
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress and perspective on chiral plasmonic nanostructures enabled by DNA programming methodology
Hao
Yang
,
Huacheng
Li
,
Pan
Tang
and
Xiang
Lan
 *
*
Xiang Lan Research Group, State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Center for Advanced Low-dimension Materials, Donghua University, No. 2999 North Renmin Str, Songjiang Dist, Shanghai, 201620, P. R. China. E-mail: xlan@dhu.edu.cn
First published on 7th October 2021
Abstract
Chirality, a fascinating property ubiquitous in nature, plays an important role in living organisms. DNA-programmed self-assembly has been extensively applied to precisely organize metal nanoparticles and/or small molecules in a chiral fashion at the nanometer scale. These assemblies exhibit significant chiroptical activities and are potential in biological detection and imaging, information processing, optical metamaterials, etc. In this review, we conclude on the DNA-enabled chiral plasmonic systems, uniquely from the perspective of the evolution of DNA programming methodology. We start by introducing a variety of natural and artificial DNA nanostructures to illustrate the fine programmability of DNA. Then we discuss the broad library of DNA-programmed chiral plasmonic nanostructures, spanning from individual nanoparticle–molecule chiral hybrids to the hierarchical chiral arrangements of nanoparticles enabled by DNA nanostructures of different complexities, which are from the primary DNA double helix to advanced architectures. Some cutting-edge applications of DNA-programmed chiral plasmonic nanostructures will also be introduced. We conclude this review with our personal perspectives on the future challenges and opportunities in this rapidly developing field. We expect this comprehensive review will inspire future studies in chiral materials at the convergence of DNA nanotechnology and chiral plasmonics.
1. Introduction
The word “chirality” comes from Greek χειρ and it means “hand”. As the name suggests, one of our hands cannot superimpose onto the other (its mirror image) through translations or rotations. In general, chirality refers to the structure attribute of an object without a mirror plane or inversion symmetry. Since Pasteur1 first separated two types of mirror-image crystals of the sodium ammonium salt of “paratartaric acid”, chirality has long been a central subject in chemistry, because of the extreme importance of chiral configurations/conformations and chirality recognition of many small molecules and biomolecules in playing their functions.The chirality of a molecule is manifested when interacting with the spin of photons of left- (LH) and right-handed (RH) circularly polarized light. Such differential interactions will lead to optical rotatory dispersion effect (ORD) and unequal optical absorption, i.e., circular dichroism (CD), which are associated with the real and imaginary part of the refractive index of the chiral molecular media, respectively. However, the chiral strength of a molecule is rather weak. According to the well-established coupled-dipole theory, it is dependent on the couplings between co-linear components of electric and magnetic dipole transitions of the molecule, which are usually very tiny.2
In contrast to small molecules, when an incident light of a specific wavelength interacts with metal nanoparticles (NPs), localized surface plasmon resonances (LSPR), that is the collective oscillation of free electrons, can be excited.3,4 Closely-spaced metal NPs generate strong plasmonic couplings and lead to a significantly enhanced local electromagnetic field in the narrow gap, called “hotspot”.5–7 Such plasmonic couplings give rise to a wealth of novel collective optical phenomena such as Fano resonances,8 surface-enhanced Raman scattering (SERS),9 optical magnetism,10 and giant chirality.11 Moreover, the LSPR properties of metal NPs are highly dependent on particle size, shape, composition and the surrounding environment,12,13 which enable a broad parameter space to modulate the emerging photonic and optoelectronic properties.
Of particular interest is the plasmonic chirality of metal nanostructures, which has so far exhibited a huge potential in various scenarios, such as ultrasensitive enantiomeric sensing,14 biomedicine,15,16 super-resolution imaging,17 photocatalysis,18 metamaterials,19,20 to name a few. Plasmonic chirality mainly originates from three different mechanisms: (a) nanocrystals with obvious chiral morphology, including chiral lattice distortion, twisted metal surface, or whole asymmetric shape.21–23 Due to their intrinsic chiral morphology and resulting asymmetric LSPR modes, these nanocrystals exhibit strong chiroptical effects; (b) the Coulomb interaction between adsorbed chiral molecules and metal NPs.7,24–27 It has been reported that Coulombic dipole coupling of a chiral molecule to an achiral plasmonic NP can enhance the molecular chiral signal and induce a new observed chiral plasmonic peak in the visible range. Because of the high polarizability of inorganic nanomaterials, such a chiral mechanism exists in many hybrid systems incorporating chiral ligands; (c) chiral arrangements of achiral NPs.28 When metal NPs are arranged into a chiral geometry, their plasmons can couple, mix and hybridize with each other, which causes resonance energy splitting under LH and RH circularly polarized light, thus exhibiting characteristic chiral responses.
Undoubtedly, in order to finely manipulate the light responses of chiral plasmonic nanostructures and enhance their optical dissymmetry, known as g-factor, reliable nanofabrication with precise control over multiple geometric parameters, such as spatial positions, orientations and even hierarchical arrangements of NPs are stringently required and have been continuously developed in recent decades. Noticeably, the field of chiral plasmonics has so far witnessed a blossom of DNA nanotechnology as a powerful nanofabrication method. Arising from the programmability, nanoscale addressability, robust designability of DNA and the ease of DNA functionalization of nanomaterials, DNA nanotechnology has evolved to be one of the state-of-the-art nanofabrication methods in the creation of unprecedented chiral plasmonic nanostructures, whose chiroptical responses can be rationally tailored and customized via DNA molecular programming.29–31
In this review, we will highlight the DNA-enabled chiral plasmonic systems, uniquely from the perspective of the evolution of DNA programming methodology. First, a brief background of DNA nanotechnology will be introduced, in order to illustrate the fine programmability of DNA molecular structures. Subsequently, a library of DNA-programmed chiral plasmonic nanostructures will be shown in order from the individual NP–molecule chiral hybrids to the hierarchical chiral arrangements of NPs. DNA origami-enabled chiral organizations of NPs will be emphasized here, as they are increasingly manifested in this field. Then, we will present some examples of the current applications of DNA-programmed chiral plasmonic nanostructures. Lastly, we will conclude this review with our personal view on the future challenges and directions of this highly cross-disciplinary research field. Note that the chiral NPs with intrinsically asymmetric morphologies are beyond the scope of this review. Also, for more theoretical understanding of the plasmonic chirality based on analytical models, we refer readers to the relevant research papers and reviews.32–35
2. A brief background of DNA nanotechnology
2.1 Natural recipes of DNA
Since Watson and Crick36 first revealed the mystery of DNA structure demonstrating the principle of complementary base-pairing in 1953, the molecular biology age had been officially opened. Deoxynucleotides are structural units of DNA and have four different DNA bases: A (adenine), C (cytosine), G (guanine) and T (thymine). The neighboring deoxynucleotides are linked by phosphodiester bonds to form the phosphate backbone. A single DNA strand of a specific sequence of bases can hybridize with the antiparallel complementary strand through hydrogen bonding, forming the Watson–Crick base-pairing (A–T, C–G). The hybridized strands twist into a double-helical structure for maximum hydrophobic stacking interactions.Apart from the canonical B-form DNA RH helix found by Watson and Crick, there are two other primary conformations of DNA helices: A-DNA, which is also RH but more rigid than B-DNA;37,38 and Z-DNA, which is surprisingly LH and found in some eukaryotic genomes.39 In addition to three helical structures, researchers have found various other DNA conformations that enriched the structural diversity of DNA, such as triplex (H-DNA),40 mini dumbbells,41 intermediate Holliday junction.42 Some nucleic acid aptamers with G-rich sequences are folded into four-stranded structures with the help of metal ions (Fig. 1A).43–45 The i-motif structure is composed of several stretches of C-rich sequences to form a quadruplex structure with C:C+ base pairs at acidic pH (Fig. 1B).46–48 TTTA or CCTG repeats were found to fold into mini dumbbells (Fig. 1C).41 To carry more genetic information, DNA molecules are compacted to constitute superstructures such as DNA supercoils.
 | ||
| Fig. 1 The native or artificial DNA structures. (A) Four-stranded G-quadruplex structure.48 Reproduced from ref. 48 with permission from the Nature Publishing Group. (B) I-motif structure.48 Reproduced from ref. 48 with permission from the Nature Publishing Group. (C) Native mini dumbbell structure.41 Reproduced from ref. 41 with permission from the American Chemical Society. (D) The synthesized 3D discrete DNA structures. From left to right: truncated octahedron,56 tetrahedron,57 bipyramid,63 biprism.64 Reproduced from ref. 56 with permission from the American Chemical Society. Reproduced from ref. 57 with permission from the AAAS. Reproduced from ref. 63 with permission from the American Chemical Society. Reproduced from ref. 64 with permission from the American Chemical Society. (E) The nanotechnology of SST.62 Reproduced from ref. 62 with permission from the AAAS. (F) The six-point-star DNA tile and the AFM image of 2D array.65 Reproduced from ref. 65 with permission from the American Chemical Society. (G), (H) and (I) are well-designed patterns or objects using DNA origami.66–68 Reproduced from ref. 66 with permission from the Nature Publishing Group. Reproduced from ref. 67 with permission from the AAAS. Reproduced from ref. 68 with permission from the Nature Publishing Group. | ||
There are numerous reports on the properties of DNA molecules and it is believed that the structural features and parameters greatly influence their biological functions, for example, gene regulation, DNA replication and repair, although some are considered to be the incentives of genetic disorders. On the other hand, the sequence-defined structural diversity and excellent physicochemical properties make DNA an ideal programmable material for the design of artificial molecular structures that will be introduced in the next section.
2.2 Artificial DNA nanostructures
In 1982, Seeman49 designed an immobile four-arm Holliday junction, which originally took DNA as construction materials and laid the foundation of the theoretical framework for the emerging research field: Structural DNA Nanotechnology. This symmetrical DNA structure, also called “DNA tile”, was expected to create periodic superlattices through hybridizations between the sequence-specific sticky ends. Inspired by this concept, a variety of DNA nanostructures with great geometric and topological complexity were fabricated.Afterwards, Fu et al.50 introduced the crossover structures between DNA double helices and created several double-crossover tiles (DX tiles) for realizing stable DNA linkages. Mao et al.51 fused four Holliday junctions together to construct rhombus DNA units, which could further self-assemble into two-dimensional (2D) DNA arrays with diamond holes. LaBean et al.52 designed a kind of triple-crossover tile (TX tile) that contains three helices formed by four strands, which was an evolution of DX tile and used for logical computation.53 Yan et al.54 reported 4 × 4 cross-shaped tiles that could periodically arrange proteins or template highly conductive metallic nanowires.
Watson–Crick base-pairing rules were also applied to the self-assembly of three-dimensional (3D) structures, as illustrated in Fig. 1D. Seeman et al.55,56 constructed covalently closed cube-like and truncated octahedral DNA nanostructures. Goodman et al.57,58 successfully assembled a series of DNA tetrahedral structures with different sizes by using four single-stranded DNA (ssDNA), which are potential to transport or release drug molecules in vivo. Mao et al.59,60 assembled DNA dodecahedral, icosahedral, and buckyball nanocages, which were composed of DNA tiles of different shapes. Shih et al.61 synthesized a reproducible DNA octahedron by folding a 1.7-kilobase ssDNA. Yin et al.62 took some identical 42-base DNA single-stranded tiles (SSTs) with four concatenated modular domains to construct DNA lattices (Fig. 1E). The basic consideration of using SST is to use as few tiles as possible to form the desired 2D structures. Subsequently, Ke et al.69 proposed the concept of “DNA brick” on the basis of SST, which enabled modular self-assembly of well-defined 3D structures from short strands. Numerous regular and irregular assembled structures with DNA tiles were developed, including various periodic arrays (Fig. 1F). However, the accompanying issues have been long exercised by researchers, such as “thermodynamic trap” problem, wide dimension distributions, and high cost.
In 2006, Rothemund66 invented a revolutionary DNA self-assembly technology to construct arbitrary 2D patterns: DNA origami (Fig. 1G). In short, a single-stranded DNA (ssDNA) scaffold, typically several thousand nucleotides long, is folded into the desired shape by hundreds of short oligonucleotide strands (staples), typically 15–60 nt long, in a manner analogous to knitting. To accurately populate the scaffold path, the scaffold commonly used is derived from the phage M13mp18 that contains 7249 bases. On the one hand, the ssDNA scaffold has neither abundant repeating sequence fragments nor large secondary stem-loop structures. On the other hand, each staple strand possesses a unique sequence to locate in the deterministic position. In contrast to DNA tiles, DNA origami is more stable and supports more complicated designs.70 Moreover, it allows researchers to ignore the pureness and relative stoichiometric ratio of DNA strands, which dramatically reduces experimental errors and time.
During the last decade, researchers have continuously improved the DNA design approach and have fabricated various DNA origami structures from simple 2D geometry of single-layer to complicated 3D structures (Fig. 1H). However, most of them are sub-100 nm dimensions limited by the length of the scaffold strand and the sequence library. In recent years, researchers have introduced the concept of hierarchical assembly of DNA origami by using similar origami blocks to build arbitrary array geometries via shape complementarity or sticky-end association, such as micrometer-scale Mona Lisa pattern (Fig. 1I),68 gigadalton-scale tube71 and unfoldable nano books.72 This multi-stage assembly produces enormous gigadalton-scale ordered structures with even over micrometer sizes, and thus brings possibilities to bridge molecular and macroscopic scales. Overall, the technology of scaffolded DNA origami shows great potentials in multi-scale material design and fabrication.
Recently, dynamic DNA nanotechnology that allows to engineer the reconfigurable DNA structures has renewed interest in this field. The most widespread dynamic regulation approach is the DNA toehold-mediated strand displacement reaction (SDR).73 The concept of toeholds was introduced by Yurke et al.74 and refers to the single-stranded (sticky) end suspended at the terminus of a DNA duplex, which generally consists of several (6–10) bases. In the process, the added “fuel strand” DNAs hybridize with the toehold region and displace the prehybridized strands through DNA branch migration. Nowadays, the ability to dynamically manipulate DNA structures has led to many applications, for example, in DNA actuators,75,76 responsive DNA hydrogels,77 DNA-controlled drug release,78 and DNA-computing devices in vitro79 and in vivo,80etc. Therefore, dynamic DNA nanotechnology offers a new dimension for material fabrications toward multiple and complex functionalities.
2.3 DNA–nanoparticle conjugates
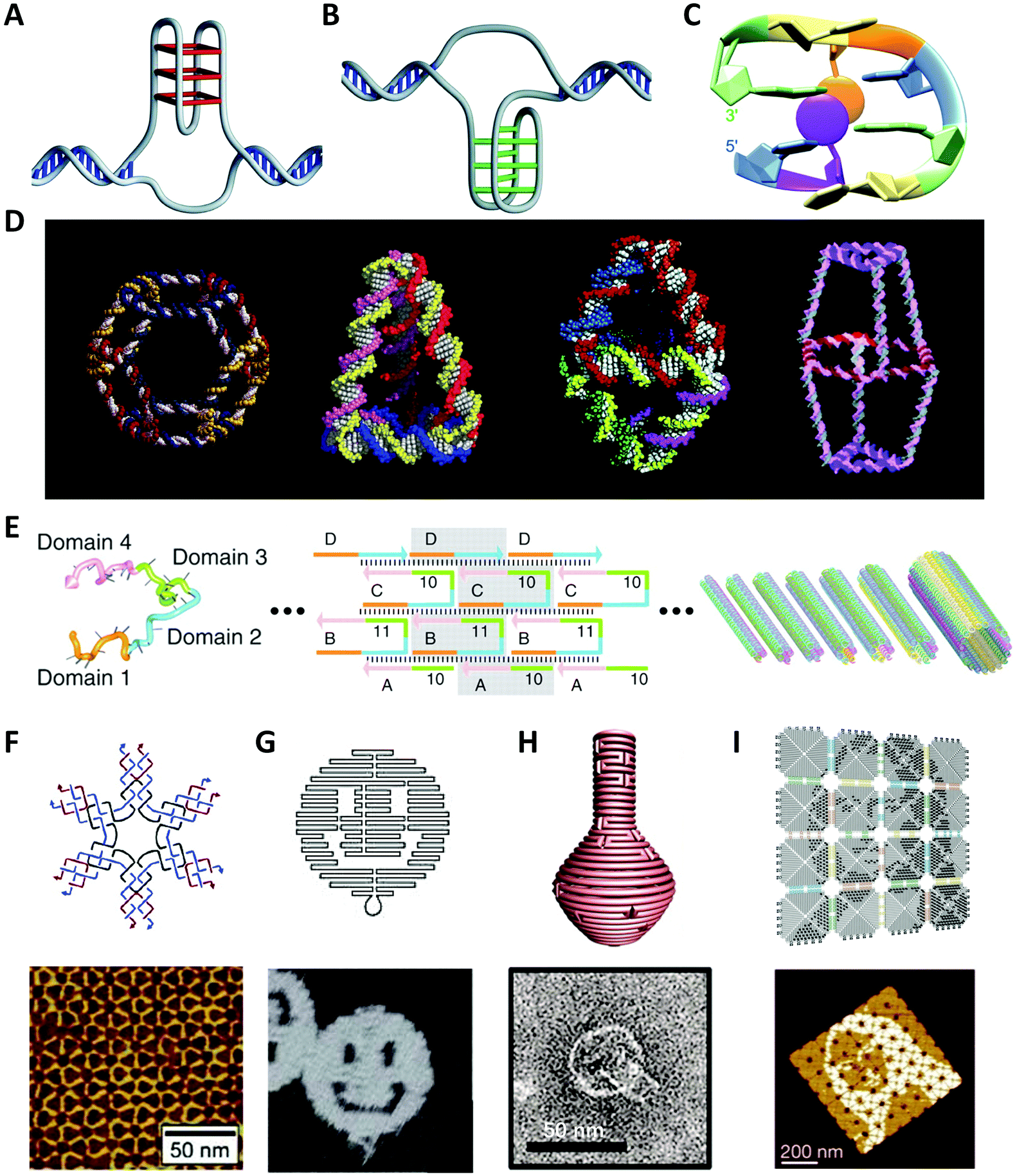
The power of DNA-programmed fabrication of nanostructures also relies on DNA's ability to encode nanoparticle surfaces via DNA sequence design and versatile DNA–nanoparticle conjugation chemistry. The first well-characterized frame was made with gold cores and DNA shells proposed by Mirkin et al.81 Specifically, the citrate capped spherical AuNPs were incubated with alkylthiol-modified oligonucleotides in a salt solution to form stable Au–S bonds (Fig. 2). It is necessary to highlight that the DNA monolayer also influences the yield of the following self-assembly. However, this seemingly simple conjugate is actually hard to synthesize in experiments. It is primarily because DNA and AuNPs are both negatively charged and the electrostatic repulsion is pervasive in the mixture, whether between DNA and AuNPs or between DNA strands.82 The initial method is the stepwise addition of NaCl salt for screening the charge repulsions and promoting DNA adsorptions.83 Despite strenuous efforts that have been made to improve such “salt-aging” method, the process is still troublesome and time-consuming and may result in irreversible aggregation of AuNPs by carelessness.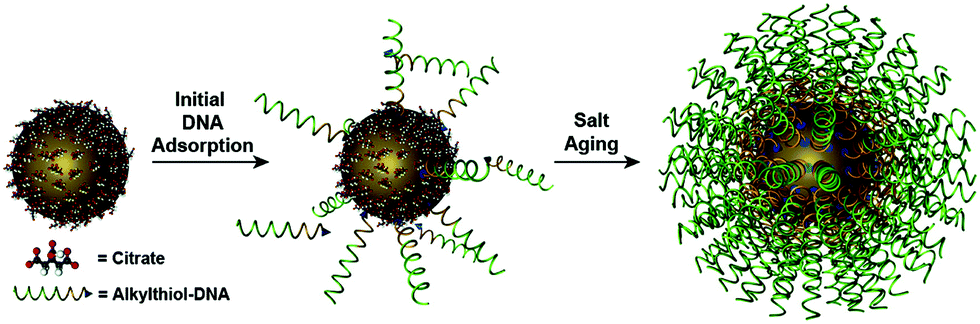 | ||
| Fig. 2 Schematic of synthesizing the spherical AuNP–thiolated DNA conjugate.82 Reproduced from ref. 82 with permission from the American Chemical Society. | ||
To overcome such constraints, Zhang et al.84 achieved fast and quantitative DNA adsorptions on AuNPs using a low pH buffer, providing a new protocol to control the DNA-loading process. Later, they further demonstrated nonthiolated DNA–AuNP conjugates with high loading capacity using the pH-assisted strategy.85 An alternative method is to insert an oligoethylene glycol (OEG) spacer between a DNA sequence and a terminal thiol group.86 The OEG spacer favored the adsorption kinetics and shielded electrostatic repulsion between DNA and AuNPs. This method is short-time, free of surfactant, and more importantly, enables the direct immobilization of thiolated molecular beacons (MBs) at physiological pH, offering a new parameter to manipulate the modification of AuNPs. Liu et al.87 suggested that low temperature (∼−20 °C) was also beneficial to the adsorption process of DNA strands, and could reduce the preparation time to a few minutes. More importantly, the post-thawing conjugates exhibited higher DNA density than typical salt-aging method (more than 20–30%). Recently, Deng et al.88 synthesized a conjugate with record-high DNA density in seconds by the so-called instant dehydration in butanol (INDEBT) method, in which the principle was butanol extraction.
There is evidence that DNA sequence will affect the overgrowth of NPs due to differential binding affinity between nucleobases and the NP surface, in particular, G > A > C > T.89 Wang et al.90–92 discovered that different DNA sequences would induce the growth of NPs into various shapes. For example, NPs would grow to six-point stars in the presence of DNA sequences of T30, while hexagonal nanoplates were formed with G20. Such a DNA-encoded scheme can also direct the overgrowth of anisotropic NPs93 and even bimetallic NPs.94 In addition to influencing the NP morphology, the affinity of DNA sequences to NP surface also provides an approach to decorate NPs with tunable multivalency. Yao et al.95,96 utilized this strategy to synthesize many atom-equivalents by programming valence bonds of AuNPs and further constructed colloidal molecules and oligomers.
3. Chiral hybrids of nanoparticle and molecules
The DNA molecule is an undoubted natural chiral material. As we mentioned, when chiral biomolecules adsorb on the surfaces of NPs, the hybrid system could exhibit chiroptical responses by Coulombic dipole–dipole interactions, even though they are off-resonance. Govorov et al.97 for the first time theoretically interpreted this inductive effect. In their theoretical framework, an oscillating dipole of the adsorbed chiral molecule induces dissipative chiral currents in the metal NP, which transfers the optical chirality from the molecular absorption band to the plasmon resonances in the visible region (Fig. 3A). Based on such a theory, Li et al.98 explained the origin of the plasmonic chirality of their assemblies, a kind of dsDNA-linked macroscopic gold nanorod (AuNR) aggregate with reversible CD responses through the thermal transition of DNA duplex. They considered the plasmonic chirality coming from the chiral currents induced by DNA rather than the indistinctive chiral arrangement of AuNRs.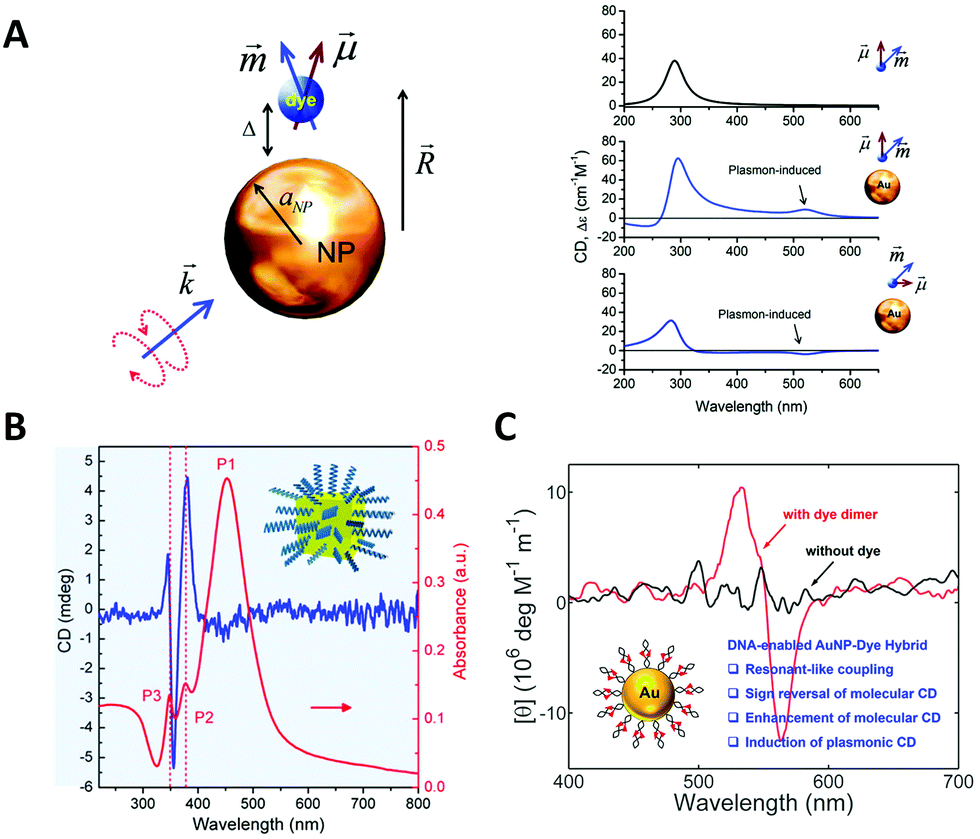 | ||
| Fig. 3 Chiral hybrids of molecules and plasmonic NPs. (A) The theoretical model to illustrate the interactions between a metal nanoparticle and a dye molecule (left) and the calculated spectra (right).97 Reproduced from ref. 97 with permission from the American Chemical Society. (B) The CD and absorbance spectra of dandelion-like DNA–NP hybrids.100 Reproduced from ref. 100 with permission from the American Chemical Society. (C) The CD spectra of AuNP–dye dimer hybrids templated by dsDNA.101 Reproduced from ref. 101 with permission from the American Chemical Society. | ||
Bagheri et al.99 investigated the conjugate of AuNR with folded G-quadruplex and found the intensity increase of CD signal in the visible region. Lu et al.100 obtained a hybrid dandelion-like structure in which ssDNAs were attached on the surface of Au@Ag core/shell nanocubes and radially arranged owing to the electrostatic repulsion (Fig. 3B). The complexes showed two orders of magnitude CD enhancement in the near-visible region, as compared to the DNA native CD signal. Lan et al.101 reported a new hybrid structure with organic chromophores directly self-assembled on the surface of AuNPs in a chiral arrangement through dsDNA hybridization mechanism (Fig. 3C). This strategy of DNA-templated molecular self-assembly allows to accurately control the hierarchical organizations of the chromophores and even their nanoscale distances to the NP surface. Although the chromophores used (cyanine dyes) were achiral, they still observed a significant molecular CD signal originating from the templating effect of DNA duplex. Moreover, they found the critical role of resonant plasmon–exciton couplings in reshaping the chiral responses of the hybrids. All these results showed new approaches to fabricate chiral plasmonic nanostructures as individual NPs with chiral adsorbates, illustrating many possibilities not only in the massive production for potential applications, but also in the study of fundamental chiral light–matter interactions, for example, the recently-emerged chiral strong coupling phenomena.102 Notably, the contribution of induced plasmonic CD to the overall optical activity is much less than the structural chirality in the chiral arrangements of NPs, so it is often neglected in those reports involving structural chirality, intentionally or accidentally.32
4. Chiral arrangements of nanoparticles based on primary DNA duplex
In 1996, Alivisatos et al.103 reported a landmark work in which they controllably assembled AuNPs into dimers or trimers with defined DNA duplex templates. This work demonstrated the feasibility and potential of using DNA molecules to organize inorganic NPs into discrete nanostructures. The dimer linked by DNA is the simplest self-assembled plasmonic nanostructure. It is a basic and convenient model to understand the complex plasmon couplings that emerged in hierarchical chiral arrangements of NPs.Note that while AuNP is generally considered to be a perfect sphere, its true shape, according to transmission electron microscopy (TEM) and 3D electron tomography, could be an ellipsoid with a small aspect ratio. In fact, Auguié et al.104 theoretically studied a twisted dimer of ellipsoidal AuNPs separated in the scale of the wavelength of the light, and interpreted the remarkable CD responses from this system as a result of the far-field scattering couplings. Experimentally, Yan et al.105 assembled twisted heterodimers with different-sized AuNPs, which were induced by sodium chloride (NaCl) or DNA (Fig. 4A). They all exhibited high optical activity, resulting from the spontaneous formation of small dihedral angles between adjacent ellipsoidal AuNPs due to electrostatic repulsion and steric hindrance. Here, near-field plasmonic couplings between closely-spaced AuNPs are responsible for their observed CD responses. Hao et al.106 fabricated DNA–directed Au NP-NR asymmetric heterodimers with different CD responses at visible wavelengths by adjusting the gaps between adjacent NPs and altering the size of components; Ma et al.107 tried to assemble AuNR dimers side by side and observed a negative dihedral angle (−7.0 to −9.0 degree) through the TEM tomography (Fig. 4B). Then, they expanded the number of AuNRs by PCR, interestingly, this angle persisted between adjacent nanorods over a long distance, leading to a whole twisted structure with high chiroptical activity.
 | ||
| Fig. 4 Chiral arrangements of NPs induced by primary DNA duplex. (A) The CD spectra of AuNP dimers induced by NaCl (black) and DNA (red), respectively.105 Reproduced from ref. 105 with permission from the The American Chemical Society. (B) The CD spectra (left) of twisted AuNR assemblies by PCR and the cryo-EM tomography images (right).107 Reproduced from ref. 107 with permission from the Nature Publishing Group. (C) The AuNP–DNA pyramids.108 Reproduced from ref. 108 with permission from the American Chemical Society. (D) The heterogeneous pyramid containing AgNP, AuNPs and quantum dots (QDs).109 Reproduced from ref. 109 with permission from the American Chemical Society. | ||
In general, a tetravalent carbon atom with four different substituents represents an original chiral geometry in stereochemistry. Analogously, Mastroianni et al.108 constructed a NP pyramid by DNA self-assembly. It was assembled with the help of four well-designed ssDNA, where each ssDNA anchored on an AuNP was partially complementary to the others. To break the symmetry, four AuNPs of different sizes were positioned on the vertices of the pyramid after DNA hybridizations forming a chiral structure (Fig. 4C). Unfortunately, they did not observe any CD signal for some reason. Chen et al.110 reported pyramidal structure mixed with other assemblies in the same year. Notably, there was no selective synthesis or separation of the enantiomers, but it is astonishing that the assemblies showed the preference of a particular chirality for unknown reasons. This is the first experimental observation of chiroptical responses stemming from the coupled plasmon resonances of NP assemblies. Later, they further assembled a heterogeneous pyramid, which contained AgNP, AuNPs and quantum dots (QDs) (Fig. 4D).109 This hybrid system exhibited obvious and multi-peak chiroptical signals owing to the high yield (80%) and multi-components with distinct optical absorption bands.
5. Chiral arrangements of nanoparticles based on scaffold-free DNA tiles
Interestingly, many naturally occurring biomolecules, such as DNA double helix and α-helix peptide, exhibit an ubiquitous chiral form, a helix. If metal NPs are helically arranged, the coupled collective plasmons show a “corkscrew”-like chiral fashion, a twist of specific handedness resembling the twisted propagation of electric field of LH/RH circularly polarized light, thus resulting in differential optical absorptions.111 Fundamentally, almost all the optically active objects are helical structures just with differences in the degree of curvature, length scale, or dimension.Sharma et al.112 reported an early example of NP helix, which existed in a mixture of tube-like NP self-assemblies (Fig. 5A). They designed an array system consisting of four types of DX DNA tiles through sticky-end associations. On one side of the 2D DNA tile array, several columns of attached AuNPs could curl up the tile arrays to tubular structures owing to the systematic steric and electrostatic repulsions. In the self-assembled products, four types of tube-like conformations co-existed including single helix and double helix, due to differences in the twisting and bending energy. It is interesting that these DNA nanotubes prefer to be in the LH state, which might result from the accumulation of supercoiling tendency of individual DNA tiles. In this work, no characteristic chiral response of NP helix was reported, which was observed until the DNA origami method was used to precisely construct the enantiomerically pure NP helix years later.113 In a similar way, Mitchell et al.114 used two kinds of DX DNA tiles to assemble a helical nanotube with additional mesoscopic structural chirality (Fig. 5B). Moreover, Sun et al.115 generated helical nanotubes with large diameter (range of 50–550 nm) using DNA bricks as repeating units (Fig. 5C). These DNA structures offered a new platform for studying chirality in a larger scale. Although no further experiment was reported, we can speculate that the inside and outside surfaces of the tube can be functionalized with NPs on account of the sufficient surface area.
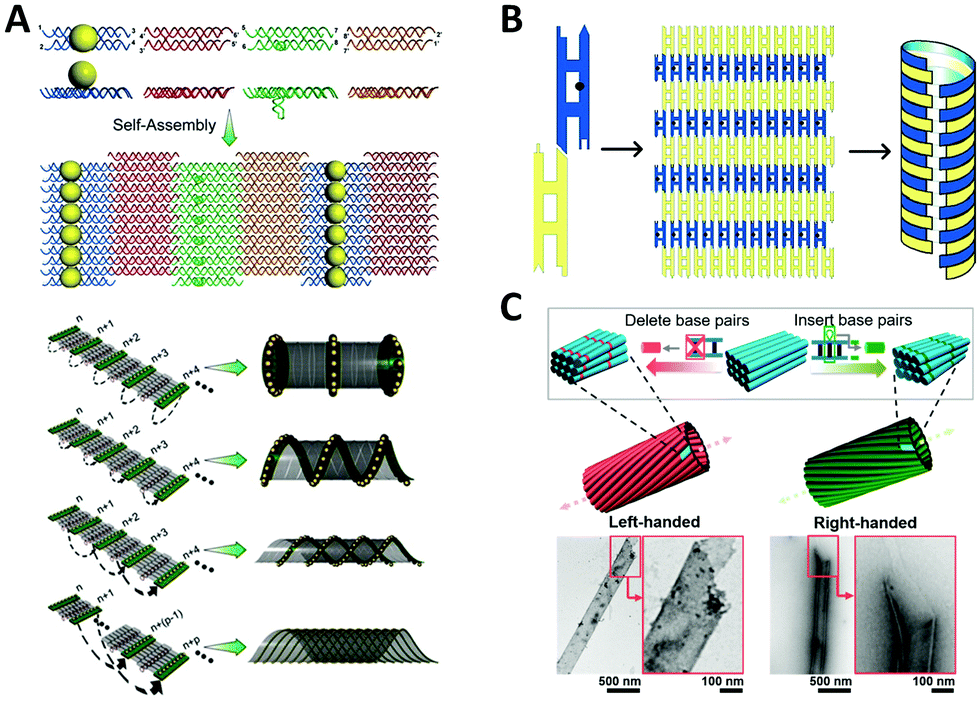 | ||
| Fig. 5 Chiral arrangement of metal NPs assisted by scaffold-free DNA tiles. (A) Schematic of the formation of AuNPs helix by four different tiles.112 Reproduced from ref. 112 with permission from the AAAS. (B) The DNA tube with structural chirality comprising two kinds of tiles.114 Reproduced from ref. 114 with permission from the American Chemical Society. (C) The large helical tubes designed by SST.115 Reproduced from ref. 115 with permission from the American Chemical Society. | ||
6. Chiral arrangements of nanoparticles based on scaffolded DNA origami
In order to enhance the chiroptical activities, many efforts are made continuously in the fabrication of helical plasmonic superstructures, as they have accumulative strong interactions with the incoming light and thus are predicted to show more prominent chiroptical signals than the low-order structures. Kuzyk et al.113 fabricated NP helices by designing DNA origami bundle for site-specific arrangements of AuNPs, and demonstrated an impressive high yield (Fig. 6A). They observed an enhancement of the g-factor to as high as 0.02 after Ag deposition on the AuNP helix, because AgNPs had much stronger plasmonic couplings between them. Shen et al.116 rolled up a rectangular DNA origami composed of 24 DNA helices to form a nanotube, then transformed two columns of AuNPs previously addressed on the DNA origami into a 3D helix (Fig. 6B). Benn et al.117 introduced the inherent twist into a DNA origami tube by adjusting the location of DNA crossovers. Besides the success of helical arrangements of AuNPs on the outside surface of DNA origami tube, such DNA tube could transport a molecular cargo along a track that was “laid” on the internal surface. Urban et al.118 engineered a plasmonic toroidal helix-like system by connecting the ends of four identical bending DNA origami bundles with a string of AuNPs winding on the surface to form a helical frame (Fig. 6C). These self-assemblies greatly enlarged the library of chiral plasmonic nanostructures with rationally-tunable and enhanced chiral responses. Some even shed new light on the single-particle studies for more mechanistic insights into the plasmonic chirality.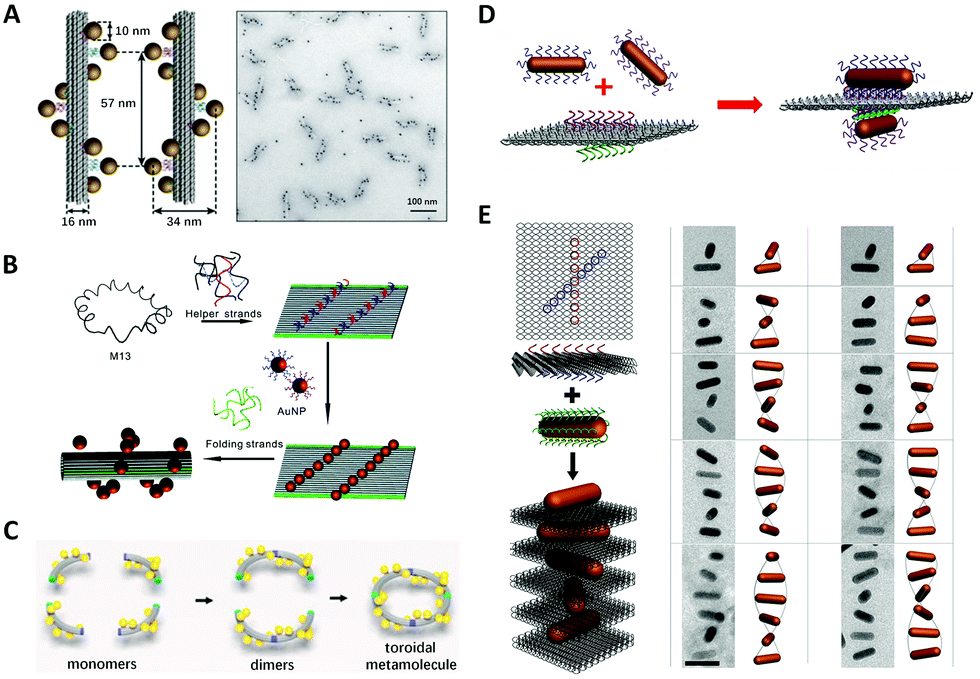 | ||
| Fig. 6 Chiral arrangements of metal NPs assisted by DNA origami. (A) and (B) Two ways to assemble DNA origami–directed AuNP helix.113,116 Reproduced from ref. 113 with permission from the Nature Publishing Group. Reproduced from ref. 116 with permission from the American Chemical Society. (C) The plasmonic toroidal structure.118 Reproduced from ref. 118 with permission from the American Chemical Society. (D) Two crossed AuNRs arranged on a bifacial rectangular DNA origami.121 Reproduced from ref. 121 with permission from the American Chemical Society. (E) The plasmonic superstructures formed by stacking the AuNRs and square DNA origami (left) and the Cryo-EM images.124 Reproduced from ref. 124 with permission from the American Chemical Society. | ||
In contrast to the spherical NPs, the characteristic plasmon resonances of anisotropic building blocks, such as metal NRs, can lead to noticeable properties, such as the light-antenna effect, by which the light can be harvested and confined at the ends or edges.119 These strong light–matter interactions of anisotropic building blocks are of significance in the context of the enhancement of g-factors of chiral plasmonic nanostructures. Therefore, after successfully addressing the problem of positional and orientational control of anisotropic NRs with the DNA origami method,120 a plethora of chiral plasmonic nanostructures with NR building blocks rapidly arose. Lan et al.121 realized the chiral arrangement of AuNRs on a DNA origami sheet (Fig. 6D). Two sets of DNA capture strands with specific sequences were extended from the opposite side of a bifacial DNA origami sheet at the specified positions and in different orientations, which eventually gave rise to 3D AuNR crossed dimers. By adjusting the 3D spatial configuration of the dimer, the unique chiroptical activities were rationally tailored. These findings were also corroborated by Shen et al.122 and they pointed out that the chiroptical activity was related to a mode splitting originating from the near-field plasmonic couplings between two closely-spaced AuNRs. Recently, Zhu et al.102 introduced such structural chirality of AuNR twisted dimer into the plasmonic–excitonic (plexcitonic) nanosystem and found an intriguing chiral strong-coupling phenomenon, which brought more possibilities to engineer the chiral plasmonic systems. Chen et al.123 assembled T-shaped AuNR trimers on 2D DNA origami and thoroughly investigated the origin of their plasmonic chirality. By analyzing the observed CD signals of the trimers and their sub-dimers, they found the chiroptical signature of the trimers was essentially a collective behavior of their total sub-dimers.
To move forward in engineering more complex chiral plasmonic nanostructures, Lan et al.124 developed a new self-assembly strategy to make plasmonic helical superstructures (Fig. 6E). They turned the locations of capture strands on their optimized square DNA origami into an “X” pattern with a ±45° angle, and after layer-by-layer self-assembly of AuNRs and DNA origami at the optimal molar ratio, they achieved an AuNR-origami alternating twisted superstructure with intense optical activity. TEM imaging revealed that the self-assembled superstructure with long-range order could contain near 20 AuNRs. These AuNR helical superstructures exhibited a high g-factor approaching 0.02, even though no Ag enhancement was employed, which demonstrated the significance of creating chiral plasmonic superstructures with anisotropic building blocks. Afterwards, Lan et al. reported a one-pot self-assembly mechanism of plasmonic helical superstructures.125 In their study, a delicate “V-shaped” DNA origami composed of two rectangular arms with four binding domains, each on the top or at the bottom, was designed. The connection between different domains between neighboring DNA origami resulted in four supramolecular DNA frameworks. Finally, the same AuNR–DNA origami conjugate units were arranged into four kinds of helical superstructures. Wang et al.126 designed a DNA origami “hashtag” in which four identical DNA double-decks were stacked. This structure had superior stiffness and their connected chain framework presented a high-fidelity even up to a microscale length. With such “hashtag” DNA origami chain, they assembled chiral AuNR polymers and found that plasmonic chirality could couple between AuNR dimers and cascade along the chain.
7. Reconfigurable chiral arrangements of nanoparticles based on dynamic DNA nanotechnology
One of the most important goals of nanotechnology is to create artificial nanomachines that can imitate natural biomolecules for performing analogous functions. However, the living cells need to continuously vary the conformation of biomolecules for accommodating the environmental change. Thus, it is highly desirable to design and fabricate environmentally responsive nanostructures with dynamically engineered functions and properties. More specifically, self-assembled chiral plasmonic nanostructures possessing flexible reconfigurability will pave the way for new materials and devices with multi-functionalities, for example, used for optical sensing, dynamic display, optical communications, etc.In 2014, Kuzyk et al.19 pioneered a tunable 3D twisted AuNR dimer, called metamolecule, and controlled its configuration via SDR (Fig. 7A). They decorated two DNA-connected bundles of a cross shape with partially complementary ssDNAs as “DNA lock”, and switched the lock on/off by adding specific fuel strands.127 Thus, the AuNR dimer could realize the reversible reconfiguration among three states: LH, RH and relaxed. Similarly, “RNA lock” was also proved feasible.128 Structural modulating by pH is highly efficient and produces no waste in comparison with SDR. Based on the previous AuNR dimer, Kuzyk et al.129 separately put the ssDNA and dsDNA part of a DNA triplex on the DNA bundles and controlled the association and dissociation of the DNA triplex by tuning the pH in the solution, ultimately re-adjusting the spatial conformation. Among lots of external stimuli, light is a unique clean energy and allows for easy operations. Azobenzene molecules are widely used in building up light-responsive dynamic systems, which is a cis-form under ultraviolet (UV) light while trans-form under visible (Vis) light. Kuzyk et al.132 incorporated azobenzene into DNA lock strands that showed a photoinduced transition of chirality of the twisted AuNR dimer, via the photoisomerization of the underlying DNA bundle. However, the process of doping azobenzene is time-consuming and expensive. Ryssy et al.133 chose the merocyanine-based photoacid (MCH+) as a new light-responsive regulator. Upon blue light illumination, MCH+ produced a ring-closing reaction along with the release of H+. In contrast, when the light was off, the closed-ring isomer opened the “ring” to recapture H+ from the environment. They dispersed the AuNR dimer incorporating the DNA triplex lock in the solution of MCH+ and switched the lock on/off with remote light control. Jiang et al.130 exploited the distance-dependent plasmonic couplings to control the chiroptical signals of chiral nanostructures (Fig. 7B). They assembled two AuNRs into an L-shape on a rhombus DNA origami where two triangular origami were linked by i-motif sequences. By controlling the pH to manipulate the i-motif DNA conformation to increase or reduce the inter-rod distances, the CD signals of AuNR dimers were dynamically tuned. They also tried different chemical stimuli to realize reversible or irreversible conformational changes, such as glutathione, restriction endonuclease and azobenzene molecules.
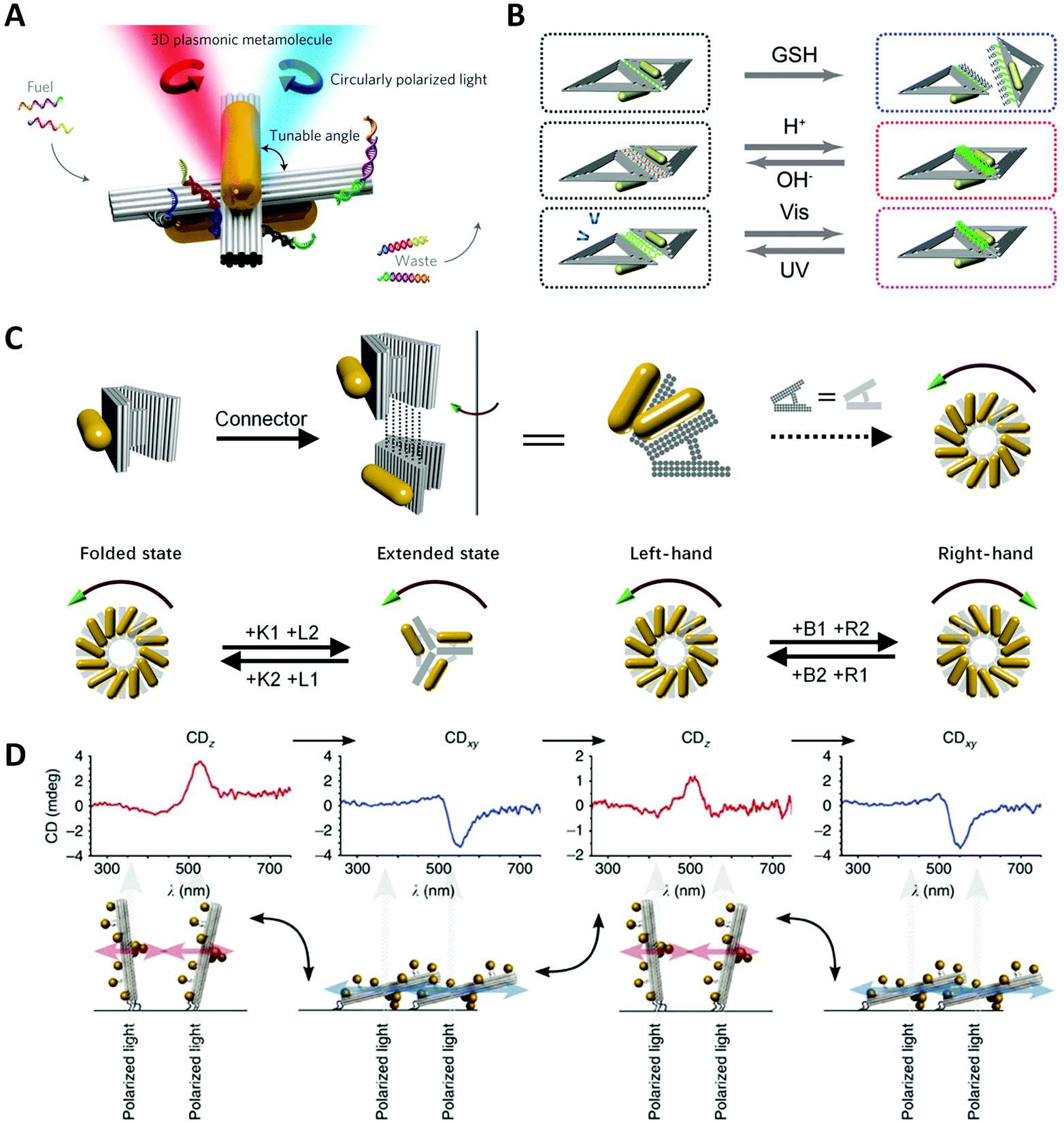 | ||
| Fig. 7 Dynamic devices based on DNA origami. (A) Reconfigurable twisted AuNR dimer templated by DNA origami.19 Reproduced from ref. 19 with permission from the Nature Publishing Group. (B) Two AuNRs on rhombus origami can reversibly or irreversibly change the distance under different stimuli.130 Reproduced from ref. 130 with permission from the American Chemical Society. (C) Reconfigurable chiral helix superstructure of AuNRs.20 Reproduced from ref. 20 with permission from the American Chemical Society. (D) The manipulation of chiroptical response by relative orientation between the incident light and the AuNP helix.131 Reproduced from ref. 131 with permission from the Nature Publishing Group. | ||
Certainly, there are many other chiral oligomeric structures containing two or three AuNRs, such as automatic nano clock,134 stepwise walker135,136 and so on. But the unchanged core of these dynamic plasmonic devices is to manipulate/amplify the (sub-)nanometer conformational changes of the stimuli-responsive molecules incorporated in the systems.
Taking a big step forward, Lan et al.20 presented a reconfigurable chiral helix superstructure of AuNRs in 2018. It must be noted that to realize a more advanced structure with reconfigurability means greater experimental challenges. In order to design such systems, researchers not only need to efficiently adjust the organizations of the structural units in accordance with the designed dynamics but also need to coordinate the stable connection between units during the transformation, not to mention considering other factors at the same time. To fulfill this goal, Lan et al. improved their previous design of a V-shaped DNA origami,125 whose arms were thickened to three layers and the spacer was replaced with a more rigid but switchable DNA helix bundle (Fig. 7C). By repeatedly adding the corresponding fuel strands, the DNA origami monomer can transform between the folded state (inter-arm angle of 30°) and the extended state (inter-arm angle of 120°). Connecting the monomer addressed with AuNRs layer-by-layer to be a twisted geometry, they assembled a reconfigurable staircase chiral superstructure. To achieve more attractive chiral inversions, they further modified the V-shaped origami into a relaxed H-shape, which could transform between states of different handedness (LH, RH and relaxed) with high efficiency. Thus, they generated a chiral superstructure with fully switchable chirality including the chiral amplitude and sign. This strategy represents an important success of chiral colloidal self-assembly, enhancing one's ability to dynamically manipulate much more complicated chiral nanostructures, and is also a great step toward the realization of reconfigurable chiroptical devices.
Wang et al.137,138 reported a plasmonic device, where a rotary bundle with extended ssDNAs was connected to a rectangular plate at the bottom via a DNA scaffold and was subsequently conjugated with two DNA-functionalized AuNRs. Two pairs of different ssDNAs were placed along the diagonal lines of the origami plate, respectively, which underwent SDR steps to switch the configuration. They stacked up such building units to fabricate a superstructure with four individual chiral centres and finally generated eight stereoisomers. As expected, the arrangement of total-left-hand (LLL) and total-right-hand (RRR) showed the strongest CD signals. For other diastereomers (RRL, RLL, etc.), their chiral responses were highly correlated with the order of chiral centres rather than their simple summation. Aside from altering the structural parameters, Schreiber et al.131 reported a dynamic chiral system by attaching the terminus of an AuNP-helix on the glass surface (Fig. 7D). Upon drying or rehydration, the helix would “lie down” or “stand upright” on the substrate with different orientations to the incident light. The obtained CD spectra of the two states were dynamically reversed because perpendicular and tangential light excitation with respect to the axis of the AuNP helix could lead to distinct plasmonic modes and thus a reversal of the CD lineshape.
8. Application
8.1 Biodetection
The chiral DNA–nanoparticle assemblies can serve as biocompatible and sensitive probes for bio-detection in vitro and in vivo. In contrast to the Förster resonance energy transfer (FRET) method, chiral plasmonic probes are based on polarization optics, diminishing the intensity variation due to fluorescent quenching effect, and thus are stable and highly sensitive. Kuang et al.139 assembled a kind of scissor-like AuNP dimer connected by an anti-bisphenol A (BPA) aptamer and its complementary fragments. In the presence of BPA targets, the dimers disassembled and the chiroptical response was reduced. The amplitude of CD signals would indicate the concentration of BPA in the sample. The limit of detection (LOD) of this aptamer-based method was as low as 0.008 ng mL−1 in the range of 0.02–5 ng mL−1. Li et al.140 constructed DNA-driven gold-upconversion nanoparticle (Au–UCNP) pyramids that could act as intracellular nanoprobes to detect miRNAs in real-time. Based on the structural association and disassociation strategy, it allows detecting the telomerase activity,141 adenosine-5′-triphosphate (ATP),142 serum alpha-fetoprotein (AFP),143 8-hydroxy-2′-deoxyguanosine (8-OHdG),144,145 Ag+ ions146 and so on. Ma et al.107 obtained chiral twisted side-by-side assemblies of AuNRs through PCR replication procedure and made possible detection of DNA with the LOD as low as 3.7 aM. The introduction of DNA origami technology could offer a reusable platform for the detection of target molecules. Huang et al.147 designed the aptamer-based “locks” into the previously introduced twisted AuNR dimer on DNA origami and switched the lock on/off by inputting the adenosine. Similar AuNR dimers were also applied to detect RNA128 and thrombin.148 Zhou et al.149 created a dual-responsive plasmonic system with DNA origami, which was modulated by both aptamer–target interactions and thermal changes, and demonstrated chiroptical detections of ATP and cocaine. Overall, those DNA-programmed chiral plasmonic probes can detect target species with high accuracy and present the subtle specific binding of molecules as significant changes of optical output.8.2 DNA computer
In addition to excellent ability for nanofabrication, the earliest function of DNA is the carrier of genetic information in biology. Nowadays, more and more people consider the use of DNA in information storage and computing, and imagine the transformative DNA computer. DNA computing devices generate, extract and test the logical results of specific problems at the molecular level through natural parallel operations and biochemical processing of a large number of DNA molecules. In fact, Adleman150 successfully used the coordination of DNA molecules with enzymes to calculate the Hamiltonian path problem as early as 1994. Subsequently, Okamato et al.151 combined digital circuits with DNA computing and designed DNA logic gates and circuits. The molecular logic gate systems utilized different efficiencies of DNA hole transport through base pairs and generated the output signal when hole transport occurred. Lately, Dong et al.152 introduced the concept of DNA computing into their previous dynamic chiral plasmonic nanostructures. The integrated system could execute a complete set of Boolean logical gates by reading elaborate DNA strands as inputs and returning plasmonic chiroptical signals as outputs. More interestingly, the logic gates could perform different logical functions at different temperatures even if inputting the same DNA strands, that is to say, they had the adaptability to execute more complicated functions.8.3 Metamaterial and metadevice
Electromagnetic metamaterials are artificially engineered micro(nano)structured materials with attractive electromagnetic properties that are not existing in natural materials. In particular, chiral metamaterials are promising to realize negative refractive index, light polarization control, chirality sensing and so on. The DNA-programmed self-assembly provides a new bottom-up method to manufacture chiral optical metamaterials or metadevices with superior resolutions. A variety of 3D chiral plasmonic assemblies could act as solution-state metamaterials, also called metafluids. The plasmonic metamolecules obtained by Kuzyk et al.19 realized reconfigurable chirality at the nanoscale, which was difficult for other micro(nano) processing methods. As mentioned above, Lan et al.126 demonstrated chiral metamaterials with distinct helical morphologies by simply connecting different domains of V-shaped DNA origami units addressed with AuNR. This self-assembly strategy also enabled chiral metamaterials with switchable chirality.20 The successful creation and manipulation of hierarchical chiral nanostructures is essential for studying the fundamental chiral light–matter interactions, and meanwhile, for constructing cross-scale metadevices. In fact, the aforementioned chiral structures, whether static or dynamic, can be regarded as the prototypes of novel optical devices. Such diverse chiral DNA–NP assemblies lay the foundation to fabricate multifunctional optical devices in the future.8.4 Other applications
Recently, Göpfrich et al.153 exploited droplet-based microfluidics to successfully encapsulate DNA-programmed chiral nanostructures inside uniform cell-sized microfluidic compartments. Such a combined system enabled filtration of plasmonic enantiomers efficiently on the microfluidic chip. This strategy inspired researchers to synthesize artificial cells by integrating biochemical molecules such as enzymes in a similar way. Attempts have also been made in the field of energy conversions. Martens et al.154 demonstrated efficient plasmon-assisted long-range chiral interactions through both experiments and simulations. In their model, spherical AuNPs were placed between two separated AuNRs to transfer plasmonic energy. More surprisingly, the simulation illustrated that the transfer effect persisted even for AuNR inter-distance up to 200 nm.155 This innovative mechanism might bring new insights into the long-distance chiral transfer and the design of new transmitter micro-devices in optical circuits. Previous studies have shown that surface photochemistry and plasmonic nanocrystal growth could be induced by hot electrons (HEs).156,157 Liu et al.158 introduced the concept of HE generation in the chiral plasmonic nanostructures. In their DNA-templated model system, the HEs were first injected into small TiO2 nanocrystals from chiral plasmonic AuNR dimers and later transferred to the reactive species. They observed a significant CD in the generation rates of HEs and considered that the chiral optical asymmetry of plasmonic nanostructure was transferred into the HE generations. Such a mechanism of the chiral photochemical effect is different from conventional chiral photochemistry and has potential for practical applications, such as polarization-sensitive photochemistry.9. Summary and perspective
We have provided a comprehensive overview of the development of chiral nanostructures enabled by DNA, from the aspect of the evolution of DNA programming methodology. Because of the excellent programmability and specific recognition ability of DNA molecules, researchers utilize DNA to program the organizations of NPs in nanoscale precision and create various chiral plasmonic nanostructures, including NP–molecule chiral hybrids, and structurally-chiral systems spanning simple static NP arrangements to sophisticated dynamic organizations at different hierarchical levels. These chiral nanostructures could bring new opportunities in nanophotonics, biodetection, catalysis, DNA computer, microfluidics, and so on.To date, top-down fabrication technologies, such as electron beam lithography (EBL),159,160 focused ion beam (FIB) lithography161 and direct laser writing,162 have proven to be successful in fabricating chiral nanostructures over a large area and with excellent reproducibility. However, these methods are time- and cost-consuming and the processing of highly complex 3D structures has always been difficult. In the realm of self-assembly, there are also successful examples of using polymers,163 peptides,164 and proteins165 to mediate the chiral assembly of NPs. However, interactions and compatibility between NPs and these templates/inducers need to be further improved to expand the NP material selections. Also, the lack of precise addressability in terms of positional and orientational control has limited the applicability of these strategies to some extent. Moreover, the designability and tunability of these templates/inducers remain to be enhanced to meet the growing demand for the fabrication of chiral nanomaterials.
In this context, DNA self-assembly methodology is manifesting as it becomes increasingly mature. The next focus would be the discovery and advancement of the properties and functions of chiral nanostructures by means of a combinatorial investigation including theoretical modeling, structural designing, advanced spectroscopic characterizations. The goals would at least include the enhancement of g-factors to improve the far-field properties and engineering the near fields to enhance the local superchiral fields. Therefore, DNA-mediated anisotropic interactions among new building blocks, which are varied in sizes, compositions, shapes, etc., should be tightly regulated, allowing precise control over the spatial orientation, quantity and inter-distance of the building blocks. It is also highly intriguing to dynamically control and/or switch the properties and functions of chiral nanostructures, for example, by manipulating the chiral self-assembly process or the resulting chiral nanostructures in space and time, with various physical, chemical and/or mechanical external stimuli. Such capability should lead to the development of chiral materials with multi-responsiveness for various applications. To solve these problems, we envision that new opportunities will arise from the convergence of DNA programming technologies with the new frontiers across fields, including symmetry-breaking NP surface engineering, hybrid nanocomposites, soft/responsive device fabrications, electromagnetic theories, etc.
Although DNA fabrication methods are still suffering from the scale-up self-assembly over micrometers with high efficiency, new solutions or possibilities could be offered using the Damascene technology with the patterned substrates for hosting and arranging the DNA-programmed nanostructures, which can indeed circumvent the size limitations for the device-scale fabrications. Such technological integration should open new avenues toward the on-chip chiral photonic/plasmonic devices for many light control applications, such as polarization filtering/conversion and chiral light sources with positioned quantum emitters. Enantiomeric-molecular sensing, an extremely important target in chemistry and biology, is also promising with the DNA programming methodology. In combination with theoretical discovery of the superchiral local field and DNA-programmed nanofabrication, optimal and precise positioning of target enantiomers in the chiral nanostructures can be expected. The single-molecular addressability of DNA can even help push the chiral sensing resolution to the limit of single-molecule level, for example, via chiral strong coupling between plasmon and molecular excitons.
In general, artificial self-assembled chiral nanostructures are still limited to the low-order chirality compared to the complex high-order chiral materials existing in nature. The chiral nanostructures obtained at present are considerably simple in both structure and function. We speculate on a new possible development stage of self-assembly of chiral nanostructures toward the integration of multi-components and multi-functionalities, device-scale applications, and intellectualization. DNA programming methodology has evolved to be a powerful modern nanofabrication technology and will boost this rapidly-developing field of chiral materials, with its own continuous advancement and convergence with other areas simultaneously.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Lan gratefully thanks for the grant support from the National Natural Science Foundation of China (No. 22004015).References
- L. Pasteur, Ann. Chim. Phys. Sér. 3, 1848, 24, 442–459 Search PubMed.
- W. Ma, L. Xu, A. F. d. Moura, X. Wu, H. Kuang, C. Xu and N. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- N. Halas, S. Lal, W.-S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- E. Ozbay, Science, 2006, 311, 189–193 CrossRef CAS PubMed.
- L. Kneer, E.-M. Roller, L. Besteiro, R. Schreiber, A. Govorov and T. Liedl, ACS Nano, 2018, 12, 9110–9115 CrossRef CAS PubMed.
- E. Severoni, S. Maniappan, L. Liz-Marzán, J. Kumar, F. J. G. d. Abajo and L. Galantini, ACS Nano, 2020, 14, 16712–16722 CrossRef CAS PubMed.
- L. Besteiro, H. B. Zhang, J. Plain, G. Markovich, Z. M. Wang and A. Govorov, Adv. Opt. Mater., 2017, 5, 1700069 CrossRef.
- F. Shafiei, F. Monticone, K. Q. Le, X. Liu, T. Hartsfield, A. Alù and X. Li, Nat. Nanotechnol., 2013, 8, 95–99 CrossRef CAS PubMed.
- X. Lan, Z. Chen, X. Lu, G. Dai, W. Ni and Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 10423–10427 CrossRef CAS PubMed.
- A. Alú and N. Engheta, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085112 CrossRef.
- X. Shen, A. Asenjo-Garcia, Q. Liu, Q. Jiang, F. J. G. d. Abajo, N. Liu and B. Ding, Nano Lett., 2013, 13, 2128–2133 CrossRef CAS PubMed.
- K. Kelly, E. Coronado, L. Zhao and G. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- L. Liz-Marzán, Langmuir, 2006, 22, 32–41 CrossRef PubMed.
- L. Xu, Z. Xu, W. Ma, L. Liu, L. Wang, H. Kuang and C. Xu, J. Mater. Chem. B, 2013, 1, 4478–4483 RSC.
- A. Kumar, S. Kumar, W. Rhim, G.-H. Kim and J. Nam, J. Am. Chem. Soc., 2014, 136, 16317–16325 CrossRef CAS PubMed.
- Y.-L. Luo, Y.-S. Shiao and Y.-F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef CAS PubMed.
- W. Li and X. Chen, Nanomedicine, 2015, 10, 299–320 CrossRef CAS PubMed.
- W. Li, J. Miao, T. Peng, H. Lv, J.-G. Wang, K. Li, Y. Zhu and D. Li, Nano Lett., 2020, 20, 2507–2513 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, H. B. Zhang, A. Govorov, T. Liedl and N. Liu, Nat. Mater., 2014, 13, 862–866 CrossRef CAS PubMed.
- X. Lan, T. Liu, Z. Wang, A. Govorov, H. Yan and Y. Liu, J. Am. Chem. Soc., 2018, 140, 11763–11770 CrossRef CAS PubMed.
- Z. Fan and A. Govorov, Nano Lett., 2012, 12, 3283–3289 CrossRef CAS PubMed.
- G. González-Rubio, J. Mosquera, V. Kumar, A. Pedrazo-Tardajos, P. Llombart, D. M. Solís, I. Lobato, E. Noya, A. Guerrero-Martínez, J. M. Taboada, F. Obelleiro, L. G. Macdowell, S. Bals and L. Liz-Marzán, Science, 2020, 368, 1472–1477 CrossRef PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- I. Lieberman, G. Shemer, T. Fried, E. Kosower and G. Markovich, Angew. Chem., 2008, 47, 4855–4857 CrossRef CAS PubMed.
- J. Slocik, A. Govorov and R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- T. Levi-Belenkova, A. Govorov and G. Markovich, J. Phys. Chem. C, 2016, 120, 12751–12756 CrossRef CAS.
- C. Rao, Z. Wang, N. Li, W. Zhang, X. Xu and B. Ding, Nanoscale, 2015, 7, 9147–9152 RSC.
- Z. Fan and A. Govorov, Nano Lett., 2010, 10, 2580–2587 CrossRef CAS PubMed.
- A. Cecconello, L. Besteiro, A. Govorov and I. Willner, Nat. Rev. Mater., 2017, 2, 17039 CrossRef CAS.
- D. Winogradoff, P.-Y. Li, H. Joshi, L. Quednau, C. Maffeo and A. Aksimentiev, Adv. Sci., 2021, 8, 2003113 CrossRef CAS PubMed.
- A. Cecconello and F. Simmel, Small, 2019, 15, e1805419 CrossRef PubMed.
- A. Govorov, J. Phys. Chem. C, 2011, 115, 7914–7923 CrossRef CAS.
- A. Govorov and Z. Fan, ChemPhysChem, 2012, 13, 2551–2560 CrossRef CAS PubMed.
- Z. Fan, H. Zhang and A. Govorov, J. Phys. Chem. C, 2013, 117, 14770–14777 CrossRef CAS.
- Z. Fan and A. Govorov, J. Phys. Chem. C, 2011, 115, 13254–13261 CrossRef CAS.
- J. Watson and F. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- V. Ivanov, L. Minchenkova, A. Schyolkina and A. Poletayev, Biopolymers, 1973, 12, 89–110 CrossRef CAS PubMed.
- A. Leslie, S. Arnott, R. Chandrasekaran and R. Ratliff, J. Mol. Biol., 1980, 143, 49–72 CrossRef CAS PubMed.
- G. Schroth, P. Chou and P. Ho, J. Biol. Chem., 1992, 267, 11846–11855 CrossRef CAS.
- H. Moser and P. Dervan, Science, 1987, 238, 645–650 CrossRef CAS PubMed.
- P. Guo and S. L. Lam, J. Am. Chem. Soc., 2016, 138, 12534–12540 CrossRef CAS PubMed.
- F. Hays, Z. Jones and P. Ho, Biochemistry, 2004, 43, 9813–9822 CrossRef CAS PubMed.
- D. Sen and W. Gilbert, Nature, 1988, 334, 364–366 CrossRef CAS PubMed.
- G. Parkinson, M. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS PubMed.
- M. D. Antonio, A. Ponjavic, A. Radzevjčius, R. T. Ranasinghe, M. Catalano, X. Zhang, J. Shen, L.-M. Needham, S. F. Lee, D. Klenerman and S. Balasubramanian, Nat. Chem., 2020, 12, 832–837 CrossRef PubMed.
- A. Phan, M. Guéron and J. Leroy, J. Mol. Biol., 2000, 299, 123–144 CrossRef CAS PubMed.
- H. Day, P. Pavlou and Z. A. E. Waller, Bioorg. Med. Chem., 2014, 22, 4407–4418 CrossRef CAS PubMed.
- M. Zeraati, D. Langley, P. Schofield, A. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. Dinger and D. Christ, Nat. Chem., 2018, 10, 631–637 CrossRef CAS PubMed.
- N. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS PubMed.
- T. Fu and N. Seeman, Biochemistry, 1993, 32, 3211–3220 CrossRef CAS PubMed.
- C. Mao, W. Sun and N. Seeman, J. Am. Chem. Soc., 1999, 121, 5437–5443 CrossRef CAS.
- T. LaBean, H. Yan, J. Kopatsch, F. Liu, E. Winfree, J. Reif and N. Seeman, J. Am. Chem. Soc., 2000, 122, 1848–1860 CrossRef CAS.
- C. Mao, T. LaBean, J. Reif and N. Seeman, Nature, 2000, 407, 493–496 CrossRef CAS PubMed.
- H. Yan, S. H. Park, G. Finkelstein, J. Reif and T. LaBean, Science, 2003, 301, 1882–1884 CrossRef CAS PubMed.
- J. Chen and N. Seeman, Nature, 1991, 350, 631–633 CrossRef CAS PubMed.
- Y. Zhang and N. Seeman, J. Am. Chem. Soc., 1994, 116, 1661–1669 CrossRef CAS.
- R. Goodman, I. Schaap, C. Tardin, C. Erben, R. Berry, C. Schmidt and A. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- R. Goodman, R. Berry and A. Turberfield, Chem. Commun., 2004, 1372–1373 RSC.
- Y. He, T. Ye, M. Su, C. Zhang, A. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS PubMed.
- C. Zhang, M. Su, Y. He, X. Zhao, P. Fang, A. Ribbe, W. Jiang and C. Mao, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10665–10669 CrossRef CAS PubMed.
- W. Shih, J. Quispe and G. Joyce, Nature, 2004, 427, 618–621 CrossRef CAS PubMed.
- P. Yin, R. Hariadi, S. Sahu, H. M. Choi, S. H. Park, T. LaBean and J. Reif, Science, 2008, 321, 824–826 CrossRef CAS PubMed.
- C. Erben, R. P. Goodman and A. Turberfield, J. Am. Chem. Soc., 2007, 129, 6992–6993 CrossRef CAS PubMed.
- F. A. Aldaye and H. Sleiman, J. Am. Chem. Soc., 2007, 129, 13376–13377 CrossRef CAS PubMed.
- Y. He, Y. Tian, A. Ribbe and C. Mao, J. Am. Chem. Soc., 2006, 128, 15978–15979 CrossRef CAS PubMed.
- P. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- G. Tikhomirov, P. Petersen and L. Qian, Nature, 2017, 552, 67–71 CrossRef CAS PubMed.
- Y. Ke, L. Ong, W. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed.
- C. Zhou, X. Duan and N. Liu, Acc. Chem. Res., 2017, 50, 2906–2914 CrossRef CAS PubMed.
- K. F. Wagenbauer, C. Sigl and H. Dietz, Nature, 2017, 552, 78–83 CrossRef CAS PubMed.
- T. Gerling, K. F. Wagenbauer, A. Neuner and H. Dietz, Science, 2015, 347, 1446–1452 CrossRef CAS PubMed.
- F. Simmel, B. Yurke and H. R. Singh, Chem. Rev., 2019, 119, 6326–6369 CrossRef CAS PubMed.
- B. Yurke, A. Turberfield, A. Mills Jr, F. Simmel and J. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- L. L. Hildebrandt, S. Preus, Z. Zhang, N. V. Voigt, K. Gothelf and V. Birkedal, J. Am. Chem. Soc., 2014, 136, 8957–8962 CrossRef CAS PubMed.
- Z. Yu, M. Centola, J. Valero, M. Matthies, P. Šulc and M. Famulok, J. Am. Chem. Soc., 2021, 143, 13292–13298 CrossRef PubMed.
- F. Topuz and O. Okay, Macromolecules, 2008, 41, 8847–8854 CrossRef CAS.
- T. Zhang, J. Nong, N. Alzahrani, Z. Wang, S. W. Oh, T. Meier, D. Yang, Y. Ke, Y. Zhong and J. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 29512–29521 CrossRef CAS PubMed.
- J.-H. Lee, S. H. Lee, C. Baek, H.-S. Chun, J.-h. Ryu, J.-W. Kim, R. Deaton and B.-T. Zhang, BioSystems, 2017, 158, 1–9 CrossRef CAS PubMed.
- L. Kari, Future Gener. Comput. Syst., 2001, 17, 823–834 CrossRef.
- C. Mirkin, R. Letsinger, R. Mucic and J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- J. Storhoff, R. Elghanian, R. C. Mucic, C. Mirkin and R. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- X. Zhang, M. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266–7269 CrossRef CAS PubMed.
- X. Zhang, B. Liu, N. Dave, M. Servos and J. Liu, Langmuir, 2012, 28, 17053–17060 CrossRef CAS PubMed.
- Q. Xu, X. Lou, L. Wang, X. Ding, H. Yu and Y. Xiao, ACS Appl. Mater. Interfaces, 2016, 8, 27298–27304 CrossRef CAS PubMed.
- B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 9471–9474 CrossRef CAS PubMed.
- Y. Hao, Y. Li, L. Song and Z. Deng, J. Am. Chem. Soc., 2021, 143, 3065–3069 CrossRef CAS PubMed.
- M. Östblom, B. Liedberg, L. Demers and C. Mirkin, J. Phys. Chem. B, 2005, 109, 15150–15160 CrossRef PubMed.
- Z. Wang, J. Zhang, J. Ekman, P. Kenis and Y. Lu, Nano Lett., 2010, 10, 1886–1891 CrossRef CAS PubMed.
- Z. Wang, L. Tang, L. H. Tan, J. Li and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 9078–9082 CrossRef CAS PubMed.
- L. H. Tan, H. Xing and Y. Lu, Acc. Chem. Res., 2014, 47, 1881–1890 CrossRef CAS PubMed.
- T. Song, L. Tang, L. H. Tan, X. Wang, N. S. R. Satyavolu, H. Xing, Z. Wang, J. Li, H. Liang and Y. Lu, Angew. Chem., Int. Ed., 2015, 54, 8114–8118 CrossRef CAS PubMed.
- N. S. R. Satyavolu, L. H. Tan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 16542–16548 CrossRef CAS PubMed.
- G. Yao, H. Pei, J. Li, Y.-z. Zhao, D. Zhu, Y. Zhang, Y. Lin, Q. Huang and C. Fan, NPG Asia Mater., 2015, 7, e159 CrossRef.
- G. Yao, J. Li, Q. Li, X. Chen, X. Liu, F. Wang, Z. Qu, Z. Ge, R. P. Narayanan, D. Williams, H. Pei, X. Zuo, L. Wang, H. Yan, B. Feringa and C. Fan, Nat. Mater., 2019, 19, 781–788 CrossRef PubMed.
- A. Govorov, Z. Fan, P. Hernandez, J. Slocik and R. Naik, Nano Lett., 2010, 10, 1374–1382 CrossRef CAS PubMed.
- Z. Li, Z. Zhu, W. Liu, Y. Zhou, B. Han, Y. Gao and Z. Tang, J. Am. Chem. Soc., 2012, 134, 3322–3325 CrossRef CAS PubMed.
- Z. Bagheri, B. Ranjbar, A. Azizi, H. Latifi, M. I. Zibaii and T. T. Moghadam, Sci. Iran., 2017, 25, 1783–1788 Search PubMed.
- F. Lu, Y. Tian, M. Liu, D. Su, H. Zhang, A. Govorov and O. Gang, Nano Lett., 2013, 13, 3145–3151 CrossRef CAS PubMed.
- X. Lan, X. Zhou, L. A. McCarthy, A. Govorov, Y. Liu and S. Link, J. Am. Chem. Soc., 2019, 141, 19336–19341 CrossRef CAS PubMed.
- J. Zhu, F. Wu, Z. Han, Y. Shang, F. Liu, H. Yu, L. Yu, N. Li and B. Ding, Nano Lett., 2021, 21, 3573–3580 CrossRef CAS PubMed.
- A. Alivisatos, K. Johnsson, X. Peng, T. Wilson, C. Loweth, M. Bruchez Jr and P. Schultz, Nature, 1996, 382, 609–611 CrossRef CAS PubMed.
- B. Auguié, J. Alonso-Gómez, A. Guerrero-Martínez and L. Liz-Marzán, J. Phys. Chem. Lett., 2011, 2, 846–851 CrossRef PubMed.
- W. Yan, W. Ma, H. Kuang, L. Liu, L. Wang, L. Xu and C. Xu, J. Phys. Chem. C, 2013, 117, 17757–17765 CrossRef CAS.
- C. Hao, L. Xu, W. Ma, L. Wang, H. Kuang and C. Xu, Small, 2014, 10, 1805–1812 CrossRef CAS PubMed.
- W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. Kotov, Nat. Commun., 2013, 4, 2689 CrossRef PubMed.
- A. J. Mastroianni, S. Claridge and A. Alivisatos, J. Am. Chem. Soc., 2009, 131, 8455–8459 CrossRef CAS PubMed.
- W. Yan, L. Xu, C. Xu, W. Ma, H. Kuang, L. Wang and N. Kotov, J. Am. Chem. Soc., 2012, 134, 15114–15121 CrossRef CAS PubMed.
- W. Chen, A. Bian, A. Agarwal, L. Liu, H. Shen, L. Wang, C. Xu and N. Kotov, Nano Lett., 2009, 9, 2153–2159 CrossRef CAS PubMed.
- H. Liu, X. Shen, Z. Wang, A. Kuzyk and B. Ding, Nanoscale, 2014, 6, 9331–9338 RSC.
- J. Sharma, R. Chhabra, A. Cheng, J. Brownell, Y. Liu and H. Yan, Science, 2009, 323, 112–116 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. Simmel, A. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- J. C. Mitchell, J. Harris, J. Malo, J. Bath and A. Turberfield, J. Am. Chem. Soc., 2004, 126, 16342–16343 CrossRef CAS PubMed.
- S. Sun, Y. Yang, D. Li and J. Zhu, J. Am. Chem. Soc., 2019, 141, 19524–19528 CrossRef CAS PubMed.
- X. Shen, C. Song, J. Wang, D. Shi, Z. Wang, N. Liu and B. Ding, J. Am. Chem. Soc., 2012, 134, 146–149 CrossRef CAS PubMed.
- F. Benn, N. E. C. Haley, A. E. Lucas, E. Silvester, S. Helmi, R. Schreiber, J. Bath and A. Turberfield, Angew. Chem., Int. Ed., 2018, 57, 7687–7690 CrossRef CAS PubMed.
- M. J. Urban, P. K. Dutta, P. Wang, X. Duan, X. Shen, B. Ding, Y. Ke and N. Liu, J. Am. Chem. Soc., 2016, 138, 5495–5498 CrossRef CAS PubMed.
- A. Guerrero-Martínez, B. Auguié, J. L. Alonso-Gómez, Z. Džolić, S. Gómez-Graña, M. Žinić, M. Cid and L. Liz-Marzán, Angew. Chem., Int. Ed., 2011, 50, 5499–5503 CrossRef PubMed.
- S. Pal, Z. Deng, H. Wang, S. Zou, Y. Liu and H. Yan, J. Am. Chem. Soc., 2011, 133, 17606–17609 CrossRef CAS PubMed.
- X. Lan, Z. Chen, G. Dai, X. Lu, W. Ni and Q. Wang, J. Am. Chem. Soc., 2013, 135, 11441–11444 CrossRef CAS PubMed.
- X. Shen, P. Zhan, A. Kuzyk, Q. Liu, A. Asenjo-Garcia, H. Zhang, F. D. d. Abajo, A. Govorov, B. Ding and N. Liu, Nanoscale, 2014, 6, 2077–2081 RSC.
- Z. Chen, C. K. K. Choi and Q. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 26835–26840 CrossRef CAS PubMed.
- X. Lan, X. Lu, C. Shen, Y. Ke, W. Ni and Q. Wang, J. Am. Chem. Soc., 2015, 137, 457–462 CrossRef CAS PubMed.
- X. Lan, Z. Su, Y. Zhou, T. Meyer, Y. Ke, Q. Wang, W. Chiu, N. Liu, S. Zou, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 14632–14636 CrossRef CAS PubMed.
- P. Wang, J.-H. Huh, H. Park, D. Yang, Y. Zhang, Y. Zhang, J. Lee, S. Lee and Y. Ke, Nano Lett., 2020, 20, 8926–8932 CrossRef CAS PubMed.
- Y. Huang, M.-K. Nguyen and A. Kuzyk, J. Visualized Exp., 2019, 145, e59280 Search PubMed.
- T. Funck, F. Nicoli, A. Kuzyk and T. Liedl, Angew. Chem., Int. Ed., 2018, 57, 13495–13498 CrossRef CAS PubMed.
- A. Kuzyk, M. J. Urban, A. Idili, F. Ricci and N. Liu, Sci. Adv., 2017, 3, e1602803 CrossRef PubMed.
- Q. Jiang, Q. Liu, Y. Shi, Z.-G. Wang, P. Zhan, J. Liu, C. Liu, H. Wang, X. Shi, L. Zhang, J. Sun, B. Ding and M. Liu, Nano Lett., 2017, 17, 7125–7130 CrossRef CAS PubMed.
- R. Schreiber, N. Luong, Z. Fan, A. Kuzyk, P. C. Nickels, T. Zhang, D. M. Smith, B. Yurke, W. Kuang, A. Govorov and T. Liedl, Nat. Commun., 2013, 4, 2948 CrossRef PubMed.
- A. Kuzyk, Y. Yang, X. Duan, S. Stoll, A. Govorov, H. Sugiyama, M. Endo and N. Liu, Nat. Commun., 2016, 7, 10591 CrossRef CAS PubMed.
- J. Ryssy, A. K. Natarajan, J. Wang, A. Lehtonen, M.-K. Nguyen, R. Klajn and A. Kuzyk, Angew. Chem., Int. Ed., 2020, 60, 5859–5863 CrossRef PubMed.
- L. Xin, C. Zhou, X. Duan and N. Liu, Nat. Commun., 2019, 10, 5394 CrossRef PubMed.
- C. Zhou, X. Duan and N. Liu, Nat. Commun., 2015, 6, 8102 CrossRef CAS PubMed.
- M. J. Urban, C. Zhou, X. Duan and N. Liu, Nano Lett., 2015, 15, 8392–8396 CrossRef CAS PubMed.
- M. Wang, J. Dong, C. Zhou, H. Xie, W. Ni, S. Wang, H. Jin and Q. Wang, ACS Nano, 2019, 13, 13702–13708 CrossRef CAS PubMed.
- M.-K. Nguyen and A. Kuzyk, ACS Nano, 2019, 13, 13615–13619 CrossRef CAS PubMed.
- H. Kuang, H. Yin, L. Liu, L. Xu, W. Ma and C. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 364–369 CrossRef CAS PubMed.
- S. Li, L. Xu, W. Ma, X. Wu, M. Sun, H. Kuang, L. Wang, N. Kotov and C. Xu, J. Am. Chem. Soc., 2016, 138, 306–312 CrossRef CAS PubMed.
- M. Sun, L. Xu, F. Pan, W. Xiaoling, H. Kuang, L. Liu and C. Xu, Adv. Funct. Mater., 2016, 26, 7352–7358 CrossRef CAS.
- P. Fu, M. Sun, L. Xu, X. Wu, L. Liu, H. Kuang, S. Song and C. Xu, Nanoscale, 2016, 8, 15008–15015 RSC.
- H. Zhao, S. Bian, Y. Yang and X. Wu, Microchim. Acta, 2017, 184, 1855–1862 CrossRef CAS.
- W. Xiuxiu, L. Xu, C. Hao, C. Xu and H. Kuang, Environ. Sci. Technol. Lett., 2018, 5, 500–507 CrossRef.
- Y. Liu, M. Wei, L. Zhang, Y. Zhang, W. Wei, L. Yin, Y. Pu and S. Liu, Anal. Chem., 2016, 88, 6509–6514 CrossRef CAS PubMed.
- Z. Xu, L. Xu, L. Liz-Marzán, W. Ma, N. Kotov, L. Wang, H. Kuang and C. Xu, Adv. Opt. Mater., 2013, 1, 626–630 CrossRef.
- Y. Huang, M.-K. Nguyen, A. K. Natarajan, V. Nguyen and A. Kuzyk, ACS Appl. Mater. Interfaces, 2018, 10, 44221–44225 CrossRef CAS PubMed.
- T. Funck, T. Liedl and W. Bae, Appl. Sci., 2019, 9, 3006 CrossRef CAS.
- C. Zhou, L. Xin, X. Duan, M. J. Urban and N. Liu, Nano Lett., 2018, 18, 7395–7399 CrossRef CAS PubMed.
- L. Adleman, Science, 1994, 266, 1021–1024 CrossRef CAS PubMed.
- A. Okamoto, K. Tanaka and I. Saito, J. Am. Chem. Soc., 2004, 126, 9458–9463 CrossRef CAS PubMed.
- J. Dong, M. Wang, Y. Zhou, C. Zhou and Q. Wang, Angew. Chem., Int. Ed., 2020, 59, 15038–15042 CrossRef CAS PubMed.
- K. Göpfrich, M. J. Urban, C. Frey, I. Platzman, J. Spatz and N. Liu, Nano Lett., 2020, 20, 1571–1577 CrossRef PubMed.
- K. Martens, F. Binkowski, L.-D. Nguyen, L. Hu, A. Govorov, S. Burger and T. Liedl, Nat. Commun., 2021, 12, 2025 CrossRef CAS PubMed.
- L. Hu, T. Liedl, K. Martens, Z. Wang and A. Govorov, ACS Photonics, 2019, 6, 749–756 CrossRef CAS.
- E. Cortés, W. Xie, J. Cambiasso, A. S. Jermyn, R. Sundararaman, P. Narang, S. Schlücker and S. Maier, Nat. Commun., 2017, 8, 14880 CrossRef PubMed.
- P. Wang, A. Krasavin, M. Nasir, W. Dickson and A. Zayats, Nat. Nanotechnol., 2017, 13, 159–164 CrossRef PubMed.
- T. Liu, L. Besteiro, T. Liedl, M. Correa-Duarte, Z. Wang and A. Govorov, Nano Lett., 2019, 19, 1395–1407 CrossRef CAS PubMed.
- J. Trevino, H. Cao and L. D. Negro, Nano Lett., 2011, 11, 2008–2016 CrossRef CAS PubMed.
- V. Valev, J. Baumberg, B. D. Clercq, N. Braz, X. Zheng, E. J. Osley, S. Vandendriessche, M. Hojeij, C. Blejean, J. Mertens, C. Biris, V. Volskiy, M. Ameloot, Y. Ekinci, G. Vandenbosch, P. Warburton, V. Moshchalkov, N. Panoiu and T. Verbiest, Adv. Mater., 2014, 26, 4074–4081 CrossRef CAS PubMed.
- A. Drezet, C. Genet, J.-Y. Laluet and T. Ebbesen, Opt. Express, 2008, 16, 12559–12570 CrossRef PubMed.
- J. K. Gansel, M. Thiel, M. Rill, M. Decker, K. Bade, V. Saile, G. v. Freymann, S. Linden and M. Wegener, Science, 2009, 325, 1513–1515 CrossRef CAS PubMed.
- G. Cheng, D. Xu, Z. Lu and K. Liu, ACS Nano, 2019, 13, 1479–1489 CAS.
- J. George and K. G. Thomas, J. Am. Chem. Soc., 2010, 132, 2502–2503 CrossRef CAS PubMed.
- R. Mout, G. Y. Tonga, L.-s. Wang, M. Ray, T. Roy and V. Rotello, ACS Nano, 2017, 11, 3456–3462 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |