
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in probing host–guest interactions with solid state nuclear magnetic resonance
Ashlea R.
Hughes
 a and
Frédéric
Blanc
*ab
a and
Frédéric
Blanc
*ab
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: frederic.blanc@liverpool.ac.uk
bStephenson Institute for Renewable Energy, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
First published on 2nd March 2021
Abstract
An overview of the recent role of solid state nuclear magnetic resonance (NMR) spectroscopy in the field of supramolecular chemistry to probe host–guest interactions is provided. Over the last few years, solid state NMR methodologies have delivered unique insights into the atomic level structure and dynamics of guest molecules adsorbed in solid materials that are not available by other approaches. This Highlight discusses the range of NMR interactions that enable access to this information and provides a number of illustrating examples that highlight their applications in a wide range of chemical systems and porous materials covering metal organic frameworks, porous molecular solids and zeolites.
 Ashlea Hughes | Ashlea Hughes obtained her MChem in Chemistry (with a year in industry) from the University of York (2014). Following a few years in industry, she returned to her studies in 2016 at the University of Liverpool to start a PhD in Frédéric Blanc's group focusing on the structure and dynamics of advanced materials by solid state NMR. |
 Frédéric Blanc | Frédéric Blanc is currently Reader in the Department of Chemistry and Stephenson Institute for Renewable Energy at the University of Liverpool. He received a M.Sc, a MChem (2004) in Physical Chemistry and Catalysis from the Lyon School of Chemistry and a PhD (2007) from the University of Lyon. Following postdoctoral work and a number of Fellowships at the State University of New York in Stony Brook, NY and at the University of Cambridge, he joined the faculty at the University of Liverpool (2012). His current research focuses on the interplay of (MAS DNP) NMR with materials chemistry, in particular energy materials and supramolecular assemblies. |
1. Introduction to host–guest interactions and supramolecular chemistry
Host–guest interactions are prevalent in nature and play important roles in many critical functions. These non-covalent interactions form supramolecular complexes between multiple molecules to occur by hydrogen bonding, hydrophobic forces, van der Waals forces, π–π interactions and electrostatic effects and are responsible for the three-dimensional structure of biomacromolecules, molecular recognition processes, self-assembly, and many further examples which would not be able to perform their function without these crucial interactions.1 These examples listed above have inspired chemists to exploit host–guest interactions for drug discovery, molecular separation and catalysis leading to the growing field of supramolecular chemistry. To the best of our knowledge, the first example of a synthetic compound capable of complexing alkali metal cations was the crown ether dibenzo-18-crown-6 (illustrated in Fig. 1), discovered by Charles J. Pedersen in 1967.2 The affinity of dibenzo-18-crown-6 for the potassium ion was found to be greater than that of the smaller sodium cation, enabling preferential molecular separation of these alkali metal cations. Leading on from this seminal work, Charles J. Pedersen along with Donald J. Cram and Jean-Marie Lehn were in 1987 awarded the Nobel Prize in Chemistry for their “development and use of molecules with structure-specific interactions of high selectivity”.3–5 The field of supramolecular chemistry was then again rewarded with the 2016 Nobel Prize in Chemistry to Jean-Pierre Sauvage, Sir J. Fraser Stoddart and Bernard L. Feringa for their design and production of molecular machines, where molecules are linked together via a mechanical bond, rather than covalent.6–8 The diversity of this research area causes it to be an ever growing and a rapidly developing field of chemistry.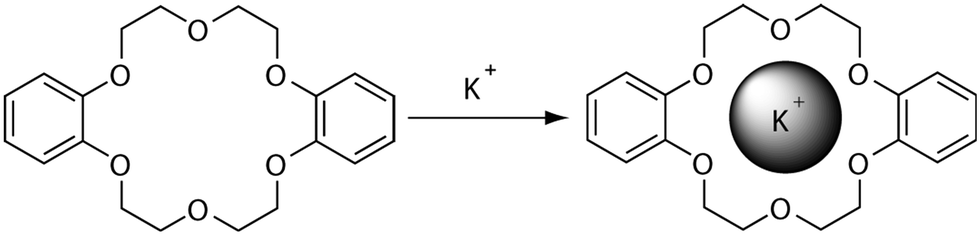 | ||
| Fig. 1 Typical host–guest chemistry example of dibenzo-18-crown-6 and K+ ions forming supramolecular complex held together by electrostatic interactions originally discovered by Charles J. Pedersen.2 | ||
The understanding of the functionality of supramolecular materials requires the knowledge of both the structure of the materials and their configuration in relation to each other. This however largely depends on the materials dynamics and flexibility, as these factors control the chemistry, adsorption or encapsulation. In this Highlight article, recent examples of host–guest interactions in supramolecular assemblies in which the understanding has been advanced by solid state NMR experiments in order to demonstrate its utility will be reviewed. This article will highlight the most recent advances since the last review on this topic in 2016 (ref. 9) and describe the interactions from the perspective of the solid state NMR techniques which have been used rather than either the types of interactions,9–11 or class of molecules.12 Each section is illustrated by brief examples which use a particular solid state NMR technique needed to gain further insights into the structure and dynamics which are of high importance in this field.
2. Widely used approaches to probe host–guest interactions
A range of experimental methods has been used to investigate host–guest systems. Fluorescence spectroscopy is widely exploited within this area, due to its ability to yield key information regarding the occurrence of complexation in the form of a spectral shift13 whilst contributing rather limited information about the strength or location of the host–guest interaction and is therefore often combined with other methods to achieve this. Calorimetry is able to provide the strength of the host–guest interaction,14 however, the location of the guest and the type of interactions are difficult to decipher without prior knowledge of potential binding sites. Diffraction-based methods such as X-ray diffraction (XRD) probe long-range order and are critical to elucidate structural host–guest features. Nevertheless, the dynamics and mobility of supramolecular assemblies are challenging to assess using these methods due to the inherent requirement to accessing short range structural information. Diffraction methods are therefore often paired with NMR to provide a more complete understanding of the interactions and dynamics at play in supramolecular assemblies.11 Additionally, density functional theory (DFT) studies are becoming more and more prevalent to procure detailed structural information regarding bonding and electronic structures (e.g. to compute NMR parameters and support spectral assignment),15–17 all providing complementary information to experimental data. Hence, in conjunction with diffraction-based methods and computational studies, solid state NMR crystallography9,11,18,19 is playing an important role in the study of host–guest systems and has strong potential to provide information regarding both structure and dynamics of supramolecular chemistry as illustrated below.3. Solid state NMR and NMR interactions as probes to study supramolecular materials.
Solid state NMR is a powerful tool for understanding the short-range order and dynamics for a range of solid materials in both crystalline and amorphous states. NMR provides site-specific and local structural information, and is therefore a robust approach for probing spatial proximities. The NMR spectra of solids are affected by a number of NMR interactions which include Zeeman splitting (interaction of the nuclear spin with the external magnetic field B0), isotropic and orientation dependent (anisotropic) chemical shifts (indirect interaction of the nuclear spin with B0via the electrons), through-space dipolar coupling (direct nuclear spin dipole–dipole coupling) and through-bond scalar J couplings (indirect nuclear spin dipole–dipole coupling via the electrons), and for nuclei with spin quantum number I > 1/2, quadrupolar interaction (interaction of the nuclear electrical quadrupolar moment with the electric field gradient (EFG)). All of these interactions affect the appearance of the NMR spectrum and provide structural and dynamics information on the local environment.The two most common methods to explore host–guest interactions by solid state NMR methods exploit chemical shieldings (directly relating to the observed chemical shifts) and dipolar interactions, both of which are highly dependent on local structure and packing arrangements. The chemical shielding interaction can be decomposed into diamagnetic and paramagnetic shielding contributions. The diamagnetic shielding results from the circulation of electrons that is induced by the external magnetic field. This motion creates a smaller magnetic field opposing (shielding) the applied field and relates directly to the ground state electron density surrounding the nucleus. The paramagnetic shielding contribution is also induced by the external magnetic field and arises from electron orbital angular momentum and the mixing of the ground state with various excited states increasing (hence deshielding) the applied field. Note that the paramagnetic shielding contribution is not be confused with the paramagnetic shift which captures the interaction between NMR-active nuclei and unpaired electrons in paramagnetic centres such as radicals and some metal ions. These paramagnetic shielding currents dominate (except for 1H where it is negligeable) and depend on both the inverse of the excitation energy and the distance between the nucleus and its electrons. These currents perturb the magnetic field that the nucleus experiences from the external magnetic field (Zeeman interaction) causing the nucleus to resonate at a different frequency and hence chemical shift. The dipolar interaction arises from the interaction between the nuclear magnetic moments of two alike (homonuclear dipolar coupling) or different (heteronuclear dipolar coupling) nuclear spins and depends on both the strength of the magnetic moments and the inverse to the cube of the distance between both spins.
Although the chemical shielding and dipolar coupling interactions provide a wealth of information regarding the local environment of the nucleus, the information can be challenging to extract due to the inherently low sensitivity, poor resolution, and broad resonances often dominating the experimental spectra which has led to the development of a range of methods to overcome these obstacles. The NMR sensitivity is largely dependent on the Boltzmann energy difference between the magnetic states of the nuclear spin which is determined by the strength of the external magnetic field and the value of the gyromagnetic ratio of the nucleus. High field NMR20–22 is therefore often used to overcome this challenge by increasing this energy gap and also often results in higher resolution by increasing chemical shift dispersion. Dynamic nuclear polarisation (DNP)23–25 and cross polarisation (CP)26 emerged as powerful approaches to significantly increase sensitivity by polarisation transfer of the high magnetisation of electron spins to nucleus spins, and of higher polarised nucleus spins (e.g.1H) to lower polarised spins, respectively. Other hyperpolarisation methods (of which DNP belongs to) that amplified the nuclear polarisation beyond that obtained by the Boltzmann distribution have also been discovered, mostly in liquid state NMR spectroscopy. Additionally, magic angle spinning (MAS)27 that approximates molecular tumbling experienced in the liquid state, is routinely used in the solid state to suppress first order anisotropic interactions such as chemical shieldings and dipolar couplings increasing both sensitivity and resolution.28 Many of these methods are exploited in the examples given below and their more comprehensive description being beyond the scope of this review, the reader is directed to explore other sources.11,18,29
3.1. Complexation induced shift
Complexation induced shifts (CIS),30 also known as chemical shift perturbation, delivers a significant advantage for structural and conformational determination in supramolecular complexes, and provides indispensable information regarding binding in supramolecular assemblies. Chemical shifts are indicative of the local environment surrounding a nucleus as described in section 3 above and complexation induced shifts probe the change in chemical shielding of the nuclei in a molecule due to electron density arising from nearby nuclei or molecules in an assembly. CISs were originally discovered in liquid state NMR due to solvent effects30 and have now been used extensively in solid state NMR to observe host–guest assemblies.31 CH–π interactions of toluene and pyridine in p-tert-butylcalix[4]arene32 have been observed using CISs combining NMR crystallography and ab initio calculations to determine the positioning of the toluene or pyridine location inside the host structures.Most notably, hydrogen bonding, which is one of the strongest driving forces for host–guest interactions in supramolecular assemblies, results in a very large change of 1H chemical shifts up to approximately 20 ppm.33 Hydrogen bonding can also be monitored via a heteronuclear spin (e.g.13C) that results in reduced electron density around the bridging proton, greater amount of deshielding experienced by this nucleus and a higher chemical shift.
Hydrogen bonding interactions are often exploited in pharmaceutical sciences to obtain amorphous solid dispersions (ASD)34 in which polyvinylpyrrolidone (PVP) is a common polymer used to stabilise amorphized active pharmaceutical ingredients (API) with increased oral bioavailability.3513C CP MAS NMR has recently been used to study the cogrinding process of the nifedipine (NIF) API with PVP and a surfactant (Fig. 2(a)) and assess improved drug dissolution properties from physical cogrinding modification.36 The 13C CP MAS NMR spectrum of the PVP NIF ASD obtained after a short period of 15 minutes of grinding (Fig. 2(b)) shows two narrow well resolved signals for the two chemically inequivalent carbonyl carbons around 170 ppm (ref. 37) and supports a crystalline NIF phase. Upon further cogrinding to 100 minutes, these sharp carbonyl peaks significantly broaden (Fig. 2(b)) and suggest complete NIF amorphization as confirmed by the absence of Bragg reflections in the XRD pattern. It was also observed that there is a small deshielded shift of the PVP carbonyl carbon during the grinding process (Fig. 2(b)) which might indicate some hydrogen bonding in these systems.38 This interaction between PVP and NIF potentially results in stabilising the amorphization, making PVP an appealing polymer to increase oral bioavailability. To strengthen the argument for hydrogen bonding, a single NMR technique is rarely used and a multinuclear approach combining homonuclear/heteronuclear correlation techniques or NMR crystallography is often used to provide further insights.39
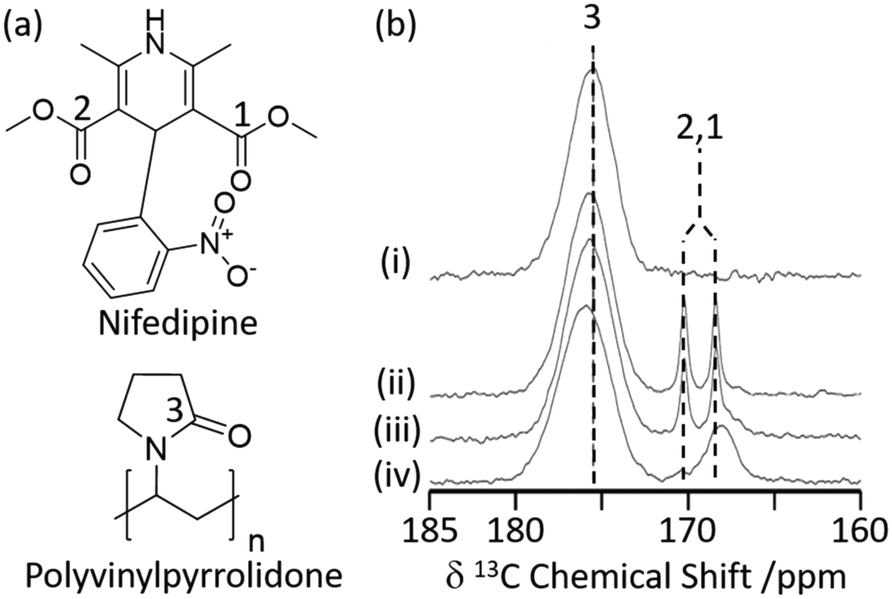 | ||
| Fig. 2 (a) Chemical structure of nifedipine (NIF) and polyvinylpyrrolidone (PVP). (b) 13C CP MAS NMR spectra of (i) PVP, a mechanically ground mixture of NIF/PVP/sodium dodecyl sulfate system ground for (ii) 15 min, (iii) 40 min, and (iv) 100 min obtained under MAS at νr = 15 kHz. The spectral assignments are shown in the figure. Reprinted (adapted) with permission from ref. 36. Copyright 2016 American Chemical Society. | ||
3.2. Lineshape analysis
Deuterium 2H (I = 1) is a quadrupolar nucleus which lineshape is dominated by broadening arising from the quadrupolar interaction. 2H NMR is a very common tool exploited for the study of molecular dynamics as its lineshape is highly sensitive to a range of different motions and their rates on the kHz–MHz timescale.40 The low natural abundance of this isotope (0.0115%) is both a drawback with the need for isotopic enrichment requiring additional synthetic steps and an advantage as selective labelling allows site specific information to be obtained. In the absence of motion, static 2H spin echo NMR experiments under non spinning conditions produce a Pake pattern which have outer horns that are symmetrically distributed towards the edges of the pattern (Fig. 3). However, the presence of motion in the 103–108 Hz frequency range causes changes in this lineshape to occur due to T2 anisotropy preventing certain crystallite orientations to refocus during the spin echo sequence.40,41 The temperature-dependence evolution of the lineshape (Fig. 3) is a function of the type of motion, geometry and reorientation rates (accessible from numerical simulations using for example the EXPRESS,42 MXET1 (ref. 43) or weblab44 software packages), thereby producing a change in lineshape characterising the overall dynamics of the motion.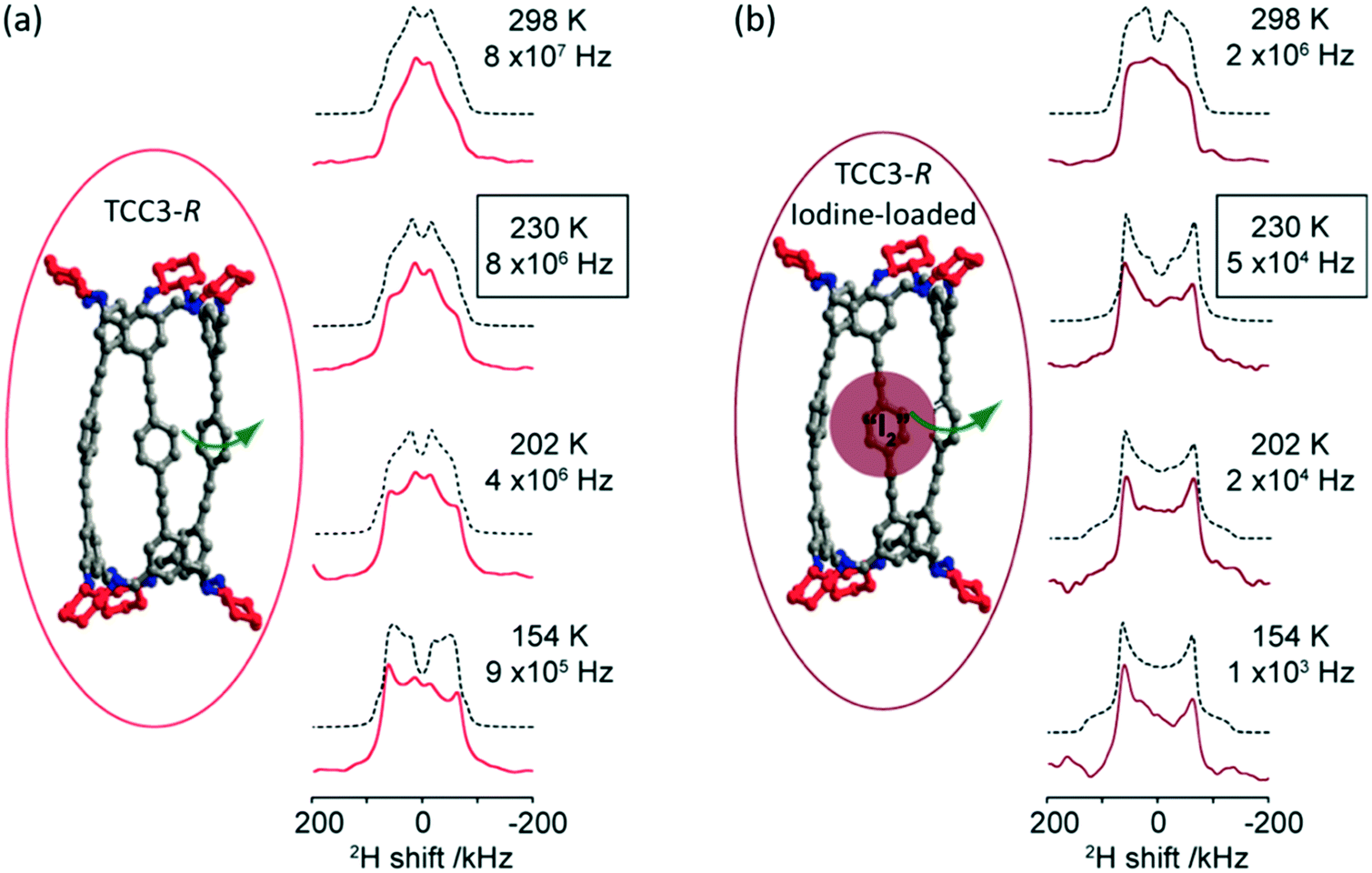 | ||
| Fig. 3 (a) TCC3-R structure (red) and variable temperature 2H static solid echo NMR spectra of [D12]TCC3-R. (b) Iodine-loaded TCC3-R structure (burgundy) and variable temperature 2H static solid echo NMR spectra of iodine loaded [D12]TCC3-R. Corresponding simulated spectra (black dashed lines) and rotational rates obtained from numerical simulations of the NMR lineshape obtained at various temperatures are also given. In the crystal structures, the cyclohexane groups are represented in red; other C, grey; N, blue; H/D omitted for clarity. Ref. 48 is an open access article distributed under the terms of the Creative Commons CC BY license. | ||
2H NMR has been used to explore the motion in molecular rotors in a range of systems45–47 and it has recently been identified that tubular covalent cages (TCC)48 are the fastest exclusively organic molecular rotors found to date. The two TCCs that possess a central phenylene ring between either the trisubstituted aromatic (TCC2) or acetylene moieties (TCC3, Fig. 3(a))49 have the potential to rotate in the solid state. Variable temperature 2H static NMR lineshape analysis of TCCs (that have been deuterated on the phenylene ring) show a typical Pake pattern in the slow motion regime at 105 K and a temperature dependent lineshape that is typical of a fast rotational 180° flip upon heating. Through numerical simulations, the 2H NMR lineshapes of TCC3-R were found to consistently have faster reorientation rates than its smaller counterpart TCC2-R and this is ascribed to facile rotation around the acetylene bonds due to a reduction in steric hindrance, showing structural dependency of phenylene motion.
Upon iodine loading (Fig. 3(b)), the change of 2H NMR lineshape indicates significantly slower reorientation rates and demonstrates that the iodine host has hindered phenylene rotation in these TCCs molecular rotors. Upon iodine release from TCCs at 353 K, faster rotational rates are obtained, indicating that these TCCs are responsive materials not only to temperature changes, but also host–guest interactions.
2H NMR lineshape analysis has also recently been used to study the unique dynamics of aromatic hydrocarbon tubular host, [4]CC, with corannulene (COR) guest (Fig. 4(a)).50 Theoretical DFT calculations gave insights into the presence of CH–π bonds interaction in this supramolecular assembly and revealed a bowl-in tube host–guest structure where, despite the presence of multiple CH–π bonds binding the assembly together, guest rotation is permissible as revealed by experimental work deploying solid state NMR. Utilising a deuterated guest ([D10]COR), 2H NMR studies examined the dynamics of the bowl shaped guest in the assembly (Fig. 4(b)) and show that a single resonance is observed in the 2H Pake pattern, indicating that the COR guest molecules are all equivalent. The quadrupolar splitting measured from the horns of the Pake pattern was found to be smaller than that expected value for motionless molecules, indicating some rotational motion with monotypic dynamics is occurring in the guest.
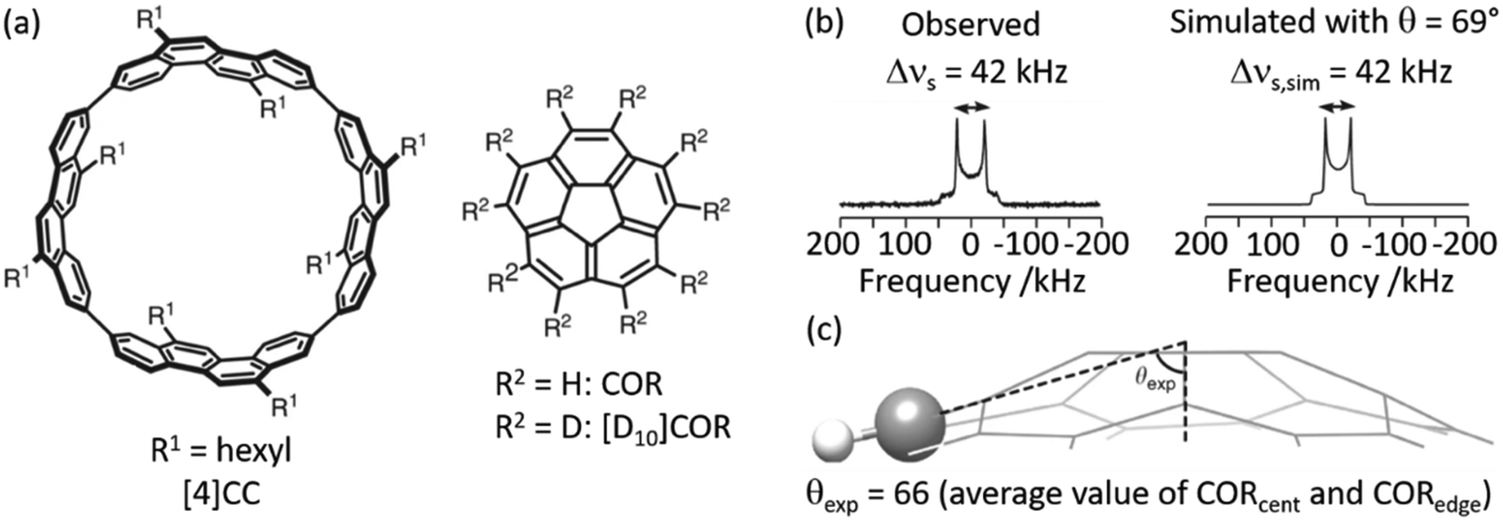 | ||
| Fig. 4 (a) Chemical structures of tubular host ([4]CC) and bowl-shaped guest (COR). (b) Observed and simulated 2H static solid echo NMR spectra of [D10]COR in [4]CC obtained at 298 K, (c) schematic of the molecular structure of COR showing a representative experimental cone angle (θexp) measured from the crystal structure. Ref. 50 is an open article distributed under the terms of the Creative Commons CC BY license. | ||
13C is obviously a popular I = 1/2 nucleus for which lineshape analysis has been extensively used to probe host and guest in supramolecular assemblies. A significant challenge is the poor natural abundance of 13C (1.1%), hence isotopic enrichment or transfer of polarisation techniques such as CP are often used to improve sensitivity.
Guest capture of CO2 is of particular interest in supramolecular chemistry45,51,52 and has recently been extensively investigated from the analysis of the 13C chemical shift anisotropy (CSA) lineshape.52,5313C labelled CO2 is often used to focus on the loaded CO2 whilst signals arising from the MOF framework linkers at natural abundance are highly decreased in intensity. One example compares two different metal centered (Al and Ga) MIL-53 metal organic frameworks (MOFs), that contain corner sharing MO4(OH)2 octahedra interconnected by benzenedicarboxylate linkers, and the related amine functionalised derivatives,54 providing an understanding of the CO2 dynamics within this family of MOFs. The experimental static 13C CSA NMR spectra could be modelled with two types of possible motion, a six- and a two-fold rotation. This analysis showed that MIL-53(Al) gave a smaller hopping angle of CO2 rotation in comparison to MIL-53(Ga), which had more mobility, and indicates that the metal centre affects the CO2 binding strength with Ga causing a weaker CO2 adsorption. Adsorption of CO2 in the NH2 functionalised benzenedicarboxylate (Al and Ga) MIL-53 was also studied and revealed similar 13C CSA lineshape for both cations, although with a general greater affinity for the CO2 in comparison to the non-functionalised MIL-53. In other work, 13C CSA lineshape analysis on Mg2(dobdc) MOF-74 (dobdc4− = 2,5-dihydroxy-1,4-benzenedicarboxylic acid)52,53 adsorbed with a range of CO2 pressure showed that the CO2 molecules have an orientation dependency on the pressure and hence the number of CO2 molecules adsorbed.
A combination of 13C CSA and pulsed field gradient (PFG) experiments has recently been used to quantitatively determine the diffusion coefficients of CO2 in Zn2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate)52 which is also a MOF-74 possessing one-dimensional hexagonal channels containing unsaturated metal centres. Static 13C NMR spectra of 13CO2 loaded Zn2(dobdc) are shown in Fig. 5(a) and do not show the expected narrow resonance expected for CO2 in the gas phase but rather exhibit a powder pattern that corresponds to different orientations of the confined CO2 molecules relative to B0. This pattern is a consequence of the reduced 13C CSA of CO2 confined in the pores due to preferred orientations of the crystal frame relative to B0 (Fig. 5(b)). Based on a number of considerations, including MAS NMR spectra, the sign of the 13C CSA, difference CO2 dose, diffusion coefficients D values and molecular dynamics (MD) simulations, the deshielded and shielded edges of the powder pattern were assigned to crystals perpendicular (D⊥) and parallel (D∥) to B0, respectively.
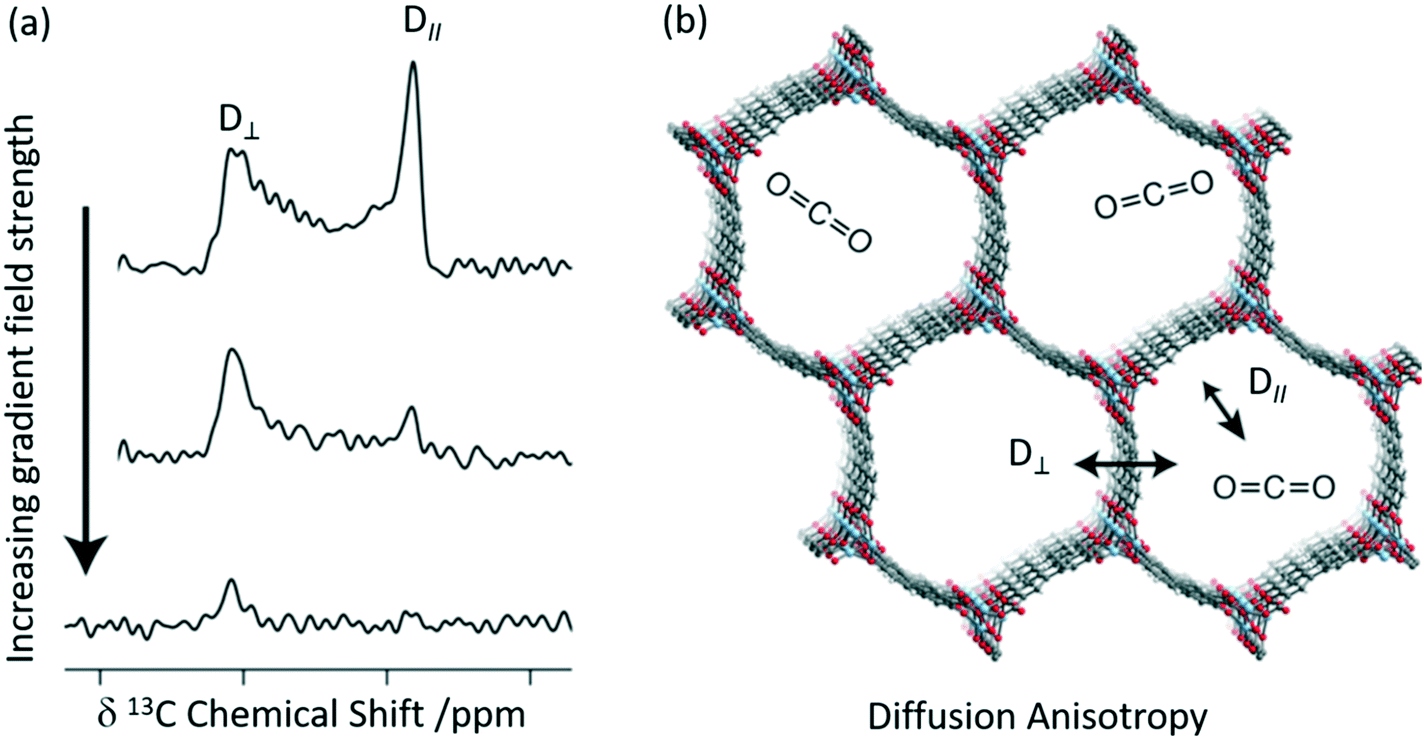 | ||
| Fig. 5 (a) 13C PFG static NMR spectra for Zn2(dobpdc) crystals at a pressure of 625 mbar 13CO2 with different applied gradient strengths and at 298 K. D∥ is the signal arising from crystals parallel to the external magnetic field at a lower chemical shift, whilst those at higher chemical shift are aligned perpendicular to the field D⊥. (b) Cross-section of the crystal structure of the Zn2(dobpdc) framework at 298 K showing the two diffusion pathways. Light blue, red, grey, and white spheres represent Zn, O, C, and H atoms, respectively. Reprinted (adapted) with permission from ref. 52. Copyright 2018 American Chemical Society. | ||
Static 13C NMR spectra of 13CO2 loaded Zn2(dobdc) recorded as a function of increasing gradient field strengths (Fig. 5(a)) have revealed faster decay of the deshielded signal than the shielded signal and enabled CO2 self-diffusion coefficients in both the parallel (D∥) and perpendicular (D⊥) directions to the hexagonal channels to be obtained. CO2 was observed to diffuse through the channels parallel to the crystallographic c axis (D∥) as expected but also and, more surprisingly, between the hexagonal channels in the crystallographic ab plane (D⊥, Fig. 5(b)) and has been attributed to defects in the MOF structure. It is worth pointing out that neither in situ XRD or MD revealed this anisotropic diffusion thereby highlighting the complementarity of these methods.
3.3. NMR relaxometry
NMR relaxometry encompasses a wide range of experiments that access the rates of relaxation (either longitudinal relaxation times in the laboratory frame T1 or rotating frame T1ρ, or transverse relaxation time T2) which measure polarisation returning to their equilibrium values in a magnetic field. Temperature dependent relaxometry measurements in particular are insightful for the study of molecular dynamics, allowing access to the correlation times of the motion and their activation energies, providing some important information about host–guest interactions.22,36The adsorption of 13C labelled CO2 in MOF-74 Zn2(dotp) (where dotp4− = 2,5-dioxidoterephthaate, Fig. 6(a)) has recently been studied using a combination of solid state NMR techniques, including 13C CSA lineshape analysis, 2D exchange spectroscopy, 13C MAS and NMR relaxometry.55 The T1 times of the CO2 guest were found to increase with increasing numbers of CO2 molecules (Fig. 6(b)), indicating that at higher pressures the CO2 mobility is hindered due to the CO2–CO2 interactions. The correlation times obtained from T1 data over the 305–363 K temperature range and assuming a CSA relaxation mechanism were calculated and yielded activation energies of 4.4 and 3.5 kJ mol−1 for two samples with different CO2 pressure of 100 and 1000 mbar, respectively (Fig. 6(c)). This slight difference supports the hypothesis that the CO2–CO2 and CO2-pores surface interactions increase with pressure. The correlation time found was also significantly longer than that of gaseous CO2, also providing the evidence for the host–guest interactions occurring at the pore surface.
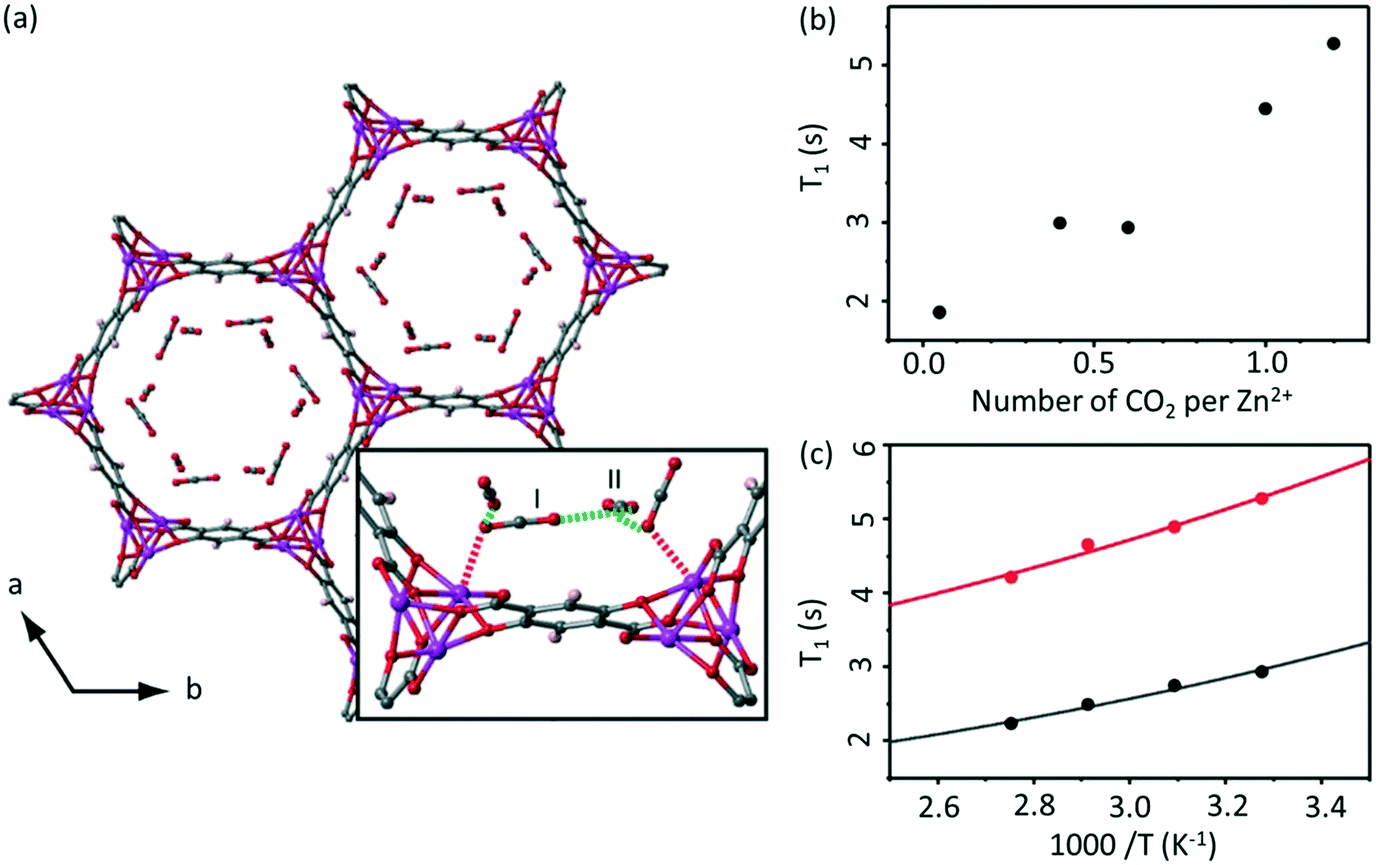 | ||
| Fig. 6 (a) MOF-74 chemical structure with adsorption sites shown. (b) Pressure and (c) temperature dependence of T1 values for the adsorbed CO2 in Zn2(dotp) MOF-74 under MAS at νr = 6 kHz, 305 K and at 100 mbar (black) and 1000 mbar (red), respectively. Experimental and calculated T1 values (based on a 13C CSA relaxation mechanism) are shown as data points and solid lines, respectively. Reproduced from ref. 55 with permission from the PCCP Owner Societies. | ||
Analysis of the T1 data in Zn2(dotp) MOF-74 also revealed the occurrence of unusually slow localized wobbling and hopping motions (at a rate of 10−8 s) that is much smaller than for the Mg-MOF-74 (10−10 s) analogue and further studies are highly anticipated to provide more information on the mobility, adsorption and separation properties of these MOFs.
3.4. Spin diffusion
Spin diffusion NMR experiments observe the transfer of nuclear magnetization via through space dipole–dipole interactions and has been used to probe host–guest interactions among a range of systems.39 As spin diffusion coefficients, D, are proportional to the cube root of the nuclei concentration and the square of the nuclei gyromagnetic ratio, nuclei with sufficiently high values (e.g.1H, 7Li, etc.) are often used in these experiments and provide qualitative and quantitative information on spatial proximities.Along with T1 relaxometry data on Zn2(dotp) MOF-74, 13C two-dimensional (2D) exchange MAS NMR data was also used to investigate the diffusion of CO2 enriched in 13C thereby increasing 13C spins concentration.55 With the possibility of multiple adsorption sites, the determination of the type of sites is crucial for the understanding of the mechanism of adsorption. Surprisingly and in contrast to previous work assessing the 13C CSA patterns of Mg-MOF-74,56 two CO2 resonances were observed in Zn-MOF-74 (ref. 55) and initially assigned to a primary adsorption site and a secondary adsorption site only accessible under greater pressure. However, the corresponding 2D exchange 13C MAS NMR spectra revealed that these two signals do not exchange (over 10–1000 ms mixing times) and it was therefore concluded that the second resonance is due to mobile CO2 located in the dead space of the NMR tube rather than a secondary adsorption site.
1H 1H spin diffusion experiments were performed on Zr6O4(OH)4(bpdc)6 MOF UiO-67 (where bpdc2− = biphenyl-4,4′-dicarboxylate) loaded with a range of light alkanes (methane, ethane and propane) to explore their interactions into the MOF pores (Fig. 7(a)).57 First principle calculations and neutron diffraction studies had previously been used to probe host–guest interaction in methane-loaded M2(dhtp) MOFs (M = Mg, Mn, Co, Ni, Zn; dhtp2− = 2,5-dihydroxyterephthalate),58 however, due to the motional dynamics and disorder in these systems, significant direct experimental evidence for the interaction at room temperature has been difficult to achieve. The 1H 1H spin diffusion spectra of the methane loaded UiO-67 are shown in Fig. 7(b) at various mixing times and revealed cross peaks correlating methane (−0.4 ppm) with aromatics proton belonging to the UiO-67 bpdc organic linker, supporting van der Waals interactions. Experiments conducted with heavier alkanes (ethane and propane) showed similar results, albeit requiring longer spin diffusion times for cross peaks to appear and suggests there is a slight preference for the smaller alkanes, and overall showing evidence that UiO-67 could be utilised for the storage of these light alkanes.
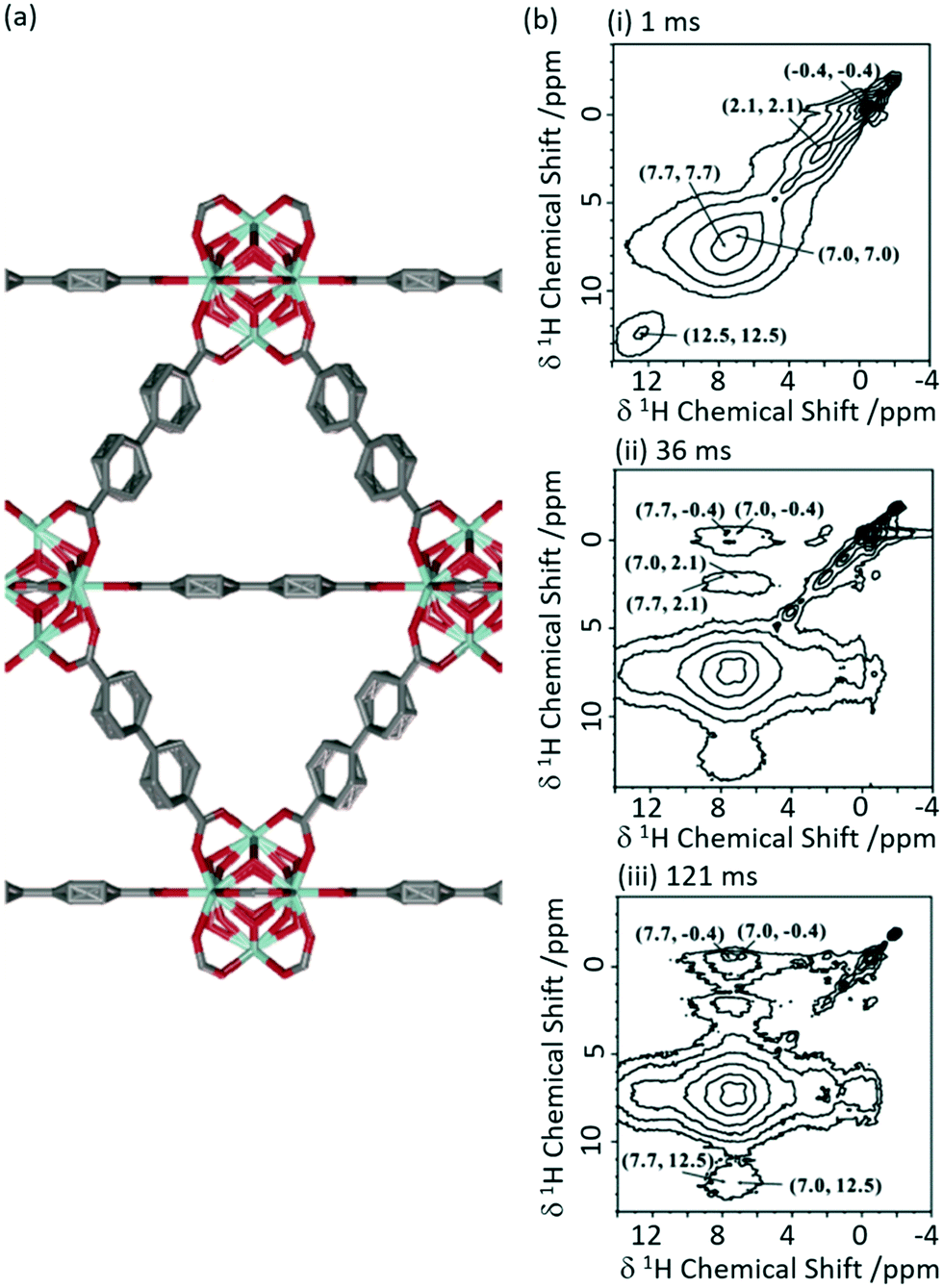 | ||
| Fig. 7 (a) Schematic structure of UiO-67. (b) 2D 1H–1H spin-diffusion spectra of methane loaded UiO-67 at mixing times of (i) 1 ms, (ii) 36 ms and (iii) 121 ms obtained at a MAS rate νr = 10 kHz. Reprinted (adapted) with permission from ref. 57. Copyright 2017 American Chemical Society. | ||
3.5. Xenon NMR
Xenon 129Xe (I = 1/2) enables detailed information about the size and shape of cavity in porous solids59 and is often used to probe supramolecular assemblies. This is largely due to the inert nature and large electron density of Xe which makes this nucleus particularly sensitive to the atomic scale.59,60 Although the high gyromagnetic ratio (−7.441 × 107 rad T−1 s−1) and natural abundance (26.4%) seem favourable for NMR detection, the reduced xenon–xenon interactions needed to promote observation of adsorption sites requires low concentration challenging detection. Hyperpolarised 129Xe NMR is therefore use to perform experiments at lower xenon concentration with significant sensitivity. Importantly, changes in the chemical shifts from free gaseous xenon (at 0 ppm) indicate host–guest interactions and result from a combination of the xenon–surface interaction and the pore volume.59129Xe has been extensively used to investigate the pore structure in solids which is largely key to the physical properties. An example of such material includes zeolites, which structural flexibility enabled by structure-directing agent leads to new catalytic applications.61,62 Recently, interlayer expansion of zeolites has been proposed to expand the current range of available zeolite topological structures and 129Xe been shown to determine the order of the interlayer structure in a new ten-membered ring (MR) functionalised skeleton zeolite (named COE-4) prepared by expansion of the 8-MR layered silicate RUB-36 zeolite precursor.63 The 129Xe spectrum of the calcined RUB-36 (RUB-37) phase shows a weak signal at 89 ppm at 213 K (Fig. 8(a)) and has been ascribed to accumulated Xe in the mesopores and not in the 8-MR channels (of size of 3.1 × 4.7 Å and 2.5 × 4.2 Å along the [010] and [001] directions, respectively) which are too small for Xe (dynamic diameter of 4.4 Å). In COE-4, the 129Xe signal appears at a larger shift of 111 ppm (Fig. 8(b)) indicating the xenon is more “trapped” and these are classed as micropores. Additionally, the 129Xe lineshape of the single resonance observed in COE-4 (Fig. 8(b)) indicates high symmetry and a homogenous distribution of the interlayer expansion. It should also be noted that the 129Xe NMR spectra of both COE-4 and ZSM-5 zeolites are very similar, indicating that the pore sizes are comparable.
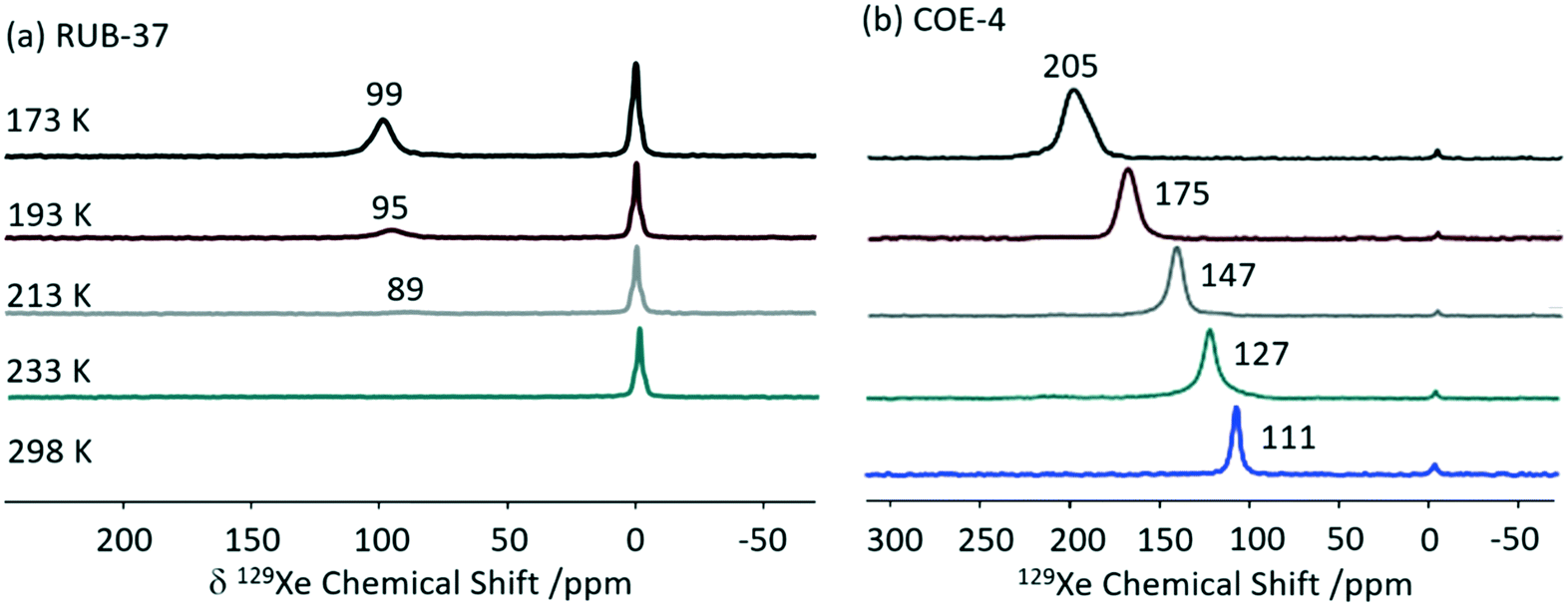 | ||
| Fig. 8 Variable temperature hyperpolarised static 129Xe NMR spectra of (a) RUB-37 and (b) COE-4. Reprinted from ref. 63: structural investigation of interlayer-expanded zeolite by hyperpolarized 129Xe and 1H NMR spectroscopy, 109555, Copyright 2019, with permission from Elsevier. | ||
Porous organic cages (POCs) are a relatively new family of supramolecular assemblies, with the above mentioned TCC3 being one of their newest members. CC3 is another POC consisting of tetrahedral cages formed by imine bonds connecting rigid aromatic rings to the more flexible cyclohexane linkers and arranges into an interconnected 3D pore structure when packed together (Fig. 9(a)). Since their discovery in 2009, POCs have been shown to possess wide applications and unprecedented performance in rare gas separation.60 The cage structure of CC3 was initially studied by 129Xe NMR and it was observed that the xenon can diffuse in three dimension throughout the crystalline pore structure, moving between the window cavities via the cage cavities and indicating interconnectivity of the pore structure.64 A follow up study60 delved into the CC3 cage and its dynamics revealing only one resonance for both the cage and window sites due to fast thermal motion causing the exchange of nuclei between these sites to occur faster than the NMR timescale (Fig. 9(b)). Upon Xe loading, it was observed that the 129Xe chemical shift increases to 210 ppm as the more shielded window sites of the cage (as opposed to the Xe nuclei located in the less shielded cage cavities which appear at 20 ppm) become more occupied. At a lower temperature of 260 K, the chemical shift is slightly reduced due to the increase in relative occupancy of the window cavities. 129Xe spin lattice relaxation rates were also measured due to their dependence on dynamics. Slower motional correlation times were obtained with increasing Xe loading, suggesting restricted xenon diffusion at high loading due to the cage and window cavities being occupied.
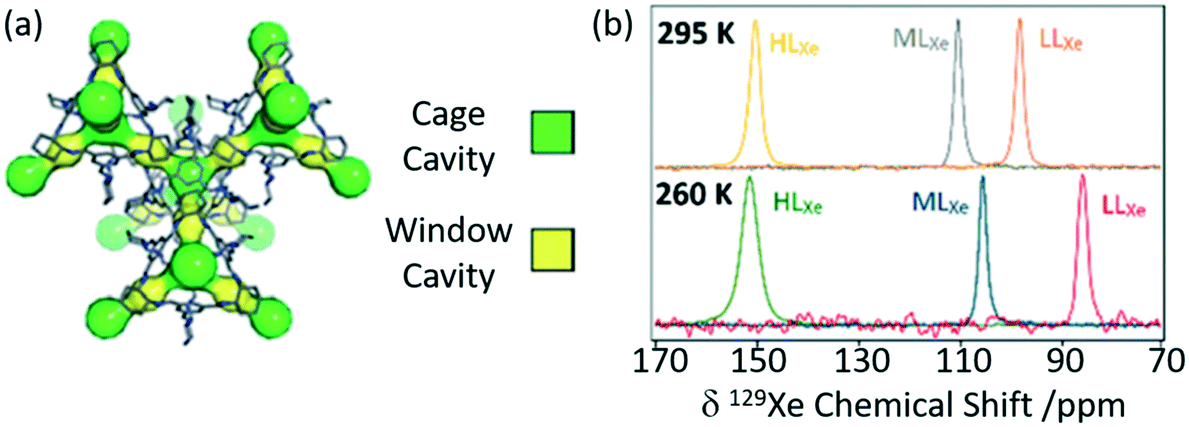 | ||
| Fig. 9 (a) Crystal structure of CC3 showing both cage cavities (yellow) and window cavities (green). (b) 129Xe NMR spectra of CC3 with varying xenon loadings (HLXe – high loading, MLXe – middle loading and LLXe – low loadings) at 295 and 260 K. Reproduced from ref. 60 with permission from the PCCP Owner Societies. | ||
3.6. Cross polarisation
CP is one of the most commonly used experiments in solid state NMR to transfer polarisation from abundant nuclei (typically 1H) to sparse nuclei (e.g.13C, 15N) in order to gain sensitivity. The polarisation transfer is largely dependent on the through space heteronuclear dipolar interactions, and hence can also be exploited to probe dynamics and measure distances including the observation of internuclear interactions and informing host–guest binding. MIL-53 has also been studied with CP and, recently, 1H 129Xe CP has been utilised for the first time in this class of supramolecular assemblies to probe the mobility of xenon (Fig. 10).65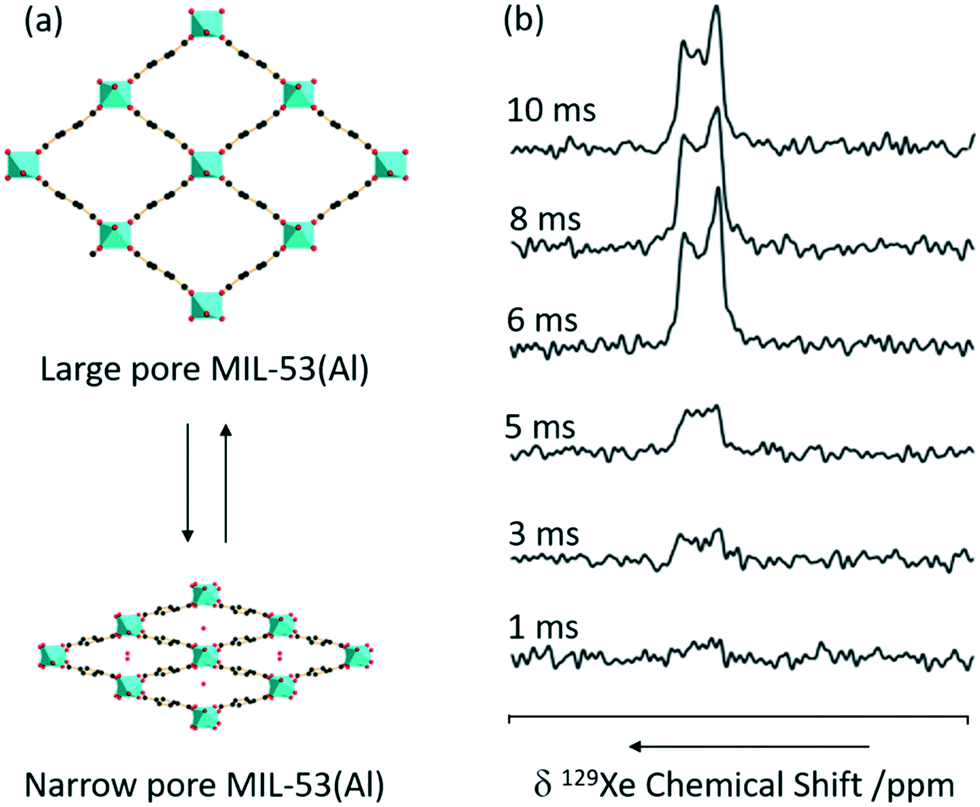 | ||
| Fig. 10 (a) 3D structure of MIL-53(Al) in exchange between the large and narrow pore form. (b) 1H 129Xe CP spectra as a function of contact time under a MAS rate νr = 8 kHz at a temperature of 153 K and a pressure of 500 mbar. Reprinted (adapted) with permission from ref. 65. Copyright 2016 American Chemical Society. | ||
No 129Xe signal is present in large-pore MIL-53 due to the high mobility completely averaging the 1H 129Xe heteronuclear dipolar interactions, and hence significantly reducing CP efficiency. The signal intensity obtained for the 129Xe located in the narrow-pore MIL-53 was found to increase with CP contact time until a maximum at 10 ms which is significantly longer than in the absence of motion (2 ms), hence concluding that the xenon atoms still possess significant motions in these pores.
Zeolite H-ZSM-5 catalysed methanol-to-hydrocarbon (MTH) process remains a highly studied industrial reaction due to its mechanistic aspects and complexity.66 Solid state NMR has already identified a range of catalytic intermediates aiding the mechanistic understandings.67 Recently, it has also been reported that Lewis acidity is not a spectator in this MTH reaction and that the incorporation of alkaline-earth metals into the zeolite results in a generation of Lewis acid site changing the reactivity of the zeolite catalyst throughout the MTH process.68 Using 1D 1H 13C CP MAS experiments performed at increasing contact times, the post reacted H-ZSM-5 and Ca-ZSM-5 zeolites have been analysed to study diffusion characteristics (Fig. 11).69 The peak at 100 ppm is assigned as an acetal (OCH2O) and its intensity increases with increasing contact time, indicating a more efficient CP transfer at longer contact times, suggesting that acetals are preferentially located within the zeolite framework. Direct spectral comparison of both zeolites shows large signal intensity in the 120–135 ppm region for H-ZSM-5 indicating a higher concentration of unsaturated HCP species (like aromatics/polyaromatics) on its surface in comparison to Ca-ZSM-5 (Fig. 11(a)). Upon increasing the CP contact times to 3 ms, the aromatic peak around 133 ppm in the H-ZSM-5 spectra (Fig. 11(b)(i)) disappears, implying the presence of polyaromatic products exclusively residing on the surface of the zeolite. However, this weak peak at 133 ppm is absent in the post-reacted Ca-ZSM-5 zeolite (Fig. 11(b)(ii)) at short contact times, which indicates the absence of any aromatic species in the calcium modified zeolite, especially on its surface.
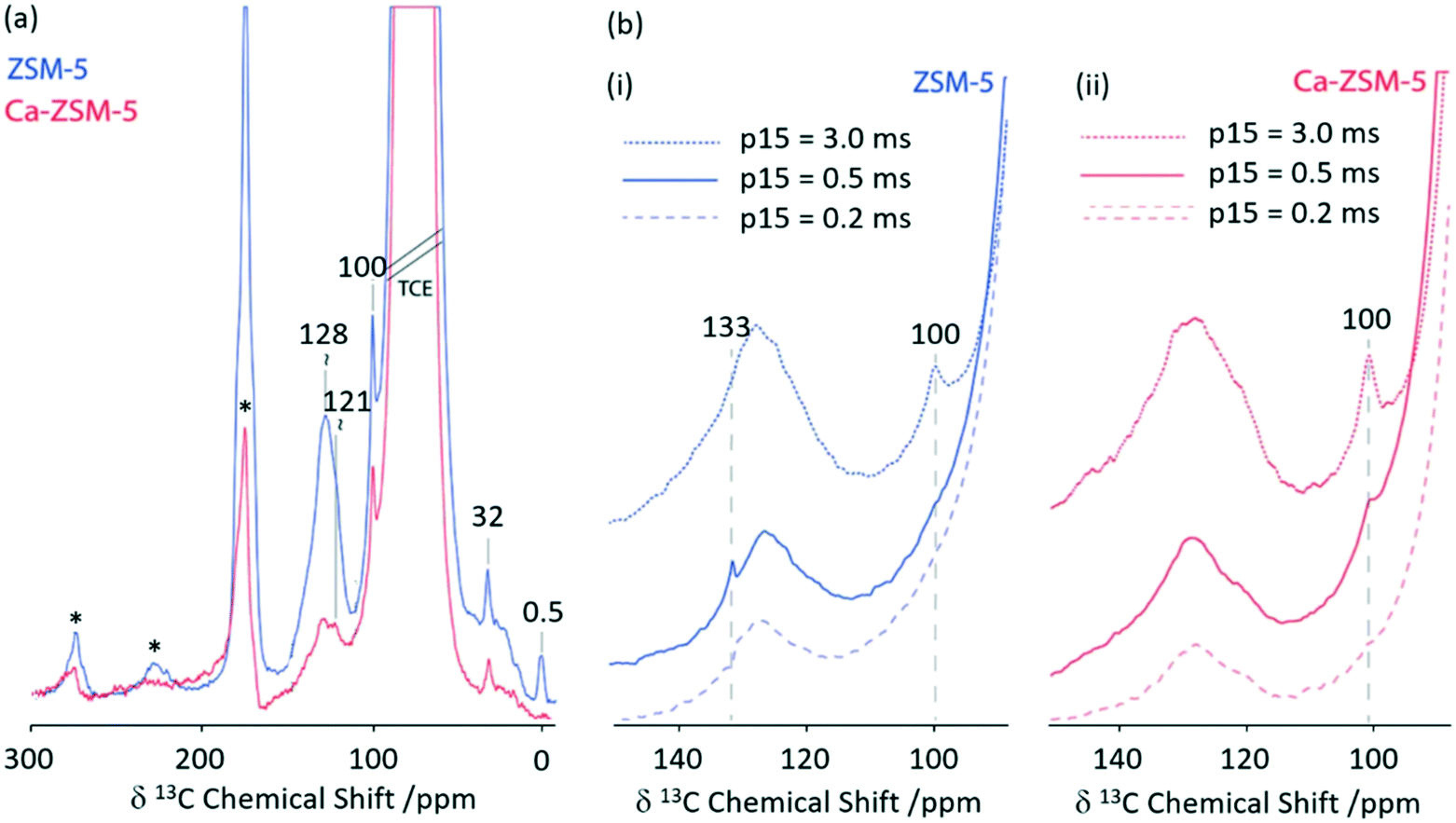 | ||
| Fig. 11 (a) 1D 1H 13C CP MAS DNP spectra of post-MTH reacted H-ZSM-5 (blue) and Ca-ZSM-5 (red) at a contact time of 2 ms. (b) 1D 1H 13C CP MAS DNP spectra at various CP contact times (p15) of (i) H-ZSM-5 (blue) and (ii) Ca-ZSM-5 (red). Samples have been formulated in a 16 mM TEKPol solution70 in 1,1,2,2-tetrachloroethane. Data were obtained with a recycle delay of 3 s and a MAS rate νr = 8 kHz. Ref. 69 – published by the Royal Society of Chemistry. | ||
3.7. Rotational echo double resonance
Driven by the need for high resolution, MAS is used extensively to average out heteronuclear dipolar interactions to zero, however, these interactions provide access to spatial proximities and hence probe host–guest interactions. Their reintroduction, whilst under MAS to still achieve the desired spectral resolution, is often achieved using recoupling pulse sequences so that the dipolar coupling is not averaged out to zero under MAS of which the rotational echo double resonance (REDOR)71 is the archetype. Spin echo experiments with (S0) and without (S′) refocusing pulses are acquired as a function of evolution time and enable access to (recoupled) heteronuclear dipolar couplings and interatomic distances. In particular, REDOR type experiments are prevalent in the study of zeolites due to their catalytic performance often being linked to host–guest interactions.Fig. 12 shows such 29Si–13C REDOR experiments to probe the confined hydrocarbon species in the H-ZSM-5 zeolite framework.72 Fitted REDOR fraction (Fig. 12(b)) returns a 29Si–13C heteronuclear coupling of 80 Hz and an internuclear distance of 4.2 Å which highlights the strong hydrocarbon – zeolite interaction and provide insights into zeolite poisoning by blockage of the channels rather than the acid sites.
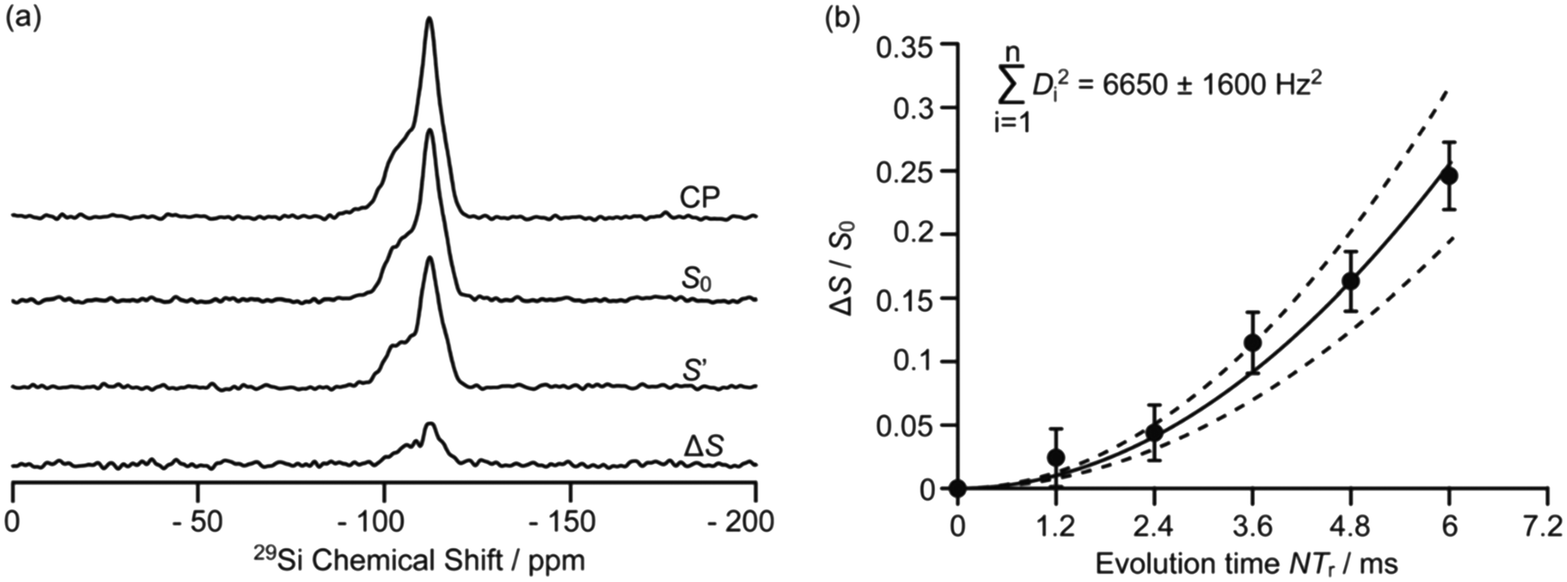 | ||
| Fig. 12 (a) 29Si CP, 29Si CP spin echo (S0), and 29Si {13C} REDOR signal reintroducing dipolar couplings (S′) and the difference ΔS signal (S0 − S′) at a MAS rate of νr = 5 kHz for H-ZSM-5 zeolite. (b) Plot of the REDOR fraction S0 − S′/S0 as a function of evolution time N/νr and corresponding best-fit curve (black line) and fit boundaries (dashed lines). Ref. 72 – published by the Royal Society of Chemistry. | ||
An adaptation of the REDOR experiment to measure dipolar interactions between spin 1/2 and quadrupolar nuclei was introduced in 2010 as symmetry-based resonance-echo saturation-pulse double-resonance (S-RESPDOR)73 and recently used to understand spatial proximities between 13C in the MTH hydrocarbon pools and 27Al in zeolites having different topologies (e.g. H-ZSM-5, HSSZ-13 and H-MOR).74 Comparison of both S0 and S′ RESPDOR signals (Fig. 13) indicate the spatial proximities of methylbenzenes (17.0 and 19.3 ppm), cyclopentenyl cations (25.2, 45.8 and 48.1 ppm), surface bound methanol (58.7 ppm) and DME (60.0 and 63.4 ppm) with a preference for the latter.
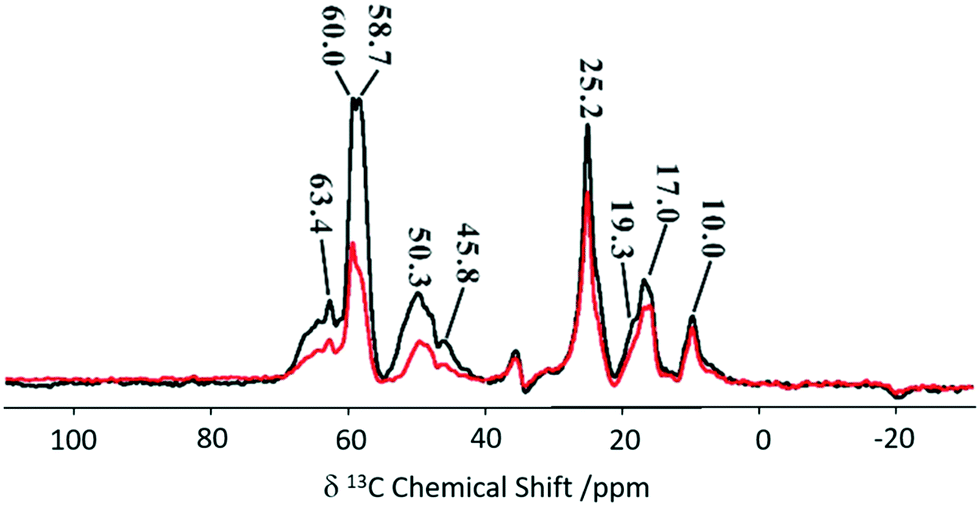 | ||
| Fig. 13 13C spectra observed with (S, black) and without (S0, red) {27Al} S-RESPDOR for the trapped products retained in H-ZSM-5 after reaction with methanol for 15 minutes at 573 K. NMR spectra obtained at a MAS rate of νr = 10 kHz. Reprinted (adapted) with permission from ref. 74. Copyright 2017 American Chemical Society. | ||
Variable temperature experiments in the 300–400 K range showed minimal change in S0 and S′ spectra for the cyclopentyl carbons suggesting the retention of close spatial proximity even at higher temperatures. This is in sharp contrast to the methylbenzene signals where ΔS becomes 0 at higher temperatures due to a reduction in the spatial interaction of methylbenzene enabling the guest to have more mobility within the framework. Comparison between three topologies indicate no significant difference for the cyclic carbocations, however, methylbenzenes present stronger interactions with H-SSZ-13 and H-MOR implying that these two zeolites may have a range of reactivity in the methanol to olefin reaction.
3.8. Separated local field NMR
Separated local field (SLF) NMR experiments correlate heteronuclear dipolar couplings with chemical shifts in two dimensional NMR spectra and provide orientation and distance dependent restraints to aid structure determination, accessing dynamics and probing host–guest interactions. These experiments were initially designed for the study of liquid crystals75,76 and now find widespread use including in supramolecular assemblies. A particular version of SLF is the dipolar chemical shift correlation (DipShift) which uses a principle similar to REDOR that recouple heteronuclear dipolar interactions with refocusing π pulses.13C–1H DipShift NMR was used to obtain the magnitude of the motionally averaged one-bond 13C 1H dipolar couplings and values of −11.1 and −8.7 kHz were obtained for the aromatic carbons of styrene and ethylbenzene loaded on MIL-53(Al), respectively (Fig. 14).77 These values are much slower than the ones expected in the absence of motion (−23 kHz) and determined from CH distances which indicate significant dynamic processes. The lower value obtained for ethylbenzene indicates higher mobility in the MIL-53 pore than for styrene and suggests that this latter aromatic has a stronger host–guest interaction with this MOF. This work has provided direct insights into the structure–selectivity relationship in MIL-53 and has contributed to further understanding of the exceptional property of this MOF to separate both aromatics which is a challenging separation. | ||
| Fig. 14 Schematic of MIL-53(Al) upon adsorption of (a) styrene and (b) ethylbenzene and their corresponding dipolar dephasing curves extracted from 13C–1H DipShift MAS NMR dataset obtained at a MAS rate of νr = 6 kHz. The CH sites at 128 and 127 ppm where chosen to capture the dynamic in styrene and ethylbenzene, respectively. Reprinted from ref. 77: host–guest interaction of styrene and ethylbenzene in MIL-53 studied by solid-state NMR, Copyright 2018, with permission from Elsevier. | ||
4. Conclusion
Solid state NMR approaches that provide detailed insights into host–guest chemistry in supramolecular assemblies have been described. These studies enable understanding of the role of dynamics, binding sites and spatial interactions and often complement other approaches e.g. diffraction-based methods and computational investigations, to deliver comprehensive knowledge. The NMR technology plays an important role in the design and development of materials with new and improved adsorption phenomena for a wide range of applications including separation, catalysis and energy conversion and storage.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the Engineering and Physical Sciences Research Council (EPSRC) for a Doctoral Training Studentship to A. R. H. is acknowledged.References
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Chichester, UK, 2nd edn, 2009 Search PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1112 CrossRef.
- J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- Y. Xu, S. A. Southern, P. M. J. Szell and D. L. Bryce, CrystEngComm, 2016, 18, 5236–5252 RSC.
- M. R. Chierotti and R. Gobetto, Solid-State NMR Studies on Supramolecular Chemistry, Wiley, Chichester, UK, 2012 Search PubMed.
- M. J. Duer, R. K. Harris and R. E. Wasylishen, NMR Crystallography, Wiley, Chichester, 2009 Search PubMed.
- M. J. Potrzebowski and S. Kazmierski, Top. Curr. Chem., 2004, 246, 91–140 CrossRef PubMed.
- M. Novo, D. Granadero and J. B. W. Al-soufi, J. Inclusion Phenom. Macrocyclic Chem., 2011, 70, 259–268 CrossRef CAS.
- F. P. Schmidtchen, in Analytical Methods in Supramolecular Chemistry, Wiley, Weinheim, 2nd edn, 2012, pp. 67–104 Search PubMed.
- K. Jie, M. Liu, Y. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921–6930 CrossRef CAS PubMed.
- S. Halder, A. Barma, C. Rizzoli, P. Ghosh and P. Roy, ACS Appl. Nano Mater., 2019, 2, 5469–5474 CrossRef CAS.
- S. E. Ashbrook and D. M. Dawson, Acc. Chem. Res., 2013, 46, 1964–1974 CrossRef CAS PubMed.
- J. A. Ripmeester and R. E. Wasylishen, CrystEngComm, 2013, 15, 8598 RSC.
- B. Elena and L. Emsley, J. Am. Chem. Soc., 2005, 127, 9140–9146 CrossRef CAS PubMed.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid-State NMR: Basic Principles and Practice, Momentum Press, New York, 1st edn, 2012 Search PubMed.
- R. W. Martin, J. E. Kelly and J. I. Kelz, J. Struct. Biol., 2019, 206, 73–89 CrossRef CAS PubMed.
- P. Hodgkinson and S. Wimperis, Phys. Chem. Chem. Phys., 2009, 11, 6875 RSC.
- A. W. Overhauser, Phys. Rev., 1953, 92, 411–415 CrossRef CAS.
- T. R. Carver and C. P. Slichter, Phys. Rev., 1956, 102, 975–981 CrossRef CAS.
- A. S. L. Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102–103, 120–195 CrossRef PubMed.
- A. Pines, M. G. Gibby and J. S. Waugh, J. Chem. Phys., 1973, 59, 569 CrossRef CAS.
- E. R. Andrew, A. Bradbury and R. G. Eades, Nature, 1958, 182, 1659 CrossRef CAS.
- A. Brinkmann and M. H. Levitt, J. Chem. Phys., 2001, 115, 357–384 CrossRef CAS.
- S. E. Ashbrook, R. G. Griffin and K. E. Johnston, Annu. Rev. Anal. Chem., 2018, 11, 485–508 CrossRef PubMed.
- A. D. Buckingham, Can. J. Chem., 1960, 38, 300–307 CrossRef CAS.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS PubMed.
- D. H. Brouwer, S. Alavi and J. A. Ripmeester, Phys. Chem. Chem. Phys., 2008, 10, 3857–3860 RSC.
- N. Asakawa, S. Kuroki, H. Kurosu, I. Ando, A. Shoji and T. Ozaki, J. Am. Chem. Soc., 1992, 114, 3261–3265 CrossRef CAS.
- A. Newman, Pharmaceutical Amorphous Solid Dispersions, John Wiley & Sons, Hoboken, New Jersey, 1st edn, 2015 Search PubMed.
- M. Teodorescu and M. Bercea, Polym.-Plast. Technol. Eng., 2015, 54, 923–943 CrossRef CAS.
- K. Ueda, K. Higashi and K. Moribe, Mol. Pharmaceutics, 2016, 13, 852–862 CrossRef CAS PubMed.
- D. C. Apperley, A. H. Forster, R. Fournier, R. K. Harris, P. Hodgkinson, R. W. Lancaster and T. Rades, Magn. Reson. Chem., 2005, 43, 881–892 CrossRef CAS PubMed.
- X. Yuan, D. Sperger and E. J. Munson, Mol. Pharmaceutics, 2014, 11, 329–337 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- L. A. O'Dell and C. I. Ratcliffe, in NMR of Quadrupolar Nuclei in Solid Materials, John Wiley & Sons, Chichester, 1st edn, 2012, pp. 213–232 Search PubMed.
- L. S. Batchelder, in Encyclopedia of Nuclear Magnetic Resonance, ed. R. K. Harris and R. E. Wasylishen, John Wiley & Sons, Chichester, 1st edn, 2007 Search PubMed.
- R. L. Vold and G. L. Hoatson, J. Magn. Reson., 2009, 198, 57–72 CrossRef CAS PubMed.
- R. R. Vold and R. L. Vold, Advances in Magnetic and Optical Resonance, Academic Press, San Diego, 1991 Search PubMed.
- V. Macho, L. Brombacher and H. W. Spiess, Appl. Magn. Reson., 2001, 20, 405–432 CrossRef CAS.
- S. Bracco, F. Castiglioni, A. Comotti, S. Galli, M. Negroni, A. Maspero and P. Sozzani, Chem. – Eur. J., 2017, 23, 11210–11215 CrossRef CAS PubMed.
- S. L. Gould, D. Tranchemontagne, O. M. Yaghi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2008, 130, 3246–3247 CrossRef CAS PubMed.
- S. Bracco, A. Comotti, P. Valsesia, B. F. Chmelka and P. Sozzani, Chem. Commun., 2008, 4798–4800 RSC.
- A. R. Hughes, N. J. Brownbill, R. C. Lalek, M. E. Briggs, A. G. Slater, A. I. Cooper and F. Blanc, Chem. – Eur. J., 2017, 23, 17217–17221 CrossRef CAS PubMed.
- A. G. Slater, M. A. Little, A. Pulido, S. Y. Chong, D. Holden, L. Chen, C. Morgan, X. Wu, G. Cheng, R. Clowes, M. E. Briggs, T. Hasell, K. E. Jelfs, G. M. Day and A. I. Cooper, Nat. Chem., 2017, 9, 17–25 CrossRef CAS PubMed.
- T. Matsuno, M. Fujita, K. Fukunaga, S. Sato and H. Isobe, Nat. Commun., 2018, 9, 3779 CrossRef.
- V. J. Witherspoon, J. Xu and J. A. Reimer, Chem. Rev., 2018, 118, 10033–10048 CrossRef CAS PubMed.
- A. C. Forse, M. I. Gonzalez, R. L. Siegelman, V. J. Witherspoon, S. Jawahery, R. Mercado, P. J. Milner, J. D. Martell, B. Smit, B. Blümich, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 1663–1673 CrossRef CAS PubMed.
- R. M. Marti, J. D. Howe, C. R. Morelock, M. S. Conradi, K. S. Walton, D. S. Sholl and S. E. Hayes, J. Phys. Chem. C, 2017, 121, 25778–25787 CrossRef CAS.
- Y. Zhang, B. E. G. Lucier and Y. Huang, Phys. Chem. Chem. Phys., 2016, 18, 8327–8341 RSC.
- M. Inukai, T. Kurihara, Y. Noda, W. Jiang, K. Takegoshi, N. Ogiwara, H. Kitagawa and K. Nakamura, Phys. Chem. Chem. Phys., 2020, 22, 14465–14470 RSC.
- L. C. Lin, J. Kim, X. Kong, E. Scott, T. M. McDonald, J. R. Long, J. A. Reimer and B. Smit, Angew. Chem., Int. Ed., 2013, 52, 4410–4413 CrossRef CAS PubMed.
- J. Li, S. Li, A. Zheng, X. Liu, N. Yu and F. Deng, J. Phys. Chem. C, 2017, 121, 14261–14268 CrossRef CAS.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2009, 131, 4995–5000 CrossRef CAS PubMed.
- E. Weiland, M. A. Springuel-Huet, A. Nossov and A. Gédéon, Microporous Mesoporous Mater., 2016, 225, 41–65 CrossRef CAS.
- P. Håkansson, M. A. Javed, S. Komulainen, L. Chen, D. Holden, T. Hasell, A. Cooper, P. Lantto and V. V. Telkki, Phys. Chem. Chem. Phys., 2019, 21, 24373–24382 RSC.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- J. Xu, Q. Wang, S. Li and F. Deng, Solid-State NMR in Zeolite Catalysis, Springer Nature, Singapore, 1st edn, 2019 Search PubMed.
- Z. Zhao, X. Li, S. Li, S. Xu, X. Bao, Y. Bilge, P. Andrei-Nicolae, M. Ulrich and W. Zhang, Microporous Mesoporous Mater., 2019, 288, 109555 CrossRef CAS.
- S. Komulainen, J. Roukala, V. V. Zhivonitko, M. A. Javed, L. Chen, D. Holden, T. Hasell, A. Cooper, P. Lantto and V. V. Telkki, Chem. Sci., 2017, 8, 5721–5727 RSC.
- R. Giovine, C. Volkringer, M. A. Springuel-Huet, A. Nossov, F. Blanc, J. Trébosc, T. Loiseau, J. P. Amoureux, O. Lafon and F. Pourpoint, J. Phys. Chem. C, 2017, 121, 19262–19268 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- I. Yarulina, S. Bailleul, A. Pustovarenko, J. R. Martinez, K. De Wispelaere, J. Hajek, B. M. Weckhuysen, K. Houben, M. Baldus, V. Van Speybroeck, F. Kapteijn and J. Gascon, ChemCatChem, 2016, 8, 3057–3063 CrossRef CAS.
- A. D. Chowdhury, I. Yarulina, E. Abou-hamad, A. Gurinov and J. Gascon, Chem. Sci., 2019, 10, 8946–8954 RSC.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196–200 CAS.
- D. Xiao, S. Xu, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2017, 8, 8309–8314 RSC.
- L. Chen, Q. Wang, B. Hu, O. Lafon, J. Trébosc, F. Deng and J. P. Amoureux, Phys. Chem. Chem. Phys., 2010, 12, 9395–9405 RSC.
- C. Wang, J. Xu, Q. Wang, X. Zhou, G. Qi, N. Feng, X. Liu, X. Meng, F. Xiao and F. Deng, ACS Catal., 2017, 7, 6094–6103 CrossRef CAS.
- V. Domenici, Liq. Cryst. Today, 2017, 26, 2–10 CrossRef.
- S. V. Dvinskikh, in Thermotropic Liquid Crystals, ed. A. Ramamoorthy, Springer, Netherlands, Dordrecht, 2007, pp. 117–140 Search PubMed.
- S. Li, J. Li, J. Tang and F. Deng, Solid State Nucl. Magn. Reson., 2018, 90, 1–6 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |