
Fusogenic porous silicon nanoparticles as a broad-spectrum immunotherapy against bacterial infections†
Byungji
Kim
 a,
Qinglin
Yang
a,
Leslie W.
Chan
b,
Sangeeta N.
Bhatia
b,
Erkki
Ruoslahti
c and
Michael J.
Sailor
*ad
a,
Qinglin
Yang
a,
Leslie W.
Chan
b,
Sangeeta N.
Bhatia
b,
Erkki
Ruoslahti
c and
Michael J.
Sailor
*ad
aMaterials Science and Engineering Program, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093, USA. E-mail: msailor@ucsd.edu
bMassachusetts Institute of Technology, Cambridge MA 02139, USA
cCancer Research Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California 92037, USA
dDepartment of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093, USA
First published on 9th February 2021
Abstract
Bacterial infections are re-emerging as substantial threats to global health due to the limited selection of antibiotics that are capable of overcoming antibiotic-resistant strains. By deterring such mutations whilst minimizing the need to develop new pathogen-specific antibiotics, immunotherapy offers a broad-spectrum therapeutic solution against bacterial infections. In particular, pathology resulting from excessive immune response (i.e. fibrosis, necrosis, exudation, breath impediment) contributes significantly to negative disease outcome. Herein, we present a nanoparticle that is targeted to activated macrophages and loaded with siRNA against the Irf5 gene. This formulation is able to induce >80% gene silencing in activated macrophages in vivo, and it inhibits the excessive inflammatory response, generating a significantly improved therapeutic outcome in mouse models of bacterial infection. The versatility of the approach is demonstrated using mice with antibiotic-resistant Gram-positive (methicillin-resistant Staphylococcus aureus) and Gram-negative (Pseudomonas aeruginosa) muscle and lung infections, respectively. Effective depletion of the Irf5 gene in macrophages is found to significantly improve the therapeutic outcome of infected mice, regardless of the bacteria strain and type.
New conceptsThe past several decades have seen a global resurgence of bacterial infections, which has led to the development new classes of small molecule-based antibiotics. However, critical challenges remain in drug resistance development and excessive immune response, which contribute significantly to patient mortality. This work presents the first RNAi-mediated therapy that is generalizable across different bacteria types (Gram-positive, Gram-negative, and antibiotic-resistant strains), achieved by engineering nanoparticles to deliver siRNA to selectively targeted macrophages in the infection site, turning off proinflammatory signaling pathways. The three new concepts needed to achieve this involved designing the porous silicon nanoparticle to carry a large quantity of nucleic acid payload, attaching a pendant peptide that specifically targets macrophages, and incorporating a fusogenic coating that delivers the payload directly to the cytosol, resulting in near 90% transfection efficiency in the targeted macrophages in vivo. |
Introduction
Since the first identification of antibiotic-resistant strains of bacteria,1 the number of resistant strains has increased while the number of effective antibiotics has remained more or less static.2 While development of antibiotics against Gram-positive bacteria remains relatively diverse and fast-paced, the dual-walled nature of Gram-negative bacteria makes it much more difficult to develop countermeasures; although clinically available antibiotics are generally able to penetrate a single bacterial cell wall to trigger their mechanism of action, they regularly fail to breach the walls (particularly the outer membrane) possessed by Gram-negative bacteria.3,4 The outer membrane consists of lipopolysaccharide (LPS) molecules, which are densely-packed due to their saturated lipid chains and electrostatic crosslinking of their phosphorylated inner core region by divalent cations. This packing results in a hydrophilic oligosaccharide brush layer on the cell wall surface that impedes the diffusion of hydrophobic small molecules.5 Thus, the drug-impermeable outer membrane contributes to innate drug resistance. While innate drug resistance limits the number of treatment options, acquired resistance mechanisms such as that in multidrug resistance strains further reduces the number of effective drugs. Currently adjuvant therapies, including peptide-based potentiators and enzymatic inhibitors, are the subject of intense research.6–10 However, there continues to be few effective options in the clinic.Because new antibiotics will inevitably face resistance over time,1 there is a need for solutions that can evolve alongside the bacteria, in order to minimize the need to develop new drugs as new pathogen strains arise. Furthermore, it is highly desirable to develop broad-spectrum approaches that can defeat bacteria regardless of their class. This work describes such a strategy, based on an immunotherapy approach that harnesses the body's immune system and its innate ability to evolve in response to emergent bacterial threats.
Immunotherapy has primarily been explored for treatment of cancer and autoimmune disorders, with little emphasis on infectious diseases.11 While the pre-antibiotic era of treatments against bacterial infections involved transient anti-serum or lasting vaccine immunotherapy, more recent efforts tend to focus on monoclonal antibodies as therapeutics.12–15 Antibody-based treatments are pathogen-specific, and few have reached clinical trials despite showing promising results in the laboratory.16,17 The approach presented in this work addresses one of the characteristics common to many lethal bacterial and viral infections – the excessive inflammatory response mounted by the immune system.18–20 In particular, here we focus on reprogramming pro-inflammatory signals in the M1 phenotype of macrophages in order to suppress their inflammatory over-response.
The approach presented in this work involves fusogenic porous silicon nanoparticles (F-pSiNPs) which are engineered to reprogram macrophages by delivering siRNA against the Irf5 gene, shutting down pro-inflammatory signals in the M1 phenotype. The nanoparticles contain three key design features that allow them to overcome the substantial barriers to in vivo gene silencing21 (Fig. 1a): (1) they are selectively targeted to activated macrophage cells in the inflammatory site via a highly effective CRV peptide22 (sequence: CRVLRSGSC), which is tethered to the nanoparticle exterior; (2) they bypass endocytosis to deliver the siRNA payload to the cellular compartment where it can be most effective – accomplished using a fusogenic lipid overcoating on the nanoparticles; and (3) they deliver a maximal quantity of nucleic acid to the cell by employing a condensation chemistry that allows the porous inorganic nanoparticles to carry 25% by mass of an siRNA payload.
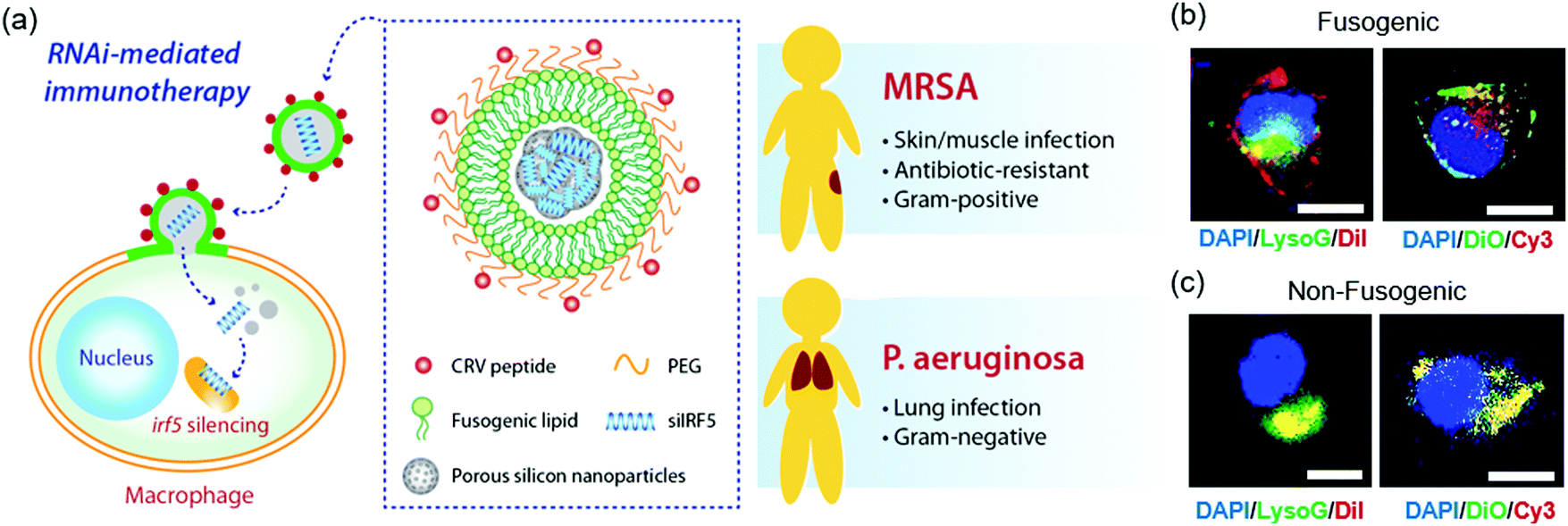 | ||
| Fig. 1 Fusogenic porous silicon nanoparticles directly deliver siIRF5 into the cell cytoplasm of macrophages. (a) Schematic depicting the peptide-targeted, fusogenic porous silicon nanoparticles (F-pSiNPs) and their mode of action in delivering and silencing the Irf5 gene in macrophages, as a broad-class strategy for treatment of Gram-positive (Methicilin-resistant Staphylococcus aureus, MRSA) and Gram-negative (Pseudomonas aeruginosa) bacterial infections. (b) Representative confocal microscope image of J774a.1 macrophage cells treated with fusogenic porous silicon nanoparticles (F-pSiNPs). Left image: Cells were treated with fusogenic pSiNPs wherein the lipid coating on the pSiNPs contained the lipophilic DiI membrane dye (red channel). The pSiNP core carried a non-labeled, non-functional siRNA payload. Lysosomes are stained with LysoTracker Green (LysoG, green channel) and cell nucleus is stained with DAPI (blue channel). Right image: Cells were treated with F-pSiNPs wherein the lipid coating contained the lipophilic DiO membrane dye (green channel). The pSiNP core carried a Cy3-labeled siRNA payload (red channel) and the cell nucleus is stained with DAPI (blue channel); (c) confocal microscope image of J774a.1 macrophage cells equivalent to (b) but using non-fusogenic porous silicon nanoparticles (NF-pSiNPs). Left image: DiI membrane stain loaded in the lipid coating of the pSiNPs (red); lysosomes stained with LysoTracker Green (green channel); DAPI nuclear stain (blue channel). Right image: DiO membrane stain loaded in the lipid coating of the pSiNPs (green channel); Cy3-siRNA loaded in the pSiNP core (red channel); DAPI nuclear stain (blue channel). Nanoparticles in (b) and (c) contained CRV targeting peptides pendant to the lipid coating of the pSiNPs. | ||
We previously demonstrated that treatment with this F-pSiNP therapeutic resulted in almost complete recovery of mice infected with otherwise fatal Staphylococcus aureus pneumonia by delivering siRNA against the Irf5 gene to macrophages in the diseased tissues, effectively inhibiting the excessive inflammatory response that contributes significantly to mortality in this infection.23 The major question we aimed to answer in the present study was if this approach – using RNAi to modulate host response to infection – was generalizable across different bacteria Gram types and infection sites to elicit broad-spectrum protection. To evaluate the system under particularly challenging conditions, we chose methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa, both of which are major contributors to the morbidity and mortality of hospital-acquired infections.24,25 We also assessed therapeutic efficacy for different infection sites – in muscle for infection of MRSA (strain: USA100) and in lungs for infection of P. aeruginosa (strain: PA01).
Results and discussion
Fusogenic nanoparticle characterization
The F-pSiNPs were nominally 200 nm in size and consisted of clusters of siRNA-loaded porous silicon nanoparticles (F-pSiNPs) in a solid core, with a more fluid targeting peptide-conjugated lipid coating as the shell (Fig. 1a).23,26 The size of the F-pSiNPs was adjusted to optimize stability, to maximize siRNA payload capacity, and to display an adequate in vivo circulation time. The core porous silicon nanoparticles (pSiNPs) were prepared by electrochemical etch of silicon wafers followed by ultrasonication, which resulted in nanoparticles nominally 50 nm in size (Fig. S1 and Table S1, ESI†). Loading of siRNA into the pSiNPs was then induced using a calcium chloride condensing agent, which forms a calcium silicate matrix in the presence of porous silicon nanoparticles.27,28 An aqueous suspension of the resulting core particles was then used to hydrate the lipid film, and the particles were mechanically extruded through a 200 nm polycarbonate membrane to form a stable core–shell structure as previously described.23,26 Fig. S1 (ESI†) shows transmission electron microscope (TEM) images of the particles before siRNA loading, after the calcium silicate loading chemistry, and after liposomal coating/cluster formation. Mass loading of siRNA in the final lipid-coated, peptide-conjugated nanoparticles was ∼25% (mass siRNA/total mass of nanoparticle + payload).The average hydrodynamic size of the final lipid-coated clusters was 220 nm (Table S1, ESI†); these F-pSiNPs were found to display sufficient circulation times to be effective in homing to the diseased site (see below). As the lipid coating process relied on extrusion to coat the lipids around the pSiNP–siRNA clusters, the size of the final nanoparticles was set somewhat by the size of the pores (200 nm) in the polycarbonate membrane used in the extruder. We found that extruding the particles through pore sizes of less than 200 nm in size resulted in significant loss of siRNA-loaded pSiNPs.
The processing conditions also influenced the surface charge of the final F-pSiNP formulation, which was slightly positive (Table S1, ESI†). Before the loading chemistry was applied, the empty pSiNPs displayed a negative zeta potential (Table S1, ESI†), attributed to the presence of surface silicon oxide. The calcium chloride condenser chemistry that was used to load siRNAs within the pSiNPs decreased this negative charge substantially, presumably due to ion pairing between Ca2+ and the negatively charged silicate and nucleic acid components. The DOTAP lipid component of the fusogenic coating is cationic, and addition of the fusogenic liposome coating therefore decreased the negative charge further, to the point that the final formulation displayed a slightly positive zeta potential (Table S1, ESI†).
Confocal microscopy was used to visualize the intracellular localization of the targeted fusogenic nanoparticles, their cellular uptake, and cytosolic delivery of siRNA in a J774a.1 macrophage cell line. For these experiments we incorporated fluorescent dyes into either the fusogenic coating (lipophilic DiI or DiO) or into the siRNA payload contained within the pSiNP core (Cy3-labeled siRNA) of the F-pSiNPs in order to independently track the fusogenic coating and the nanoparticle payload. We additionally used the fluorescent indicator LysoTracker Green, which selectively stains lysosomes, in order to assess whether or not the nanoparticle components became co-localized with lysosomes. The left panel of Fig. 1b shows a representative cell treated with fusogenic nanoparticles where the fusogenic coating included lipophilic DiI (red) and the payload was not labelled. The signal from the red DiI dye stained the cell's plasma membrane, indicating successful fusion of the F-pSiNPs which resulted in transfer of DiI from the particle's lipid coating to the cell's plasma membrane, as has been reported previously.29 The fusogenic mechanism is further supported by the observation that the red DiI label associated with the fusogenic coating did not co-localize with the lysosomes (labelled green), indicating that the uptake pathway does not involve endosomal capture. The right panel on Fig. 1b shows a cell treated with similar fusogenic nanoparticles, where the siRNA payload was labelled with Cy3 (red) and the fusogenic coating was labelled with lipophilic DiO (green). Consistent with the above DiI experiments and prior results,29 the green DiO signal stained the plasma membrane. The Cy3-labelled siRNA payload was found in the cytoplasm as punctate spots in the perinuclear region, indicating that the fusogenic coating was shed upon entry into the cell.
The same set of experiments was then performed using non-fusogenic particles, which were prepared with a structure identical to the fusogenic nanoparticles but differing only in the composition of the lipid shell. These experiments were executed to validate the performance of the fusogenic formulation, as the non-fusogenic lipid coating used was that of a more conventional liposome that typically is endocytosed by cells.23,26,30,31 The left panel on Fig. 1c shows an experiment similar to that in the left panel of Fig. 1b except using non-fusogenic nanoparticles: a cell treated with nanoparticles labelled with DiI (red) in their lipid coating and in which the lysosomes were stained with LysoTracker Green. The red DiI and green lysosome signals co-localized, and there was no visible staining of the plasma membrane, which indicates endocytic uptake. Similarly, the right panel on Fig. 1c used non-fusogenic lipids and can be directly compared to the right panel on Fig. 1b which used fusogenic lipids. In this latter case, a cell treated with non-fusogenic nanoparticles containing Cy3-labelled siRNA (red) and lipophilic DiO (green) in the lipid coating showed co-localization of the DiO and Cy3-labelled siRNA signals, with no visible plasma membrane staining, indicating entrapment of the lipid-coated particles with their siRNA payload in the lysosomes, as expected for an endocytosis uptake pathway. This data supports the hypothesis that fusogenic coatings should be more effective at delivering the siRNA payload to the cells, as literature shows that 70% of endocytosed siRNA is excreted out of the cell, with the majority of the remaining 30% degrading within the acidic lysosomal compartments.32–34 Therefore, we expected that delivery of siRNA using the fusogenic nanoparticles would result in a higher gene silencing effect.
In vitro and in vivo gene silencing efficiency
With the confirmation of the cellular fusion behavior of the particles, we next quantified their in vitro gene silencing effect using quantitative real time-polymerase chain reaction (qRT-PCR). Fig. 2a shows that fusogenic nanoparticles that were loaded with siRNA against the Irf5 gene (siIRF5) and conjugated with the activated macrophage-targeting peptide, CRV (F-siIRF5-CRV), attained 86% silencing effect in J774a.1 macrophages in vitro, comparable to the degree of silencing (80%) that was obtained when the commercially available transfection agent Lipofectamine® 2000 was used to transfect the cells with siIRF5 (LF-siIRF5). On the other hand, non-fusogenic particles containing the same siIRF5 payload and the CRV homing peptide (NF-siIRF5-CRV) achieved only ∼45% gene silencing, which was significantly less effective than the fusogenic counterpart (p = 0.001).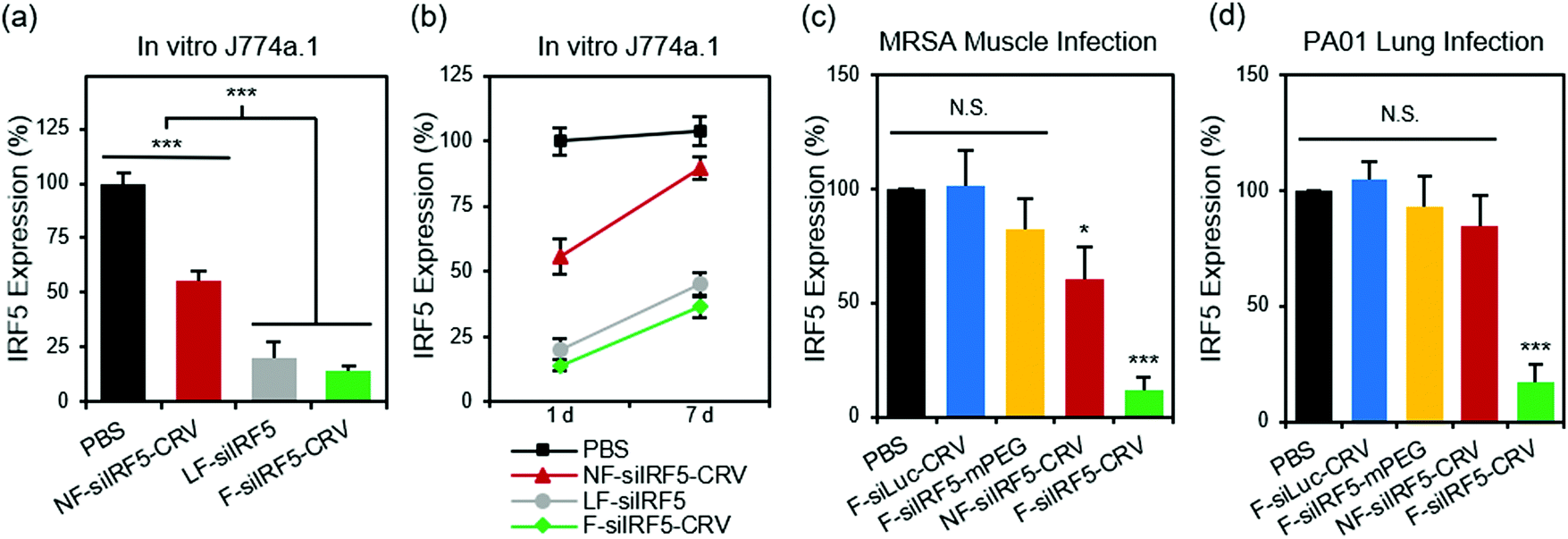 | ||
| Fig. 2 In vitro and in vivo gene silencing efficiency of fusogenic nanoparticles. (a) In vitro knockdown of irf5 gene expression in J774a.1 macrophages treated with PBS, non-fusogenic pSiNPs loaded with siIRF5 and conjugated with CRV peptide (NF-siIRF5-CRV), Lipofectamine 2000 with siIRF5 (LF-siIRF5), or fusogenic pSiNPs loaded with siIRF5 and conjugated with CRV peptide (F-siIRF5-CRV). Bars represent standard deviation with n = 6; (b) irf5 gene expression change from 1 day to 7 days post-transfection of J774a.1 macrophages with PBS, NF-siIRF5-CRV, LF-siIRF5, or F-siIRF5-CRV. Bars represent standard deviation with n = 6; (c) irf5 gene expression in purified macrophages from the MRSA muscle infection site of mice that were intravenously injected with PBS, fusogenic pSiNPs loaded with siLuc and conjugated with CRV peptide (F-siLuc-CRV), fusogenic pSiNPs loaded with siIRF5 without peptide conjugation (F-siIRF5-mPEG), NF-siIRF5-CRV, or F-siIRF5-CRV. Bars represent standard deviation with n = 6; (d) irf5 gene expression in purified macrophages from the PA01 lung infection of mice that were intravenously injected with PBS, F-siLuc-CRV, F-siIRF5-mPEG, NF-siIRF5-CRV, or F-siIRF5-CRV. Bars represent standard deviation with n = 6; N.S. represents no statistical significance and ***represents p < 0.01 from one-way ANOVA with Tukey's post hoc analyses. | ||
To determine the duration of the silencing effect, we maintained cultures of J774a.1 macrophages for a week post-transfection, and conducted another qRT-PCR analysis of Irf5 gene expression (Fig. 2b). While the degree of gene expression in NF-siIRF5-CRV-treated cells recovered from 55% to approximately 90% in 7 days, the F-siIRF5-CRV particle-treated cells maintained levels of expression well below 50% throughout the same time period, with the level increasing from 14% to 37%. The result was comparable to that observed with the benchmark Lipofectamine approach (LF-siIRF5), which showed recovery of gene expression from 20% to 45%. This result suggests that the F-siIRF5-CRV formulation used as a therapeutic might require less frequent dosing compared with conventional endocytic formulations (liposomal or polymer nanoparticle delivery systems) and small molecule drugs.21
We next determined the cytotoxicity of the nanoformulations compared to the Lipofectamine® 2000 standard, which is not suitable for in vivo use due to toxicity and becomes increasingly cytotoxic in vitro with increasing incubation time. Fig. S2 (ESI†) compares equivalent doses of siRNA in J774a.1 macrophages. Lipofectamine® 2000 (LF-siIRF5) decreased cell viability beginning after 4 h of incubation; cell viability decreased to 22% after 24 h of incubation (p < 0.03). In contrast, cells treated with the particle formulations NF-siIRF5-CRV, F-siIRF5-mPEG, and F-siIRF5-CRV all retained >80% cell viability for at least 6 h of incubation, and ∼60% viability was retained after 24 h of incubation (pF-siIRF5-CRV < 0.035; pNF-siIRF5-CRV < 0.048; pF-siIRF5-mPEG < 0.039). Thus, the nanoformulation is more biocompatible for in vitro transfection compared to the benchmark Lipofectamine method.
To determine if the superior gene silencing efficiency and biocompatibility of the fusogenic nanoparticles translated to in vivo models of infection, we established a MRSA muscle infection mouse model. While prior work had established the approach for treatment of a non-resistant strain (S. aureus subsp. aureus Rosenbach), here we chose to test MRSA because of its antibiotic resistance, in order to provide a more rigorous test of whether or not reprogramming of macrophages could be an effective treatment. The muscle infection model was chosen to address the problem of infection of the skeletal muscles (pyomyositis). Though a relatively rare occurrence compared to superficial skin or other types of infections, cases of muscle infection are a growing concern, particularly in patients with pre-existing conditions or with deep wound injuries.35–37 For the mouse model we chose direct intramuscular injection of the bacteria, as we found systemic introduction of bacteria to be uncontrolled and too fatal to generate reliable and consistent results. Thus, MRSA colonies were intramuscularly injected in the right hind thigh of each animal, and the abscess was allowed to form over 3 days before intravenously injecting the treatment formulations. After 24 h, we harvested the right popliteal lymph node and the right hind thigh muscles for homogenization and macrophage purification. The Irf5 gene expression was quantified in the purified macrophages using qRT-PCR. Fig. 2c shows that the fusogenic, peptide-targeted F-siIRF5-CRV treatment obtained a dramatic 89% knockdown (p = 0.001) of Irf5 gene expression in macrophages isolated from the infection site. Controls involving PBS, a fusogenic formulation using the CRV homing peptide but containing siRNA against luciferase (F-siLuc-CRV), and a fusogenic formulation containing siIRF5 but with no homing peptide (F-siIRF5-mPEG) showed negligible gene silencing effects, while a control using the non-fusogenic NF-siIRF5-CRV treatment resulted in ∼40% knockdown (p = 0.023). These results demonstrate that a fusogenic coating and an effective targeting peptide on the exterior of the nanoparticle are both necessary to obtain high knockdown efficiency from the nanoparticles in vivo.
We next explored the species specificity (Gram-positive vs. Gram-negative) of macrophage targeting and knockdown. For our Gram-negative infection model, we established lung infection in mice by intratracheal delivery of Pseudomonas aeruginosa via a catheter. 24 h post-infection, we intravenously injected the treatment formulations for 24 h circulation. Animals were sacrificed, bronchoalveolar lavage fluid (BALF) was collected, and the lungs were then harvested for homogenization. Macrophages were purified from both the BALF and the lung homogenates, and Irf5 gene expression was quantified using qRT-PCR. Fig. 2d shows that only the F-siIRF5-CRV formulation induced a significant knockdown (83%, p = 0.001) of Irf5 gene expression in the macrophages associated with this pneumonia model.
In vivo infection homing to MRSA and PA01 infection
While the above results established that the fusogenic nanoparticles are able to induce a stronger RNAi effect relative to either non-fusogenic particles or non-targeted particles in vivo, the nanoparticles must also home to specific cell types to minimize off-target effects. Thus, we next evaluated the targeting efficiency of CRV-conjugated particles to infected tissues. The CRV peptide targets the retinoid X receptor beta (RXRB), which at least partially shifts from the intracellular localization to the cell surface upon macrophage activation.22
Fig. 3a shows DiI-loaded particle localization in a MRSA muscle infection model. Infected mice were intravenously injected with the fusogenic nanoparticle formulation containing an siIRF5 payload and the CRV targeting peptide (F-CRV). Injections of PBS, a fusogenic formulation containing siIRF5 but with no homing peptide (F-mPEG), and a non-fusogenic formulation containing siIRF5 and the CRV targeting peptide (NF-CRV) were used as controls. The test mice along with healthy control mice were sacrificed at either 1 h or 24 h timepoints post-injection, and the hind thigh muscles [both the contralateral (C) muscle on the opposing side of the infection, and the ipsilateral (I) infected muscle] were harvested for ex vivo imaging. The qualitative images showed that all nanoparticle formulations accumulated to some degree in the infected muscle relative to the uninfected muscle; this is consistent with previous observations that inflamed tissues are passively targeted by nanoparticles.38–40 Whereas the NF-CRV and F-mPEG formulations showed moderately low levels of accumulation, accumulation of the F-CRV formulation was quite strong. Quantification of these images (Fig. 3b) showed significant accumulation to the ipsilateral muscle compared to the contralateral muscle for the F-mPEG (p1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h = 0.038; p24h = 0.032), NF-CRV (p1h = 0.046; p24h = 0.001), and F-CRV (p1h = 0.001; p24h = 0.001) groups, and the accumulation of the F-CRV formulation was significantly greater (p = 0.001) compared to the control formulations at both 1 h and 24 h timepoints.
h = 0.038; p24h = 0.032), NF-CRV (p1h = 0.046; p24h = 0.001), and F-CRV (p1h = 0.001; p24h = 0.001) groups, and the accumulation of the F-CRV formulation was significantly greater (p = 0.001) compared to the control formulations at both 1 h and 24 h timepoints.
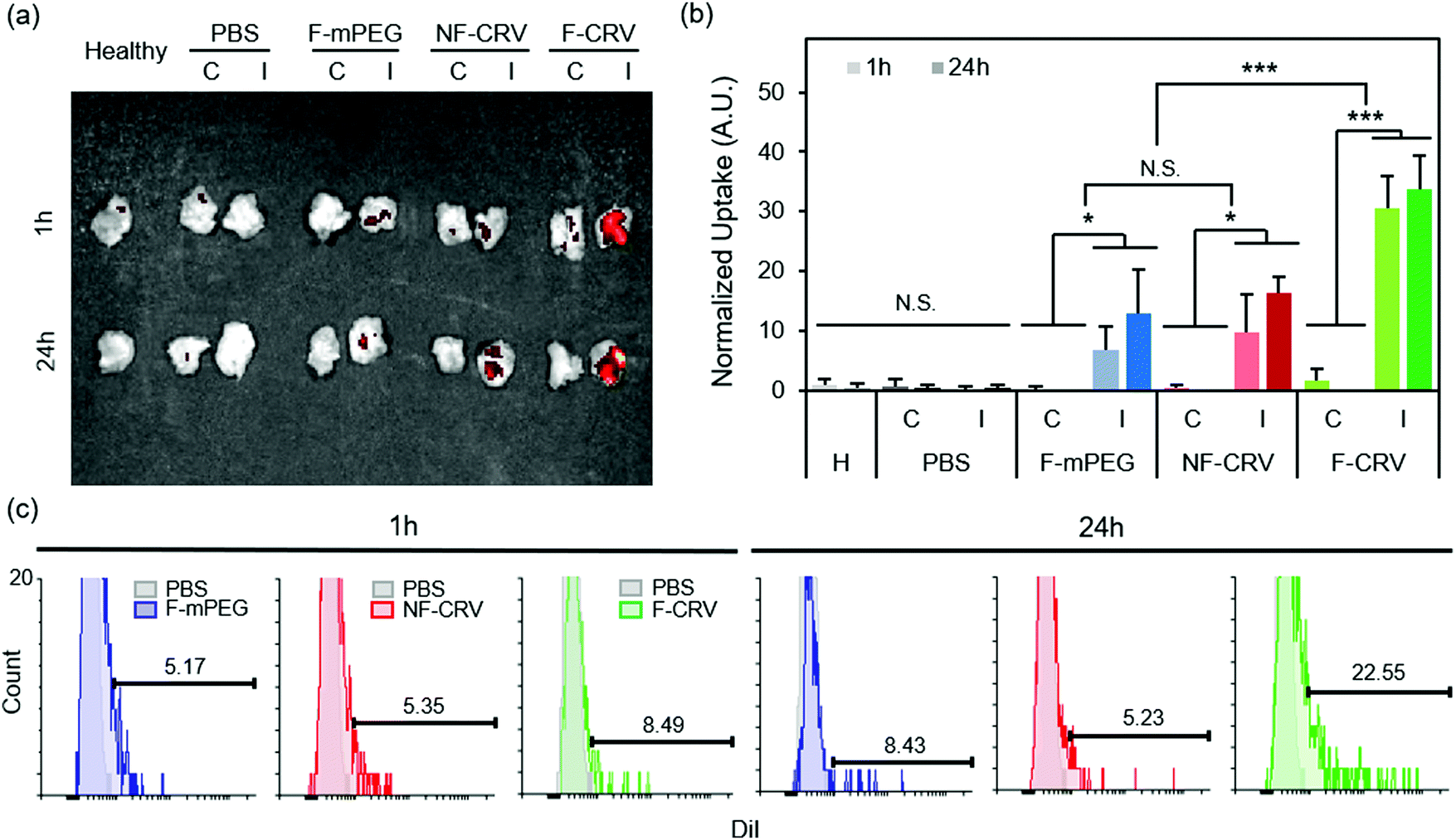 | ||
| Fig. 3 In vivo infection homing to MRSA and PA01 infection. (a and b) DiI signal accumulation in hind leg muscles (‘C’ represents the contralateral muscle to the infected side, and ‘I’ represents ipsilateral muscles with the infection) of healthy (H) mice, and MRSA-infected mice. Mice were intravenously injected with PBS, DiI-loaded fusogenic pSiNPs loaded with siIRF5 without peptide conjugation (F-mPEG), DiI-loaded non-fusogenic pSiNPs loaded with siIRF5 and conjugated with CRV peptide (NF-CRV), or DiI-loaded fusogenic pSiNPs loaded with siIRF5 and conjugated with CRV peptide (F-CRV); (a) representative image obtained with IVIS 200 imaging system; (b) ImageJ quantification of the DiI accumulation signals from IVIS 200 images (n = 3). Bars represent standard deviation with; N.S. indicates no significance, *indicates p < 0.05 and ***indicates p < 0.01 from one-way ANOVA with Tukey's HSD post hoc analyses; (c) DiI signal quantification using flow cytometry of macrophages purified from the bronchoalveolar lavage (BAL) fluid and the infected lung homogenates. Samples were harvested from mice intravenously injected with DiI loaded F-mPEG, NF-CRV, or F-CRV. Data are representative of n = 3. | ||
We quantified CRV-mediated particle accumulation to PA01 pneumonia by assaying purified macrophages from the BAL fluid and from the lung homogenates of PA01-infected mice. For this we measured the DiI signal (the dye associated with the lipid coat on the nanoparticles) by flow cytometry (Fig. 3c). At 1 h post-intravenous injection of the F-CRV, and the PBS, DiI-loaded F-mPEG, and NF-CRV control formulations, there was a negligible difference in DiI accumulation in the macrophages between any of the formulations. However, 24 h post-injection, the F-CRV formulation showed a substantial increase in accumulation; 23% of macrophages showed positive DiI signals. By contrast, the F-mPEG and NF-CRV control formulations showed no substantial changes in accumulation in this same time period.
The data here are consistent with findings in the previous report regarding S. aureus pneumonia models, where both the CRV peptide and the fusogenic coating were required to obtain macrophage targeting and uptake into those cells.23 The lack of notable accumulation of non-fusogenic particles in that model and in the MRSA muscular infection and PA01 pneumonia models of the present work implies that simply docking the nanoparticle to the cell surface (via the CRV-to-cell-surface receptor interaction) is not sufficient to see strong accumulation in macrophages. All three models confirm that the CRV-targeted, fusogenic nanoparticles provide superior accumulation in macrophages associated with infected tissues.
In vivo therapeutic efficacy in MRSA muscle infection
Having established the ability of the targeted fusogenic nanoparticles to selectively home to macrophages at the infection site and effectively silence the Irf5 gene, we next investigated the therapeutic efficacy of this treatment. Fig. S3 (ESI†) shows photographic tracking of MRSA-induced abscess in the hind thigh. Mice were intravenously injected with the CRV peptide-targeted, fusogenic formulation containing siIRF5 (F-siIRF5-CRV). Controls included PBS, 145 mg kg−1 vancomycin (the typical therapeutic dose based on the human dose of approximately 7 mg kg−141,42), a fusogenic formulation containing the CRV homing peptide but delivering siRNA against luciferase (F-siLuc-CRV, negative control), and the non-fusogenic formulation containing siIRF5 and the CRV peptide (NF-siIRF5-CRV). At 3, 7, and 14 days after the injection, mice were sacrificed for abscess observation. While PBS- and F-siLuc-CRV injected mice showed large abscess formation over time, the vancomycin- and NF-siIRF5-CRV injected mice showed a marked reduction in the abscess size, although the infection visibly persisted over the 14-day test period. On the other hand, the F-siIRF5-CRV treatment group showed a more substantial reduction in the abscess size within 3 days of treatment, and by 7 days, the abscess was unidentifiable by eye, and showed no visible difference from the healthy control animals (Fig. S3, ESI†).
The abscess formation and bacterial clearance was further studied using histopathology of the infected muscles. At day 7 post-treatment, mice were sacrificed, and the infected muscles were harvested and fixed in 4% PFA to be sectioned for qualitative analyses using hematoxylin and eosin (H&E) and Gram stains (arrows indicate spherical Gram-positive bacteria in purple dots). The H&E stains in Fig. 4a show that while PBS, vancomycin, F-siLuc-CRV, and NF-siIRF5-CRV treatment groups resulted in large abscess formations (purple) within the muscle tissue (pink), the F-siIRF5-CRV treatment resulted in only a light and mild abscess formation on the right hand side of the tissue, and the majority of the section maintained a pink fibrous structure similar to that seen in the healthy control.
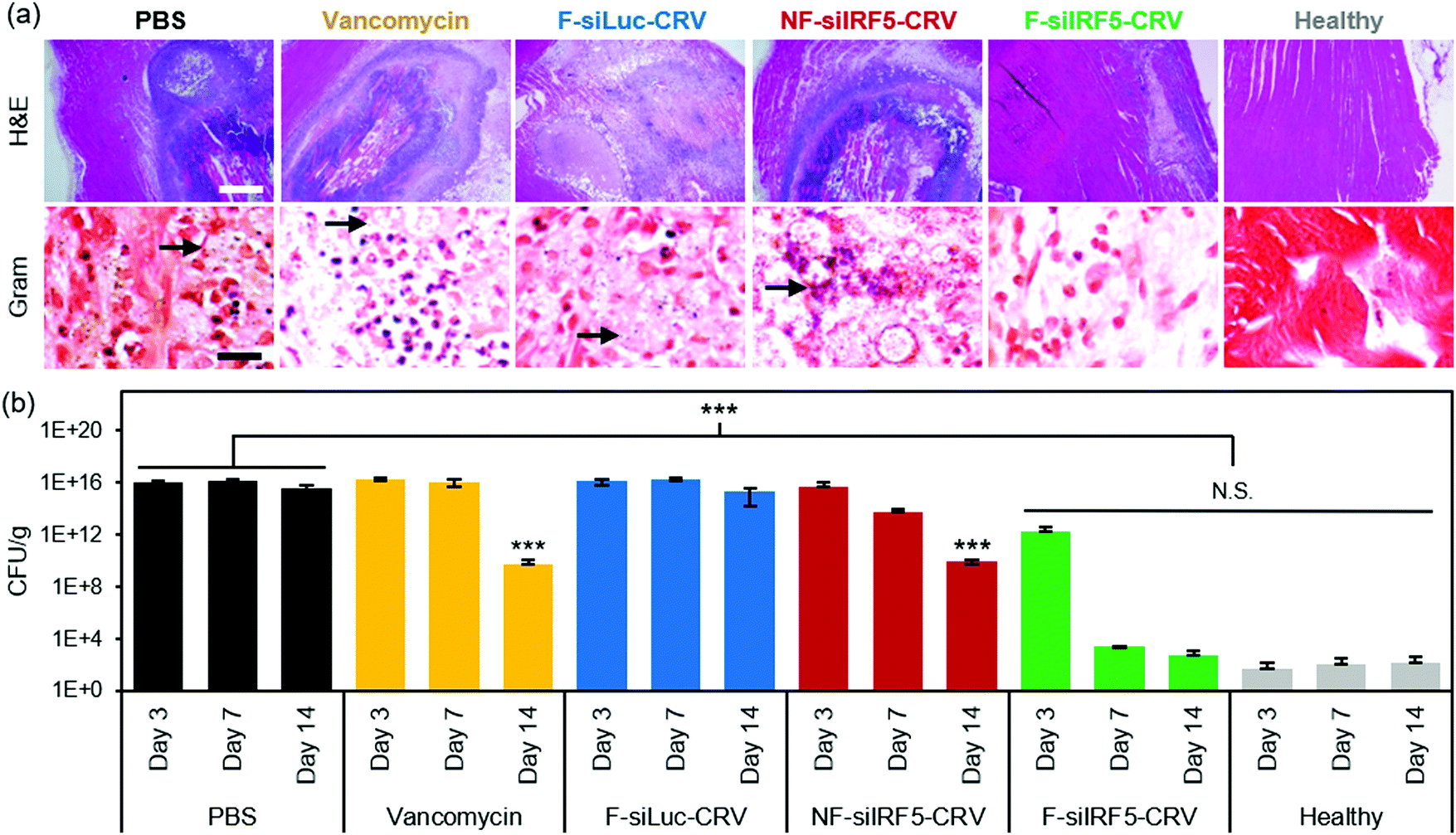 | ||
| Fig. 4 In vivo therapeutic efficacy in MRSA muscle infection. (a) H&E (top) and Gram (bottom) stains of infected muscle tissues from mice 7 days after treatment with PBS, vancomycin, non-fusogenic, targeted pSiNPs containing siIRF5 (NF-siIRF5-CRV), fusogenic, CRV-targeted pSiNPs containing siRNA against luciferase, as a negative control for siIRF5 (F-siLuc-CRV), and fusogenic, CRV-targeted pSiNPs containing siIRF5 (F-siIRF5-CRV); scale bar represents 1 mm (top row) and 100 μm (bottom row); (b) bacterial titer (CFU per mass of tissue) from muscles of healthy and MRSA-infected mice intravenously injected with the treatment formulations as indicated. Animals were infected on day 0 and therapeutic or control injections were given on day 1. Mice were sacrificed for titer counts at days 3, 7, and 14 days post-intravenous injection. Bars indicate standard deviation with n = 6. N.S. represents no significance and ***represents p < 0.01 relative to the PBS group from one-way ANOVA with Tukey's HSD post hoc analyses. | ||
In order to quantifiably determine the therapeutic effect, we homogenized the thighs at each time point (n = 6 animals per group) and conducted a serial dilution for agar plating. The number of colony forming units (CFUs) grown on the agar plate from the homogenate dilutions were counted and the thighs were weighed and are reported in Fig. 4b. Similar to the photographic findings, the PBS and F-siLuc-CRV treatments resulted in over 1 × 1015 CFU per g in the thigh throughout the 14 days, while the vancomycin and NF-siIRF5-CRV treatments resulted in a moderate decrease in the bacterial titer to >1 × 109 CFU per g by day 14. In contrast, the F-siIRF5-CRV treatment resulted in <6 × 102 CFU per g by day 14, which was not statistically different from the healthy control counts (p > 0.9). Thus, the F-siIRF5-CRV therapeutic is able to recover the MRSA-infected thigh to a healthy state within one week of administration.
In vivo therapeutic efficacy in PA01 lung infection
Finally, we tested therapeutic effects of the fusogenic particles in the PA01 pneumonia model. First, we compared a non-fusogenic formulation containing siIRF5 and CRV targeting peptide (NF-siIRF5-CRV) with the fusogenic formulation containing siIRF5 and CRV peptide (F-siIRF5-CRV) intravenously injected in mice with PA01 pneumonia. Healthy and infected mouse groups with PBS injections were used as controls. At the end-point [7 days post-injection for healthy and F-siIRF5-CRV groups, and ad mortem (under 7 days) for infected and NF-siIRF5-CRV groups], mouse lungs were inflated and harvested for histopathological analyses (Fig. 5a). Similar to the infected control group with no treatment, the NF-siIRF5-CRV lung showed a trend of excessive inflammation with neutrophil infiltration, and presence of Gram-negative rods in the Gram stain (Fig. 5a, NF-siIRF5-CRV inset with arrows). On the other hand, the F-siIRF5-CRV treatment group showed a normal morphology with expanded alveoli and no presence of Gram-negative bacteria.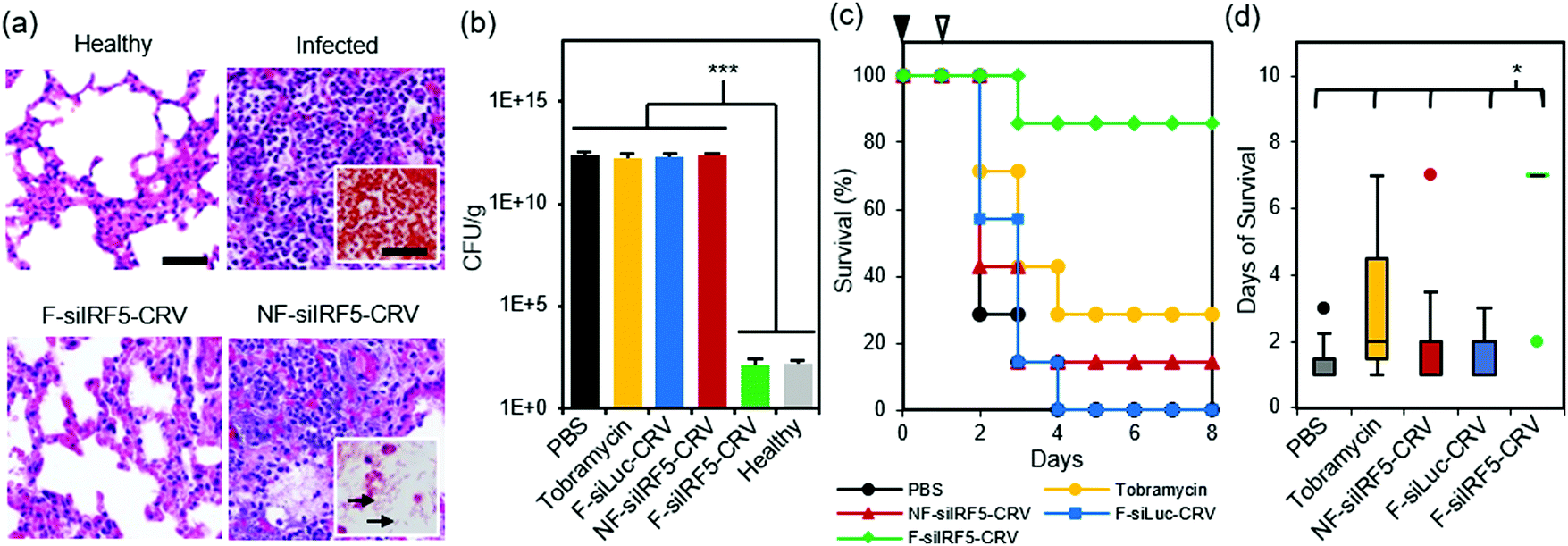 | ||
| Fig. 5 In vivo therapeutic efficacy in PA01 lung infection. (a) H&E stained sections of mouse lungs subjected to histopathological analyses. Top left panel shows healthy mouse with no treatment, top right panel shows lung of infected mouse with no treatment (insets shows Gram stain), bottom left panel shows lung of infected mouse treated with the fusogenic, CRV-targeted pSiNP formulation F-siIRF5-CRV, and bottom right panel shows lung of infected mouse treated with the non-fusogenic, targeted pSiNP formulation NF-siIRF5-CRV control (inset shows Gram stain of rod-shaped PA01 populations (arrow) in the lung); (b) bacterial titers from lungs of PA01-infected mice intravenously injected with: PBS; tobramycin; fusogenic, CRV-targeted pSiNPs containing siRNA against luciferase as a negative control for siIRF5 (F-siLuc-CRV); non-fusogenic, CRV-targeted pSiNPs containing siIRF5 as a negative control for fusogenic component (NF-siIRF5-CRV); and fusogenic, CRV-targeted pSiNPs containing siIRF5 (F-siIRF5-CRV). Titers from healthy mice shown for comparison. Titers were analyzed at either ad mortem (PBS, Tobramycin, F-siLuc-CRV, NF-siIRF5-CRV) or at 7 days post-injection (F-siIRF5-CRV). Bars indicate standard deviation with n = 4; (c) mouse survival post-infection (at day 0; black arrowhead) and post-therapeutic injection (at day 1; white arrowhead) of PBS, Tobramycin, NF-siIRF5-CRV, F-siLuc-CRV, or F-siIRF5-CRV, as indicated. Each group has n = 7 mice. (d) Average days of survival of mice from (c) post-infection (at day 0) and post-therapeutic injection (at day 1). ***represents p < 0.01 and *represents p < 0.05 from one-way ANOVA with Tukey's HSD post hoc analyses. | ||
A similar trend was observed when we quantified bacterial titers in lung homogenates at the same end-points (Fig. 5b; ad mortem for mice that were moribund under 7 days post-injection, and the 7-day point for surviving mice). While controls involving PBS, 100 mg kg−1 tobramycin, a fusogenic formulation containing siRNA against luciferase and pendant CRV homing peptides (F-siLuc-CRV), and the non-fusogenic formulation containing siIRF5 and pendant CRV peptides (NF-siIRF5-CRV) all showed >1 × 1012 CFU per g, the F-siIRF5-CRV formulation significantly decreased the titer to approximately 1 × 102 CFU per g in 7 days (p < 0.005), which was not significantly different from the healthy control (p = 0.9). The tobramycin control was used in these experiments as a benchmark because it is a common therapeutic used to treat Pseudomonas and other Gram-negative infections.43–47
Lastly, we tallied the survival of PA01 pneumonia-carrying mice that were intravenously administered with PBS, 100 mg kg−1 tobramycin, NF-siIRF5-CRV, F-siLuc-CRV, or F-siIRF5-CRV. Mice were infected on day 0, and the therapeutics were intravenously injected on day 1, and mice were observed for the following 7 days. Fig. 5c shows that while the PBS, NF-siIRF5-CRV, and F-siLuc-CRV groups yielded less than 20% survival rate, the tobramycin treatment group yielded a moderate 30% survival. The mediocre result attained by the standard antibiotic benchmark may be due to the fact that tobramycin is clinically administered at 1 mg kg−1 three times daily in humans,43 while the intravenous dosing range of tobramycin in mouse models reported in the literature ranges from 10–400 mg per kg per day.44–47 The treatment regimen used in the present study used only a single administration of 100 mg kg−1 at 24 h post-infection to match the immunotherapy nanoformulations, which require only single-doses. In contrast, a single administration of the F-siIRF5-CRV formulation rescued 6 out of 7 mice in its cohort to complete recovery within 7 days of treatment, which was significantly more effective than all other treatment groups (p < 0.024) (Fig. 5d). Thus, the F-siRF5-CRV treatment in mice was found to be an effective immunotherapy against MRSA muscular and PA01 lung infection models.
Conclusions
Bacterial infection has returned as an increasing threat in the era of antibiotic-resistance, and solutions to reduce its threat have become a high priority globally. While there have been increasing research into antibiotics development and FDA clearance of novel classes of antibiotics to combat the prevailing “superbugs”, it is inevitable that further resistance development will occur.1,2,4,11,48–52 Moreover, Gram-negative bacteria have remained a challenging target to treat because of their dual cell wall. Thus, the present study aimed to develop a solution that is independent of small molecule antibiotics, by modulating the innate immune system.The fusogenic porous silicon nanoparticles first emerged as a potential immunotherapy platform when they showed outstanding in vivo homing to activated macrophages and significant gene silencing that led to an Irf5-depleted anti-inflammatory immune response. This effect helped focus the immune system in clearing out the bacteria and minimizing auto-immune damage caused by excessive inflammation and fibrosis.23 As immunotherapy offers a solution that should operate regardless of the pathogen type, we explored its effect in antibiotic-resistant (MRSA) and Gram-negative (P. aeruginosa) bacterial infections in deep tissues (muscle and lungs) that are difficult-to-reach by standard oral or dermal administrations. The fusogenic nanoparticles carrying siIRF5 and the CRV homing peptide demonstrated potent therapeutic potential in both MRSA muscular and PA01 lung infections, demonstrating its broad-spectrum protection against Gram-positive, Gram-negative, and antibiotic-resistant bacteria.
The approach presented in this study serves as a generalizable anti-inflammatory therapeutic, which may have potential in other diseases characterized by excessive immune response (e.g. autoimmune disorders, inflammatory bowel disease, atherosclerosis, etc.) and for other types of infections (such as viral) where the pathogens themselves may not be the primary contributors to patient mortality.53 Moreover, the modular approach presented here implies that simple exchange of the targeting peptide and the siRNA payload will allow treatment of other diseases that could respond to gene modulation. For example, using the appropriate combination of siRNA and targeting peptide, cancer cells and tumor-associated macrophages have also been reprogrammed for a positive therapeutic outcome in mouse tumor models.29 Despite the recent successful translation of siRNA therapy into the clinic,54 the deployment of an effective delivery system for RNAi therapeutics remains a formidable challenge.21 As siRNA and RNAi-mediated gene silencing induces a transient yet relatively lasting (>7 days) effect, the fusogenic nanosystem deployed in this work presents a promising platform technology for gene therapy.
Experimental
Materials
Highly boron-doped p-type silicon wafers [∼1 mΩ cm resistivity, polished on the (100) face] were obtained from Virginia Semiconductor, Inc or Siltronix, Inc. Hydrofluoric acid (48% aqueous, ACS grade) was obtained from Fisher Scientific. Anhydrous calcium chloride was obtained from Spectrum Chemicals (Gardena, CA). Deionized (18 mΩ) water was used for all aqueous dilutions. For lipids, 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)]-2000 (DSPE-mPEG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG-maleimide), and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) were purchased from Avanti Polar Lipids (Alabaster, AL) and stored at −4 °C. Fluorescent dyes hydrophobic 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, Life Technologies) and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO, Life Technologies) were used, and LysoTracker™ Green DND-26 and Lipofectamine® 2000 Transfection Reagent were obtained from Thermo Fisher Scientific. Custom siRNAs were purchased from Dharmacon (Lafayette, CO), and primers were purchased from IDT DNA (San Diego, CA). Macrophage-targeting peptide (CRV) was custom synthesized by CTC Scientific (Sunnyvale, CA). For in vitro studies, Raw 264.7 and J774a.1 cells were purchased from ATCC (Manassas, VA) within 6 months prior to all experiments. DMEM cell media was purchased from GE Healthcare Life Sciences (HyClone, Pittsburg, PA), with supplemental fetal bovine serum (HyClone, Pittsburg, PA) and penicillin/streptomycin (HyClone, Pittsburg, PA). Staphylococcus aureus subsp. aureus Rosenbach (ATCC® 25923™) was purchased from ATCC (Manassas, VA) within 6 months prior to all experiments, and 6 week-old male Balb/C were purchased from Envigo (Placentia, CA). Tobramycin was purchased from Sigma Aldrich (St. Louis, MO). Vancomycin was purchased from Cayman Chemical Company (Ann Arbor, MI).Preparation of porous silicon nanoparticles
Porous silicon (pSi) samples were prepared by electrochemical etching of silicon wafers in an electrolyte consisting of 3:1 (v:v) of 48% aqueous HF:ethanol (CAUTION: HF is highly toxic and proper care should be exerted to avoid contact with skin or lungs). A silicon working electrode with an exposed area of 8.6 cm2 was contacted on the back side with aluminum foil and mounted in a Teflon cell. The silicon wafer was then anodized in a two-electrode configuration with a platinum counter electrode, by applying an alternating current of square waveform, with lower current density of 50 mA cm−2 for 0.6 s and high current density of 400 mA cm−2 for 0.36 s repeated for 500 cycles. Then the porous layer is lifted off by etching at a constant current density of 3.7 mA cm−2 for 250 s in a 1:20 (v:v) of 48% aqueous HF:ethanol solution, to be sonicated in deionized water for 12 h into nanoparticles. Fluorescent dye and siRNA payloads were loaded into the pSiNPs by pore sealing by calcium silicate formation; the calcium silicate sealing chemistry has demonstrated high efficiency in loading anionic payloads previously.27 For siRNA loading, we used siIRF5 (IRF5, sense 5′-dTdT-CUG CAG AGA AUA ACC CUG A-dTdT-3′ and antisense 5′-dTdT UCA GGG UUA UUC UCU GCA G dTdT-3′) and siLuc (luciferase, 5′-CUU ACG CUG AGU ACU UCG A dTdT-3′ and antisense 5′-UCG AAG UAC UCA GCG UAA G dTdT-3′). The relevant siRNA was dissolved in RNAse-free water to 150 μM and pipetted gently with 150 μL of pSiNP and 700 μL of 2 M calcium chloride under ultrasonication for 15 min for loading.
Liposomal coating
Fusogenic (F) and non-fusogenic (NF) coatings were prepared from DMPC, DSPE-PEG, and DOTAP at the molar ratio of 76.2:3.8:20 and 96.2:3.8:0, respectively. The lipid films were prepared by evaporating the organic solvent, with 725.5 μg of DMPC, 151.6 μg of DSPE-PEG (methoxy or maleimide terminated), and 196.3 μg of DOTAP (F) or 916.0 μg of DMPC and 151.6 μg of DSPE-PEG (methoxy or maleimide terminated) (NF). The DiI or DiO-incorporated films were added with 26.3 μg of DiI/DiO (1.25 mg mL−1 in 100% ethanol). The films were then hydrated with payload-pSi solution and prepared by film hydration/extrusion; the pSi-hydrated lipid was heated to 40 °C with constant magnetic stirring for 10 min. Then the mixture was extruded through a 200 nm polycarbonate membrane 20 times. CRV (C-Ahx-CRVLRSGSC) was conjugated to maleimide-terminated PEG by mixing 100 μL of 1 mg mL−1 CRV (in deionized water) in 1 mg mL−1 of the liposomal pSi (by lipid mass) overnight at 4 °C. Particles were washed three times at each step by centrifugation in Microcon-30 kDa Centrifugal Filter Unit (EMD Millipore) by spinning at 5000g at 25 °C. The loaded siRNA concentration was quantified with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, ND-2000) after each step of particle formation by checking the ultraviolet absorption of the supernatant and pellet of each wash. Nanoparticle size and zeta-potential were measured by dynamic light scattering (DLS, Zetasizer ZS90, Malvern Instruments), and structural morphology were visualized by JEOL 1200 EX II TEM. Samples were prepared by dropping 5 μL of the sample on the TEM grid, drying off excess solvent after 1 min, and dropping 5 μL of uranyl acetate for negative staining.
In vivo infection model
All animal work was conducted using 6–8-week old male Balb/C mice. For MRSA muscle infection model, the bacteria underwent a 16 h incubation in CAMHB. Then, MRSA was sub-cultured at 1:100, 1:250, and 1:400 dilutions in 5 mL of fresh broth for 2 h to reach growth phase. The optical density at 600 nm was measured using a cuvette spectrometer with the broth set as the blank. 5 mL of bacterial culture at OD600 ≈ 0.5 was centrifuged, the bacteria were washed by centrifugation in PBS three times, and re-suspended in 500 μL of PBS for inoculation, resulting in OD600 ≈ 2.25. Each mouse was intramuscularly injected in the right hind thigh with 50 μL of the MRSA stock (equating to approximately 1.25 × 108 CFU per mouse). For the PA01 pneumonia model, the bacteria underwent a 16 h incubation in brain heart infusion broth. Then, PA01 was sub-cultured at 1:100, 1:250, and 1:400 dilutions in 5 mL of fresh BH broth for 2.5 h to reach growth phase. The optical density at 600 nm was measured using a cuvette spectrometer with the broth set as the blank. 5 mL of bacterial culture at OD600 ≈ 0.7 was centrifuged, the bacteria were washed by centrifugation in PBS three times and re-suspended in 1 mL of PBS for inoculation. Each mouse was infected by intratracheal catheter injection of approximately 1 × 109 CFU of bacteria in 50 μL of PBS. All treatment-injections were performed 3 d (MRSA) or 24 h (PA01) after inoculation of the bacteria.
Conflicts of interest
MJS is a scientific founder, a member of the board of directors, and holds an equity interest in Spinnaker Biosciences, Inc., and Cend Therapeutics. He also has a financial interest (as a consultant, shareholder, scientific advisor, and/or board member) with Bejing ITEC Technologies, Illumina, Matrix Technologies, NanoVision Bio, Pacific Integrated Energy, TruTag Technologies, and Well-Healthcare Technologies. MJS is a Guest Professor at Zhejiang University, China. Although one or more of the grants that supported this research has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to these companies, the research findings included in this particular publication may not necessarily relate to their interests. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. SNB and ER are also founders and hold an equity interest in Cend Therapeutics, which has an option to license some of the technology in this paper.Acknowledgements
BK and QY designed, synthesized, and characterized nanoparticles, performed in vitro and in vivo experiments. BK, QY, LC, SB, ER, MJS discussed and analyzed data. BK, QY, LC, SB, ER, MJS conceived the project and wrote the manuscript. All authors read and approved the manuscript. We thank K. Osborn for histopathology evaluations and I. Andersen for technical assistance. This work was supported in part by the National Science Foundation (NSF CBET-1603177), by the UC San Diego Materials Research Science and Engineering Center (NSF DMR-2011924), and by the National Institutes of Health, (NIH R01 AI132413-01, R01 CA188883 and R56 CA207839). The electron micrographs were obtained in the Cellular and Molecular Medicine Electron microscopy core facility, supported in part by National Institutes of Health (NIH S10 OD023527), and in the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, supported by the National Science Foundation (NSF ECCS-1542148). The work was also partially funded by the University of California San Diego's Dissertation Fellowship. Tissue Technology Shared Resource is supported by a National Cancer Institute Cancer Center Support Grant (CCSG Grant P30CA23100).References
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS.
- C. L. Ventola, P T, 2015, 40, 277–283 Search PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS.
- I. Nikolaidis, S. Favini-Stabile and A. Dessen, Protein Sci., 2014, 23, 243–259 CrossRef CAS.
- L. A. Clifton, F. Ciesielski, M. W. Skoda, N. Paracini, S. A. Holt and J. H. Lakey, Langmuir, 2016, 32, 3485–3494 CrossRef CAS.
- C. González-Bello, Bioorg. Med. Chem. Lett., 2017, 27, 4221–4228 CrossRef.
- D. Corbett, A. Wise, T. Langley, K. Skinner, E. Trimby, S. Birchall, A. Dorali, S. Sandiford, J. Williams, P. Warn, M. Vaara and T. Lister, Antimicrob. Agents Chemother., 2017, 61, e00200–17 CrossRef CAS.
- P. C. Dewan, A. Anantharaman, V. S. Chauhan and D. Sahal, Biochemistry, 2009, 48, 5642–5657 CrossRef CAS.
- D. Uppu, M. M. Konai, P. Sarkar, S. Samaddar, I. C. M. Fensterseifer, C. Farias-Junior, P. Krishnamoorthy, B. R. Shome, O. L. Franco and J. Haldar, PLoS One, 2017, 12, e0183263 CrossRef.
- P. Bernal, C. Molina-Santiago, A. Daddaoua and M. A. Llamas, Microb. Biotechnol., 2013, 6, 445–449 CrossRef.
- A. Manohar, J. Ahuja and J. K. Crane, Immunol. Invest., 2015, 44, 731–737 CrossRef CAS.
- D. Roux, G. B. Pier and D. Skurnik, J. Antimicrob. Chemother., 2012, 67, 2785–2787 CrossRef CAS.
- R. Babb and L. A. Pirofski, Virulence, 2017, 8, 1055–1058 CrossRef.
- B. D. Navalkele and T. Chopra, Biologics, 2018, 12, 11–21 CAS.
- C. Saylor, E. Dadachova and A. Casadevall, Vaccine, 2009, 27(Suppl 6), G38–G46 CrossRef CAS.
- M. H. Wilcox, D. N. Gerding, I. R. Poxton, C. Kelly, R. Nathan, T. Birch, O. A. Cornely, G. Rahav, E. Bouza, C. Lee, G. Jenkin, W. Jensen, Y. S. Kim, J. Yoshida, L. Gabryelski, A. Pedley, K. Eves, R. Tipping, D. Guris, N. Kartsonis and M. B. Dorr, N. Engl. J. Med., 2017, 376, 305–317 CrossRef CAS.
- S. O. Ali, X. Q. Yu, G. J. Robbie, Y. Wu, K. Shoemaker, L. Yu, A. DiGiandomenico, A. E. Keller, C. Anude, M. Hernandez-Illas, T. Bellamy, J. Falloon, F. Dubovsky and H. S. Jafri, Clin. Microbiol. Infect., 2019, 25, 629.e621 Search PubMed.
- J. M. Bennett, G. Reeves, G. E. Billman and J. P. Sturmberg, Front. Med., 2018, 5, 316 CrossRef.
- C. K. Lin and B. I. Kazmierczak, J. Innate Immun., 2017, 9, 250–261 CrossRef CAS.
- M. Merad and J. C. Martin, Nat. Rev. Immunol., 2020, 20, 355–362 CrossRef CAS.
- B. Kim, J.-H. Park and M. J. Sailor, Adv. Mater., 2019, 31, 1903637 CrossRef CAS.
- T. Tang, Y. Wei, J. Kang, Z. G. She, D. Kim, M. J. Sailor, E. Ruoslahti and H. B. Pang, J. Controlled Release, 2019, 301, 42–53 CrossRef CAS.
- B. Kim, H. B. Pang, J. Kang, J. H. Park, E. Ruoslahti and M. J. Sailor, Nat. Commun., 2018, 9, 1969 CrossRef.
- S. DeLeon, A. Clinton, H. Fowler, J. Everett, A. R. Horswill and K. P. Rumbaugh, Infect. Immun., 2014, 82, 4718–4728 CrossRef.
- E. R. Sydnor and T. M. Perl, Clin. Microbiol. Rev., 2011, 24, 141–173 CrossRef CAS.
- B. Kim and M. J. Sailor, JoVE, 2019, e59440, DOI:10.3791/59440.
- J. Kang, J. Joo, E. J. Kwon, M. Skalak, S. Hussain, Z.-G. She, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2016, 28, 7962–7969 CrossRef CAS.
- E. Choi, J. Lee, I. C. Kwon and S. Kim, Biomaterials, 2019, 209, 126–137 CrossRef CAS.
- B. Kim, S. Sun, J. A. Varner, S. B. Howell, E. Ruoslahti and M. J. Sailor, Adv. Mater., 2019, 31, 1902952 CrossRef.
- J. Kim, O. A. Santos and J.-H. Park, J. Controlled Release, 2014, 191, 98–104 CrossRef CAS.
- J. Lee, J. Kim, M. Jeong, H. Lee, U. Goh, H. Kim, B. Kim and J.-H. Park, Nano Lett., 2015, 15, 2938–2944 CrossRef CAS.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653–658 CrossRef CAS.
- Y. Wang and L. Huang, Nat. Biotechnol., 2013, 31, 611–612 CrossRef CAS.
- N. F. Crum-Cianflone, Clin. Microbiol. Rev., 2008, 21, 473–494 CrossRef.
- L. C. Chiedozi, Am. J. Surg., 1979, 137, 255–259 CrossRef CAS.
- N. F. Crum, Am. J. Med., 2004, 117, 420–428 CrossRef.
- E. M. Collnot, H. Ali and C. M. Lehr, J. Controlled Release, 2012, 161, 235–246 CrossRef CAS.
- W. Ulbrich and A. Lamprecht, J. R. Soc., Interface, 2010, 7(Suppl. 1), S55–S66 CAS.
- A. Lamprecht, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 195–204 CrossRef CAS.
- H. K. Kim, S. M. Choi, G. Kang, K. H. Park, D. G. Lee, W. B. Park, S. J. Rhee, S. Lee, S. I. Jung and H. C. Jang, Yonsei Med. J., 2020, 61, 301–309 CrossRef CAS.
- P. N. K. Wijesekara, W. W. Kumbukgolla, J. A. A. S. Jayaweera and D. Rawat, Vet. Sci., 2017, 4, 6 CrossRef.
- M. J. Rybak, S. C. Boike, D. P. Levine and S. R. Erickson, J. Antimicrob. Chemother., 1986, 17, 115–120 CrossRef CAS.
- L. De Leo, N. Di Toro, G. Decorti, N. Malusa, A. Ventura and T. Not, Antimicrob. Agents Chemother., 2010, 54, 1644–1646 CrossRef CAS.
- A. Louie, W. Liu, S. Fikes, D. Brown and G. L. Drusano, Antimicrob. Agents Chemother., 2013, 57, 2788–2792 CrossRef CAS.
- G. Potel, J. Caillon, B. Fantin, J. Raza, F. Le Gallou, J. Y. Lepage, P. Le Conte, D. Bugnon, D. Baron and H. Drugeon, Antimicrob. Agents Chemother., 1991, 35, 111–116 CrossRef CAS.
- J. S. Wold and S. A. Turnipseed, Rev. Infect. Dis., 1981, 3(Suppl.), S224–S229 CrossRef.
- J. M. Conly and B. L. Johnston, Can. J. Infect. Dis. Med. Microbiol., 2005, 16, 159–160 CrossRef.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- C. Garzoni and W. L. Kelley, Trends Microbiol., 2009, 17, 59–65 CrossRef CAS.
- J. Rai, G. K. Randhawa and M. Kaur, Int. J. Appl. Basic Med. Res., 2013, 3, 3–10 CrossRef.
- D. M. Shlaes, D. Sahm, C. Opiela and B. Spellberg, Antimicrob. Agents Chemother., 2013, 57, 4605–4607 CrossRef CAS.
- J. L. Forbester, M. Clement, D. Wellington, A. Yeung, S. Dimonte, M. Marsden, L. Chapman, E. L. Coomber, C. Tolley, E. Lees, C. Hale, S. Clare, I. Udalova, T. Dong, G. Dougan and I. R. Humphreys, J. Virol., 2020, 94, e00121-20 CrossRef.
- K. Garber, Nat. Biotechnol., 2018, 36, 777–778 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00624f |
| This journal is © The Royal Society of Chemistry 2021 |