
Forced gradient copolymerisation: a simplified approach for polymerisation-induced self-assembly†
Sihao
Xu
,
Nathaniel
Corrigan
and
Cyrille
Boyer
 *
*
Centre for Advanced Macromolecular Design and Australian Centre for NanoMedicine, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: cboyer@unsw.edu.au
First published on 20th July 2020
Abstract
In this work, a novel and versatile gradient copolymerisation approach to simplify polymeric nanoparticle synthesis through polymerisation-induced self-assembly (PISA) is reported. In contrast with the commonly performed two-step PISA process, which involves chain-extension of a pre-synthesised stabiliser (or solvophilic block), this work demonstrates a one pot PISA approach via the formation of gradient copolymers through a gradual injection of the core-forming monomer in the presence of a solvophilic monomer. To demonstrate this concept, two model PISA systems were tested using a methacrylate monomer pair and an acrylamide pair. PISA using dimethylacrylamide (DMA) and diacetone acrylamide (DAAm) was first established to form a range of nanoparticle morphologies (spheres and worms), and importantly, a pure worm phase was observed without the addition of a co-solvent or a second solvophilic monomer during the polymerisation of the core-forming monomer. To demonstrate the gradient approach can be applied to other PISA monomer pairs, this methodology was applied to a system using oligo(ethylene glycol)methyl ether methacrylate (OEGMA300) and 2-hydroxypropyl methacrylate (HPMA) as monomers. PISA of this monomer pair resulted in the formation of nanoparticles with various morphologies, including spheres, worms and vesicles. More interestingly, the nanoparticles formed using these gradient copolymers presented thermoresponsive behaviour, exhibiting a sphere-to-worm transition with an increase in temperature from 25 °C to 40 °C. Thus, this facile gradient copolymerisation approach was shown to simplify the PISA process into a single step approach with easily tuneable solvophilic block length and copolymer composition, and additionally provide nanoparticle structures that afford unique properties.
Introduction
Polymeric nanoparticles, including spherical micelles, worm-like micelles and vesicles, among others, have attracted significant interest over the last few decades due to their wide range of applications in drug delivery, catalysis, cosmetics, etc.1–8 Polymeric micelles are formed by the assembly of block copolymers, which are made of polymer chains with distinct segments of differing solubility.9 Due to their numerous applications, a range of approaches have been developed for the synthesis of polymeric nanoparticles with diverse morphologies, including traditional self-assembly of pre-synthesised block copolymers via solvent manipulations,7,10–15 post-polymerisation modification,16–19 polymerisation-induced self-assembly (PISA),20–35 and polymerization-induced electrostatic self-assembly (PIESA).31,36–38 Recently, PISA has emerged as a more economic and ecofriendly method allowing the rapid in situ production of polymeric nanoparticles.23–26,31,39–58 In comparison with traditional self-assembly by solvent manipulation, PISA generally involves a two-step process. A solvophohilic polymer is first synthesised via controlled/living polymerisation and purified, and subsequently chain-extended in the presence of solvophobic monomers, resulting in the in situ formation of nanoparticles. The composition of the block copolymer, solid content, and chain length of each block affect the size and morphology of the resulting nanoparticles.20,59–63 Using the PISA process, spherical and worm-like micelles, vesicles, and interestingly, rare and complex inverse mesophasic morphologies,105 have all been observed. With the purpose of developing more versatile nanoparticles, stimuli-responsive nanoparticles prepared by PISA have also been extensively studied. Using temperature,64–69 pH70,71 or light49,72 as external stimuli, the physical and chemical properties of the nanoparticles can be manipulated, resulting in morphological changes or dissociation of the nanoparticles.In addition to block copolymers, other classes of copolymers can be used for self-assembly based on solubility differences between distinct segments in the polymer chain. Notably, gradient copolymers are well-suited in this regard, as they show a gradual change in composition throughout the polymer chain. Gradient copolymers can be considered inexpensive alternatives to block copolymers as their syntheses can be realised in a one pot operation without the need of intermediate purification. Gradient copolymers can be synthesised using monomer pairs with different reactivity ratios, which results in a composition drift along a chain upon polymerisation.73–75 Alternatively, for monomers with similar or otherwise unsuitable reactivity ratios, a “forced” gradient strategy can be employed, where a monomer is slowly fed to the polymerisation mixture, changing the instantaneous monomer composition.76 Interestingly, block-like copolymers can also be synthesised through the “starve-feed” method by feeding secondary monomer into the initial polymerization mixture when the conversion of the first monomer is high. However, in comparison to such copolymers, true gradient copolymers can often exhibit more interesting properties. For instance, using the forced gradient approach, the structure of the gradient copolymer can be precisely manipulated according to the feeding rate to yield gradient copolymers containing more precisely tailored domains. Furthermore, with different ratios of each domain presented in the gradient copolymer, the glass transition temperature (Tg) can be designed accordingly.77 Despite their interesting properties, in contrast to the block copolymers, gradient copolymers have been rarely used and investigated in copolymer self-assembly to prepare polymeric nanoparticles. For example, Harrisson and co-workers have successfully synthesised Poly(styrene-grad-acrylic acid) amphiphilic gradient copolymers by exploiting the significantly different reactivity ratios of these monomers, i.e., 0.082 and 0.21 for rAA and rSt, respectively.78 These gradient copolymers were self-assembled to form polymeric micelles which exhibited interesting properties compared to conventional block copolymer self-assembly, such as pH responsiveness.79–87 Chen and coworkers have also reported the successful synthesis of forced-gradient copolymers via the metered addition of 2,2,2-trifluroethyl methacrylate into a mixture of acrylic acid and poly(acrylic acid), which induced self-assembly to form spherical micelles with good pH stability.88
With the foreseeable advantages that gradient copolymerisation can provide, it raised the question of whether such approaches can be applied for PISA. Choi and coworkers have reported the use of gradient copolymers in ring-opening metathesis polymerisation (ROMP) to prepare nanoparticles via the in situ nanoparticlisation of conjugated polymers (INCP) method.89–91 More recently, our group has applied gradient copolymers synthesised by reversible addition–fragmentation chain transfer (RAFT) polymerisation in a PISA process through the copolymerisation of disparate monomer reactivity ratios (i.e., oligo(ethylene glycol)methyl ether methacrylate (OEGMA500, 500 g mol−1) and diacetone acrylamide (DAAm)) in water. As OEGMA presents a higher reactivity than DAAm, a P(OEGMA) rich block is formed at the beginning of the polymerisation, which provides colloidal stability and allows the polymerisation of hydrophobic DAAm polymers. Spherical and cylindrical micelles were successfully obtained via this one-single step PISA approach, without the need to pre-synthesise solvophilic homopolymers.92
Inspired by these earlier works, herein, we report a new method of applying gradient copolymerisation to the PISA process using monomers with similar reactivity ratios. To illustrate the versatility of this approach, two common PISA systems were investigated using this single-step process. The core-forming monomer was gradually injected in the reaction mixture to yield polymeric nanoparticles without the need to prepare and purify an initial stabilising block (Fig. 1). Morphologies, such as spherical, worm-like micelles and vesicles were directly synthesised by this single-step approach through altering the total copolymer degree of polymerisation (DPn) and solvophilic monomer composition. Finally, P(OEGMA300-grad-HPMA) copolymers and their corresponding polymeric nanoparticles were prepared and were shown to exhibit unique thermoresponsive behaviour. Spherical micelles were formed at room temperature, which were reorganised into cylindrical micelles by increasing the temperature to 40 °C, resulting in the formation of gels. Interestingly, the analogous POEGMA300-b-PHPMA block copolymer did not exhibit such behaviour, which highlights the utility of the gradient copolymerisation approach (Fig. 1).
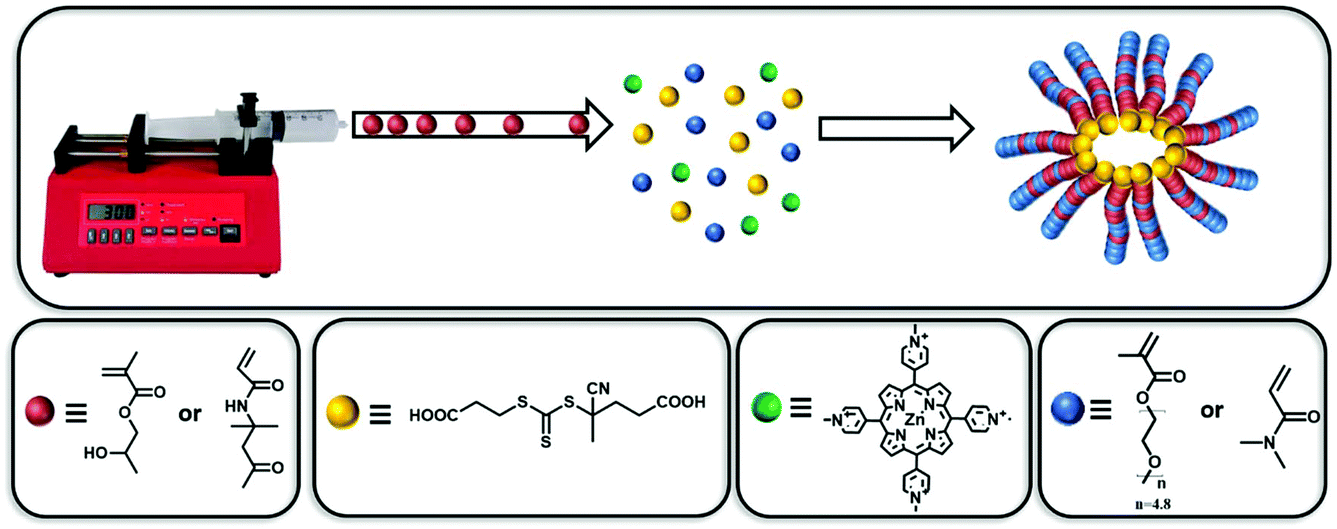 | ||
| Fig. 1 Representative scheme of the photoinduced electron/energy transfer – reversible addition–fragmentation chain transfer (PET-RAFT) forced-gradient copolymerisation in water under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using zinc meso-tetra(N-methyl-4-pyridyl) porphine tetrachloride (ZnTMPyP) as photocatalyst. | ||
Results and discussion
Preliminary kinetics of PET-RAFT dispersion forced gradient copolymerisation
In our previous study,92 we demonstrated that nanoparticles can be formed through a simple one-pot approach by selecting a pair of monomers which have vastly different reactivity ratios to form gradient copolymers. During that process, the solvophilic monomer was rapidly polymerised due to its reactivity ratio with the solvophobic monomer, while the solvophobic monomer was incorporated to a large extent later in the reaction. As the solvophilic monomer was polymerised to a larger extent in the early stages of the copolymerisation, the resulting solvophilic polymer provides adequate colloidal stability to the nanoparticles which prevented macroscopic phase separation. However, this approach is limited to the selection of the monomers with favourable reactivity ratios. To overcome this disadvantage, we hypothesised that the same effect could be achieved by the gradual addition of solvophobic monomer during a PISA process, resulting in gradient copolymers via a starve-feed (forced gradient) approach. To test this hypothesis, dimethylacrylamide (DMA) and diacetone acrylamide (DAAm) were selected as monomers for the solvophilic block and core-forming block, respectively. A water soluble RAFT agent 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CTCPA) and a water soluble photocatalyst zinc meso-tetra(N-methyl-4-pyridyl) porphine tetrachloride (ZnTMPyP) were selected to allow the polymerisation to be performed in water. ZnTMPyP is an efficient photocatalyst capable of activating photoinduced electron/energy transfer – reversible addition–fragmentation chain transfer (PET-RAFT) polymerisation (Fig. 2A).50 Additionally, CTCPA was selected as the RAFT agent due to its capability to control the polymerisation of both acrylate and acrylamide monomers. As preliminary tests, kinetics studies were carried out to optimise the formation of the gradient copolymer by using a ratio of [DMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DAAm]:[CTCPA]:[ZnTMPyP] = 30:270:1:0.02 (10 mol% DMA) at a total solid content of 20 wt% under orange light irradiation. To synthesise gradient copolymers, DAAm was slowly added into the reaction mixture by a syringe pump (Fig. S1†) following a specific injection addition protocol (Fig. 2B). The total injection time was optimised to minimise precipitation and was set to 80 minutes.
:[DAAm]:[CTCPA]:[ZnTMPyP] = 30:270:1:0.02 (10 mol% DMA) at a total solid content of 20 wt% under orange light irradiation. To synthesise gradient copolymers, DAAm was slowly added into the reaction mixture by a syringe pump (Fig. S1†) following a specific injection addition protocol (Fig. 2B). The total injection time was optimised to minimise precipitation and was set to 80 minutes.
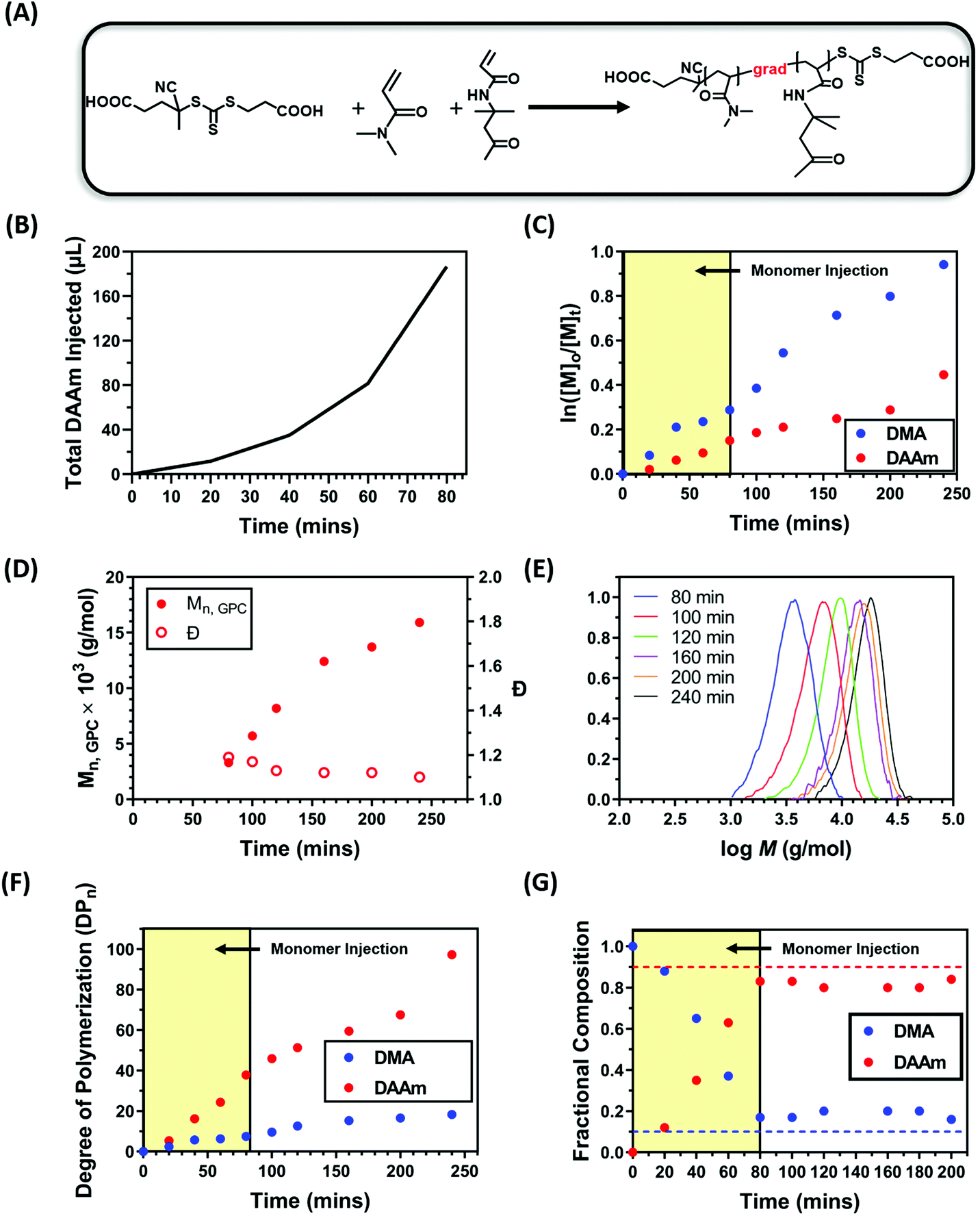 | ||
| Fig. 2 (A) Representative scheme of the PET-RAFT gradient copolymerisation of DMA and DAAm in water. (B) Injection profile showing the total DAAm injected vs addition time. (C) Aqueous PET-RAFT forced gradient copolymerisation kinetic plots of DMA and DAAm. (D) Evolution of GPC-derived molecular weight and dispersity with time and (E) corresponding molecular weight distributions. (F) Evolution of degree of polymerisation (DPn) versus time and (G) variation in monomer composition with reaction time. Note: polymerisations were conducted using ZnTMPyP as photocatalyst under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) at a total solids content of 20 wt% ([DMA]:[DAAm]:[CTCPA]:[ZnTMPyP] = 30:270:1:0.02). | ||
Kinetic studies revealed that ∼50% of the DMA was polymerised within 120 minutes while only ∼25% of the DAAm was incorporated (Fig. 2C and F). This suggests the successful formation of a block-like gradient copolymer through the starve fed monomer approach. The linear evolution of ln([M]0/[M]t) with time, as well as the unimodal and symmetric molecular weight distributions and low dispersities (Đ ∼ 1.10) obtained by gel permeation chromatography (GPC) indicated a well-controlled RAFT polymerisation process (Fig. 2C–E). Additionally, the monomer composition within the copolymer backbone at different time points was calculated using NMR. As expected, DMA was incorporated to a larger extent during the initial stages of the copolymerisation, resulting in a copolymer that was initially rich in DMA (Fig. 2G). The slow injection of DAAm gradually changed the copolymer composition by increasing the content of DAAm. After 80 min of polymerisation, the monomer composition in the copolymer remained relatively constant at 18:82 DMA:DAAm.
Gradient copolymerisation through PET-RAFT dispersion polymerisation with DMA/DAAm
Encouraged with the initial kinetic results, an investigation of the PET-RAFT PISA process was investigated with the DMA and DAAm monomer pair using the forced gradient approach. Firstly, the effect of varying the total targeted DPn of DMA and DAAm, as well as the initial monomer feed ratio (DAAm:DMA) on the polymeric nanoparticle formation was investigated (Table 1). All polymerisations were carried to high monomer conversion (α > 80%) within 24 hours of irradiation, resulting in milky solutions which contained precipitates in some cases. In contrast to conventional PISA performed by chain extension of a pre-synthesised macroRAFT, in this study, the composition and the total DPn of the diblock copolymers were easily tuned by changing DMA and DAAm feed, allowing the preparation of a phase-diagram.93–96 A 2D phase diagram was plotted with the overall (solvophilic and solvophobic) DPn on the x-axis, and the total mol percentage of the solvophilic monomer on the y-axis.
| Entry | [DAAm]:[DMA] |
Overall DP | DMA composition (mol%) | Overall conversion (%) | M n, Theo (g mol−1) | M n, GPC (g mol−1) | Đ | Morphology (major/minor) |
|---|---|---|---|---|---|---|---|---|
|
a Conditions: polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using [CTCPA]:[ZnTMPyP] = 1:0.02. S = sphere, W = worm, P = precipitation. n/r = not run.
|
||||||||
| PISA-1 | 210:90 |
300 | 30 | 97 | 43400 |
31100 |
1.14 | S |
| PISA-2 | 225:75 |
300 | 25 | 87 | 39900 |
27700 |
1.12 | W/S |
| PISA-3 | 240:60 |
300 | 20 | n/r | n/r | n/r | n/r | P |
| PISA-4 | 255:45 |
300 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-5 | 270:30 |
300 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-6 | 285:15 |
300 | 5 | n/r | n/r | n/r | n/r | P |
| PISA-7 | 280:120 |
400 | 30 | 90 | 53600 |
47800 |
1.12 | W |
| PISA-8 | 300:100 |
400 | 25 | 82 | 51100 |
39900 |
1.12 | W |
| PISA-9 | 320:80 |
400 | 20 | n/r | n/r | n/r | n/r | P |
| PISA-10 | 340:60 |
400 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-11 | 360:40 |
400 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-12 | 380:20 |
400 | 5 | n/r | n/r | n/r | n/r | P |
| PISA-13 | 350:150 |
500 | 30 | 85 | 63200 |
50300 |
1.19 | W/S |
| PISA-14 | 375:125 |
500 | 25 | 81 | 61700 |
48200 |
1.14 | S |
| PISA-15 | 400:100 |
500 | 20 | n/r | n/r | n/r | n/r | P |
| PISA-16 | 425:75 |
500 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-17 | 450:50 |
500 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-18 | 475:25 |
500 | 5 | n/r | n/r | n/r | n/r | P |
Interestingly, different morphological domains were observed, including pure worm-like micelles and spherical micelles for some compositions, however, macroscopic precipitation was noted when the DMA composition was less than 20 mol% (Fig. 3). In these cases, the precipitation was attributed to the inadequate length of the solvophilic block, which was unable to stabilise the solvophobic core. It is interesting to note that a pure worm phase was only observed when the total DPn was 400. More surprisingly, higher DPn resulted in spherical and worm like mixtures. We hypothesise that at higher total DPn, the longer solvophilic block limited the morphological transition of the nanoparticles and therefore trapped the nanoparticle transition at the spherical shape. However, to our surprise, a pure worm phase is uncommon in DAAm PISA systems (Fig. 3).97,98 Due to the high Tg of PDAAm, nanoparticles which contain DAAm as the core-forming block typically display a rigid nanoparticle core, which kinetically traps the nanoparticle morphology in a spherical shape. Sumerlin and coworkers proposed the idea of copolymerising a solvophilic monomer such as DMA during the core-forming process in order to reduce the core rigidity of the nanoparticles.99 Alternatively, the same effect can be achieved through the addition of a co-solvent such as DMF which improves the mobility of the nanoparticle core. In our approach, the solvophilic monomer DMA is also present in the nanoparticle core during the core-forming stage which results in higher chain mobility. This finding is also consistent with our previous study on one-pot single-step gradient copolymerisation of DAAm and OEGMA500 with varying reactivity ratios, where wider worm phases were also present without the addition of a secondary solvophilic monomer.92 The copolymerisation of DMA and DAAm resulted in narrow molecular weight distribution (Đ < 1.2) and good control of molecular weights (Fig. 4).
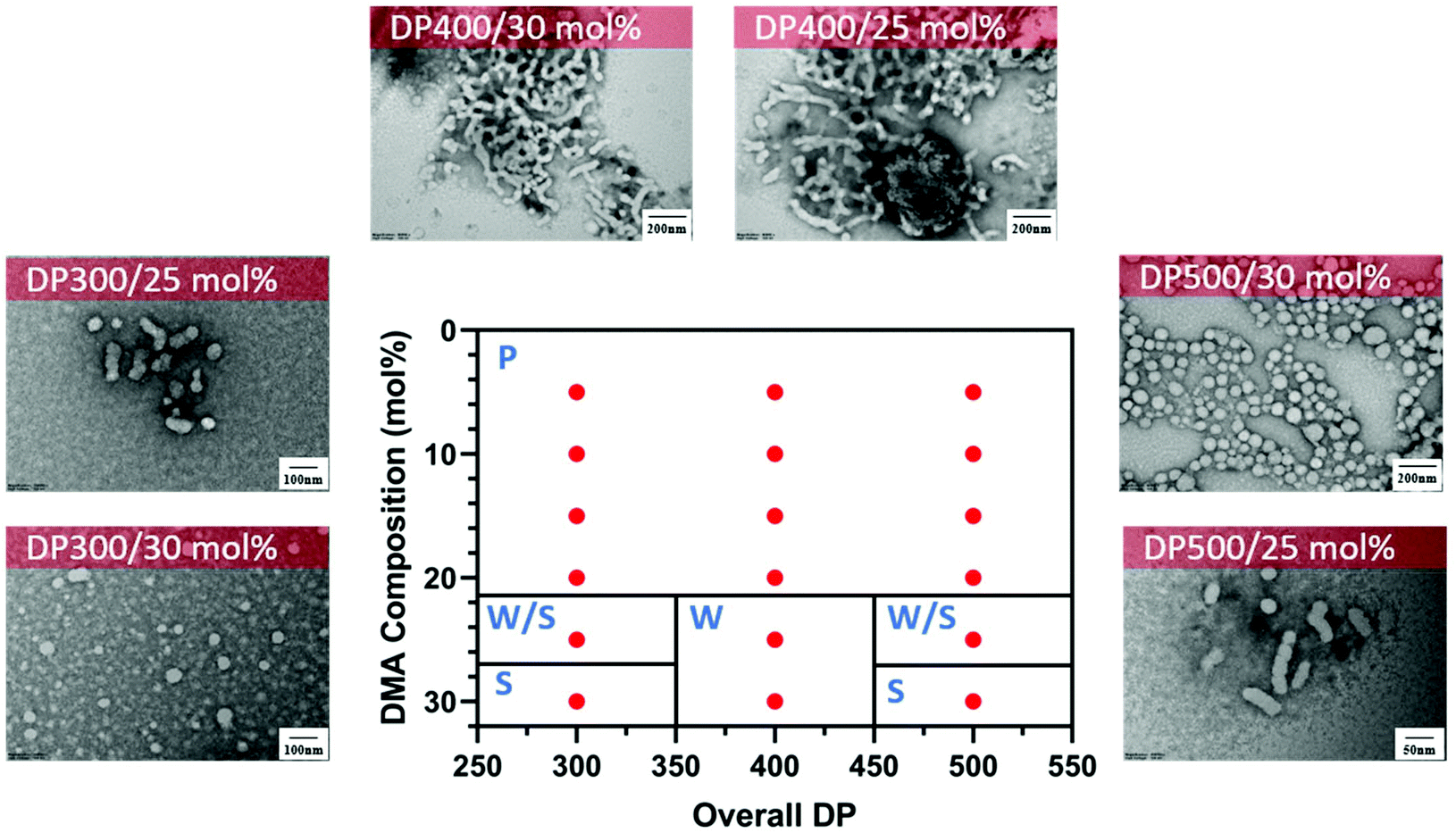 | ||
| Fig. 3 Representative TEM micrographs and corresponding phase diagram for a series of nanoparticles synthesised using PET-RAFT forced-gradient dispersion polymerisation of DMA and DAAm with varying monomer ratios and target DPn at a total solids content of 20 wt%. Polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using [CTCPA]:[ZnTMPyP] = 1:0.02. S = Sphere, W = Worm, P = Precipitation. See ESI, Table S1† for additional experimental and characterisation data. | ||
 | ||
| Fig. 4 Molecular weight distributions from the PET-RAFT forced-gradient dispersion polymerisation of DMA and DAAm with varying monomer ratios and target DPn at a total solids content of 20 wt%. Polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using [CTCPA]:[ZnTMPyP] = 1:0.02. | ||
In addition, we performed a morphological study at lower solids content to determine the effect on the formation of the nanoparticles and the presence of macroscopic precipitation (Table S1†). As expected, at lower solids content (15 wt%), macroscopic precipitation only occurred when the DMA composition was lower than 20 mol%. Furthermore, the pure worm phase was observed at lower DMA composition with increasing total DPn (DPn = 500) (Fig. 5). Despite the change in morphological transitions, a lower solids content had no noticeable influence on the control of the polymerisation as demonstrated by narrow molecular weight distributions (Đ < 1.3) and good control of the molecular weights (Fig. S3†).
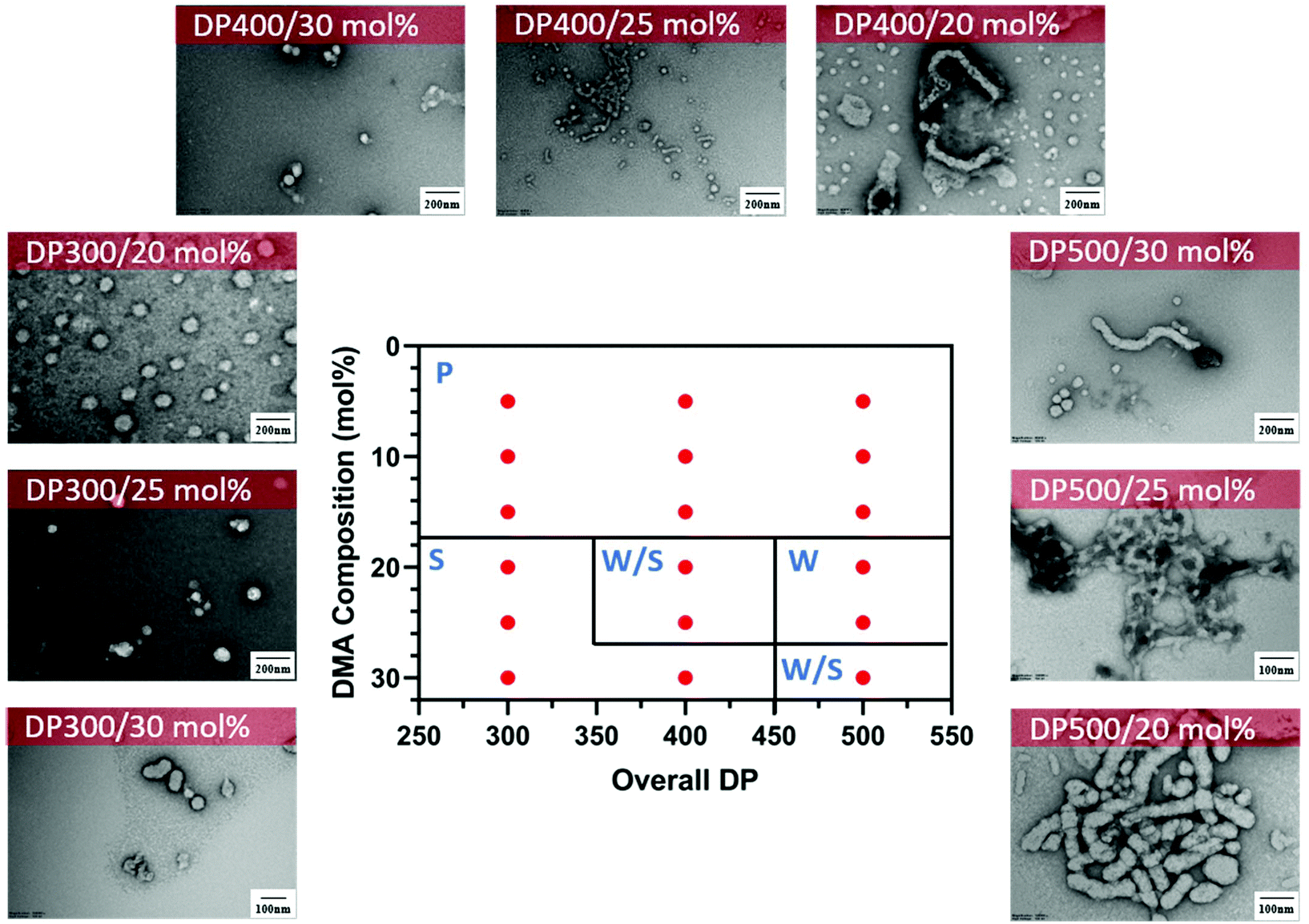 | ||
| Fig. 5 Representative TEM micrographs and corresponding phase diagram for a series of nanoparticles synthesised using PET-RAFT gradient dispersion polymerisation of DMA and DAAm with varying monomer ratios and target DPns at a total solids content of 15 wt%. Polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using [CTCPA]:[ZnTMPyP] = 1:0.02. S = Sphere, W = Worm, J = Jellyfish, P = Precipitation. See ESI, Table S1† for additional experimental and characterisation data. | ||
Kinetics of PET-RAFT dispersion gradient copolymerisation with OEGMA300/HPMA
With the initial success of the gradient copolymerisation of DMA/DAAm systems for PISA, we decided to test our methodology with another monomer system to show the versatility of this approach. In this case, oligo(ethylene glycol)methyl ether methacrylate (OEGMA300, 300 g mol−1) and 2-hydroxypropyl methacrylate (HPMA) were selected as the solvophilic and solvophobic monomer, respectively. HPMA is commonly used in PISA to afford nanoparticles through RAFT dispersion polymerisation.100–103 Polymerisations were conducted using a ratio of [OEGMA300]:[HPMA]:[CTCPA]:[ZnTMPyP] = 80:320:1:0.02 (20 mol% OEGMA300) at a total solid content of 20 wt%. Due to the faster polymerisation rate of OEGMA300 and HPMA compared with the DMA and DAAm system, the injection rate of HPMA was increased accordingly, and the total injection time was set to 28 minutes (Fig. 6B). Fig. 6E shows the evolution of monomer composition, which again supported the formation of a polymer rich in OEGMA300 at the beginning of the polymerisation; whereas 60% of the OEGMA300 was polymerised within the first 40 minutes of the polymerisation, only 40% of the HPMA was consumed (Fig. 6C and D). This confirmed the successful formation of gradient copolymers through the starve feed injection approach.
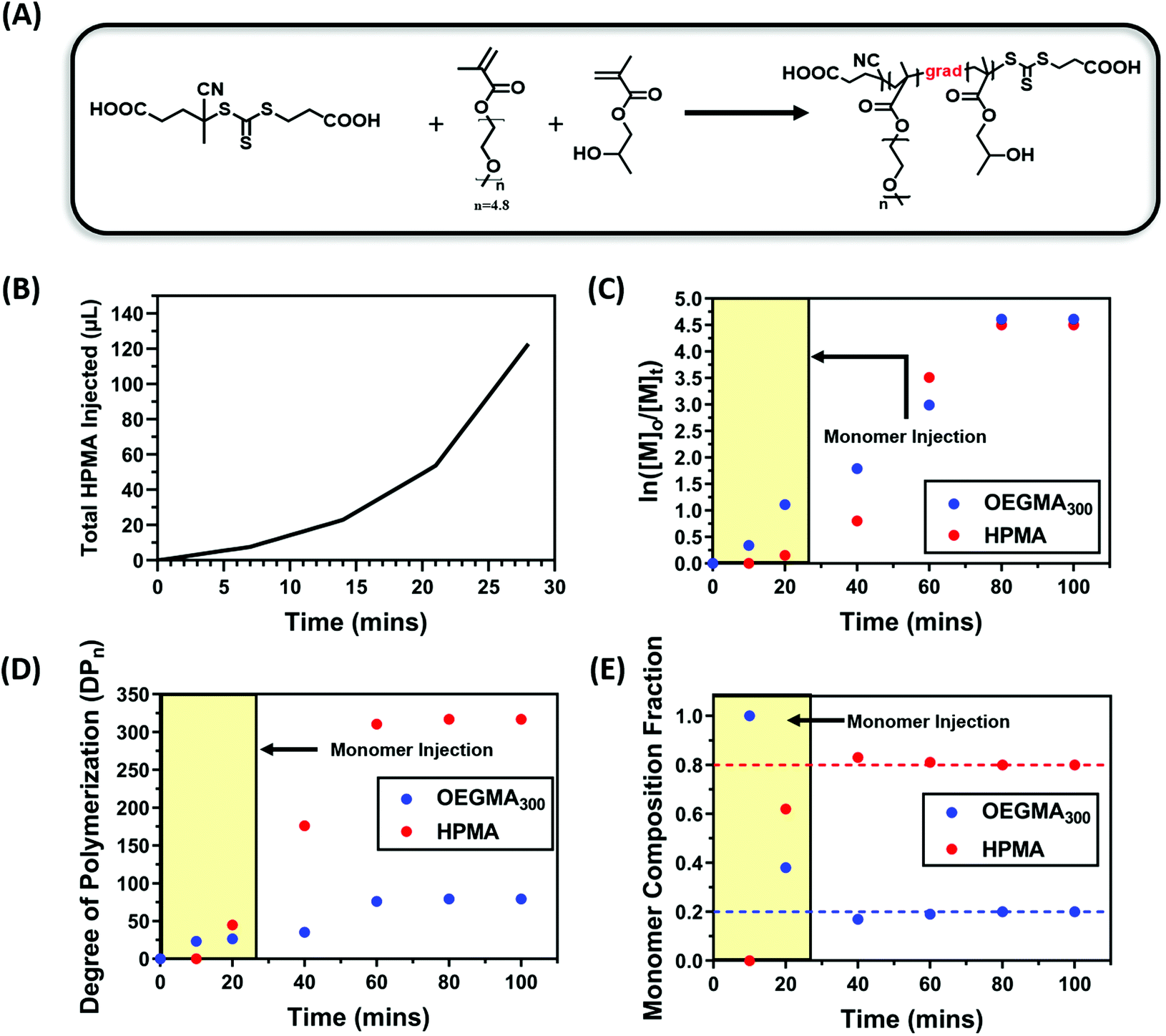 | ||
| Fig. 6 (A) Scheme of the PET-RAFT gradient copolymerisation of OEGMA300 and HPMA in water under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using ZnTMPyP as photocatalyst. (B) Injection profile showing the total HPMA injected vs addition time (C) Aqueous PET-RAFT gradient copolymerisation kinetics of OEGMA300 and HPMA using a [OEGMA300]:[HPMA]:[CTCPA]:[ZnTMPyP] = 80:320:1:0.02. (D) Evolution of OEGMA300 and HPMA DPn with time. (E) Evolution of polymer composition with reaction time. Polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) at a total solids content of 20 wt%. | ||
Gradient copolymerisation through PET-RAFT dispersion polymerisation
Encouraged with the successful formation of gradient copolymers using OEGMA300 and HPMA in water, we decided to investigate the nanoparticle formation in these systems by TEM and DLS. A 2D phase diagram was constructed for a changing total DPn and OEGMA300 composition (Table 2). All polymerisations were carried to near full monomer conversion (α > 95%) within 3 hours of irradiation. The morphologies of the nanoparticles were examined via TEM for the stable samples. Fig. 7 shows the phase diagram at 20 wt% solids content prepared by varying the composition of OEGMA300 from 5 to 30 mol% and the overall DPn from 300 to 600. It is worth noting that at lower OEGMA300 composition (<20 mol%), the nanoparticles were not stable which led to the formation of precipitates. In comparison with the previous reports of stable PPEGMA-b-PHPMA nanoparticles synthesised via photo-PISA by Tan et al.104 the nanoparticles observed in our system were often less stable at high HPMA:OEGMA300 ratios. This behaviour attributed to the different solvophilicity of the gradient copolymers in our system, compared with the block copolymers in the earlier work. The inclusion of small amount of solvophobic monomers in the solvophilic polymeric stabiliser reduced the overall solvophilicity in our system, and thus slightly reduced the ability to stabilise the resulting polymeric nanoparticles. Similarly, at high overall DPn, a higher OEGMA300 content was required in order to stabilise the nanoparticles. Regardless, diverse and stable nanoparticles were still obtained in the current system when using higher ratios of solvophilic:solvophobic monomers. This behaviour highlights the property changes that can occur through the use of gradient copolymers.
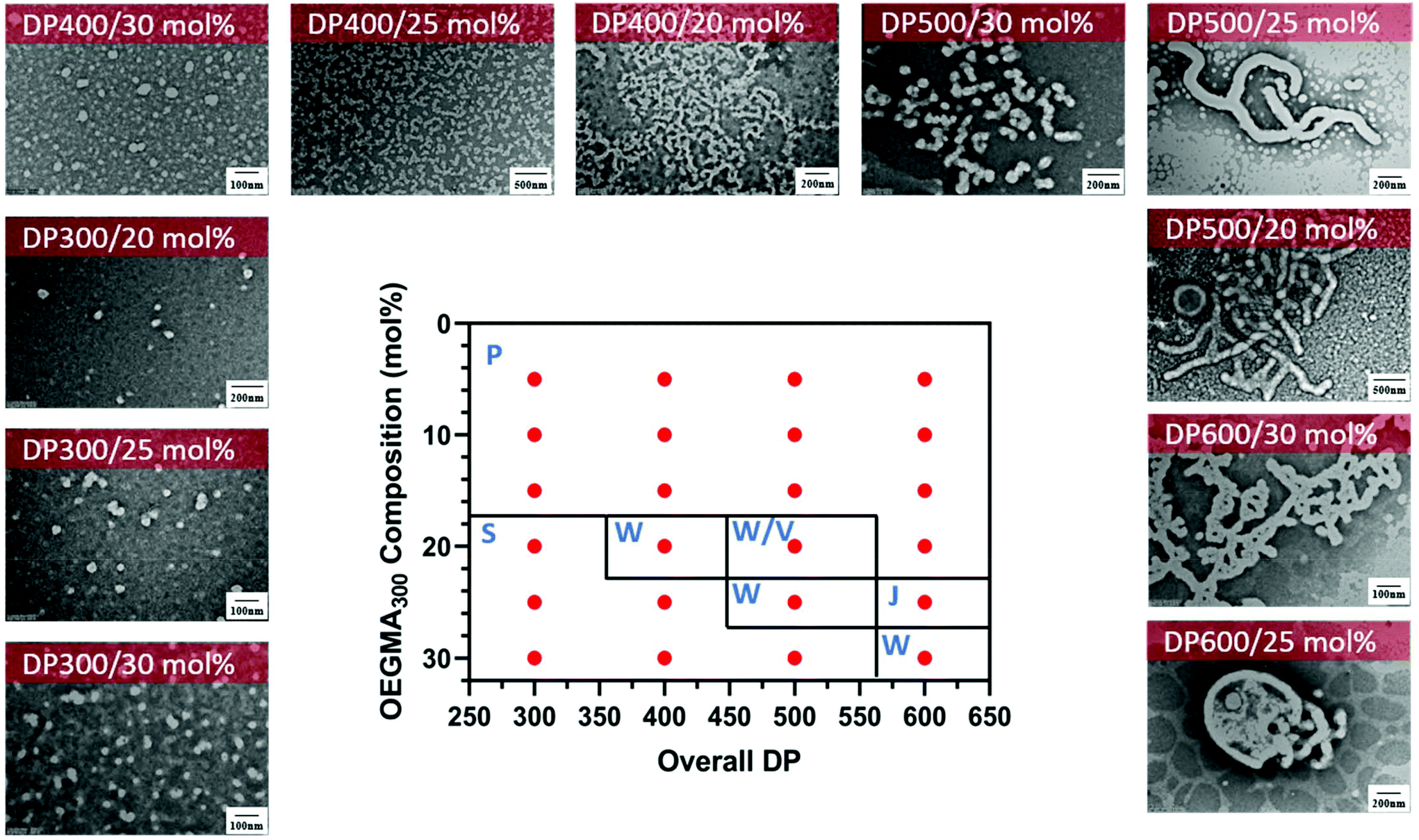 | ||
| Fig. 7 Representative TEM micrographs and the corresponding phase diagram for a series of nanoparticles synthesised using PET-RAFT gradient dispersion polymerisation of OEGMA300 and HPMA with varying monomer ratios and target DPns at a total solids content of 20 wt%. Polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using [CTCPA]:[ZnTMPyP] = 1:0.02. S = sphere, W = worm, J = jellyfish, P = precipitation. | ||
| Entry | [HPMA]:[OEGMA300] |
Overall DP | OEGMA300 composition (mol%) | Overall conversion (%) | M n, Theo (g mol−1) | M n, GPC (g mol−1) | Đ | Morphology (major/minor) |
|---|---|---|---|---|---|---|---|---|
|
a Note: Note: Conditions: polymerisations were conducted under orange light irradiation (λmax = 595 nm, 10.2 mW cm−2) using [CTCPA]:[ZnTMPyP] = 1:0.02. S = sphere, W = worm, J = jellyfish, P = precipitation, V = vesicles. n/r = not run.
|
||||||||
| PISA-37 | 210:90 |
300 | 30 | 95 | 54700 |
51600 |
1.66 | S |
| PISA-38 | 225:75 |
300 | 25 | 98 | 54100 |
50400 |
1.66 | S |
| PISA-39 | 240:60 |
300 | 20 | 93 | 49197 |
49600 |
1.41 | S |
| PISA-40 | 255:45 |
300 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-41 | 270:30 |
300 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-42 | 285:15 |
300 | 5 | n/r | n/r | n/r | n/r | P |
| PISA-43 | 280:120 |
400 | 30 | 98 | 75100 |
73000 |
1.85 | S |
| PISA-44 | 300:100 |
400 | 25 | 96 | 70600 |
63800 |
1.77 | S |
| PISA-45 | 320:80 |
400 | 20 | 99 | 70400 |
71100 |
2.02 | W |
| PISA-46 | 340:60 |
400 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-47 | 360:40 |
400 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-48 | 380:20 |
400 | 5 | n/r | n/r | n/r | n/r | P |
| PISA-49 | 350:150 |
500 | 30 | 99 | 95600 |
124400 |
1.77 | S |
| PISA-50 | 375:125 |
500 | 25 | 99 | 91900 |
139200 |
1.94 | W |
| PISA-51 | 400:100 |
500 | 20 | 99 | 88000 |
97900 |
1.75 | W/V |
| PISA-52 | 425:75 |
500 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-53 | 450:50 |
500 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-54 | 475:25 |
500 | 5 | n/r | n/r | n/r | n/r | P |
| PISA-55 | 420:180 |
600 | 30 | 90 | 94900 |
83700 |
1.64 | W |
| PISA-56 | 450:150 |
600 | 25 | 98 | 108000 |
128600 |
1.64 | J |
| PISA-57 | 480:120 |
600 | 20 | n/r | n/r | n/r | n/r | P |
| PISA-58 | 510:90 |
600 | 15 | n/r | n/r | n/r | n/r | P |
| PISA-59 | 540:60 |
600 | 10 | n/r | n/r | n/r | n/r | P |
| PISA-60 | 570:30 |
600 | 5 | n/r | n/r | n/r | n/r | P |
Despite some macroscopic precipitation, the TEM micrographs show successful formation of higher order morphologies. Worm-like micelles, vesicles and jellyfish-like micelles were observed through various total DPn and OEGMA300 composition, however, pure vesicles were not observed. To compare nanoparticle morphologies formed via the gradient approach and a conventional block copolymer PISA system (i.e., POEGMA300-b-PHPMA), a conventional two-step PISA process was performed. For this comparison, POEGMA300 was pre-synthesised by thermally initiated RAFT polymerisation with CTCPA and purified, followed by chain extension with HPMA in water. Unexpectedly, only a mixture of spherical micelles and short worm-like micelles were observed with different DPn (Fig. S7†). This may be due to the lengthy stabiliser, which increases the solvophilicity and therefore kinetically traps the micelles, hence preventing the evolution to higher order morphologies.59 We then moved on to compare the gradient PISA copolymerisation with a well-studied RAFT dispersion system; in this system, the macroRAFT PEG113-CDTPA was synthesised through esterification of PEG113 with CDTPA, followed by chain extension with HPMA. The phase diagram revealed the successful formation of pure vesicles at higher solids contents (>10 wt%) and DPn > 500 (ESI, Fig. S8†). Our hypothesis of the absence of pure vesicles in our gradient system was attributed to the copolymerisation of the OEGMA300 during the core-forming phase which affected the assembly of these gradient copolymers. The solvophilic monomers being incorporated in the core of the nanoparticles allows the solvent to penetrate deeper into the nanoparticles therefore resulting in an inadequate packing parameter to allow vesicle formation.24,106 Somewhat surprisingly, the gradient copolymerisation system showed a relatively broad molecular weight distribution (Đ = 1.66–1.94) (Fig. S5†). We attributed this relatively high Đ to the presence of dimethacrylate in HPMA which causes the broad molecular weight distribution; this was supported by the presence of RAFT agent at high molecular weights, as evidenced by UV-GPC traces (Fig. S6†).106–108 This is also evident in similar conventional two-step PISA systems where block copolymers have shown extremely broad molecular weight distributions (Đ > 5).
Thermoresponsive behaviour of forced-gradient P(OEGMA300-grad-HPMA) nanoparticles
It is known that in gradient copolymer nanoparticles, the interfacial area per chain is larger due to the wide distribution of the solvophobic monomer within the polymer chain. This causes the interfacial polymer chains to form loops which increases the maximum solvophilic group concentration and reduces the concentration of solvophobic groups in the core of the nanoparticles. Once the solvent condition changes, more solvophobic chains would be brought into the core of the nanoparticles thus affecting the nanoparticle transition. This phenomenon is known as the “reel in” effect. By contrast, the same phenomenon can also be observed in more block like gradient copolymers, however, as less solvophobic groups are capable of being brought into the core of the nanoparticles, the “reel-in” effect is less significant. Hence, in some gradient copolymer nanoparticles, stimuli-responsiveness can be observed. However, unlike thermoresponsive block copolymer-based nanoparticles, in which the morphological transition tends to occur at a specific point, for gradient copolymers the diffusion of the solvophobic chain occurs through a wider temperature range.73,109,110With the support of the theory, we sought to test the thermoresponsive behaviour of the P(OEGMA300-grad-HPMA) gradient copolymer nanoparticles. During our research, we visually observed that nanoparticles synthesised through the gradient PISA approach formed a free-flowing liquid at room temperature (25 °C), and a worm-like copolymer gel at higher temperatures (40 °C). This process was also reversible and therefore gel samples were easily converted back to free-flowing liquids by cooling the sample down. To demonstrate this thermoresponsive mechanism, DLS studies were carried out on a worm sample (PISA-15) at low concentration (0.01 wt%). It is interesting to note that according to the DLS data, the nanoparticles have an average diameter of 50 nm at 25 °C which is consistent with a spherical micelle morphology. Comparatively, at 40 °C, the average sphere-equivalent diameter was observed at around 150 nm which is significantly larger than the size at 25 °C. A nanoparticle average size of 150 nm is often associated to a worm-like micelle in PISA systems.111 To further confirm the morphological transition, TEM micrographs were carried out at different temperatures, which show the formation of spherical micelles at 25 °C and worm-like micelles at 40 °C (Fig. 8A and B). Such unique temperature responsive behaviour is attributed to the gradient structure of the nanoparticles.
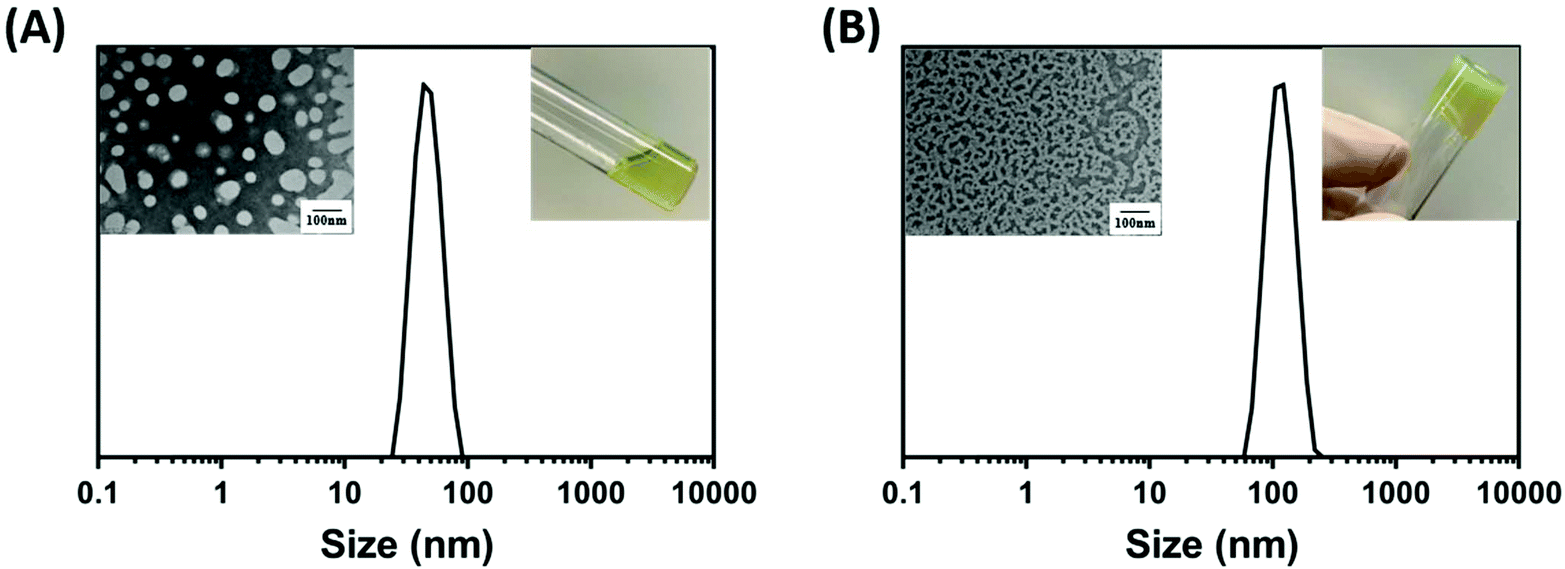 | ||
| Fig. 8 (A) Intensity-based size distributions determined by DLS, TEM micrographs and digital photograph of gradient self-assembled POEGMA300-grad-PHPMA nanoparticles (PISA-51) at 25 °C and (B) P(OEGMA300-grad-HPMA) nanoparticles (PISA-51) at 40 °C. | ||
To test that the gradient architecture of P(OEGMA300-grad-HPMA) confers this property, we decided to prepare POEGMA-b-PHPMA nanoparticles through a conventional two-step PISA. Interestingly, the nanoparticles that were prepared by two-step PISA which had similar DPn and overall solvophobic/solvophilic composition did not show any visual change with increasing temperature (ESI, Fig. S9†). These results further support the hypothesis that the block copolymers have a fixed interfacial area, and the solvophobic chains are incapable of diffusion into the nanoparticle core. Therefore, with the change in solvent condition that occurs on increasing temperature from 25 to 40 °C, there is no morphological transition. This is also confirmed in the DLS and TEM studies where the average sizes remain the same at 25 °C and at 40 °C, while TEM micrographs displayed no apparent change in morphology. These combined data support that the successful synthesis of the thermoresponsive nanoparticles due to the gradient architecture of the copolymer.
Conclusion
In conclusion, we have developed a versatile one-pot, single step PISA approach using a gradual injection of core-forming monomer, resulting in the formation of gradient copolymers. One benefit of this approach is the suppression of the synthesis of the stabilising block; the composition of the copolymer can be easily tailored through the injection of the solvophobic monomer which can award unique properties. This approach was implemented for aqueous PISA of DMA and DAAm, where successful nanoparticle formation was observed. Furthermore, worm phases were obtained without the addition of a co-solvent or any secondary solvophilic monomers. Hence, the gradient approach can not only be carried out using different conventional PISA monomers, but can also aid in morphological transitions of core-forming monomers with glassy nature. Aqueous PISA dispersion polymerisation of OEGMA300 and HPMA was also successfully achieved and a variety of morphologies such as spherical, worm-like, vesicle and jellyfish micelles were formed, which was supported by the construction of the phase diagram. Due to the gradient architecture of the copolymers, we designed a thermoresponsive P(OEGMA300-grad-HPMA) nanoparticle where a reversible sphere-to-worm morphological transition occurs with a modest temperature increase from 25 °C to 40 °C. Although block copolymers can also be synthesised through the starve-feed approach, gradient copolymers were chosen due to their advantages. In the DMA/DAAm system, worm-like morphologies were formed due to the gradient nature of the copolymer which allows the presence of DMA in the core of the nanoparticles. Furthermore, thermoresponsive behaviour was observed in OEGMA300/PHPMA system owing to the gradient architecture of the copolymer. Thus, the gradient PISA approach appears to be compatible to a range of monomers and significantly simplifies the PISA process with tuneable solvophilic block length, high efficiency and reproducibility. This system should allow the PISA process to be applied to commercial settings for large-scale manufacturing of polymeric nanoparticles with unique properties.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
CB acknowledges the Australian Research Council (ARC) for his Future Fellowship (FT12010096).References
- F. Masood, Mater. Sci. Eng., C, 2016, 60, 569–578 CrossRef CAS.
- G. Moku, B. Layek, L. Trautman, S. Putnam, J. Panyam and S. J. C. Prabha, Cancers, 2019, 11, 491 CrossRef CAS PubMed.
- K. M. El-Say and H. S. El-Sawy, Int. J. Pharm., 2017, 528, 675–691 CrossRef CAS PubMed.
- M. Shokeen, E. D. Pressly, A. Hagooly, A. Zheleznyak, N. Ramos, A. L. Fiamengo, M. J. Welch, C. J. Hawker and C. J. Anderson, ACS Nano, 2011, 5, 738–747 CrossRef CAS PubMed.
- S. J. Lam, E. H. H. Wong, C. Boyer and G. G. Qiao, Prog. Polym. Sci., 2018, 76, 40–64 CrossRef CAS.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- J. Allouche, in Nanomaterials: A Danger or a Promise? A Chemical and Biological Perspective, ed. R. Brayner, F. Fiévet and T. Coradin, Springer London, London, 2013, pp. 27–74, DOI:10.1007/978-1-4471-4213-3_2.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 1199–1208 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- J.-F. Gohy, B. G. G. Lohmeijer, A. Alexeev, X.-S. Wang, I. Manners, M. A. Winnik and U. S. Schubert, Chem. Eur. J., 2004, 10, 4315–4323 CrossRef CAS PubMed.
- S. G. Jang, D. J. Audus, D. Klinger, D. V. Krogstad, B. J. Kim, A. Cameron, S.-W. Kim, K. T. Delaney, S.-M. Hur, K. L. Killops, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 6649–6657 CrossRef CAS PubMed.
- J.-M. Williford, J. L. Santos, R. Shyam and H.-Q. Mao, Biomater. Sci., 2015, 3, 894–907 RSC.
- Y. Yu and A. Eisenberg, J. Am. Chem. Soc., 1997, 119, 8383–8384 CrossRef CAS.
- D. Moinard-Chécot, Y. Chevalier, S. Briançon, L. Beney and H. Fessi, J. Colloid Interface Sci., 2008, 317, 458–468 CrossRef PubMed.
- E. Allémann, R. Gurny and E. Doelker, Int. J. Pharm., 1992, 87, 247–253 CrossRef.
- U. Bilati, E. Allémann and E. Doelker, Pharm. Dev. Technol., 2003, 8, 1–9 CrossRef CAS PubMed.
- M. F. Zambaux, F. Bonneaux, R. Gref, P. Maincent, E. Dellacherie, M. J. Alonso, P. Labrude and C. Vigneron, J. Controlled Release, 1998, 50, 31–40 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid. Commun., 2017, 38, 1600528 CrossRef PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- X. Wang and Z. An, Macromol. Rapid. Commun., 2019, 40, 1800325 CrossRef.
- M. A. Touve, C. A. Figg, D. B. Wright, C. Park, J. Cantlon, B. S. Sumerlin and N. C. Gianneschi, ACS Cent. Sci., 2018, 4, 543–547 CrossRef CAS PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS.
- J.-L. Six and K. Ferji, Polym. Chem., 2019, 10, 45–53 RSC.
- Q. Yu, Y. Ding, H. Cao, X. Lu and Y. Cai, ACS Macro Lett., 2015, 4, 1293–1296 CrossRef CAS.
- Y. Ma, P. Gao, Y. Ding, L. Huang, L. Wang, X. Lu and Y. Cai, Macromolecules, 2019, 52, 1033–1041 CrossRef.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS.
- J. Sarkar, A. W. Jackson, A. M. van Herk and A. Goto, Polym. Chem., 2020, 11, 3904–3912 RSC.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- L. Huang, Y. Ding, Y. Ma, L. Wang, Q. Liu, X. Lu and Y. Cai, Macromolecules, 2019, 52, 4703–4712 CrossRef CAS.
- Y. Ding, M. Cai, Z. Cui, L. Huang, L. Wang, X. Lu and Y. Cai, Angew. Chem., Int. Ed., 2018, 57, 1053–1056 CrossRef CAS PubMed.
- L. Cao, Q. Zhao, Q. Liu, L. Ma, C. Li, X. Wang and Y. Cai, Macromolecules, 2020, 53, 2220–2227 CrossRef CAS.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef CAS PubMed.
- D. E. Mitchell, J. R. Lovett, S. P. Armes and M. I. Gibson, Angew. Chem., Int. Ed., 2016, 55, 2801–2804 CrossRef CAS PubMed.
- C. Chen, F. Richter, C. Guerrero-Sanchez, A. Traeger, U. S. Schubert, A. Feng and S. H. Thang, ACS Macro Lett., 2020, 9, 260–265 CrossRef CAS.
- F. Zhou, R. Li, X. Wang, S. Du and Z. An, Angew. Chem., Int. Ed., 2019, 58, 9479–9484 CrossRef CAS PubMed.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- J. Tan, X. Dai, Y. Zhang, L. Yu, H. Sun and L. Zhang, ACS Macro Lett., 2019, 8, 205–212 CrossRef CAS.
- D. Liu, W. Cai, L. Zhang, C. Boyer and J. Tan, Macromolecules, 2020, 53, 1212–1223 CrossRef CAS.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- S. Xu, G. Ng, J. Xu, R. P. Kuchel, J. Yeow and C. Boyer, ACS Macro Lett., 2017, 6, 1237–1244 CrossRef CAS.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS.
- K. Ferji, P. Venturini, F. Cleymand, C. Chassenieux and J.-L. Six, Polym. Chem., 2018, 9, 2868–2872 RSC.
- Y. Jiang, N. Xu, J. Han, Q. Yu, L. Guo, P. Gao, X. Lu and Y. Cai, Polym. Chem., 2015, 6, 4955–4965 RSC.
- D. Liu, J. He, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 1660–1669 CrossRef CAS.
- C. Bergerbit, F. Baffie, A. Wolpers, P.-Y. Dugas, O. Boyron, M. Taam, M. Lansalot, V. Monteil and F. D'Agosto, Angew. Chem., 2020, 59, 10385–10390 CrossRef CAS.
- T. Lückerath, K. Koynov, S. Loescher, C. J. Whitfield, L. Nuhn, A. Walther, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Angew. Chem. Int. Ed. Engl, 2020, DOI:10.1002/anie.201916177.
- T. N. Tran, S. Piogé, L. Fontaine and S. Pascual, Macromol. Rapid. Commun., 2020, 2000203 CrossRef CAS PubMed.
- J. Wan, B. Fan, Y. Liu, T. Hsia, K. Qin, T. Junkers, B. M. Teo and S. H. Thang, Polym. Chem., 2020, 11, 3564–3572 RSC.
- S. Parkinson, S. T. Knox, R. A. Bourne and N. J. Warren, Polym. Chem., 2020, 11, 3465–3474 RSC.
- J. Rieger, Macromol. Rapid. Commun., 2015, 36, 1458–1471 CrossRef CAS.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- J. Lesage de la Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef CAS.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- A. B. Lowe, Polymer, 2016, 106, 161–181 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8, 9915–9922 RSC.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- K. Ren and J. Perez-Mercader, Polym. Chem., 2017, 8, 3548–3552 RSC.
- J. He, J. Cao, Y. Chen, L. Zhang and J. Tan, ACS Macro Lett., 2020, 533–539, DOI:10.1021/acsmacrolett.0c00151.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- S. L. Canning, T. J. Neal and S. P. Armes, Macromolecules, 2017, 50, 6108–6116 CrossRef CAS PubMed.
- J. Tan, X. Zhang, D. Liu, Y. Bai, C. Huang, X. Li and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1600508 CrossRef.
- Y. Zhang, J. He, X. Dai, L. Yu, J. Tan and L. Zhang, Polym. Chem., 2019, 10, 3902–3911 RSC.
- C. Zheng, Soft Matter, 2019, 15, 5357–5370 RSC.
- J. Kuldova, P. Kosovan, Z. Limpouchova and K. Prochazka, Macromol. Theory Simul., 2013, 22, 61–70 CrossRef CAS.
- G. Pandav, V. Pryamitsyn, K. C. Gallow, Y.-L. Loo, J. Genzer and V. Ganesan, Soft Matter, 2012, 8, 6471–6482 RSC.
- S. Saubern, X. Nguyen, V. Nguyen, J. Gardiner, J. Tsanaktsidis and J. Chiefari, Macromol. React. Eng., 2017, 11, 1600065 CrossRef.
- C. L. H. Wong, J. Kim and J. M. Torkelson, Macromol. React. Eng., 2007, 45, 2842–2849 CAS.
- S. Harrisson, F. Ercole and B. W. Muir, Polym. Chem., 2010, 1, 326–332 RSC.
- A. I. Buzin, M. Pyda, P. Costanzo, K. Matyjaszewski and B. Wunderlich, Polymer, 2002, 43, 5563–5569 CrossRef CAS.
- J. Kim, M. M. Mok, R. W. Sandoval, D. J. Woo and J. M. Torkelson, Macromolecules, 2006, 39, 6152–6160 CrossRef CAS.
- M. M. Mok, J. Kim and J. M. Torkelson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 48–58 CrossRef CAS.
- Y. N. Zhou, J. J. Li and Z. H. Luo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3052–3066 CrossRef CAS.
- M. M. Mok, C. J. Ellison and J. M. Torkelson, Macromolecules, 2011, 44, 6220–6226 CrossRef CAS.
- W. Yuan, M. M. Mok, J. Kim, C. L. H. Wong, C. M. Dettmer, S. T. Nguyen, J. M. Torkelson and K. R. Shull, Langmuir, 2010, 26, 3261–3267 CrossRef CAS PubMed.
- C. Zheng, H. Huang and T. He, Macromol. Rapid Commun., 2013, 34, 1654–1661 CrossRef CAS PubMed.
- C. Zheng, H. Huang and T. He, Macromol. Rapid Commun., 2014, 35, 309–316 CrossRef CAS.
- Y. Zhao, Y.-W. Luo, B.-G. Li and S. Zhu, Langmuir, 2011, 27, 11306–11315 CrossRef CAS PubMed.
- Y. Chen, W. Luo, Y. Wang, C. Sun, M. Han and C. Zhang, J. Colloid Interface Sci., 2012, 369, 46–51 CrossRef CAS PubMed.
- E.-H. Kang, S. Yang, S. Y. Yu, J. Kim and T.-L. Choi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3058–3066 CrossRef CAS.
- S. Shin, M.-L. Gu, C.-Y. Yu, J. Jeon, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 475–482 CrossRef CAS PubMed.
- S. Shin, K.-Y. Yoon and T.-L. Choi, Macromolecules, 2015, 48, 1390–1397 CrossRef CAS.
- S. Xu, T. Zhang, R. P. Kuchel, J. Yeow and C. Boyer, Macromol. Rapid Commun., 2020, 41, e1900493 CrossRef PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- J. Zhou, W. Zhang, C. Hong and C. Pan, Polym. Chem., 2016, 7, 3259–3267 RSC.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 CrossRef CAS.
- J. Tan, X. Li, R. Zeng, D. Liu, Q. Xu, J. He, Y. Zhang, X. Dai, L. Yu, Z. Zeng and L. Zhang, ACS Macro Lett., 2018, 7, 255–262 CrossRef CAS.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495–499 CrossRef CAS.
- C. A. Figg, R. N. Carmean, K. C. Bentz, S. Mukherjee, D. A. Savin and B. S. Sumerlin, Macromolecules, 2017, 50, 935–943 CrossRef CAS.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- D. Zhou, R. P. Kuchel, S. Dong, F. P. Lucien, S. Perrier and P. B. Zetterlund, Macromol. Rapid Commun., 2019, 40, e1800335 CrossRef PubMed.
- K. Wang, Y. Wang and W. Zhang, Polym. Chem., 2017, 8, 6407–6415 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- J. Tan, Y. Bai, X. Zhang and L. Zhang, Polym. Chem., 2016, 7, 2372–2380 RSC.
- F. Lv, Z. An and P. Wu, Nat. Commun., 2019, 10, 1397 CrossRef PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- A. M. I. Ali, P. Pareek, L. Sewell, A. Schmid, S. Fujii, S. P. Armes and I. M. Shirley, Soft Matter, 2007, 3, 1003–1013 RSC.
- V. S. Kravchenko and I. I. Potemkin, J. Phys. Chem. B, 2016, 120, 12211–12217 CrossRef CAS PubMed.
- S. Okabe, K.-i. Seno, S. Kanaoka, S. Aoshima and M. Shibayama, Macromolecules, 2006, 39, 1592–1597 CrossRef CAS.
- J. Yeow, S. Shanmugam, N. Corrigan, R. P. Kuchel, J. Xu and C. Boyer, Macromolecules, 2016, 49, 7277–7285 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and characterisation data (Fig. S1–S9 and Table S1-PISA19-36). See DOI: 10.1039/d0py00889c |
| This journal is © The Royal Society of Chemistry 2021 |