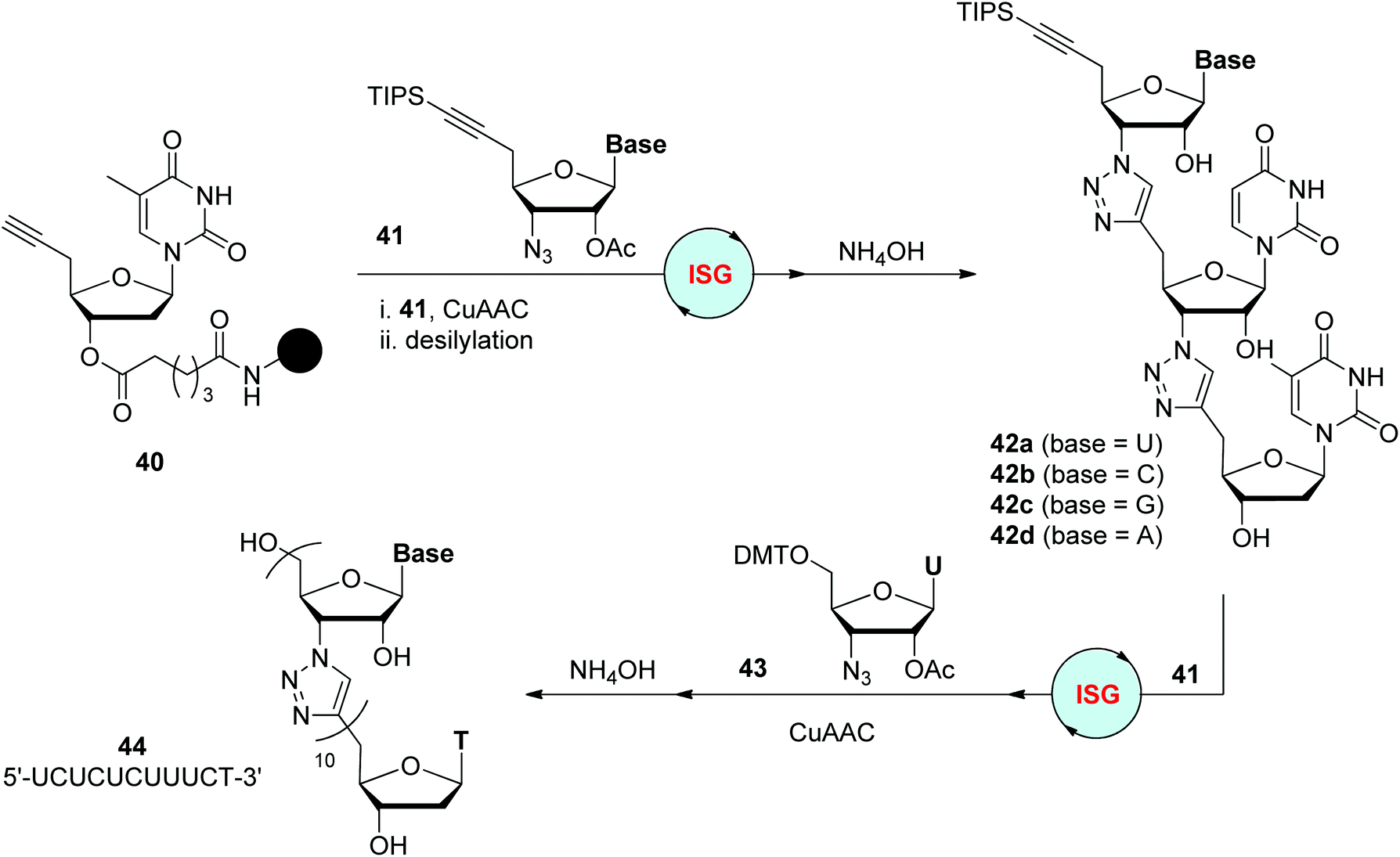 | ||
| Scheme 9 Preparation of triazole-linked analogues of RNA.32–34 TIPS: triisopropylsilyl. | ||
2.4. Triazole-linked analogues of peptide nucleic acids
As analogues of natural nucleic acids, the nucleobases in peptide nucleic acids (PNAs) are anchored to a neutral and achiral backbone bearing amide bonds.2 With the replacement of amide bonds with triazole moieties, Ganesh and Devi achieved the construction of triazole-based analogues of PNAs in 2010 (Scheme 10a).35 The functionalized monomer 46 was synthesized to start this iterative sequential growth route, the terminal alkynyl group of which could first react with the azide group on the solid support, and the Boc-protected amino group could be released and subsequently converted to the azide group for the next step. Creation of 6-mer 47 was accomplished by repeating this two-step iterative cycle. In comparison with other PNAs in which the amide bonds are not totally replaced by triazole rings, this fully modified analogue showed a higher self-ordering property. The Chaleix group designed a similar monomer 49 featuring an alkynyl group for CuAAC reaction and a chloride group for further azidation (Scheme 10b).36 Starting from azide-modified thymidine 48, the triazole-linked trimer 50 was successfully obtained as a PNA analogue via the repetition of this CuAAC/azidation cycle.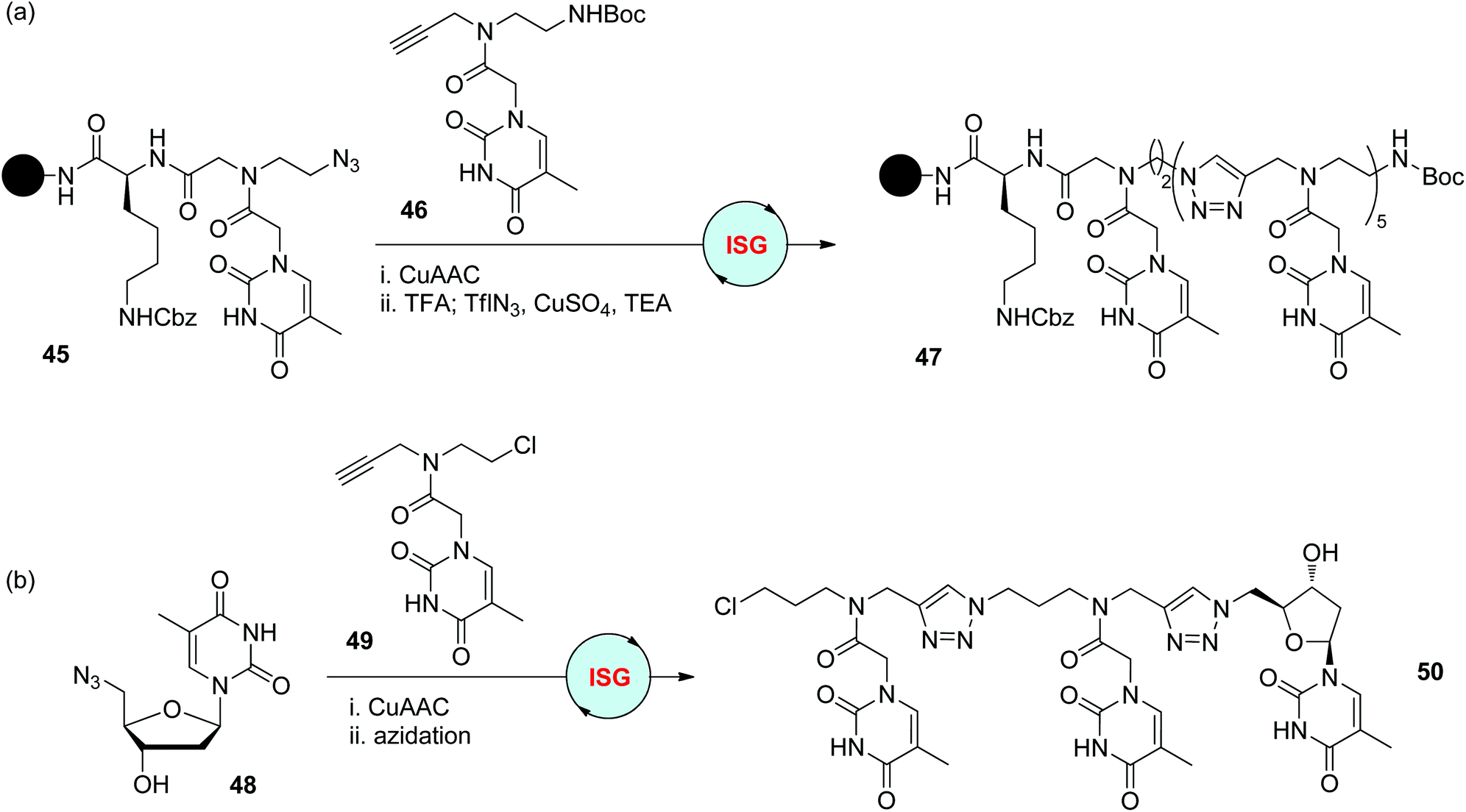 | ||
| Scheme 10 Synthesis triazole-based analogues of PNAs 47 (a) and 50 (b).35,36 | ||
2.5. Triazole-linked analogues of oligosaccharides
The replacement of the glycosidic oxygen atoms in oligo- and polysaccharides with other atoms or groups to create carbohydrate analogues is one important component in the discovery of bioactive compounds. In 2006, the Dondoni and Marra group reported the first synthesis of triazole-linked analogues of oligosaccharides through a two-reaction iterative protocol involving CuAAC and azidation (Scheme 11a).37 Mannose was selected as the sugar precursor for the preparation of alkyl 6-azido-α-C-mannoside 51 and ethynyl α-C-mannoside 52 as monomers to initiate the CuAAC-based ligation. The azidation step was realized by the direct transformation of the free hydroxyl group into the azido group in the presence of diphenyl phosphoryl azide (DPPA) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), allowing the efficient construction of 5-mer 53 in an overall 29% yield after four iterative cycles. The oligomannose analogue 54 with free hydroxyl groups was further obtained by the deprotection of MOM and benzyl groups in 53, which are supposed to be suitable for biological tests. 3-Hydroxy-3-methylbutynyl 6-azidomannoside 55 and ethynyl 6-deoxymannoside 56 were introduced as two new monomers in their later research for the fabrication of analogues of oligomannosides bearing a different number of monomers via diverse synthetic strategies (Scheme 11b).38 The CuAAC reaction of 55 with 56 and subsequent deacetonation under basic conditions generates a dimer featuring an ethynyl group, which could react with more units of 55 by repeating this two-reaction cycle. Through this strategy, one hexamannoside with a capping 6-deoxymannose fragment and an ethynyl group was synthesized, which was ligated with another hexamannoside bearing an azide group (derived from 53) to afford a decamannoside analogue. By using a convergent synthetic protocol, a range of triazole-linked 1,6-oligomannosides with up to 16 mannose units were further created from these four monomers (51, 52, 55 and 56).39 Bioactivity studies revealed the potential of free hydroxyl oligomannosides 57 as inhibitors of Mycobacterium tuberculosis cell wall synthetase.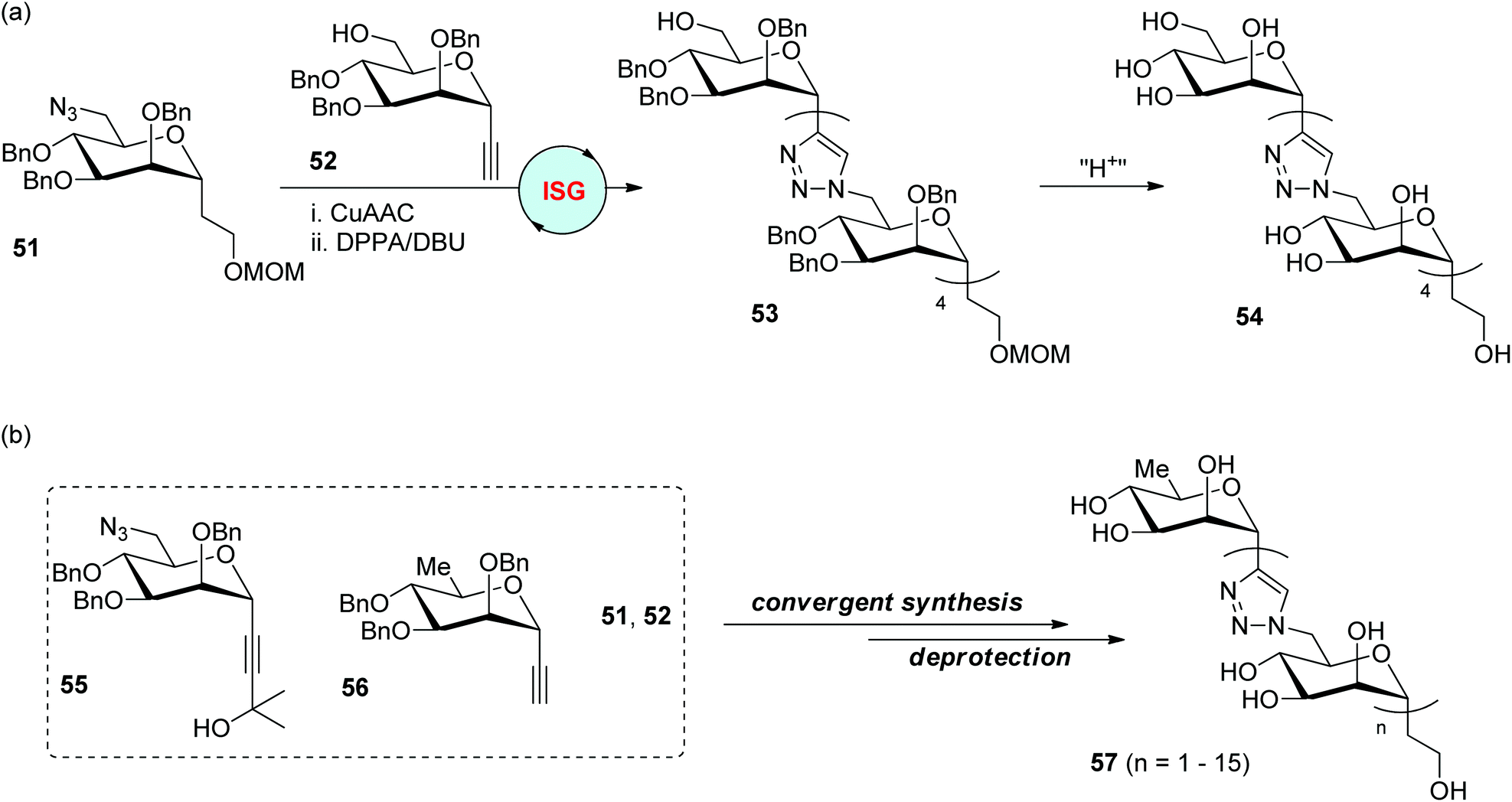 | ||
| Scheme 11 Preparation of triazole-linked oligomannoses via ISG (a) and convergent synthesis (b).37–39 Bn: benzyl. MOM: methoxymethyl. DPPA: diphenyl phosphoryl azide. DBU: diazabicyclo[5.4.0]undec-7-ene. | ||
Similarly, the Ye group prepared propargylic α-sialoside 58 and trimethylsilylated-propargyl 9-azido-α-sialoside 59 from sialic acid as two building blocks for the synthesis of triazole-tethered analogues of oligosialic acids in 2014 (Scheme 12).40 Mild CuAAC conditions were established by the authors for the coupling of 58 with 59, which was crucial for stabilizing the silylalkyne and avoiding CuAAC-based polymerizations. The desilylation step was then easily realized under conditions involving AgNO3. A series of oligosialic acids 60 featuring free hydroxyl and carboxylic acid groups were obtained by repeating different times of CuAAC/desilylation cycles and final deprotection.
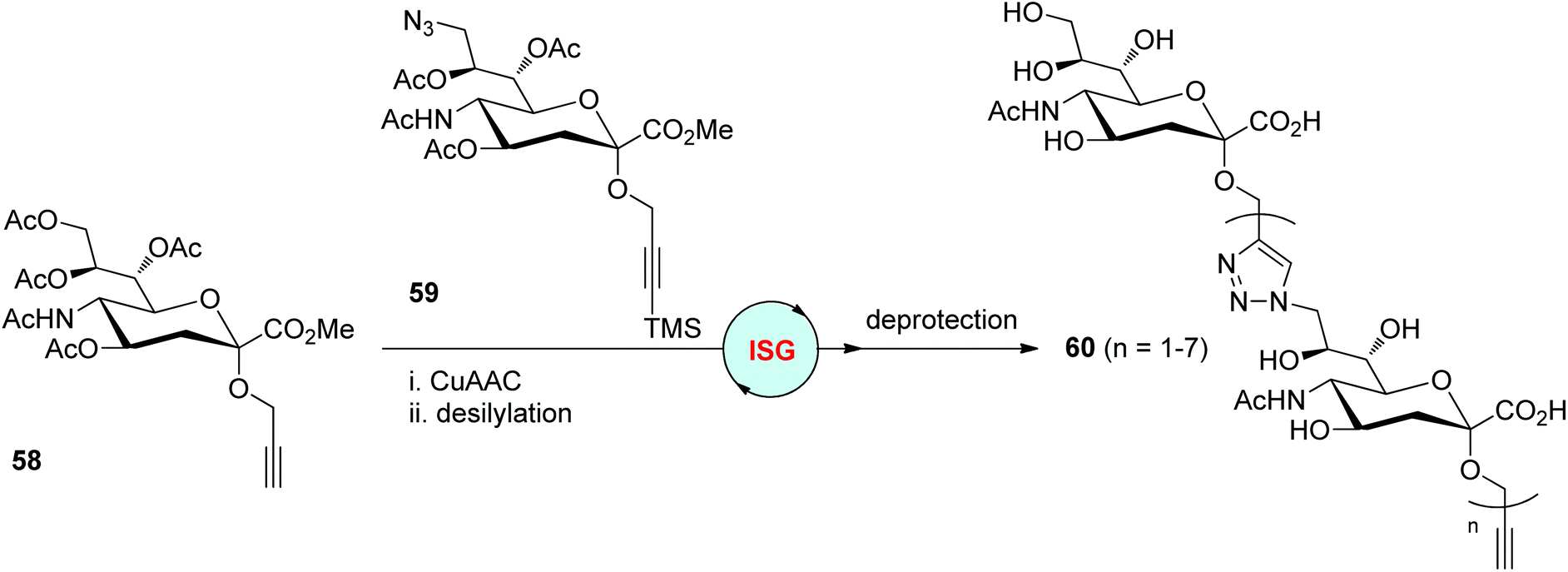 | ||
| Scheme 12 Construction of triazole-linked analogues of oligosialic acids.40 Ac: acetyl. | ||
In 2018, Hotha and Vangala released the assembly of triazole-linked oligosaccharide 67 as an analogue of Neisseria meningitids A capsular polysaccharide (Scheme 13).41 Both of the precursors 61 and 62 were derived from commercially available methyl α-D-glucopyranoside via a tedious synthetic route due to the introduction of the 2-acetamido group. In the absence of protection for the ethynyl group in 61, the CuAAC reaction of 61 with 62 or 63 only provided the related desired products 64 or 65 in poor yields.
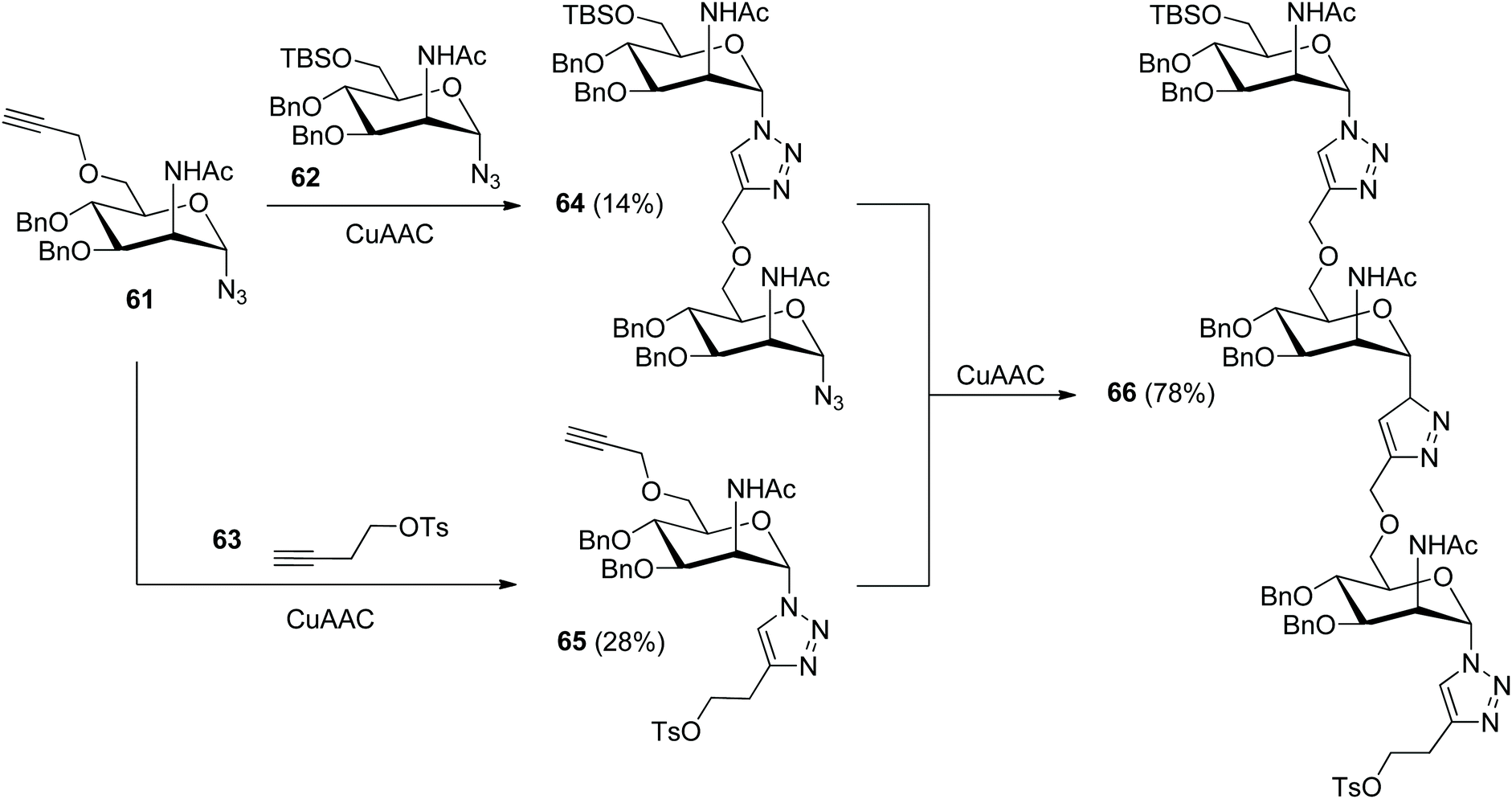 | ||
| Scheme 13 Fabrication of triazole-linked analogue of Neisseria meningitidis A capsular polysaccharide.41 | ||
The examples described in this section demonstrate the feasibility and fidelity of the triazole ring in tethering natural monomers to provide biomimetic triazole-linked oligomers. Azido- and alkynyl building blocks are mainly prepared from natural monomers, including amino acids, nucleosides and carbohydrates, for the construction of triazole rings under CuAAC or RuAAC conditions. The two-step iterative sequential growth strategy is the major pathway used in these cases. Convergent or IEG synthetic strategies established on the basis of CuAAC are more efficient in building longer oligomers. The variation of side chains was less investigated. Tedious preparation of precursors might be one reason for this.
3. Non-biomimetic sequence-defined polytriazoles
Besides the triazole-linked analogues of biopolymers discussed in section 2, diverse pure synthetic sequence-defined polytriazoles have also been constructed, which are primarily driven by two reasons: the high efficiency and simplicity of metal-catalyzed azide–alkyne cycloaddition reactions in constructing triazole motifs, and the convenient introduction and protection of azide and alkynyl groups in the preparation of monomers featuring different side chains. On the basis of these “click” reactions, various effective synthetic strategies, including facile two-step orthogonal ISG protocols, and IEG strategies involving highly efficient coupling and protection–deprotection steps, have been established to assemble various functionalized building blocks into desired sequence-defined polytriazoles, which will be discussed in this section.3.1. CuAAC-based ISG strategies
The advantages of CuAAC, such as exclusive regioselectivity, high yield, fast kinetics, and tolerance of a wide range of reaction conditions and reagent structures, enable it to be an ideal reaction in the development of robust, efficient, and simple iterative sequential growth strategies involving two orthogonal reactions for sequence-defined polymer synthesis. The CuAAC-based ISG protocols described in section 2, including CuAAC/azidation, CuAAC/desilylation, and CuAAC/amidification, were also used to build pure synthetic sequence-defined polytriazoles. More orthogonal approaches, including CuAAC/SuFEx (sulfur(VI) fluoride exchange) and CuAAC/Menschutkin reaction, have been developed in recent years for the synthesis of diverse sequence-regulated polymers.In early 2006, the Burgess group achieved the assembly of triethylene glycol molecules by using the CuAAC/azidation ISG protocol (Scheme 14).42 The phthalyl group-featured azide 67 and alkyne–tosylate 68 were prepared as the initial substrates, from which the triazole-tethered oligomer 69 was synthesized in a 30% overall yield by repeating the ISG cycle three times. The defined length of this kind of oligomeric compound, as well as the functional groups equipped in the two ends, make them highly desirable linkers in drug discovery and other areas.
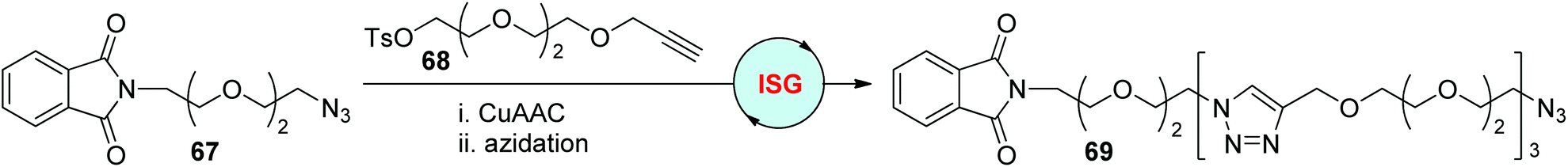 | ||
| Scheme 14 Construction of sequence-defined polymers through the ISG strategy involving CuAAC/azidation.42 | ||
The ISG strategy involving alternating CuAAC and desilylation processes relies on the sluggishness of internal 1-silylalkynes in CuAAC reactions. In 2006, by using this protocol, Hughes and coworkers realized the construction of triazole-linked sequence-defined polymers 73 (x = y = 0, such as 73a) from propargyl alcohol 70 and protected trialkylsilylpropargyl azides 71 (Scheme 15).43 The variation of the R2 group in 71 is facile to achieve on account of the convenient preparation of 71 from numerous commercially available aldehydes and terminal 1-silylalkynes. In 2010, the same group designed the synthesis of protected chiral trialkylsilylhomopropargyl azides 71 (x = 0, y = 1) from (S)-α-amino acids, and further finished the fabrication of a series of chiral sequence-defined polymers bearing different side chains (such as 73b) through this efficient and easily handled protocol.44
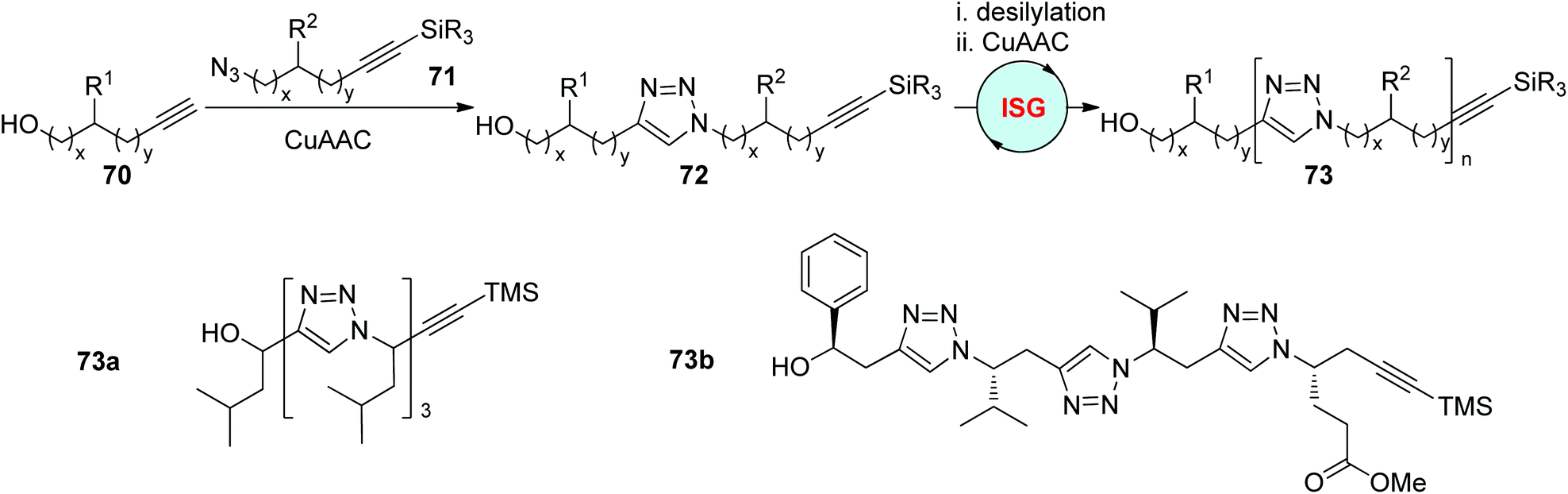 | ||
| Scheme 15 Construction of sequence-defined polymers through the ISG strategy involving CuAAC/desilylation.43,44 | ||
The practicability of this ISG strategy involving CuAAC/desilylation makes it a feasible technique to construct diverse functional sequence-defined polymers. In 2018, on the basis of their study on the supported trifunctional Cu/TEMPO/NMI-catalyzed aerobic oxidation of alcohols to aldehydes,45 Fernandes and coworkers created the functional oligomer 76 from monomers 74 and 75 by repeating the CuAAC and desilylation reactions (Scheme 16; TEMPO: (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl. NMI: 1-methylimidazole).46 This oligomer with precisely regulated side chains was further immobilized to azide-functionalized mesoporous silica particles to afford the supported catalyst 77, which showed superiority over the unfixed catalyst 76/CuI in catalyzing the aerobic oxidation of benzyl alcohol. The sequence of side chains was also proved to be crucial to its catalytic activity, as lower turnover frequencies were observed when the positions of TEMPO and NMI were exchanged.
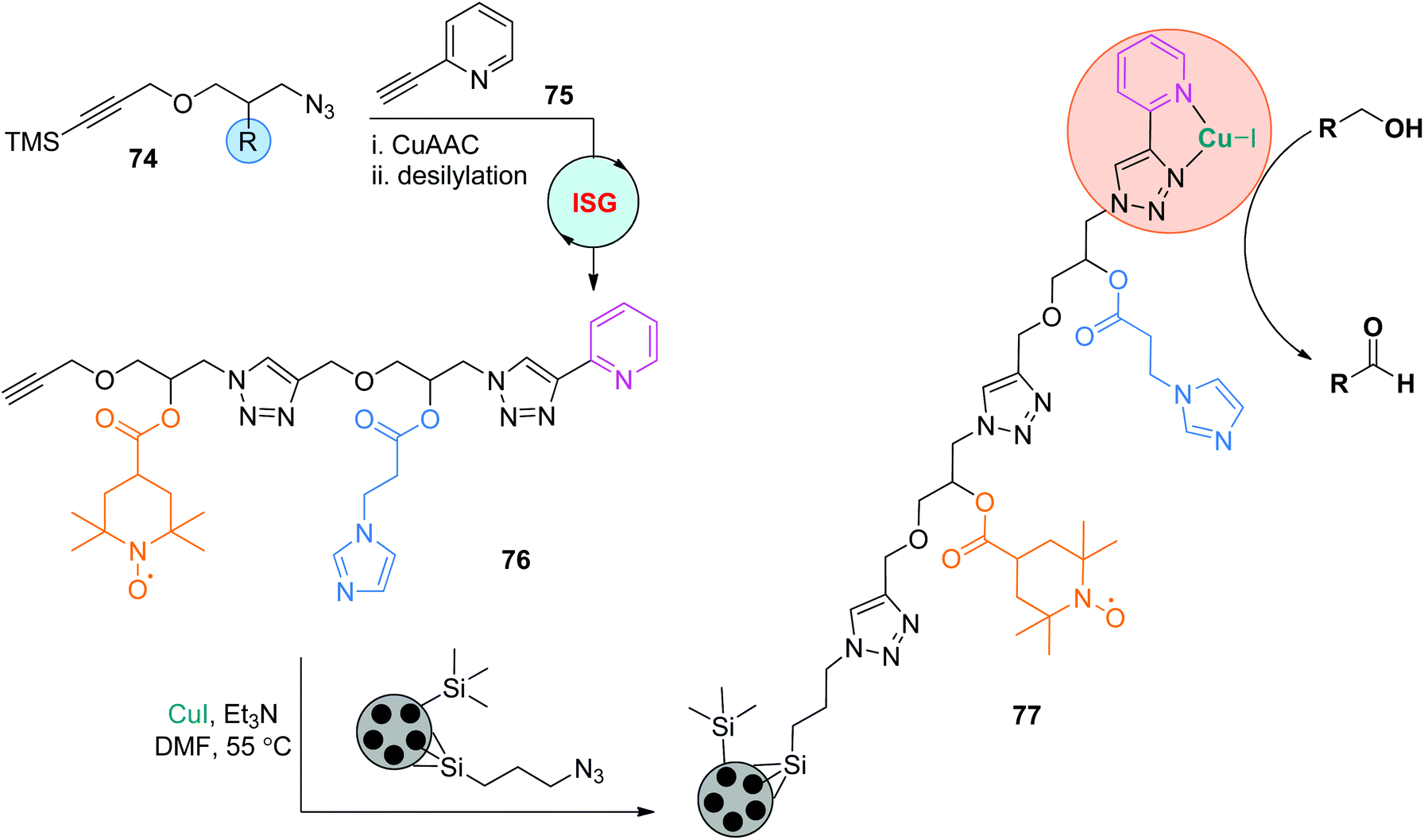 | ||
| Scheme 16 Application of functional sequence-defined polytriazoles in heterogeneous catalysis.46 | ||
CuAAC exhibited high efficiency in tethering fragments to produce synthetic sequence-defined polymers in DNA-templated polymerizations.47 One remarkable templating system was introduced by the Liu group, through which diverse alkyne- or azide-functionalized building blocks, including polyethylene and α- and β-peptides, could be effectively assembled under CuAAC conditions to afford sequence-regulated polymers with molecular weights of up to 26 kDa.47c In 2019, the Hunter group reported the construction of oligotriazoles through a covalent template-directed protocol (Scheme 17).48 The template polytriazole 81 was first obtained from the iterative assembly of 1-silylalkynyl and azido group-equipped monomers 79via a CuAAC-based ISG strategy. Desilylated monomers 82 featuring a carboxyl or hydroxyl group at the R position could be regularly attached to 81 through the esterification of carboxylic acid with relative phenols to form oligomer 83, which could be further transformed into duplex 84 by CuAAC coupling of the neighbouring alkynyl and azido groups. This biomimetic template-directed protocol for linear oligomer synthesis was accomplished by the final cleavage step, resulting in the formation of a complementary copy of template 81.
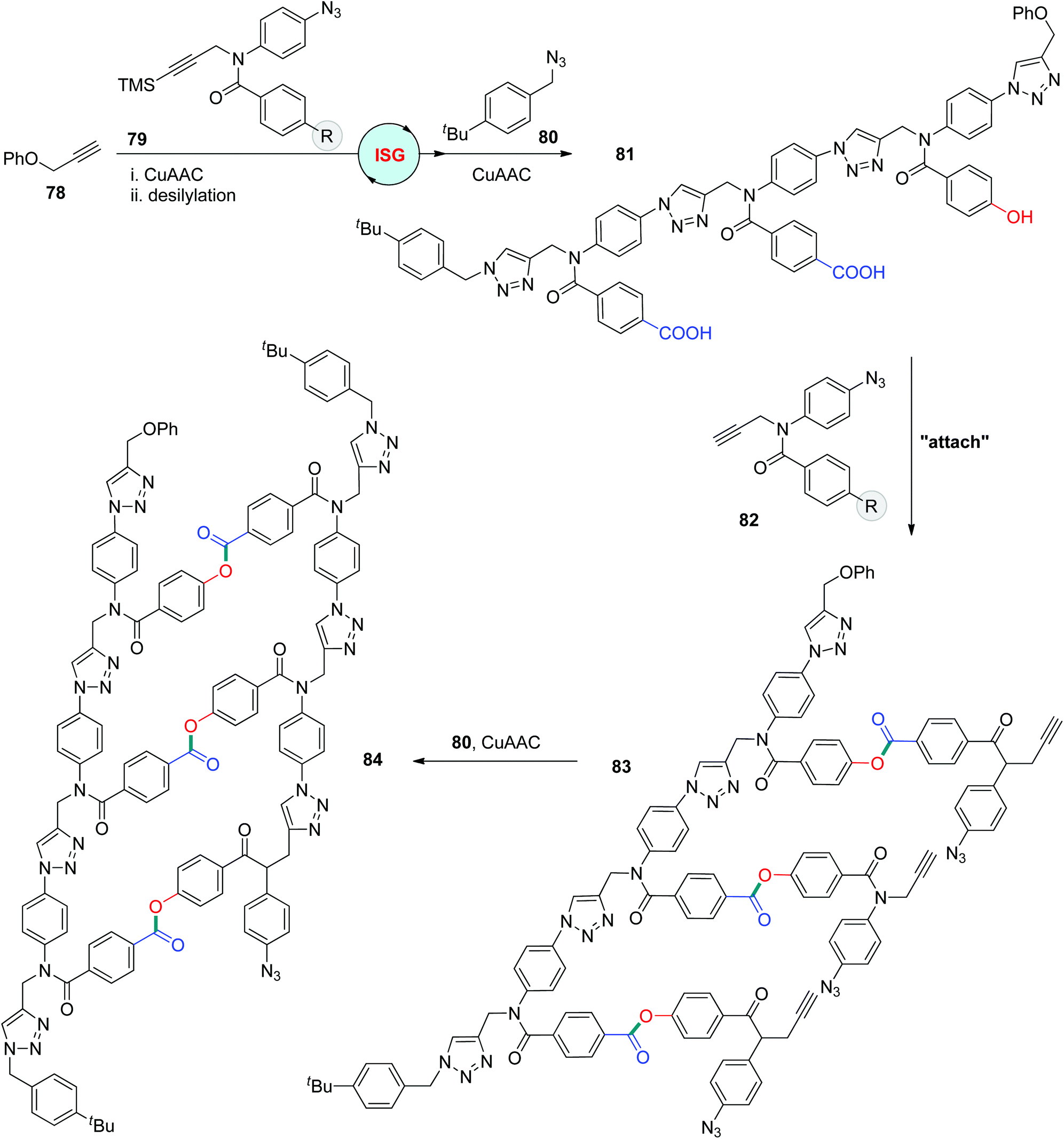 | ||
| Scheme 17 Utilization of sequence-defined polytriazoles as a template to direct the synthesis of linear oligomers.48 | ||
Amidification is a crucial step in Merrifield solid-phase peptide synthesis. As demonstrated in section 2.1, this efficient chemical reaction was also utilized in combination with CuAAC or RuAAC to produce triazole-based peptidomimetics. In 2009, the Lutz group reported the liquid-phase construction of the sequence-defined segment 88 on a soluble polystyrene support 87 by alternatingly repeating the amidification and CuAAC reaction between two types of difunctionalized monomers 85 and 86a (Scheme 18a).49 During this synthetic process, the extended polymer after each step could be quantitatively isolated by facile precipitation, which allows the simple and efficient purification manipulation in this protocol. This soluble polymer-supported chemoselective “AB + CD” ISG strategy was further applied to the construction of information-containing oligomers by the same group in 2015 (Scheme 18b).50 5-Hexynoic acids featuring different groups at the 3-position (86b & 86c) were selected as coding building blocks. Instead of the stepwise assembly of three monomers (85, 86b & 86c), four dyad-encoded oligomers 89a–89d were first prepared, which could be further ligated through the same strategy to form information-containing macromolecules 90 in a rapid and efficient manner.
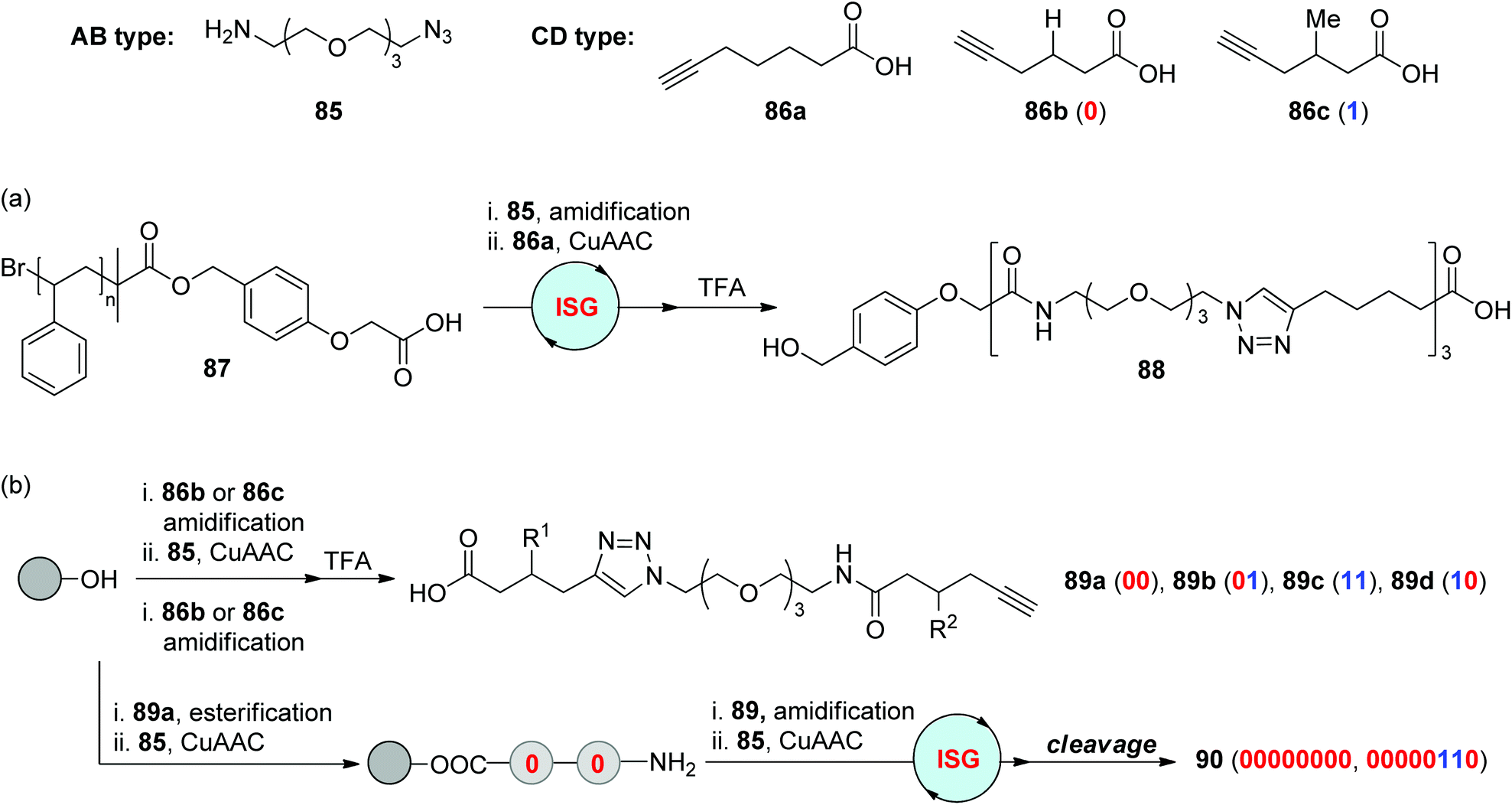 | ||
| Scheme 18 Synthesis of triazole-linked sequence-defined polymers (a) and digital polymers (b) through the strategy involving CuAAC/amidification.49,50 | ||
In 2018, the Niu group reported one novel ISG strategy involving orthogonal SuFEx and CuAAC reactions for the synthesis of sequence-defined polymers (Scheme 19).51 The SuFEx reaction, the abbreviation of sulphur–fluoride exchange reaction, was introduced by the Sharpless group in 2014 as one new click reaction with high efficiency, rapid kinetics and excellent tolerance toward functional groups.52 Two types of difunctionalized monomers 92 and 94 were prepared according to the requirements of SuFEx and CuAAC click reactions, allowing the synthesis of sequence-defined polymers through the protecting-group-free “AB + CD” ISG strategy introduced by the Lutz group. Starting from the silyl ether-functionalized resin 91, monodisperse oligomers with up to nine precisely regulated units (95 & 96) were facilely constructed by the iterative coupling of monomers 92 and 94 under orthogonal SuFEx and CuAAC conditions.
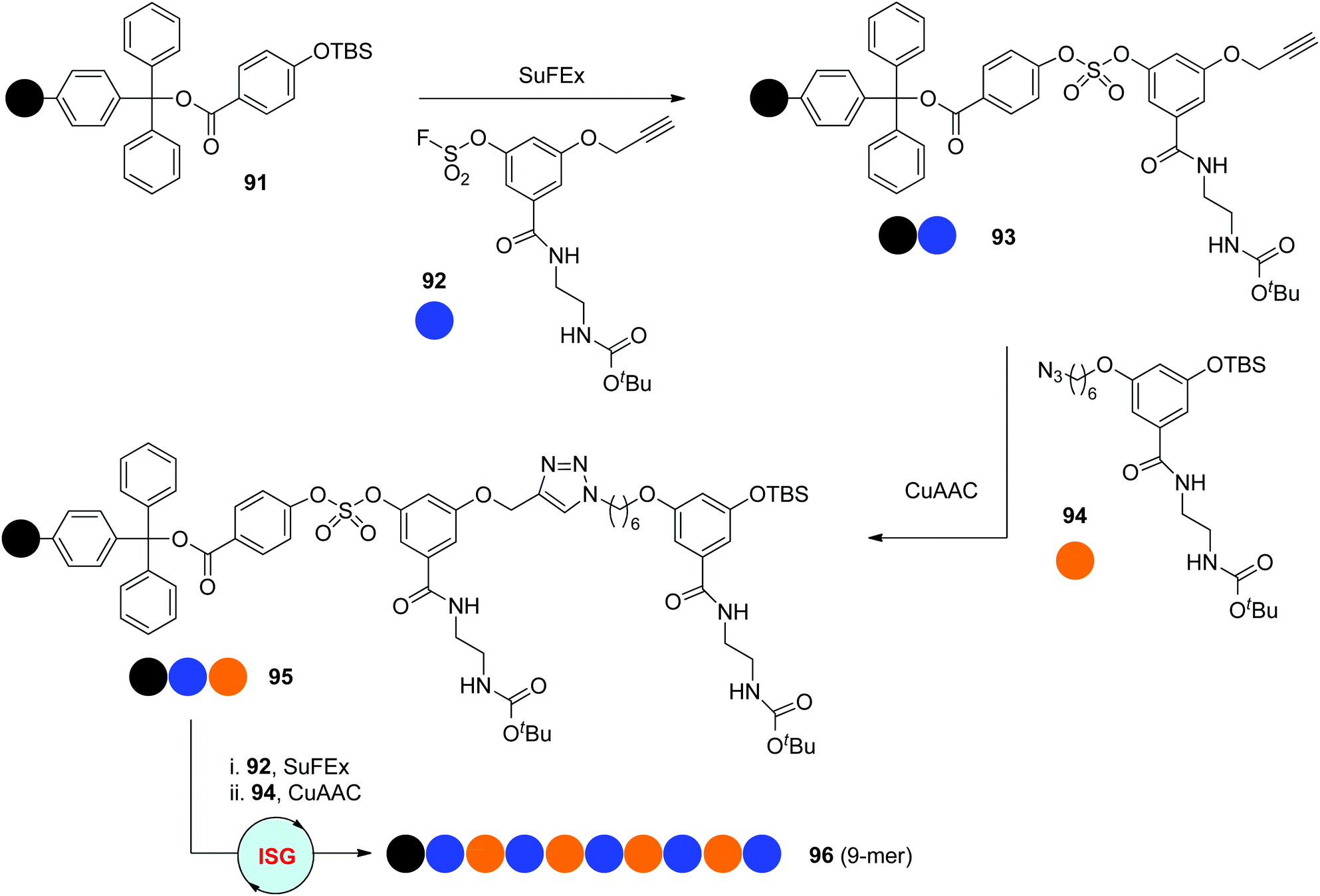 | ||
| Scheme 19 Synthesis of sequence-defined polytriazoles through ISG strategies involving orthogonal SuFEx and CuAAC reactions.51 | ||
The reaction of tertiary amines with alkyl halides, known as Meshutkin reaction, is one efficient coupling reaction for the preparation of charged quaternary ammonium salts. In 2019, the Gao group reported the construction of cationic sequence-defined polymers through the iteratively alternating processes of Menschutkin reaction and CuAAC reaction in the liquid phase (Scheme 20).53 This strategy exhibited several advantages, such as the convenient preparation of nitrine-tertiary amine monomers 97 featuring diverse side groups from simple synthons, the simple but highly efficient Menschutkin reaction and CuAAC reaction conditions, and the quantitative isolation of pure products by facile precipitation from the liquid phase, all of which could contribute to the creation of desired sequence-defined polymers on a large scale. As shown in Scheme 20, starting with 2-(dimethylamino)ethanol 98, the precisely programmed oligomer 101 with a quaternary ammonium backbone was obtained in an overall yield of 67% after running the ISG cycle 4 times.
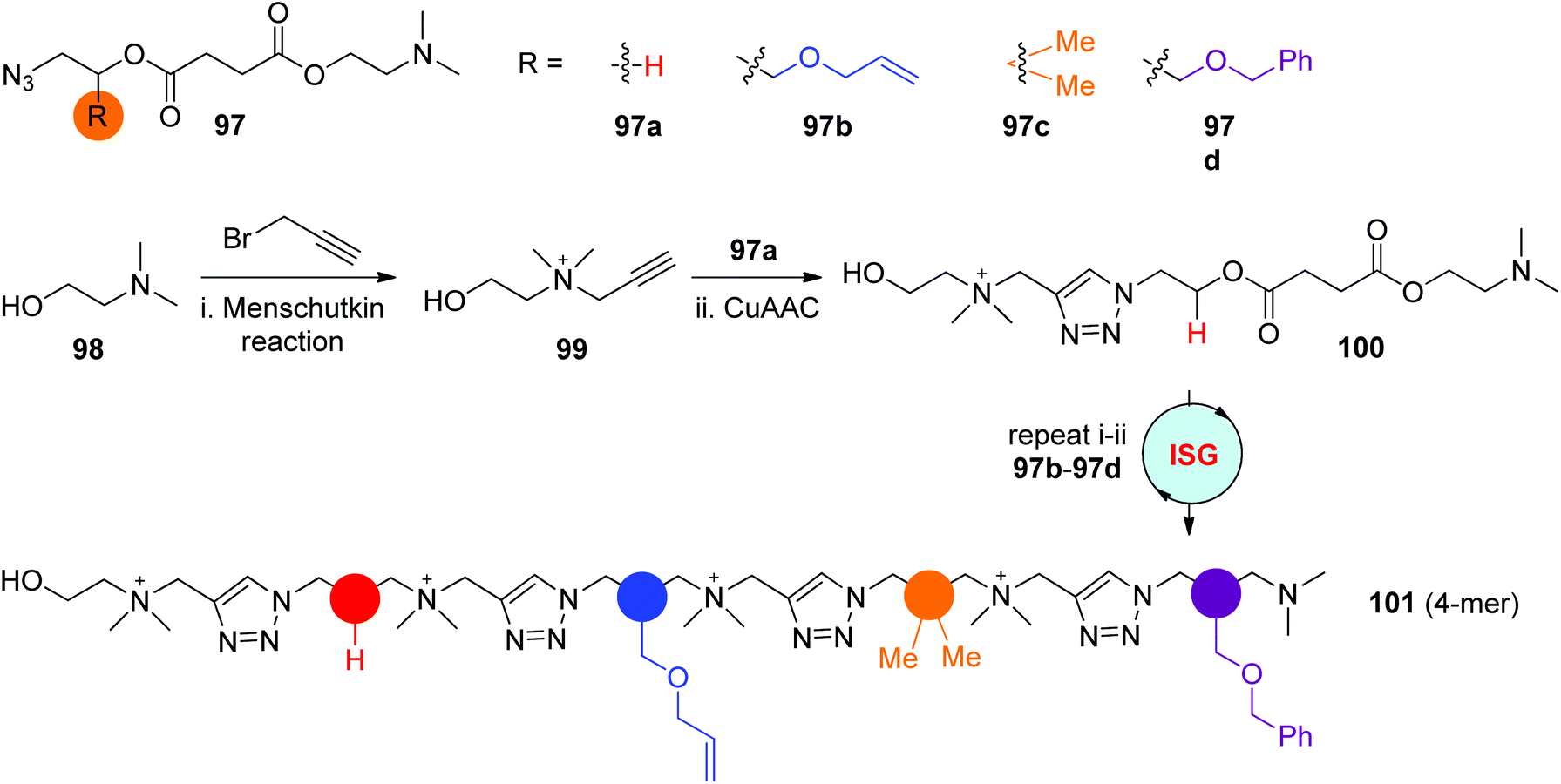 | ||
| Scheme 20 Synthesis of positively charged sequence-defined polymers by alternately performing Menschutkin reaction and CuAAC reaction.53 | ||
3.2. CuAAC-based IEG strategies
Ever since the pioneering report by Whiting and coworkers,54 the iterative exponential growth (IEG) strategy has evolved as a powerful method for the preparation of long-chain monodisperse polymers. One successful IEG cycle usually involves three efficient reactions: two separate deprotection reactions to generate two complementary monomers from one common original molecule, and one coupling reaction to tether these two halves to form a new molecule in an exponential manner. CuAAC has proved its high specificity and efficiency in coupling terminal alkynes and azides. This offers opportunities to elongate various monomers equipped with protected alkynyl and azido groups at two ends through CuAAC-based IEG strategies. The first case using this concept was released by the Drockenmuller group in 2009 (Scheme 21).55 The α-TIPS-ω-Cl-diprotected oligo(ethylene glycol) monomer 102, which was easily prepared from the reaction of 2-[2-(2-chloroethoxy)ethoxyl]ethanol with triisopropylsilyl propargyl bromide, could be selectively converted into 103 by desilylation, or 104 by the replacement of chloride with an azide group. Subsequent CuAAC coupling of 103 and 104 offered the corresponding α-TIPS-ω-Cl-diprotected dimer 105 in nearly quantitative yield, which could then be facilely extended to higher-generation oligomers 106 through this CuAAC-based exponential chain-growth protocol.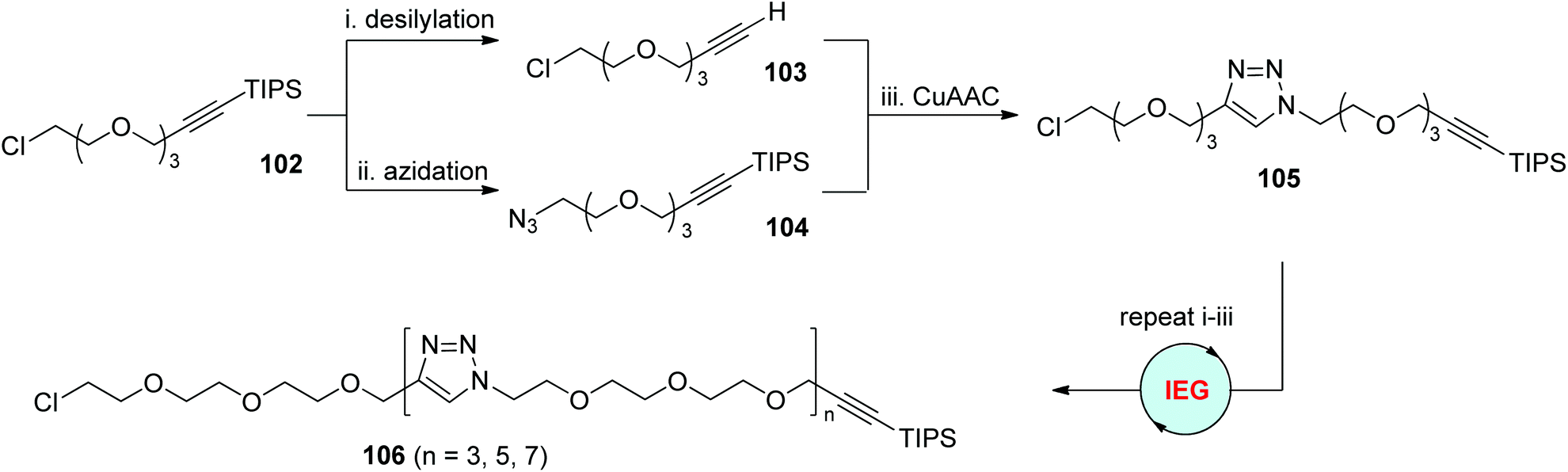 | ||
| Scheme 21 Synthesis of tri(ethylene glycol)-based oligomers through the CuAAC-based IEG strategy.55 | ||
On the basis of their eminent research on flow chemistry, Johnson and Jamison developed one CuAAC-based Flow-IEG system for the rapid and scalable synthesis of sequence-defined polymers in a semiautomatic fashion.56 This system is not only suitable for the construction of oligomers from one sole monomer, but also efficient in linking different monomers. As shown in Scheme 22, the cross-coupling of monomers 107 and 102 could afford dimer 108 in an overall yield of 73% after the performance of three reactions and an in-line purification in a total residence time of around 10 minutes. The (ABAB)n sequence-regulated 16-mer 109 was successfully obtained in a 39% total yield by repeating this Flow-IEG cycle 3 more times, indicating the potential and efficiency of this process in producing more structurally complicated sequence-defined polymers.
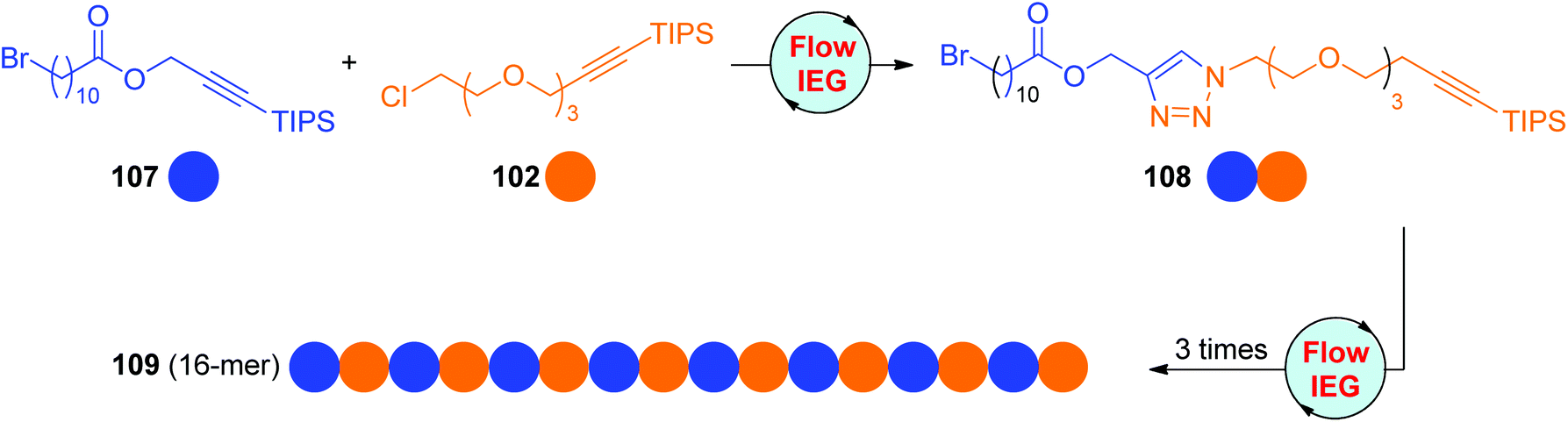 | ||
| Scheme 22 Scalable construction of sequence-defined polymers through the CuAAC-based flow-IEG strategy.56 | ||
In 2015, the Johnson group reported an iterative exponential growth plus side-chain functionalization (IEG+) strategy, through which different side-chain functional groups could be introduced after each IEG cycle (Scheme 23).57 Chiral TBS-protected epoxy-alkynes 110, which could be easily prepared from propargyl alcohol and chiral epichlorohydrin, were designed for this IEG+ protocol. The side-chain decoration was realized in the process of fabricating the azide partner 111 through the nucleophilic opening of the epoxide ring with an azide anion and subsequent functionalization of the derived hydroxyl group. Dimer 113 from the CuAAC reaction of 111 with 112 was successfully extended to a series of novel stereocontrolled sequence-defined polymers with up to 32 units (such as 114) by the repetition of steps i–iv that constitute this IEG+ cycle. Selective deprotection of the side-chain acetyl groups to yield water-soluble SDPs was also demonstrated by the authors to probe the potential biomedical applications of these monodisperse linear polymers.
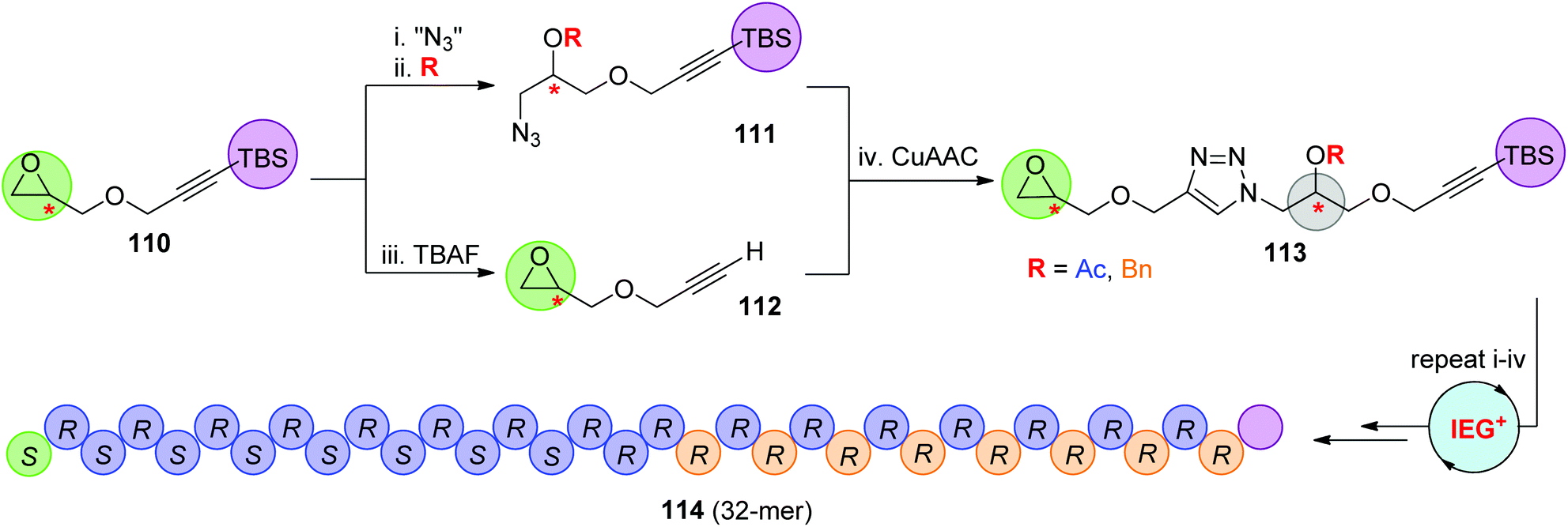 | ||
| Scheme 23 Synthesis of stereo- and sequence-defined polymers through the CuAAC-based IEG+ strategy.57 | ||
Sequence-defined polymers bearing decoratable side-chain groups were further synthesized via the IEG+ strategy by Johnson and coworkers for the construction of functional macromolecules by post-modification (Scheme 24).58 The first generation dimer 117 featuring two allyl groups was created from the deprotected monomer 112 by running one IEG+ cycle, during which the allyl group was installed at both of the azide monomer 115 and the alkyne monomer 116. The elongation of 117 to 16-mer 118 was achieved through the previously demonstrated CuAAC-based IEG strategy. A series of unimolecular stereoisomeric diblock copolytriazoles (119 & 120) were realized through post-functionalization of these 16-mers with hydrophilic or hydrophobic side chains via efficient thiol–ene addition reactions and subsequent coupling by CuAAC reaction. Both of the side chains and the stereochemical sequence were found to have an influence on the self-assembly behaviours of these diblock copolymers.
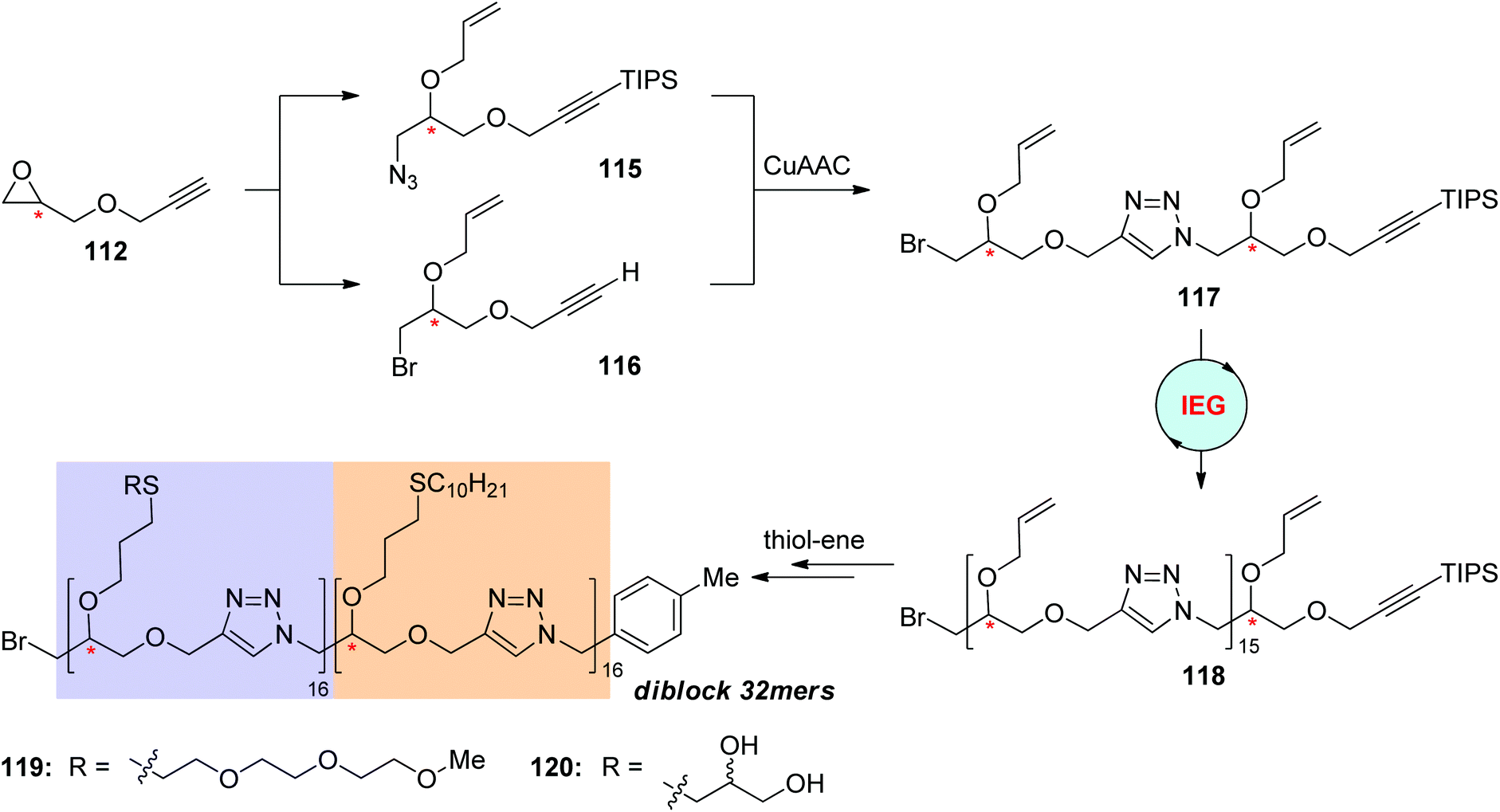 | ||
| Scheme 24 Preparation of triazole-linked uniform diblock copolymers through the click reaction-based IEG strategy and post-modification.58 | ||
Instead of anchoring functional groups at the hydroxyl group of 121 to derive azide partners, Fernandes et al. designed the functionalized monomers 122 through the CuAAC reaction of 121 with terminal alkynes (Scheme 25).59 The α-TBS-ω-N3 monomer 123 was smoothly synthesized by successive treatments with p-nitrophenyl chloroformate and 3-azidopropylamine, in which the newly introduced carbamate group was found to be more stable than ester groups. To enrich the diversity of side groups, dimer 125 was assembled from two different monomers 122 rather than from one. More complicated sequence-regulated oligomers with up to 8 units and various triazole-linked functional side groups (such as 126) were successfully built through the same IEG-based cross-coupling strategy. Similar to the Johnson group's research, the stereochemical structure was well controlled during this elongation process. Structural studies were also carried out by the authors through molecular dynamics (MD) simulations, the results of which disclosed the importance of π-stacking interactions in affecting the folding and aggregation performances of these oligomers.
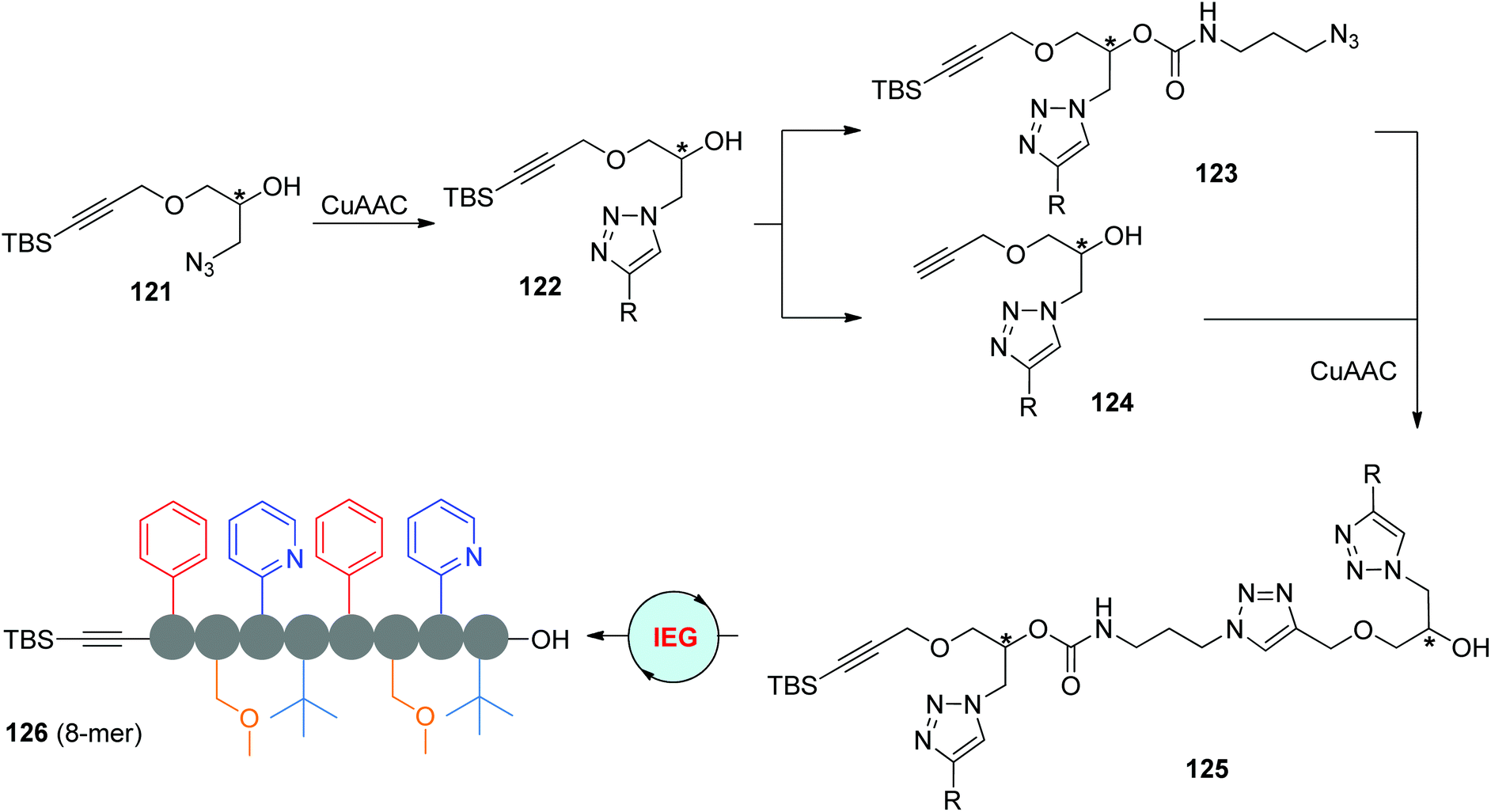 | ||
| Scheme 25 Synthesis of multifunctional stereocontrolled sequence-defined oligomers through the CuAAC-based IEG strategy.59 | ||
The diphenyl phosphorazidate (DPPA) and 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU) method is an efficient protocol for the direct transformation of hydroxyl groups to azides, which has been used by the Dondoni group in their iterative sequential synthesis of triazole-linked analogues of oligosaccharides (Scheme 11a).37 In 2016, the Monteiro group reported ISG and IEG strategies relying on the DPPA/DBU method for the facile construction of diverse sequence-defined polymers (Scheme 26).60 Near quantitative conversion of the designed α-TIPS-ω-OH monomer 127 to α-TIPS-ω-N3 monomer 128 was achieved under the conditions of DPPA/DBU in DMF at 50 °C. Instead of building macromolecules from one monomer, the α-TIPS-ω-OH trimer 130 was first assembled through the iterative coupling of TIPS-protected monomer 127 with different propargyl monomers 129, which was then elongated to diverse side group-featured 12-mer 131via the DPPA/DBU-based IEG strategy. In consideration of the significance of side chains in the behaviour of sequence-defined polymers, comprehensive utilization of ISG and IEG strategies would be an important way to create functionalized macromolecules.
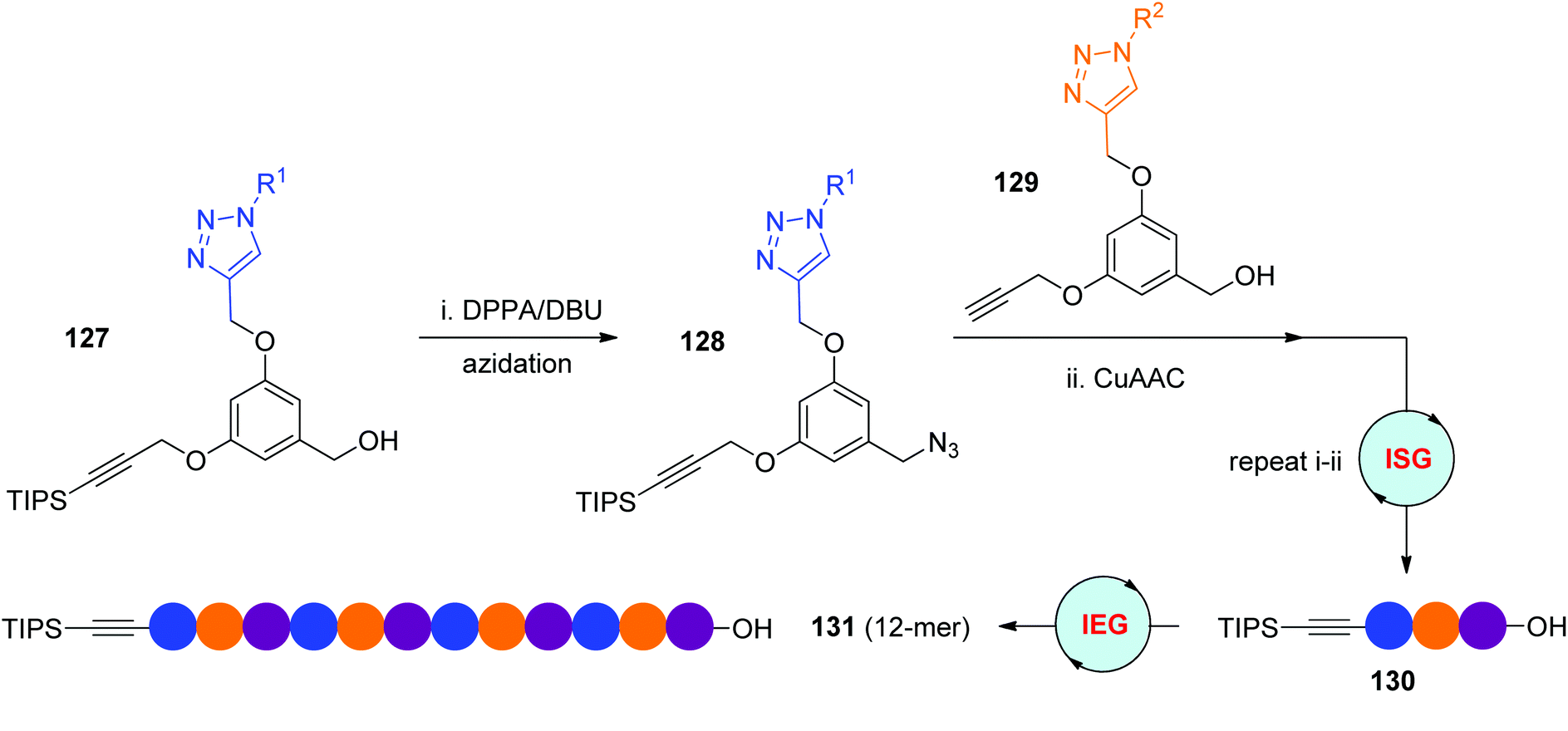 | ||
| Scheme 26 Utilization of direct azidation of alcohols in CuAAC-based ISG and IEG strategies.60 | ||
In the above CuAAC-based IEG strategies, three major reactions were usually included: desilylation, azidation and CuAAC, and the exponential elongations could be achieved from one monomer. Very recently, the Zhang group developed a novel IEG strategy on the basis of three click reactions involving CuAAC, SuFEx and Ugi-4CR, which enabled the construction of triazole-tethered sequence-defined polymers in a protecting-group-free fashion.61 As shown in Scheme 27, three different monomers bearing two orthogonal clickable terminals (132–134) are required to initiate this extension. Their orthogonal couplings occurred via CuAAC, SuFEx and Ugi-4CR with parallelly generated three resultant dimers 135–137 with two exchanged end groups. Notably, two external side groups could be easily introduced in the Ugi-4CR coupling reaction. Three types of sequence-defined polymers with up to 32 units were successfully created by further iterative application of these three coupling reactions in a parallel manner.
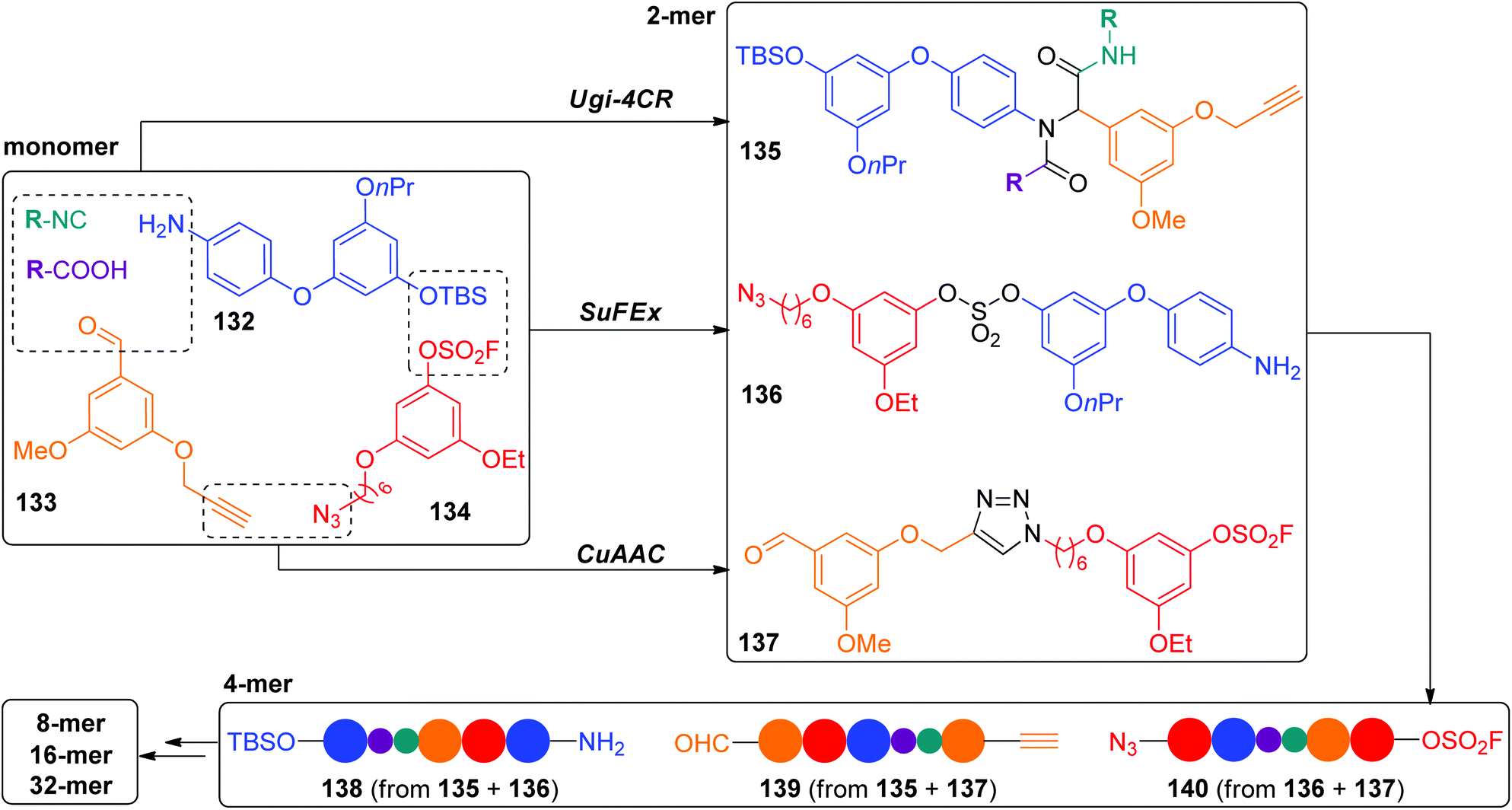 | ||
| Scheme 27 Synthesis of sequence-defined polymers by the protecting-group-free IEG+ strategy.61 | ||
3.3. IrAAC-based strategies
The iridium-catalyzed cycloaddition of organic azides with internal 1-thioalkynes (IrAAC) developed by the Sun group offered a direct and effective approach to 1,4,5-trisubstituted triazoles with exclusive regioselectivity.62 In 2020, Ding and coworkers established diverse IrAAC-based strategies for the construction of sequence-defined polytriazoles, in which the functional side groups are sequentially installed at the C-5 position of triazole rings (Scheme 28).63 The two-step ISG strategy involving IrAAC and azidation allows the facile synthesis of oligomers from the desired initial azides in the liquid phase. For instance, hexamer 144 was smoothly built in an overall yield of 34% from the iterative reaction of modifiable Csp2–Br-featured benzyl azide 141 with thioalkynes involving six different functional OTs groups 142. The α-TMS-ω-OTBS trimer 145 was obtained as well through the repetition of this two-step process, which could be further extended to long-chain polymers with up to 31 blocks (148) by using the CuAAC-based IEG strategy. It is noteworthy that in this case the hydrogen atom at the C-5 position of 1,4-isomers derived from CuAAC could also act as an important side-chain variation, which enables the chain length to be elongated in a 2n + 1 manner. In addition, one more complicated monodisperse polymer was fabricated by the authors through the CuAAC coupling of an ISG-generated oligomer with an IEG-generated segment.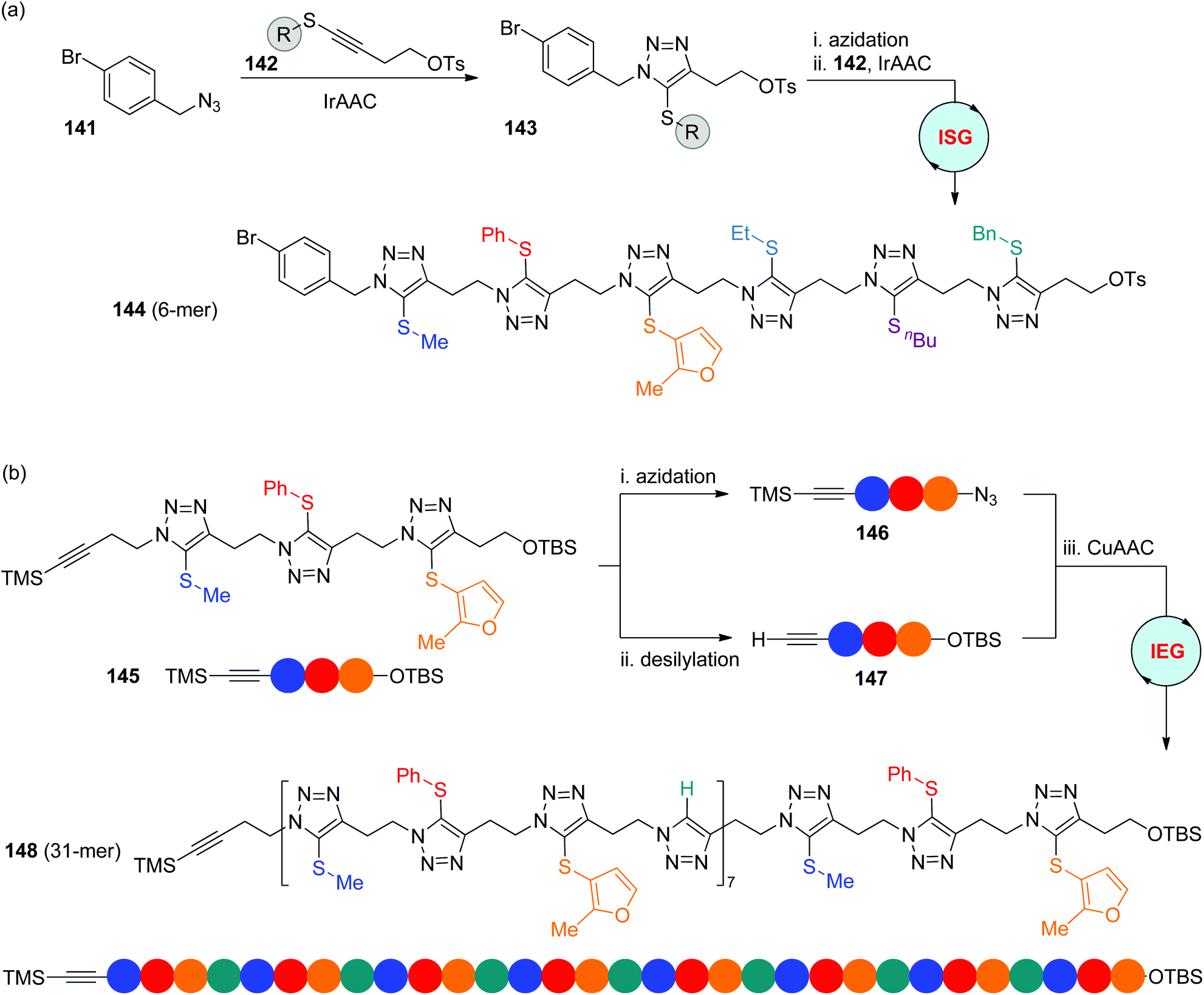 | ||
| Scheme 28 Synthesis of sequence-defined polytriazoles through IrAAC-based ISG (a) and IEG (b) strategies.63 | ||
4. Conclusion
This review summarizes the progress in triazole-based sequence-defined polymers, which were primarily obtained by the high efficiency of metal-catalyzed azide–alkyne cycloaddition reactions in the regioselective construction of triazole skeletons. A variety of CuAAC-based ISG strategies have been successfully developed for the stepwise connection of natural or artificial monomers in the solid- or liquid-phase. Effective IEG protocols involving CuAAC were likewise established on the basis of the sluggishness of 1-silylalkynes in CuAAC reactions. RuAAC-based IEG strategies were mainly utilized in the fabrication of triazole-linked sequence-defined peptidomimetics due to their specificity in providing 1,5-disubstituted triazole motifs. While these disubstituted triazole rings derived from CuAAC and RuAAC are limited as linkers to tether different components, the 1,4,5-fully substituted triazole blocks from IrAAC could also act as the carrier of side chains, allowing the synthesis of sequence-defined polytriazoles equipped with theoretically unlimited variable backbones and side groups. In consideration of the unique properties of the triazole architecture and recent advances in transition metal-catalyzed cycloaddition reactions of azides with internal alkynes, more efficient MAAC-based strategies for the creation of functional triazole-linked sequence-defined polymers are highly desirable. We hope that this review could not only help with the design of novel sequence-defined polymers, but also stimulate the corresponding structural and functional studies, which are still under-developed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21702015 and 21871023) and Big Science Project from Beijing University of Chemical Technology (XK180301).References
- C. Yang, K. B. Wu, Y. Deng, J. Yuan and J. Niu, ACS Macro Lett., 2021, 10, 243 CrossRef CAS.
- (a) P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CrossRef CAS PubMed; (b) P. Wittung, P. E. Nielsen, O. Buchardt, M. Egholm and B. Nordén, Nature, 1994, 368, 561 CrossRef CAS PubMed.
- (a) R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646 CrossRef CAS; (b) A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665 CrossRef CAS PubMed.
- (a) D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071 CrossRef CAS; (b) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed.
- (a) T. T. Trinh, C. Laure and J.-F. Lutz, Macromol. Chem. Phys., 2015, 216, 1498 CrossRef CAS; (b) J. W. Lehmann, D. J. Blair and M. D. Burke, Nat. Rev. Chem., 2018, 2, 0115 CrossRef PubMed; (c) Chapter 6 in ref. 7b.
- (a) O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc., Chem. Commun., 1982, 1165 RSC; (b) S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drokenmuller, Macromol. Rapid Commun., 2011, 32, 147 CrossRef CAS PubMed.
- (a) J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, Sequence-controlled polymers: synthesis, self-assembly, and properties, American Chemical Society, Washington, DC, 2014 Search PubMed; (b) J.-F. Lutz, Sequence-controlled polymers, Wiley-VCH, Weinheim, 2018 Search PubMed; (c) J.-F. Lutz, Polym. Chem., 2010, 1, 55 RSC; (d) J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed; (e) M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2019, 31, 1806027 CrossRef PubMed; (f) P. Nanjan and M. Porel, Polym. Chem., 2019, 10, 5406 RSC; (g) S. L. Perry and C. E. Sing, ACS Macro Lett., 2020, 9, 216 CrossRef CAS.
- (a) P. Espeel and F. E. Du Prez, Macromolecules, 2015, 48, 2 CrossRef CAS; (b) S. Martens, J. O. Holloway and F. E. Du Prez, Macromol. Rapid Commun., 2017, 38, 1700469 CrossRef PubMed; (c) Chapter 13 in ref. 7b.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- (a) W.-Q. Fan and A. R. Katritzky, Comprehensive Heterocyclic Chemistry II, 1996, vol. 4, pp. 1–126 Search PubMed; (b) A. C. Tomé, in Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, ed. R. C. Storr and T. L. Gillchrist, Thieme, Stuttgart, 2004, vol. 13, pp. 415–601 Search PubMed.
- J. Ma and S. Ding, Asian J. Org. Chem., 2020, 9, 1872 CrossRef CAS.
- (a) N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716 CrossRef CAS PubMed; (b) A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645 RSC; (c) M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974 CrossRef CAS PubMed; (d) P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907 CrossRef CAS PubMed; (e) M. S. Taylor, Coord. Chem. Rev., 2020, 413, 213270 CrossRef CAS; (f) R. Hein, P. D. Beer and J. J. Davis, Chem. Rev., 2020, 120, 1888 CrossRef CAS PubMed; (g) J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS; (h) É. M. Foyle and N. G. White, Chem. – Asian J., 2021, 16, 575 CrossRef PubMed.
- (a) Y. L. Angell and K. Burgess, Chem. Soc. Rev., 2007, 36, 1674 RSC; (b) J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325 RSC; (c) D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2399 CrossRef CAS.
- I. E. Valverde and T. L. Mindt, Chimia, 2013, 67, 262 CrossRef CAS PubMed.
- N. G. Angelo and P. S. Arora, J. Am. Chem. Soc., 2005, 127, 17134 CrossRef CAS PubMed.
- N. G. Angelo and P. S. Arora, J. Org. Chem., 2007, 72, 7963 CrossRef CAS PubMed.
- A. L. Jochim, S. E. Miller, N. G. Angelo and P. S. Arora, Bioorg. Med. Chem. Lett., 2009, 19, 6023 CrossRef CAS PubMed.
- Z. Zhang and E. Fan, Tetrahedron Lett., 2006, 47, 665 CrossRef CAS.
- Z. Ke, H.-F. Chow, M.-C. Chan, Z. Liu and K.-H. Sze, Org. Lett., 2012, 14, 394 CrossRef CAS PubMed.
- L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS PubMed.
- M. R. Krause, R. Goddard and S. Kubik, J. Org. Chem., 2011, 76, 7084 CrossRef CAS PubMed.
- J. R. Johansson, E. Hermansson, B. Nordén, N. Kann and T. Beke-Somfai, Eur. J. Org. Chem., 2014, 2703 CrossRef CAS.
- A. S. Stålsmeden, A. J. Paterson, I. Cs. Szigyártó, L. Thunberg, J. R. Johansson, T. Beke-Somfai and N. Kann, Org. Biomol. Chem., 2020, 18, 1957 RSC.
- O. Kracker, J. Góra, J. Krzciuk-Gula, A. Marion, B. Neumann, H.-G. Stammler, A. Nieß, I. Antes, R. Latajka and N. Sewald, Chem. – Eur. J., 2018, 24, 953 CrossRef CAS PubMed.
- D. Althuon, F. Rönicke, D. Fürniss, J. Quan, I. Wellhöfer, N. Jung, U. Schepers and S. Bräse, Org. Biomol. Chem., 2015, 13, 4226 RSC.
- A. Nuzzi, A. Massi and A. Dondoni, QSAR Comb. Sci., 2007, 26, 1191 CrossRef CAS.
- H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Org. Lett., 2008, 10, 3729 CrossRef CAS PubMed.
- T. Fujino, N. Yamazaki, A. Hasome, K. Endo and H. Isobe, Tetrahedron Lett., 2012, 53, 868 CrossRef CAS.
- (a) T. Fujino, N. Yamazaki and H. Isobe, Tetrahedron Lett., 2009, 50, 4101 CrossRef CAS; (b) T. Fujino, K. Yasumoto, N. Yamazaki, A. Hasome, K. Sogawa and H. Isobe, Chem. – Asian J., 2011, 6, 2956 CrossRef CAS PubMed; (c) H. Isobe, N. Yamazaki, A. Asano, T. Fujino, W. Nakanishi and S. Seki, Chem. Lett., 2011, 40, 318 CrossRef CAS; (d) H. Isobe and T. Fujino, Chem. Rec., 2014, 14, 41 CrossRef CAS PubMed.
- R. Lucas, R. Zerrouki, R. Granet, P. Krausz and Y. Champavier, Tetrahedron, 2008, 64, 5467 CrossRef CAS.
- T. Fujino, K. Endo, N. Yamazaki and H. Isobe, Chem. Lett., 2012, 41, 403 CrossRef CAS.
- T. Fujino, T. Suzuki, K. Okada, K. Kogashi, K. Yasumoto, K. Sogawa and H. Isobe, J. Org. Chem., 2016, 81, 8967 CrossRef CAS PubMed.
- T. Fujino, T. Suzuki, T. Ooi, K. Ikemoto and H. Isobe, Chem. – Asian J., 2019, 14, 3380 CrossRef CAS PubMed.
- G. Devi and K. N. Ganesh, Artif. DNA: PNA XNA, 2010, 1, 68 CrossRef PubMed.
- J. Vergnaud, P.-A. Faugeras, V. Chaleix, Y. Champavier and R. Zerrouki, Tetrahedron Lett., 2011, 52, 6185 CrossRef CAS.
- P. Cheshev, A. Marra and A. Dondoni, Org. Biomol. Chem., 2006, 4, 3225 RSC.
- M. L. Conte, A. Chambery, A. Marra and A. Dondoni, Synlett, 2009, 2679 Search PubMed.
- M. L. Conte, A. Marra, A. Chambery, S. S. Gurcha, G. S. Besra and A. Dondoni, J. Org. Chem., 2010, 75, 6326 CrossRef PubMed.
- D.-C. Xiong, Y. Zhou, Y. Cui and X.-S. Ye, Tetrahedron, 2014, 70, 9405 CrossRef CAS.
- M. Vangala and S. Hotha, J. Carbohydr. Chem., 2018, 37, 393 CrossRef CAS.
- G. Lu, S. Lam and K. Burgess, Chem. Commun., 2006, 1652 RSC.
- O. D. Montagnat, G. Lessene and A. B. Hughes, Tetrahedron Lett., 2006, 47, 6971 CrossRef CAS.
- O. D. Montagnat, G. Lessene and A. B. Hughes, J. Org. Chem., 2010, 75, 390 CrossRef CAS PubMed.
- A. E. Fernandes, O. Riant, K. F. Jensen and A. M. Jonas, Angew. Chem., Int. Ed., 2016, 55, 11044 CrossRef CAS PubMed.
- P. Chandra, A. M. Jonas and A. E. Fernandes, J. Am. Chem. Soc., 2018, 140, 5179 CrossRef CAS PubMed.
- (a) H. Liu, T. Torring, M. Dong, C. B. Rosen, F. Besenbacher and K. V. Gothelf, J. Am. Chem. Soc., 2010, 132, 18054 CrossRef CAS PubMed; (b) X. Peng, H. Li and M. Seidman, Eur. J. Org. Chem., 2010, 4194 CrossRef CAS PubMed; (c) J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282 CrossRef CAS PubMed.
- (a) M. Ciaccia, D. Núñez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2019, 141, 10862 CrossRef CAS PubMed; (b) D. Núñez-Villanueva, M. Ciaccia, G. Ladevaia, E. Sanna and C. A. Hunter, Chem. Sci., 2019, 10, 5258 RSC.
- S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195 CrossRef CAS PubMed.
- T. T. Trinh, L. Oswald, D. Chan-Seng, L. Charles and J.-F. Lutz, Chem. – Eur. J., 2015, 21, 11961 CrossRef CAS PubMed.
- C. J. Yang, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2018, 57, 16194 CrossRef CAS PubMed.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430 CrossRef CAS PubMed; (b) J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466 CrossRef CAS PubMed.
- B. Zhao, Z. Gao, Y. Zheng and C. Gao, J. Am. Chem. Soc., 2019, 141, 4541 CrossRef CAS PubMed.
- O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc., Chem. Commun., 1982, 1165 RSC.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654 CrossRef CAS PubMed.
- F. A. Leibfarth, J. A. Johnson and T. F. Jamison, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10617 CrossRef CAS PubMed.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810 CrossRef CAS PubMed.
- (a) Y. Jiang, M. R. Golder, H. V.-T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369 CrossRef CAS PubMed; (b) M. R. Golder, Y. Jiang, P. E. Teichen, H. V.-T. Nguyen, W. Wang, N. Milos, S. A. Freedman, A. P. Willard and J. A. Johnson, J. Am. Chem. Soc., 2018, 140, 1596 CrossRef CAS PubMed.
- J. Li, M. Leclercq, M. Fossepré, M. Surin, K. Glinel, A. M. Jonas and A. E. Fernandes, Polym. Chem., 2020, 11, 4040 RSC.
- F. Amir, Z. Jia and M. J. Monteiro, J. Am. Chem. Soc., 2016, 138, 16600 CrossRef CAS PubMed.
- Z. Li, X. Ren, P. Sun, H. Ding, S. Li, Y. Zhao and K. Zhang, ACS Macro Lett., 2021, 10, 223–230 CrossRef CAS.
- S. Ding, G. Jia and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 1877 CrossRef CAS PubMed.
- C. Ju, C. Meng, J. Ma, X. Zhang and S. Ding, Chem. Commun., 2020, 56, 3955 RSC.
| This journal is © The Royal Society of Chemistry 2021 |