
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence5-(Diarylimino)- and 5-(sulfoximido)dibenzothiophenium triflates: syntheses and applications as electrophilic aminating reagents†
Zhen
Li‡
,
Gonela
Vijaykumar‡
 ,
Xiangdong
Li
,
Christopher
Golz
and
Manuel
Alcarazo
*
,
Xiangdong
Li
,
Christopher
Golz
and
Manuel
Alcarazo
*
Institut für Organische und Biomolekulare Chemie Georg-August-Universität Göttingen Tammannstr 2, 37073 Göttingen, Germany. E-mail: manuel.alcarazo@chemie.uni-goettingen.de
First published on 16th March 2021
Abstract
The one-pot synthesis of well-defined 5-(diarylimino) and 5-(sulfoximido)dibenzothiophenium triflates, respectively from diarylimines or sulfoximines, is reported and the structures of a series of these compounds are elucidated by X-ray crystallography. In analogy to their hypervalent I(III) analogues, the iminoyl and sulfoximidoyl groups of these compounds can be selectively transferred to organic substrates. Specifically, the uncatalyzed imination of thiols or sulfinates proceeds with good yields, while under the mild reaction conditions offered by visible light photoredox catalysis, the radical amination of hydrazones or the sulfoximidation of benzylic, allylic and propargylic C–H bonds takes place satisfactorily.
Introduction
The reactivity of S-(aryl/alkyl) sulfonium salts shows many similarities with that of hypervalent I(III) reagents of an analogue substitution pattern.1 Both types of compounds do react with low valent metals such as Ni(0) or Pd(0) via oxidative addition at the C–S or C–I bond, respectively, making these compounds suitable partners for cross coupling reactions.2 In addition, the highly electrophilic nature of the central heteroatom in these species facilitates one-electron reduction of both sulfonium salts and hypervalent I(III) reagents, which ultimately leads to the homolytic fragmentation of one of the C–E (E = heteroatom) bonds and generates a carbon-centred radical.3 This fragmentation pattern paves the way to the use of both compounds as radical precursors; however, the synthetic utility of this methodology can only be fully exploited if the chemoselective cleavage of only one of the C–E bonds is achieved.4 Finally, the uncatalyzed reaction of both species with suitable nucleophiles is also possible. This last process can be seen as an attack of the nucleophile on the central heteroatom followed by reductive elimination of a Nu–R moiety (Scheme 1a).5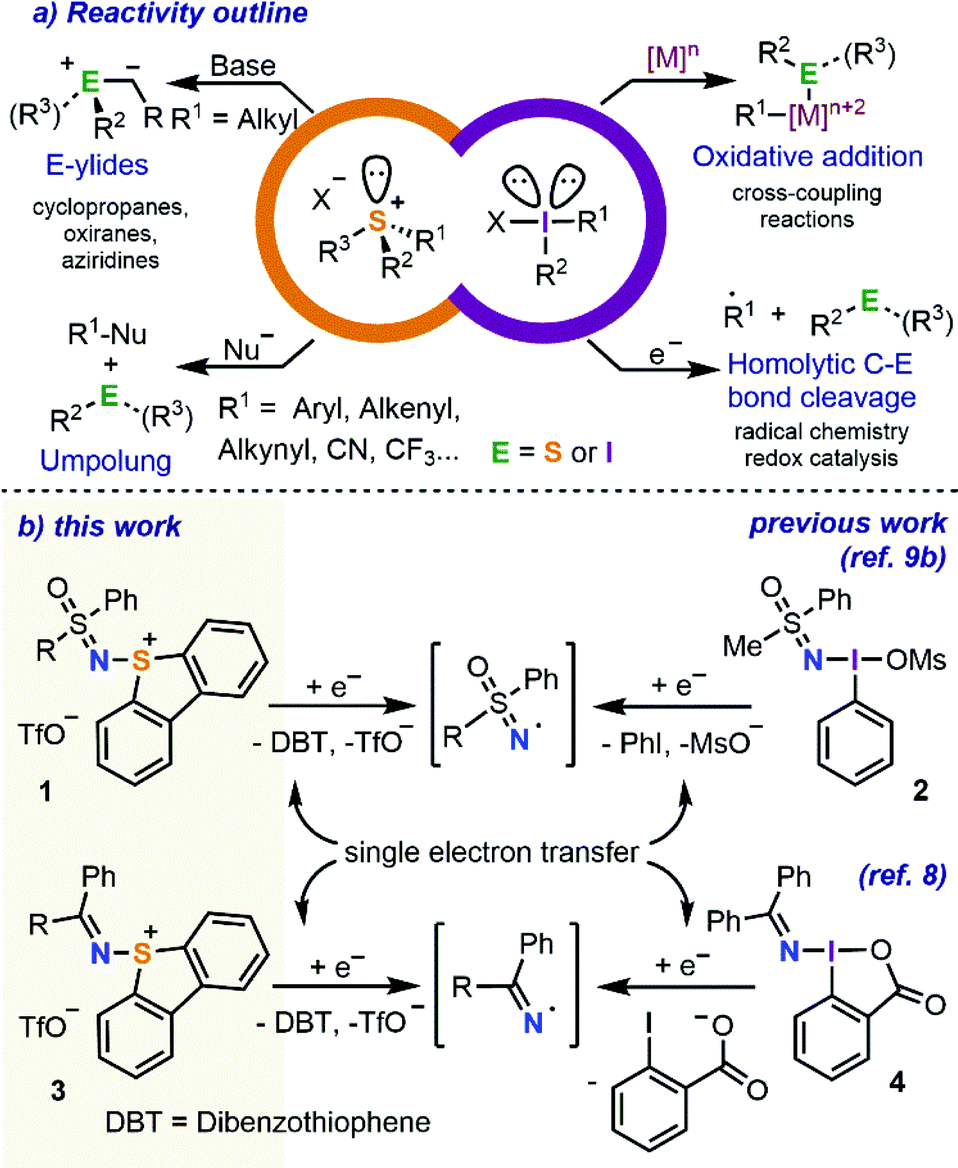 | ||
| Scheme 1 Reactivity overview and previous and current work on the use of hypervalent iodine and sulfonium salts as electrophilic sulfoximide/imine transfer reagents. | ||
Much more obscure is the chemistry of sulfonium salts bearing nitrogen-based substituents. Specifically, while many hypervalent iodine reagents containing transferable nitrogen functional groups such as azido,6 phthalimido,7 diarylimino,8 or sulfoximido are available,9 only a few S-analogues of these compounds have been reported to date and the studies regarding their reactivity are even more scarce.10 We speculated however that they should also share similarities with their iodine counterparts and hypothesized that imino- and sulfoximido-substituted sulfonium salts might potentially function as electrophilic aminating reagents or efficient sources of imine/sulfoximide radicals (Scheme 1b).
With this idea in mind, we report herein the multigram-scale syntheses of a series of sulfonium salts containing sulfoximido 1 and diarylimino 3 functional groups and preliminarily evaluate their use in the electrophilic amination of thiols and organic sulfonites. The generation of sulfoximidoyl and iminyl radicals from the same reagents under photochemical conditions and their subsequent reaction with hydrazones and benzylic C–H bonds to deliver N-(amino)amidines and N-benzylic sulfoximides, respectively, are also studied.
Results and discussion
Synthesis and structure of S-imino- and S-sulfoximido-substituted sulfonium salts
Our initial efforts focused on the synthesis of the parent sulfonium salts 1, which have a sulfoximidoyl moiety attached to the dibenzothiophene platform (Scheme 2). While the preparation of structurally analogous sulfonium salts has been described by the reaction of N-halosulfilimines with dialkyl/arylsulfides,10e,11 we decided to explore alternative methods, which employ less elaborated starting materials and minimize the number of synthetic steps. Hence, the direct reaction of diphenylsulfoximide with Tf2O-activated dibenzothiophene S-oxide was evaluated.12 Gratifyingly, the desired reaction took place at −50 °C in the presence of pyridine as a base, affording 1a as a beige crystalline solid in 83% yield. Scaling the reaction up to multigram quantities is possible without a drop in the isolated yield (see the ESI for details†). This simple synthetic protocol seems to be general and it has been successfully extended to the preparation of sulfonium salts 1b and 1c, containing two different sulfoximidoyl moieties (Scheme 2a). Following a very similar methodology, but just replacing the sulfoximide with benzophenone imines 5, one obtains diarylimino-substituted sulfonium salts 3a–c. Compounds 3d–f, which employ 2,8-difluorodibenzothiophene, phenoxathiin and thianthrene units as sulfonium platforms, are also prepared from the respective sulfoxides through the same route.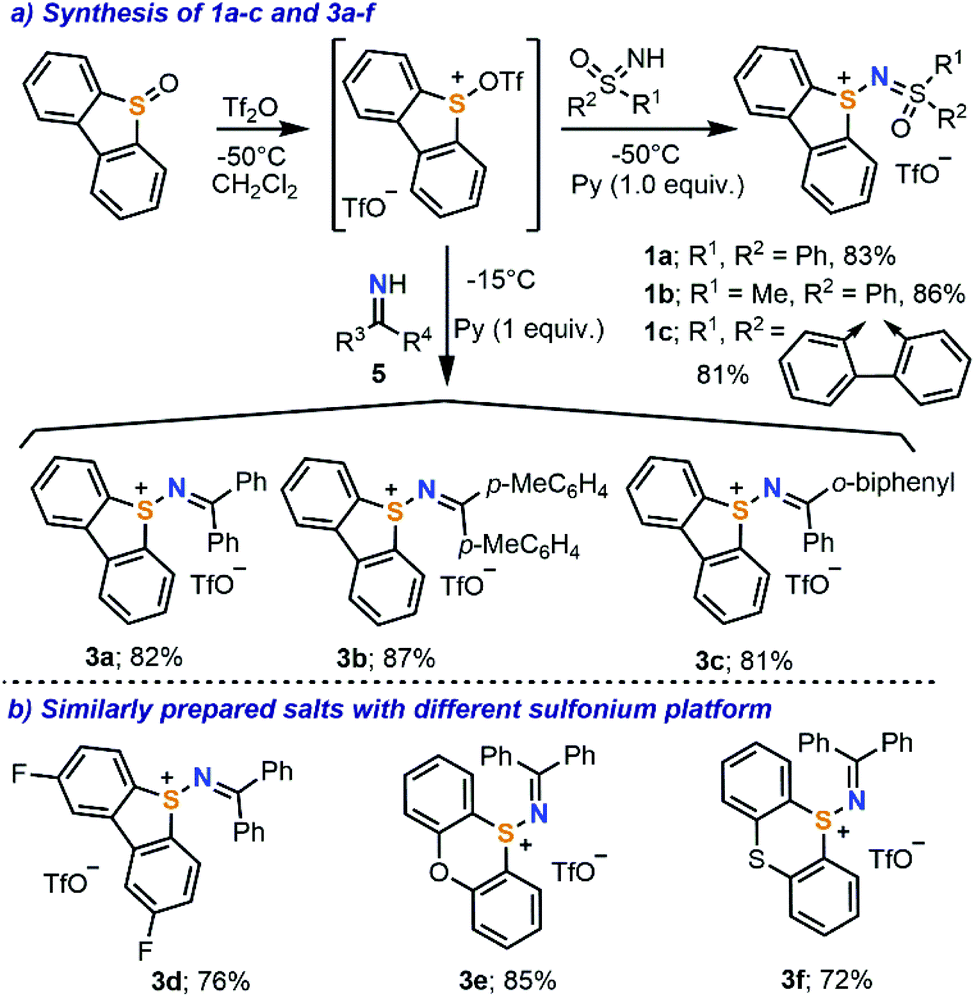 | ||
| Scheme 2 Synthesis of target sulfonium salts and scope of the transformation. | ||
X-ray diffraction analysis of single crystals of 1a–c and 3a–f confirmed the expected connectivity of the newly prepared compounds (see Fig. 1 for the structures of 1b, 1c, 3a and 3e and the ESI for the others†). In 1c, the sulfur atom from the dibenzothiophenium unit (S1) remains in the plane defined by that heterocycle and adopts a trigonal-pyramidal bond geometry, the sum of the bond angles around S1 being 304.6°. The S1–N1 bond distance of 1.652(1) Å falls in between the ranges for S–N single bonds and double bonds, while the N1–S2 length for the sulfoximide moiety (1.597(1) Å) gets closer to the typical S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(sp2) double bond distance of neutral sulfoximides (1.517 Å).13 Although the general bonding situation in 3a is relatively similar to that in 1c, two differences are of note. An S1–N1 bond distance of 1.705(1) Å was determined, which more clearly indicates that a single bond connects both atoms. Moreover, in 3a, the sum of the bond angles around S1 decreases to 297.9°. Both facts can be attributed to the lack of significant π-interaction between S1 and N1 in this molecule; this is also confirmed by the N1–C13 bond distance, 1.304(2) Å, which is just slightly longer than the typical one for CN double bonds in imines (1.297 Å).13
N(sp2) double bond distance of neutral sulfoximides (1.517 Å).13 Although the general bonding situation in 3a is relatively similar to that in 1c, two differences are of note. An S1–N1 bond distance of 1.705(1) Å was determined, which more clearly indicates that a single bond connects both atoms. Moreover, in 3a, the sum of the bond angles around S1 decreases to 297.9°. Both facts can be attributed to the lack of significant π-interaction between S1 and N1 in this molecule; this is also confirmed by the N1–C13 bond distance, 1.304(2) Å, which is just slightly longer than the typical one for CN double bonds in imines (1.297 Å).13
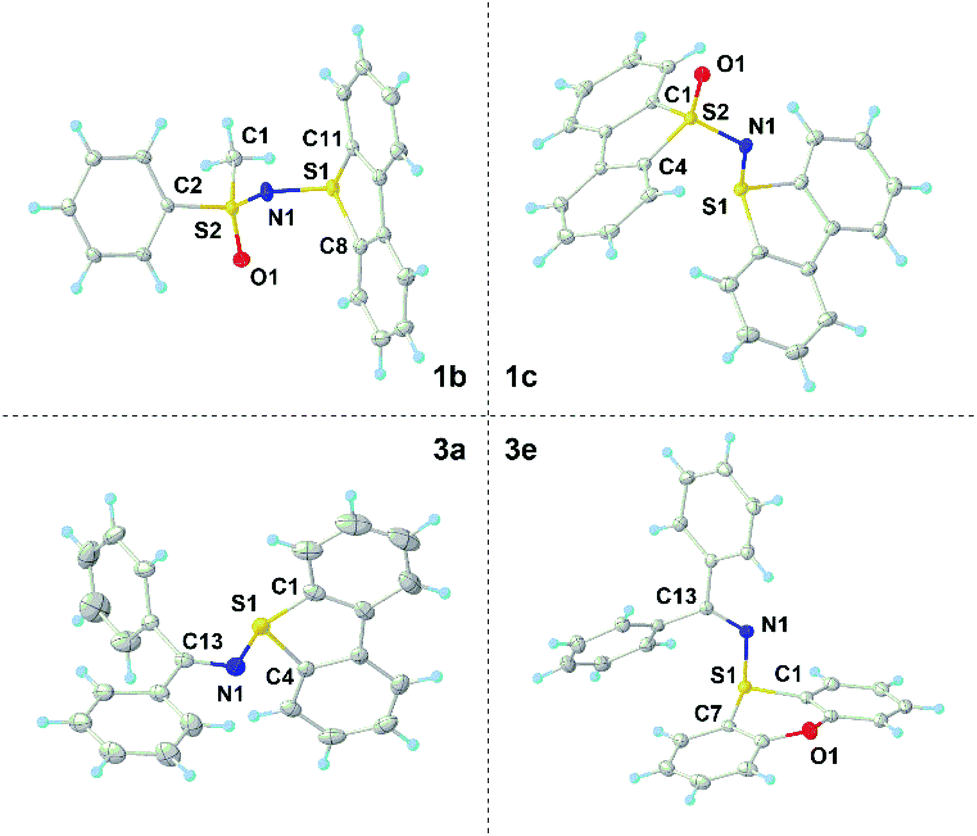 | ||
| Fig. 1 Molecular structures of compounds 1b (top left), 1c (top right), 3a (bottom left) and 3e (bottom right) in the solid state. Anisotropic displacement shown at the 50% probability level; triflate anions and hydrogen atoms are omitted for clarity.14 | ||
In an attempt to better understand the bonding situation just described, the geometry of each compound was optimized at the PBE/def2TZVP/univ-JFIT level of theory and an IBO (Intrinsic Bond Orbital) analysis was carried out for 1c and 3a (see Fig. 2 for the most important IBOs and the ESI for full details†). In line with our interpretation of the crystallographic data, a weak π-interaction (2e; 3c) is detected between the central nitrogen and the two flanking sulfur atoms in 1c, which may account for the reduced bond length in between these atoms. Conversely, in 3a the bond situation is better represented by a clear CN π-bond. Non-shared electron pairs are found at S1 and N1 in both compounds.
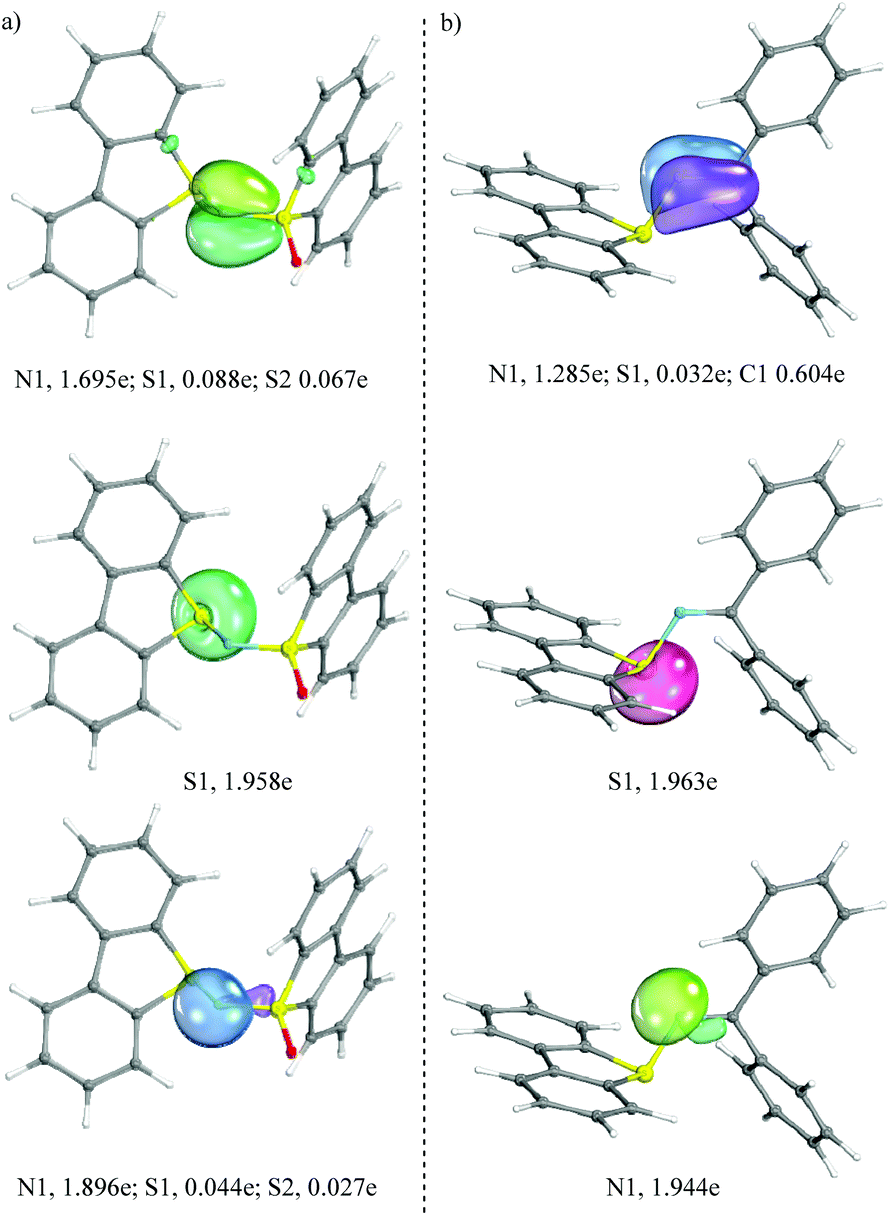 | ||
| Fig. 2 Selected IBO plots for complexes 1c (a) and 3a (b); assigned partial charges for the given IBOs to the individual atoms. Threshold value for printing: 80. | ||
Along these lines, natural population analysis (B3LYP/6-31G*) in 1c and 3a indicates that the sulfur atoms belonging to the dibenzothiophene unit (S1) bear an entire positive charge in both compounds (+1.050e, 1c, and +1.021e, 3a, respectively), while the nitrogen atoms significantly differentiate from each other. N1 bears a negative charge in 1c (−1.060e) while this atom is only partially charged in 3a (−0.613e). Wiberg bond indices for the S1–N1 interaction are 1.023 and 0.966 for 1c and 3a, respectively, 1.043 for N1–S2 in 1c and 1.672 for the N1C13 bond in 3a. This analysis, together with the X-ray structures, points to a slightly stronger S1–N1 interaction in 1c than in 3a.
Inspection of the frontier orbitals reveals that in both 1c and 3a the very low-lying LUMO+1 largely corresponds to a mixture between the π*-system of the dibenzothiophene platform and the σ*(S1–N1) bond (see the ESI†). This distribution of orbital coefficients suggests that any donation of electron density to this orbital weakens the S1–N1 bond. Hence, one might expect that the amination of appropriately selected nucleophiles might occur via the formation of a sulfurane intermediate and subsequent reductive elimination from the S-center. In addition, single electron transfer to 1 or 3 should deliver dibenzothiophene and N-center radicals, as in the case of the analogous I(III)-compounds.8,9
Reactivity studies
Once compounds 1a–c and 3a–f were completely characterized, their potential as electrophilic amination reagents was preliminarily examined. Our study started with an attempt to prepare sulfenylimines 6 by direct reaction of 3a–f with thiols under mildly basic conditions. While several methods are already available for the synthesis of these products,15 to the best of our knowledge, the disconnection approach we report here is new. Moreover, most protocols start from disulfides, and the only one described employing thiol substrates requires metal catalysis.16 This could be attributed to the facile oxidation of thiols, resulting in undesired products.4-(t-Butyl)benzenethiol 7a was initially used as a model substrate (Scheme 3). Treatment of that thiol with salts 3a,d–f smoothly afforded the desired sulfenylimine 6a, with reagent 3a being the one that delivered the desired product with better yields. Employing 3a, the reaction indistinctly works with electron-rich or electron-poor thiols, albeit all yields are moderate. On the other hand, the functional group tolerance of the transformation is quite broad; ether, ester, amide and nitro groups as well as a range of heterocycles did not disturb. As a side reaction, the formation of 5–10% of the corresponding disulfide was systematically observed in all experiments. When TEMPO (1.0 equiv.) was added to the imination reaction, the formation of 6a was completely suppressed. It seems therefore reasonable to believe that this reaction takes place via one-electron oxidation of the thiolate by 3a followed by coupling of the iminyl and thiyl radicals.
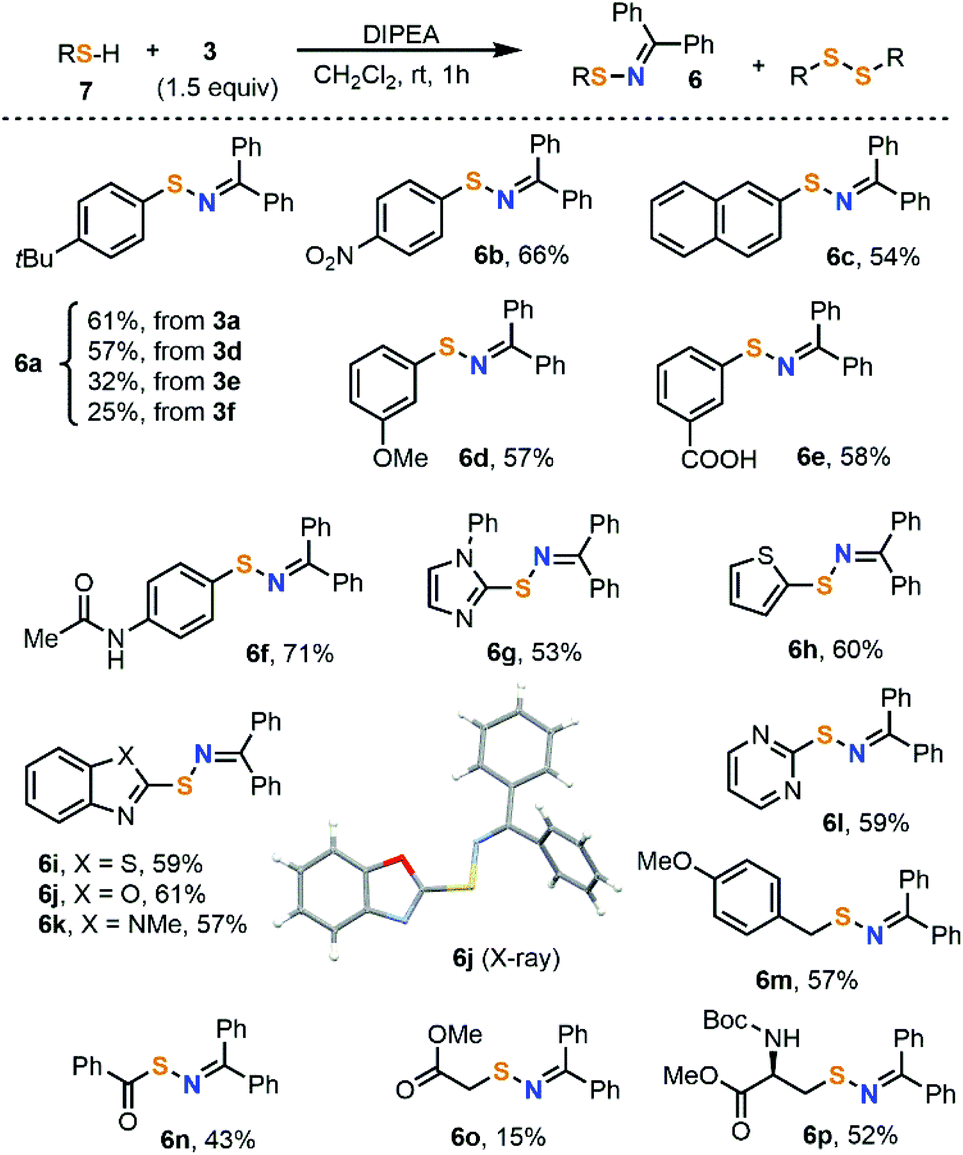 | ||
| Scheme 3 Substrate scope of the electrophilic amination of thiols. The reaction was conducted at room temperature. Unless specified, all reactions were performed using reagent 3a (1.5 equiv.). All reactions were quenched after 1 h and yields are of the isolated products. | ||
The electrophilic amination could also be extended to sulfinates 8 to obtain sulfonamides 9. In this case, bisimine 10 is often observed as a homocoupled side product. Moreover, the addition of TEMPO (1.0 equiv.) to the reaction mixture significantly reduced the isolated yield of 9b (23%). For these reasons we also believe that the aryl sulfinate salts, which have low oxidation potentials (E1/2 = −0.37 V vs. SCE for sodium phenylsulfinate), undergo SET oxidation in the presence of 3a (E1/2 = −0.46 V vs. SCE) to furnish sulfonyl and iminyl radicals, which recombine to form 9 and small amounts of 10 (Scheme 4).17
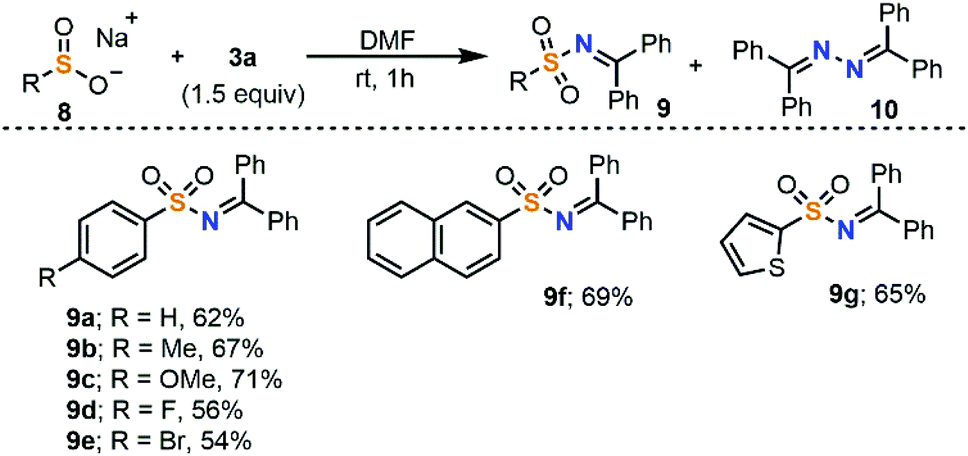 | ||
| Scheme 4 Substrate scope of the electrophilic amination of sodium sulfinates with 3a. All reactions were conducted at room temperature and quenched after 1 h. Yields are of isolated products. | ||
After this initial contact with the reactivity of salts with the general formula of 3, and given the number of precedents in which sulfonium salts are reduced via single electron transfer under photochemical conditions to produce radicals of different nature,2 it was hypothesized that under these soft activation conditions, 3a also might be able to generate iminyl radicals. An experiment often used to determine the feasibility of that process consists of the evaluation of fluorescence intensity quenching of the photoexcited state of a photocatalyst (Stern–Volmer) in the presence of the desired substrate. In fact, this analysis revealed an effective oxidative quenching of the photoexcited Ru catalyst (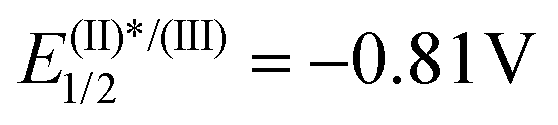 vs. SCE) in the presence of 3a (Ered = −0.46 V vs. SCE). The formation of the envisaged iminyl radical was subsequently evidenced by a radical scavenger experiment. Hence, irradiation of 3c, bearing an o-biphenyl substituent, in the presence of [Ru(bpy)3]Cl2 (1 mol%) quantitatively afforded the product of radical trapping, phenanthridine 11 (Scheme 5).18
vs. SCE) in the presence of 3a (Ered = −0.46 V vs. SCE). The formation of the envisaged iminyl radical was subsequently evidenced by a radical scavenger experiment. Hence, irradiation of 3c, bearing an o-biphenyl substituent, in the presence of [Ru(bpy)3]Cl2 (1 mol%) quantitatively afforded the product of radical trapping, phenanthridine 11 (Scheme 5).18
 | ||
| Scheme 5 (a) Stern–Volmer plot of photocatalyst luminescence quenching by 3a and (b) intramolecular radical trap experiment employing substrate 3c. | ||
Having established the conditions to form iminyl radicals from 3a, we set about determining the potential of this photocatalytic system on the C(sp2)–H amination of aldehyde-derived hydrazones 12 into hydrazonamides 13, which would represent the most step-economical method to carry out this transformation.19 We were pleased to see that the desired transformation actually proceeded, leading to the formation of C(sp2)–H aminated products; the yields however are moderate and they drop dramatically if the hydrazones used as substrates are not derived from electron-rich aromatic aldehydes (Scheme 6).
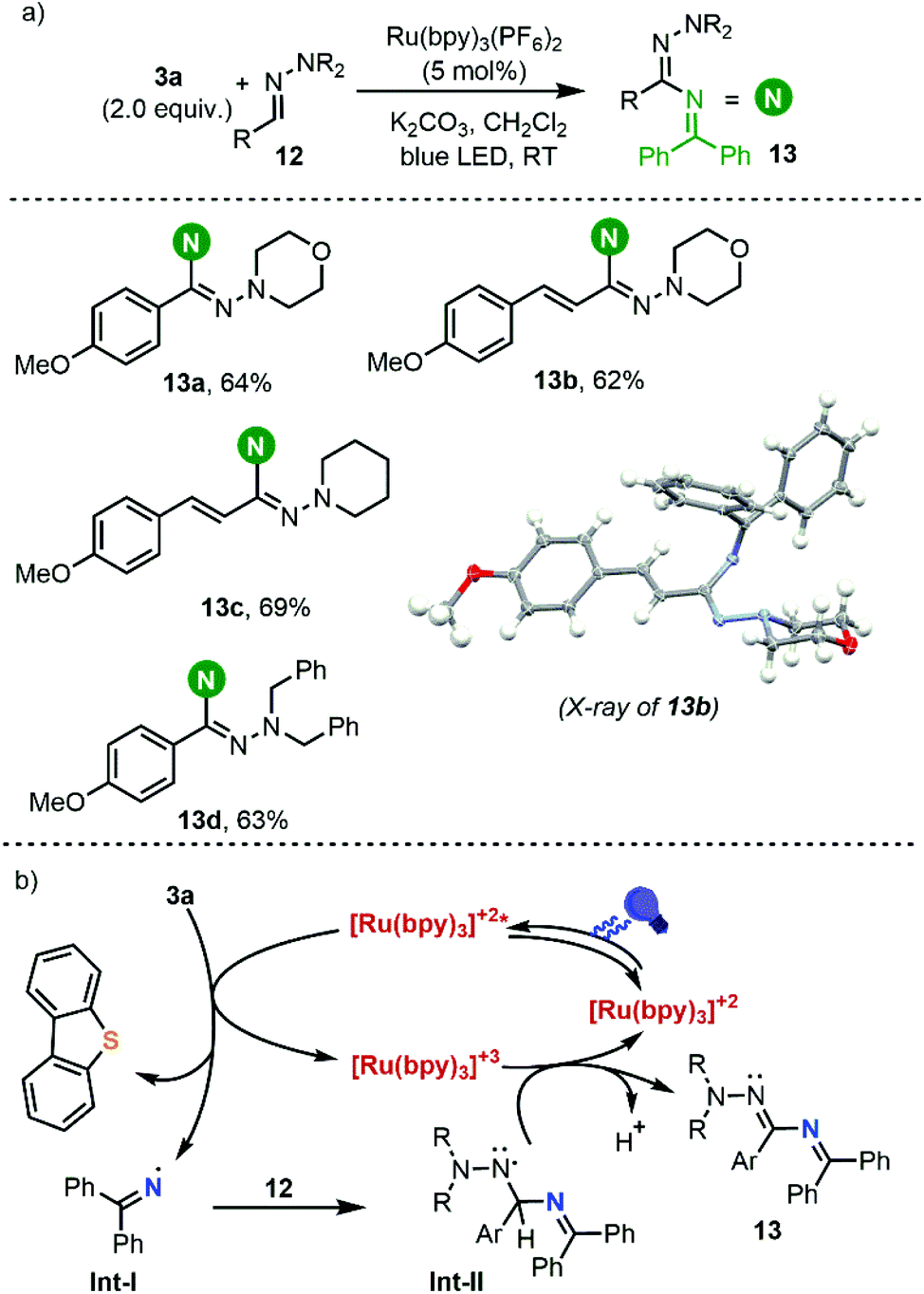 | ||
| Scheme 6 (a) Synthesis of hydrazonamides 13 and (b) plausible reaction mechanism. | ||
Based on these results and literature precedents, we proposed the reaction pathway shown in Scheme 6b. Initial irradiation of [Ru(bpy)3]2+ generates photoexcited ([Ru(bpy)3]2+)*, which reduces reagent 3avia single-electron transfer and generates the transient iminyl radical (Int-I) via homolytic S–N bond cleavage. Addition of the N-centered radical to the azomethine carbon of 12 delivers an aminyl radical intermediate (Int-II), which is stabilized by the adjacent N atom. At this stage, Int-II can be oxidized by [Ru(bpy)3]3+ (E(III)/(II)1/2 = 1.29 V vs. SCE) to produce a diazenium cation with regeneration of the photocatalyst, followed by deprotonation to deliver 13.20 Alternatively, deprotonation of Int-II may occur first leading to the formation of the corresponding radical anion, which would undergo oxidation to 13.21
Finally, we also checked the reactivity of salt 1a in the sulfoximidation of benzylic C–H bonds. A closely related transformation to this one has been already described by Bolm and co-workers, also under photochemical conditions, but employing hypervalent I(III) reagent 2.9c These authors report the formation of the desired sulfoximides in moderate to good yields. Mechanistic studies suggest the formation of a sulfoximidoyl radical as a key intermediate, which is able to abstract a benzylic hydrogen atom from the substrate delivering the N–H sulfoximine and a benzyl radical. Oxidation of this radical by the oxidized photocatalyst delivers a benzylic carbocation, which reacts with the sulfoximine to afford the N-functionalized products. Hence, if sulfoximidoyl radicals could be generated from sulfonium salts 1a–c, then, similar reactivity could be expected.
We initially evaluated this possibility by cyclic voltammetry. These experiments showed an irreversible reduction with Ered = −0.462 V and −0.606 V vs. SCE for 1a and 1c, respectively; values that are considerably less negative than that determined for the photoexcited state of [Ru(bpy)3][PF6] (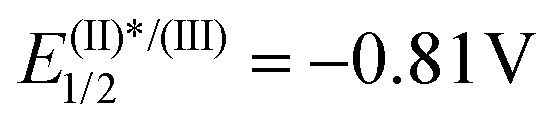 vs. SCE) indicate the feasibility of the necessary SET event. Moreover, a Stern–Volmer experiment confirms that 1c effectively quenches the excited state of [Ru(bpy)3][PF6] (Fig. 3a). A final confirmation of the generation of sulfoximidoyl radicals by one-electron reduction of 1a was provided by the reaction of that compound with Cp2Co (Ered = −0.93 vs. SCE) in the presence of N-(t-butyl)phenylnitrone (PBN) as a radical trap. The EPR spectrum of the reaction mixture shows a well-resolved splitting as a result of hyperfine coupling with a N- and a H-atom (aN = 14.61; aH = 2.22 G), a pattern that fits with that expected for radical 14.
vs. SCE) indicate the feasibility of the necessary SET event. Moreover, a Stern–Volmer experiment confirms that 1c effectively quenches the excited state of [Ru(bpy)3][PF6] (Fig. 3a). A final confirmation of the generation of sulfoximidoyl radicals by one-electron reduction of 1a was provided by the reaction of that compound with Cp2Co (Ered = −0.93 vs. SCE) in the presence of N-(t-butyl)phenylnitrone (PBN) as a radical trap. The EPR spectrum of the reaction mixture shows a well-resolved splitting as a result of hyperfine coupling with a N- and a H-atom (aN = 14.61; aH = 2.22 G), a pattern that fits with that expected for radical 14.
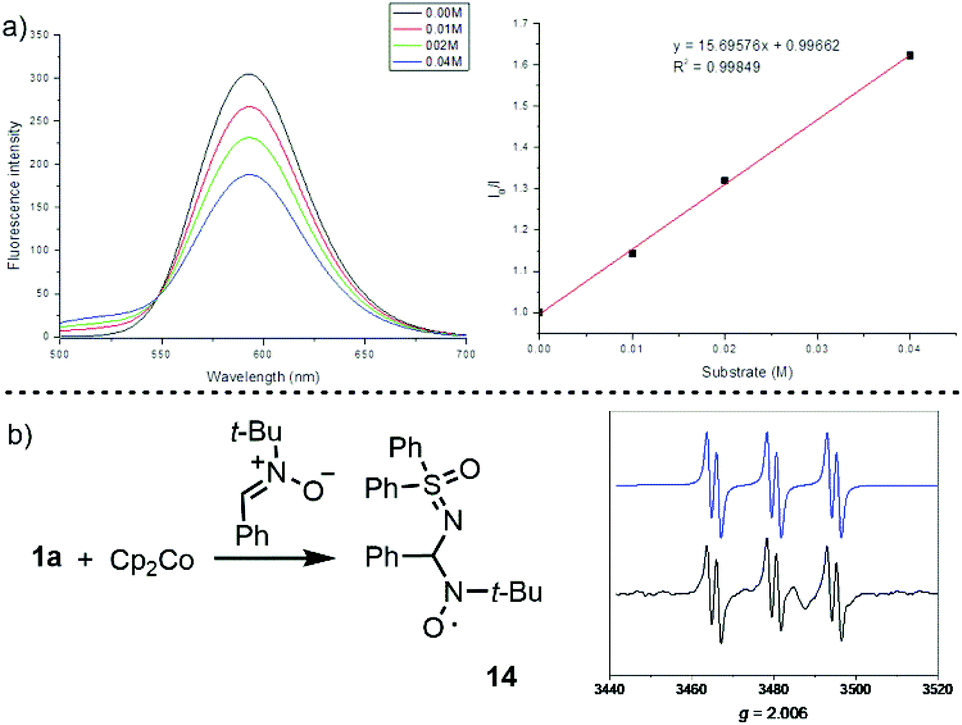 | ||
| Fig. 3 Mechanistic studies: (a) Stern–Volmer plot of photocatalyst luminescence quenching by 1a and (b) simulated (blue) and experimental (black) EPR spectra of 14. | ||
Once the generation of the desired sulfoximidoyl radical was confirmed, the amination of benzylic C–H bonds was evaluated. In fact, we could perform this reaction with a scope very similar to that reported by Bolm,9c which includes not only benzylic positions 16a–aa, but also allylic 16ab–ag and propargylic ones 16ah–aj (Scheme 7).22 A light on/off experiment confirmed the necessity of continuous irradiation for the reaction to proceed, and the quantum yield of the reaction to form 16a (Φ = 0.044) indicates that a radical chain process cannot be a predominant pathway.
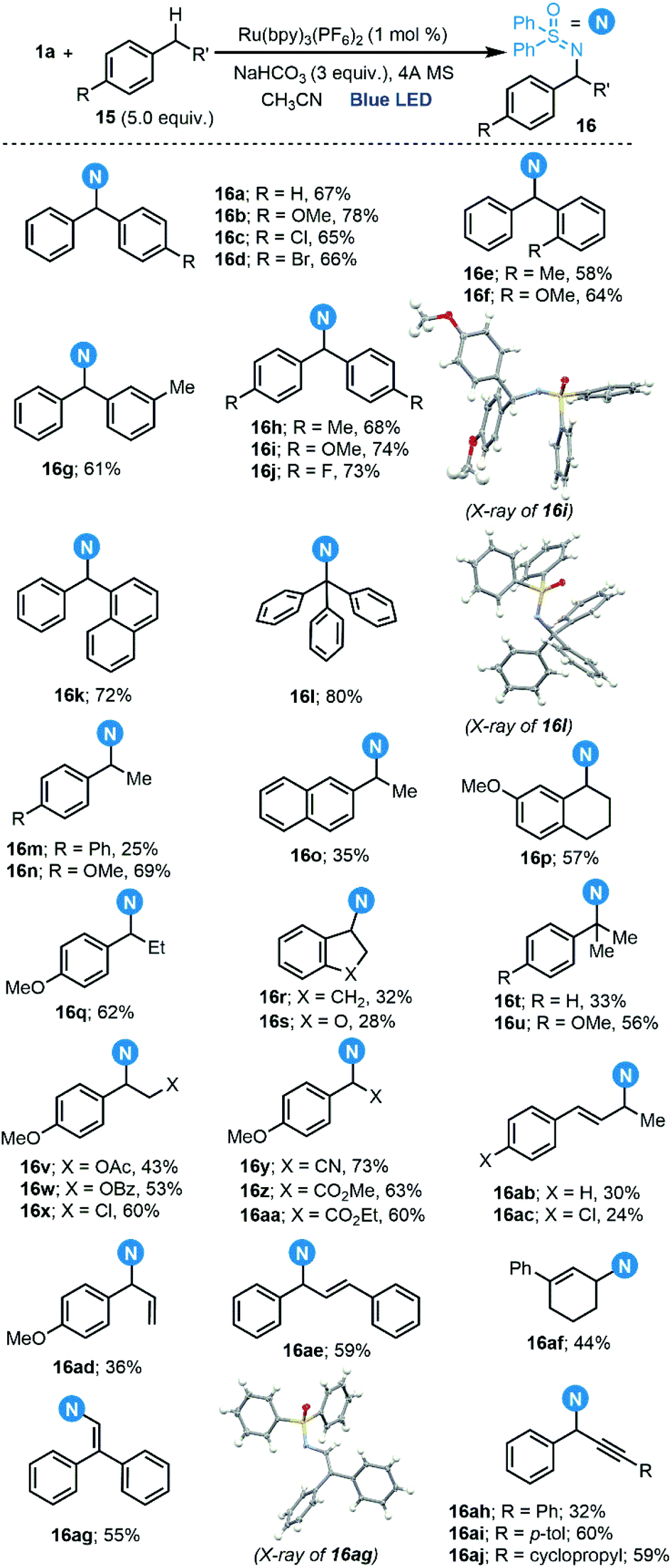 | ||
| Scheme 7 Reaction scope for the sulfoximidation of benzylic C–H bonds. | ||
Conclusions
In summary, we report herein the straightforward syntheses of sulfonium salts containing imino and sulfoximido substituents and demonstrated that under photochemical conditions, these compounds are effective sources of iminyl and sulfoximidoyl radicals, respectively, which can be subsequently employed in the imination of hydrazones or the sulfoximidation of benzylic C–H positions. The straightforward synthesis, easy handling and safe profile of compounds 1 and 3 confirm these sulfonium salts as convenient surrogates of iodonium salts of analogue structure and reactivity.Experimental section
Synthesis of 1a
Triflic anhydride (1.4 g, 5.0 mmol, 1.0 equiv.) was dropwise added to a solution of dibenzothiophene-S-oxide (1.00 g, 5.0 mmol) in dry DCM (40 mL) at −50 °C. After stirring the resulting mixture for 1 hour, pyridine (403 μL, 5.0 mmol) and diphenylsulfoximide (1.09 g, 5.0 mmol) dissolved in DCM (10 mL) were sequentially added dropwise, and the mixture was further stirred at −50 °C for 6 additional hours. Then, the cooling system was removed, and the obtained solution was allowed to reach room temperature. After this, an aqueous K2CO3 solution was added, upon which the phases separated and the organic one was dried with anhydrous MgSO4 and evaporated in vacuo. The thus obtained residue was washed with dry Et2O (3×) and finally dried under vacuum to deliver 1a as a beige solid (2.28 g, 83%). 1H NMR (400 MHz, CD3CN) δ = 8.15–8.06 (m, 2H), 8.04–7.95 (m, 6H), 7.89–7.77 (m, 4H), 7.74–7.55 (m, 6H). 13C NMR (101 MHz, CD3CN) δ = 138.6, 136.6, 135.8, 135.2, 133.6, 131.2, 130.6, 128.7, 128.0, 124.0, 121.2 (q, J = 321.0 Hz); IR (neat): 3566, 3088, 1709, 1577, 1448, 1362, 1258, 1224, 1152, 1088, 1029, 1005, 969, 758, 738, 707, 685, 637, 613, 589, 573, 541, 518, 422 cm−1. HRMS calculated m/z for C24H18NOS2+ [M-TfO]+: 400.0824, found 400.0831. Mp: 152–153 °C.Synthesis of 3a
Triflic acid anhydride (0.62 g, 2.2 mmol, 1.1 equiv.) was slowly added at −50 °C to a suspension of dibenzothiophene-S-oxide (0.40 g, 2.0 mmol, 1.0 equiv.) in dry dichloromethane (16 mL). After stirring the reaction mixture for 30 min at that temperature, pyridine (0.17 g, 2.2 mmol, 1.1 equiv.) and benzophenone imine (0.39 g, 2.2 mmol, 1.1 equiv.) were added as a solution in dichloromethane (3 mL). After this, the reaction mixture was slowly warmed to −15 °C and stirred for another 6 h at the same temperature. Then the resulting mixture was washed with aq. K2CO3 and extracted with dichloromethane. The solvent was evaporated in vacuo and the residue was washed with dry Et2O (2 × 10 mL) and dried in vacuo to obtain the desired product as a pale-yellow solid (0.84 g, 82%). 1H NMR (400 MHz, CDCl3) δ = 8.03 (d, J = 7.8 Hz, 2H), 7.88 (d, J = 7.3 Hz, 2H), 7.84–7.75 (m, 5H), 7.68 (d, J = 8.0 Hz, 2H), 7.57–7.53 (m, 5H), 7.35 (t, J = 7.8 Hz, 2H) ppm; 13C NMR (101 MHz, CDCl3) δ = 190.7, 139.5, 135.9, 135.5, 135.3, 134.6, 132.9, 132.5, 131.6, 131.3, 130.4, 129.7, 129.0, 128.9, 124.1, 121.0 (q, J = 322.2 Hz) ppm; 19F NMR (377 MHz, CDCl3) δ = −78.11 ppm; IR (ATR): 3061, 1710, 1657, 1582, 1526, 1482, 1446, 1259, 1223, 1155, 1029, 999, 955, 761, 707, 637, 572, 517 cm−1; HRMS calculated m/z for C25H18NS+ [M-OTf]+ : 364.1154, found 364.1156; Mp: 122–123 °C.Author contributions
Zhen Li synthesized compounds 1a–c and studied their reactivity, Gonela Vijaykumar synthesized 3a–f and studied their reactivity, Xiangdong Li performed the mechanistic studies, Christopher Golz performed the calculations and determined the X-ray structures, and Manuel Alcarazo supervised the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the European Research Council (ERC CoG 771295) and the DFG (INST 186/1237-1 and INST 186/1324-1) is gratefully acknowledged. We also thank Mr M. Simon for assistance with preparative HPLC and Dr S. I. Kozhushkov for proof checking this manuscript.Notes and references
-
(a) S. I. Kozhushkov and M. Alcarazo, Eur. J. Inorg. Chem., 2020, 26, 2486–2500 CrossRef PubMed
; (b) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed
-
(a) S. Karreman, S. B. H. Karnbrock, S. Kolle, C. Golz and M. Alcarazo, Org. Lett., 2021 DOI:10.1021/acs.orglett.1c00087
- For seminal work see:
(a) T. J. Van Bergen, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 44, 4953–4962 CrossRef CAS
-
(a) J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56–62 CrossRef CAS PubMed
-
(a) X.-X. Ming, Z.-Y. Tian and C.-P. Zang, Chem. – Asian J., 2019, 14, 3370–3379 CrossRef CAS PubMed
-
(a) S. Alazet, J. Preindl, R. Simonet-Davin, S. Nicolai, A. Nanchen, T. Meyer and J. Waser, J. Org. Chem., 2018, 83, 12334–12356 CrossRef CAS PubMed
-
(a) J. Bergès, B. García and K. Muñiz, Angew. Chem., Int. Ed., 2018, 57, 15891–15895 CrossRef PubMed
- K. Kiyokawa, D. Okumatsu and S. Minakata, Angew. Chem., Int. Ed., 2019, 58, 8907–8911 CrossRef CAS PubMed
-
(a) C. Wang, Y. Tu, D. Ma and C. Bolm, Angew. Chem., 2020, 59, 14134–14137 CrossRef CAS PubMed
-
(a) R. Reck and J. C. Jochims, Chem. Ber., 1982, 115, 1494–1508 CrossRef CAS
- K. Akutagawa, N. Furukawa and S. Oae, Bull. Chem. Soc. Jpn., 1984, 57, 518–524 CrossRef CAS
- Cyano-, alkynyl-, alkenyl-, and aryl-substituted sulfonium salts have been prepared through this method. Selected examples:
(a) X. Li, C. Golz and M. Alcarazo, Angew. Chem., 2019, 58, 9496–9500 CrossRef CAS PubMed
-
(a)
A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, Appendix A: Typical Interatomic Distances in Organic Compounds and Organometallic Compounds and Coordination Complexes of the d- and f-block metals, ed. H.-B. Burgi and J. D. Dunitz, Structure Correlations, vol 2, VCH Publishers, Weinheim, 1994 Search PubMed
- Deposition numbers 2062172–2062181 and 2063032–2063039 contain the supplementary crystallographic data for this paper.†.
-
(a) P. Barman, S. K. Bhattacharjee and T. Bhattacharjee, Synth. Commun., 2011, 41, 5089–5094 CrossRef
-
(a) W. Long, W. Qiu, C. Li, L. Song, G. Bai, G. Zhang and H. He, RSC Adv., 2016, 6, 40945–40952 RSC
- A. U. Meyer, S. Jäger, D. P. Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050–2054 CrossRef CAS
-
(a) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289–13292 CrossRef CAS PubMed
- M. Zhang, Y. Duan, W. Li, P. Xu, J. Cheng, S. Yu and C. Zhu, Org. Lett., 2016, 18, 5356–5359 CrossRef CAS PubMed
- The oxidation of hydrazone-derived aminyl radical intermediates has been proposed for similar transformations. See, for example:
(a) B. Huang, X.-S. Bu, J. Xu, J.-J. Dai, Y.-S. Feng and H.-J. Xu, Asian J. Org. Chem., 2018, 7, 137–140 CrossRef CAS
- For a mechanistic proposal following this path, see: B. Janhsen and A. Studer, J. Org. Chem., 2017, 82, 11703–11710 CrossRef CAS PubMed
- Compounds 16ab and 16ac are obtained as mixtures of products in which the amino substituent is incorporated at the allylic or the benzylic position. See the ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2062172–2062181; 2063032–2063039. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob00285f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |