
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the mechanical flexibility of organic molecular crystals by polymorphism for flexible optical waveguides†
Torvid
Feiler
 ab,
Biswajit
Bhattacharya
*a,
Adam A. L.
Michalchuk
a,
Seon-Young
Rhim
c,
Vincent
Schröder
cd,
Emil
List-Kratochvil
*cd and
Franziska
Emmerling
*ab
ab,
Biswajit
Bhattacharya
*a,
Adam A. L.
Michalchuk
a,
Seon-Young
Rhim
c,
Vincent
Schröder
cd,
Emil
List-Kratochvil
*cd and
Franziska
Emmerling
*ab
aBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: biswajit.bhattacharya@bam.de; franziska.emmerling@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
cDepartment of Chemistry, Department of Physics, Humboldt-Universität zu Berlin, IRIS Adlershof, Zum Großen Windkanal 2, 12489 Berlin, Germany. E-mail: emil.list-kratochvil@hu-berlin.de
dHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Hahn-Meitner-Platz 1, 14109, Berlin, Germany
First published on 28th July 2021
Abstract
The ability to selectively tune the optical and the mechanical properties of organic molecular crystals offers a promising approach towards developing flexible optical devices. These functional properties are sensitive to crystallographic packing features and are hence expected to vary with polymorphic modification. Using as a model system the photoluminescent material 4-bromo-6-[(6-chloropyridin-2-ylimino)methyl]phenol (CPMBP), we herein demonstrate the simultaneous tuning of mechanical flexibility and photoluminescence properties via polymorphism. Two new polymorphic forms of CPMBP were obtained from a solution and fully characterised using a combination of experiments and density functional theory simulations. These polymorphic forms exhibit remarkably distinct mechanical properties and an order of magnitude difference in photoluminescence quantum yield. The mechanically plastic form has a higher quantum yield than the brittle polymorphic form. However, their photoluminescence emission profile is largely unaffected by the observed polymorphism, thereby demonstrating that the optical properties and bulk mechanical properties can in principle be tuned independently. By distinguishing between active (involving absorption and emission) and passive (involving no absorption) light propagation, the waveguiding properties of the plastic form of CPMBP (form II) were explored using the straight and bent crystals to highlight the potential applications of CPMBP in designing flexible optical devices. Our results demonstrated that polymorph engineering would be a promising avenue to achieve concurrent modulation of the optical and mechanical properties of photoluminescent molecular crystals for next-generation flexible optical device applications.
Introduction
Soft amorphous materials such as organic polymers and elastomers have been studied extensively in academia and industry due to their high degree of mechanical adaptivity.1–5 In contrast, crystalline molecular materials are often brittle and prone to breaking upon external mechanical forces. This fragility of crystalline materials poses challenges for their applications in next-generation technologies, including as sensors, synthetic tissues, and advanced optoelectronics. Correspondingly, the recent discovery of mechanical flexibility in single crystals6 of molecular materials7–16 and coordination polymers17–20 opens new opportunities to design next-generation flexible technologies. Numerous applications of flexible molecular crystals have been already demonstrated including their use as optical waveguides,21–26 micro-optoelectronic devices,27,28 sensors,29,30 and biomimetics.31,32 While a growing number of mechanically flexible crystalline molecular materials are being reported, they remain scarce and their preparation is largely serendipitous.Based on their qualitative mechanical behavior, the flexibility of molecular crystals can be classified as plastically (irreversible) and elastically (reversible) bendable. Plastic bending in molecular crystals has been rationalized based on anisotropic molecular arrangements and the existence of facile slip planes where molecular layers interact via weak dispersive intermolecular interactions such as halogen bonding, π⋯π stacking, or weak van der Waals interactions.7,15 In contrast, elastic bending arises from energetically isotropic molecular packing with weak interactions.9,33 These weak interactions facilitate the displacement of molecules without disrupting the stabilizing forces that maintain the structural integrity of the crystal. The success of these structural criteria for elastic and plastic bending demonstrates that the bulk mechanical compliance of molecular materials is rooted in their crystal packing and non-covalent interactions (NCIs).
The most obvious way to tune NCIs in crystalline materials is to modify the chemical functionality. This approach has been used widely in molecular and coordination polymers for altering the mechanical compliance.7,18,34 However, this approach is drastic and can change significantly the critical functional properties of the molecules. To overcome this limitation, crystal engineering strategies have been developed which allow NCIs to be selectively tuned by introducing multiple components in the crystal packing, cocrystallisation and salt formations.35–38 For example, Nath et al. recently reported how the plasticity of probenecid crystals could be infused into brittle 4,4′-bipyridine via cocrystallisation.35 Although this crystal engineering approach does not require modification of the chemical functionality, the inclusion of multiple chemical entities into the material can impose restrictions on material compatibility. For example, where the material application requires biological or environmental compatibility, the inclusion of a toxic coformer would render the material unusable.39 To avoid any chemical modification whatsoever, NCIs can also be tuned by controlling polymorphism. Polymorphism stems from the ability of a molecule to crystallise in more than one solid form, each having distinct arrangements or conformations of the constituent molecules.40 Tuning the mechanical flexibility by controlling polymorphism of a molecular crystal of 6-chloro-2,4-dinitroaniline was first reported by Reddy and Desiraju in their pioneering work.41 More examples of tuning the mechanical response of molecular crystals through polymorph screening have been since reported.42–46
The polymorphism of molecular crystals which show simultaneously mechanical flexibility and useful optical properties is rare. Single crystals of π-conjugated organic chromophores are frequently used as active micro-optical components such as optical waveguides,47 lasers,48 circuits,49 and field-effect transistors50 in designing advanced optoelectronic devices. However, these devices are predominantly prepared by using the brittle forms of single crystals. By coupling mechanical flexibility to optical properties, a variety of optoelectronic applications can be envisioned. For example, by making use of elastically flexible materials, reversible optical sensors become feasible, whereas shapable nano-optical devices can be envisioned through the use of plastically bendable crystals.
In recent years, researchers have successfully identified mechanically flexible luminescent molecular materials. For example, significant efforts have been devoted to designing mechanically flexible optical waveguides.20–26 These materials have the properties of bending the path of light, thereby offering potential applications in nano-optical devices. Similarly, various materials have been reported whose optical properties can be tuned by the extent of bending.29,30 More recently, advanced optical properties have also been demonstrated to be tunable by mechanical bending including the polarization of plane polarized light.51 Thus, fine-tuning the interplay between optical and mechanical properties has significant potential for next-generation functional materials. Only recently has polymorphism been revealed as a potential design strategy for simultaneously modifying both the optical and mechanical properties of molecular crystals. Although the strategy seems promising, only limited number of examples have been reported. Further studies are required to explore the breadth of this phenomenon before its full potential can be realized.
Polymorphism in molecular crystals is ubiquitous.40 For the purpose of this study, we opt to explore the polymorphism of 4-bromo-6-[(6-chlorolpyridin-2-ylimino)methyl]phenol (CPMBP). This molecule was selected as salicylaldehyde-derived Schiff bases are well known to be highly photoluminescent and often show polymorphism in the solid state.52 Moreover, the chlorine and the bromine atom on the backbone of the molecule can be expected to introduce mechanical compliance in crystal forms. This molecule has significant potential for studying the simultaneous effect of polymorphism on the optical and mechanical properties.
Experimental
Materials
2-Amino-6-chloropyridine (98%; CAS No: 45644-21-1) and 5-bromosalicylaldehyde (98%; CAS No: 1761-61-1) were purchased from Fluorochem and Alfa Aesar, respectively, and used as received.Synthesis and crystal growth
2-((E)-(6-Chloropyridin-2-ylimino)methyl)-4-bromophenol (CPMBP) was synthesised by liquid-assisted mechanochemical grinding (LAG) with methanol (100 μL) of an equimolecular mixture of 5-bromosalicylaldehyde (201 mg, 1 mmol) and 2-amino-6-chlropyridine (129 mg, 1 mmol) with a mortar and pestle. Single crystals were grown by dissolving the ground powder in DCM, followed by slow evaporation or anti-solvent crystallization in a beaker or tube.Powder X-ray diffraction (PXRD)
The ground crystals were packed into borosilicate capillaries (inner diameter: 0.5 mm). PXRD data were collected on a Bruker D8 Discover diffractometer (Bruker AXS, Karlsruhe, Germany) equipped with a LYNXEYE XE detector and using Cu-Kα1 radiation (λ = 1.506 Å). Data were collected from 2θ = 3° to 50° with a step size of 0.009° and 6 s per step.Single-crystal X-ray diffraction
Single crystal X-ray diffraction (SCXRD) data of both polymorphs were collected using a Bruker D8 Venture diffractometer (Bruker AXS, Karlsruhe, Germany) using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Data reduction was performed using the Bruker AXS SAINT53 and SADABS54 software packages. Both structures were solved by SHELXT 2018 (ref. 55) using direct methods, followed by successive Fourier and difference Fourier syntheses. Full-matrix least-squares refinements were done on F2 using SHELXL 2018,56 including anisotropic displacement parameters for all non-hydrogen atoms. Hydrogen atoms bonded to oxygen were located from the electron density maps, and all hydrogen atoms bonded directly to carbon atoms were fixed at their ideal positions. Data collection, structure refinement parameters, and crystallographic data for the cocrystals are summarized in Table S1.†Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) measurements were performed with a TGA/DSC 3+ (Mettler Toledo, Greifensee, Switzerland). The measurements were performed in open aluminum crucibles under a continuous N2–air flow. The experiments were conducted at a heating rate of 10 K min−1 over a range of 25 °C to 550 °C.Fourier-transform infrared spectroscopy (FTIR)
FTIR spectra of the powdered samples were measured using a Nicolet FT-IR NEXUS (Thermo Fisher, Dreieich, Germany) spectrometer equipped with a Diamond-ATR-Golden Gate unit and a DTGS KBr detector. Each spectrum was collected in a range of 200–4000 cm−1 with a spectral resolution of 4 cm−1.Photoluminescence spectroscopy
Photoluminescence measurements were performed with an Edinburgh Instruments (Livingston, UK) FLS 980 fluorescence spectrometer. All photoluminescence spectra were measured using an integrating sphere. The samples were excited with a 450 W ozone-free xenon arc lamp. The detector was a R928P PMT, electrically cooled to −20 °C.Waveguide
The use of CPMBP as a waveguide material was tested by using LED light sources (Thorlabs Inc.). For the measurements, the CPMBP crystals were placed on a silicon wafer with a 3 μm thick SiO2 layer, and the light was coupled into the crystals with a microscope objective (100× magnification, 0.8 numeric aperture (NA), Nikon Inc.) using the end-fire coupling method. Coupling the light in the direction of the optical axis of the waveguide facilitates the investigation of propagation behavior. At the other end of the crystal, the outcoupled light was collected perpendicular to the optical axis of the waveguide by applying another objective (10× magnification, 0.25 numeric aperture). The collected light was subsequently guided via fibers to a CCD detector (Kymera 328i, Oxford Instruments Inc.). Since CPMBP shows photoluminescence, we differentiate between active and passive waveguiding. Active waveguiding describes the propagation of the emitted light caused by the photoluminescence, providing that the wavelength of the LED source is in the excitation range. The light propagation without any interaction is denoted as passive waveguiding. A schematic picture of the measurement setup can be found in the ESI,† Scheme S1.Computational methods
Initial structures were taken from the experimentally determined single crystal X-ray diffraction data, as described above. Plane wave density functional theory (pw-DFT) calculations were performed using Quantum ESPRESSO v6.4.57,58 Structure relaxation was performed using the generalized gradient approximation (GGA) exchange–correlation functional of Perdew–Burke–Ernzerhof (PBE)59 with the exchange-dipole moment (XDM)60 dispersion correction. The nuclear–electron interactions were approximated using the projector augmented-wave method,61 and the electronic wave function was expanded in plane waves to a kinetic energy cut-off of 80 Ry. Convergence of the electronic wave function was accepted with change in energy <10−10 Ry, and forces were considered to be converged when <10−5 Ry a.u−1. The Brillouin zone was sampled on a Monkhorst–Pack grid62 with 0.06 Å−1 for form I and 0.05 Å−1 for form II, respectively.Intermolecular interaction energies were calculated using the CLP-PIXEL method63,64 and associated wrapper MrPIXEL,65 using a condensation level of 4 and a cluster radius of 15 Å. PIXEL calculations were based on ab initio charge densities (grid size: 0.06 Å) produced at the B3LYP/6-31G(d,p) level of theory with Gaussian v16,66 with hydrogen positions normalized to conventional neutron data.
Results and discussion
The compound, CPMBP, was synthesized by liquid-assisted mechanochemical grinding (LAG) with methanol of an equimolecular mixture of 5-bromosalicylaldehyde and 2-amino-6-chlropyridine, Fig. 1a. Slow evaporation of a saturated CH2Cl2 solution of CPMBP at room temperature in a beaker yielded flake-shaped orange crystals. When the crystals were handled with a metal needle, some crystals displayed brittle nature and others showed plastic bending and remained deformed even after removal of the stress, Fig. 1b and c, suggesting the concomitant formation of two polymorphic forms. In contrast, crystallization of CPMBP in pure DCM or using an anti-solvent in glass tubes produced pure mechanically distinct flake-shaped crystals, presumably owing to the different crystallisation kinetics as compared with crystallisation from a wide-mouthed beaker. Crystals obtained from DCM with n-hexane were brittle (form I) over both the long crystallographic faces. In contrast, crystals obtained from pure DCM deformed plastically (form II) over the major faces but appeared brittle over the other faces. Powders of each of the pure crystal forms exhibited a single endothermic peak in their differential scanning calorimetric profiles, corresponding to their melting point (Fig. S1†): 153.8 °C and 158.0 °C for form I and form II, respectively. The phase purity is also consistent with the unique PXRD diffraction pattern of the powders of the crystals (as compared with the simulated PXRD pattern from single crystal data, Fig. S2†) and FT-IR spectra (Fig. S3†). The FT-IR spectra of the two polymorphs are nearly identical. For example, the aromatic C–Br stretching vibration in both forms is at 1072 cm−1. The C–H deformation vibration of the 2,6-substituted pyridines is also at the same position, 792 and 730 cm−1, respectively.67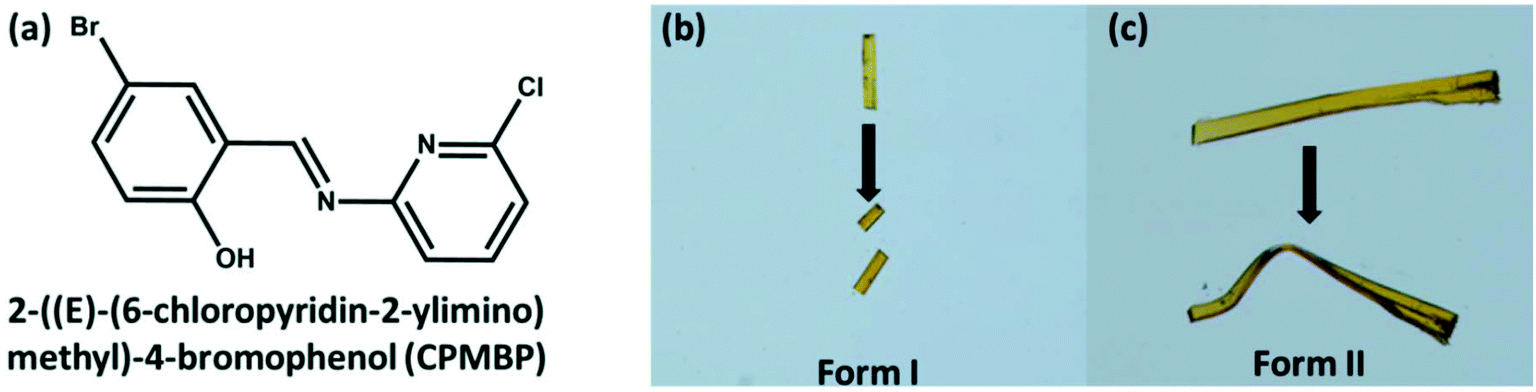 | ||
| Fig. 1 (a) Chemical structure of the title compound, CPMBP. (b and c) Optical images showing the brittle and plastic deformation behaviour of form I and form II, respectively. | ||
To understand the intermolecular interactions and molecular packing motifs responsible for the observed distinct mechanical flexibility of the two different crystalline forms of CPMBP, the crystal structures of both forms were determined by SCXRD. Form I (brittle) crystallizes in the orthorhombic space group Pca21 with one CPMBP molecule in the asymmetric unit. The cell parameters are a = 25.13 Å, b = 3.87 Å and c = 11.98 Å (Table S1†). The CPMBP molecules adopt a near-planar conformation with a dihedral angle of 8.4° between the pyridyl and phenolic rings, driven by the intramolecular O–H⋯N hydrogen bonds (Fig. S4a;† H⋯A: 1.86 Å, θ: 146.1°). Crystal face indexing by SCXRD indicates that the two long crystallographic faces are the (001)/(00![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (010)/(00) faces, whereas (100)/(00) are the top faces (Fig. 2a and S5a†). In the crystal structure, each molecule is connected to two other molecules via C–H⋯O (2.62 Å, 147.8°) and C–H⋯Cl (2.89 Å, 146°) interactions to form a corrugated ribbon (Fig. 2b). The molecules in the ribbon are slip-stacked along the crystallographic b-axis via π⋯π interactions (3.87 Å), which are further stabilized via C–H⋯Br (2.95 Å, 148°) interactions (Fig. 2c). The ribbons are connected via C–Cl⋯π interactions along the crystallographic a-axis. Together, these interactions lead to the formation of a mechanically interlocked 3D network structure, preventing the molecules from moving in response to mechanical stress (Fig. 2d). Hence, the crystal structure is consistent with the observed brittle fracture (Fig. 2e and S6a†).16,44
) and (010)/(00) faces, whereas (100)/(00) are the top faces (Fig. 2a and S5a†). In the crystal structure, each molecule is connected to two other molecules via C–H⋯O (2.62 Å, 147.8°) and C–H⋯Cl (2.89 Å, 146°) interactions to form a corrugated ribbon (Fig. 2b). The molecules in the ribbon are slip-stacked along the crystallographic b-axis via π⋯π interactions (3.87 Å), which are further stabilized via C–H⋯Br (2.95 Å, 148°) interactions (Fig. 2c). The ribbons are connected via C–Cl⋯π interactions along the crystallographic a-axis. Together, these interactions lead to the formation of a mechanically interlocked 3D network structure, preventing the molecules from moving in response to mechanical stress (Fig. 2d). Hence, the crystal structure is consistent with the observed brittle fracture (Fig. 2e and S6a†).16,44
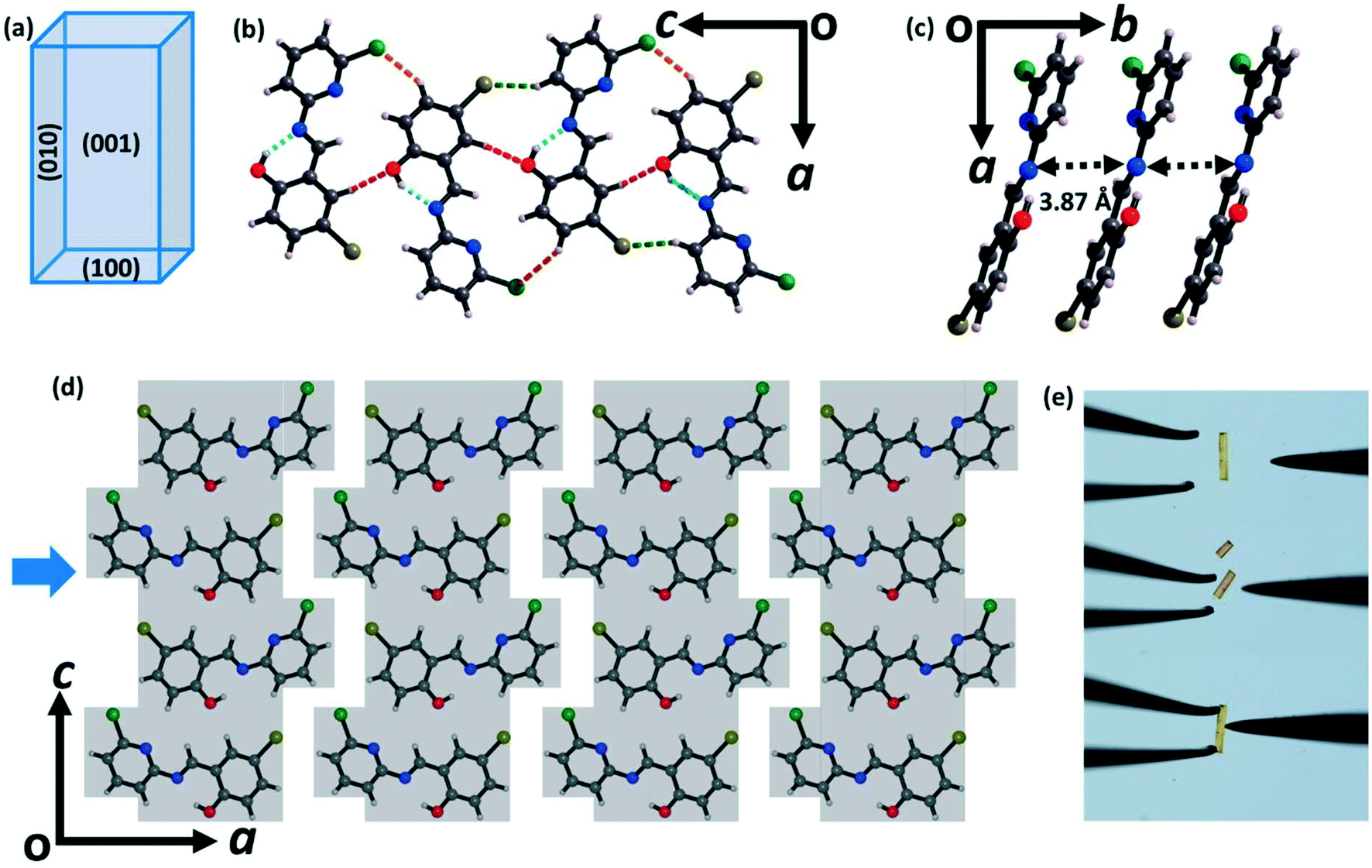 | ||
| Fig. 2 Molecular packing in the crystal structure of form I of CPMBP. (a) Schematic diagram of the crystal face indices determined by single-crystal X-ray diffraction. (b) The 1D ribbon is formed via C–H⋯O, C–H⋯Cl and C–H⋯Br interactions along the c-axis (C–H⋯O interactions: red dotted lines, O–H⋯N interactions: cyan dotted lines, C–H⋯Cl interactions: orange dotted lines and C–H⋯Br interactions: green dotted lines). (c) Slip-stacked arrangement of the CPMBP molecules along the [010] direction through π⋯π interactions. (d) Mechanically interlocked 3D network structures along the [010] plane. The blue arrow indicates the direction in which the mechanical force was applied during bending. (e) Optical microscope photographs of the brittle fracture during the three-point bending experiment of the single crystals of form I. | ||
Form II (plastic) crystallizes in the monoclinic space group P21/c with cell parameters a = 4.44 Å, b = 19.01 Å, c = 13.90 Å and β = 90.18° (Table S1†). Like in form I, there is also one CPMBP molecule in the asymmetric unit that adopts a near-planar conformation with a dihedral angle between the pyridyl and phenolic rings of 3.3° due to the intramolecular O–H⋯N hydrogen bonds (Fig. S4b;† H⋯A: 1.85 Å, θ: 146.2°). The two larger faces of the crystals of form II are the (010)/(00) and (001)/(00) faces, with the minor face being (100)/(00) (Fig. 3a and S5b†). In the crystal structure, the molecules are connected to dimers by the C–H⋯O (2.64 Å, 148.4°) bonds (Fig. 3b). These dimers are slip-stacked via π⋯π interactions (3.3 Å) along the growth axis of the crystal (a-axis) to construct a columnar assembly (Fig. 3c). These stacked columns are close-packed in the orthogonal direction (b-axis) through the weak hydrophobic –Cl groups (Fig. 3d). This feature provides slip planes or weak interaction planes along the [001] direction. The structure of form II is overall anisotropic with low energy slip planes, which is the conventional crystal engineering criterion for plastic bending (Fig. 3e and S6b†). Thus, upon mechanical stress perpendicular to the slip planes, the (010) face results in plastic deformation in the crystal of form II.7 These low energy slip-planes may also contribute to the formation of extended defects which can influence mechanical properties.68,69
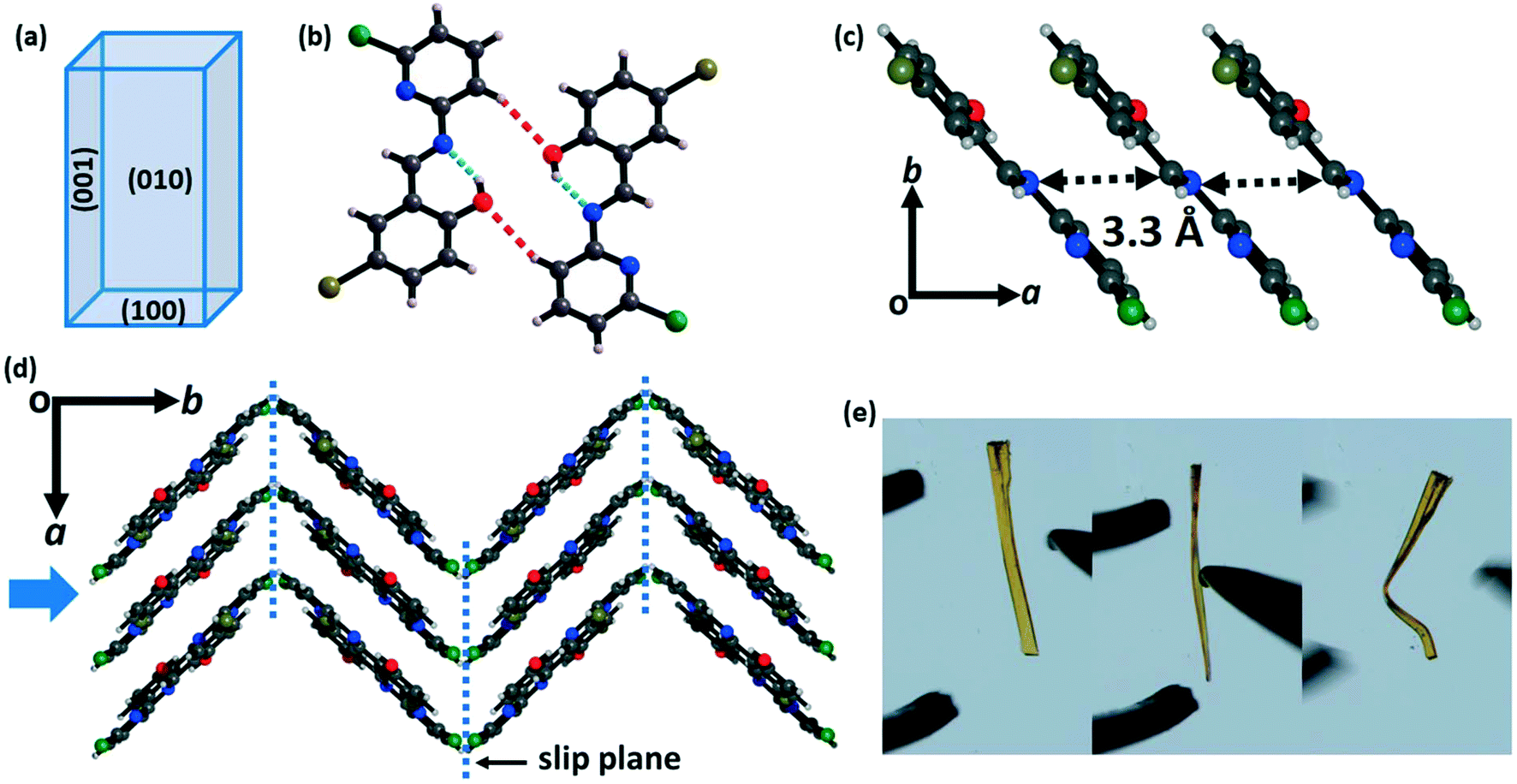 | ||
| Fig. 3 Crystal packing of form II of CPMBP. (a) Schematic diagram of the crystal face indices determined by single-crystal X-ray diffraction. (b) The dimers are formed via C–H⋯O interactions along the c-axis (C–H⋯O interactions: red dotted lines and O–H⋯N interactions: cyan dotted lines). (c) Slip-stacked arrangement of the CPMBP molecules along the [001] direction through π⋯π interactions. (d) Molecular packing with slip planes along the (001) plane (cyan dotted lines). The blue arrow shows the direction in which the mechanical force is applied during bending. (e) Optical microscope photographs of the plastic bending upon mechanical stress along the (010) plane of the single crystals of form II. | ||
The intermolecular interactions in both polymorphs were investigated via Hirshfeld surface analysis using Crystal Explorer 17.5.70 The contribution from all interactions in both the polymorphs is shown in Fig. S7,† which reveals the higher number of specific interactions in form I. Quantitative analysis of the intermolecular interactions in both polymorphic forms was conducted using the Coulomb–London–Pauli (CLP) PIXEL method for energy decomposition from quantum mechanical charge densities.63,64 This method has been widely applied to the study of intermolecular interactions in molecular solids.71–73 In brittle form I, the strongest interactions are those formed by π⋯π interactions along the b-axis (−37.9 kJ mol−1). In contrast, the interactions along the crystallographic c-axis (dominated by C–H⋯O, C–H⋯Cl, and C–H⋯Br interactions) average ca. −27.5 kJ mol−1, with those along the crystallographic a-axis (comprising no specific interactions) being much weaker at ca. −7.8 kJ mol−1. Consistent with prevailing design strategies for molecular flexibility, the intermolecular interaction energies for the plastic form II are more anisotropic than those for form I. The π⋯π interactions are somewhat stronger (−40.7 kJ mol−1), reflecting the shorter interplanar spacing along the π stacked axis. Interactions along the c-axis (C–H⋯O interactions) are again stronger, −21.8 kJ mol−1, with the b-axis C–H⋯π interactions having energies of ca. −12.8 kJ mol−1.
Following from the interesting effects of polymorphism on the mechanical properties, we sought to explore whether the polymorphism also affected the optical properties. To collect the solid-state photoluminescence (PL) spectra, the PL excitation (PLE) spectrum for each polymorph was first measured, Fig. S8.† The respective maximum of each PLE spectrum was used as the excitation wavelength for the PL emission measurements. Both crystalline forms of CPMBP exhibited different emission properties, presumably owing to their distinct intermolecular interactions and molecular packing in the solid state, Fig. 5. A powdered sample of form I exhibited very weak orange emission at 605 nm (λex = 425 nm; Φ = 0.4%) compared to powdered form II, which showed bright yellow emission at 585 nm (λex = 425 nm; Φ = 8.7%). The band gap for both forms is bound by predominantly carbon-based 2pz states (see Fig. 6, S9 and S10†). We can therefore suggest that the red-shift of the form I emission spectrum (as compared with form II) presumably stems from its weaker π–π stacking interactions (see Fig. 4).
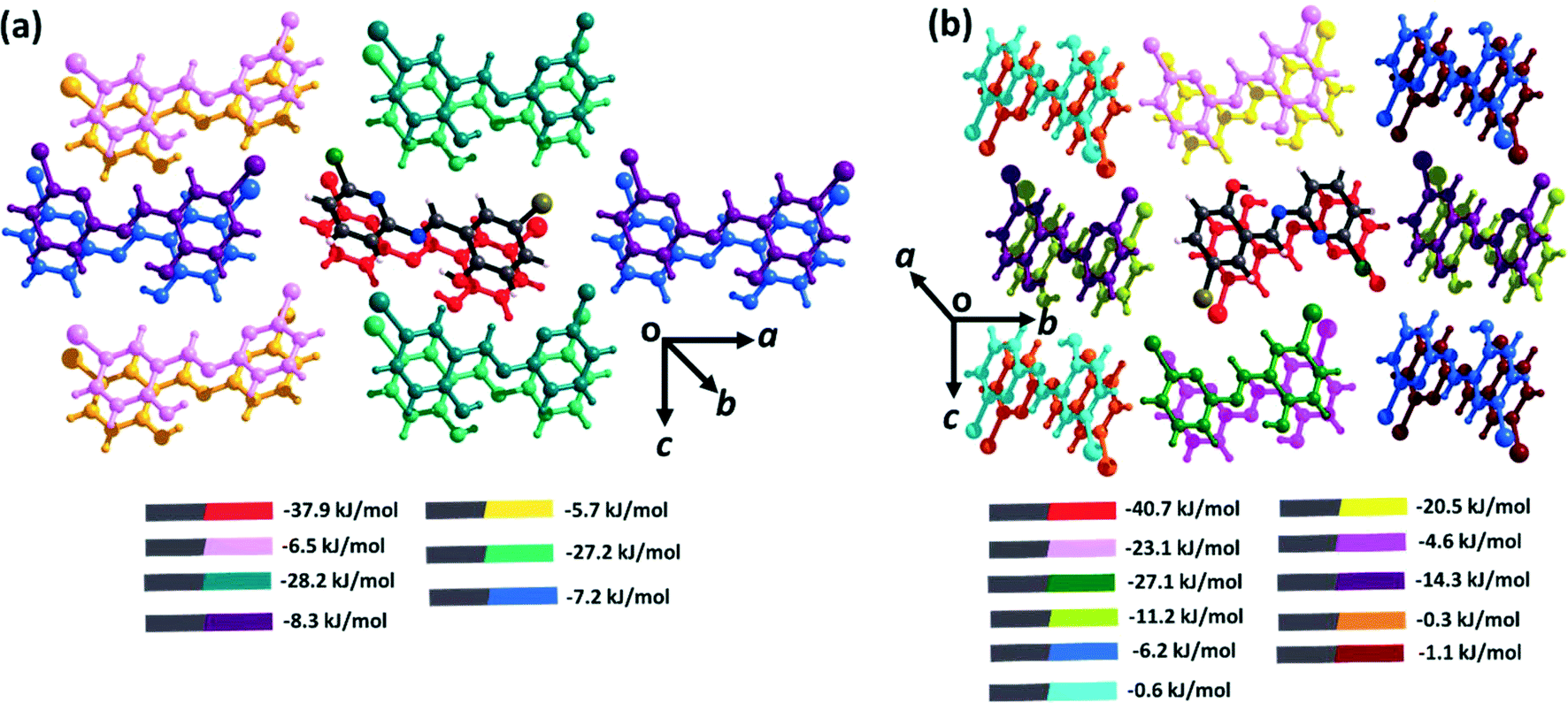 | ||
| Fig. 4 Intermolecular interaction energies for (a) form I and (b) form II. All energies are described between the element-coloured molecule (denoted as grey in the legend) in the centre, and its coloured pair, as indicated in the legend. Full energy decomposition tables are provided in ESI† Tables S2–S4. | ||
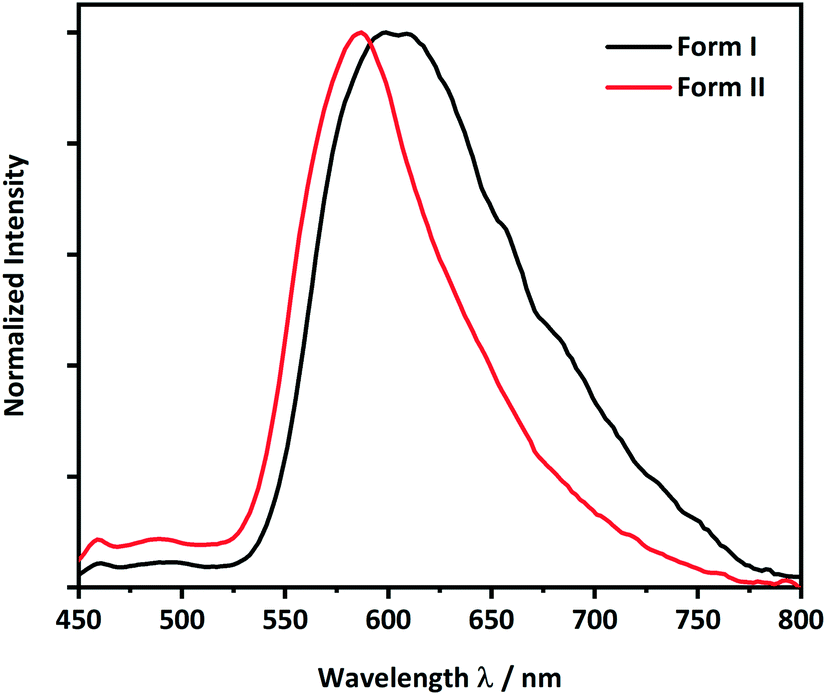 | ||
| Fig. 5 Solid-state photoluminescence spectra of the brittle (form I, black line) and plastic (form II, red line) forms of CPMBP (λex = 425 nm). | ||
The photoluminescence quantum yield,
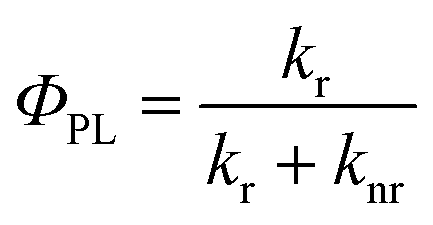
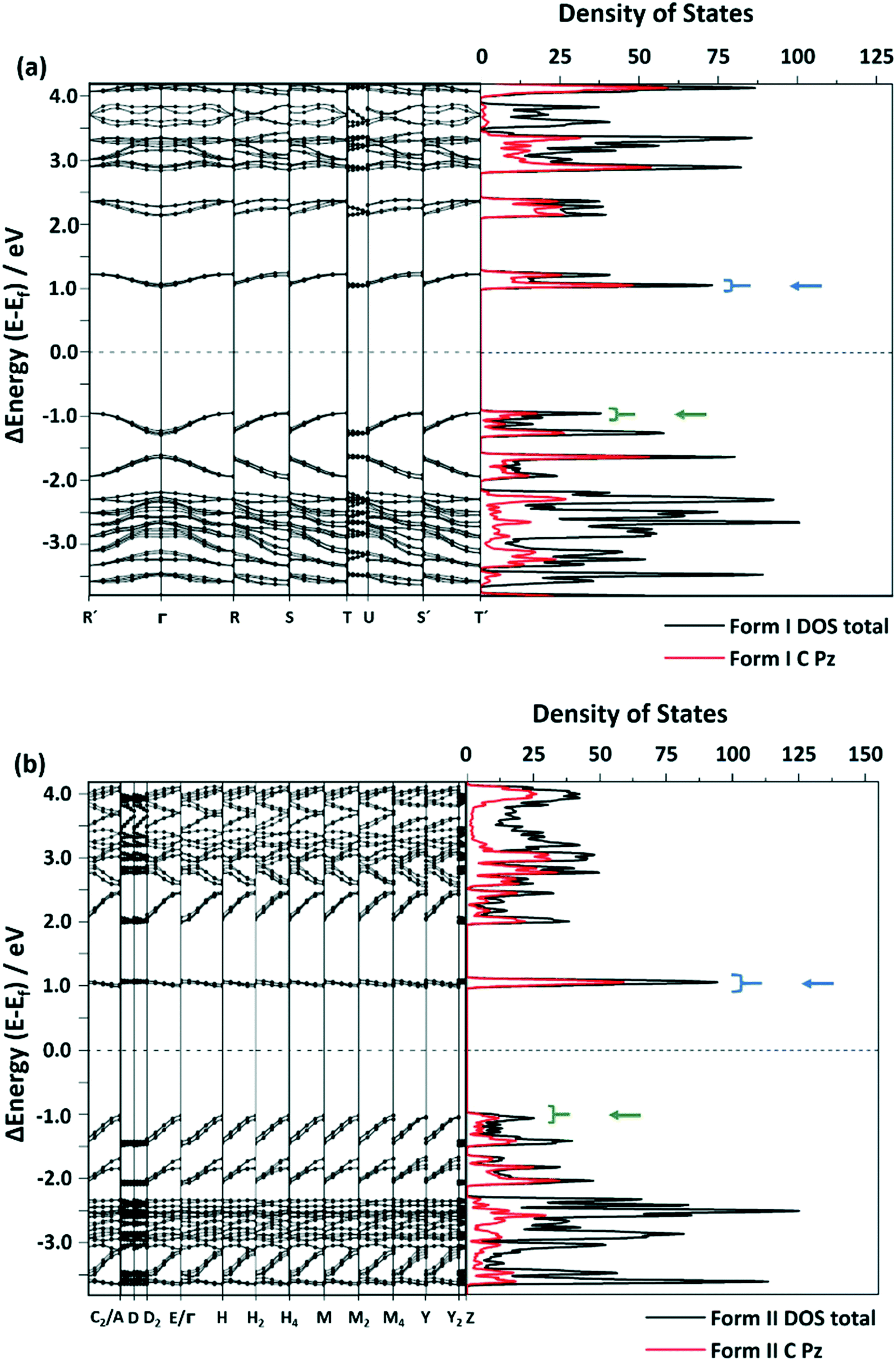 | ||
| Fig. 6 Selected areas of the band structure and density of states (DOS) of (a) form I and (b) form II. The black line shows the total DOS and the red line represents the DOS projected onto C-based atomic 2pz orbitals. The blue arrow indicates the conduction band and the green arrow indicates the valence band. The complete band structure is provided in the ESI,† Fig. S8 and S9. | ||
Under the highly idealized assumption that dipolar coupling is conserved in both polymorphic forms, decay can be roughly approximated with the number of available states in the conduction band (∈f) and the number of states in the valence band (∈i) via kr ∝ ∫dkδ(∈f(k) − ∈i(k) + ℏω), where ℏω is the photon energy. By integrating over the corresponding states for each polymorphic form (Fig. 6), we observe kr (form II) > kr (form I), consistent with the experimental observations. Hence, stemming largely from the carbon-based 2pz states, this further suggests that the different optical properties in the crystals stem from their unique π⋯π interactions.
Waveguiding properties
To test the CPMBP single crystals as potential optical waveguides, incident light was focused through a microscope objective onto the (100) face of the crystals, Fig. 7. Emitted light was subsequently collected perpendicular to the optical axis of the waveguide. Two different LED photon sources were considered for incident light; a 455 nm source was used to test the active transport (i.e. where absorption and re-emission are possible) while a 730 nm source was used to test for the passive transport (i.e. where absorption is not possible), Fig. 7b and c. In both cases, the guided light was observed to leak through defects along the crystal.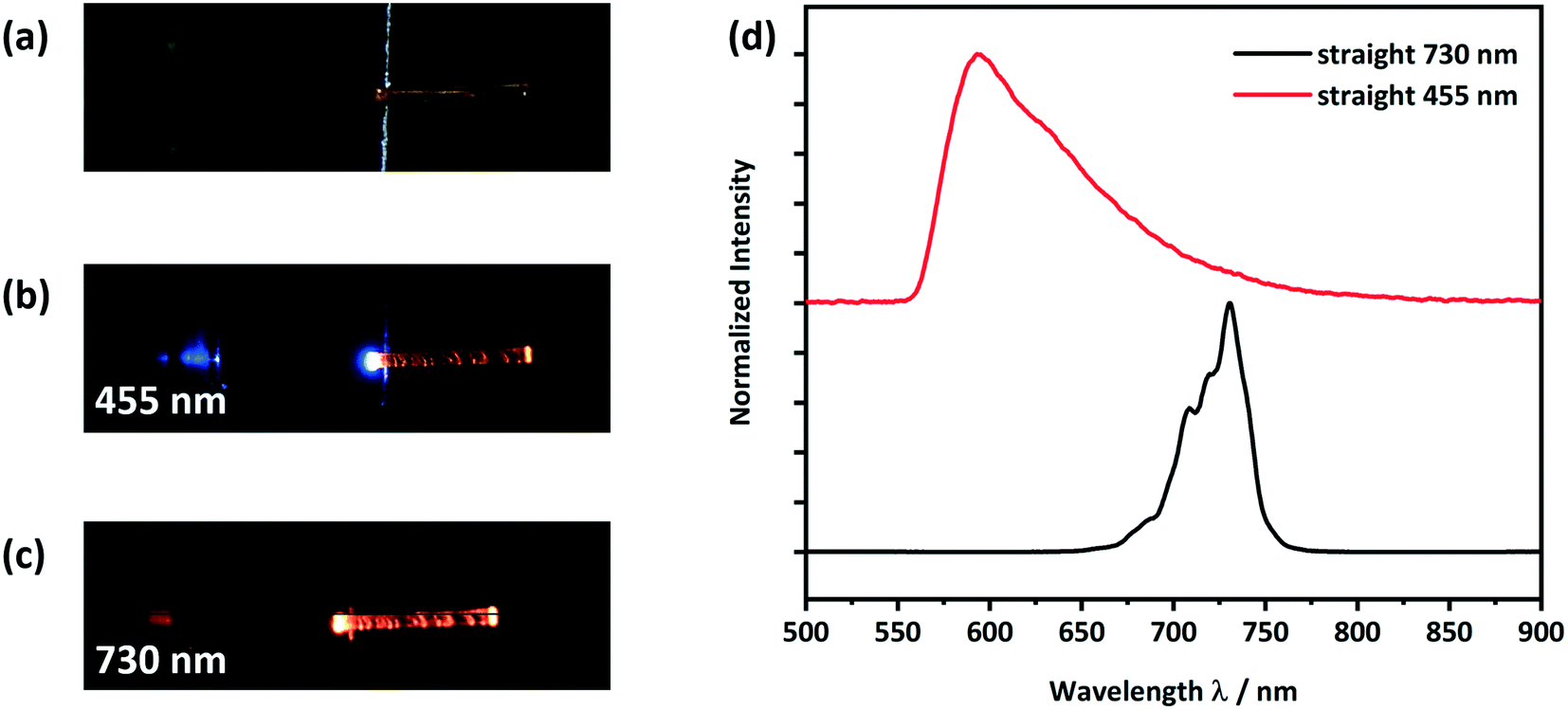 | ||
| Fig. 7 Optical microscope images of a) a form II straight crystal placed on a silicon wafer with a 3 μm thick SiO2 layer as shown in Scheme S1,† b) showing light with a wavelength of 455 nm being coupled at the left end of the crystal and c) showing light with a wavelength of 730 nm being coupled at the left end of the crystal; d) detected PL spectra at the right end of the crystal. The spectra of the LED light sources, PLE and PL are given in Fig. S11 and S12.† | ||
When using the 455 nm incident light source, the emission spectrum collected at the end of the crystal was very similar to the conventional solid-state PL spectrum (see Fig. 7d and 5). However, the high energy part of the guided emission spectrum is filtered out due to the self-absorption of the CPMBP crystal. The ability of CPMBP to absorb its own photoluminescence on account of a small Stokes shift suggests it may be a promising candidate as a flexible wavelength-division multiplexing material.22,23,27 This effect will be the focus of follow-up investigation. In contrast, the guided emission spectrum from the 730 nm incident light source is unaffected by the crystal, Fig. 7d. The whole spectra of the used LED light sources, PLE and PL can be found in the ESI,† Fig. S11 (455 nm) and S12 (730 nm).
To observe the effect of plastic bending on the waveguiding properties of form II CPMBP, guided emission spectra from both 455 nm and 730 nm incident light sources were again measured on the bent single crystals, Fig. 8. As compared with the straight crystals of form II (Fig. 7), no difference in the shape or intensity of the emission spectra at either incident energy was observed. This suggests that form II CPMBP conserves its waveguiding functionality upon deformation.
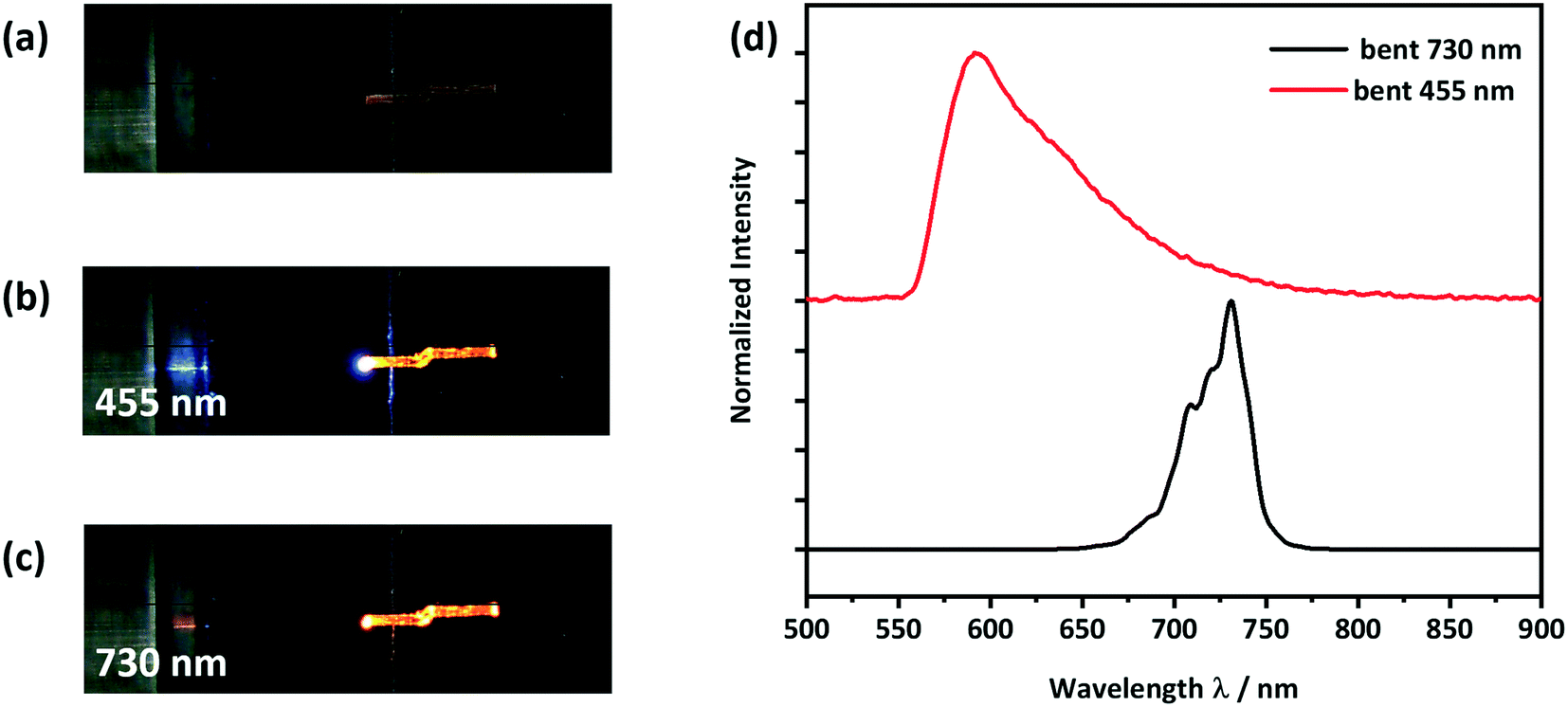 | ||
| Fig. 8 Optical microscope images of (a) the bent crystal placed on a silicon wafer with a 3 μm thick SiO2 layer, (b) showing light with a wavelength of 455 nm being coupled at the left end of the crystal and (c) showing light with a wavelength of 730 nm being coupled at the left end of the crystal; (d) detected PL-spectra at the right end of the crystal. | ||
The promising optical behavior of CPMBP single crystals suggests significant technological potential. The combination of self-absorption and flexibility can be exploited to simplify light coupling into micron-scale waveguide systems.27,28 Since the CPMBP molecules form single crystals, it can be assumed that the structure affects the polarization of the propagating light. These characteristics of the CPMBP waveguide crystal can be applied in photonic systems for specific uses, which makes the CPMBP molecule an interesting material regarding photonic circuits.
Conclusions
We report here two polymorphic forms of a photoluminescent, 4-bromo-6-[(6-chloropyridin-2-ylimino)methyl]phenol with distinctly different optical and mechanical behaviors (Scheme 1). The two polymorphs showed a similar morphology and colour and could be obtained as pure forms by controlled crystallization using different solvent mixtures. Form I adopts a mechanically interlocked 3D network structure with various weak non-covalent interactions. In contrast, form II exhibits anisotropic crystal packing with low energy slip planes. Hence, form I was found to be mechanically brittle, whereas form II showed plastic deformation upon mechanical stress. Photoluminescence spectroscopy revealed that brittle form I is only weakly emissive whereas the emission of plastically flexible form II is comparatively bright. Only a minor red shift in the emission spectrum of form I is observed as compared with that of form II, which can be correlated with the stronger π–π stacking interactions in the crystal packing of form I. Moreover, due to the shapable plastic flexibility combined with bright luminescence properties, form II exhibits the properties of bending the path of light, i.e., flexible optical waveguide. Both active and passive waveguiding were observed. We therefore demonstrate how the mechanical properties of organic luminophores can be tuned by controlling polymorphism instead of chemical modification. We expect polymorph engineering to become an important direction for designing mechanically flexible crystals with multifunctional applications in flexible optoelectronics.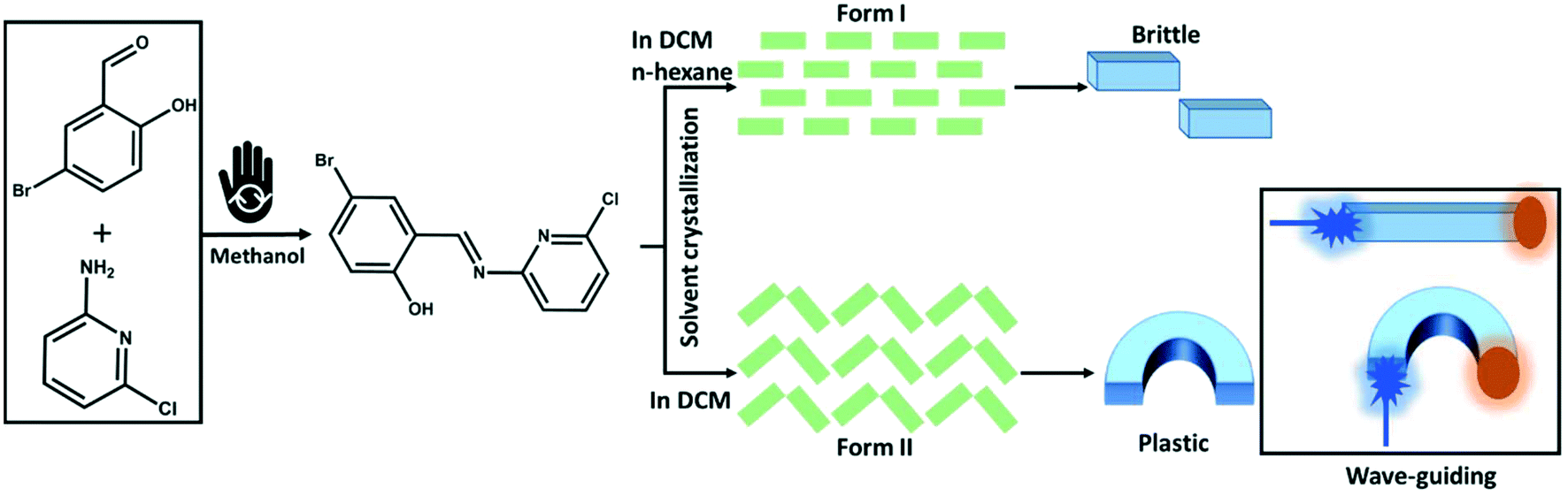 | ||
| Scheme 1 Schematic representation of the polymorphic engineering of the mechanically flexible optical materials. The mechanochemical pictogram is taken from ref. 77. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank BAM IT for access to computational resources. ELK and VS acknowledge the support from the Helmholtz Energy Materials Foundry (HEMF) and PEROSEED (ZT-0024) project. This work was carried out in the framework of the Joint Lab GEN_FAB. The authors are grateful to Prof Simon Parsons (University of Edinburgh) for access to the MrPIXEL software. BB acknowledges funding from Deutsche Forschungsgemeinschaft, Project No. 450137475.References
- Y. Su, G. Sui, J. Lan and X. Yang, Chem. Commun., 2020, 56, 11625–11628 RSC.
- M. Wang, P. Baek, A. Akbarinejad, D. Barker and J. Travas-Sejdic, J. Mater. Chem. C, 2019, 7, 5534–5552 RSC.
- H. R. Allcock, Soft Matter, 2012, 8, 7521–7532 RSC.
- B. Kang, E. Song, S. B. Lee, B.-G. Chae, H. Ahn and K. Cho, Chem. Mater., 2018, 8, 6353–6360 CrossRef.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741–1744 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- S. Ghosh and C. M. Reddy, Angew. Chem., 2012, 5 Search PubMed.
- S. Ghosh, M. K. Mishra, S. B. Kadambi, U. Ramamurty and G. R. Desiraju, Angew. Chem., 2015, 127, 2712–2716 CrossRef.
- N. P. Thekkeppat, M. Lakshmipathi, A. S. Jalilov, P. Das, A. M. P. Peedikakkal and S. Ghosh, Cryst. Growth Des., 2020, 20, 3937–3943 CrossRef CAS.
- P. Gupta, T. Panda, S. Allu, S. Borah, A. Baishya, A. Gunnam, A. Nangia, P. Naumov and N. K. Nath, Cryst. Growth Des., 2019, 19, 3039–3044 CrossRef CAS.
- P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., Int. Ed., 2018, 5 Search PubMed.
- S. Hayashi, T. Koizumi and N. Kamiya, Cryst. Growth Des., 2017, 17, 6158–6162 CrossRef CAS.
- E. Ahmed, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2018, 10 Search PubMed.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
- C. M. Reddy, G. Rama Krishna and S. Ghosh, CrystEngComm, 2010, 12, 2296 RSC.
- B. Bhattacharya, A. A. L. Michalchuk, D. Silbernagl, M. Rautenberg, T. Schmid, T. Feiler, K. Reimann, A. Ghalgaoui, H. Sturm, B. Paulus and F. Emmerling, Angew. Chem., Int. Ed., 2020, 59, 5557–5561 CrossRef CAS PubMed.
- M. Đaković, M. Borovina, M. Pisačić, C. B. Aakeröy, Ž. Soldin, B.-M. Kukovec and I. Kodrin, Angew. Chem., Int. Ed., 2018, 57, 14801–14805 CrossRef PubMed.
- L. Mei, S. An, K. Hu, L. Wang, J. Yu, Z. Huang, X. Kong, C. Xia, Z. Chai and W. Shi, Angew. Chem., 2020, 8 Search PubMed.
- N. K. Nath, P. Gupta, P. J. Hazarika, N. Deka, A. Mukherjee and G. K. Dutta, Cryst. Growth Des., 2019, 19, 6033–6038 CrossRef CAS.
- H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 RSC.
- H. Liu, Z. Lu, Z. Zhang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8448–8452 CrossRef CAS PubMed.
- H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 RSC.
- L. Catalano, D. P. Karothu, S. Schramm, E. Ahmed, R. Rezgui, T. J. Barber, A. Famulari and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 17254–17258 CrossRef CAS PubMed.
- S. Hayashi, S. Yamamoto, D. Takeuchi, Y. Ie and K. Takagi, Angew. Chem., Int. Ed., 2018, 57, 17002–17008 CrossRef CAS PubMed.
- J. Cao, H. Liu and H. Zhang, CCS Chem., 2020, 2, 2569–2575 CrossRef.
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 CrossRef CAS PubMed.
- M. Annadhasan, D. P. Karothu, R. Chinnasamy, L. Catalano, E. Ahmed, S. Ghosh, P. Naumov and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13821–13830 CrossRef CAS PubMed.
- B. Bhattacharya, D. Roy, S. Dey, A. Puthuvakkal, S. Bhunia, S. Mondal, R. Chowdhury, M. Bhattacharya, M. Mandal, K. Manoj, P. K. Mandal and C. M. Reddy, Angew. Chem., 2020, 6 Search PubMed.
- S. Hayashi, F. Ishiwari, T. Fukushima, S. Mikage, Y. Imamura, M. Tashiro and M. Katouda, Angew. Chem., Int. Ed., 2020, 7 Search PubMed.
- E. M. Horstman, R. K. Keswani, B. A. Frey, P. M. Rzeczycki, V. LaLone, J. A. Bertke, P. J. A. Kenis and G. R. Rosania, Angew. Chem., Int. Ed., 2017, 56, 1815–1819 CrossRef CAS PubMed.
- L. Adler-Abramovich, Z. A. Arnon, X. Sui, I. Azuri, H. Cohen, O. Hod, L. Kronik, L. J. W. Shimon, H. D. Wagner and E. Gazit, Adv. Mater., 2018, 30, 1704551 CrossRef PubMed.
- S. Ghosh and M. K. Mishra, Cryst. Growth Des., 2021, 21, 2566–2580 CrossRef CAS.
- P. Gupta, S. A. Rather, B. K. Saha, T. Panda, D. P. Karothu and N. K. Nath, Cryst. Growth Des., 2020, 6 Search PubMed.
- N. K. Nath, M. Hazarika, P. Gupta, N. R. Ray, A. K. Paul and E. Nauha, J. Mol. Struct., 2018, 1160, 20–25 CrossRef CAS.
- U. B. R. Khandavilli, M. Lusi and P. J. Frawley, IUCrJ, 2019, 6, 630–634 CrossRef CAS PubMed.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 13 CAS.
- S. Saha and G. R. Desiraju, Chem. Commun., 2016, 52, 7676–7679 RSC.
- B. Puschner, R. H. Poppenga, L. J. Lowenstine, M. S. Filigenzi and P. A. Pesavento, J. Vet. Diagn. Invest., 2007, 19, 616–624 CrossRef PubMed.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- C. M. Reddy, S. Basavoju and G. R. Desiraju, Chem. Commun., 2005, 2439–2441 RSC.
- S. Saha and G. R. Desiraju, Chem. Commun., 2018, 54, 6348–6351 RSC.
- M. K. Mishra and C. C. Sun, Cryst. Growth Des., 2020, 20, 4764–4769 CrossRef CAS.
- K. B. Raju, S. Ranjan, V. S. Vishnu, M. Bhattacharya, B. Bhattacharya, A. K. Mukhopadhyay and C. M. Reddy, Cryst. Growth Des., 2018, 18, 3927–3937 CrossRef CAS.
- X. Chu, Z. Lu, B. Tang, B. Liu, K. Ye and H. Zhang, J. Phys. Chem. Lett., 2020, 11, 5433–5438 CrossRef CAS PubMed.
- B. Liu, H. Liu, H. Zhang, Q. Di and H. Zhang, J. Phys. Chem. Lett., 2020, 11, 9178–9183 CrossRef CAS PubMed.
- N. Mitetelo, D. Venkatakrishnarao, J. Ravi, M. Popov, E. Mamonov, T. V. Murzina and R. Chandrasekar, Adv. Opt. Mater., 2019, 7, 1801775 CrossRef.
- W. Zhang, J. Yao and Y. S. Zhao, Acc. Chem. Res., 2016, 49, 1691–1700 CrossRef CAS PubMed.
- K. Takazawa, J. Inoue, K. Mitsuishi and T. Kuroda, Adv. Funct. Mater., 2013, 23, 839–845 CrossRef CAS.
- V. Coropceanu, J. Cornil and J.-L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- H. Zhang, H. Liu, Z. Lu, B. Tang, C. Qu and Z. Zhang, Angew. Chem., 2020, 132, 13044–13050 CrossRef.
- M. Mathivanan, B. Tharmalingam, C.-H. Lin, B. V. Pandiyan, V. Thiagarajan and B. Murugesapandian, CrystEngComm, 2020, 22, 213–228 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, SADABS, University of Göttingen, Germany, 2002.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Gavezzotti, New J. Chem., 2011, 1360–1368 RSC.
- A. Gavezzotti, Mol. Phys., 2008, 106, 1473–1485 CrossRef CAS.
- M. G. Reeves, P. A. Wood and S. Parsons, J. Appl. Crystallogr., 2020, 53, 1154–1162 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.01, 2016.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, New York, 2001 Search PubMed.
- I. A. Olson, A. G. Shtukenberg, B. Kahr and M. D. Ward, Rep. Prog. Phys., 2018, 81, 096501 CrossRef PubMed.
- M. M. Kuklja and A. B. Kunz, J. Appl. Phys., 2001, 89, 10 CrossRef.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1–6 CrossRef PubMed.
- F. P. A. Fabbiani, D. R. Allan, S. Parsons and C. R. Pulham, CrystEngComm, 2005, 7, 179 RSC.
- N. P. Funnell, C. L. Bull, C. J. Ridley, S. Parsons and J. P. Tellam, Chem. Commun., 2020, 56, 6428–6431 RSC.
- N. Giordano, C. M. Beavers, K. V. Kamenev, W. G. Marshall, S. A. Moggach, S. D. Patterson, S. J. Teat, J. E. Warren, P. A. Wood and S. Parsons, CrystEngComm, 2019, 21, 4444–4456 RSC.
- C. Deng, Y. Niu, Q. Peng, A. Qin, Z. Shuai and B. Z. Tang, J. Chem. Phys., 2011, 135, 014304 CrossRef PubMed.
- J. Collins, ECS J. Solid State Sci. Technol., 2016, 5, R3170–R3184 CrossRef CAS.
- J. M. Crowley, J. Tahir-Kheli and W. A. Goddard, J. Phys. Chem. Lett., 2016, 7, 1198–1203 CrossRef CAS PubMed.
- A. A. L. Michalchuk, E. V. Boldyreva, A. M. Belenguer, F. Emmerling and V. V. Boldyrev, Front. Chem., 2021, 9, 685789 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082067 and 2082068. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00642h |
| This journal is © The Royal Society of Chemistry 2021 |