
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in photochemical and electrochemical incorporation of sulfur dioxide for the synthesis of value-added compounds
Stephan P.
Blum†
a,
Kamil
Hofman†
b,
Georg
Manolikakes
 *b and
Siegfried R.
Waldvogel
*a
*b and
Siegfried R.
Waldvogel
*a
aDepartment of Chemistry, Johannes Gutenberg University Mainz, D-55128 Mainz, Germany. E-mail: waldvogel@uni-mainz.de; Web: https://www.aksw.uni-mainz.de/
bDepartment of Chemistry, Technical University Kaiserslautern, D-67663 Kaiserslautern, Germany. E-mail: manolikakes@chemie.uni-kl.de; Web: https://www.chemie.uni-kl.de/en/manolikakes/
First published on 16th July 2021
Abstract
Organic photochemistry and electrochemistry currently receive tremendous attention in organic synthesis as both techniques enable the reagent-less activation of organic molecules without using expensive and hazardous redox reagents. The incorporation of SO2 into organic molecules is a relatively modern research topic, which likewise gains immense popularity since the discovery of the SO2 surrogate DABSO. Sulfur-containing organic molecules are omnipresent in pharmaceuticals and agrochemicals. This review covers the recent progress in electrochemical and photochemical methodologies for the incorporation and uses of SO2 in the synthesis of value-added compounds. Additionally, different work techniques are demonstrated for the synthetic application of SO2.
 Stephan P. Blum | Stephan P. Blum received his BSc and MSc degrees from the Ludwig-Maximilians University of Munich. During his master studies, he participated in a student exchange at the City University of Hong Kong and completed an industrial internship at Boehringer Ingelheim in medicinal chemistry. His master thesis project was in organometallic chemistry supervised by Josep Cornellà at Max Planck Institute for Coal Research. In 2019, he started his PhD project on the electrochemical upcycling of sulfur dioxide to value-added products under the supervision of Prof. Dr Siegfried R. Waldvogel at Johannes Gutenberg University Mainz. |
 Kamil Hofman | Kamil Hofman was born in Frankfurt/Main (Germany) in 1990. He graduated with BSc and MSc degrees in chemistry from the Goethe-University Frankfurt (Germany). Since 2017, he has been working on his PhD thesis in the group of Georg Manolikakes. His current field of research focuses on novel multi-component reactions for the synthesis of sulfonamides. |
 Georg Manolikakes | Georg Manolikakes studied chemistry at the Ludwig-Maximilians University (LMU) Munich. At the LMU, he joined the group of Prof. Paul Knochel and received his PhD in 2009 in the field of organometallic chemistry. After a postdoctoral stay with Prof. Phil S. Baran at the Scripps Research Institute, he started his independent career at the Goethe-University Frankfurt in 2010. In 2017, he was appointed as an associate professor at the Technical University Kaiserslautern. His research interests cover multi-component and one-pot reactions, the synthesis of sulfonyl-containing molecules, asymmetric synthesis and medicinal chemistry. |
 Siegfried R. Waldvogel | Siegfried R. Waldvogel studied chemistry in Konstanz and received his PhD in 1996 from University of Bochum/Max Planck Institute for Coal Research with Prof. Dr M. T. Reetz as supervisor. After Postdoctoral research at Scripps Research Institute in La Jolla, CA (Prof. Dr J. Rebek, Jr), he started his own research career in 1998 at the University of Münster. After his professorship in 2004 at the University of Bonn, he became a full professor for organic chemistry at Johannes Gutenberg University Mainz in 2010. His research interests are novel electro-organic transformations including bio-based feedstocks from electrosynthetic screening to scale-up. In 2018, he cofounded ESy-Labs GmbH, which provides custom electrosynthesis and contract R&D services. |
Introduction
Reagent-less activation of organic molecules became increasingly popular in the past few decades. Photochemistry and the 21st century technique organic electrochemistry in particular have attracted growing interest,1,2 which is attributed to numerous advantages in comparison to conventional synthetic approaches with expensive and hazardous redox reagents.3 Inexpensive electricity or light,2 derived from renewable energy sources,4 is capable of activating organic molecules and beyond that achieving even novel and highly innovative reactivities.5 From this follows waste reduction,6 which is crucial during times of climate change,1 and a decrease of synthetic steps for the target molecule.7 Additionally, photochemistry and electrochemistry increase the safety as the reaction can be easily interrupted by switching off the power and the reactions are mostly carried out under mild reaction conditions.8 Overall, applying photochemistry9 and electrochemistry10 enables a more sustainable or “green” synthesis11 of organic chemicals.The feedstock chemical sulfur dioxide (SO2) is annually generated in vast amounts by the incineration of sulfur and sulfur-containing waste, roasting of metal sulfides and subsequent washing of flue gases.12,13 In the food industry, SO2 is widely used as a preservative (E220) for nutritional products and beverages such as wines and dried fruits. Indeed, its preserving effect has already been utilized in the ancient world, e.g., by the incineration of sulfur in wine vessels.13,14 SO2 is utilized as a bleaching agent for paper and cloth, and in gypsum production, corn processing, water and waste treatment, ore refining, oil extraction, metal and glass processes, and sulfonylation of oils and aromatic compounds.13,15 However, the majority of the SO2 produced annually is used for the synthesis of sulfuric acid production by the contact process, which has tremendous importance in the chemical industry.16 In nature, SO2 is released by volcanic eruptions. In the atmosphere, it is present in one part by parts per billion per volume (1 ppbv). Atmospheric SO2 mainly stems from the combustion of sulfur-containing fossil fuels and is one of the major air pollutants with severe negative impacts on health and the environment.17–19 Atmospheric oxidation of SO2 to sulfuric acid is responsible for the formation of acid rain.20 In industrialized countries, SO2 emissions have been constantly reduced in the past few decades, whereas in developing countries, these emissions have currently increased significantly. Flue gas desulfurization (FGD) is commonly used for SO2 removal from industrial exhausts.17 Different processes, such as the wet sulfuric acid process (WSA), enable sulfur recovery from flue gas in the form of sulfuric acid.21 The Wellman–Lord process recovers SO2 from flue gas upon reaction with Na2SO3 to obtain the SO2 surrogate Na2S2O5.12 From exhaust gases in water, collected SO2 gas can be oxidized to H2SO4 by Br2 leading to the formation of HBr. The latter can be recycled to Br2 electrochemically. Alternatively, SO2 can be oxidized electrocatalytically to SO3 on Co phthalocyanines with gas diffusion electrodes.22
Currently, there is intense research interest in sulfur chemistry and the direct incorporation of SO2 into organic molecules,18,23–35 which originates from the broad bioactivity of sulfur-containing molecules making them extraordinarily interesting in drug discovery, agrochemicals, and medicinal chemistry.24,36 Among the different approaches, radical processes are especially well-suited for the direct fixation of SO2. Additionally, SO2 is a very good trap for all kinds of carbon-based radicals and thereby the formed sulfonyl radicals are highly versatile intermediates for the construction of all kinds of SO2 functionalities. As carbon-based radicals are commonly prepared from suitable precursors via single-electron-transfer (SET) processes,26,33 photochemistry and electrochemistry are obvious choices for new SO2 binding procedures. In the past few years, various photochemical methodologies for the fixation of SO2 have been reported.27,29 Surprisingly, this trend has not reached organic electrosynthesis yet. In this review, we give an overview of different working techniques for using SO2 and the recent advances in its application in the electrochemical and photochemical preparation of organic products.
Working techniques for SO2
SO2 is a colorless, toxic, noxious and corrosive gas (boiling point: −10 °C; melting point: −75.5 °C). Due to its low boiling point, SO2 can be easily liquified. Interestingly, SO2 has a low vapor pressure (3.3 bar at 20 °C) and a high enthalpy of vaporization (5960 cal mol−1 (∼24.9 kJ mol−1) at approx. −10 °C).13,37,38 Therefore, it is not only simple to liquify SO2, but also to keep it in its liquid state, either by cooling or using pressure vessels. Indeed, SO2 is well-known as a dipolar aprotic solvent in organic synthesis.15,37,39 Liquid SO2 readily dissolves organic and inorganic salts due to its high dipole moment of 1.61 D with a dielectric constant that varies from 15.6 at 0 °C to 17.3 at −30 °C. At low temperatures, liquified SO2 can be safely transferred using precooled syringes. Additionally, this gas is easy to recover/recycle. SO2 is commercially available. The cost of high-purity SO2 gas (99.98%; water content: ≤50 ppm; costs: ∼5 € per kg) is relatively low compared to many other organic solvents at the same level of purity, such as acetonitrile (MeCN), dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF) or tetrahydrofuran (THF).13,37Beyond that, SO2 proved to be an excellent solvent in electrochemical oxidation studies as a molecule with pronounced low nucleophilicity, and it poorly solvates cations. It is reported to offer the widest known anodic regime for the electrochemical studies of highly oxidized species.40,41 The potential window of SO2 with the supporting electrolyte CsAsF6 is displayed in Fig. 1,40 which indicates that SO2 is hard to oxidize but facile to reduce to SO2 anion radicals. However, as a toxic, foul-smelling and corrosive gas, the use of SO2 in a typical academic laboratory setup is associated with safety issues and the necessity for specialized equipment. For this reason, various solid, easy-to-handle SO2 surrogates have been introduced (Fig. 2). The most prominent example is the SO2 surrogate DABSO (1,4-diazabicyclo[2.2.2]octane-bis(sulfur dioxide) adduct), which has led to a significant boost in organic SO2 chemistry in the past decade.18,31,32,42 However, it has certain drawbacks such as the high cost (Sigma Aldrich: 31.20 € per g)43 or significantly reduced overall atom economy.44 Other rather inexpensive SO2 surrogates are sodium and potassium metabisulfite. In particular, K2S2O5 has been applied in various transformations.30,45 However, solubility issues occur in organic solvents so that the application of inorganic SO2 surrogates is rather limited and elevated temperatures are mostly required.30 Just recently, a novel surrogate (SOgen) has been designed, which is based on the Diels–Alder reaction of 4-methylstyrene, and 1.2 eq. of SOgen (Fig. 2) generates only 1 eq. of SO2ex situ in a two-compartment system at elevated temperatures.25 Although this system has been well-proven in various transformations, its atom economy is very low. Alternatively, SO2 can be generated ex situ from Na2SO3 and sulfuric acid with a subsequent introduction of gas into the reaction mixture.46
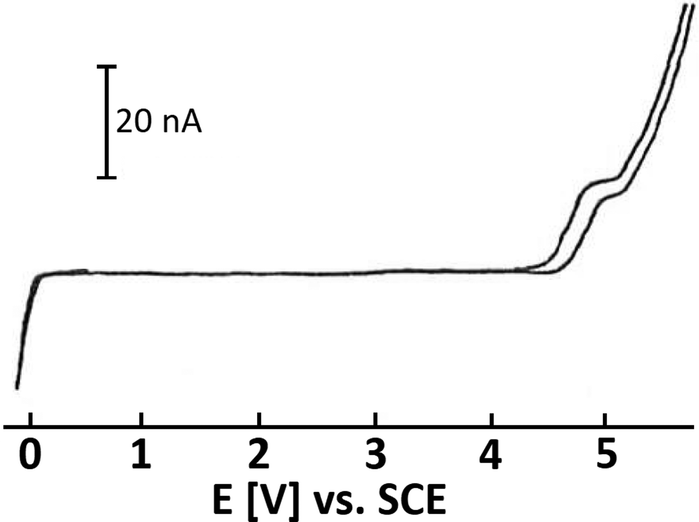 | ||
| Fig. 1 Cyclic voltammogram of liquid SO2 and CsAsF6 (∼4 mM); conditions: Pt working electrode, T = 203 K; SCE = saturated calomel electrode. Figure adopted from Bard (refurbished).40 Depicted upon permission from John Wiley & Sons. | ||
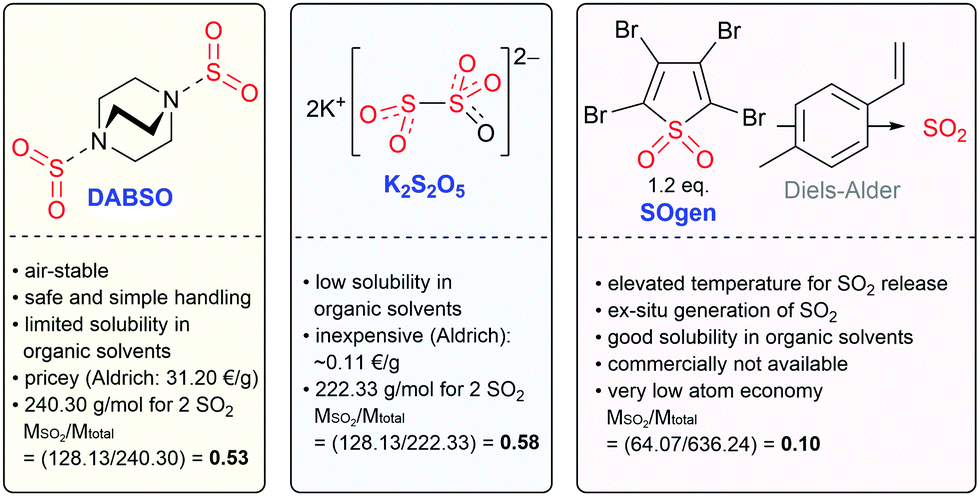 | ||
| Fig. 2 Selection of SO2 surrogates in organic synthesis.25,30,31,43 | ||
However, this technique seems rather non-practicable and reproducibility might be challenging as the SO2 concentration of the reaction mixture cannot be determined. Other SO2 surrogates are sodium dithionite (Na2S2O4) and thiourea dioxide, which have been used in various transformations in the incorporation of SO2 into organic molecules. Na2S2O4 in solution disproportionates into the SO2 anion radical and thiourea dioxide gives SO22− (upon reaction with hydroxide), both of which are considered as reduced SO2 species.34,35 However, sodium dithionite can be considered as an ex-cell electrochemical product of SO2. Consequently, these surrogates comprise different reactivities in comparison to classical SO2 surrogates shown in Fig. 2, which release gaseous SO2.
Stock solutions provide another opportunity for the safe handling of SO2 in an academic laboratory. SO2 readily dissolves in various organic solvents47 and some stock solutions are even commercially available (SO2 in THF and SO2 in CH2Cl2).48 However, THF is not the solvent of choice in electrochemical reactions due to its low oxidation potential.49 The most prominent example of SO2 stock solutions is arguably the Karl–Fischer reagent, a 15–20% solution of SO2 in MeOH (albeit also containing pyridine as the base).50 Although DABSO has been successfully employed in various photochemical processes, it is unfortunately unsuitable for electrosynthesis due to its quite low oxidation potential for 1,4-diazabicyclo[2.2.2]octan (DABCO), which will lead to competitive oxidation pathways in anodic processes.51 In photochemistry, the limited solubility of DABSO in organic solvents can cause further challenges, as the resulting suspensions drastically interfere with irradiation (scattering, absorption, etc.). Such stock solutions in dipolar aprotic solvents (e.g. MeCN) provide a superior alternative, which additionally drastically increases the atom economy of the desired reaction. The SO2 concentration can be easily determined by iodometry52 and the handling proved to be simple and safe.44,53,54 For instance, such stock solutions have been used in MeCN, DMSO or DMF in cathodic SO2 reduction studies or the synthesis of sulfones from alkyl halides.55–60 It was determined that a 6 M saturation of SO2 in MeCN/0.1 M NEt4Br at room temperature can be reached (Fig. 3, NEt4Br was used as the supporting electrolyte and can be added optionally).60 For simple and safe handling of these stock solutions, Waldvogel et al. recommended maintaining the SO2 concentration in acetonitrile below the saturation point (for example, 3–4 M in pure MeCN) and storing the stock solution at around +4 °C.44
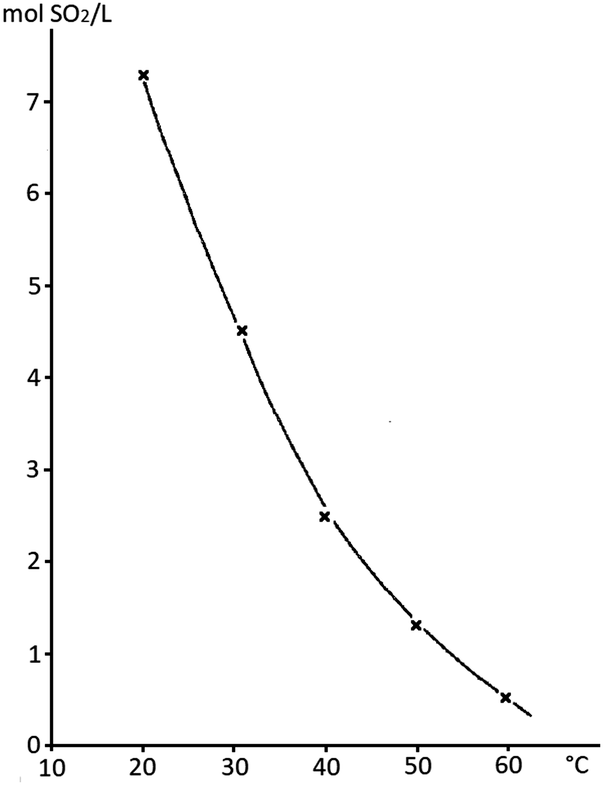 | ||
| Fig. 3 Saturation points of SO2 in mol SO2/L at different temperatures. System: 0.1 M NEt4Br in acetonitrile. Figure adopted from Knittel (refurbished).60 Depicted upon permission from Springer Nature. | ||
Factors increasing the solubility of SO2 in organic solvents are the high basicity and polarity of the medium of choice. However, the cohesion effects of the solvent lower the dissolution of SO2 due to energy consumption in forming a void within the liquid. The formation of donor–acceptor complexes is decisive for the absorption of SO2 in organic solvents. Surprisingly, aromatic hydrocarbons comprise relatively high dissolution of SO2 possibly due to the formation of complexes between the electron acceptor SO2 and the π-electron system.47,61
Overall, different alternatives for the utilization of SO2 in organic synthesis exist. The direct use of SO2 gas itself is straightforward and 100% atom-economic. Although safe handling of SO2 gas should not pose any problem for a trained chemist, issues and concerns associated with this toxic and corrosive gas can impede its use in a typical academic or medicinal chemistry laboratory. In such cases, solid, bench-stable SO2 surrogates provide a safe and easy-to-handle alternative, albeit connected with a lower atom-economy. In addition, these surrogates can contain additional components, which might interfere with the desired process. Stock solutions of SO2 in typical organic solvents, preferably available from commercial suppliers, can offer a good balance between atom-economy and safety. However, in the end, one has to evaluate and balance all factors and risks. The ideal reagent for a safe and efficient introduction of SO2 will always depend on the envisioned transformation, the available experimental setup and also the scale of the planned process.
Electrochemistry – anodic reactions with SO2
Electrochemistry with SO2 can be divided into anodic and cathodic processes. Cathodic reductions feature the formation of SO2 anion radicals59,62–65 and the synthetic exploitation thereof. Anodic processes imply the incorporation of SO2 into electrochemically activated organic molecules as oxidation of SO2 is practically impossible. However, such conversions have not been the center of attention so far. This could be due to the fact that the cathodically generated SO2 anion radicals can interfere in the anodic process due to the possible reoxidation and interference of this species or the consumption of SO2 in an undivided cell. Waldvogel and coworkers reported the first anodic reaction with SO2 as the reactant in the electrochemical synthesis of alkyl arylsulfonates and sulfonamides (Scheme 1). Both chemical motifs are highly based on bioactive molecules and therefore have tremendous importance in medicinal chemistry. Initially, the electrolysis was conducted in undivided cells but the application of the divided cells resulted in higher overall yields. SO2 was used from a stock solution in acetonitrile, which significantly increases the atom economy. Further highlights are the mild reaction conditions and the requirement of not needing any additional supporting electrolytes (Scheme 1A and B). Various electron-rich arenes were successfully converted with yields up to 73% (1) for the sulfonate esters and yields up to 85% (5) for the sulfonamides (Scheme 1C and D). Primary and secondary alcohols (2, 41%) as well as fluoroalcohols (3, 33%) are eligible for this protocol. Secondary and primary amines gave the corresponding sulfonamides, although primary amines resulted in significantly lower yields (6, 29%). Heterocyclic structures, such as 1,3-benzodioxole (7, 42%), are also suitable for this protocol. Most importantly, halogen substituents are tolerated offering complementarity to transition metal-catalyzed reactions (4, 40%; 8a, 50% and 8b, 20%). The electrochemical synthesis of sulfonamides proved to be scalable as a 13-fold scale-up reaction of 5 resulting in 85% yields, which is slightly higher than that at the smaller scale. The reaction mechanism (Scheme 1E) of both conversions is based on the reaction of a nucleophile (alcohol or amine) with SO2 in the presence of an organic base rendering in the formation of monoalkyl sulfites or amidosulfinates, respectively. Both species provide excellent electrical conductivity and therefore are well suited for electrochemical synthesis. Initial anodic oxidation of the arene forms the corresponding radical cation, with a subsequent nucleophilic attack of the in situ formed monoalkyl sulfite or amidosulfinate. Hydrogen bonding between the O-atom of these species with HFIP is considered to promote the sulfur-directed nucleophilic attack. Finally, a second anodic oxidation step accomplishes the desired product.53,54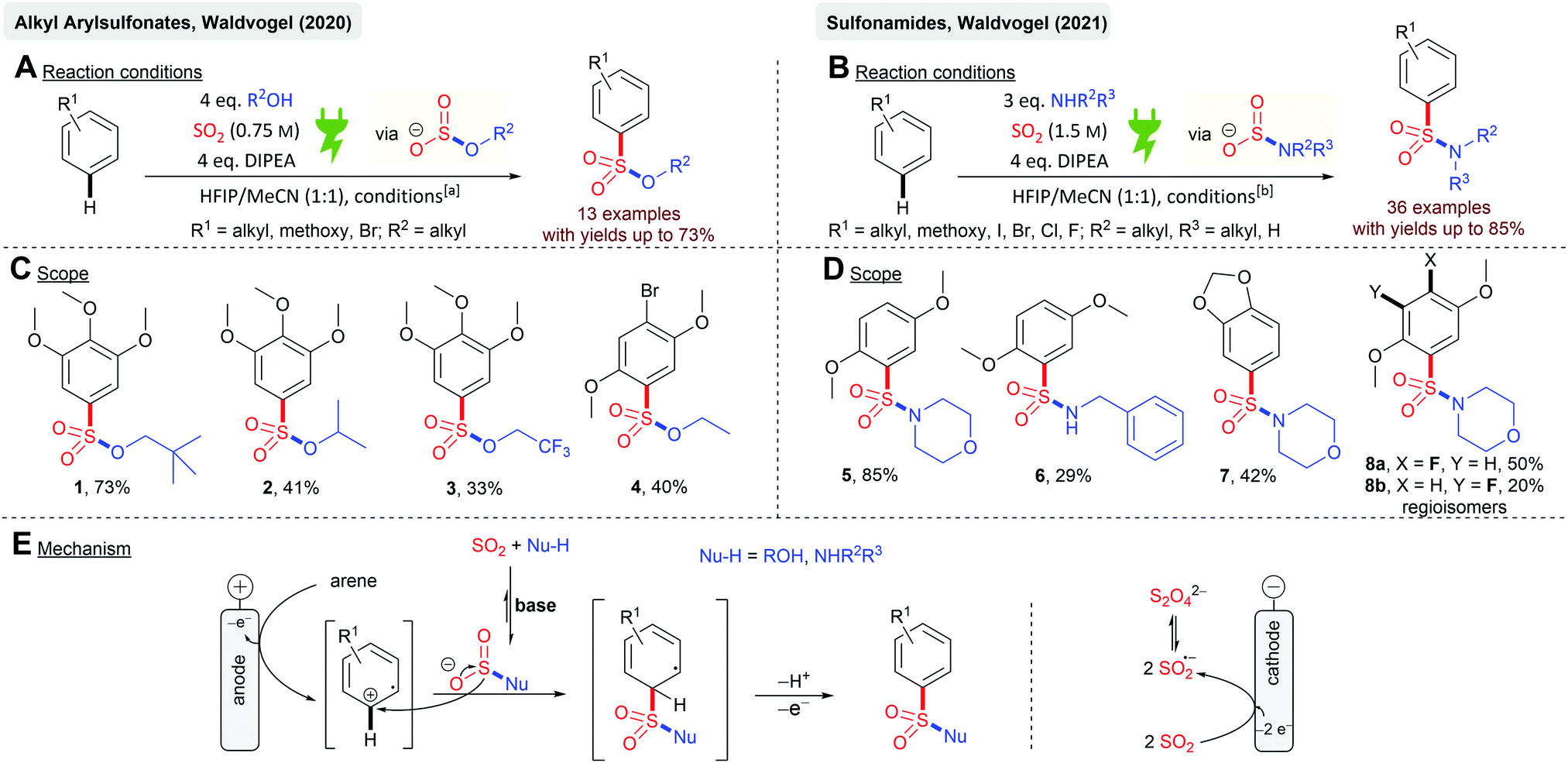 | ||
| Scheme 1 Electrochemical synthesis of alkyl arylsulfonates (left) and electrochemical synthesis of sulfonamides (right); DIPEA = N,N-diisopropylethylamine; HFIP = 1,1,1,3,3,3-hexafluoropropan-2-ol; BDD = boron-doped diamond; [a]: BDD electrodes, r.t., 14 h, 11.25 mA cm−2 (galvanostatic), 3.5 F, divided cell (glass frit); [b]: BDD electrodes, r.t., 14 h, 12 mA cm−2 (galvanostatic), 3.5 F, divided cell (glass frit). | ||
Just recently, the first electrochemical synthesis of sulfamides mediated by catalytic amounts of iodide44 has been reported (Scheme 2) with yields up to 93% (9). Sulfamides are an emerging functionality in drug design and medicinal chemistry due to their versatile biological activities. Platinum electrodes in a divided cell (glass frit) were utilized with SO2 in acetonitrile. Stoichiometric amounts of DIPEA and HFIP are required for the success of this reaction (Scheme 2A). HFIP is considered to weaken the interaction between DIPEA and SO2 as these form charge transfer complexes. The reaction comprises broad functional group tolerance to numerous anilines with excellent yields. For example, the local anesthetic agent benzocaine resulted in 10 in 86% yield (Scheme 2B), but the sterically hindered mesidine gave 11 in moderate yield (52%). The mechanistic concourse (Scheme 2C) is postulated to proceed via the formation of amidosulfinates from the aniline, SO2 and the sterically hindered base DIPEA in an equilibrium reaction. The electrochemically generated iodine is ionized by DIPEA. The subsequent reaction of the amidosulfinate with the in situ generated iodonium ion forms the sulfamoyl iodide, which is most likely stabilized by HFIP by hydrogen bonding. A second nucleophilic attack finally results in the formation of sulfamide. As a cathodic reaction, SO2 reduction to the SO2 anion radical was identified by cyclovoltammetry studies even though platinum electrodes were applied, which actually exhibit a low overvoltage for H+ discharging to hydrogen gas.66
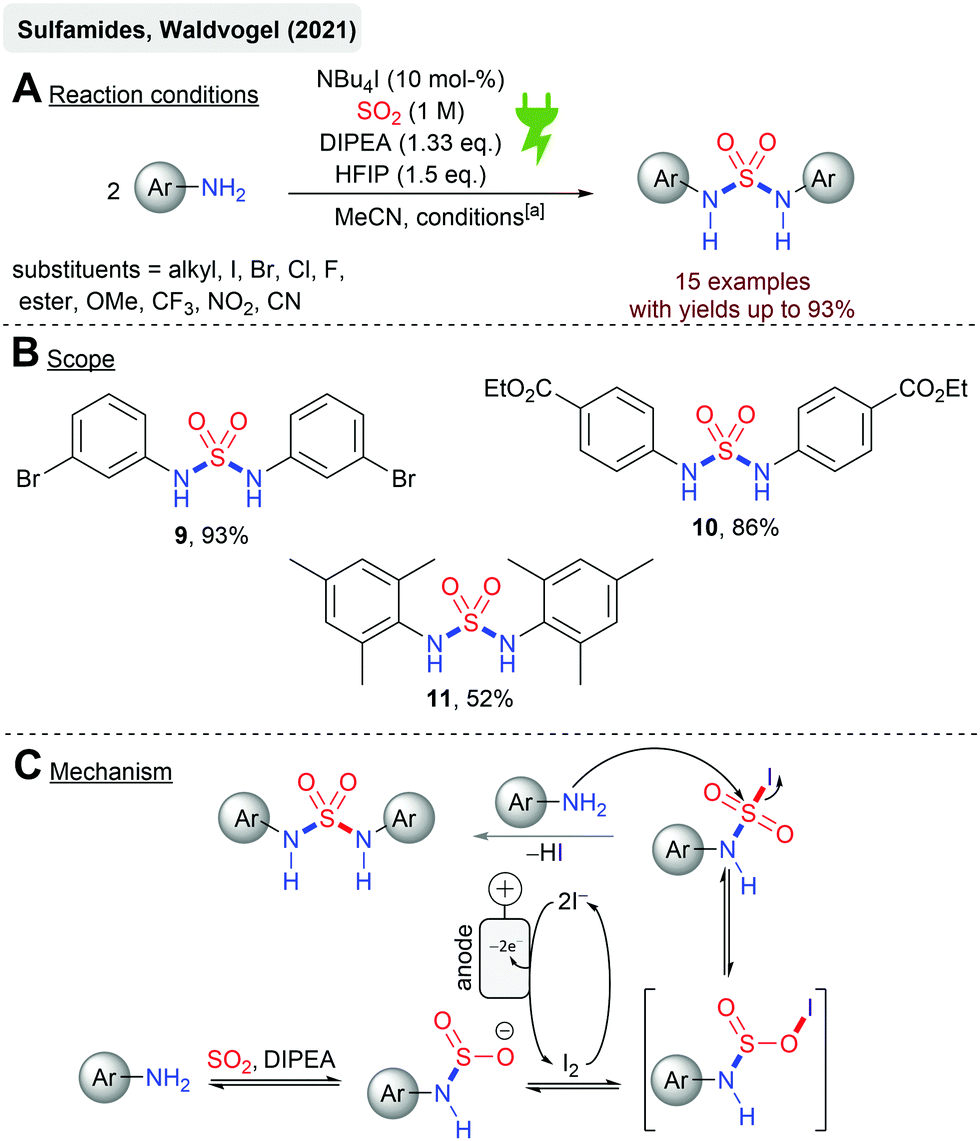 | ||
| Scheme 2 Electrochemical synthesis of symmetric sulfamides; [a]: Pt electrodes, r.t., 14 h, 7.5 mA cm−2 (galvanostatic), 2.5 F, divided cell (glass frit). | ||
Liao and coworkers reported the electrochemical cyclization reaction of N-aryl cyanamides with the terminal alkene to N-sulfonylimines (Scheme 3A) via trifluoromethylation and SO2 insertion by using Langlois’ reagent (CF3SO2Na) as the CF3 and SO2 sources. A series of rather electron-rich N-arylcyanamides were successfully converted to the corresponding products with moderate yields, although electron-withdrawing substituents (CF3, CO2Et) on the arene moiety resulted in no product formation. The highest yields were achieved with the model substrate giving 12 (64%, Scheme 3B). Halogen substituents are tolerated and remarkably, indole derivative 13 was isolated in 47% yield. Finally, several spirocyclic structures were obtained as sole diastereomers (14, 32%) from N-aryl cyanamides containing cyclopentene or cyclohexene moieties. The proposed reaction mechanism (Scheme 3C) suggested the initial anodic oxidation of CF3SO2Na under the cleavage of SO2 to form the CF3 radical, which undergoes radical addition to the terminal alkene. A subsequent intramolecular cyclization renders the iminyl radical, followed by SO2 capture and a second cyclization step on the aromatic ring. A second anodic oxidation step results in the corresponding N-sulfonylimine.67,68 The authors have not investigated this reaction in a divided cell set-up. Eventually, higher yields could have been achieved as this system suggests SO2 reduction as a cathodic side reaction, which could be one reason for the moderate yields.
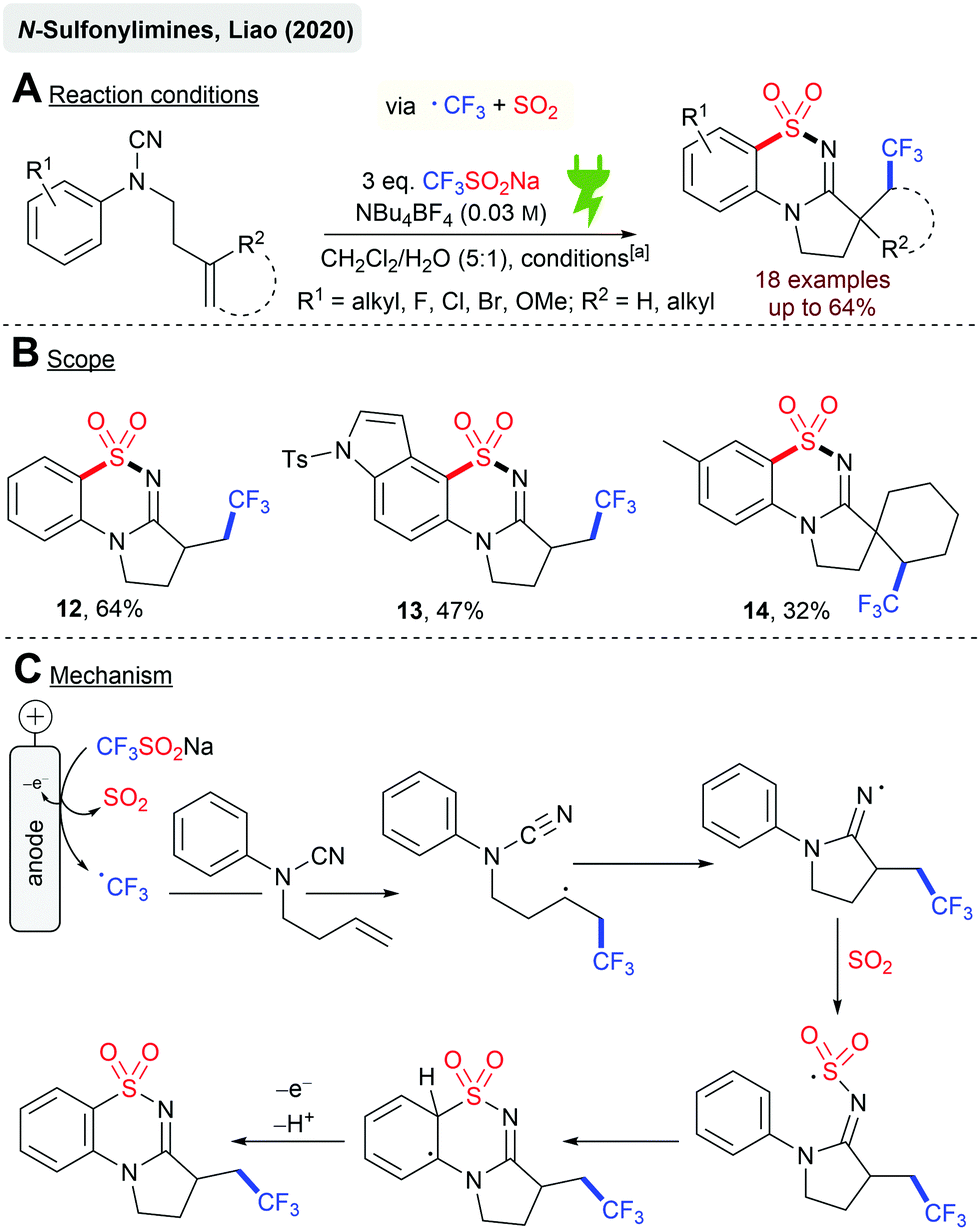 | ||
| Scheme 3 Electroctrochemical synthesis of cyclic N-sulfonylimines; [a]: Cgr||Pt (anode||cathode), 30 °C, 6 h, 2.2–3.7 mA cm−2 (galvanostatic), 2.2–3.7 F, 14 h, undivided cell. | ||
Electrochemistry – cathodic reductions of SO2
The reduction of SO2 in dipolar aprotic solvents (MeCN, DMF, DMSO) to the SO2 radical anion is considered as quite stable, and is well described in the literature.59,62–64 The synthetic exploitation of this electrochemically generated species has been first described by Knittel and coworkers in 1973 in their protocol for the synthesis of symmetric sulfones in the presence of organic halides (Scheme 4). Cyclic voltammetry experiments suggested that the SO2 reduction occurs at −0.7 V vs. Ag/AgCl (in MeCN). A galvanostatic and potentiostatic protocol has been established (Scheme 4A), whereas in the latter cathode, fouling was observed. This problem was avoided under galvanostatic conditions in combination with the replenishing of the reactants periodically. Several alkyl, allyl, benzyl, and acyl halides were successfully transformed to their corresponding sulfone in good yields (Scheme 4B). Diallylsulfone (15) even gave 88% yield. Bromo substituents on aromatic moieties were tolerated in 16 (73%) and remarkably an intramolecular reaction resulted in sultine 17a with sulfone 17b as the side product (71% combined yield). It is noteworthy that previous studies with sodium dithionite (Na2S2O4) and alkyl halides resulted in significantly lower yields (10–15%), which demonstrates that the electrochemical synthesis in this example is superior. Cyclic voltammetry experiments and SO2 anion radical titration experiments (with the alkyl halide) suggest the nucleophilic attack of the SO2 anion radical to the alkyl halide as the rate-determining step resulting in the R–SO2 radical according to the mechanism depicted in Scheme 4C. An electron transfer from another equivalent of the SO2 anion radical renders the sulfinate anion, which undergoes another S-directed nucleophilic substitution reaction with R–X yielding the symmetric sulfone.55,57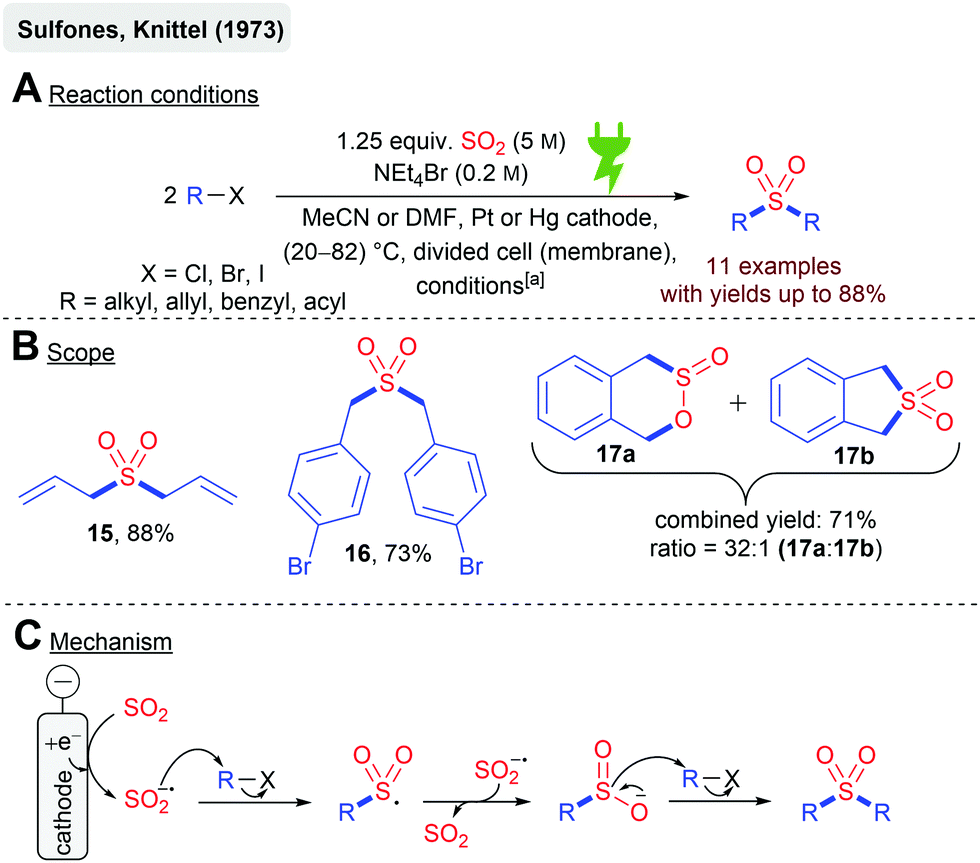 | ||
Scheme 4 Electrochemical synthesis of symmetric sulfones; [a]: potentiostatic conditions: −0.7 V vs. Ag/AgCl, 1 F mol−1 or galvanostatic conditions: 300 mA cm−2 and 500 mA cm−2, 2000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 C. 000 C. | ||
This protocol has been refined in 1980 to a paired electrolysis process with SO2, propyl alcohol and NEt4Br by the generation of propyl bromide in the anodic compartment via the formation of HBr from the electrochemically generated Br2 with SO2 and H2O.58 In 1982, this methodology was further developed by the synthesis of various sulfur-containing heterocycles, such as oxathiolane-, oxathiane-, thiane- and thiepane-oxides upon the reaction of the SO2 anion radical with 1,ω-dihalides.56
Knittel further investigated the chemical behaviour of the electrochemically generated SO2 anion radicals in MeCN or DMF towards reducible substrates (Scheme 5). It is postulated that the SO2 anion radical is in equilibrium with the S2O4 anion radical, whereas the former is the reactive species. Several molecules bearing different reducible functional groups were investigated (Scheme 5B), such as nitrosobenzenes, which resulted in the reduction to the corresponding aniline (18, 58%) in the presence of acetic acid. The author claims that the SO2 anion radical is moderately suitable for the reduction of nitrobenzene, as the hydroxylaminosulfonate intermediate gets formed. Elevated temperatures are required, which is in conflict with the solubility of SO2 in MeCN (Fig. 3). However, aniline (19) was obtained in 63% at 30 °C. The reduction of aldehydes to alcohols is unsuitable due to the formation of hydroxysulfonates (20, 67%). Dehalogenation of α-bromoketones occurred readily (21, 71%) in the presence of H2O.60
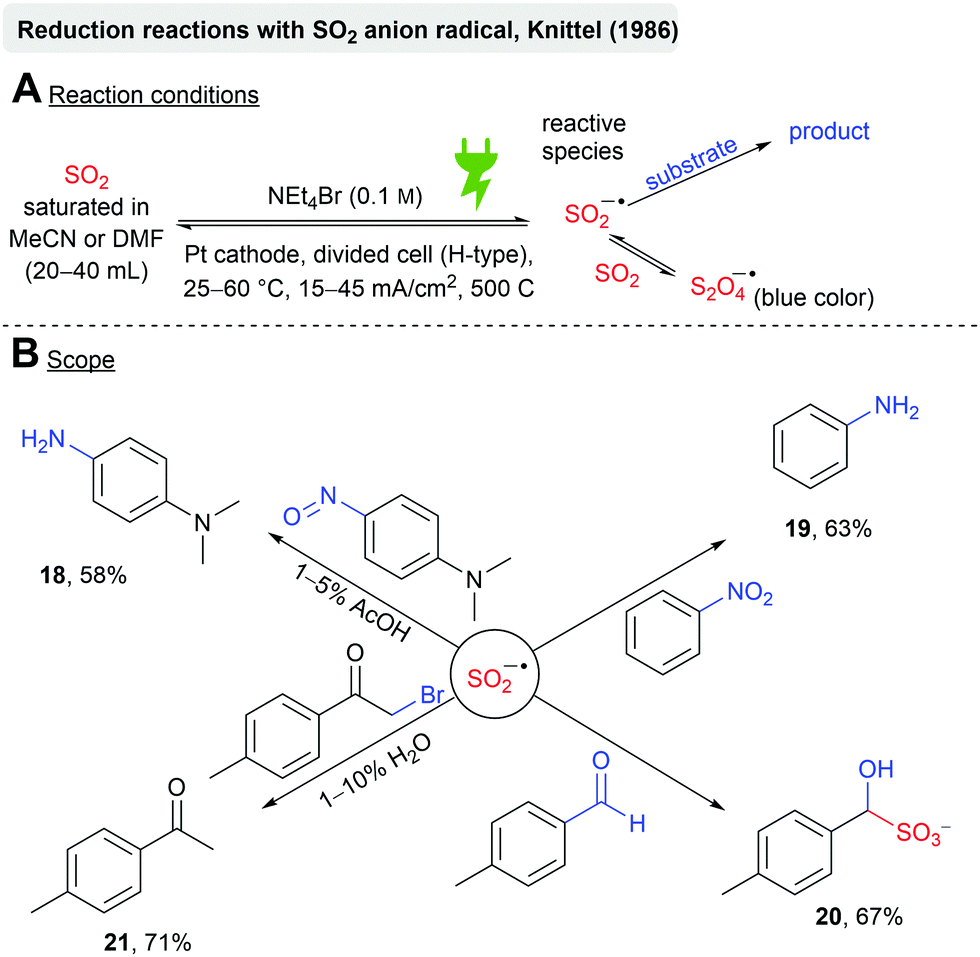 | ||
| Scheme 5 Reactivity of the electrochemically generated SO2 anion radical towards reducible substrates. | ||
In 1995, Koshechko and coworkers reported the electrochemical synthesis of trifluoroalkyl sulfides from CF3Br and several 4-substituted thiophenols in an electrocatalytic fashion (Scheme 6).69 The electrochemical reduction of SO2 to the SO2 anion radical lowers the activation barrier for CF3Br reduction to form the trifluoromethyl radical under bromide abstraction, which has been described earlier.70 Radical addition to the thiolate anion, followed by an electron transfer to SO2, gives the desired product and regenerates the SO2 anion radical. The carbamate substituent resulted in the highest yields (22, 94%), whereas strongly electron-withdrawing substituents, such as the nitro-group, diminished the yield significantly (23, 24%).68,69,71
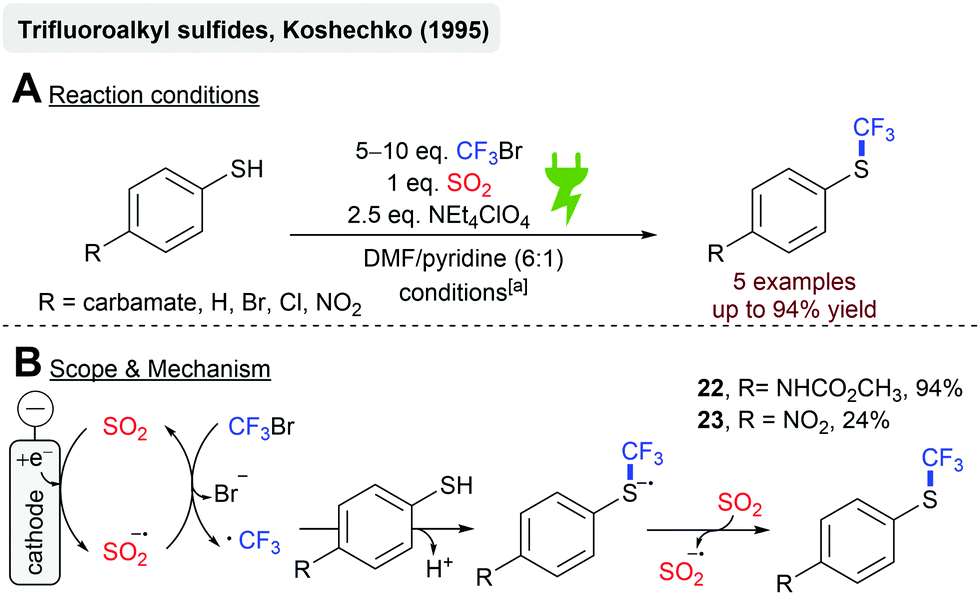 | ||
| Scheme 6 Electrochemical synthesis of trifluoroalkyl sulfides catalyzed by the SO2 anion radical; [a]: r.t., Pt||Pt, divided cell (glass frit), −0.9 to −1.0 V (potentiostatic) vs. Ag/AgNO3, 0.125–1 F. | ||
The electrochemical synthesis of perfluoroalkylsulfinic acids from perfluoroiodoalkanes and SO2 was reported in 1988 by Commeyras and coworkers (Scheme 7A). Cyclic voltammetry studies suggested the simultaneous cathodic reduction of the model substrate C6F13I (−1.38 V vs. SCE) to the corresponding perfluoroalkyl radical and the reduction of SO2 (−1.4 V vs. SCE) to the SO2 anion radical with a subsequent radical recombination (Scheme 7B). Subsequent acidic work-up gives the perfluoroalkysulfinic acids 24 (from C6F13I) and 25 (from I(CF2)4I) with 95% calculated NMR yield, respectively. Lower water content resulted in the formation of carbonic acids upon the reaction with DMF when I(CF2)4I was used.72 The initial reduction of perfluoroiodoalkanes prior to SO2 seems unlikely when considering the studies of Knittel (reduction potential for SO2 at −0.7 V vs. Ag/AgCl).55 A SO2 mediated process could be more likely.
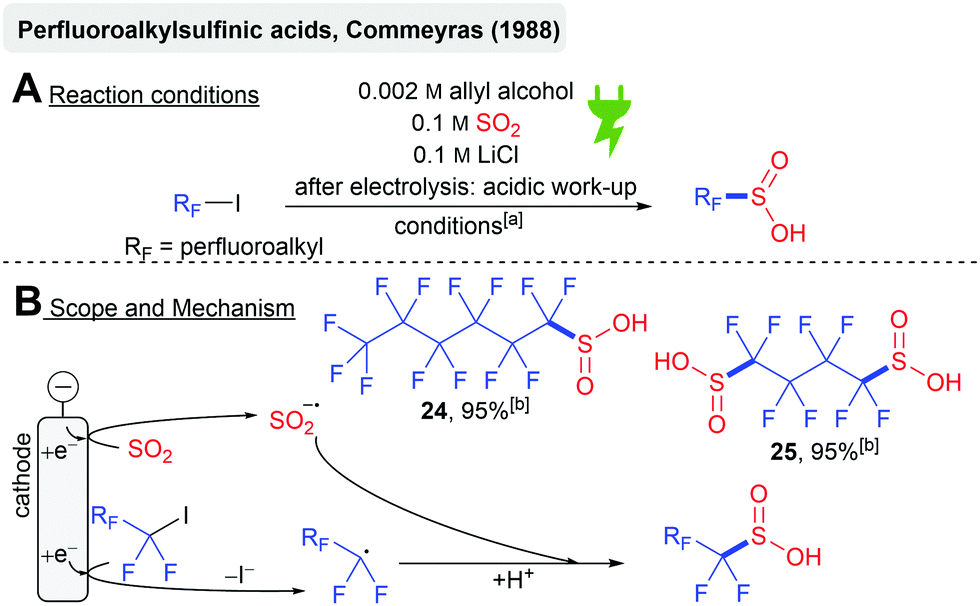 | ||
| Scheme 7 Electrochemical synthesis of perfluoroalkylsulfinic acids; [a]: DMF/H2O (9:1), 20 °C, carbon fiber electrodes, divided cell (glass frit), 1–12 mA cm−2 (galvanostatic), ∼1.05 F; [b]: yield determined by NMR integration (19F NMR). | ||
Photochemistry – early examples
The photochemical fixation of SO2 into organic molecules dates back to the development of the Reed sulfochlorination of simple alkanes in the 1930s (Scheme 8).73 This reaction proceeds via a radical chain mechanism initiated by a UV-light induced homolysis of chlorine. Similar to the classical free-radical halogenation, the chlorine atom abstracts hydrogen from the alkane. The formed alkyl radical is intercepted by SO2 leading to the corresponding sulfonyl radical. The reaction of the sulfonyl radical with Cl2 delivers the sulfonyl chloride product and a new chlorine atom, which can take part in another chain propagation step. As with other free-radical halogenation reactions, the Reed sulfochlorination only tolerates a small number of functional groups. In most cases, a mixture of regioisomers is formed. Nevertheless, the Reed process has been employed on a large scale for the production of sulfonic acid-based detergents and chlorosulfonated polyethylene.74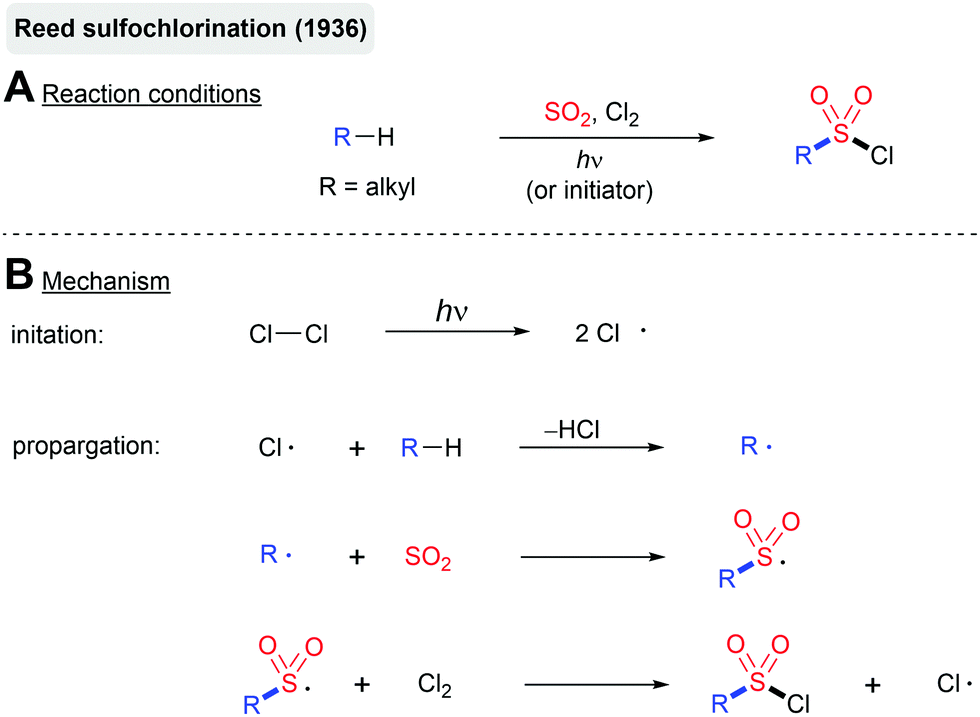 | ||
| Scheme 8 Reed sulfochlorination. | ||
Another noteworthy example is the decarboxylative sulfonylation of so-called Barton esters reported by Zard and Barton (Scheme 9).75 Photolysis of the labile N–O bond of the thiohydroxamic ester using UV irradiation affords an acyloxy radical. Extrusion of CO2 furnishes an alkyl radical, which is again intercepted by SO2. The formed sulfonyl radical reacts with another molecule of the Barton ester, leading to the formation of the thiosulfonate product and the propagation of the radical chain. The obtained products can be readily transformed into sulfones, sulfonyl chlorides or sulfonamides. The reaction proceeds through the generation of an aryl radical, trapping of this radical with SO2 and a final back electron transfer.
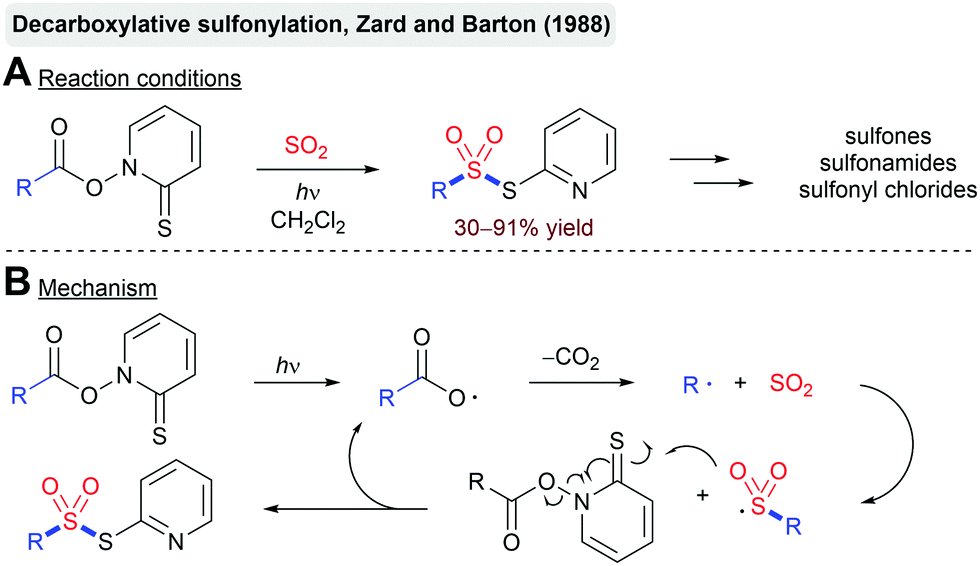 | ||
| Scheme 9 Photochemical decarboxylative sulfonylation by Barton. | ||
Photochemistry using UV irradiation
Although the photochemical fixation of SO2 has been known for almost 100 years, its application in organic synthesis has been sparsely studied.76 Only in the last ten years, the photochemical insertion of SO2 into organic molecules has gained more attention. In 2016, Wu and coworkers reported a UV-light mediated synthesis of N-aminosulfonamides from aryl or alkyl halides, DABSO and hydrazines (Scheme 10).77 Mechanistic investigations indicate the formation of a sulfonyl radical as the key intermediate. Later on, the same group extended this methodology to the construction of oxindole scaffolds.78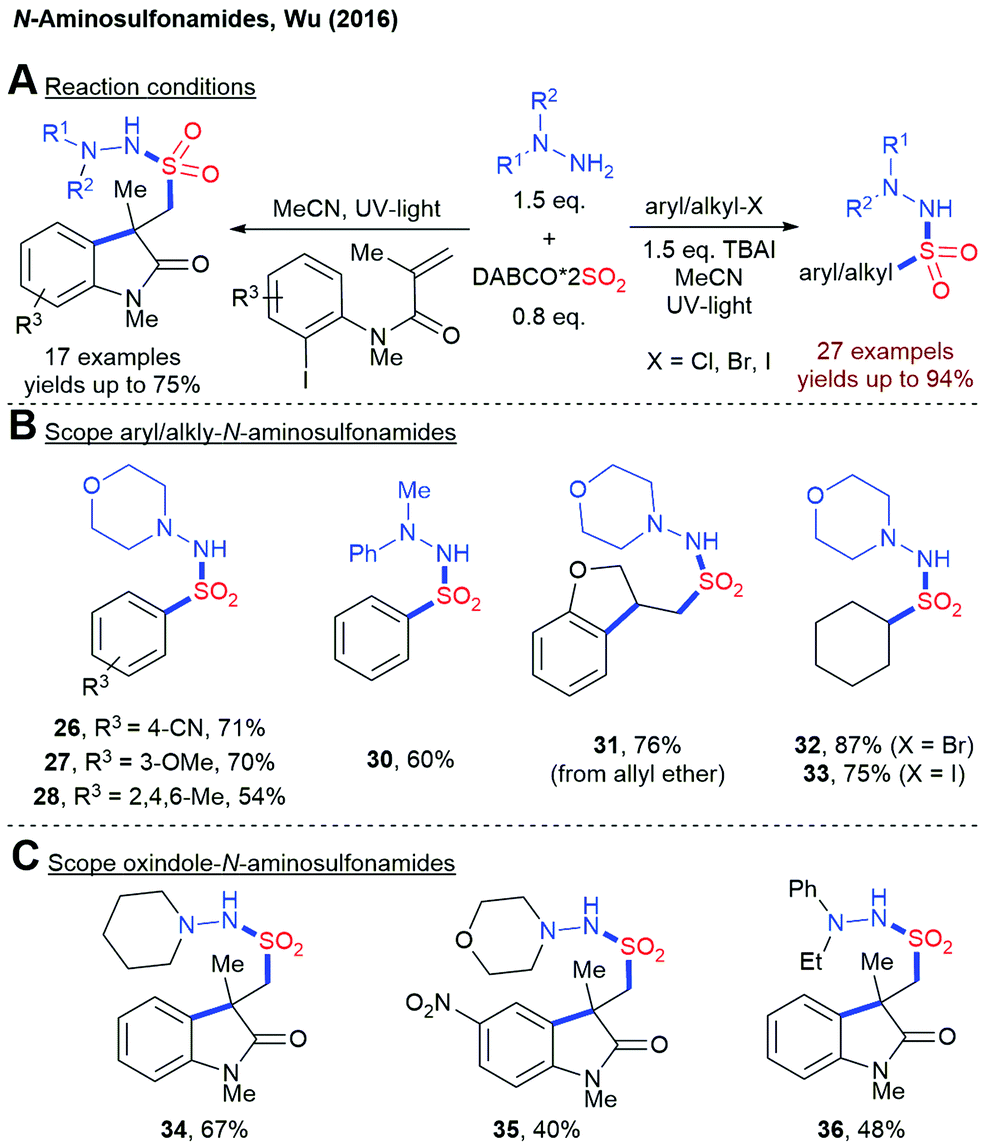 | ||
| Scheme 10 UV-light mediated insertion of SO2 into organic halides, TBAI = tetrabutylammonium iodide. | ||
The same group also described a couple of other reactions for the synthesis of sulfones exploiting a UV-light mediated insertion of SO2.79 However, the use of high-energy UV-light in organic synthesis is associated with several disadvantages. It can lead to undesirable side reactions or decomposition of the product. UV-light only constitutes a minor part of natural sunlight, necessitating the use of specialized equipment for such transformations.
Photochemistry using visible light
The Manolikakes group described the synthesis of sulfonylated coumarins from arylpropinoates, DABSO and diaryliodonium salts (Scheme 11).80 Interestingly, this transformation is solely driven by visible light and proceeds in the absence of any external photosensitizer. Presumably, the excitation of a charge transfer complex between the iodonium salt and DABSO initiates this radical transformation and the generation of a sulfonyl radical.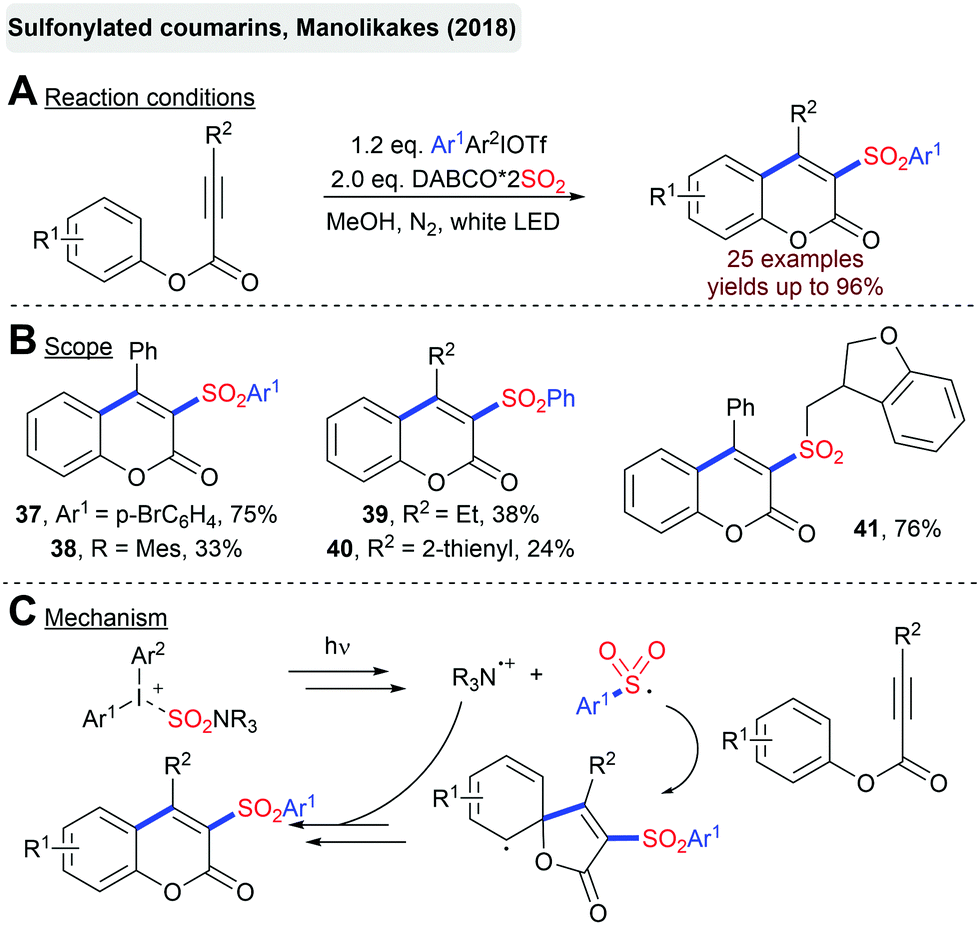 | ||
| Scheme 11 Photoinitiated synthesis of sulfonylated coumarins. | ||
This combination of diaryliodonium salt and DABSO has been utilized for a visible-light mediated synthesis of sulfonylated oxindoles and azaspiro[4,5]-trienone using either N-arylacrylamide or N-arylpropiolamides as the third reaction partner (Scheme 12).81 So far, the direct utilization of visible light for the fixation of SO2 in the absence of any external photosensitizer is limited to the reactions employing diaryliodonium salts as radical precursors. Other substrate classes have not been utilized successfully by now.
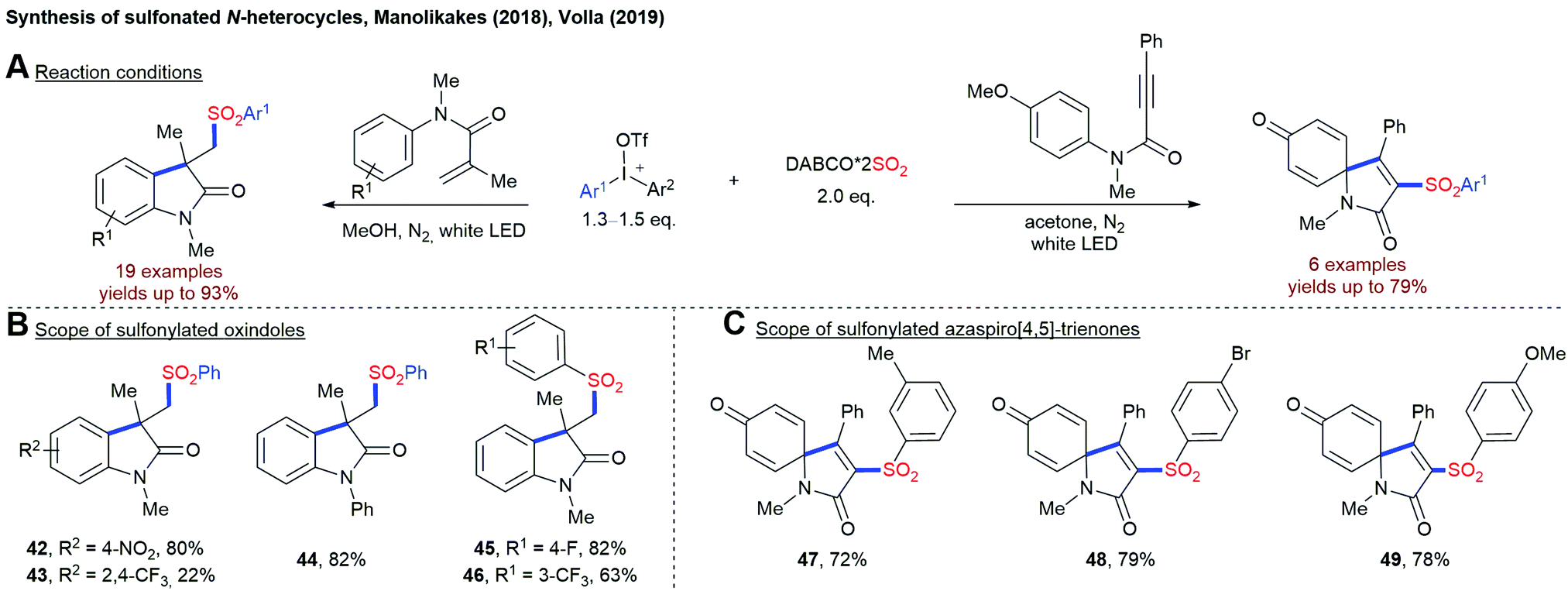 | ||
| Scheme 12 Synthesis of sulfonylated oxindole and azaspiro[4,5]-trienone using either N-arylacrylamide or N-arylpropiolamides. | ||
Photoredox-catalysis using visible light
The recent advent of photoredox-catalysis has opened intriguing possibilities to directly use (low-energy) visible-light for the activation of a plethora of different substrates.82 Inevitably, other methods for the construction of C–S bonds mediated by visible-light have also attracted considerable attention.83 The first examples employed sulfinates or sulfonyl chlorides as precursors for the generation of sulfonyl radicals.84 Later on, the in situ generation of these reactive intermediates from an organic building block and SO2 mediated by photoredox-catalysis emerged as a highly attractive opportunity for the fixation of SO2 in any type of sulfonyl functionality.27,28,30 Herein, we will highlight some selected pioneering examples as well as the most recent advances, from the last few years, in the photoredox-catalyzed synthesis of sulfonyl halides, sulfones and sulfonamides from SO2 or a suitable surrogate.Jacobi von Wangelin and coworkers described a Ru(bpy)3Cl2-catalyzed chlorosulfonylation of arenediazonium salts mediated by visible light (Scheme 13).85 Interestingly, both SO2 and the required HCl are generated by the hydrolysis of thionyl chloride (SOCl2) in the reaction.
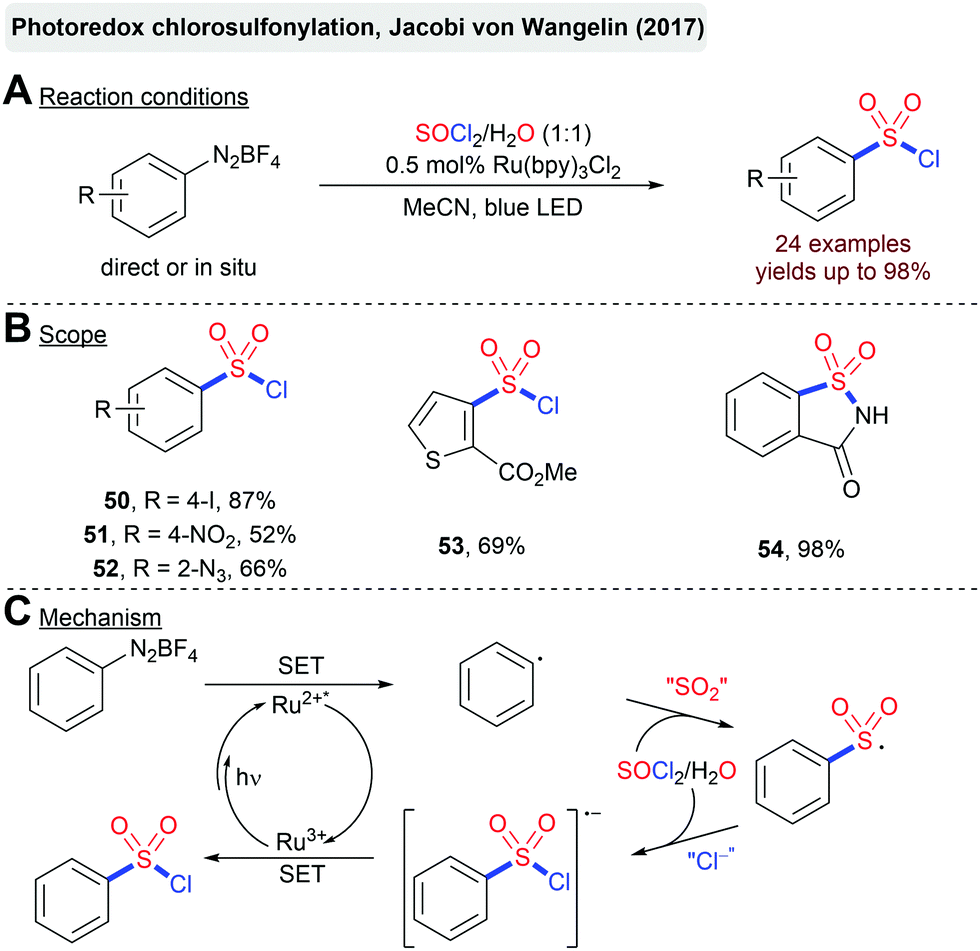 | ||
| Scheme 13 Photoredox-catalyzed synthesis of sulfonyl chlorides. | ||
Just recently, Tlili's group reported a light mediated synthesis of arylsulfonyl fluorides from arenediazonium salts and DABSO-catalyzed by an organic photoredox-catalyst (Scheme 14).86 Interestingly, DABSO plays a twofold role in this reaction. On the one hand, it serves as the SO2-source to trap the formed aryl radical. On the other hand, the thereby-released DABCO regenerates the active photocatalyst in its ground state and takes part in the generation of a highly electrophilic sulfonium salt. Although better yields are obtained with the addition of KHF2, the BF4− counterion can serve as a fluoride source itself.
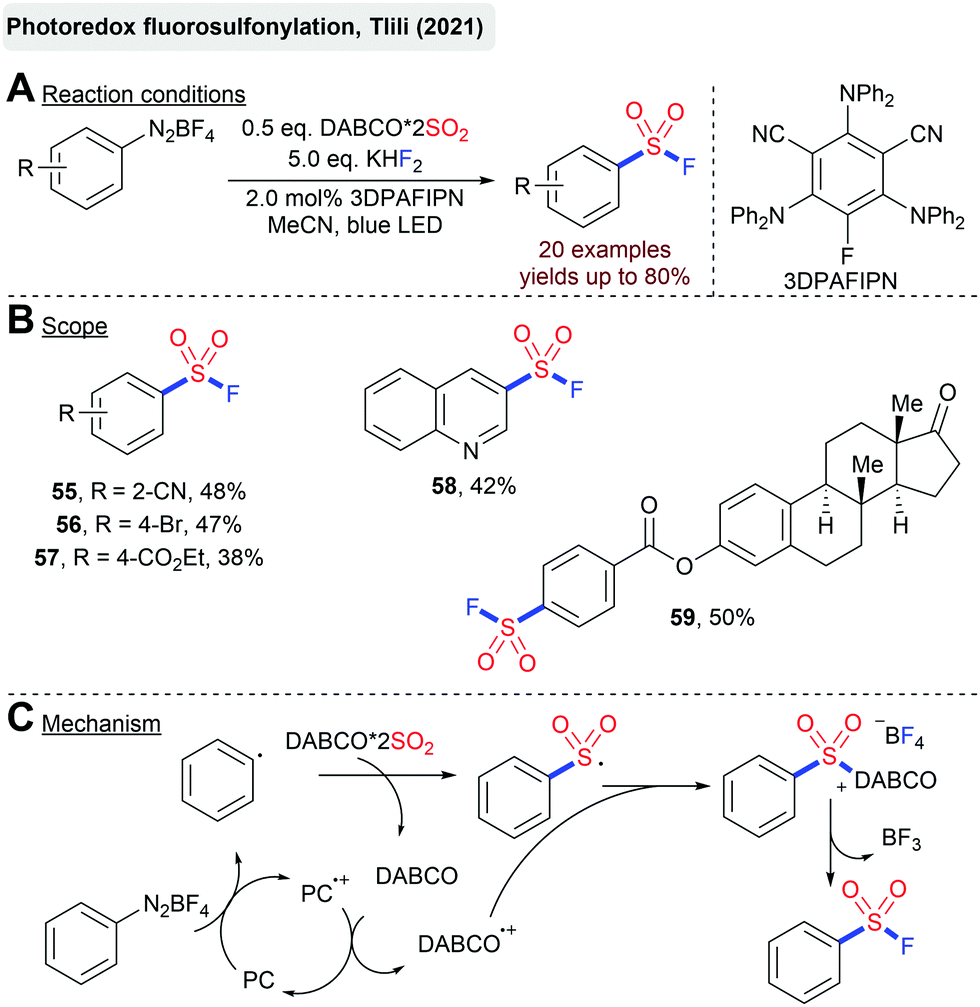 | ||
| Scheme 14 Photoredox-catalyzed synthesis of sulfonyl fluorides. | ||
In 2020, Wu and coworkers disclosed two closely related methods for the construction of alkynyl sulfones from alkynyl bromides, Na2S2O5 and alkyltrifluoroborates or 4-alkyl Hantzsch esters, respectively (Scheme 15).87 A sulfonyl radical is postulated as the key intermediate in both processes.
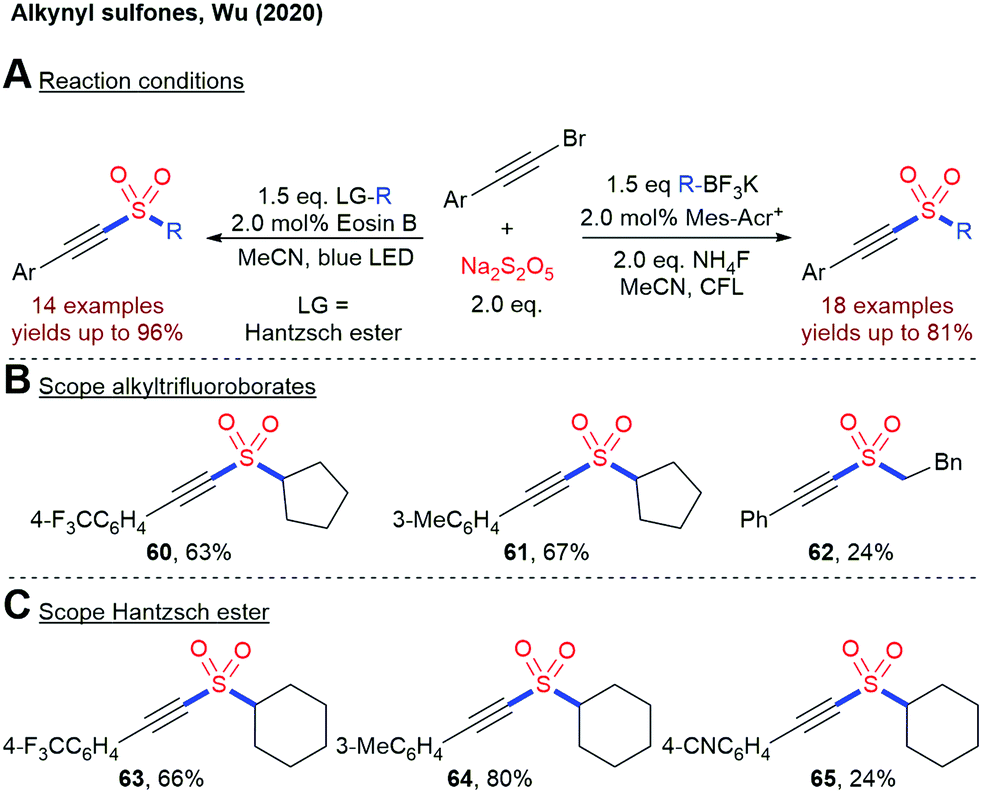 | ||
| Scheme 15 Photoredox-catalyzed insertion of SO2 into alkynyl sulfones; CFL = compact fluorescent lamp. | ||
The same group described a photoredox-catalyzed synthesis of β-ketosulfones from silylenolethers, K2S2O5 and alkylpyridinium salts as radical precursors (Scheme 16).88 Sulfonyl radicals are generated by the SET reduction of pyridinium salts, followed by the homolytic fragmentation and addition of the generated alkyl radical to SO2. Later on, Wu and coworkers described an analogous method with difluorinated silyl enol ethers.89
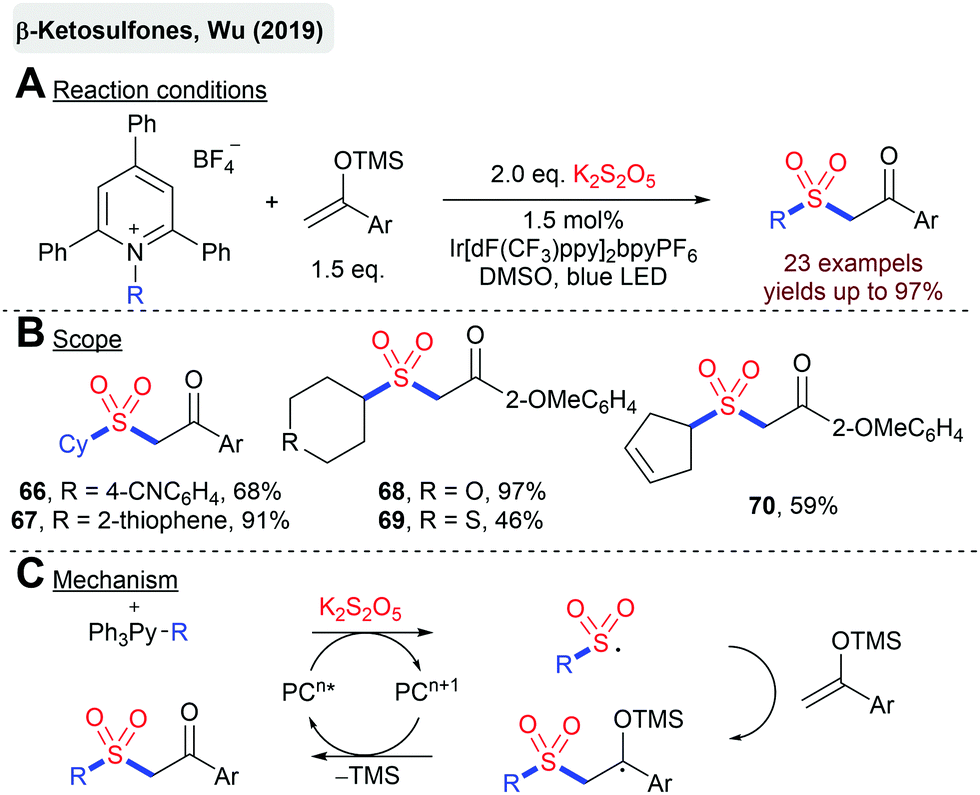 | ||
| Scheme 16 β-Ketosulfones from silylenolethers and alkylpyridinium salts. | ||
Piguel et al. reported the C–H sulfonylation of imidazoheterocycles with DABSO and diaryliodonium salts catalyzed by the organic dye Eosin Y (Scheme 17).90 This transformation provides an attractive opportunity for a direct installation of a sulfonyl moiety onto a non-functionalized heterocyclic scaffold.
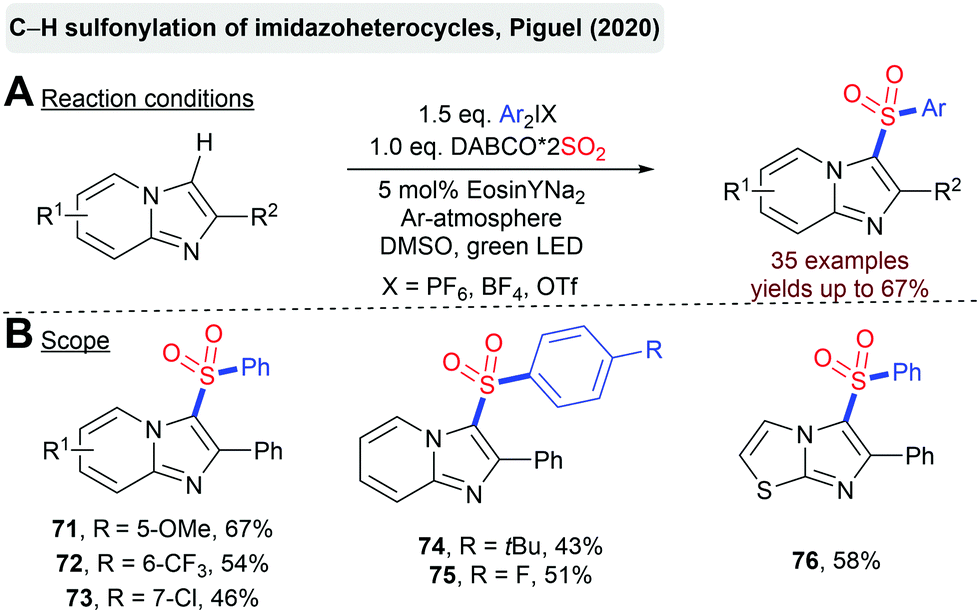 | ||
| Scheme 17 Direct sulfonylation of imidazoheterocycles. | ||
Several groups described a ring-opening approach for the visible-light mediated fixation of SO2.91 The SET reduction of cyclobutanone oxime with an excited photocatalyst leads to a ring-opening and a cyanoalkyl radical. After the addition to SO2, the formed sulfonyl radical can be intercepted by various trapping agents such as alkenes, methylenecyclopropanes or alkynoates (Scheme 18).
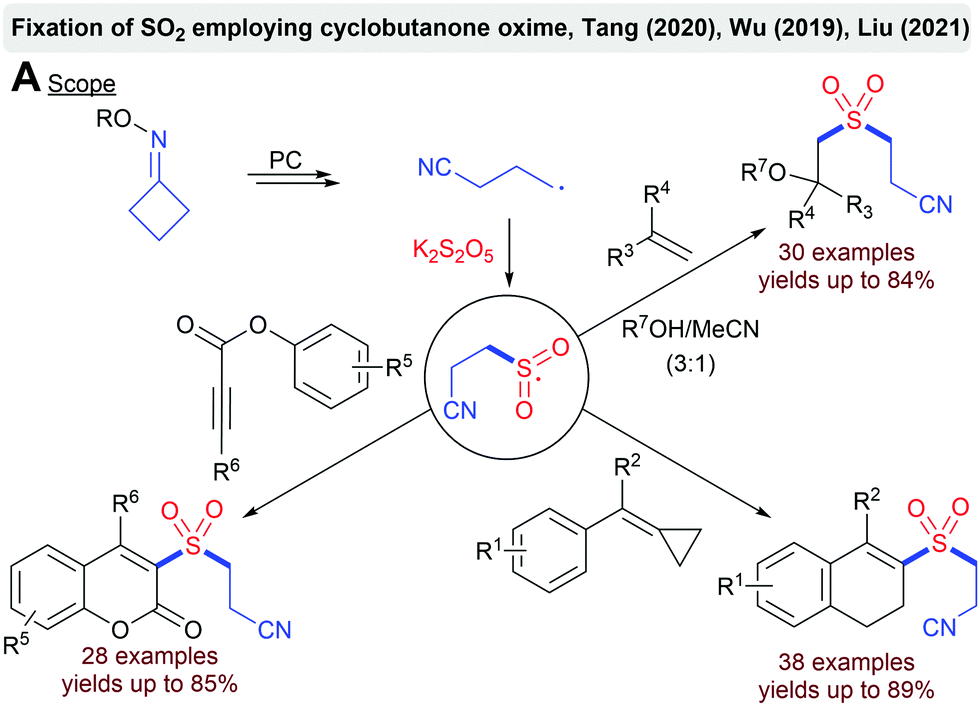 | ||
| Scheme 18 Photoredox-catalyzed fixation of SO2 employing cyclobutanone oxime. | ||
In general, radical cascades involving sulfonyl radicals enable the facile synthesis of (hetero)cyclic scaffolds bearing a sulfonyl functionality. In Scheme 19, two transformations for a photoredox-catalyzed construction of sulfone-functionalized dibenzazepines and cyclopenta[gh]phenanthridines are depicted.92 In both cases, sulfonyl radicals are generated from aryldiazonium salts and a solid SO2 surrogate. The Manolikakes group described a photoredox-catalyzed synthesis of N-aminosulfonamides from diaryliodonium salts, in-situ generated SO2 and hydrazines (Scheme 20).93 Mechanistic investigations indicate that contrary to previous reactions, aryl sulfonyl radicals are not formed. Instead, the SET oxidation of a hydrazine–SO2 adduct affords an S-centered radical as the key intermediate. Unfortunately, this method is limited to hydrazines and the synthesis of aminosulfonamides. Reactions with simple amines failed.
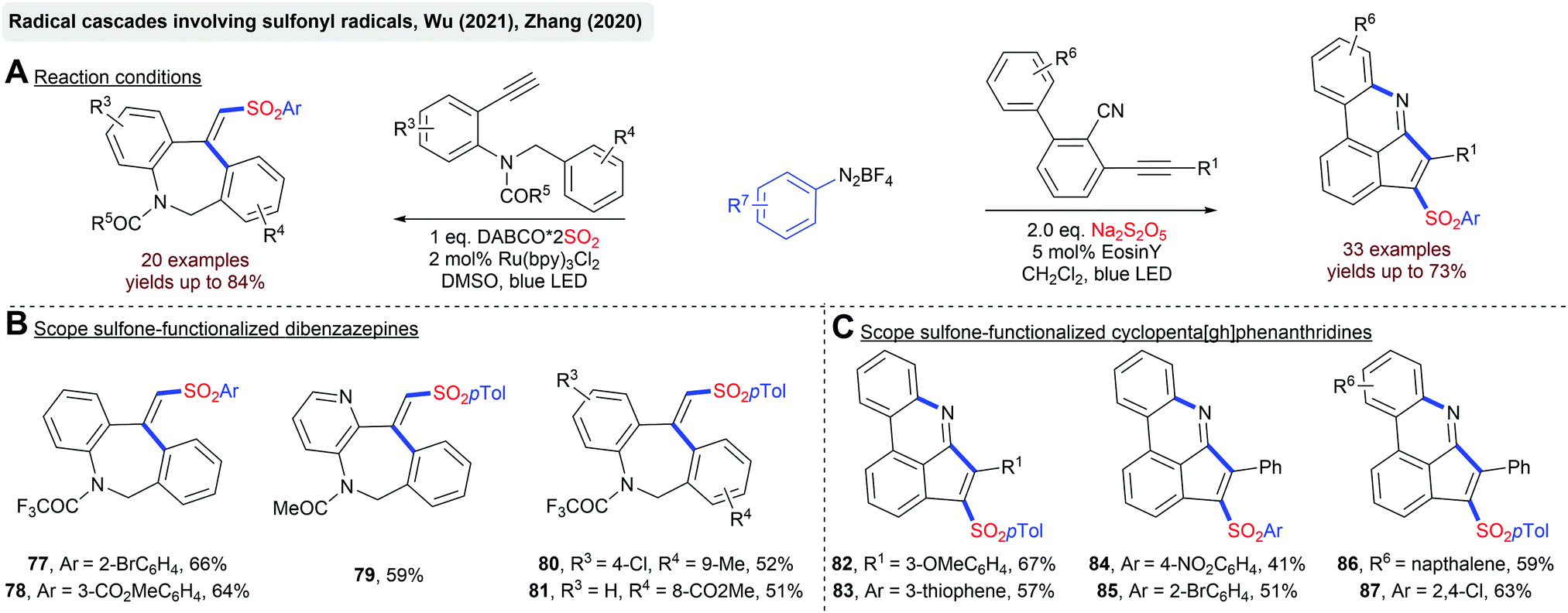 | ||
| Scheme 19 Photoredox-catalyzed cascades towards sulfonylated heterocycles. | ||
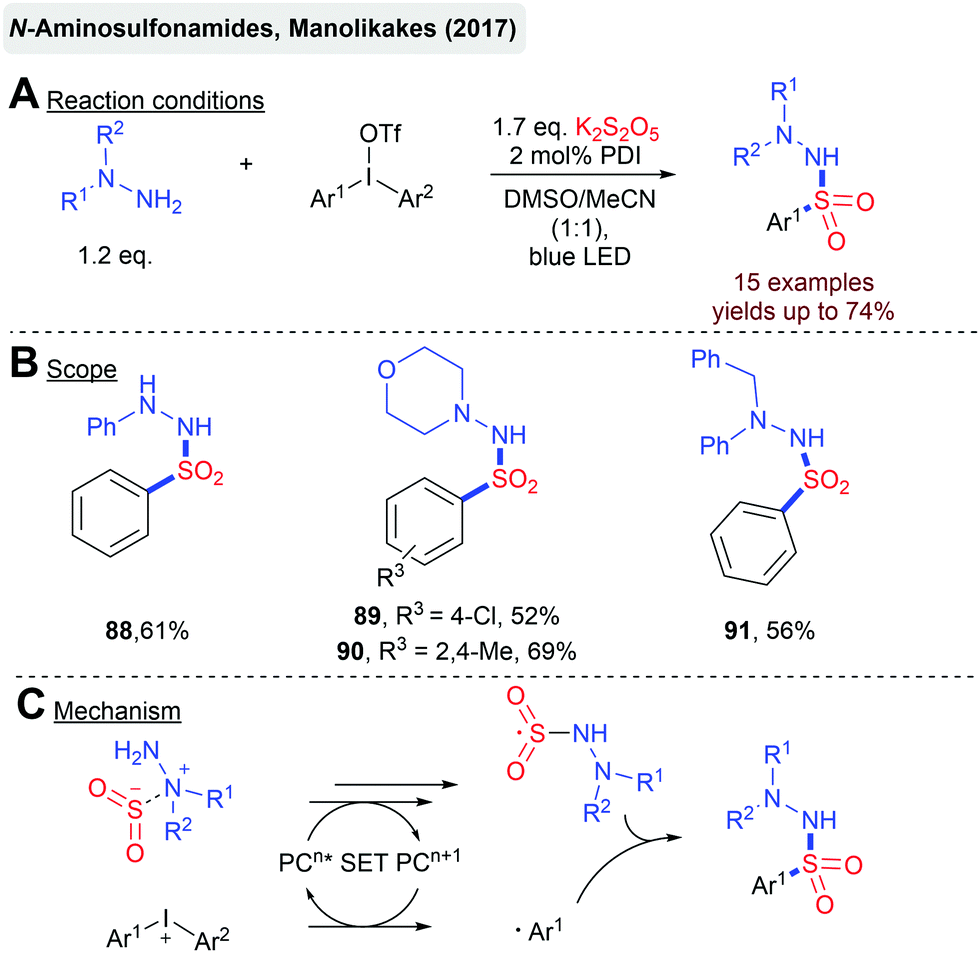 | ||
| Scheme 20 Photoredox-catalyzed synthesis of N-aminosulfonamides. | ||
In 2021, Larinov and coworkers reported a decaboxylative aminosulfonylation of free carboxylic acids using DABSO, a hydroxylamine derivative and a dual catalyst system consisting of an acridine photocatalyst and CuOTf (Scheme 21).94 By using anilines or NaN3 together with tBuO2Bz as terminal oxidant, sulfonamides and sulfonyl azides can be accessed directly. This method is also amenable to the late-stage functionalization of natural products.
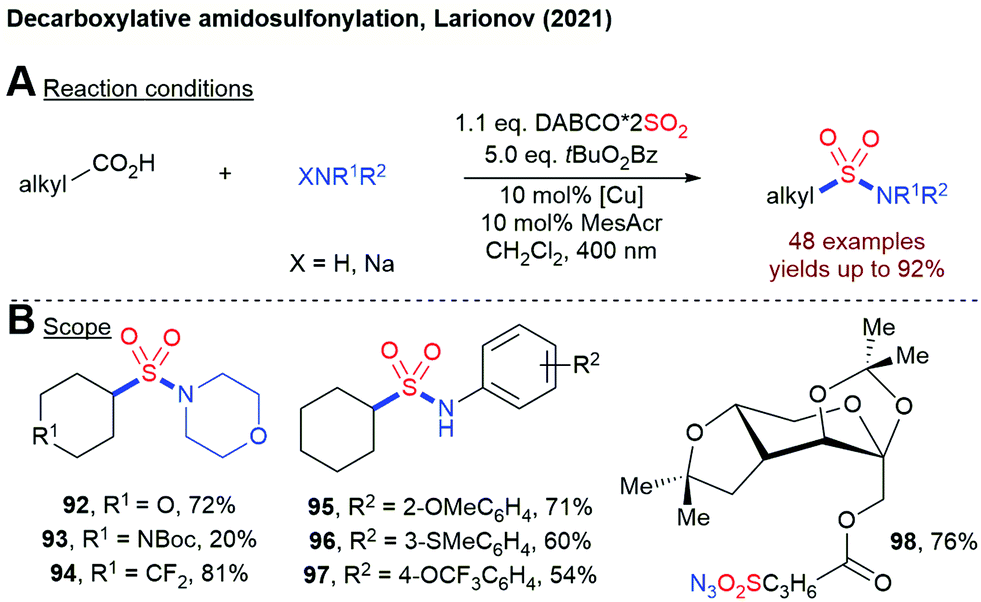 | ||
| Scheme 21 Decarboxylative aminosulfonylation of free carboxylic acids using a dual catalyst system. | ||
Conclusion and outlook
The photochemical incorporation of SO2 into value-added compounds has been used for almost 100 years. Although all the first methods, such as the Reed chlorosulfonylation, relied on the use of UV-light, the recent achievements in the field of photoredox-catalysis have led to significant advances towards the fixation of SO2 with visible-light mediated in the last few years. This field has become an active area of research and new methods have been developed at an astonishing rate. However, further progress towards the utilization of more common building blocks, in particular from renewable resources, is still needed. In particular, the lack of efficient methods for the synthesis of highly relevant sulfonamide motifs has to be addressed. On the other hand, the electrochemical incorporation of SO2 is a very new and modern research topic, although Knittel and coworkers did some pioneering work in 1973 featuring the electrochemical synthesis of symmetrical sulfones. Anodic processes involving SO2 are scarce. This could be due to the fact that it is hard to find a suitable system as the cathodically formed SO2 anion radical might interfere in anodic oxidations/reactions. Currently, divided cells are an elegant solution to circumvent this problem. Cathodic reductions of SO2 in a mediated fashion are rather established in electroorganic synthesis although not much work has been reported in this field in the past. We expect a significant increase of electrochemical methodologies involving SO2 incorporation in the near future. Other future research topics could be paired electrolysis systems involving SO2 reduction coupled with anodic oxidation processes in the synthesis of value-added products.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
Support from the Forschungsinitiative des Landes Rheinland-Pfalz in frame of SusInnoScience and NanoCat is highly appreciated.Notes and references
- S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed.
- (a) J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, E42 CrossRef; (b) J. L. Röckl, D. Pollok, R. Franke and S. R. Waldvogel, Acc. Chem. Res., 2020, 53, 45–61 CrossRef PubMed.
- (a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (c) S. Arndt, D. Weis, K. Donsbach and S. R. Waldvogel, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 CrossRef CAS PubMed; (d) X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507 CrossRef CAS PubMed; (e) S. Lips, A. Wiebe, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 10872–10876 CrossRef CAS PubMed; (f) A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef CAS PubMed; (g) A. Wiebe, B. Riehl, S. Lips, R. Franke and S. R. Waldvogel, Sci. Adv., 2017, 3, eaao3920 CrossRef PubMed.
- (a) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC; (b) A. Shatskiy, H. Lundberg and M. D. Kärkäs, ChemElectroChem, 2019, 6, 4067–4092 CrossRef CAS.
- (a) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (b) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- (a) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (b) Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- (a) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (b) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- H. Müller, Sulfur Dioxide, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 35th edn, 2000 Search PubMed.
- E. Poujol, in Gases in Agro-Food Processes, ed. R. Cachon, P. Girardon and A. Voilley, Elsevier, Academic Press, 2019, pp. 75–85 Search PubMed.
- (a) Advances in Food Research 6, ed. E. M. Mrak and G. F. Stewart, Elsevier, Academic Press, 1955 Search PubMed; (b) B. J. Freedman, Br. J. Dis. Chest, 1980, 74, 128–134 CrossRef CAS PubMed; (c) A. C. Roberts and D. J. McWeeny, Int. J. Food Sci. Technol., 1972, 7, 221–238 CrossRef CAS.
- P. Vogel, M. Turks, L. Bouchez, D. Marković, A. Varela-Álvarez and J. Á. Sordo, Acc. Chem. Res., 2007, 40, 931–942 CrossRef CAS PubMed.
- M. Alhanif, G. Sanyoto and W. Widayat, Front. Heat Mass Transfer, 2020, 15, 6 Search PubMed.
- Q. Zhong, H. Shen, X. Yun, Y. Chen, Y. Ren, H. Xu, G. Shen, W. Du, J. Meng, W. Li, J. Ma and S. Tao, Environ. Sci. Technol., 2020, 54, 6508–6517 CrossRef CAS PubMed.
- A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 2701–2710 Search PubMed.
- P. Grennfelt, A. Engleryd, M. Forsius, Ø. Hov, H. Rodhe and E. Cowling, Ambio, 2020, 49, 849–864 CrossRef CAS PubMed.
- K. Craig, Rev. Environ. Contam. Toxicol., 2019, 246, 33–64 CrossRef CAS PubMed.
- A. Jawad and A. Al-Dallal, Al-Khawarizmi Eng. J., 2013, 9, 58–69 Search PubMed.
- S. Trasatti, Int. J. Hydrogen Energy, 1995, 20, 835–844 CrossRef CAS.
- (a) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393–5398 RSC; (b) G. Pelzer, J. Herwig, W. Keim and R. Goddard, Russ. Chem. Bull., 1998, 47, 904–912 CrossRef CAS; (c) Q. Tang, X. Yin, R. R. Kuchukulla and Q. Zeng, Chem. Rec., 2021, 21, 893–905 CrossRef CAS PubMed; (d) N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 1939–1973 CAS; (e) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592–1599 RSC; (f) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691–705 RSC; (g) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561–12569 RSC.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- X. Jia, S. Kramer, T. Skrydstrup and Z. Lian, Angew. Chem., Int. Ed., 2021, 60, 7353–7359 CrossRef CAS PubMed.
- K. Hofman, N.-W. Liu and G. Manolikakes, Chem. – Eur. J., 2018, 24, 11852–11863 CrossRef CAS PubMed.
- Y. Li and J. Wu, Chem. Lett., 2020, 49, 1066–1070 CrossRef CAS.
- G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405–10414 RSC.
- S. Ye, X. Li, W. Xie and J. Wu, Eur. J. Org. Chem., 2020, 1274–1287 CrossRef CAS.
- S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013–1019 RSC.
- E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef CAS.
- M. C. Willis, Phosphorus Sulfur Relat. Elem., 2019, 194, 654–657 CrossRef CAS.
- K. Suta and M. Turks, Chem. Heterocycl. Compd., 2018, 54, 584–586 CrossRef CAS.
- M. Wang and X. Jiang, Chem. Rec., 2021, 21 DOI:10.1002/tcr.202000162.
- D. Zeng, M. Wang, W.-P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956–3966 RSC.
- (a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed; (b) P. Devendar and G.-F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed.
- J. Lugiņina, J. Uzuleņa, D. Posevins and M. Turks, Eur. J. Org. Chem., 2016, 1760–1771 CrossRef.
- W. F. Giauque and C. C. Stephenson, J. Am. Chem. Soc., 1938, 60, 1389–1394 CrossRef CAS.
- (a) P. J. Elving and J. M. Markowitz, J. Chem. Educ., 1960, 37, 75–81 CrossRef CAS; (b) D. Posevins, K. Suta and M. Turks, Eur. J. Org. Chem., 2016, 1414–1419 CrossRef CAS; (c) H. Mayr, G. Gorath and B. Bauer, Angew. Chem., Int. Ed. Engl., 1994, 33, 788–789 CrossRef.
- C. Jehoulet and A. J. Bard, Angew. Chem., Int. Ed. Engl., 1991, 30, 836–838 CrossRef.
- (a) P. Ceroni, F. Paolucci, C. Paradisi, A. Juris, S. Roffia, S. Serroni, S. Campagna and A. J. Bard, J. Am. Chem. Soc., 1998, 120, 5480–5487 CrossRef CAS; (b) P. Ceroni, F. Paolucci, S. Roffia, S. Serroni, S. Campagna and A. J. Bard, Inorg. Chem., 1998, 37, 2829–2832 CrossRef CAS PubMed; (c) J. B. Chlistunoff and A. J. Bard, Inorg. Chem., 1992, 31, 4582–4587 CrossRef CAS; (d) J. B. Chlistunoff and A. J. Bard, Inorg. Chem., 1993, 32, 3521–3527 CrossRef CAS; (e) E. Garcia and A. J. Bard, J. Electrochem. Soc., 1990, 137, 2752–2759 CrossRef CAS; (f) E. Garcia, J. Kwak and A. J. Bard, Inorg. Chem., 1988, 27, 4377–4382 CrossRef CAS; (g) J. G. Gaudiello, P. G. Bradley, K. A. Norton, W. H. Woodruff and A. J. Bard, Inorg. Chem., 1984, 23, 3–10 CrossRef CAS; (h) J. G. Gaudiello, P. R. Sharp and A. J. Bard, J. Am. Chem. Soc., 1982, 104, 6373–6377 CrossRef CAS; (i) M. Delamar, P. C. Lacaze, J.-Y. Dumousseau and J. Dubois, Electrochim. Acta, 1982, 27, 61–65 CrossRef CAS; (j) A. C. McDonald, F. R. F. Fan and A. J. Bard, J. Phys. Chem., 1986, 90, 196–202 CrossRef CAS; (k) P. R. Sharp and A. J. Bard, Inorg. Chem., 1983, 22, 2689–2693 CrossRef CAS; (l) P. R. Sharp and A. J. Bard, Inorg. Chem., 1983, 22, 3462–3464 CrossRef CAS; (m) L. A. Tinker and A. J. Bard, J. Am. Chem. Soc., 1979, 101, 2316–2319 CrossRef CAS; (n) L. A. Tinker and A. J. Bard, J. Electroanal. Chem. Interfacial Electrochem., 1982, 133, 275–285 CrossRef CAS.
- H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876–4878 CrossRef CAS PubMed.
- Reagent price based on Sigma-Aldrich (accessed April 28th, 2021).
- S. P. Blum, L. Schäffer, D. Schollmeyer and S. R. Waldvogel, Chem. Commun., 2021, 57, 4775–4778 RSC.
- (a) Y. Li, M. Wang and X. Jiang, Chin. J. Chem., 2020, 38, 1521–1525 CrossRef CAS; (b) Y. Meng, M. Wang and X. Jiang, Angew. Chem., Int. Ed., 2020, 59, 1346–1353 CrossRef CAS PubMed; (c) M. Yingying, W. Ming and J. Xuefeng, CCS Chem., 2021, 3, 17–24 Search PubMed; (d) M. Wang, S. Chen and X. Jiang, Org. Lett., 2017, 19, 4916–4919 CrossRef CAS PubMed; (e) M. Wang, Q. Fan and X. Jiang, Green Chem., 2018, 20, 5469–5473 RSC; (f) M. Wang, J. Zhao and X. Jiang, ChemSusChem, 2019, 12, 3064–3068 CrossRef CAS PubMed.
- G. Y. Chung Leung, B. Ramalingam, G. Loh and A. Chen, Org. Process Res. Dev., 2020, 24, 546–554 CrossRef.
- R. G. Makitra, S. D. Kal’muk, D. V. Bryk and I. P. Polyuzhin, Russ. J. Inorg. Chem., 2010, 55, 1322–1329 CrossRef CAS.
- Reagent price based on TCI Chemicals (accessed April 28th, 2021).
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- (a) L. Martial and L. Bischoff, Org. Synth., 2013, 90, 301–305 CrossRef CAS; (b) A. G. Jones, Analyst, 1951, 76, 5–12 RSC.
- S. F. Nelsen and P. J. Hintz, J. Am. Chem. Soc., 1972, 94, 7114–7117 CrossRef CAS.
- M. S. Yogendra Kumar, M. D. Gowtham, Mahadevaiah and G. Agendrappa, Anal. Sci., 2006, 22, 757–761 CrossRef CAS PubMed.
- S. P. Blum, T. Karakaya, D. Schollmeyer, A. Klapars and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 5056–5062 CrossRef CAS PubMed.
- S. P. Blum, D. Schollmeyer, M. Turks and S. R. Waldvogel, Chem. – Eur. J., 2020, 26, 8358–8362 CrossRef CAS PubMed.
- D. Knittel and B. Kastening, J. Appl. Electrochem., 1973, 3, 291–296 CrossRef CAS.
- D. Knittel, Monatsh. Chem., 1982, 113, 37–41 CrossRef CAS.
- H. J. Wille, B. Kastening and D. Knittel, J. Electroanal. Chem. Interfacial Electrochem., 1986, 214, 221–235 CrossRef CAS.
- H. J. Wille, D. Knittel, B. Kastening and J. Mergel, J. Appl. Electrochem., 1980, 10, 489–494 CrossRef CAS.
- D. Knittel, J. Electroanal. Chem. Interfacial Electrochem., 1985, 195, 345–356 CrossRef CAS.
- D. Knittel, Monatsh. Chem., 1986, 117, 359–367 CrossRef CAS.
- L. J. Andrews and R. M. Keefer, J. Am. Chem. Soc., 1951, 73, 4169–4172 CrossRef CAS.
- B.-S. Kim and S.-M. Park, J. Electrochem. Soc., 1995, 142, 26–33 CrossRef CAS.
- R. P. Martin and D. T. Sawyer, Inorg. Chem., 1972, 11, 2644–2647 CrossRef CAS.
- E. Potteau, E. Levillain and J.-P. Lelieur, J. Electroanal. Chem., 1999, 476, 15–25 CrossRef CAS.
- Y. Geronov, R. V. Moshtev and B. Puresheva, J. Electroanal. Chem. Interfacial Electrochem., 1980, 108, 335–346 CrossRef.
- M. Jaksic, J. New Mater. Electrochem. Syst., 2000, 3, 167–182 Search PubMed.
- Z. Li, L. Jiao, Y. Sun, Z. He, Z. Wei and W.-W. Liao, Angew. Chem., Int. Ed., 2020, 59, 7266–7270 CrossRef CAS PubMed.
- C. M. Kisukuri, V. A. Fernandes, J. A. C. Delgado, A. P. Häring, M. W. Paixão and S. R. Waldvogel, Chem. Rec., 2021, 21 DOI:10.1002/tcr202100065.
- V. G. Koshechko, L. A. Kiprianova, L. I. Fileleeva and Z. Z. Rozhkova, J. Fluorine Chem., 1995, 70, 277–278 CrossRef.
- C. P. Andrieux, L. Gelis and J. M. Saveant, J. Am. Chem. Soc., 1990, 112, 786–791 CrossRef CAS.
- V. G. Koshechko and L. A. Kiprianova, Theor. Exp. Chem., 1999, 35, 18–36 CrossRef CAS.
- S. Benefice-Malouet, H. Blancou, P. Calas and A. Commeyras, J. Fluorine Chem., 1988, 39, 125–140 CrossRef CAS.
- (a) F. Asinger, F. Ebeneder and E. Böck, Ber. dtsch. Chem. Ges. A/B, 1942, 75, 42–48 CrossRef; (b) F. Asinger and F. Ebeneder, Ber. dtsch. Chem. Ges. A/B, 1942, 75, 344–349 CrossRef; (c) F. Asinger, W. Schmidt and F. Ebeneder, Ber. dtsch. Chem. Ges. A/B, 1942, 75, 34–41 CrossRef; (d) C. F. Reed and C. L. Horn, US Pat., US2046090A, 1936 Search PubMed.
- (a) J. Texter, Reactions and synthesis in surfactant systems, Marcel Dekker, New York, 2001, vol. 100 CrossRef; (b) M. A. Smook, E. T. Pieski and C. F. Hammer, Ind. Eng. Chem., 1953, 45, 2731–2737 CrossRef CAS.
- D. H. Barton, B. Lacher, B. Misterkiewicz and S. Z. Zard, Tetrahedron, 1988, 44, 1153–1158 CrossRef CAS.
- (a) R. M. Wilson and S. W. Wunderly, J. Am. Chem. Soc., 1974, 96, 7350–7351 CrossRef CAS; (b) P. Bougeard, M. D. Johnson and G. M. Lampman, J. Chem. Soc., Perkin Trans. 1, 1982, 849 RSC.
- Y. Li, D. Zheng, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 574–578 RSC.
- K. Zhou, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 865–869 RSC.
- (a) X. Gong, Y. Ding, X. Fan and J. Wu, Adv. Synth. Catal., 2017, 359, 2999–3004 CrossRef CAS; (b) J. Zhang, K. Zhou, G. Qiu and J. Wu, Org. Chem. Front., 2019, 6, 36–40 RSC; (c) K. Zhou, J.-B. Liu, W. Xie, S. Ye and J. Wu, Chem. Commun., 2020, 56, 2554–2557 RSC; (d) S. Ye, K. Zhou, P. Rojsitthisak and J. Wu, Org. Chem. Front., 2020, 7, 14–18 RSC.
- Z. Chen, N.-W. Liu, M. Bolte, H. Ren and G. Manolikakes, Green Chem., 2018, 20, 3059–3070 RSC.
- (a) A. M. Nair, I. Halder, S. Khan and C. M. R. Volla, Adv. Synth. Catal., 2020, 362, 224–229 CrossRef CAS; (b) N.-W. Liu, Z. Chen, A. Herbert, H. Ren and G. Manolikakes, Eur. J. Org. Chem., 2018, 5725–5734 CrossRef CAS.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) Y. Lee and M. S. Kwon, Eur. J. Org. Chem., 2020, 6028–6043 CrossRef CAS.
- (a) J. Zhu, W.-C. Yang, X. Wang and L. Wu, Adv. Synth. Catal., 2018, 360, 386–400 CrossRef CAS; (b) W. Guo, K. Tao, W. Tan, M. Zhao, L. Zheng and X. Fan, Org. Chem. Front., 2019, 6, 2048–2066 RSC; (c) A. Wimmer and B. König, Beilstein J. Org. Chem., 2018, 14, 54–83 CrossRef CAS PubMed.
- (a) A. U. Meyer, S. Jäger, D. Prasad Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050–2054 CrossRef CAS; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed; (c) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999–7002 CrossRef CAS PubMed.
- M. Májek, M. Neumeier and A. Jacobi von Wangelin, ChemSusChem, 2017, 10, 151–155 CrossRef PubMed.
- D. Louvel, A. Chelagha, J. Rouillon, P.-A. Payard, L. Khrouz, C. Monnereau and A. Tlili, Chem. – Eur. J., 2021, 27, 8704–8708 CrossRef CAS PubMed.
- (a) X. Gong, M. Yang, J.-B. Liu, F.-S. He, X. Fan and J. Wu, Green Chem., 2020, 22, 1906–1910 RSC; (b) X. Gong, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Org. Chem. Front., 2020, 7, 938–943 RSC.
- X. Wang, Y. Kuang, S. Ye and J. Wu, Chem. Commun., 2019, 55, 14962–14964 RSC.
- F.-S. He, Y. Yao, W. Xie and J. Wu, Chem. Commun., 2020, 56, 9469–9472 RSC.
- C. Breton-Patient, D. Naud-Martin, F. Mahuteau-Betzer and S. Piguel, Eur. J. Org. Chem., 2020, 6653–6660 CrossRef CAS.
- (a) Y. Liu, Q.-L. Wang, Z. Chen, H. Li, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Chem. Commun., 2020, 56, 3011–3014 RSC; (b) J. Zhang, X. Li, W. Xie, S. Ye and J. Wu, Org. Lett., 2019, 21, 4950–4954 CrossRef CAS PubMed; (c) P. Chen, Z. Chen, B.-Q. Xiong, Y. Liang, K.-W. Tang, J. Xie and Y. Liu, Org. Biomol. Chem., 2021, 19, 3181–3190 RSC.
- (a) N. Zhou, M. Wu, K. Kuang, S. Wu and M. Zhang, Adv. Synth. Catal., 2020, 362, 5391–5397 CrossRef CAS; (b) Y. Yao, Z. Yin, F.-S. He, X. Qin, W. Xie and J. Wu, Chem. Commun., 2021, 57, 2883–2886 RSC.
- N.-W. Liu, S. Liang and G. Manolikakes, Adv. Synth. Catal., 2017, 359, 1308–1319 CrossRef CAS.
- V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, H. D. Arman and O. V. Larionov, Chem. Sci., 2021, 12, 6429–6436 RSC.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |