
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multifunctional divergent scaffold to access the formal syntheses of various sesquiterpenoids†
Vera P.
Demertzidou
,
Maria
Kourgiantaki
and
Alexandros L.
Zografos
 *
*
Aristotle University of Thessaloniki, Department of Chemistry, Laboratory of Organic Chemistry, University Campus, Chemistry Old Building, Thessaloniki 54124, Greece. E-mail: alzograf@chem.auth.gr
First published on 20th September 2021
Abstract
The development of a divergent scaffold able to access an array of diverse natural sesquiterpenoids is described. The route unifies the scope of previously reported plans of our group to allow the scalable synthesis of 8,12-furo and lactone sesquiterpenoid carbocyclic cores of elemanes, germacranes, guaianes, cadinanes, lindenanes and myliols. The formal syntheses of furogermenone, methyl-curdionolide, zedoarol, qweicurculactone, lindenene and sarcandralactone A are reported.
Introduction
The power of divergent synthesis is undeniably highlighted in its limits by Nature, where divergent scaffolds are built as essential intermediates of the primary metabolism of an organism and utilized to produce a largely diverse pool of secondary metabolism.1 The most impressive paradigm of this process is well emphasized by the essential unlimited diversity of terpenoids. Although divergent synthesis is probably the best machinery of evolution, its implementation in laboratory scale synthesis is still problematic due to the usual inability of organic chemists to recognize and efficiently synthesize divergent scaffolds.2Spotting this challenge, our group is focusing on developing divergent scaffolds of maximum potential.3 During the past years, we have reported a series of total syntheses for 8,12-sesquiterpenoids relying on two common furo-elemane scaffolds bearing either 2-propenyl substitution (1; Scheme 1) for accessing germacrene, guaiane and cadinene complexity3b,c or propargylic moiety (2; Scheme 1) for lindenane and myliol carbocyclic cores.3d Despite their undeniable success, furo-elemane common scaffolds suffer from their poor scalability and their inability to be oxidized to biological important α-methylene-γ-butenolide sesquiterpenoids (SQLs) (α-methylene-γ-butenolide moiety is depicted in green colour; Scheme 1).4 To overcome these drawbacks divergent scaffold 3 was designed bearing a protected α-methylene-γ-butenolide ring at 8,12-positions instead of the furan ring. The proposed 3 was projected to serve not only as a reliable alternative for the synthesis of furo- and α,β-unsaturated sesquiterpenoids that reported earlier by our group,3b–d but also as a cornerstone for the future access of α-methylene-γ-butenolide SQLs of both 8,12- and 6,12-complexity. The formal syntheses of natural products bearing the carbocyclic cores of germacranes, guaianes and lindenanes at furan and α,β-unsaturated lactone oxidation states were set as threshold for the developed divergent scaffold.
 | ||
| Scheme 1 The development of a new divergent scaffold (3) to access the formal syntheses of several furan and α,β-unsaturated sesquiterpenoids. | ||
Results and discussion
To address the poor scalability of our prior plans, a completely new synthetic strategy was designed for the synthesis of 3. Retrosynthetically, compound 3 (Scheme 1) was projected to derive from protected lactone 4, to allow the consequent successive stereoselective alkylations for the introduction of the two tertiary stereocenters. The necessity for protection of α-methylene-γ-butenolide functionality was expected to be counterbalanced by the selection of a biocompatible cell-cleaved protection.5 Lastly, a syn-direct lactonization was envisioned from the known natural product carvonic acid (5). Early introduction of the lactone moiety would supposedly induce firstly the stereoselective introduction of the vinyl group through the stereoelectronic effects produced by the cis-8,12-junction and secondly the alkynylation that follows.The synthesis began by preparing carvonic acid (5) following reported procedures.6 Attempts to introduce the cis-8,12-lactone functionality was found extremely challenging using various methods (see inset of Scheme 2). Utilization of NCS, NIS and iodine under ionic and radical conditions provided complex mixture of products deriving from the polyhalogenation and halolactonization of alkenes (ex. compound 11), while interestingly the addition of Br2 produced cis-6,12-lactone 12. The same was also evidenced when CH-lactonization was attempted following White's protocol7 or when LHMDS was used in the presence of CuCl2 or FeCl3,8 albeit in lower yields. Although product 12 was not the desired, these results show the ability of carvonic acid to selectively access 6,12-lactone, something that is beneficial in cases where 6,12-sesquiterpenoids are targeted. On the other hand, careful addition of NBS under radical conditions with AIBN as initiator allowed the synthesis of cis-8,12-lactone 6, in rather low yield (35%) besides the extensive optimization, due to its facile oxidation to aromatic component 13. With lactone 6 in hands, we next set the stage for the introduction of the tertiary vinyl group. As expected, the notorious lability of α-methylene-γ-butenolide in 6 towards nucleophiles prohibited its direct alkylation. Inspired by the concept of pharmacophore-directed retrosynthesis,9 utilization of a biocompatible protection group was selected to allow the biological screening of all prepared precursors. Following literature precedence,5 reversible protections by amines, alcohols, and thiols were tested. Numerous experiments were run to differentiate the two Michael acceptors in 6 (Scheme 2; Table 1). Typical conditions of a nucleophile employment (MeOH, thiophenol or amines) with or without the presence of K2CO3 and DMAP failed to provide clean protection of methylene group, due to the interference of the conjugated ketone and/or the cleavage of the lactone moiety. Gratifyingly, addition of DMF as a co-solvent with MeOH in the presence of equimolar amount of K2CO3 allowed the introduction of methoxy functionality to provide 4 in 71% yield.
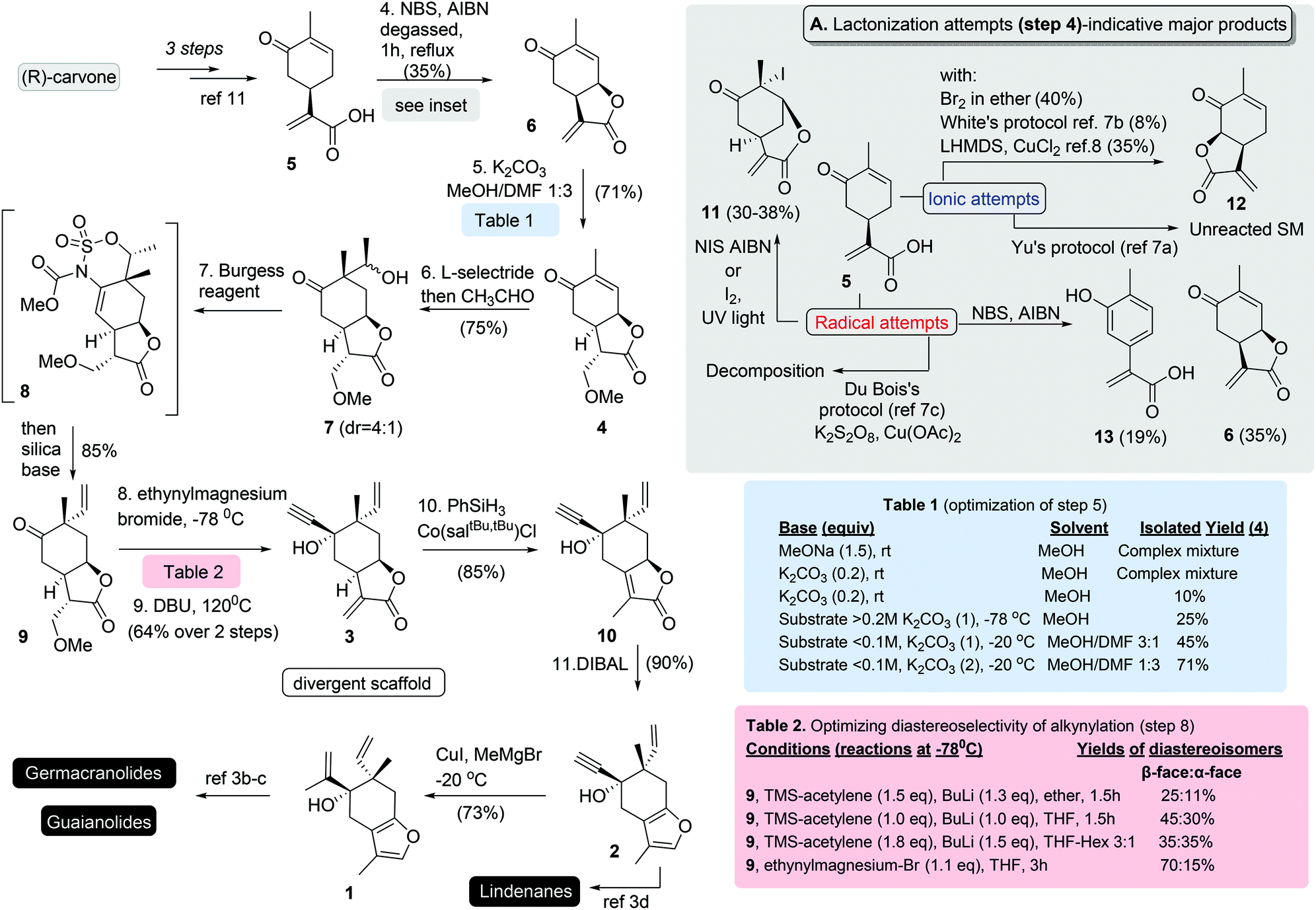 | ||
| Scheme 2 Synthetic route to furan common scaffolds 1 and 2. Attempted reactions and optimization for crucial steps included. | ||
Successful reductive stereoselective alkylation of 4 by L-selectride and acetaldehyde followed.10 Alkylation provided a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of alcohols 7 favouring the Felkin–Anh product without witnessing any alkylation from the β-face of the cyclohexane ring. The latter is directly attributed to the incorporation of rigidity from the syn-lactone to the cyclohexane ring.10 Dehydration by Burgess reagent was unencumbered affording compound 9 in 65% yield, along with Burgess-substrate hybrid 8 in 20% yield. The latter was transformed into the desired product by subjection to basic conditions, affording 9 in 85% overall yield.
:1 mixture of alcohols 7 favouring the Felkin–Anh product without witnessing any alkylation from the β-face of the cyclohexane ring. The latter is directly attributed to the incorporation of rigidity from the syn-lactone to the cyclohexane ring.10 Dehydration by Burgess reagent was unencumbered affording compound 9 in 65% yield, along with Burgess-substrate hybrid 8 in 20% yield. The latter was transformed into the desired product by subjection to basic conditions, affording 9 in 85% overall yield.
Alkylation of ketone 9 by lithium TMS-acetylene prepared by TMS-acetylene and n-BuLi was found productive but it resulted in poor diastereoselectivities for the desired β-isomer (2:1 ratio over its epimer) (Scheme 2; Table 2). To this end several conditions were tried to find that ethynylmagnesium bromide can allow the synthesis of desired diastereoisomer in a 3.5:1 ratio over its epimer. Subsequent DBU deprotection of methoxy group provided compound 3 in 64% yield over the two steps (Scheme 2). The isomerization of α-methylene-γ-butenolide 3 to α,β-unsaturated-γ-butenolide 10 was found difficult to achieve and was only accomplished by catalytic hydrogen atom transfer to the alkene, a protocol that was developed by Shenvi's group.11 Finally, the direct reduction of 10 to furan moiety by DIBAL furnishes the known furo-elemane 2, allowing the formal synthesis of epi-lindenene and sarcandralactone A.3d
Access to the formal syntheses of germacranes: furanogermenone and methylcurdionolide and to guaianes: zedoarol and gweicurculactone (Scheme 1) and other differentiated scaffolds can be obtained by selective alkylation of acetylene functionality of 2 to unify for the first time all synthetic plans. After extensive experimentation was found that methyl cupration of alkyne can be accomplished with copper iodide followed by slow addition of methylmagnesium bromide12 allowing the synthesis of 2-propenyl-intermediate 1 as the parent compound for the known oxy-Cope reaction that can build the 10-member macrocycle or the oxy-Cope/ene reaction that serves for obtaining guaiane carbocyclic core. All formal syntheses were accomplished in the same number of steps as in the previously reported plan (11 steps for common scaffold 2 from R-carvone) in slightly better yields, nonetheless with the capacity for current to deliver gram scale quantities for all synthetic intermediates.
Conclusions
In conclusion, we report a new synthetic route for the synthesis of 8,12-sesquiterpenoids. The synthetic plan employs a highly stereoselective access to both furo-elemanes 2 and 1 for the first time allowing the unified divergent synthesis of germacranes, guaianes and lindenanes among other carbocycles by taking advantage of the stereoelectronic effects of an early introduced protected α-methylene-γ-butenolide core for the stereoselective introduction of two tertiary centers. The newly developed divergent scaffold is expected to serve as cornerstone for the synthesis of biologically intriguing α-methylene-γ-butenolide lactone sesquiterpenoids. This work is currently underway and will be reported in due course.Author contributions
V. P. D. and A. L. Z. were responsible for the conceptualization of the work in every step of the project. M. K. was responsible for scaling-up some experimental procedures.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We would like to thank Prof. Ryan Shenvi from The Scripps Research Institute for helpful discussions and for providing us complex Co(saltButBu)Cl for the isomerization of 3. We thank ELIDEK agency for scholarship of V. P. D. This work was also supported by the project ‘OPENSCREEN-GR’ (MIS 5002691) which is implemented under the Action ‘Reinforcement of the Research and Innovation Infrastructure’, funded by the Operational Program ‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund). Funding is also provided by Research-Create-Innovate program (project code T1EDK-01161) which is implemented in collaboration with Pharmathen S.A. and co-financed by Greece and the European Union.Notes and references
- A. L. Zografos, in From Biosynthesis to Total Synthesis: Strategies and Tactics for Natural Products, Wiley & Sons, Hoboken, 2016 Search PubMed.
- Seminar report on divergent synthesis by: (a) C. E. Brotherthon and D. L. Boger, Total synthesis of azafluoranthene alkaloids: rufescine and imeluteine, J. Org. Chem., 1984, 49, 4050–4055 CrossRef For strategies and tactics for the synthesis of divergent scaffolds or common scaffolds can be found on: (b) E. E. Anagnostaki and A. L. Zografos, “Common synthetic scaffolds” in the synthesis of structurally diverse natural products, Chem. Soc. Rev., 2012, 41, 5613–5625 RSC; (c) J. Shimokawa, Divergent strategy in natural product total synthesis, Tetrahedron Lett., 2014, 55, 6156–6162 CrossRef CAS; (d) L. Li, Z. Chen, X. Zhang and Y. Jia, Divergent Strategy in Natural Product Synthesis, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed; (e) A. M. Szpilman and E. M. Carreira, Probing the biology of natural products: molecular editing by diverted total synthesis, Angew. Chem., Int. Ed., 2010, 49, 9592–9628 ( Angew. Chem. , 2010 , 122 , 9786–9823 ) CrossRef CAS PubMed.
- (a) A. D. Fotiadou and A. L. Zografos, Accessing the Structural Diversity of Pyridone Alkaloids: Concise Total Synthesis of Rac-Citridone A, Org. Lett., 2011, 13, 4592–4598 CrossRef CAS PubMed; (b) E. E. Anagnostaki and A. L. Zografos, Non-natural Elemane as the “Stepping Stone” for the Synthesis of Germacrane and Guaiane Sesquiterpenes, Org. Lett., 2013, 15, 152–155 CrossRef CAS PubMed; (c) E. E. Anagnostaki, V. P. Demertzidou and A. L. Zografos, Divergent pathways to furosesquiterpenes: first total syntheses of (+)-zedoarol and (Rac)-gweicurculactone, Chem. Commun., 2015, 51, 2364–2367 RSC; (d) V. P. Demertzidou and A. L. Zografos, Platinum-catalyzed cycloisomerizations of a common enyne: a divergent entry to cyclopropane sesquiterpenoids. Formal synthesis of sarcandralactone A, Org. Biomol. Chem., 2016, 14, 6942–6946 RSC . For depiction of the synopsized synthetic route leading to diverse furan and α,β-unsaturated lactone sesquiterpenoids please see ESI.†.
- (a) S. Zhang, Y.-K. Won, C.-N. Ong and H.-M. Shen, Anti-Cancer Potential of Sesquiterpene Lactones: Bioactivity and Molecular Mechanisms, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 239–249 CrossRef CAS PubMed; (b) S. B. Christensen, D. M. Skytte, S. R. Denmeade, C. Dionne, J. V. Moller, P. Nissen and J. T. Isaacs, A Trojan Horse in Drug Development: Targeting of Thapsigargins Towards Prostate Cancer Cells, Anti-Cancer Agents Med. Chem., 2009, 9, 276–294 CrossRef CAS PubMed; (c) I. Nakase, B. Gallis, T. Takatani-Nakase, S. Oh, E. Lacoste, N. P. Singh, D. R. Goodlett, S. Tanaka, S. Futaki, H. Lai and T. Sasaki, Transferrin receptor-dependent cytotoxicity of artemisinin-transferrin conjugates on prostate cancer cells and induction of apoptosis, Cancer Lett., 2009, 274, 290–298 CrossRef CAS PubMed; (d) A. Ghantous, H. Gali-Muhtasib, H. Vuorela, N. A. Saliba and N. Darwiche, What made sesquiterpene lactones reach cancer clinical trials?, Drug Discovery Today, 2010, 15, 668–678 CrossRef CAS PubMed; (e) M. R. Kreuger, S. Grootjans, M. W. Biavatti, P. Vandenabeele and K. D'Herde, Sesquiterpene lactones as drugs with multiple targets in cancer treatment: focus on parthenolide, Anticancer Drugs, 2012, 23, 883–896 CrossRef PubMed; (f) M. Chadwick, H. Trewin, F. Gawthrop and C. Wagstaff, Sesquiterpenoid Lactones: Benefits to plants and people, Int. J. Mol. Sci., 2013, 14, 12780–12805 CrossRef PubMed; (g) Y. Ren, J. Yu and A. D. Kinghorn, Development of Anticancer Agents from Plant-derived Sesquiterpene Lactones, Curr. Med. Chem., 2016, 23, 2397–2420 CrossRef CAS PubMed.
- Methoxy-protected butenolides can be readily cleaved in cells, in accordance to similar results for amino-protected sesquiterpenoids: J. R. Woods, H. Mo, A. A. Bieberich, T. Alavanja and D. A. Colby, Amino-derivatives of the sesquiterpene lactone class of natural products as prodrugs, MedChemComm, 2013, 4, 27–33 RSC.
- (a) W. Engel, In Vivo Studies on the Metabolism of the Monoterpenes S-(+)- and R-(-)-Carvone in Humans Using the Metabolism of Ingestion-Correlated Amounts (MICA) Approach, J. Agric. Food Chem., 2001, 49, 4069–4075 CrossRef CAS PubMed; (b) D. W. Lee, C. F. Manful, J. R. Gone, Y. Ma and W. A. Donaldson, Reactivity of acyclic (pentadienyl)iron(1+) cations with phosphonate stabilized nucleophiles: application to the synthesis of oxygenated metabolites of carvone, Tetrahedron, 2016, 72, 753–759 CrossRef CAS.
- For CH-lactonizations please advise: (a) Z. Zhuang and J.-Q. Yu, Nature, 2020, 577, 656–659 CrossRef CAS PubMed; (b) K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032–9033 CrossRef CAS PubMed; (c) S. Sathyarmoorthi and J. Du Bois, Org. Lett., 2016, 18, 6308–6311 CrossRef PubMed.
- M. P. DeMartino, K. Chen and P. S. Baran, Intermolecular Enolate Heterocoupling: Scope, Mechanism, and Application, J. Am. Chem. Soc., 2008, 130, 11546–11560 CrossRef CAS PubMed.
- (a) D. Romo, Bioactivity-guided retrosynthesis: “Upping the ante” for natural product synthesis, Planta Med., 2015, 81 Search PubMed; (b) J. R. Hudlicky and G. A. Sulikowski, Simple start for complex products, Nat. Chem., 2019, 11, 297–298 CrossRef CAS PubMed; (c) M. E. Abbasov, R. Alvarino, C. M. Chaheine, E. Alonso, J. A. Sanchez, M. L. Conner, A. Alfonso, M. Jaspars, L. M. Botana and D. Romo, Simplified immunosuppressive and neuroprotective agents based on gracillin A, Nat. Chem., 2019, 11, 342–350 CrossRef CAS PubMed.
- It is known that direct alkylation of R-carvone results sole alkylation from the β-face of the cyclohexane ring. Please see: J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakakallio and P. S. Baran, J. Am. Chem. Soc., 208, 130, 17938–17954 CrossRef PubMed.
- S. W. M. Crossley, F. Barabé and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 16788–16791 CrossRef CAS PubMed.
- K. A. Parker and Q. Xie, Org. Lett., 2008, 10, 1349–1352 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details for all prepared compounds. Copies of NMR H1 and NMR C13. See DOI: 10.1039/d1ob01716k |
| This journal is © The Royal Society of Chemistry 2021 |