
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible and NIR emitting Yb(III) and Er(III) complexes sensitized by β-diketonates and phenanthroline derivatives†
Gabriela Brito-Santosa,
Beatriz Gil-Hernández ab,
Inocencio R. Martínbc,
Ricardo Guerrero-Lemusbc and
Joaquín Sanchiz*ab
ab,
Inocencio R. Martínbc,
Ricardo Guerrero-Lemusbc and
Joaquín Sanchiz*ab
aDepartamento de Química, Facultad de Ciencias, Universidad de La Laguna, Tenerife 38206, Spain. E-mail: jsanchiz@ull.edu.es
bInstituto Universitario de Materiales y Nanotecnología, Universidad de La Laguna, Tenerife 38206, Spain
cDepartamento de Física, Facultad de Ciencias, Universidad de La Laguna, Tenerife 38206, Spain
First published on 24th July 2020
Abstract
Five new complexes namely, [Er(bta)3(me-phen)] (1), [Yb(bta)3(me-phen)] (2), [Gd(bta)3(me-phen)] (3), [Yb(bta)3(pyz-phen)] (4), and [Er(tta)3(pyz-phen)] (5) have been prepared with the fluorinated β-diketone ligands Hbta and Htta (Hbta = benzoyltrifluoroacetone and Htta = 2-thenoyltrifluoroacetone) combined with the azacyclo phenanthroline-derivatives, 5-methyl-1,10-phenanthroline (me-phen) and pyrazino[2,3-f][1,10]phenanthroline (pyz-phen). The crystal structures of 2, 4 and 5 have been solved by single-crystal X-ray diffraction. PXRD patterns show that 1–3 are isostructural. All the compounds exhibit a molecular structure with the metal atom in an eight-coordination geometry. The photophysical processes involved in the photoluminescence of the complexes are investigated; as a result, the radiative lifetimes (τLn), the 4f–4f emission quantum efficiencies (ΦLn) and the energy-levels diagram are calculated.
Introduction
Lanthanide complexes exhibiting visible and NIR luminescence constitute a class of materials which receive increasing interest because of their potential applications in bio-sensing, multimodal luminescence imaging,1,2 data transmission by optical fibres and enhancing the conversion efficiency of solar cells.3–8 In particular, Yb(III) hybrid organic–inorganic materials are employed in bio-imaging,1,9 whereas the silica optical telecommunication window uses Er(III) doped materials.10,11 Moreover, the integration of down-shifting and down-conversion into metal–organic frameworks using Gd(III),12 and Yb(III) enhance the spectral conversion in VIS and NIR of solar cells.13 Despite its low molar absorption coefficient, the combination of the lanthanoid ions with organic ligands that exert the so-called antenna effect considerably enhance the photoluminescence (PL) by a down-shifting process (DS), and this is especially intense in complexes formed with β-diketonates and derivatives of phenanthroline.14–16It is generally accepted that the down-shifting process comprises the absorption of the radiation by the organic chromophores and their transition from its ground singlet-state to an excited singlet-state. Afterwards, an intersystem crossing (ISC) with a triplet state may occur. Subsequently, the ligand transfers the energy to the lanthanoid ion (ET), and the return of the lanthanoid ion to its ground-state produces the emission by the radiative deactivation.1,17
The fluorinated β-diketones, such as benzoyltrifluoroacetone (Hbta) and 2-thenoyltrifluoroacetone (Htta), are widely used ligands as antennas due to the high efficiency in all the steps of the process, Scheme 1. Their suitability as DS-ligands lays in their high absorption coefficient and the low energy losses by thermal vibration of the C–F bonds. Also, the conjugated bonds give rigidity to the entire molecule and the direct bond to the metal ion favours the energy transfer. The derivatives of phenanthroline, such as 5-methyl-1,10-phenanthroline (me-phen) and pyrazino[2,3-f][1,10]phenanthroline (pyz-phen) are also very efficient for their rigidity and the direct bond of the nitrogen atoms to the metal ion when the complex is formed. Moreover, the combination of both ligands produces a high coordination number that excludes the solvent molecules from the coordination sphere of the metal ions minimising the deactivation by non-radiative processes. Additionally, the singlet and triplet states of these ligands have the right energy for an effective energy transfer that results in high lifetimes and quantum yields.18–21
 | ||
| Scheme 1 Structure of Hbta, Htta, me-phen and pyz-phen. | ||
For a complete understanding of the photoluminescence process, the purity of the samples and the knowledge of the molecular structure of the complexes is crucial.2,15,16,22–24 In this sense, in this publication, we present the synthesis, the crystal structures determined by X-ray diffraction together with the photoluminescence study of five new complexes which combine β-diketonates and derivatives of the phenanthroline, namely [Er(bta)3(me-phen)] (1), [Yb(bta)3(me-phen)] (2), [Gd(bta)3(me-phen)] (3), [Yb(bta)3(pyz-phen)] (4), and [Er(tta)3(pyz-phen)] (5). Their PL properties and the commercial availability of the ligands make these compounds very suitable for photovoltaics and NIR imaging, as well as for data transmission.
Experimental
Materials and instrumentation
Er(NO3)3·5H2O, Yb(NO3)3·5H2O, Gd(NO3)3·6H2O, Hbta, Htta, 5-methyl-1,10-phenanthroline, triethylamine, pyrazino[2,3-f][1,10]phenanthroline, ethanol, acetonitrile and n-pentane were commercially available and used without further purification unless otherwise stated. All synthetic procedures were performed under an N2 atmosphere to avoid oxidation of the reagents. Elemental analyses (of C, H, N, S) and FT-IR (as KBr disks, between 400 cm−1 and 4000 cm−1) were recorded on a FLASH EA 1112 CHNS–O microanalyser and on a Thermo NicolletAvatar 360 FT-IR spectrometer, respectively. ESI-MS measurements were performed by electrospray ionisation technique (ESI) on a Micromass AutoSpec Mass Spectrometer. UV-visible spectra between 220 nm and 800 nm, were recorded on a Varian Cary 50 bio UV-visible Spectrophotometer with samples dissolved in ethanol. The emission spectra were measured with a 300–1100 nm ANDOR Newton Detector with a DU920P CCD. The samples were excited with a BrixX 375-200 HP OMICRON diode laser at 375 nm. The PL emissions in the IR region were measured with an 800–1800 nm ANDOR Newton Detector with also a DU920P CCD. Lifetime measurements were recorded on a LeCroy WS 424 Oscilloscope. An EKSPLA/NT342/3/UVE Optical Parametric Oscillator (OPO) pulsed laser was used to excite the sample.Synthesis of complexes 1–5
Characterization of the complexes
X-ray diffraction data acquisition and analysis
Single-crystal X-ray diffraction data were collected at 293(2) K with an Agilent SuperNova diffractometer with micro-focus X-ray on Cu-Kα radiation (λ = 1.5418 Å) for compounds 2 and 4 and on Mo-Kα radiation (λ = 0.71073 Å) for compound 5. CrysalisPro software was used to collect, index, scale and apply analytical absorption correction.25For compound 2, the hydrogen atoms of the CH3 group of the 5-methyl-1,10-phenanthroline were positioned geometrically (C–H = 0.98 Å) and refined using a riding model (AFIX 137) with Uiso(H) = 1.5Ueq(C). SIMU restraints were used to model the slight disorder displayed by the ligand pyrazino[2,3-f][1,10]phenanthroline in compound 4. One thienyl group of compound 5 (ligand D) is disordered in two possible orientations related by a 180° rotation with respect to the C–C bond in the ring. This disorder was modelled using PART and SADI commands. Furthermore, RIGU restraints were necessary to restrain all anisotropic ADPs on ligands B and C.
Powder X-ray diffraction (PXRD) patterns were collected with a PANanalytical Empyrean X-ray diffractometer with Cu Kα radiation = 1.54184 Å at room temperature.
Result and discussion
Preparation of the compounds
The compounds were prepared under a dinitrogen atmosphere in order to avoid oxidation of the reagents. The aim to use only commercial products is to obtain low-cost species easily available and reproducible in a large scale fabrication process. Instead of using water, where the reagents and products have poor solubility, ethanol was used as solvent and triethylamine as a base for the deprotonation of the β-diketones to form the β-diketonates. The reaction is completed after 3 h at 60 °C and the addition of water produces the precipitation of the crude product in high yield. The compounds were crystallised and purified by a vapour-diffusion method with acetonitrile as solvent and vapour of heptane as precipitating agent. The recrystallized products were used for all the measurements and characterisations, namely, IR and UV-vis spectroscopy, elemental analysis, powder diffraction and single-crystal X-ray diffraction, this latter when suitable crystals were obtained.Description of the structures
Selected crystal data and structure refinement parameters are shown in Table 1 for 2, 4 and 5 complexes. Powder diffraction pattern analyses, Fig. S4,† reveal that complexes 1–3 are isostructural. We have solved the crystal structure of 2 by single-crystal X-ray diffraction and the description given for the structure of 2 is also valid for 1 and 3, with the obvious replacement of Yb(III) by Er(III) and Gd(III), respectively. The [Yb(bta)3(me-phen)] complex (2) crystallises in the monoclinic P21/c space-group and has a molecular structure in which the Yb(III) ion is bound to six oxygen atoms of three β-diketonate ligands and also bound to two nitrogen atoms of one 5-methyl-phenanthroline ligand, Fig. 1. We have labelled the diketonate ligands as B, C and D for a better description of the structure (see Fig. S1, ESI†).| a R1 = [∑(||Fo|−|Fc||)/∑|Fo|].b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | |||
|---|---|---|---|
| Complex | 2 | 4 | 5 |
| Empirical formula | C43H28F9N2O6Yb | C44H26F9N4O6Yb | C38H20ErF9N4O6S3 |
| M/g mol−1 | 1012.71 | 1050.73 | 1063.02 |
| Temperature/K | 293 | 293 | 293 |
| λ/Å | 1.54184 | 1.54184 | 0.71073 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P21/c | Pna21 | Pbca |
| a/Å | 10.1647 (3) | 21.3482 (3) | 21.0780 (3) |
| b/Å | 37.6790 (9) | 10.96337 (12) | 18.3155 (2) |
| c/Å | 10.9281 (3) | 18.2233 (2) | 21.2034 (2) |
| α/° | 90 | 90 | 90 |
| β/° | 91.586 (2) | 90 | 90 |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 4183.8 (2) | 4265.12 (9) | 8185.66(17) |
| Z | 4 | 4 | 8 |
| Dcalc/g cm−3 | 1.608 | 1.636 | 1.725 |
| μ/mm−1 | 4.93 | 4.87 | 2.30 |
| Theta range/° | 4.2–73.9 | 4.2–67.1 | 1.8–28.5 |
| No. of measured, independent, and observed reflections | 29578, 8356, 7347 | 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 621, 7614, 7493 621, 7614, 7493 |
106005, 9984, 7529 |
| Rint | 0.036 | 0.032 | 0.029 |
| GOF on F2 | 1.15 | 1.05 | 1.03 |
| R1[I > 2σ(I)]a | 0.060 | 0.033 | 0.040 |
| wR2[I > 2σ(I)]b | 0.1247 | 0.0925 | 0.1026 |
| Flack parameter | −0.027 (4) | ||
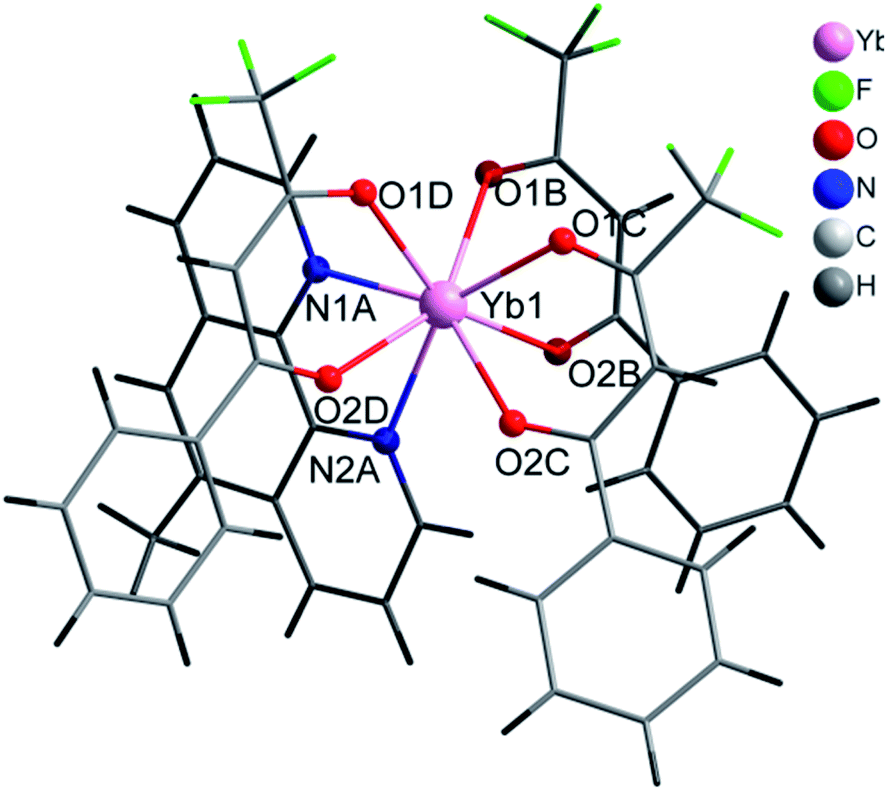 | ||
| Fig. 1 Coordination environment of Yb(III) and molecular structure of 2. | ||
The central Yb atom exhibits an eight-coordination with a distorted square antiprismatic geometry. The atoms N1A, N2A, 01B and O2B; and 01C, 02C, O1D and O2D give rise to the square planes (Fig. 2). The Yb–N and Yb–O distances span in the range 2.5239(1)–2.4907(1) Å and 2.2633(1)–2.3188(1) Å, respectively, which are within the expected range in this kind of complexes (Tables 2 and S4, ESI†).28–30 The atoms of the diketonate group and the aromatic ring of the bta− ligands adopt a highly-flat structure with a small twist of the phenylene ring.
 | ||
| Fig. 2 Local coordination geometry of the Yb(III) and Er(III) ions in 2, 4 and 5 complexes. | ||
| [Yb(bta)3(me-phen)] (2) | [Yb(bta)3(pyz-phen)] (4) | [Er(tta)3(pyz-phen)] (5) | |||
|---|---|---|---|---|---|
| a The number in parenthesis is the standard deviation in the last digit. | |||||
| Yb1–N1A | 2.491(5)a | Yb1–N1A | 2.512(7) | Er1–N1A | 2.543(4) |
| Yb1–N2A | 2.524(5) | Yb1–N2A | 2.499(7) | Er1–N2A | 2.518(4) |
| Yb1–O1B | 2.319(4) | Yb1–O1B | 2.277(5) | Er1–O1B | 2.311(3) |
| Yb1–O2B | 2.283(4) | Yb1–O2B | 2.264(5) | Er1–O2B | 2.306(3) |
| Yb1–O1C | 2.282(4) | Yb1–O1C | 2.278(4) | Er1–O1C | 2.320(3) |
| Yb1–O2C | 2.271(4) | Yb1–O2C | 2.274(5) | Er1–O2C | 2.270(3) |
| Yb1–O1D | 2.286(4) | Yb1–O1D | 2.288(5) | Er1–O1D | 2.290(3) |
| Yb1–O2D | 2.263(4) | Yb1–O2D | 2.263(4) | Er1–O2D | 2.311(3) |
This feature maximises the conjugation of the double bonds in the molecule and results in a very efficient antenna effect.7,17,31 In the D molecule of bta−, the deviation is only of 2.92° whereas in B and C the twisting angle of the aromatic ring respect to the diketonate group is of 28.50° and 21.33°, respectively (Fig S1, ESI†). Intra and intermolecular interactions are responsible for the different orientations of the phenylene rings. In the case of the D ligand, we observe an intramolecular hydrogen bonding between an oxygen atom of the diketonate group and a carbon atom of the phenylene ring, C6D–H6D⋯O2D with a distance of 2.701 Å, that retains the planarity of the molecule.
On the other hand, intermolecular π–π stacking interactions between the aromatic ring of the B molecule with a neighbouring 5-methyl-phenanthroline deviates the phenylene ring of the B molecule 28.50° from the planarity (Table S1, ESI†). All the bta− ligands in this complex show the same orientation of the –CF3 groups which point all of them to the same direction (Fig. S1, ESI†). The me-phen ligand shows its typical coordination mode with its two donor nitrogen atoms directly bound to the Ln(III) ion. The rest of the intermolecular interactions are rather weak and favour the solubility of the complex in solvents such as CH2Cl2 of CH3CN. The voluminous character of the ligands and the high coordination number of the complex prevents the entry of water molecules into the coordination sphere of the metal ion which would deactivate the molecule by non-radiative vibrating processes7 and also excludes crystallisation solvent molecules.
The complex [Yb(bta)3(pyz-phen)] (4) crystallises in the orthorhombic Pna21 space-group and similar to 2 the compound has a molecular structure with the Yb atom surrounded by three bta− β-diketonate ligands (labelled as B, C and D, Fig. S2, ESI†) and one pyrazino[2,3-f][1,10]phenanthroline ligand, Fig. 3. The compound is isostructural with the previously reported analogous Er(III) complex.28 The eight-coordination number is achieved with two nitrogen atoms from the derivative of the phenanthroline and the six oxygen atoms from the three bta− ligands. In this complex, the atoms that form the two planes of the square antiprism around the metal ion are N1A, N2A, O1D and O2D; O1B, O2B, O1C and O2C, Fig. 2. The Yb–N distances are 2.5132(1) Å and 2.4991(1) Å whereas the Yb–O distances span in the range 2.2624(1)–2.2889(1) Å which is comparatively slightly shorter than those observed in complex 1, Table 2. The bta− ligands also adopt a highly-flat structure with deviations from the planarity of the aromatic ring respect to the diketonate group of 18.61°, 13.46° and 7.63° for B, C and D respectively. The smaller deviation occurs for D, which exhibits an intramolecular hydrogen bonding between an adjacent carbon atom of the phenylene ring and an oxygen atom of the diketonate, C6D–H6D⋯O2D with a distance of 2.698 Å. B and C molecules exhibit π–π stacking interactions between their phenylene groups and neighbouring pyz-phen ligands, resulting in more significant deviations (Table S1, ESI†). One structural difference between 2 and 4 is the relative orientation of the –CF3 groups of the bta− ligands in both complexes. In 2 all these groups point to the same direction. In contrast, in 3 they are alternated, with two of them with the same orientation and the central one contrary oriented respect the other two (Fig. S1 and S2, ESI†). The pyz-phen group adopts its normal coordination mode in these complexes.23 Another feature of this compound is its chirality. The bta− and pyz-phen ligands are non-chiral, but when they form the complex, the phenylene rings deviate from the planarity leading to a chiral molecule. The crystallisation of the chiral molecules in a non-centrosymmetric space group results in a chiral compound.
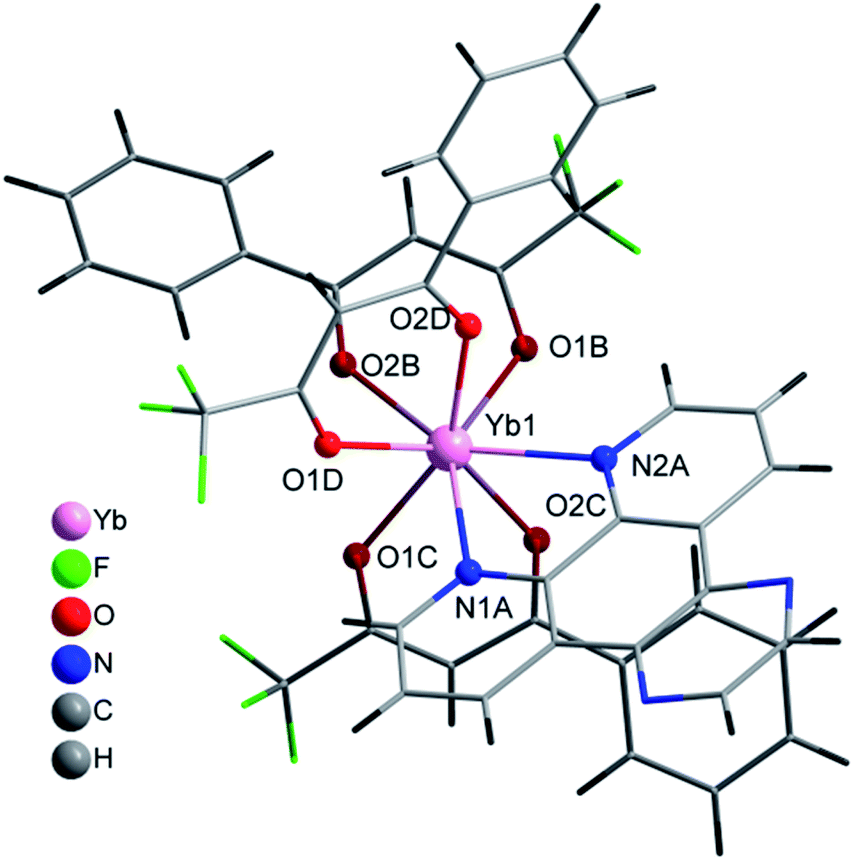 | ||
| Fig. 3 Coordination environment of Yb(III) and molecular structure of 4. | ||
Compound 5 crystallises in the orthorhombic Pbca space-group and its structure consist of molecules in which the Er(III) ions are surrounded by three tta− β-diketonate ligands (labelled as B, C and D) and one pyz-phen ligand, Fig. 4. The environment of the metal ion, with an eight-coordination, displays a distorted square-prismatic geometry with N1, N2, O1B and O2B; and O1C, O2C, O1D and O2D, respectively forming the planes, Fig. 2. The Er–N distances of 2.5444(1) Å and 2.5235(1) Å are slightly longer than those of 2 and 4, Table 2. Whereas the Er–O distances are not significantly longer than those of 2 but longer than those of 4 and span in the range from 2.2677(1) Å to 2.3246(1) Å. In this complex we observe a smaller deviation of the thienyl rings respect of the plane of the diketonate groups than in previous compounds and take values of 1.60°, 7.53° and 7.80° for C, B and D, respectively. Table S3, ESI,† shows the π–π staking interactions among the thienyl rings and the neighbouring pyz-phen molecules. In this complex, we also find an alternation in the relative orientation of the –CF3 groups of the tta− ligands (Fig S3, ESI†). The thiophene group is bound to the diketonate by a single bound, and therefore with free rotation which results in two possible orientations and some disorder is observed. The coordination mode and the geometry of the pyz-phen ligand are as expected.23
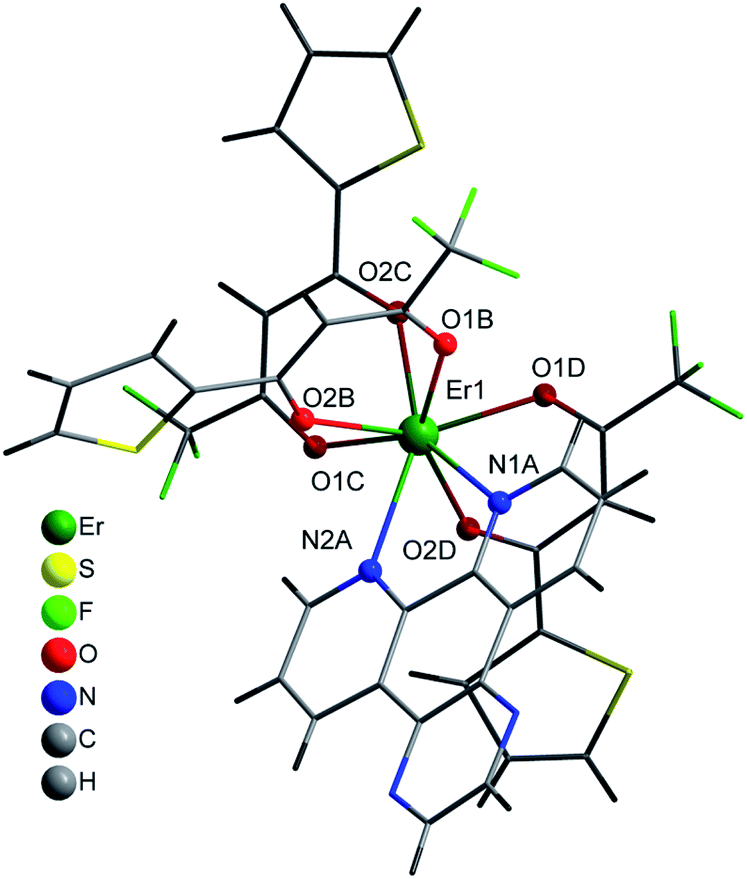 | ||
| Fig. 4 Coordination environment of Er(III) and molecular structure of 5. | ||
X-ray powder diffraction
The positive match of the PXRD patterns of 1–3 indicates their isostructural character. Moreover, the simulated PXRD patterns of 2, 4 and 5 correspond to their experimental diffractograms which supports the correspondence between the single-crystals and the bulk polycrystalline material, Fig. S4–S6, ESI.†UV-vis spectra
The UV-vis absorption spectra of the complexes is dominated by that of the bta− and tta− ligands, but considerably enhanced since the complexes contain three molecules of the diketonate ligands plus the phenanthroline derivative.In this sense, the pattern of the absorption of 1–4 complexes is very similar around 320 nm since all the compounds contain the same diketonate ligand and the contribution of the phenanthroline derivative is expressed at lower wavelengths. 5 has a higher absorption coefficient at higher wavelengths due to the more intense absorption of the tta− ligands at higher wavelengths. All the complexes have little absorption in the visible region, and diluted solutions are almost colourless. The maximum in absorbance of the plots can be attributed to the singlet–singlet (1π–π*) absorptions of the ligands in the complexes and gives the energy of the singlet excited states that will be used for the construction of the energy levels diagram. The values obtained are 26700 cm−1 and 23500 cm−1 for the bta− and tta− ligands, respectively, which are values in agreement with those previously reported.5,28 The shoulder observed for pyz-phen at 310 nm and the band found for me-phen at 290 nm correspond also to the singlet–singlet (1π–π*) absorptions and their values correspond to those previously reported.28,32
Solid state photoluminescence of complexes 1–5
The photophysical measurements were performed with the samples as solids in order to avoid solvent effects. Fig. 6 shows the emission spectra of 1–5 complexes under excitation at 375 nm in order to investigate all the steps in the DS process. Complexes [Gd(bta)3(me-phen)] (3), and [Er(tta)3(pyz-phen)] (5) exhibit visible emission, whereas [Er(bta)3(Me-phen)] (1), [Yb(bta)3(me-phen)] (2), [Yb(bta)3(pyz-phen)] (4), and also [Er(tta)3(pyz-phen)] (5) display NIR-emission. The energy of the excited levels of Gd(III) in 3 is above the energy of the excitation radiation and what we observe is the phosphorescence of the ligands as a consequence of the T1 → So transition (T1 is the lowest-energy excited triplet state of the ligands and S0 the singlet ground state), Fig. 6c. The visible emission of complex 5 corresponds to the phosphorescence of the ligands in addition to a small emission due to the 4S3/2 → 4I15/2 transition (about 550 nm) of the Er(III) showing some structure associated with the Stark components of the involved levels, Fig. 6a. This emission range is especially attractive for enhancing the conversion efficiency of standard Si-based solar cells with very low external quantum efficiency (EQE) in the UV spectral range.3–5,8 The same potential application can be considered for the complex 3, as it shows a wide emission band in the 550 nm spectral range. The NIR-emission observed for complexes 1 and 5 corresponds to the 4I13/2 → 4I15/2 transition of the Er(III), Fig. 6b. It is important to note that it is an intense emission, which is interesting for optical telecommunication processes,33 and it shows a relevant structure due to the Stark component of the involved levels. Fig. 6d, shows PL spectra of complexes 2 and 4, which consist of an intense NIR-emission centred at 980 nm that corresponds to the typical 2F5/2 → 2F7/2 transition for Yb(III). The emission band centred at 980 nm is also attractive for the application of these compounds in solar cells, as the EQE in standard Si-based solar cells is usually higher about 980 nm than in most of the UV spectral range of the sun irradiation.Fig. 7 shows the decay curves of the photoluminescence obtained under excitation at 375 nm and detecting the emission of the respective lanthanide ions (1550 nm for 1 and 5; and 980 nm for 2 and 4). All the curves have a characteristic behaviour with an initial fast rise time and long decay time. Initially, under direct excitation in the ligand at 375 nm, there are fast processes due to the intersystem crossing (ISC), relaxation of the ligand and the ligand to lanthanide energy transfer process (ET). After that, the excited lanthanide relaxes with a lifetime τLn. The following equation relates the temporal evolution of the intensity with the ligand and Ln lifetimes:
| I(t) = K[e−t/τLn − e−t/τOrg] | (1) |
However, the absence of emissions from the ligands in the samples 1, 2 and 4, and the fast-rising time observed in Fig. 7, indicates that the ligand to lanthanide rate transfer is very high and that the ET is highly efficient.
The decay component of the plots is related to the intrinsic relaxation of the lanthanide ions. All the curves have an exponential character with lifetimes values (τLn) given in Table 3, compared with those of related complexes. The excellent match of the data to the exponential decay indicates the occurrence of a unique site for the lanthanide ions in the complexes, in agreement with the structural analysis. The NIR emission obtained from the lanthanide ions is very interesting for optical applications (telecommunication and optical amplification), therefore is interesting to obtain the intrinsic quantum yield of the emission given by:
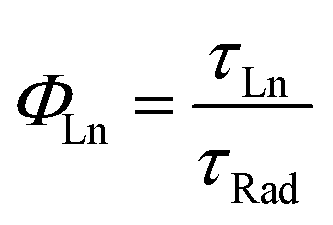 | (2) |
000 μs, and for the emission at 980 nm of Yb(III) ions, this value is about 2000 μs.28,42,43 Respect to the Er(III) ions, the obtained values for the intrinsic quantum yield are similar to values obtained in other azaciclo–diketonate complexes.28,29,31,38–40,43 However, for the Yb(III) the values span in a wider range. Nevertheless, the values obtained for our complexes compare well with previously reported and make them very suitable for optical amplifiers working at about 1000 nm.10
| Complexes | Lifetime (μs) | ΦLn (10−4) | Ref. |
|---|---|---|---|
| a Htta- = 2-thenoyltrifluoroacetone; Hbta = 1-benzoyl-3,3,3-trifluoroacetone; Hdbm = dibenzoylmethane; Htfac = 1,1,1-trifluoro-2,4-pentanedione; Htpm = 1,1,1-trifluoro-5,5-dimethyl-2,4-hexanedione; Htfod = 6,6,7,7,8,8,8-heptafluoro-2,2-dimethyl-3,5-octanedione; phen = 1,10-phenanthroline; mephen = 5-methyl-1,10-phenanthroline; pyz = 2,3-pyrazin-1,10-phenanthroline; bath = 4,7-diphenyl-1,10-phenanthroline; 5-NO2phen = 5-nitro-1,10-phenanthroline; bpy = 2,2′-bipyridine; dppz = dipyrido[3,2-a:2′,3′-c]phenazine. Htfnb = 4,4,4-trifluoro-1-(2-naphthyl)-1,3-butanedione, Htfa = 4,4,4-trifluoro-1-(2-furyl)-1,3-butanedione, Htfac = 1,1,1-trifluoro-2,4-pentanedione, Htpm 1,1,1-trifluoro-5,5-dimethyl-2,4-hexanedione, Hfhd = 1,1,1,5,5,6,6,7,7,7-decafluoro-2,4-heptanedione, Hh = 2,4-hexanedione, Hdmh = 2,6-dimethyl-3,5-heptanedione.28,29,31,36–41 | |||
| [Yb(bta)3(pyz)] | 6.1 | 31 | This work |
| [Yb(bta)3(me-phen)] | 7.2 | 36 | This work |
| [Yb(dbm)3(phen)] | 11.3 | 57 | 35 |
| [Yb(pfnp)3(phen)] | τ1 = 0,85; τ2 = 4.87 | — | 36 |
| [Yb(tta)3(phen)] | 12.0 | 74 | 37 |
| [Yb(tfnb)3(5NO2phen)] | 7.2 | 36 | 29 |
| [Yb(tfa)3(5NO2phen)] | 5.8 | 29 | 29 |
| [Yb(tfac)3(5NO2phen)] | 4.7 | 24 | 29 |
| [Yb(tpm)3(5NO2phen)] | 3.6 | 18 | 29 |
| [Yb(fhd)3(5NO2phen)] | 3.3 | 17 | 29 |
| [Yb(h)3(5NO2phen)] | 5.2 | 26 | 29 |
| [Yb(dmh)3(5NO2phen)] | 5.2 | 26 | 29 |
|
|||
| [Er(bta)3(me-phen)] | 1.61 | 1.15 | This work |
| [Er(tta)3(pyz)] | 1.30 | 0.93 | This work |
| [Er(Hbta)3(H2O)2] | 1.17 | 0.83 | 28 |
| [Er(Hbta)3(bpy)] | 1.19 | 0.85 | 28 |
| [Er(Hbta)3(phen)] | 2.06 | 1.47 | 28 |
| [Er(Hbta)3(pyz)] | 3.27 | 2.33 | 28 |
| [Er(Hbta)3(dppz)] | 4.53 | 3.24 | 28 |
| [Er(tpm)3(5-NO2phen)] | 1.53 | 1.09 | 29 |
| [Er(tpm)3(bipy)] | 1.77 | 1.26 | 29 |
| [Er(tpm)3(bath)] | 1.55 | 1.11 | 29 |
| [Er(tfac)3(bpy)] | 1.65 | 1.18 | 31 |
| [Er(tfac)3(bath)] | 1.40 | 1.00 | 31 |
| [Er(tfa)3(bipy)] | 1.24 | 0.89 | 38 |
| [Er(tfnb)3(bipy)] | 1.53 | 1.09 | 39 |
| [Er(tfac)3(bipy)] | 1.65 | 1.18 | 31 |
| [Er(tfac)3(5-NO2phen)] | 1.33 | 0.95 | 31 |
| [Er(tfod)3(bipy)] | 1.50 | 1.07 | 40 |
| [Er(fod)3(bath)] | 1.39 | 1.07 | 40 |
We can also observe in Table 3 that Yb(III) complexes have higher quantum yields than those Er(III) ones. This is in agreement with the energy gap law, that says that those lanthanide ions with larger energy gaps exhibit smaller non-radiative rate constants and, therefore, higher emission quantum efficiencies.44 Indeed, the 2F5/2 → 2F7/2 magnetic dipole transition of Yb(III), with ∼10200 cm−1, is more energetic than the 4I13/2 → 4I15/2 transition of Er(III) with 6500 cm−1.
Energy level diagram and DS process
The combined study of the UV-vis absorption and photoluminescence spectra of 1–5 complexes allows estimating the energy of the relevant states in the energy absorption and energy transfers (ET) of the DS processes, Fig. 8. The energy of the singlet excited states of the ligands can be obtained from the absorbance UV-vis spectra (Fig. 5), and for the me-phen and pyz-phen ligands are found to be 32300 cm−1 and 34500 cm−1, which are in agreement with the literature values and somewhat higher than that of the unsubstituted 1,10-phenanthroline, 31000 cm−1.28,32 The energy of the triplet state of the pyz-phen has been taken from the literature as 21200 cm−1.28,32 We prepared the [Gd(bta)3(me-phen)] complex, and from its phosphorescence spectrum, we obtained the energy of the triplet state of the me-phen as 18400 cm−1, Fig. 6c. The energy of the excited states of the Gd(III) are well above those of the triplet states of the ligands and they cannot be deactivated by the metal ion leading to the phosphorescence which allows obtaining the energy of the lower energy triplet state of me-phen in 3. The excitation of the pure me-phen gives a very similar value, but a shift to higher wavelengths is found in compound 3, Fig S7, ESI.†
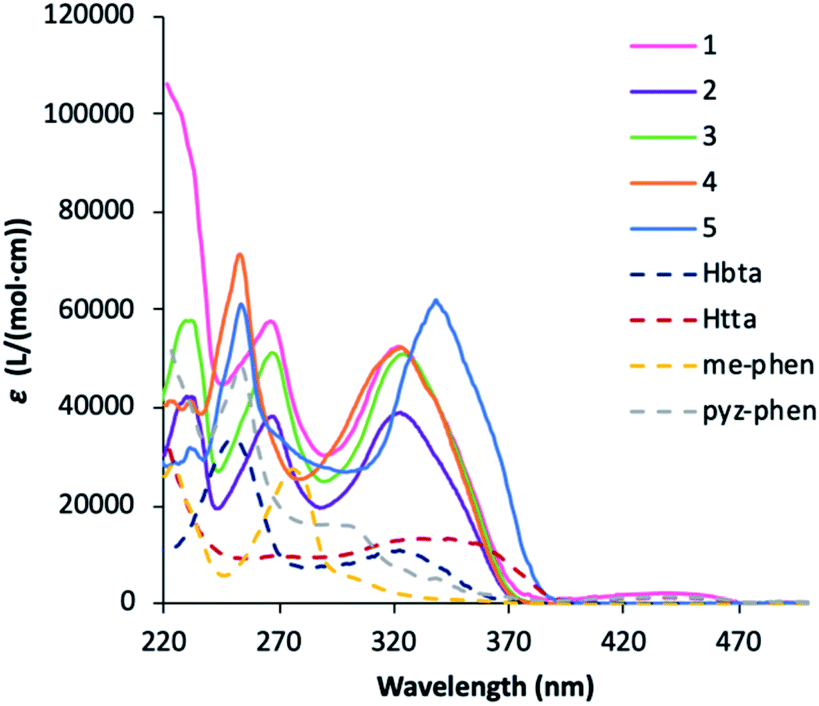 | ||
| Fig. 5 UV-vis spectra of the Hbta, Htta, me-phen and pyz-phen ligands together with 1–5 complexes in ethanol at c ≈ 10−5 M. | ||
 | ||
| Fig. 6 Emission spectra of complexes 1–5 in the visible and NIR regions excited at 375 nm. | ||
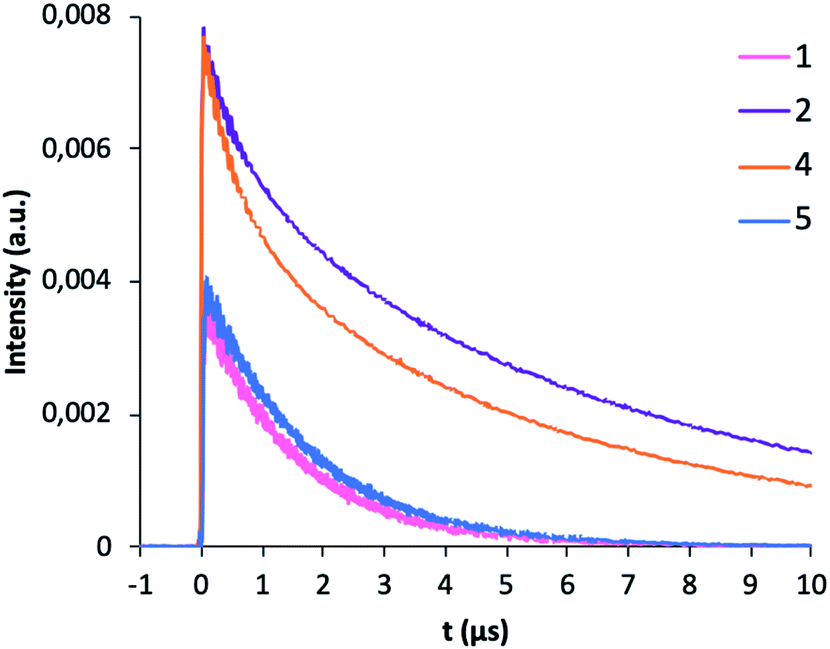 | ||
| Fig. 7 Room-temperature photoluminescence profile decay for complexes 1, 2, 4 and 5 excited at 375 nm. | ||
With all these energy values together with the energy levels of the Yb(III) and Er(III), we constructed the energy levels diagram shown in Fig. 8 which will allow a complete understanding of the DS process. The energy of the triplet states of the me-phen, pyz-phen, tta− and bta− ligands is higher than that of the 2F5/2 state of the Yb(III), and we observe the emission centred at 980 nm corresponding to the 2F5/2 → 2F7/2 transition. Therefore, there is an efficient energy transfer from the ligands to the Yb(III) ion in 2 and 4. In the same way, there is an efficient ET from the triplet states of the ligands to the 4I13/3 state of the Er(III) and its characteristic emission in the NIR centred at 1550 nm is observed in 1 and 5. However, the energy of the triplet state of the me-phen is not high enough to populate the 4S3/2 state, and complex 1 has no emission in the visible region. On the other hand, the higher energy of the triplet states of the pyz-phen ligand in 5 results in an efficient ET to the 4S3/2 state and the emission in the visible region occurs for this complex.
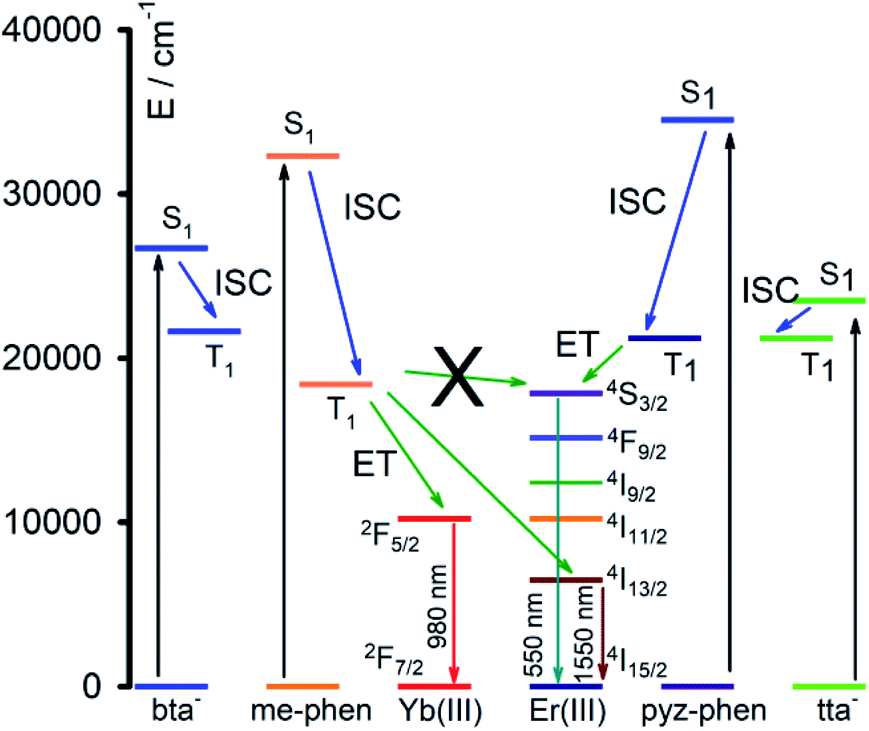 | ||
| Fig. 8 Scheme of the energy transfer mechanism and photoluminescence process. | ||
Conclusions
The complex formation by a combination of β-diketonate ligands, such as Hbta and Htta, with azacyclo ligands, such as pyz-phen and me-phen, with the Er(III) and Yb(III) ions afforded mixed-ligands visible-NIR light-emitting mononuclear species. Their structures were solved by single-crystal X-ray diffraction and showed that all the compounds are eight-coordinated. The voluminous character of the ligands avoids the occurrence of water molecules in the coordination sphere of the Ln(III) ions preventing the non-radiative deactivation by the coordinating water or other solvent molecules. The direct bond of the ligands through the nitrogen and oxygen donor atoms, the fluorinations of the diketonate ligands together with the high-enough energy of the excited states of the sensitizers, ensures an efficient energy transfer to the metal ion which results in high quantum yields, long lifetimes and strong NIR luminescence. The high intensity of the emission of the Er(III) complexes anticipates possible applications in optical amplifiers and the intense luminescence of the Yb(III) complexes may lead to applications in bio-imaging. Emissions bands centred in 550 nm and 980 nm for the different complexes of this study can be also attractive for increasing the conversion efficiency of solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the Spanish Ministerio de Ciencia, Innovación y Universidades (RTI2018-095563-B-I00 and MAT-2016-75586-C4-4-P). We thank SEGAI-SIDIX staff for their valuable technical support.References
- G.-Q. Jin, Y. Ning, J.-X. Geng, Z.-F. Jiang, Y. Wang and J.-L. Zhang, Inorg. Chem. Front., 2020, 7, 289–299 RSC.
- S. Dasari, S. Singh, S. Sivakumar and A. K. Patra, Chem.–Eur. J., 2016, 22, 17387–17396 CrossRef CAS PubMed.
- S. González-Pérez, J. Sanchiz, V. D. Rodríguez, D. Cañadillas-Ramallo, J. González-Platas, D. Borchert, B. González-Díaz, C. Hernández-Rodríguez and R. Guerrero-Lemus, J. Lumin., 2018, 201, 148–155 CrossRef.
- R. Guerrero-Lemus, J. Sanchiz, M. Sierra, I. R. Martín, C. Hernández-Rodríguez and D. Borchert, Sol. Energy Mater. Sol. Cells, 2018, 185, 312–317 CrossRef CAS.
- R. Guerrero-Lemus, J. Sanchiz, M. Sierra-Ramos, I. R. Martín, C. Hernández-Rodríguez and D. Borchert, Sens. Actuators, A, 2018, 271, 60–65 CrossRef CAS.
- H. Q. Ye, Z. Li, Y. Peng, C. C. Wang, T. Y. Li, Y. X. Zheng, A. Sapelkin, G. Adamopoulos, I. Hernandez, P. B. Wyatt and W. P. Gillin, Nat. Mater., 2014, 13, 382–386 CrossRef CAS PubMed.
- F. Artizzu, M. L. Mercuri, A. Serpe and P. Deplano, Coord. Chem. Rev., 2011, 255, 2514–2529 CrossRef CAS.
- B. González-Díaz, M. Sierra-Ramos, J. Sanchiz and R. Guerrero-Lemus, Sens. Actuators, A, 2018, 276, 312–319 CrossRef.
- S. V. Eliseeva and J. C. Bunzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- A. Polman and F. C. J. M. van Veggel, J. Opt. Soc. Am. B, 2004, 21, 871 CrossRef CAS.
- K. Kuriki, Y. Koike and Y. Okamoto, Chem. Rev., 2002, 102, 2347–2356 CrossRef CAS PubMed.
- Y. Yu, L. Lan and H. Cai, J. Phys. Chem. C, 2018, 122, 96–104 CrossRef CAS.
- Z. Hou, C. Liu, G. Feng, Z. Li and Y. Wang, Colloid Polym. Sci., 2016, 294, 1495–1501 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- O. T. Alexander, R. E. Kroon, A. Brink and H. G. Visser, Dalton Trans., 2019, 48, 16074–16082 RSC.
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 9408–9414 CrossRef CAS.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- S.-i. Kishimoto, T. Nakagawa, T. Kawai and Y. Hasegawa, Bull. Chem. Soc. Jpn., 2011, 84, 148–154 CrossRef CAS.
- A. P. Bassett, S. W. Magennis, P. B. Glover, D. J. Lewis, N. Spencer, S. Parsons, R. M. Williams, L. De Cola and Z. Pikramenou, J. Am. Chem. Soc., 2004, 126, 9413–9424 CrossRef CAS PubMed.
- F. M. Cabral, D. A. Gálico, I. O. Mazali and F. A. Sigoli, Inorg. Chem. Commun., 2018, 98, 29–33 CrossRef CAS.
- A. Gavriluta, T. Fix, A. Nonat, A. Slaoui, J.-F. Guillemoles and L. J. Charbonnière, J. Mater. Chem. A, 2017, 5, 14031–14040 RSC.
- XCalibur CCD System, CRISALISPRO Software System, Version 1.171.36.24 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hubschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- Q. Sun, P. Yan, W. Niu, W. Chu, X. Yao, G. An and G. Li, RSC Adv., 2015, 5, 65856–65861 RSC.
- P. Martin-Ramos, P. S. Pereira da Silva, V. Lavin, I. R. Martin, F. Lahoz, P. Chamorro-Posada, M. R. Silva and J. Martin-Gil, Dalton Trans., 2013, 42, 13516–13526 RSC.
- S. Dasari, S. Singh, P. Kumar, S. Sivakumar and A. K. Patra, Eur. J. Med. Chem., 2019, 163, 546–559 CrossRef CAS PubMed.
- P. Martin-Ramos, C. Coya, V. Lavin, I. R. Martin, M. R. Silva, P. S. Silva, M. Garcia-Velez, A. L. Alvarez and J. Martin-Gil, Dalton Trans., 2014, 43, 18087–18096 RSC.
- A. Hussain, D. Lahiri, M. S. Ameerunisha Begum, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2010, 49, 4036–4045 CrossRef CAS PubMed.
- S. Tanabe, J. Alloys Compd., 2006, 408–412, 675–679 CrossRef CAS.
- L. Y. Zhang, Y. J. Hou, M. Pan, L. Chen, Y. X. Zhu, S. Y. Yin, G. Shao and C. Y. Su, Dalton Trans., 2015, 44, 15212–15219 RSC.
- L. Abad Galan, B. L. Reid, S. Stagni, A. N. Sobolev, B. W. Skelton, E. G. Moore, G. S. Hanan, E. Zysman-Colman, M. I. Ogden and M. Massi, Dalton Trans., 2018, 47, 7956–7964 RSC.
- J. Feng, J. B. Yu, S. Y. Song, L. N. Sun, W. Q. Fan, X. M. Guo, S. Dang and H. J. Zhang, Dalton Trans., 2009, 2406–2414 RSC.
- L. N. Puntus, K. J. Schenk and J.-C. G. Bünzli, Eur. J. Inorg. Chem., 2005, 2005, 4739–4744 CrossRef.
- M. R. Silva, P. Martin-Ramos, J. T. Coutinho, L. C. Pereira, V. Lavin, I. R. Martin, P. S. Silva and J. Martin-Gil, Dalton Trans., 2015, 44, 1264–1272 RSC.
- P. Martín-Ramos, C. Coya, Á. L. Álvarez, M. Ramos Silva, C. Zaldo, J. A. Paixão, P. Chamorro-Posada and J. Martín-Gil, J. Phys. Chem., 2013, 117, 10020–10030 Search PubMed.
- P. Martín-Ramos, M. Ramos Silva, F. Lahoz, I. R. Martín, P. Chamorro-Posada, M. E. S. Eusebio, V. Lavín and J. Martín-Gil, J. Photochem. Photobiol., A, 2014, 292, 16–25 CrossRef.
- H.-H. Chen, D.-F. Wu, Y.-Y. Duan, L. Li, Y.-J. Wang, X.-M. Zhang, J.-Z. Cui and H.-L. Gao, New J. Chem., 2020, 44, 2561–2570 RSC.
- S. I. Klink, L. Grave, D. N. Reinhoudt, F. C. J. M. van Veggel, M. H. V. Werts, F. A. J. Geurts and J. W. Hofstraat, J. Phys. Chem. A, 2000, 104, 5457–5468 CrossRef CAS.
- M. H. V. Werts, J. W. Verhoeven and J. W. Hofstraat, J. Chem. Soc., Perkin Trans. 2, 2000, 433–439 RSC.
- G. Stein and E. Würzberg, J. Chem. Phys., 1975, 62, 208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, additional figures and tables. CCDC for complexes 2, 4 and 5 1983885–1983887. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05539e |
| This journal is © The Royal Society of Chemistry 2020 |