 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of linker functionalisation on the catalytic properties of Cu nanoclusters embedded in MOFs in direct CO and CO2 reduction by H2†
Cornelia Elizabeth (Lisette)
Pompe
a and
Petra Ágota
Szilágyi
 *b
*b
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
bSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Campus, E1 4NS, London, UK. E-mail: p.szilagyi@qmul.ac.uk
First published on 10th March 2021
Abstract
Metal–organic frameworks are promising host supporting matrices for catalytically active guests. In particular, their crystallinity renders them desirable as their pores may act as atom-precise templates for the growth of nanoparticles or nanoclusters therein. This is very advantageous for studying the fundamentals of heterogeneous catalytic processes, especially for site-selective ones, in which the catalyst particle size and shape are of import. Furthermore, metal–organic frameworks may be decorated with organic functional groups, which affect a number of the frameworks’ physical and chemical properties, while remaining isotopological, i.e. having identical structural features and thus templating characteristics. Effectively, this allows for the independent tuning of the Brønsted and Lewis acidity of the substrate while the geometry of the catalytically active guest remains identical. In this study, we systematically study the effect of amino functionalisation of UiO-66(Zr) as a supporting matrix for Cu nanoclusters on the direct reduction of carbon dioxide with hydrogen. In particular, we have found that through modulation of the acidity of the inorganic nodes, framework functionalisation effectively controls the reaction selectivity.
Introduction
The catalytic conversion of carbon dioxide (CO2) into simple chemicals such as formic acid, dimethyl ether, methanol, and carbon monoxide is critical not only for the management of greenhouse gases and thus for the mitigation of climate change, but also for the implementation of a zero carbon energy cycle. Methanol production is of particular importance as methanol is regarded as a suitable liquid energy carrier under ambient conditions, while it can also be used for the synthesis of other industrially relevant products, such as olefins, formalin, etc. Consequently, the development of high-performance and selective catalysts for the conversion of CO2 into methanol by direct hydrogenation is highly desirable, but remains a challenge nevertheless. The reason for this is that CO2 is a highly stable molecule both thermodynamically and structurally, hindering the ease of conversion reactions.Both CO and CO2 can be hydrogenated directly on a Cu catalyst, however it has been shown that it is CO2 whose conversion yields the vast majority of methanol molecules;1,2 on the other hand, CO undergoes the water–gas shift (WGS)2,3 reaction, sustaining low concentrations of H2O and CO2 in the reaction mixture. This is advantageous, as even though from an environmental perspective, the direct hydrogenation of CO2 would be desirable, catalyst stability would be compromised4 because of the high water concentrations unmitigated by WGS.5
It should be noted that the direct conversion of CO2 to methanol through hydrogenation is structure sensitive, i.e. catalytic properties are strongly influenced by the dimension of the catalytically active particles, and the geometry and composition of the catalyst–substrate interface.6,7 In particular, strong metal–support interactions (SMSIs) and confinement effects have previously been shown to be applicable to stabilising Cu nanoparticle (NP) catalysts for the direct catalytic hydrogenation of CO2.8–14 For example, zirconia supports engage in stronger interactions with Cu NPs than alumina ones, leading to better Cu NP dispersion, and thus to increased catalyst stability.15 Furthermore, porous materials, such as mesoporous zeolites have also been successfully applied to inhibit NP growth through nanoconfinement.11,16,17
Metal–organic frameworks (MOFs), built up of inorganic nodes (cations or metal oxide clusters) interconnected by organic linkers (acids or heterocycles) are typically porous crystalline solids. As the pores are part of the crystal structure, they have shown promise as supports for highly active catalysts.18–23 Organic functionalities on the MOF linkers can further stabilise NPs and tune their surface structures by specific SMSIs.24–26 Furthermore, MOFs offer proximity of their inorganic nodes and compositional tunability while possibly controlling the dimensions of the catalyst particles, which could enable a systematic study of the catalytic interface.8,24,27–30 Consequently, MOFs can combine both geometry control through nanoconfinement within pores and possibly SMSIs with their building blocks to enable new Cu-based CO2 hydrogenation catalysts.
In particular, Cu@MOF core@shell structures of 18 nm Cu NPs have been reported to catalyse CO2 hydrogenation with high selectivity.8 While much smaller, ca. 1–1.5 nm, Cu nanoclusters (NCs) have also demonstrated excellent catalytic properties in the direct hydrogenation of CO2.31 It should be emphasised that this good performance can be attributed to the strong promotion effect arising from the Cu/ZnOx SMSIs, as Cu particles smaller than 8 nm would be expected to display much poorer performance on account of the site sensitivity of the catalytic reaction.7 This highlights that strong interactions with the supporting matrix may have an enabling effect on Cu NCs for the direct hydrogenation of carbon dioxide. We have previously shown that functionalisation of UiOs may result in enhanced interaction with embedded Pd NCs, and in a potential change in the catalytic performance.32,33 In particular, the effect of amino groups is worth exploring as (i) they can form strong interactions with metal NCs embedded in the MOF’s pores, potentially mimicking SMSIs,32–34 and furthermore (ii) they have been shown to interact strongly with CO2 and thus their presence may be favourable for its activation and conversion.35 At the same time, linker functionalisation may also have a long-range effect on the inorganic node (i.e. an effect going beyond the immediate surroundings of the functional group),36 an idea which has recently been explored for CO2 conversion on Cu@MOFs.37
In order to test the effect of linker functionalisation on the catalytic properties of Cu nanoclusters embedded in MOFs in direct CO and CO2 reduction by H2, we therefore selected UiO-66(Zr) and NH2–UiO-66(Zr) as matrices, which both provide pores as templates for Cu NC growth of up to 1.1 nm particle diameter when embedded in the pores. At the same time, we assumed that the presence of the amino groups might change both the interactions between the Cu NCs and the MOF (SMSI-like behaviour) and those between the framework and CO2, while they would definitely alter the Lewis acidity of the zirconia-like inorganic nodes, an effect that has been shown to affect the catalyst selectivity of zirconia-supported Cu NPs.38
Results and discussion
As the performance of supported Cu catalysts towards the direct hydrogenation of CO2 is dependent on both the particle size and the supporting substrate, if the effect of the MOF functional groups on the reaction is to be determined, it is imperative that the catalyst particles are of uniform size distribution and of quasi-identical size for the pristine and functionalised frameworks. As UiO-66(Zr) and NH2–UiO-66(Zr) are isotopological frameworks, their pore sizes are also quasi-identical. It follows that as long as the Cu particles fill their pores, i.e. they are embedded therein, the size effect in the two MOF matrices should be identical and, accordingly, all changes pertaining to the catalytic performance would be a consequence of the effect of the functional group. This effect may be either direct, i.e. a result of an interaction between the functional group and the Cu particles, or indirect, such as an effect of the changes in the acidity of the inorganic node.Therefore, in order to ascertain that the Cu particle sizes are equal in the two different supporting scaffolds, we have acquired TEM images of them (Fig. 1).
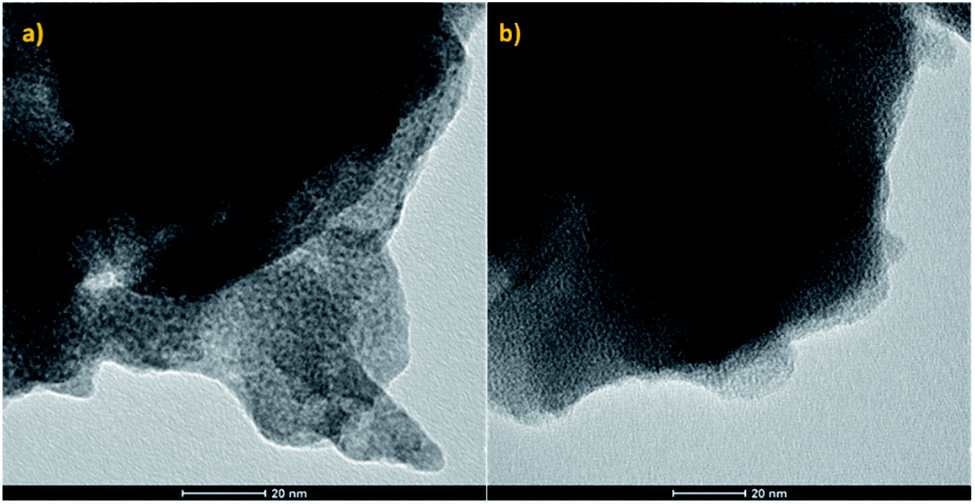 | ||
| Fig. 1 TEM micrographs of (a) pristine UiO-66(Zr) and (b) NH2–UiO-66(Zr) loaded with Cu nanoclusters. | ||
Evidently, our TEM micrographs show no evidence of Cu nanoparticles larger than the framework pore sizes (0.7 and 1.1 nm (ref. 39)). This demonstrates that the embedding of Cu nanoclusters in the selected frameworks’ pores was successful, and as the Cu nanocluster size is determined by the templating effect of the frameworks, their sizes are equal in both the pristine and functionalised MOFs. It is interesting to note that in previous work, wherein Pd nanoclusters were embedded in the pores of the same frameworks, only the functionalised framework stabilised the Pd nanoclusters.32 This suggests higher thermodynamic stability of Cu nanoclusters when compared with Pd. Strong host–guest interactions, analogous to SMSIs, have been observed in the case of Pd and NH2–UiO-66, and were proposed to be responsible for stabilising Pd nanoclusters. In the case of Cu, however, this might not be the case as Cu nanoclusters were also immobilised in the pores of pristine UiO-66, which was incapable of forming strong host–guest interactions with Pd.
To explore this possibility, i.e. to probe the existence of SMSIs, we have analysed the chemical state of Cu embedded in UiO-66(Zr) and NH2–UiO-66(Zr) using X-ray photoelectron spectroscopy (XPS, Fig. 2).
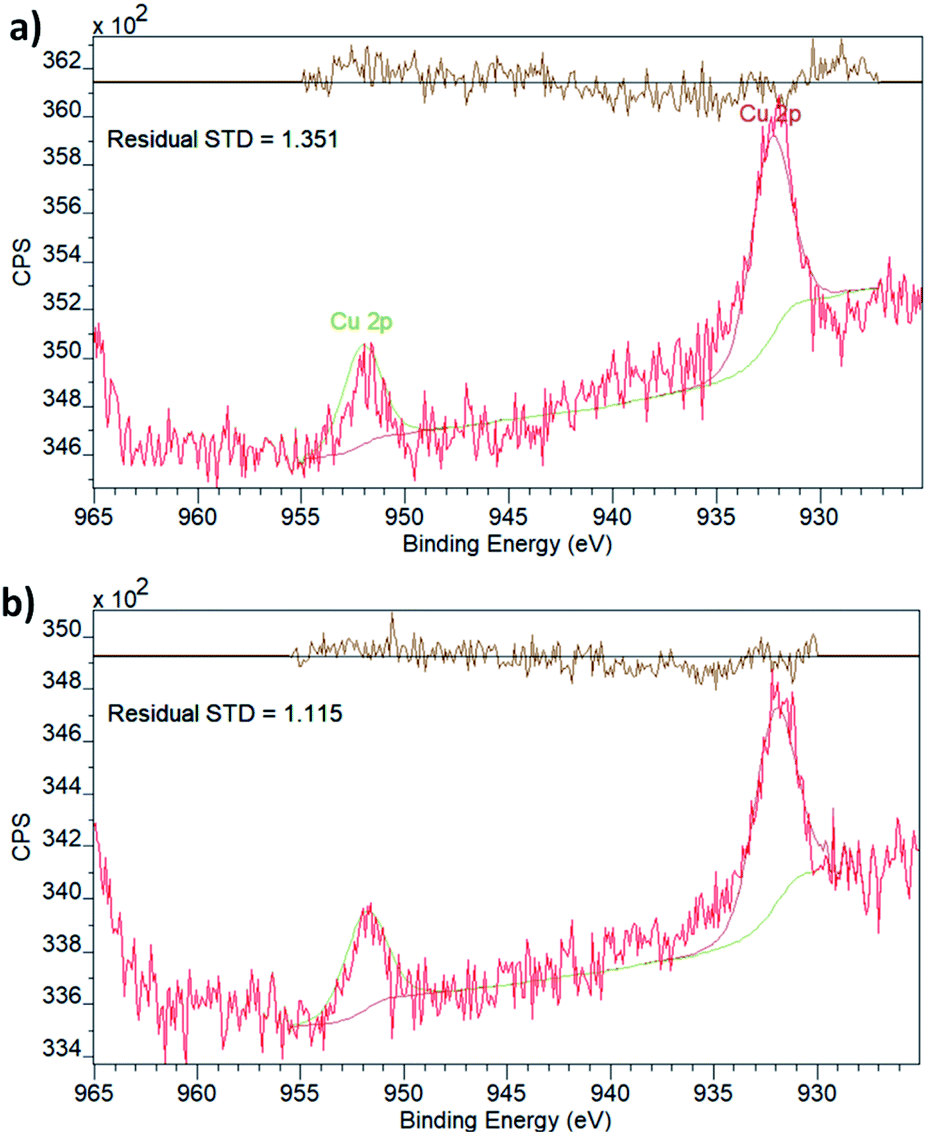 | ||
| Fig. 2 XPS spectra of Cu-laden (a) UiO-66(Zr) and (b) NH2–UiO-66(Zr). | ||
Both spectra display two peaks in the Cu 2p region, corresponding to the two different spin–orbit interactions of 2p orbitals. The binding energies (Table 1) are in effect identical for the two samples and in good agreement with what is expected for metallic Cu.40 It is, however, important to point out that the 2p1/2 and 2p3/2 binding energies of Cu do not vary significantly with the oxidation state, which is best evaluated by the existence and prevalence of the relevant satellite peaks.41 Though neither of the spectra display any obvious satellite peaks, in line with the oxidation state being zero, in principle, a small charge transfer between the linker functionalities and the Cu nanoclusters could still exist. This has been observed in the case of Pd embedded in the pores of NH2–UiO-66(Zr) by evaluating both the Pd and N chemical states.32 As the Cu XPS spectrum could not provide unequivocal evidence, we also evaluated the chemical state of N in the amino functional group of the linker in NH2–UiO-66(Zr).
| E b (eV) | Cu 2p | N 1s | Zr 3d | C 1s | ||
|---|---|---|---|---|---|---|
| 2p1/2 | 2p3/2 | 3d3/2 | 3d5/2 | |||
| UiO-66 | — | — | 185.2 | 182.9 | 284.7 (86%) | |
| 288.4 (6%) | ||||||
| 286.0 (8%) | ||||||
| Cu in UiO-66 | 952.8 | 933.1 | — | 185.2 | 182.9 | 284.7 (76%) |
| 288.7 (10%) | ||||||
| 286.1 (14%) | ||||||
| NH2–UiO-66 | — | 399.4 (70%) | 185.2 | 182.8 | 284.7 (78%) | |
| 400.8 (30%) | 288.7 (8%) | |||||
| 285.9 (13%) | ||||||
| Cu in NH2–UiO-66 | 952.9 | 933.1 | 399.2 (68%) | 185.1 | 182.7 | 284.7 (81%) |
| 400.5 (32%) | 288.5 (9%) | |||||
| 286.0 (10%) | ||||||
The XPS spectrum in the N 1s region (Fig. 3) of empty NH2–UiO-66(Zr) is in good agreement with previous results,32 and in fact, the spectrum of Cu-laden NH2–UiO-66(Zr) is virtually identical, revealing that no significant interactions exist between the Cu nanoclusters and the nitrogen atom of the amino group in the linker. This is an intriguing finding, as in the case of Pd-laden NH2–UiO-66(Zr), the existence of a significant charge transfer between the Pd and N atoms was observed.32 The reason for this difference can be attributed to two different effects: (i) on the one hand, this may be a consequence of the different valence electronic configurations of Cu and Pd, as in the case of Cu, the d orbitals capable of dative bonding are not the outermost ones; (ii) on the other hand, the reason for this difference might originate from the different stabilities of the metal nanoclusters. We have seen that Pd nanoclusters of ca. 1 nm are unstable unless anchored or immobilised in the pores of MOFs via strong host–guest interactions; conversely, Cu nanoclusters of 1 nm can even be observed and thus stabilised in the pores of pristine UiO-66(Zr), a framework incapable of forming strong interactions with guest particles due to the lack of available functional groups on the linkers.
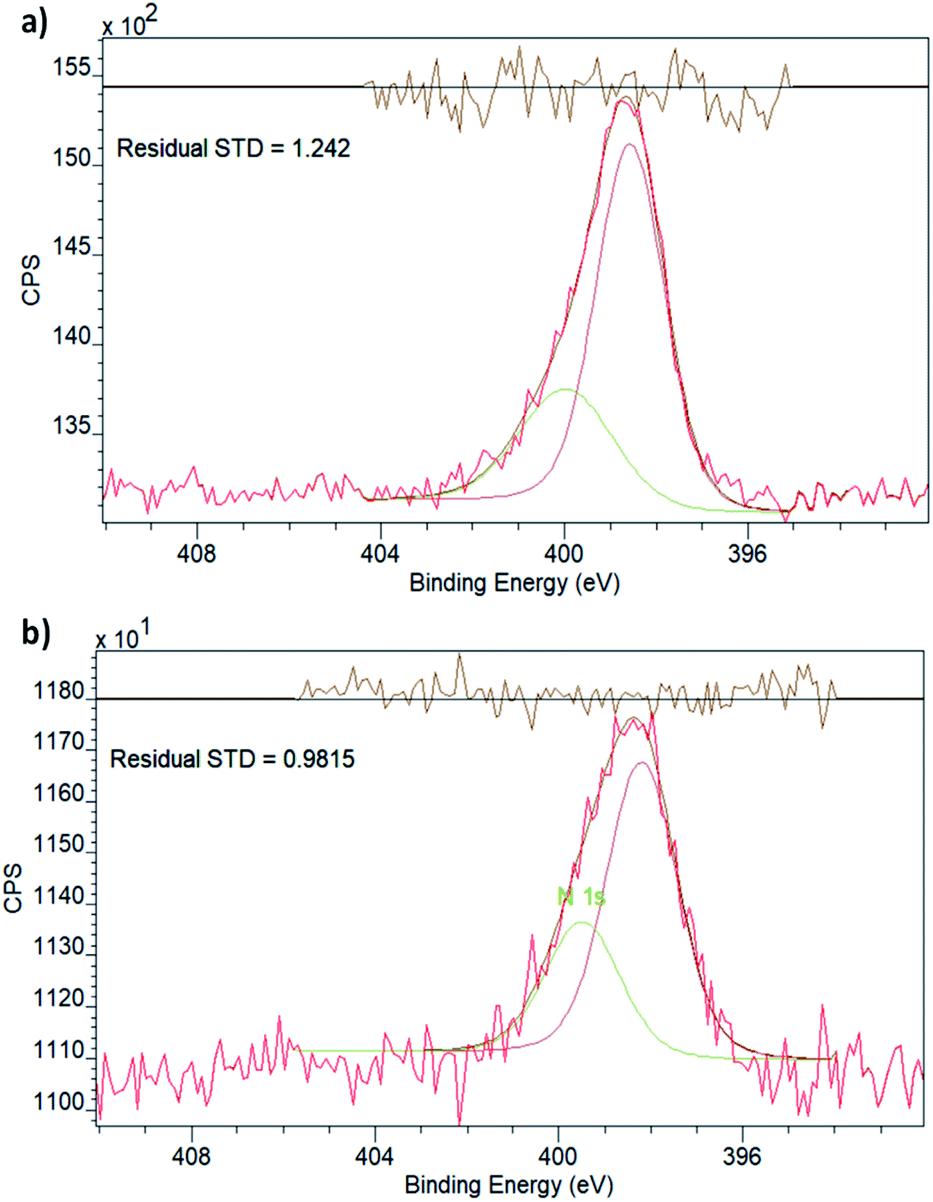 | ||
| Fig. 3 XPS spectra in the N 1s region for (a) empty NH2–UiO-66(Zr) and (b) Cu-laden NH2–UiO-66(Zr). | ||
Analysing the Cu contents of the two samples, it is also apparent that they are practically identical: 1.5 wt% Cu was loaded in UiO-66 while the Cu loading in NH2–UiO-66 was 1.4 wt%. Considering the slight difference in the framework molar mass, this is a likely consequence of no specific interaction between the guest and the host framework, leading to low loading governed by the surface area.
We conclude that Cu nanoclusters were successfully embedded in the pores of both UiO-66(Zr) and NH2–UiO-66(Zr), without any evidence for strong host–guest interactions. Therefore, the existence of SMSIs can be safely excluded in both Cu-laden UiO-66(Zr) and Cu-laden NH2–UiO-66(Zr). Crucially, this is not only valid for strong interactions with the functional groups of the organic linker but also for interactions with the inorganic nodes as the chemical state of Zr does not change significantly when Cu nanoclusters are embedded in the MOFs’ pores (Table 1). This observation is in contrast with suggestions made previously for larger Cu nanoparticles,37,42 in which a shift in the Zr binding energies was observed on the incorporation of Cu nanoparticle cores into MOF shells. The composition, structure, and strain effects are obviously different for nanocluster guest@MOF core@shell particles when compared with guest nanoclusters embedded in the pores of MOFs, which merits further investigations, particularly with respect to any lattice strain effect on the inorganic nodes of the MOF shell. From the lack of SMSIs, which could trigger an electronic promotion effect, we therefore expect that the catalytic performance of the Cu nanoclusters embedded in UiO-66(Zr) and NH2–UiO-66(Zr) will be poorer than what was observed for larger particles and promoted ones, in accordance with the site sensitivity of the reaction.
Considering that the catalytically active Cu particle sizes are quasi-identical in the two Cu-laden specimens, the Cu contents of the two samples are comparable, and that neither of the supporting scaffolds afford SMSIs, we can safely hypothesise that all differences in the catalytic performance of the two samples in the direct hydrogenation of CO and CO2 can only be an indirect effect of the presence of the amino functional groups in the case of the NH2–UiO-66(Zr) matrix. Such an effect may be a consequence of increased CO2 uptake and a potential contribution to CO2 activation through the –NH2 groups, or related to the differences in the acidity of the inorganic nodes of the two frameworks. In the first case, the catalytic activity of Cu-laden NH2–UiO-66(Zr) would be expected to eclipse that of Cu-laden UiO-66(Zr), whereas in the second case, Cu-laden UiO-66(Zr) should perform better as the presence of –NH2 groups has been shown to decrease the Lewis acidity of the inorganic nodes of MOFs,36 while the presence of a Lewis acid is beneficial for the direct reductive conversion of carbon dioxide on copper.38
In order to establish whether the linker functionality has such a secondary effect and what the effect consists of, we have assessed both the activity and selectivity of the Cu-laden MOF catalysts.
Upon inspecting the catalyst testing results, we noted that our assumption that Cu nanoclusters not engaging in SMSIs would display a poorer catalyst performance than their larger and/or promoted counterparts was correct. Cu NP@MOF core@shell particles of >10 nm already displayed almost complete selectivity towards methanol production in the 175–220 °C temperature range,37,42 while Cu NCs supported on Zn-functionalised UiO-bpy(Zr), partaking in SMSIs with the Zn moieties, also produced methanol at only 250 °C.31 Cu-laden UiO-66(Zr) and NH2–UiO-66(Zr), however, still showed low catalytic activity above 250 °C (Fig. S2†), which is in good agreement with previous studies on the site selectivity of the direct CO2 hydrogenation on Cu,7 unless strongly promoted, like in the case of Zn-functionalised UiO-bpy(Zr).31
It is most apparent that the activity of the Cu catalyst embedded in MOF pores did not improve with framework functionalisation, i.e. the –NH2 groups did not contribute significantly to the catalytic reaction through activation of the CO2 molecule or through enhanced gas adsorption. Considering the high temperature at which Cu-laden NH2–UiO-66(Zr) begins to show any activity for CO2 conversion, this is unsurprising, as the heats of CO2 adsorption on NH2- and NO2-functionalised MOFs have been determined to be in the 25–35 kJ mol−1 range,43 too weak to significantly change the course of this reaction.
On the other hand, it must be pointed out that Cu-laden UiO-66(Zr) shows very different selectivity from its amino-functionalised counterpart (Fig. 4). Specifically, the conversion reaction favours the formation of dimethyl ether at higher temperatures on pristine Cu-laden UiO-66(Zr). At 280 °C, the selectivity towards DME is up to 50% of converted CO and CO2. This is highly interesting and differs significantly from the catalyst selectivities observed for Cu supported on metal–organic frameworks, which typically tend to favour the formation of methanol.31,37,42 Considering the stark difference in selectivity when compared with Cu-laden NH2–UiO-66(Zr), and the fact that both Cu nanocluster size and surface chemistry are quasi-identical in these samples, this difference must be attributed to the changes in Lewis acidity of the inorganic nodes on functionalisation. In particular, amino-functionalised UiO-66(Zr) nodes have significantly lower acid character. The preference for ether formation must therefore be a direct contribution of the acid sites available on the inorganic nodes of UiO-66(Zr). Acidic sites are known to dehydrogenate methanol and induce the formation of DME. DME is considered a promising economical transportation fuel because it can be used as an efficient H2 carrier for fuel cell applications, a fuel source for diesel engines, and a substitute for Liquefied Petroleum Gas (LPG).44–46 As DME is typically produced via methanol dehydration, its direct synthesis is desirable.47 Herewith, we showed that this is possible on Cu nanoclusters by the judicious selection of a metal–organic framework supporting matrix. Most importantly, this result highlights that attention needs to be paid to all building blocks in a MOF when designing its catalytic applications, to develop understanding of their roles and possible functions. Furthermore, the selectivity we observed exceeds 50%, which is greater than typical values (around 7–12%).48
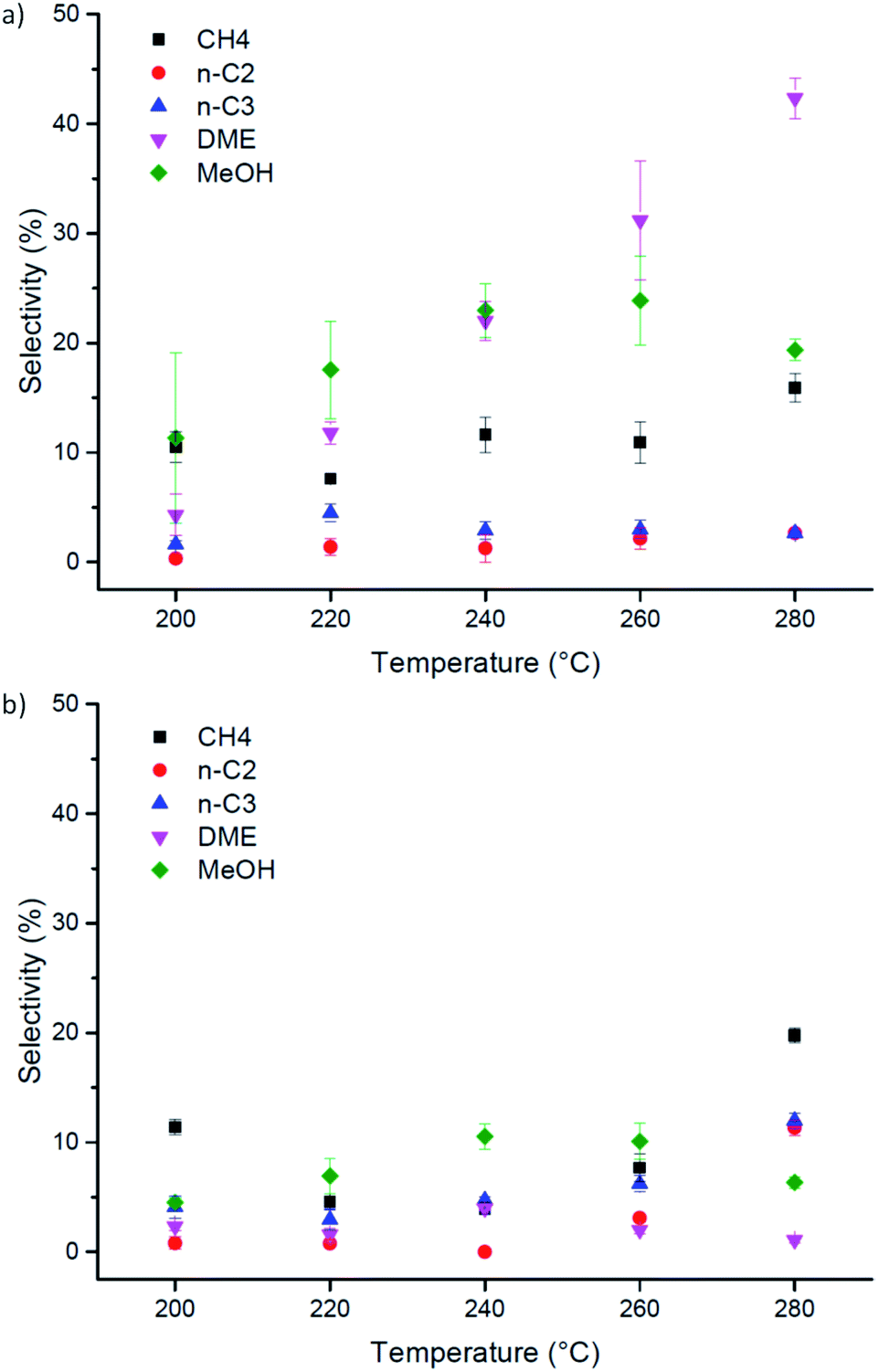 | ||
| Fig. 4 Performance of Cu-laden (a) UiO-66(Zr) and (b) NH2–UiO-66(Zr) in the direct hydrogenation of CO2 as a function of reaction temperature. | ||
Finally it is noteworthy that the samples demonstrated poorer thermal stability under catalytic conditions than in an oxidative atmosphere, as according to thermal analyses (Fig. S1, ESI†), both of these samples are stable up to well above 300 °C in an oxidative atmosphere, yet under reaction conditions they already started to show signs of degradation at 280 °C. This was later confirmed by acquiring post-catalysis TEM micrographs (Fig. S3, ESI†), which showed large Cu particles on the MOF surfaces. We attribute this decrease in stability to the evolution of water as a reaction by-product. Although both UiO-66(Zr) and NH2–UiO-66(Zr) demonstrate good water resistance under ambient conditions, the conditions are significantly different when the catalytic reactions occur, under which conditions the reactivity of the inorganic nodes is expected to change. This in turn leads to framework degradation, specifically to the removal of linkers, which in turn no longer separate the MOF pores and thus inhibit the formation of larger Cu particles. On the other hand, this very observation also highlights the ability and effectiveness of MOFs to hinder the agglomeration of metal nanoclusters embedded in their pores, provided their structural integrity is preserved.
Experimental
Synthesis
Reagents were purchased from Sigma Aldrich and used without further purification.Characterisation
The extent of Cu loading was determined from XPS survey spectra, wherein integration of the Cu and Zr peaks enabled the determination of the Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratio, which was then further used to calculate the Cu loading. In each case the averages of at least 5 spectra were used.
:Zr ratio, which was then further used to calculate the Cu loading. In each case the averages of at least 5 spectra were used.
Before catalysis. Powdered samples were dispersed in anhydrous THF in an Ar glove box, and drop-cast on holey carbon Cu TEM grids. In order to minimise contact with air, they were transported in an airtight desiccator. Micrographs were acquired with a JEOL JEM-2100F instrument operated at an accelerating voltage of 200 kV. To stabilise the samples in the beam, they were beam-showered for 30 minutes prior to imaging.
After catalysis. Transmission Electron Microscopy (TEM) was performed using a Tecnai 12 microscope, operating at 120 kV; the microscope was equipped with a CCD camera. Suspensions of the samples in ethanol were drop cast on carbon/formvar 300 mesh Cu grids, and after drying they were imaged in the microscope.
Catalyst testing
The activity and selectivity of the samples were tested in a two parallel reactor set-up from Autoclave Engineers. 100–200 mg of each sample (425–630 μm grains), diluted with 1 or 2 mL SiC, was loaded into the reactor and was supported in the middle of the reactor tube by a hollow steel tube and quartz wool. Prior to syngas conversion the samples were reduced in situ at 250 °C (heating rate of 2 °C min−1) for 2.5 hours using a 110 mL min−1 flow of 20% H2 in Ar. After the reduction, the reactor was cooled down before syngas was introduced. The reactor was flushed with syngas for 30 minutes and subsequently pressurized to 40 bar. A premixed syngas was used with the composition: 23% CO, 7% CO2, 60% H2 and 10% Ar. The Ar in the syngas feed acted as an internal standard for the GC. Carbonyls were removed from the mixture by a carbonyl trap placed before the reactor inlet, which contained zeolite-Y and activated carbon. The carbonyl trap was heated to 50 °C. After pressurisation three blank chromatograms were recorded to determine the CO/Ar and CO2/Ar signals of the feed stream, and subsequently the reactor was heated to 200 °C at a rate of 2 °C min−1. The temperature was increased in steps of 20 °C (with a heating rate of 2 °C min−1) to 280 °C to screen the activity and selectivity.The products were analysed by an on-line Varian 450 GC. This GC was equipped with two parallel channels; the first channel consisted of two series-connected HAYESEP Q (0.5 m × 1/8′′) packed columns and a MOLSIEVE 13X (1.5 m × 1/8′′) molecular sieve column connected to a TCD to determine the activity from the relative decrease in CO/Ar and CO2/Ar signals. The second channel consisted of a CP-SIL 8CB FS capillary column connected to an FID to determine the selectivity.
Conclusions
In this study, we systematically study the effect of amino functionalisation of UiO-66(Zr) as a supporting matrix for Cu nanoclusters on the direct reduction of carbon monoxide and carbon dioxide with hydrogen. Both of the Cu-laden MOF samples are less active than commercial catalysts for the direct hydrogenation of carbon dioxide, and they have different selectivity. This means that the zirconia-like nodes are not sufficient to promote and stabilise the required sites of these small Cu nanocluster catalysts.When comparing the two Cu-laden MOF samples, they did not display significantly different catalyst activity. However, the selectivity of the two catalysts differed substantially. For Cu in NH2–UiO-66(Zr), the main conversion product was CH4, which is less desirable than methanol. On the other hand, Cu in UiO-66(Zr) catalysed the conversion of CO and CO2 into dimethyl ether (DME), possibly through a tandem reaction with the inorganic node Lewis acid sites. This is highly desirable as when methanol is used as a platform chemical, it normally is first transformed into DME, so our method offers simple, direct DME production. This is attributed to the effect of the amino-functionalisation on the inorganic nodes, decreasing their Lewis acidity, which was found to be an advantage.
Author contributions
Sample syntheses and characterisation were done and overseen by Dr Szilágyi; catalyst testing and post-catalysis characterisation were performed by Dr Pompe. All authors partook in discussions regarding data analysis and contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was partly funded through the Royal Society’s International Exchanges programme, grant no. IE161747. We thank Prof. Petra de Jongh (Utrecht University) for her support and discussions, Dr Catriona McGilvery (Imperial College London) for the TEM imaging of the samples prior to catalysis testing, the NEXUS facility for XPS experiments, and Mr Omotoyosi Phillips for the original attempts at sample synthesis.Notes and references
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- Y. Yang, C. A. Mims, D. H. Mei, C. H. F. Peden and C. T. Campbell, J. Catal., 2013, 298, 10–17 CrossRef CAS.
- O. Martin and J. Pérez-Ramírez, Catal. Sci. Technol., 2013, 3, 3343–3352 RSC.
- J. Wu, M. Saito, M. Takeuchi and T. Watanabe, Appl. Catal., A, 2001, 218, 235–240 CrossRef CAS.
- R. Dalebout, N. L. Visser, C. E. L. Pompe, K. P. de Jong and P. E. de Jongh, J. Catal., 2020, 392, 150–158 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- R. van den Berg, G. Prieto, G. Korpershoek, L. I. van der Wal, A. J. van Bunningen, S. Lægsgaard-Jørgensen, P. E. de Jongh and K. P. de Jong, Nat. Commun., 2016, 7, 13057 CrossRef CAS.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- J. Weigel, R. A. Koeppel, A. Baiker and A. Wokaun, Langmuir, 1996, 12, 5319–5329 CrossRef CAS.
- G. Bonura, M. Cordaro, L. Spadaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., B, 2013, 140–141, 16–24 CrossRef CAS.
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS.
- M. Müller, S. Hermes, K. Kähler, M. W. E. van den Berg, M. Muhler and R. A. Fischer, Chem. Mater., 2008, 20, 4576–4587 CrossRef.
- K. Li and J. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef CAS.
- J. Wang, S.-m. Lu, J. Li and C. Li, Chem. Commun., 2015, 51, 17615–17618 RSC.
- M. Zhong, X. Zhang, Y. Zhao, C. Li and Q. Yang, Green Chem., 2015, 17, 1702–1709 RSC.
- L. Pan, H. Liu, X. Lei, X. Huang, D. H. Olson, N. J. Turro and J. Li, Angew. Chem., Int. Ed., 2003, 42, 542–546 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS.
- C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- G. Cai and H.-L. Jiang, Angew. Chem., Int. Ed., 2017, 56, 563–567 CrossRef CAS PubMed.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- K. M. Choi, K. Na, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 7810–7816 CrossRef CAS PubMed.
- T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed.
- J.-M. Yan, Z.-L. Wang, L. Gu, S.-J. Li, H.-L. Wang, W.-T. Zheng and Q. Jiang, Adv. Energy Mater., 2015, 5, 1500107 CrossRef.
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- P. Hu, J. V. Morabito and C.-K. Tsung, ACS Catal., 2014, 4, 4409–4419 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- K. Na, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2014, 14, 5979–5983 CrossRef CAS PubMed.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS.
- D. E. Coupry, J. Butson, P. S. Petkov, M. Saunders, K. O’Donnell, H. Kim, C. Buckley, M. Addicoat, T. Heine and P. Á. Szilágyi, Chem. Commun., 2016, 52, 5175–5178 RSC.
- J. King, L. Zhang, S. Doszczeczko, O. Sambalova, H. Luo, F. Rohman, O. Phillips, A. Borgschulte, M. Hirscher, M. Addicoat and P. Á. Szilágyi, J. Mater. Chem. A, 2020, 8, 4889–4897 RSC.
- P. Á. Szilágyi, D. M. Rogers, I. Zaiser, E. Callini, S. Turner, A. Borgschulte, A. Züttel, H. Geerlings, M. Hirscher and B. Dam, J. Mater. Chem. A, 2017, 5, 15559–15566 RSC.
- J. Ethiraj, E. Albanese, B. Civalleri, J. G. Vitillo, F. Bonino, S. Chavan, G. C. Shearer, K. P. Lillerud and S. Bordiga, ChemSusChem, 2014, 7, 3382–3388 CrossRef CAS.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- H. Kobayashi, J. M. Taylor, Y. Mitsuka, N. Ogiwara, T. Yamamoto, T. Toriyama, S. Matsumura and H. Kitagawa, Chem. Sci., 2019, 10, 3289–3294 RSC.
- A. T. Bell, in Studies in Surface Science and Catalysis, ed. E. Iglesia, J. J. Spivey and T. H. Fleisch, Elsevier, 2001, vol. 136, pp. 13–19 Search PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database 20, Version 4.1 Last Update to Data Content, 2012, DOI:10.18434/T4T88K.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- B. Rungtaweevoranit, J. Baek, J. R. Araujo, B. S. Archanjo, K. M. Choi, O. M. Yaghi and G. A. Somorjai, Nano Lett., 2016, 16, 7645–7649 CrossRef CAS.
- Z. H. Rada, H. R. Abid, H. Sun, J. Shang, J. Li, Y. He, S. Liu and S. Wang, Prog. Nat. Sci.: Mater. Int., 2018, 28, 160–167 CrossRef CAS.
- K. Sato, Y. Tanaka, A. Negishi and T. Kato, J. Power Sources, 2012, 217, 37–42 CrossRef CAS.
- T. H. Fleisch, A. Basu and R. A. Sills, J. Nat. Gas Sci. Eng., 2012, 9, 94–107 CrossRef CAS.
- G. Thomas, B. Feng, A. Veeraragavan, M. J. Cleary and N. Drinnan, Fuel Process. Technol., 2014, 119, 286–304 CrossRef CAS.
- T. Witoon, T. Permsirivanich, N. Kanjanasoontorn, C. Akkaraphataworn, A. Seubsai, K. Faungnawakij, C. Warakulwit, M. Chareonpanich and J. Limtrakul, Catal. Sci. Technol., 2015, 5, 2347–2357 RSC.
- Y. Zhang, D. Li, Y. Zhang, Y. Cao, S. Zhang, K. Wang, F. Ding and J. Wu, Catal. Commun., 2014, 55, 49–52 CrossRef CAS.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Thermogravimetric analysis, productivity plots, and post-catalysis TEM. See DOI: 10.1039/d1fd00012h |
| This journal is © The Royal Society of Chemistry 2021 |