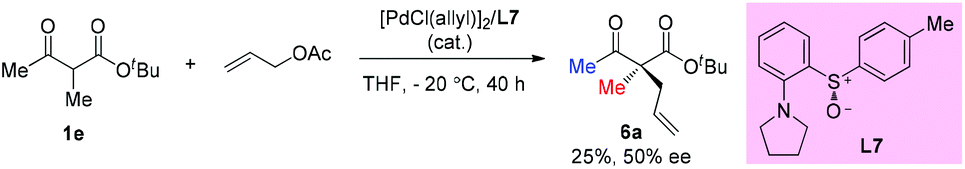DOI:
10.1039/D0QO00673D
(Review Article)
Org. Chem. Front., 2020,
7, 3557-3577
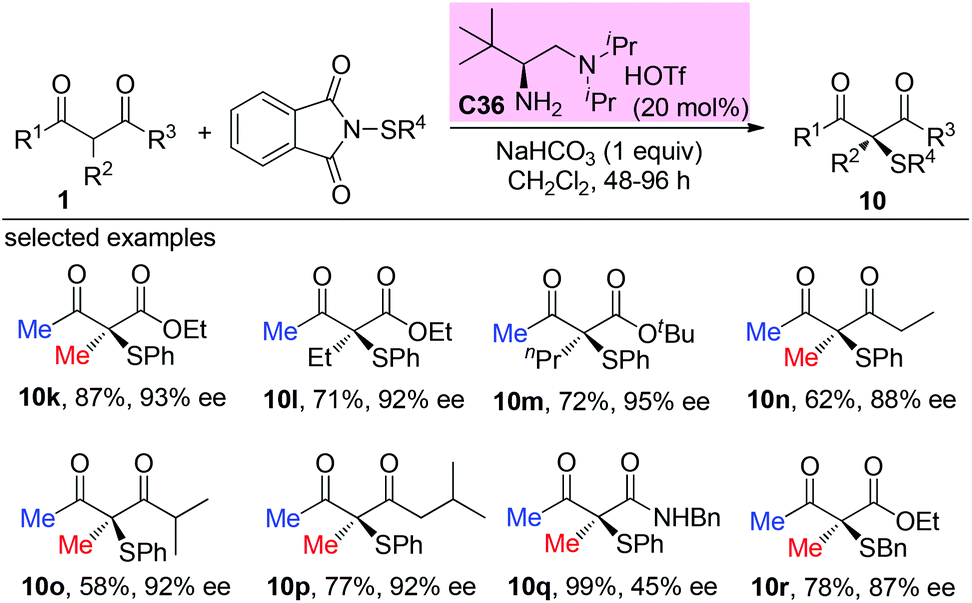
Asymmetric catalytic construction of fully substituted carbon stereocenters using acyclic α-branched β-ketocarbonyls: the “Methyl Rule” widely exists
Received
8th June 2020
, Accepted 20th August 2020
First published on 20th August 2020
Abstract
The catalytic asymmetric construction of tetrasubstituted carbon stereocenters constitutes one of the most challenging research topics in organic synthesis, which further plays vital roles in diverse fields including medicinal and materials chemistry. In this context, the α-functionalization of β-ketocarbonyl compounds serves as one of the most frequently used strategies to address the issue. Compared to cyclic β-ketocarbonyls, the catalytic asymmetric α-functionalization of acyclic β-ketocarbonyls is more difficult but has gained enough progress over the last several decades. This review illustrates the recent advances in this field, including asymmetric fluorination with acyclic α-branched β-ketocarbonyl participation, Michael addition, amination, aldol reaction, α-alkylation, α-alkynylation, α-oxygenation, Mannich reaction, etc. Furthermore, a thorough survey of all these reactions indicates the existence of a general principle which is called the “Methyl Rule”. Alternative methods that can complement the deficiency of reports of the use of direct asymmetric catalysis are also presented.
1. Introduction
Asymmetric catalysis mediated by α-substituted β-ketocarbonyl compounds plays an important role in organic synthesis, because the functionalization of the title compounds at the α-position constitutes one of the most frequently employed strategies for the construction of fully substituted carbon centers, and the corresponding enantioenriched products serve as valuable building blocks in a large number of further transformations for the synthesis of complicated molecules such as natural products and pharmaceuticals. However, compared with cyclic β-ketocarbonyls, acyclic β-ketocarbonyls are structurally more flexible and relatively less reactive, which bring about more challenges in achieving the highly stereoselective α-functionalization of acyclic β-ketocarbonyls. Nevertheless, the recent two decades have witnessed significant progress in addressing the issue via the development of various asymmetric catalytic systems. Therefore, a critical review summarizing the progress is currently necessary. Although some reactions described in this paper have appeared in the reviews discussing different topics such as asymmetric creation of quaternary carbon centers and chiral primary amine catalysis,1 a special review focusing on the utility of α-branched β-ketocarbonyls remains absent.
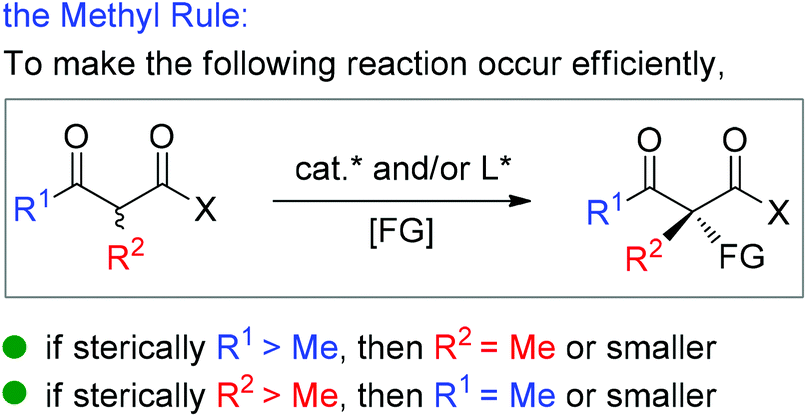
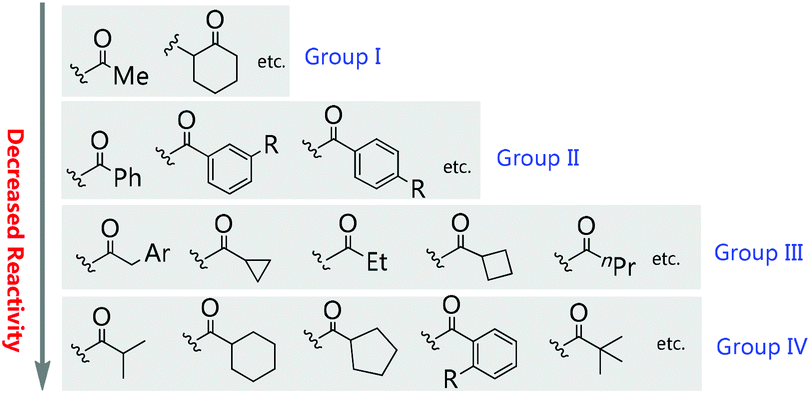
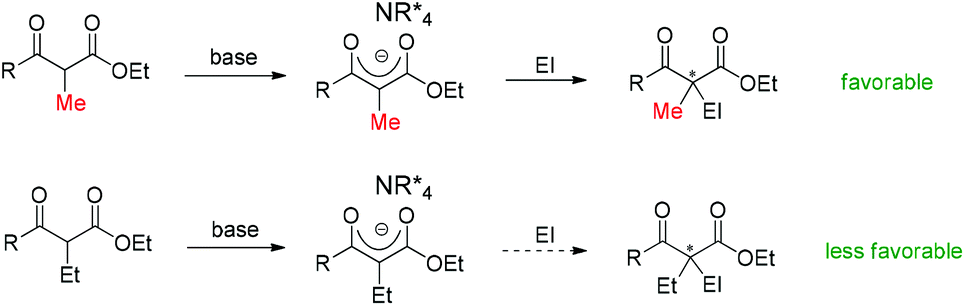
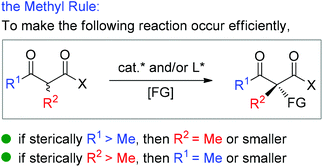
Moreover, during the preparation of the review and our recent studies on these substrate-mediated transformations, we found an interesting empirical rule, which we call the Methyl Rule. In more detail and as shown in Scheme 1, for a successful asymmetric catalytic reaction mediated by an α-substituted β-ketocarbonyl compound, when the α-substituent R2 is sterically bulkier than the Me group, the ketone substituent R1 can only be Me or a “smaller” group; correspondingly, when ketone substituent R1 is bulkier than Me, then R2 can only be Me or a “smaller” one. In short, at least one group of R1 and R2 needs to be Me or a “smaller” one; otherwise, the reaction would be reluctant to occur.
 |
| | Scheme 1 A general profile of the Methyl Rule. | |
In this review, we summarize the reports of asymmetric catalysis using α-substituted β-ketocarbonyls according to the reaction types, highlight the application of the Methyl Rule, and also emphasize the exceptional examples. A preliminary analysis of the origin of the rule is presented, and alternative methods that can complement the deficiency led by this rule to produce α,α-disubstituted β-ketocarbonyls with two bulky groups are also presented. We hope that this review can be useful for new reaction design, to evaluate the feasibility of a synthetic plan, and to inspire new catalyst and ligand discovery to break the Methyl Rule.
2. Asymmetric fluorination of β-ketocarbonyls
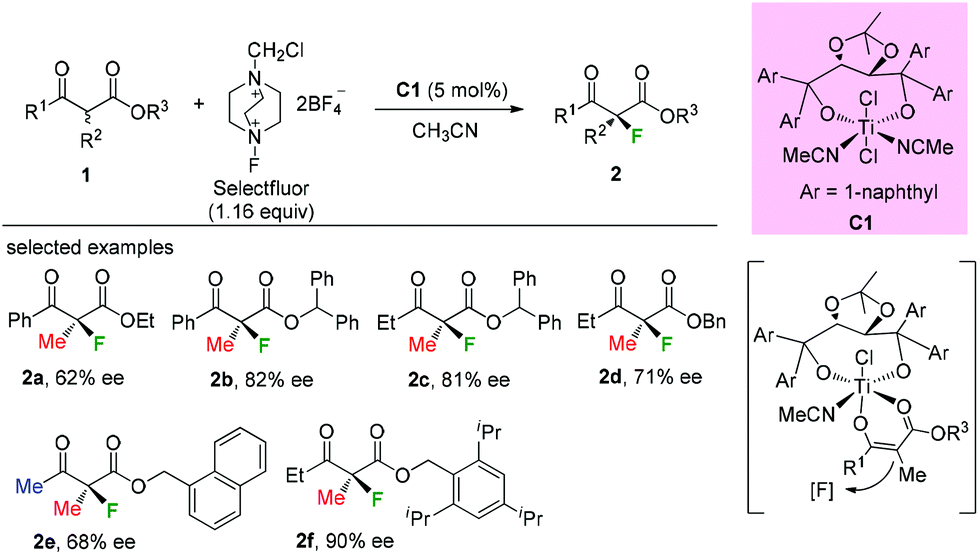
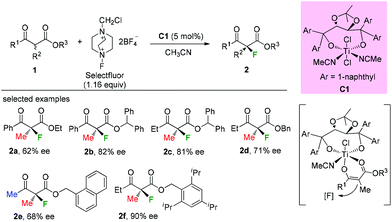
Early in 2000, Togni and co-workers developed for the first time the catalytic fluorination of β-ketoesters (Scheme 2).2a A titanium complex C1 played a vital role in obtaining products with good to high enantioselectivity. Only α-methyl-substituted β-ketoesters were tested, and the nature of the ester group was found to influence the enantioselectivity. A coordinated complex between Ti and the enol form of ketoesters was proposed, and the computational and experimental studies suggested that a single electron transfer (SET) mechanism may be involved in this process.2b
 |
| | Scheme 2 Asymmetric fluorinations of α-methyl-β-ketoesters with a Ti-complex. | |
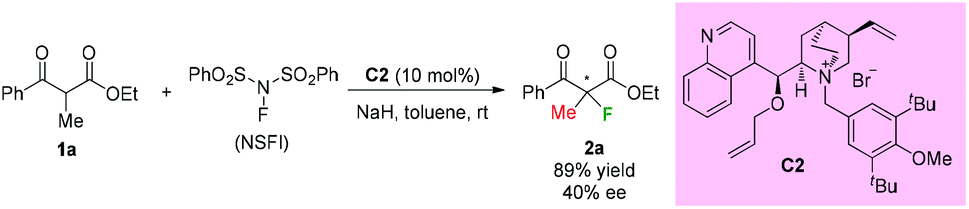
In the same year, a catalytic enantioselective α-fluorination of β-ketoesters was reported by Kim and co-workers using a phase transfer catalyst (PTC) C2 (Scheme 3).3 Although most of the substrates were cyclic ketoesters, they showed one example using ethyl benzoylpropanoate 1a, and the corresponding product 2a was generated in 89% yield with 40% ee.
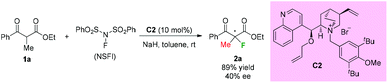 |
| | Scheme 3 Enantioselective fluorination of ethyl benzoylpropanoate using PTC C2. | |
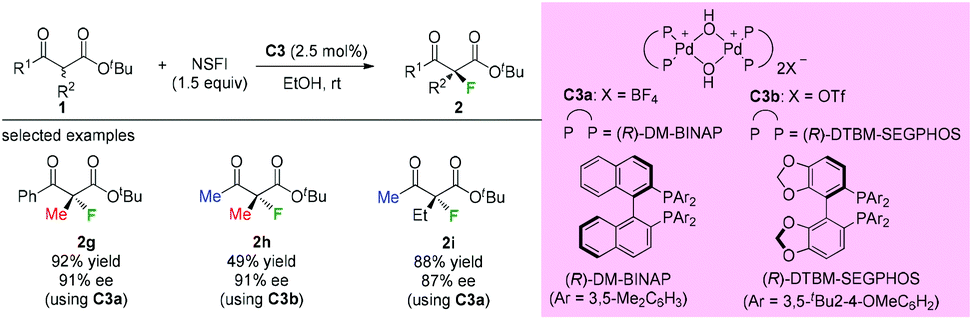
Subsequently, Sodeoka's group disclosed the highly enantioselective α-fluorination of various β-ketoesters catalyzed by chiral palladium complexes C3 (Scheme 4).4 Three acyclic substrates were examined, and 87–91% ee values were observed. It can be seen that the methyl rule is obeyed again in this study, and there must be at least one Me group within the substrates. Later studies from the same group showed that the catalysts could be reused when an ionic liquid was employed,5 and the system could be expanded to β-ketophosphonates and β-ketoamides, but again the α-substituent was limited to the Me group.6,7
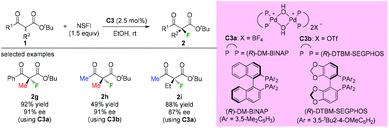 |
| | Scheme 4 Pd-Complex catalyzed asymmetric α-fluorination of β-ketoesters. | |
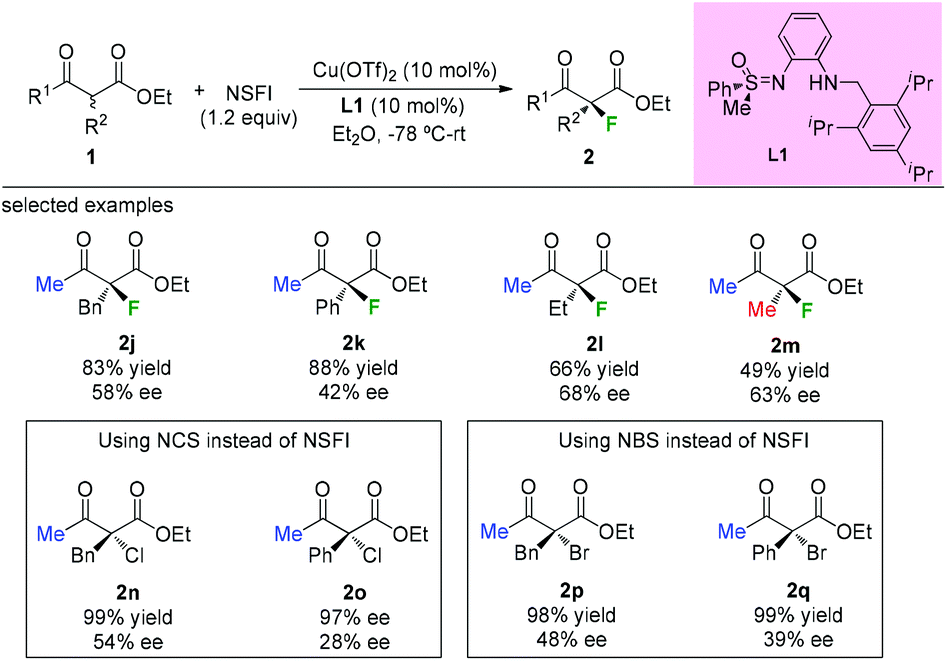
In 2009, the Bolm group developed a C1-symmetric amino sulfoximine chiral ligand L1 and used it in copper-catalyzed asymmetric fluorination of β-ketoesters (Scheme 5).8 In all cases, substrates bearing a methyl ketone moiety were tested, and diverse levels of enantioselectivities of the products were detected (2j to 2m). The same catalytic system can also be applied in chlorination and bromination reactions using methyl keto-esters (2n to 2q).
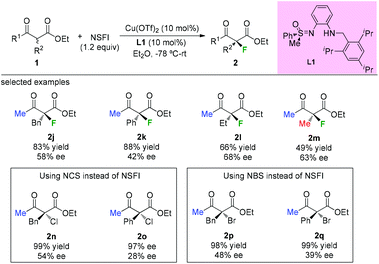 |
| | Scheme 5 Asymmetric halogenations of β-ketoesters with a copper complex. | |
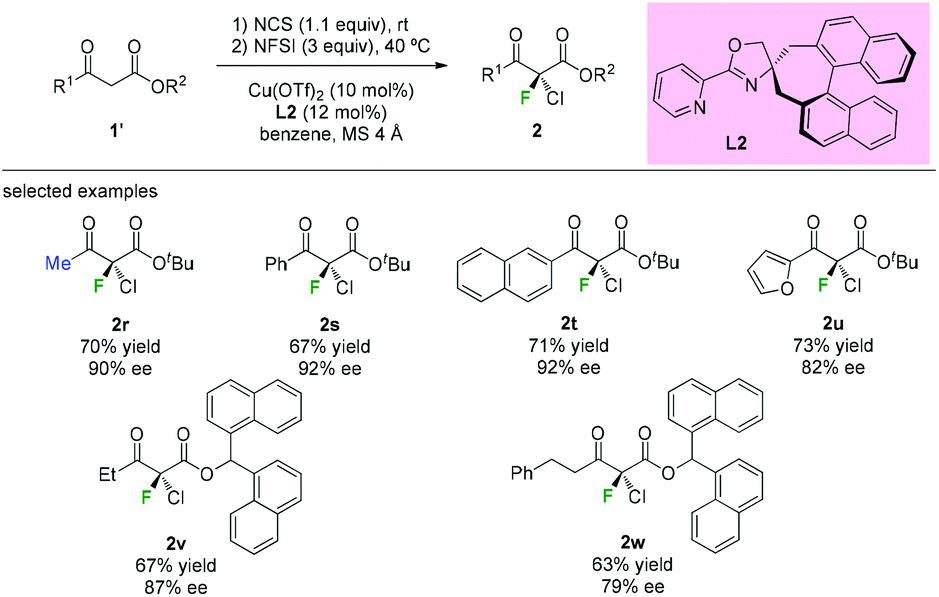
The highly enantioselective gem-chlorofluorination of an active methine compound catalyzed by a copper(II) complex of a chiral spiro pyridyl monooxazoline ligand L2 was demonstrated by Shibatomi and co-workers (Scheme 6).9a This approach could provide the chiral α-chloro-α-fluoro-β-ketoesters by a sequential double halogenation in one-pot operation. Several substrates, including aliphatic, aromatic, and heterocyclic β-ketoesters, were examined to furnish the corresponding dihalogenated products with good to high optical purity (79–92% ee) in this work. Using similar conditions, asymmetric chlorination was also achieved by the same group.9b
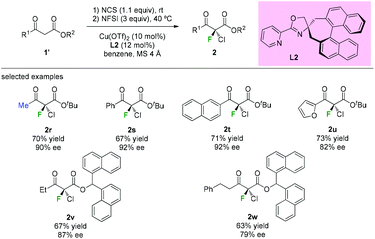 |
| | Scheme 6 One-pot asymmetric gem-chlorofluorinations of β-ketoesters. | |
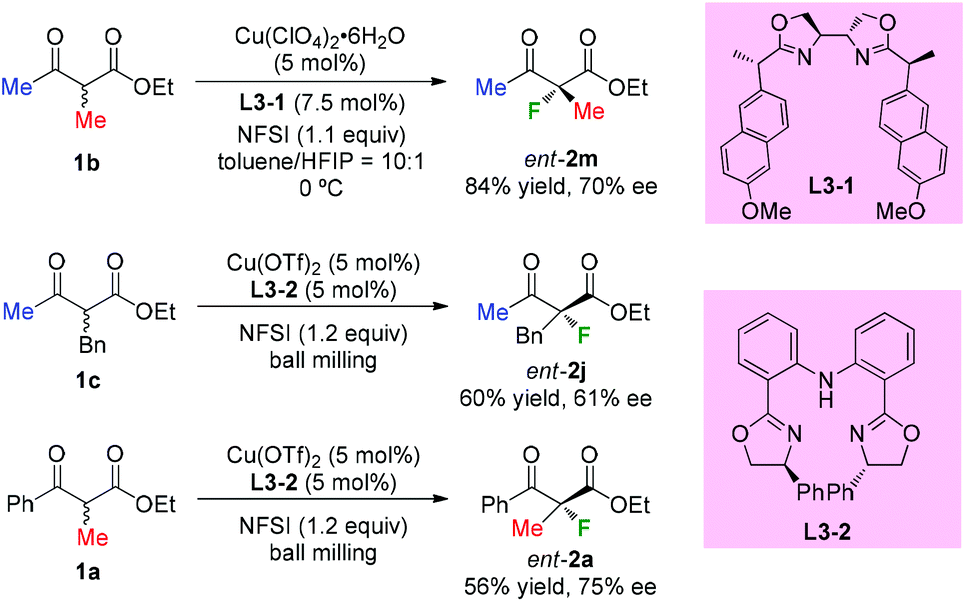
In 2013, Kesavan's group developed an enantioselective fluorination of β-ketoesters using a tartrate derived bidentate bioxazoline ligand L3-1-Cu(II) complex (Scheme 7).10a While most examples used cyclic ketoesters, there was one example using acyclic methyl-ketoester 1b, and the fluorination product ent-2m was obtained with 70% ee. Xu and co-workers reported in 2017 copper-catalyzed asymmetric fluorination under ball milling conditions. Ligand L3-2 was used, and moderate ee values were observed when acyclic products were surveyed (Scheme 7, ent-2j and ent-2a).10b
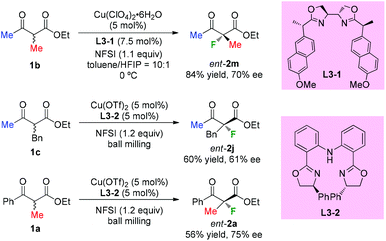 |
| | Scheme 7 Copper-catalyzed enantioselective α-fluorination of β-ketoesters. | |
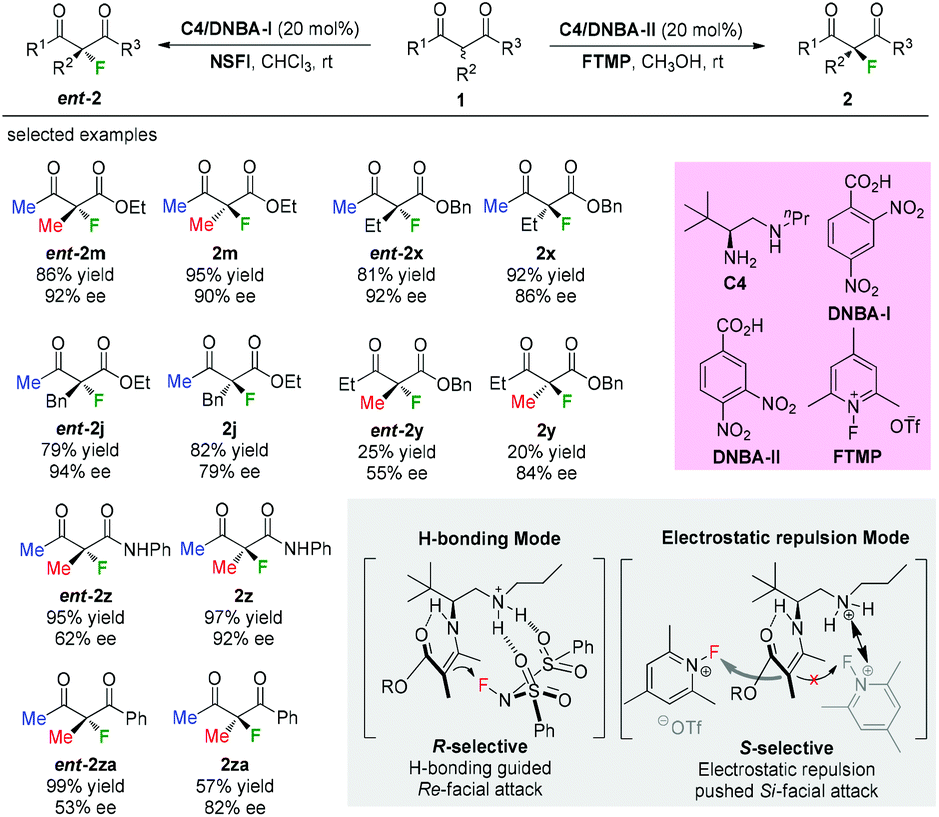
In 2016, Luo and co-workers disclosed an elegant primary amine catalyzed enantioselectivity switchable asymmetric fluorination of β-ketocarbonyls. Through a simple swap of the fluorination reagents, both enantiomers of the products could be obtained with moderate to high enantioselectivities (Scheme 8).11 Mechanistic studies indicated dual H-bonding and electrostatic stereocontrol modes for the catalysis. The Methyl Rule is clearly demonstrated in this study, and either the α-substituent or the ketone group needs to be Me, or both of them should be Me. It is noteworthy that products 2y and ent-2y were obtained in very low yields, indicating the relatively low reactivity when R1 is “bigger” than Me.
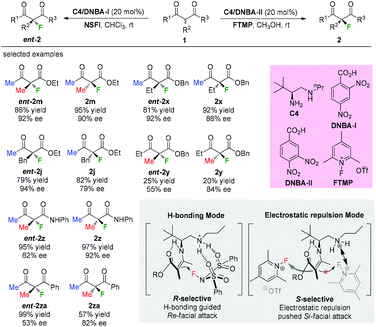 |
| | Scheme 8 Asymmetric fluorinations of β-ketocarbonyls by primary amine C4. | |
3. Michael additions mediated by acyclic α-substituted-β-ketocarbonyls
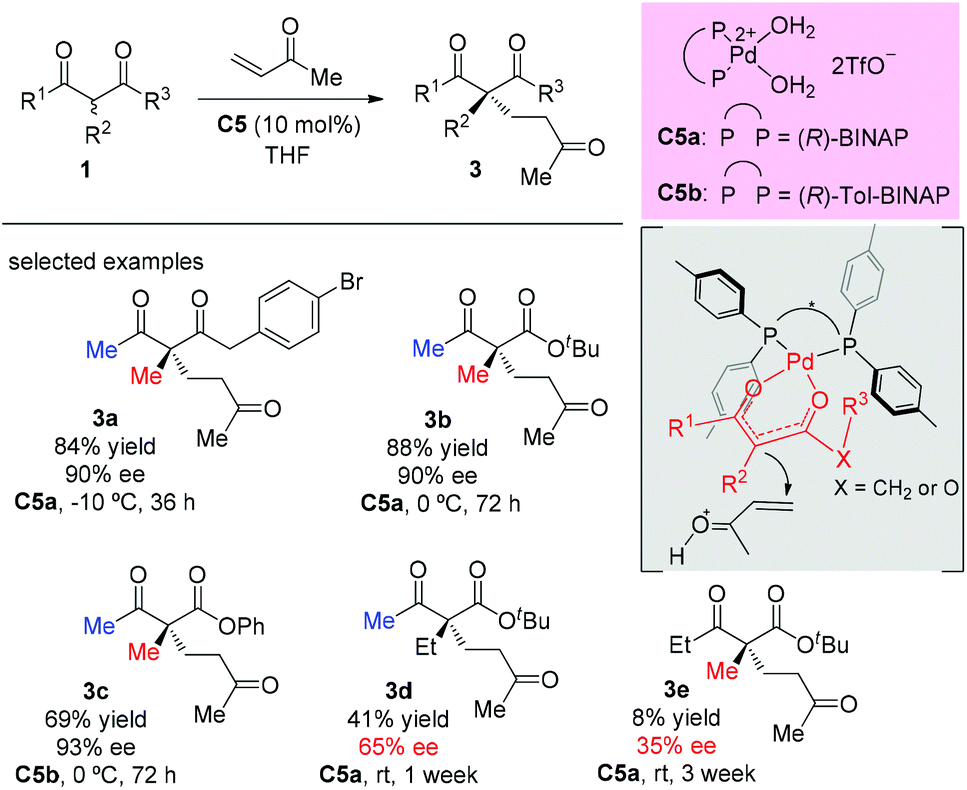
A palladium complex catalyzed asymmetric Michael addition between 1,3-dicarbonyls and vinyl ketones was disclosed by the Sodeoka's group in 2002 (Scheme 9).12,13 Mechanistically, the palladium complex allows successive supply of a Brønsted base and a Brønsted acid. The former activates the 1,3-dicarbonyl compounds to give the chiral palladium enolate, and the latter activates the enone. Among the acyclic dicarbonyls examined, all α-methyl-substituted methyl ketone substrates (3a to 3c) gave good yields and excellent ee (90–93%), but once one of the methyl groups was replaced by an ethyl group (3d and 3e), both the yields and the ee values dropped sharply (41% yield, 65% ee and 8% yield, 35% ee).
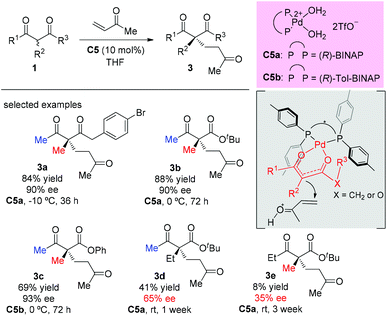 |
| | Scheme 9 Pd-Catalyzed asymmetric addition of 1,3-dicarbonyls to vinyl ketones. | |
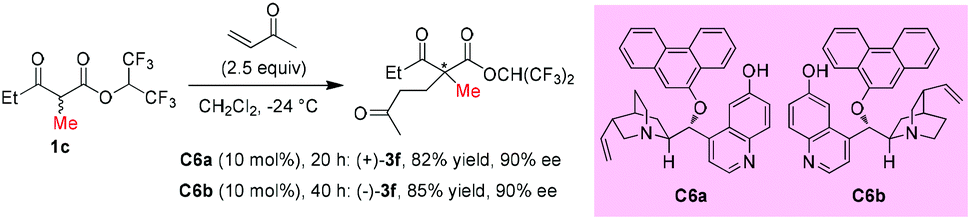
In 2006, Deng and co-workers described a 6′-hydroxy cinchona alkaloid catalyzed reaction that could provide direct access to a wide variety of 1,4-adducts between α-substituted β-ketoesters and α,β-unsaturated ketones in high yields with excellent enantioselectivities. The work showed one example using acyclic methyl ketoester to provide the 1,4-adducts (+)-3f and (−)-3f in 82% and 85% yields with 90% ee by using C6a and C6b, respectively, as the catalysts (Scheme 10).14
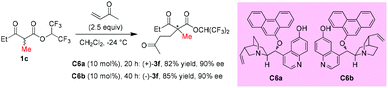 |
| | Scheme 10 Asymmetric additions catalyzed by cinchona alkaloids C6a and C6b. | |
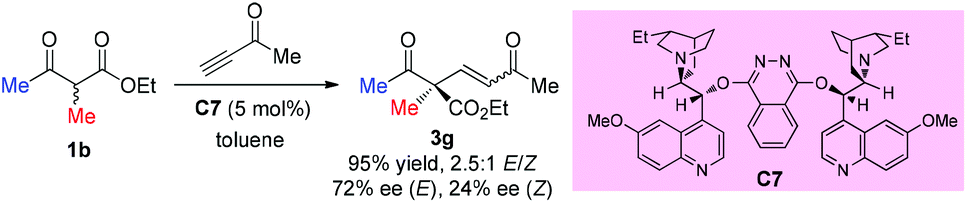
In 2004, Jørgensen et al. reported the first organocatalytic enantioselective conjugate addition of β-dicarbonyl compounds to alkynones (Scheme 11).15 The protocol works well for cyclic dicarbonyl compounds, producing products with a quaternary carbon center and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, which is useful for further transformations. One example is shown for ethyl 2-methyl-3-oxobutanoate, delivering the corresponding addition product 3g in high yield with moderate ee. A retro-Michael addition may account for the low ee of Z-3g.
C bond, which is useful for further transformations. One example is shown for ethyl 2-methyl-3-oxobutanoate, delivering the corresponding addition product 3g in high yield with moderate ee. A retro-Michael addition may account for the low ee of Z-3g.
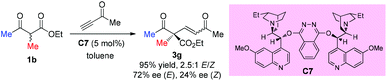 |
| | Scheme 11 Asymmetric 1,4-addition of β-ketoester to alkynone using catalyst C7. | |
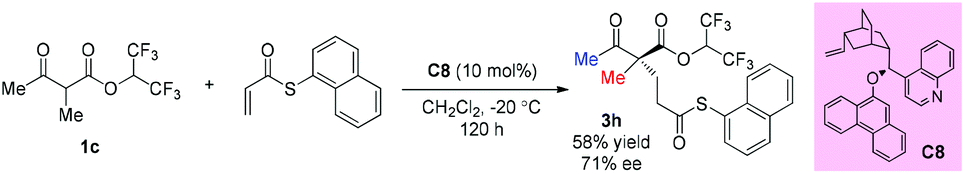
In 2008, Dixon's group reported an asymmetric Michael addition reaction with a β-ketoester as the pro-nucleophile catalyzed by a cinchona alkaloid derived bifunctional organocatalyst C8. Only one example using acyclic methyl ketoester was examined and the corresponding product was obtained in moderate yield with 71% ee (Scheme 12).16
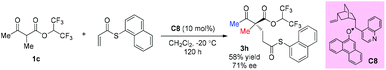 |
| | Scheme 12 Asymmetric addition of β-ketoester to vinyl thioester catalyzed by C8. | |
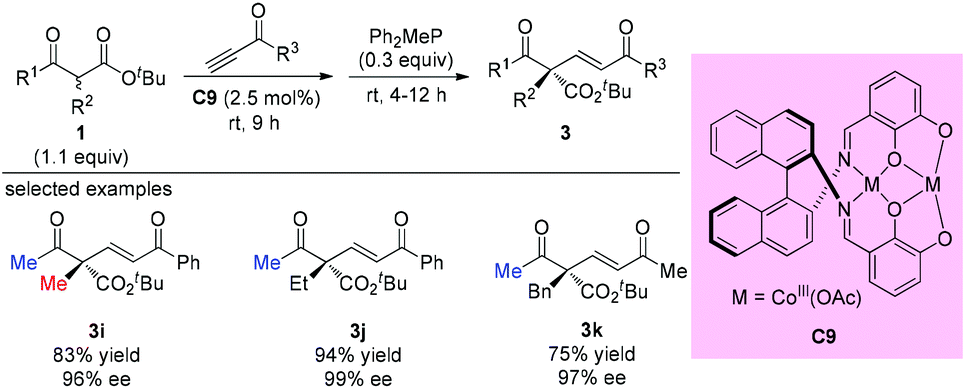
One year later, Matsunaga, Shibasaki, and co-workers reported a stable homodinuclear biscobalt(III)-Schiff base complex (C9) for the catalytic asymmetric 1,4-addition of β-ketoesters to alkynones. The reaction proceeds giving high yields and high enantioselectivities at room temperature under neat conditions without air or moisture sensitivity. All three samples using acyclic β-ketoesters contain methyl ketone units, and the α-substituents can be Me, Et, and Bn groups (3i to 3k) (Scheme 13).17a Subsequently, the same group employed C9 and the corresponding Ni-complex in asymmetric 1,4-additions of β-keto esters to nitroalkenes, and the corresponding adducts were obtained with good to excellent ee.17b,c
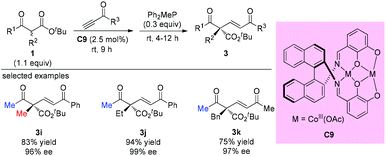 |
| | Scheme 13 Asymmetric addition of β-ketoesters to alkynones using a Co-Schiff base. | |
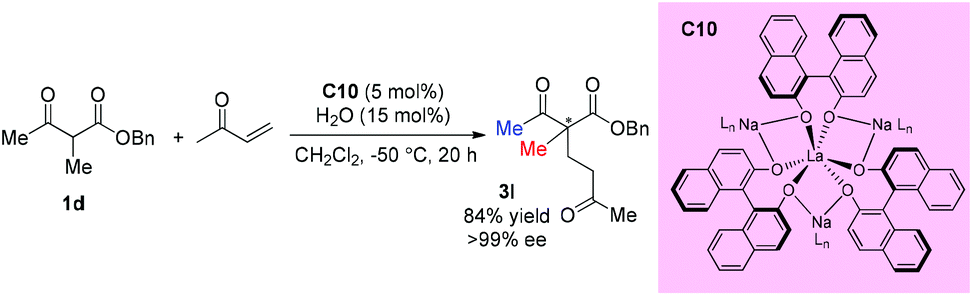
In 2014, Walsh and co-workers reported the self-assembly of novel hydrogen-bonded rare earth metal BINOLate complexes that serve as bench-stable precatalysts for Shibasaki's heterobimetallic complexes (REMB catalysts). The incorporation of hydrogen-bonded guanidinium cations in the secondary coordination sphere leads to improved stability toward moisture in solution and in the solid state. The catalyst can be used in diverse reactions including Michael additions, aza-Michael additions, and direct aldol reactions. The real catalyst C10 can be formed in situ, and one acyclic ketoester 1d was tested, and the corresponding Michael addition product was produced in good yield with excellent 99% ee (Scheme 14).18
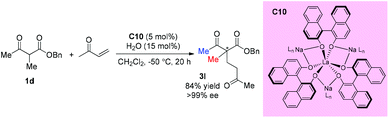 |
| | Scheme 14 Asymmetric Michael addition catalyzed by La complex C10. | |
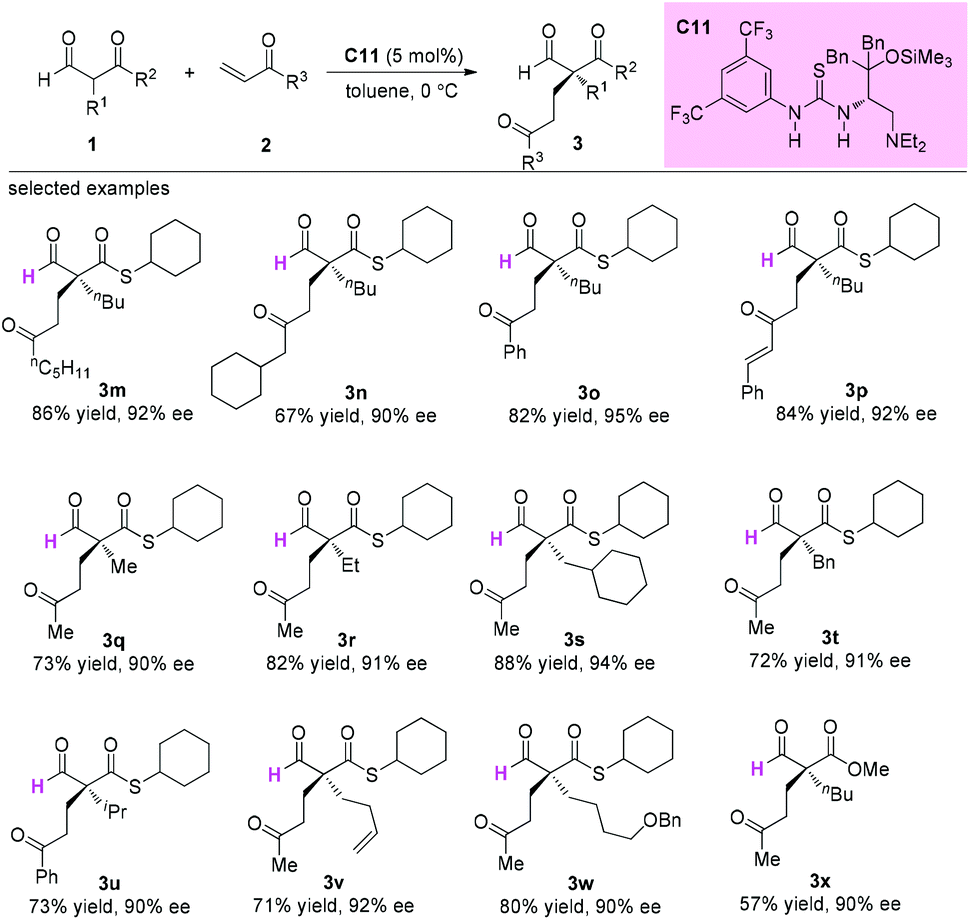
In 2015, Sugimura and co-workers developed an asymmetric 1,4-addition reaction of 2-formyl(thio)esters to vinylketones using a newly developed thiourea-tertiary amine catalyst C11 involving a quaternary carbon stereocenter construction in an acyclic system. In all cases, excellent enantioselectivities were observed for the products (Scheme 15).19
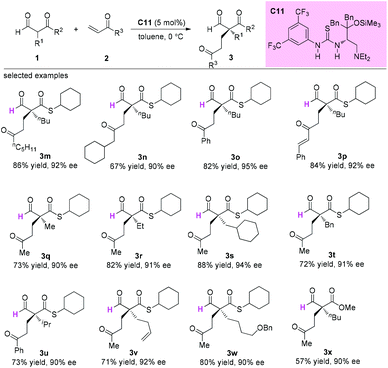 |
| | Scheme 15 Asymmetric addition of 2-formyl(thio)esters to vinyl ketones. | |
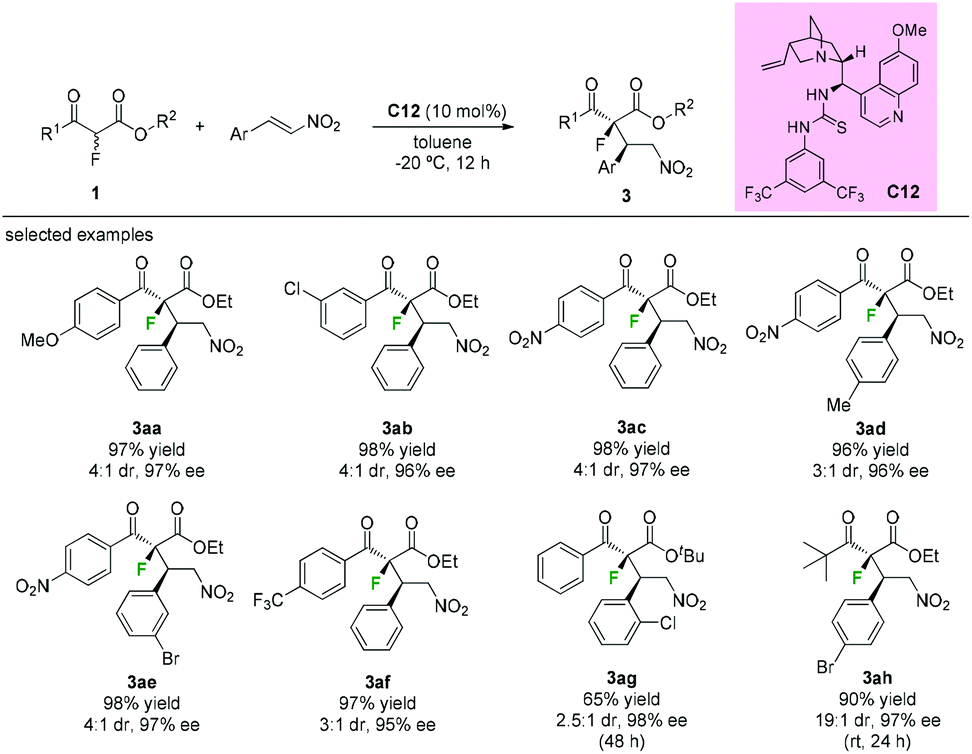
Besides fluorinations at the α-branched 1,3-dicarbonyls, asymmetric additions using α-fluorine β-ketoesters as nucleophiles are also efficient protocols to construct the fluorinated tetrasubstituted carbons adjacent to tertiary stereocenters. In 2009, Lu's group disclosed thiourea bifunctional organocatalyst C12-catalyzed asymmetric Michael reactions of α-fluoro-β-ketoesters with nitroolefins. A series of aromatic keto-esters furnished the desired adducts (3aa to 3ag) with moderate diastereoselectivities (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–4:1 dr) and excellent enantioselectivities (95–98% ee), and the use of a tert-butyl ketone group could improve the dr values to 19:1 while retaining the high enantioselectivities (97% ee) (Scheme 16).20
:1–4:1 dr) and excellent enantioselectivities (95–98% ee), and the use of a tert-butyl ketone group could improve the dr values to 19:1 while retaining the high enantioselectivities (97% ee) (Scheme 16).20
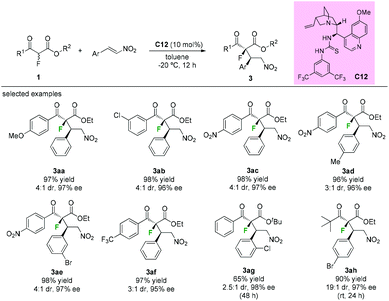 |
| | Scheme 16 Lu's construction of a fluorinated tetrasubstituted carbon stereocenter. | |
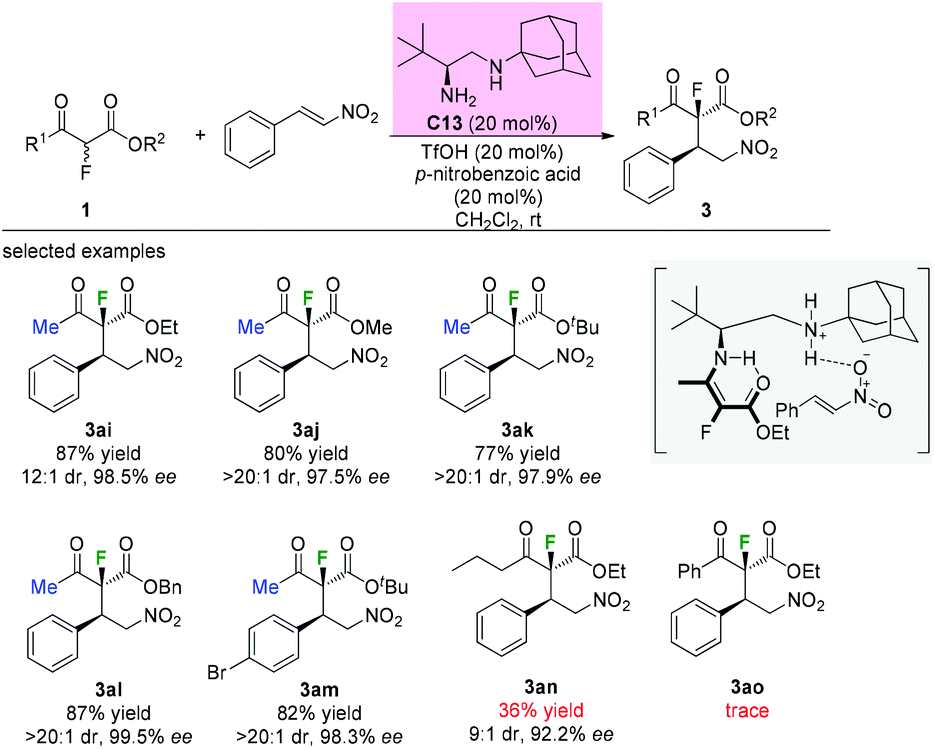
A similar addition reaction was reported by Zou and co-workers in 2015 using a different primary–secondary diamine catalyst. Diamine catalyst C13 (20 mol%), TfOH (20 mol%) and p-nitrobenzoic acid (20 mol%) in CH2Cl2 were used as the standard conditions. It is noteworthy that only the substrates having methyl ketone units could furnish the adducts in good yields and with high dr and ee values. When a long carbon chain or a phenyl group was utilized, a sharp decrease in the yield was observed (Scheme 17, 3an and 3ao).21 Mechanistically, a transition state involving an enamine intermediate was proposed to illustrate the catalysis model of the reaction.
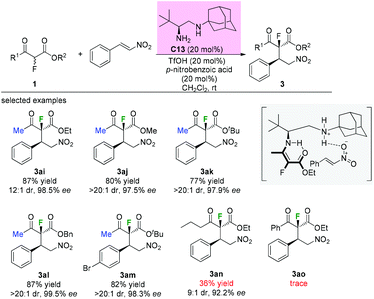 |
| | Scheme 17 Asymmetric reactions of α-fluoro-β-ketoesters with nitroethylenes catalyzed by C13. | |
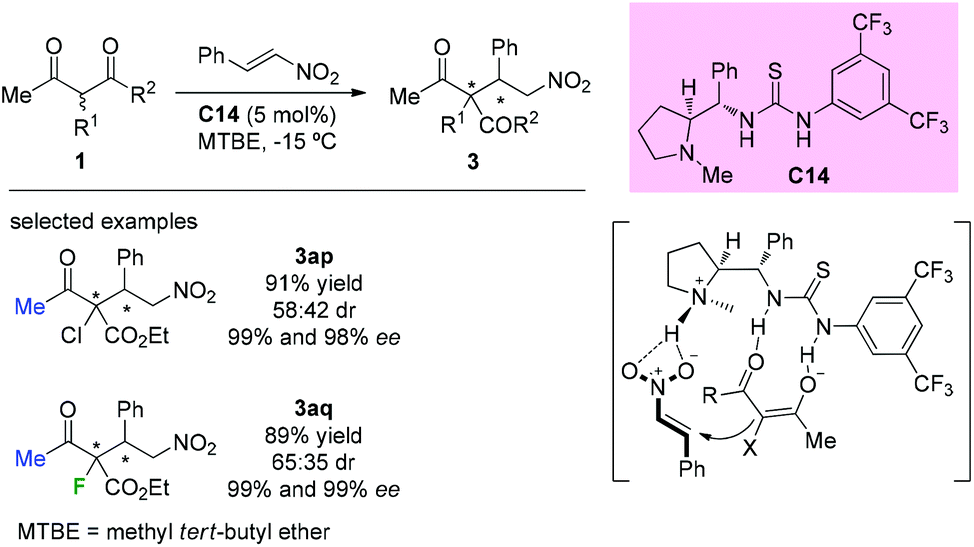
In 2014, Kesavan et al. disclosed the use of L-proline derived novel bifunctional thiourea catalyst C14 in conjugate additions of 1,3-dicarbonyls to nitroolefins. Although low diastereoselectivity was achieved by using acyclic substituted β-ketoesters containing Cl or F at the α-position, the corresponding products (3ap and 3aq) were obtained in good yields and with high enantioselectivities (Scheme 18).22 The catalyst is considered to play dual roles in activating both the ketoesters and nitroolefins.
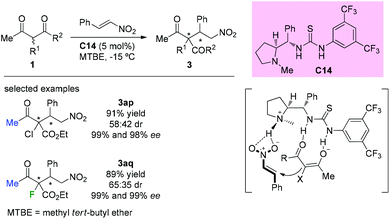 |
| | Scheme 18 Asymmetric conjugate additions catalyzed by thiourea catalyst C14. | |
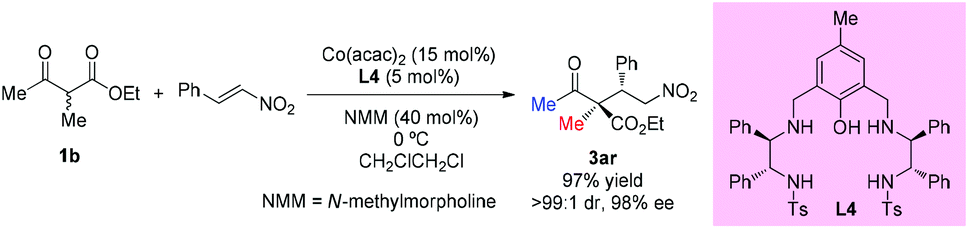
Zhou's group reported that the combination of Co(acac)2 with the aminophenol sulfonamide ligand (L4) displayed effective catalytic performance in the asymmetric additions of β-ketoesters to nitroolefins. One example of acyclic ethyl 2-methyl-3-oxobutanoate was selected to be surveyed to give the addition adduct 3ar in 97% yield with excellent dr (>99:1) and ee (98%) (Scheme 19).23
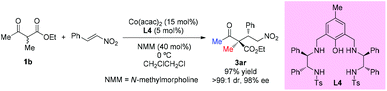 |
| | Scheme 19 One example of asymmetric Michael addition catalyzed by a Co-complex. | |
4. Amination of β-ketoesters
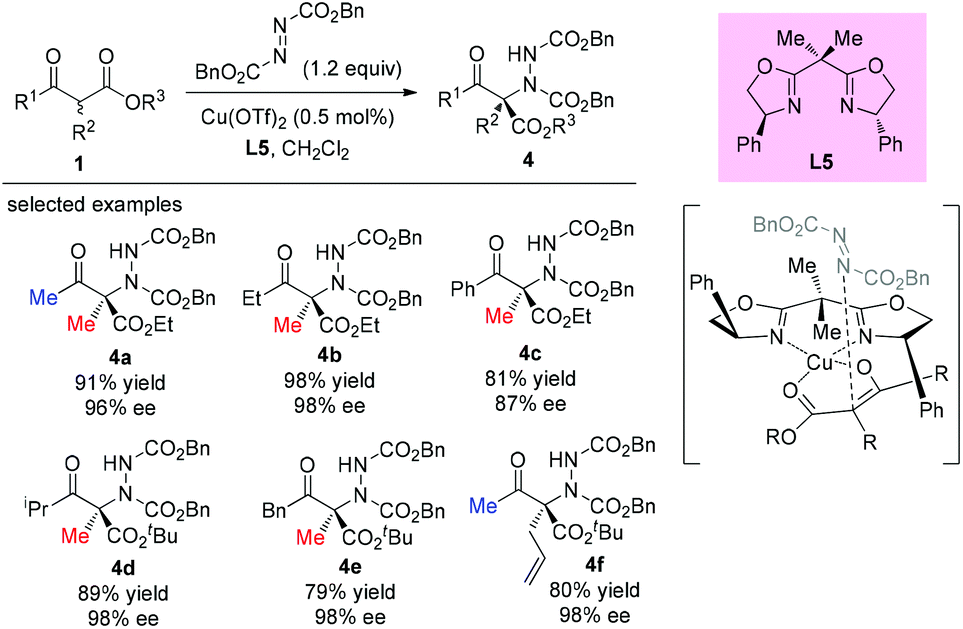
The electrophilic amination of an activated methine compound with azo species was a direct and general strategy to construct a nitrogen-containing tetrasubstituted carbon stereocenter. A number of methodologies have been developed employing different catalytic systems. In 2003, the Jørgensen group revealed the first direct α-amination of α-substituted β-ketoesters catalyzed by a chiral copper(II)-bisoxazoline complex with azodicarboxylates as the nitrogen fragment source.24 0.5 mol% of the catalyst was found to be sufficient to give the desired products in high yields with excellent enantiomeric excesses (Scheme 20). A survey of the acyclic β-ketoesters used in this work indicates that the Methyl Rule is still applicable, because at least one methyl group is required in the substrates. A copper-enolate complex was proposed to demonstrate the stereochemical outcome of the reaction.
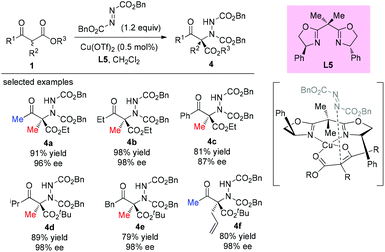 |
| | Scheme 20 The aminations of β-ketoesters catalyzed by Cu(OTf)2 with bisoxazoline ligand L5. | |
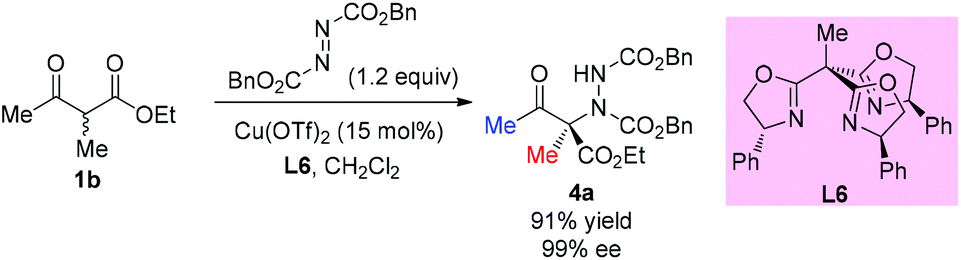
Later, a similar reaction was used to survey the potential of a series of C3-chiral 1,1,1-tris(oxazolinyl)ethane ligands in copper Lewis acid catalysis, and the results showed that similar outcomes can be achieved using this type of C3 ligand such as L6 (Scheme 21).25
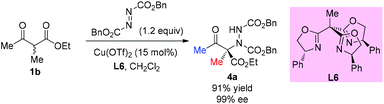 |
| | Scheme 21 Access to aminated β-ketoester catalyzed by a Cu-complex. | |
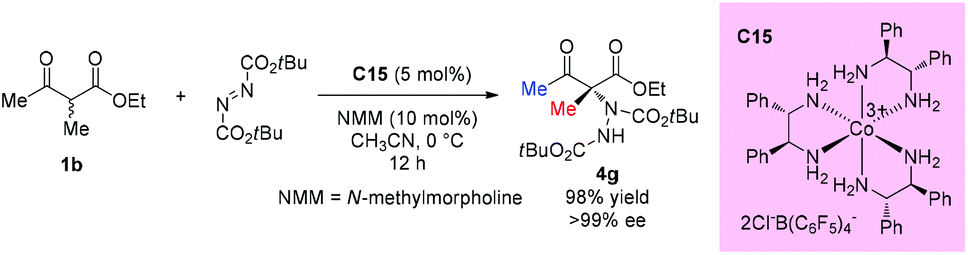
Another catalytic enantioselective amination via such addition was developed by Gladysz and his co-workers in 2016. In this protocol, an enantiopure salt Δ-[Co((S,S)-dpen)3]3+2Cl−B(C6F5)4− (C15) was employed as the hydrogen bond donor catalyst. All substrates, including a series of cyclic 1,3-dicarbonyl substrates, α-cyanocyclopentanone, and one example of acyclic β-ketoester, were examined in this work. The amination of ethyl 2-methyl-3-oxobutanoate proceeded giving high yield (98%) with excellent enantioselectivity (>99% ee) (Scheme 22).26
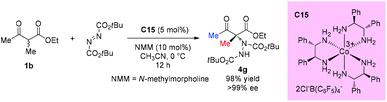 |
| | Scheme 22 Catalytic amination catalyzed by a cobalt salt. | |
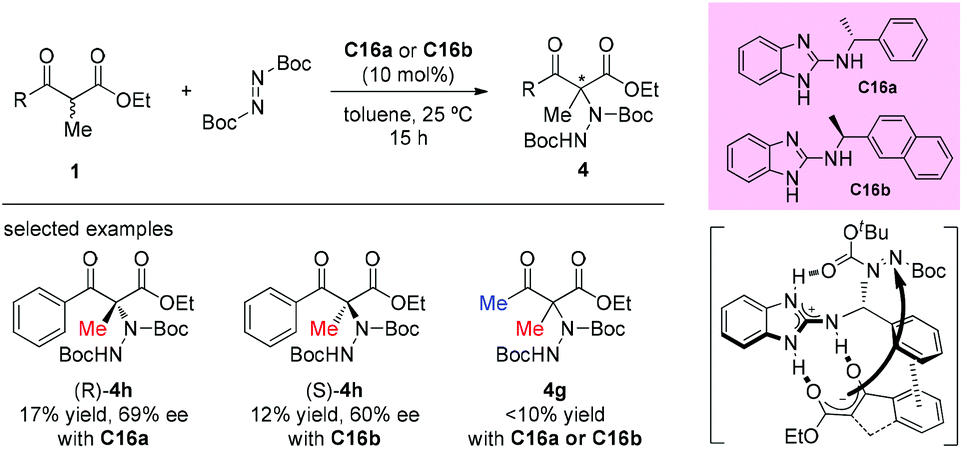
In 2017, Baeza's group reported the synthesis of a set of new chiral guanidines derived from benzimidazoles, and the applications of this type of catalyst in the asymmetric addition of 1,3-dicarbonyl compounds by using di-t-butylazodicarboxylate as an aminating agent. The scope of β-ketoesters was examined; open-chained substrates did not work well in this catalytic system, especially the methyl 3-oxo-2-methylbutanoate which failed to undergo the transformation (4g). A π–π stacking is thought to be involved in the transition state (Scheme 23).27
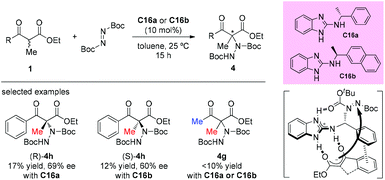 |
| | Scheme 23 Aminations of acyclic β-ketoesters according to Baeza's process. | |
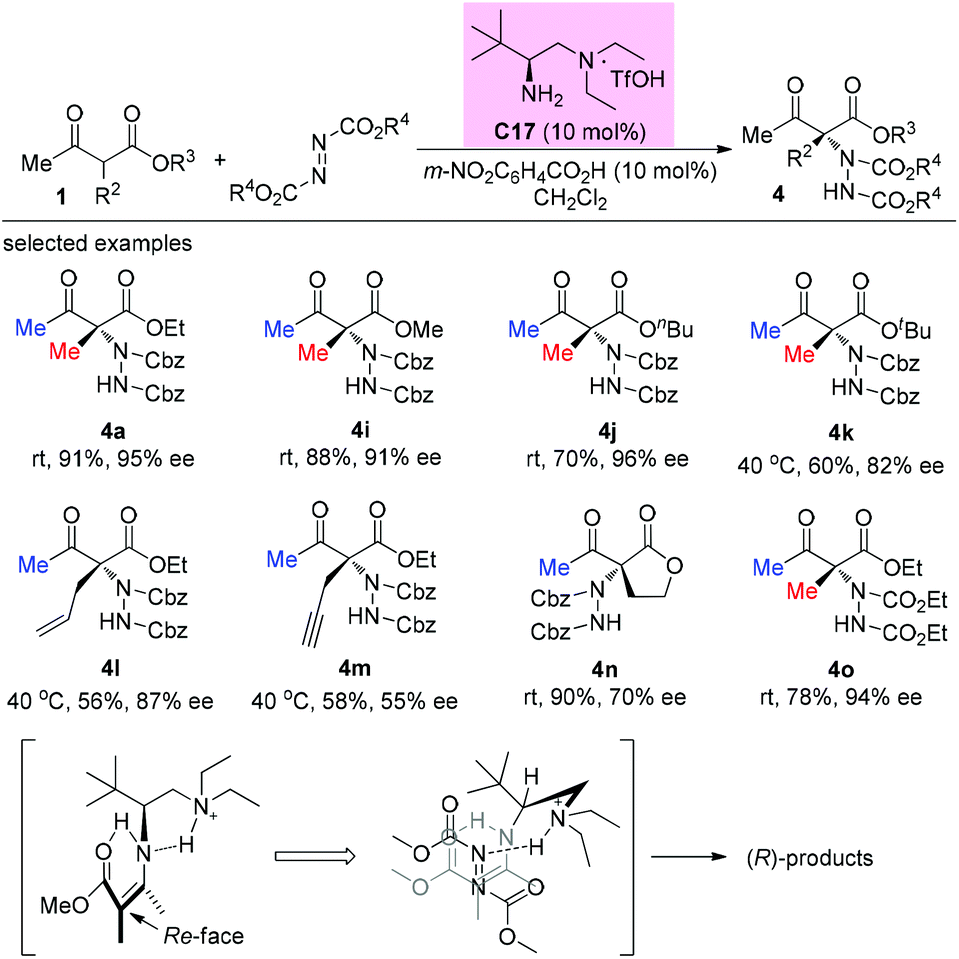
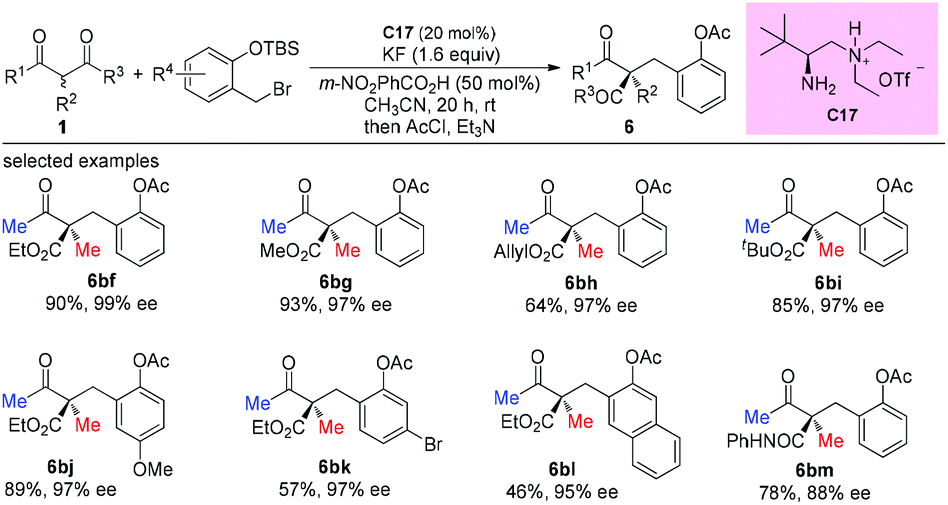
In 2014, the Luo group revealed a chiral primary amine catalyzed α-hydrazination of β-ketoesters. The joint use of a strong acid TfOH and a weak acid m-nitrobenzoic acid is critical to facilitate the catalytic turnover and to tune the selectivity. In all cases, only methyl ketoesters were tested, and the reaction proceeded giving moderate to high yields, producing the products with 55–96% ee. A Re-face addition of the enamine intermediate is favored to deliver (R)-products (Scheme 24).28
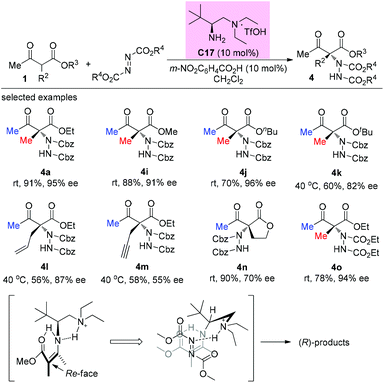 |
| | Scheme 24 Asymmetric amination catalyzed by C17. | |
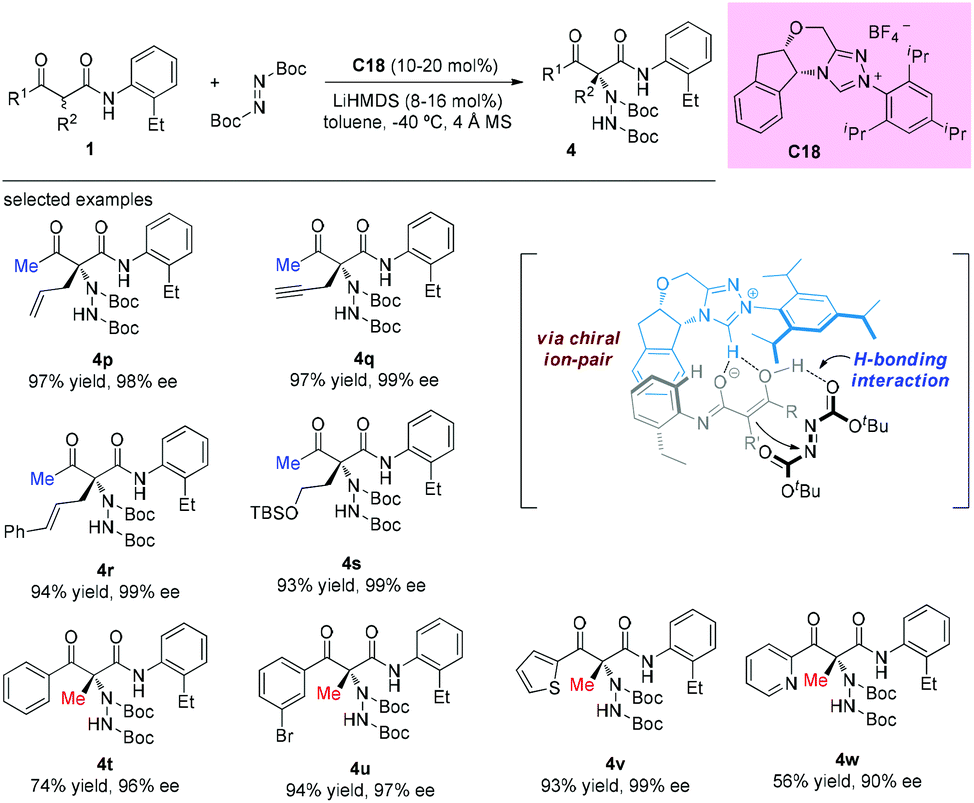
The catalytic asymmetric α-amination of α-substituted acyclic 1,3-ketoamides and 1,3-amidoesters using a chiral N-heterocyclic carbene (NHC) catalyst was disclosed by the Guin group recently. The reaction was considered to proceed through a deprotonation of the acidic N–H present in the substrate with chiral NHC to render a chiral ion pair comprising the enolate and the azolium ion. Then the activated chiral enolate reacted with azodicarboxylate to furnish the open-chained 1,3-dicarbonyls containing an N-substituted fully substituted stereocenter. A large amount of the amination products were obtained in satisfactory yields (38–98%) with high enantioselectivities (76–99% ee) (Scheme 25).29 It is still evident that the outcomes were in accordance with the principle of the Methyl Rule.
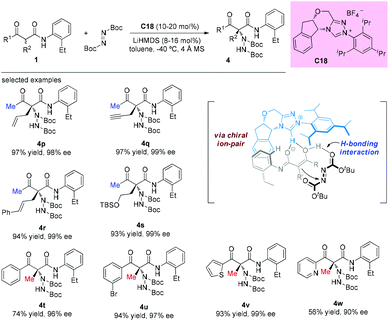 |
| | Scheme 25 Protocol for asymmetric α-amination of acyclic 1,3-ketoamides. | |
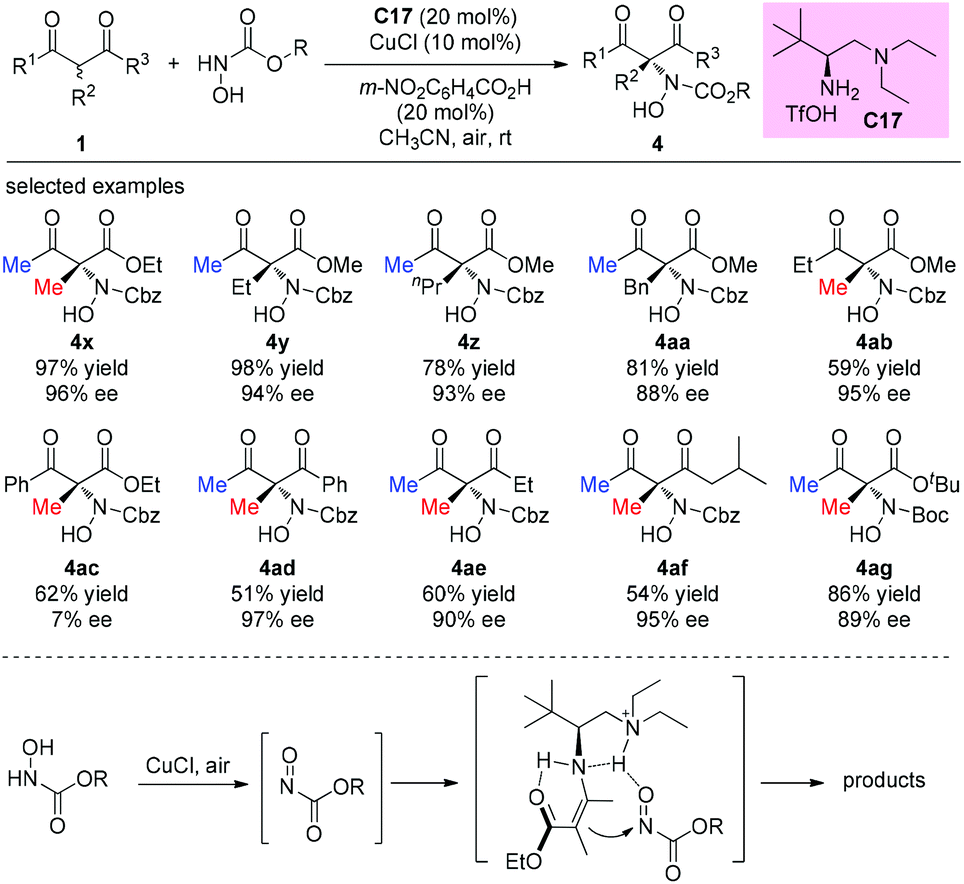
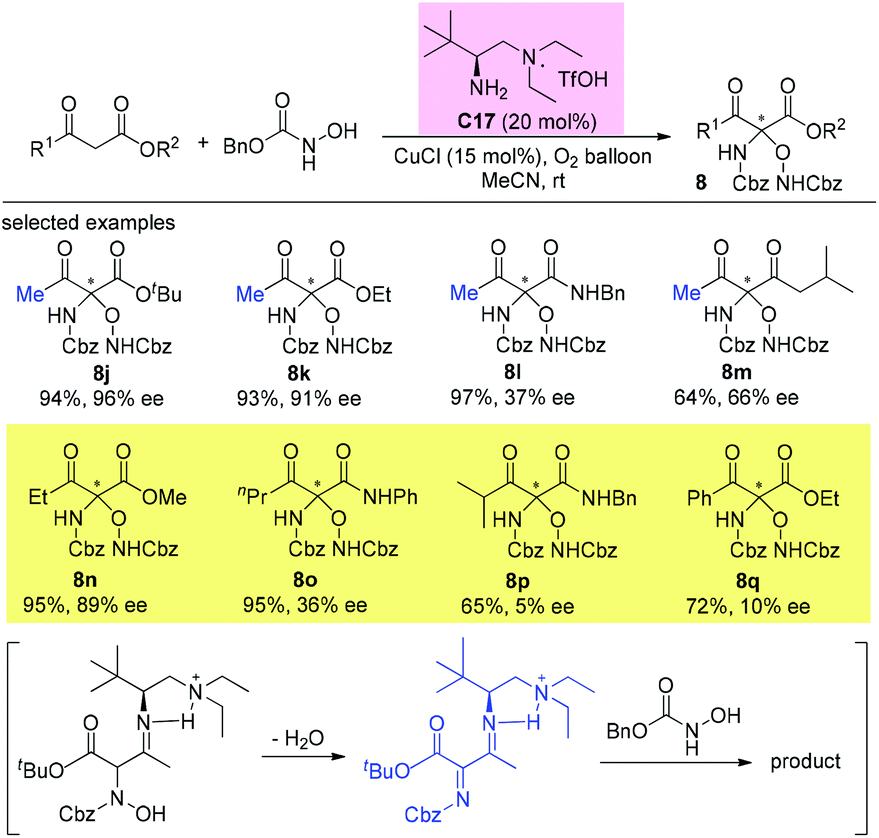
An alternative α-amination of β-ketocarbonyls was developed by the Luo group using a simple primary amine catalyst (C17) through the coupling of a catalytic enamine intermediate and a nitrosocarbonyl generated in situ from N-hydroxy carbamate. The protocol is compatible for a series of β-ketoesters and 1,3-diketones. The authors listed nineteen examples using acyclic β-ketocarbonyls, and all of them still adhere to the Methyl Rule. A further careful survey shows that β-ketoesters are generally more efficient than 1,3-diketones with respect to the yields of the products. Furthermore, β-ketoesters bearing ethyl ketone (4ab) and phenyl ketone (4ac) moieties are less effective than those with methyl ketone units (Scheme 26).30 It is worth mentioning that in de Alaniz's copper catalyzed non-asymmetric α-amination of β-ketoesters using N-hydroxycarbamates, the Methyl Rule is still the effective underlying principle.31
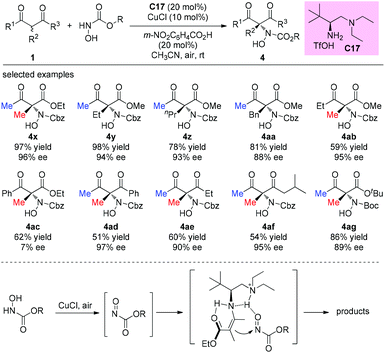 |
| | Scheme 26 Asymmetric amination with N-hydroxy carbamate. | |
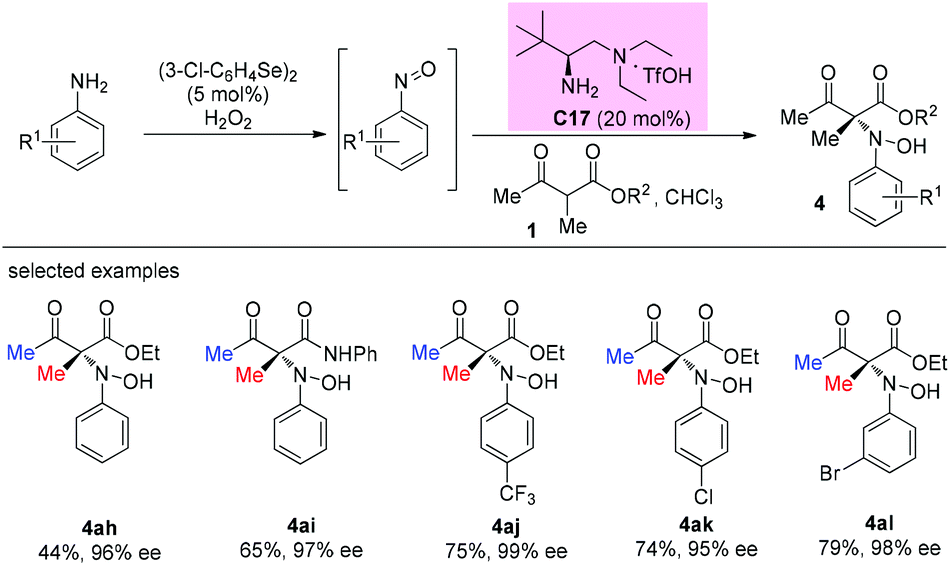
An enantioselective primary amine C17-catalyzed N-selective addition reaction was achieved through the oxidation of primary aromatic amines to the corresponding nitrosoarenes catalyzed by selenium reagents and H2O2. The protocol provides a facile and highly efficient access to α-hydroxyamino carbonyls bearing chiral tetrasubstituted carbon centers under mild conditions (Scheme 27).32
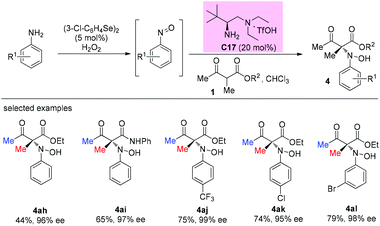 |
| | Scheme 27 Asymmetric amination using in situ generated nitrosoarenes. | |
5. Aldol reaction mediated by β-ketoesters
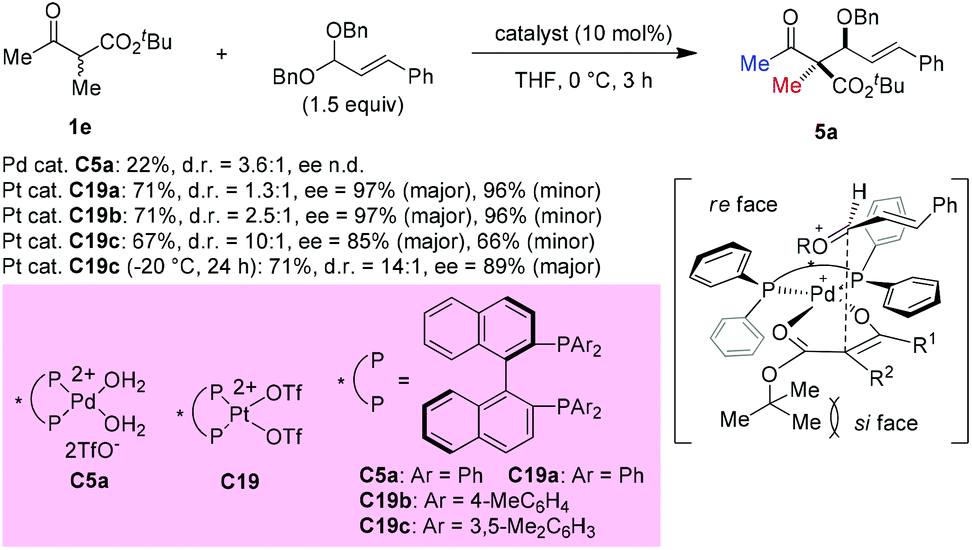
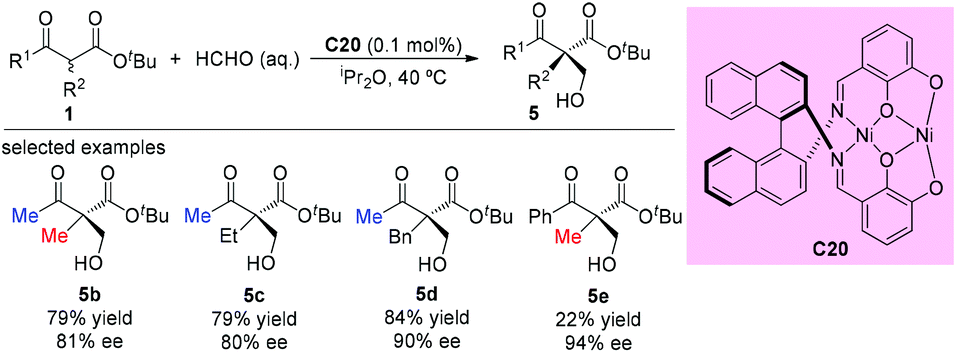
In 2008, Sodeoka et al. disclosed a catalytic asymmetric aldol-type reaction of β-ketoesters with acetals. In the presence of chiral Pd(II)-bisphosphine complexes (C5a), the reaction between five- or six-membered cyclic β-ketoesters and acetals could proceed giving moderate to good yields with almost perfect ee values. However, the reactivity decreased noticeably for the acyclic β-ketoester. To address this issue, the Pt(II)-bisphosphine complexes (C19a–C19c) were developed and utilized in the aldol reactions of tert-butyl 2-methyl-3-oxobutanoate with cinnamaldehyde dibenzyl acetal, giving the desired adduct (5a) in moderate yields with satisfactory stereoselectivities. A transition state favoring Re-face addition was proposed (Scheme 28).33
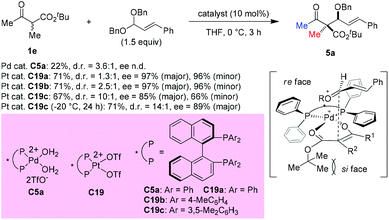 |
| | Scheme 28 Aldol reaction of an acyclic β-ketoester with Pt-complex catalysis. | |
The asymmetric aldol reaction between β-ketoesters and formaldehyde was reported by Shibasaki and co-workers in 2009, using a homodinuclear Ni2-Schiff base complex (C20). The resulting hydroxyethylated adducts can be obtained with good to excellent ee values (Scheme 29). It can be concluded that Me-ketone substrates are more reactive, and a Ph-ketone substrate gave only 22% yield, albeit with 94% ee (5e).34
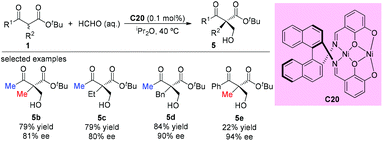 |
| | Scheme 29 Ni2-Schiff base catalyzed aldol reaction of β-ketoesters with HCHO. | |
6. α-Alkylation of β-ketocarbonyls
Early in 1997, Suzuki and co-workers developed a series of chiral β-amino sulfoxide ligands and used them in palladium-catalyzed asymmetric allylic alkylation. The best result was observed when ligand L7 was used and 50% ee of the product was obtained. In this case, only methyl-substituted methyl ketoester was tested (Scheme 30).35
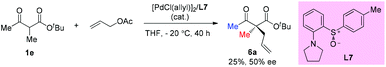 |
| | Scheme 30 Asymmetric allylation of 1evia palladium catalysis. | |
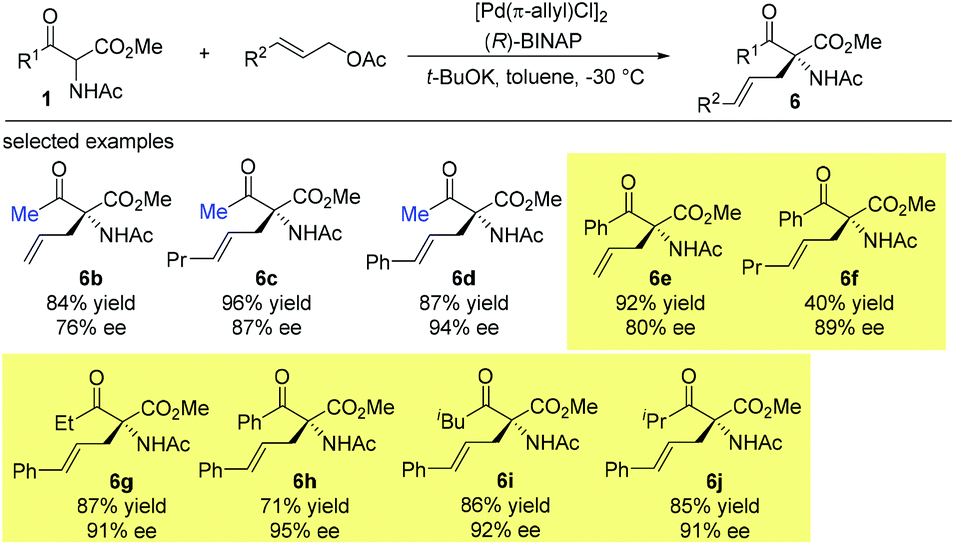
Then later in 1999, the Ito group reported a highly enantioselective allylation of a series of α-acetamido-β-ketoesters catalyzed by the chiral BINAP-palladium complex. The reactions proceed smoothly, affording the corresponding products with 76–95% ee. It is noteworthy that the Methyl Rule is not followed in this work, and the NHAc group may play a crucial role in overcoming the inherent low reactivity of the nucleophiles (Scheme 31).36
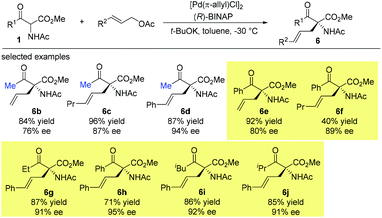 |
| | Scheme 31 Asymmetric allylation of α-acetamido-β-ketoesters. | |
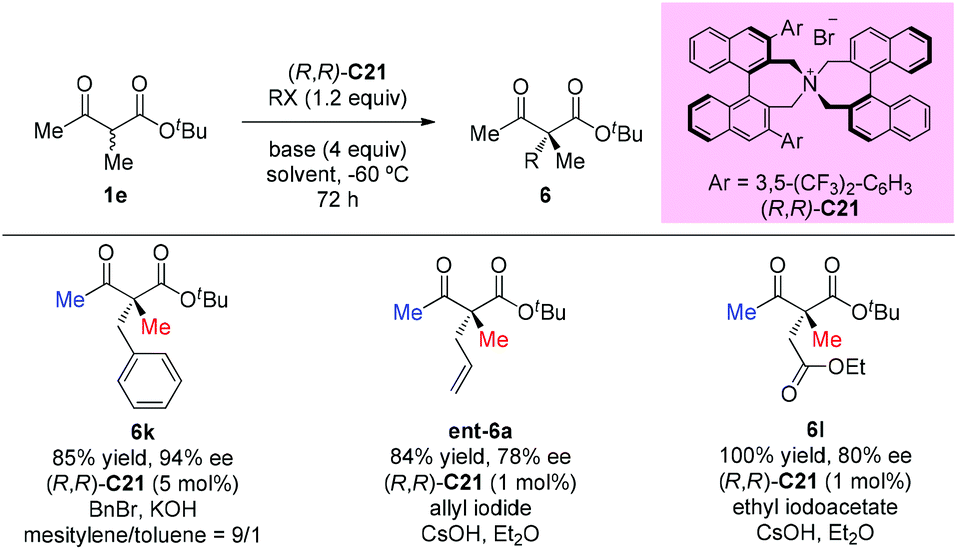
In 2009, Itoh et al. disclosed a phase-transfer-catalyzed asymmetric alkylation of α-substituted acetoacetates with alkyl halides using an N-spiro chiral quaternary ammonium salt ((R,R)-C21). This work provided a straightforward protocol to construct an all-carbon quaternary center. Among the β-ketoester substrates, tert-butyl 2-methyl-3-oxobutanoate having methyl groups at both α- and β-positions was tested as a unique example to react with three alkyl halides, giving the corresponding products under each suitable condition in good yields with high enantioselectivities (Scheme 32).37
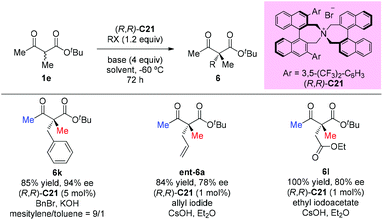 |
| | Scheme 32 Asymmetric alkylation of acetoacetates with phase-transfer catalysis. | |
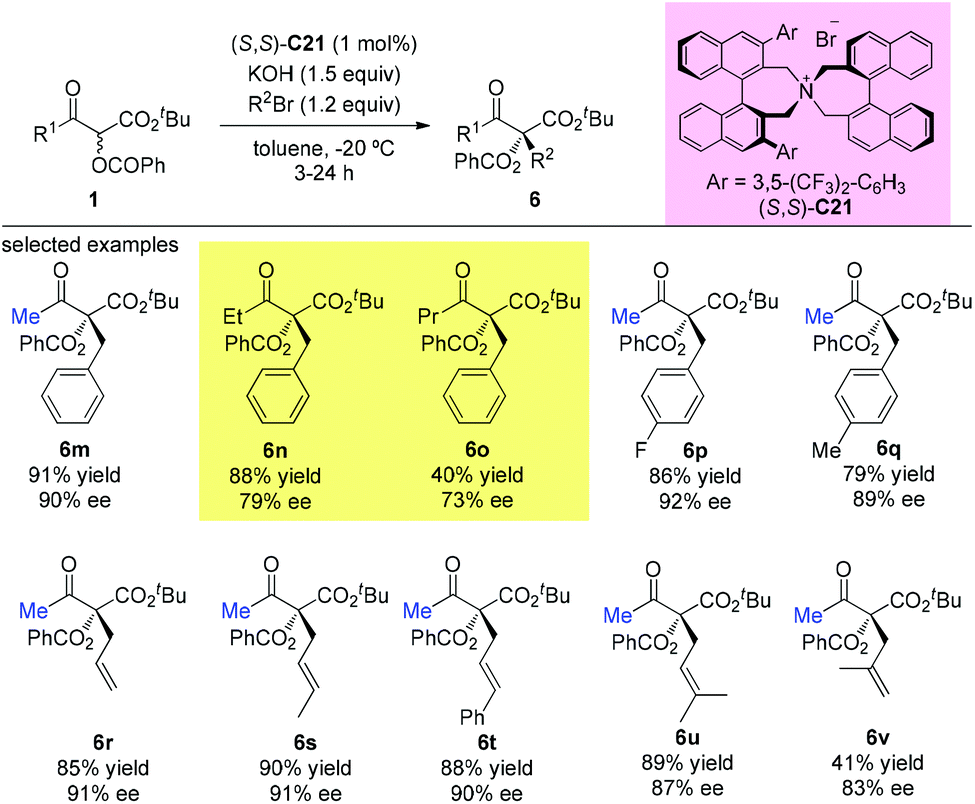
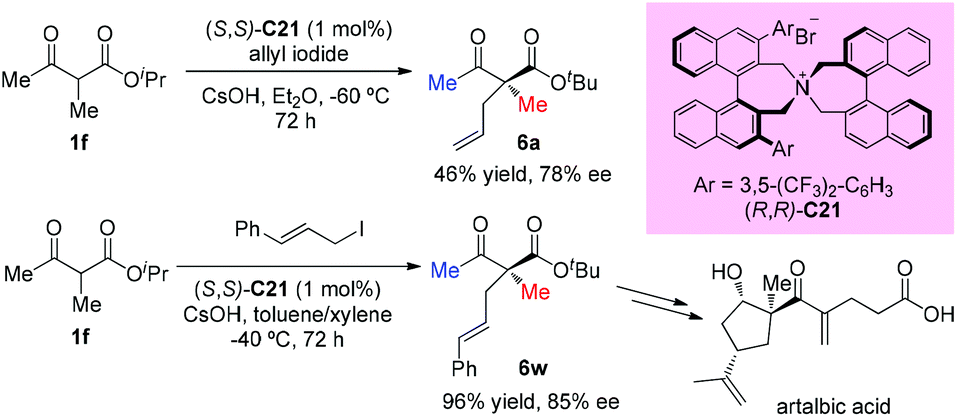
In the subsequent year, the oppositely configured catalyst (S,S)-C21 was utilized to promote the enantioselective substitutions of α-branched β-ketoesters by Maruoka et al. The α-benzoyloxy-β-ketoesters were subjected to reaction with a number of benzylic and allylic bromides, furnishing the products in at least moderate yields (40–91%) with satisfactory enantioselectivities (73–92%) (Scheme 33). Two exceptional examples with respect to the Methyl Rule are shown in this work using ethyl or propyl ketone substrates, but the reactions showed decreased efficiencies and enantioselectivities (6n and 6o).38
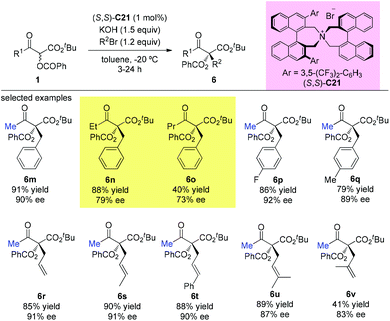 |
| | Scheme 33 Phase-transfer-catalyzed alkylations of α-benzoyloxy-β-ketoesters. | |
In 2016, Ito and co-workers reported the first total synthesis of (+)-artalbic acid using asymmetric allylation of an acetoacetate derivative with a phase-transfer catalyst. This synthetic work was completed in 12 steps from isopropyl acetoacetate with high stereocontrol (Scheme 34).39
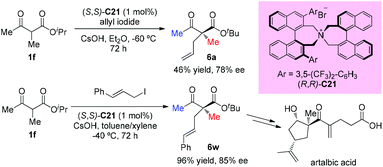 |
| | Scheme 34 Asymmetric allylation catalyzed by PTC C21. | |
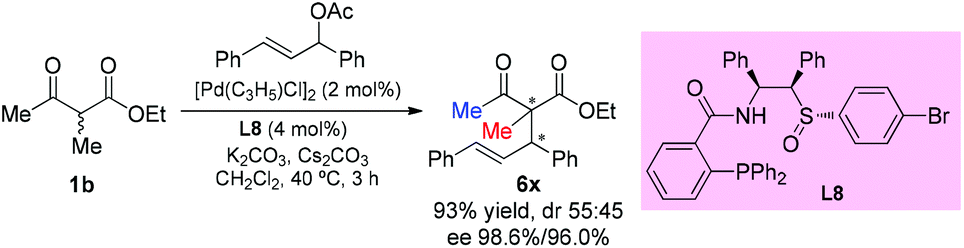
In 2014, the Xiao group developed a new type of chiral sulfoxide-phosphine ligand by a rational combination of two privileged scaffolds for Pd-catalyzed asymmetric allylic alkylation reactions. Generally high yields and excellent enantioselectivities were obtained. Only one example using an acyclic β-ketoester was tested, and the product was formed in excellent yield with a high level of enantioselectivity (Scheme 35).40
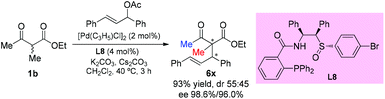 |
| | Scheme 35 Palladium-catalyzed asymmetric allylation using L8 as the ligand. | |
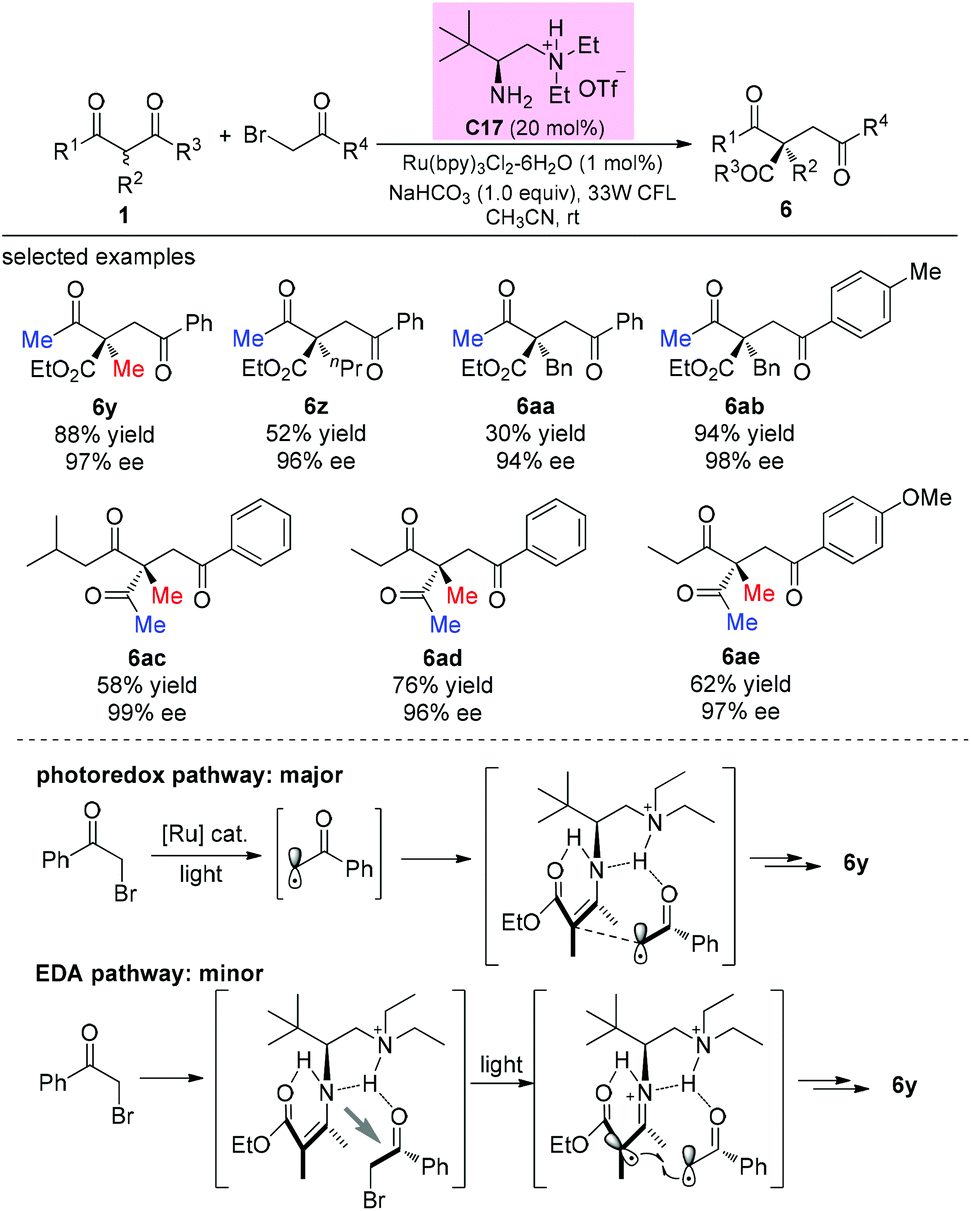
In 2014, an elegant enantioselective α-photoalkylation of β-ketocarbonyls by merging photoredox catalysis with chiral primary amine catalysis was reported by Luo and co-workers. The reaction enables the creation of all-carbon stereocenters with excellent enantioselectivities and encompasses a broad range of substrates including acyclic 1,3-diketones (Scheme 36). It can be seen that the α-substituents of the β-ketoesters greatly influence the yields (6y–6aa), and in all these cases, the methyl group at either the α-position or the ketone moiety of the β-ketocarbonyls is necessary. Mechanistically, a photoredox pathway is considered as the major one, and a minor electron donor–acceptor (EDA) pathway may also coexist.41a Later, a similar reaction but without the use of a photocatalyst was also reported by the same group.41b
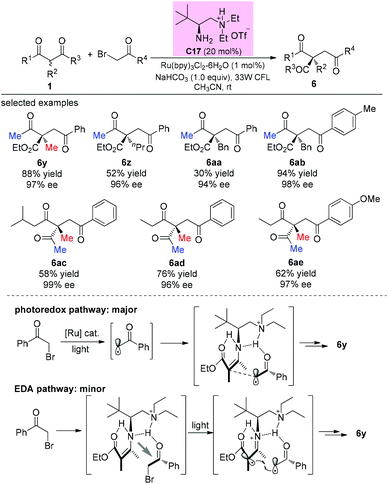 |
| | Scheme 36 Asymmetric alkylation of β-ketocarbonyls with photoredox catalysis. | |
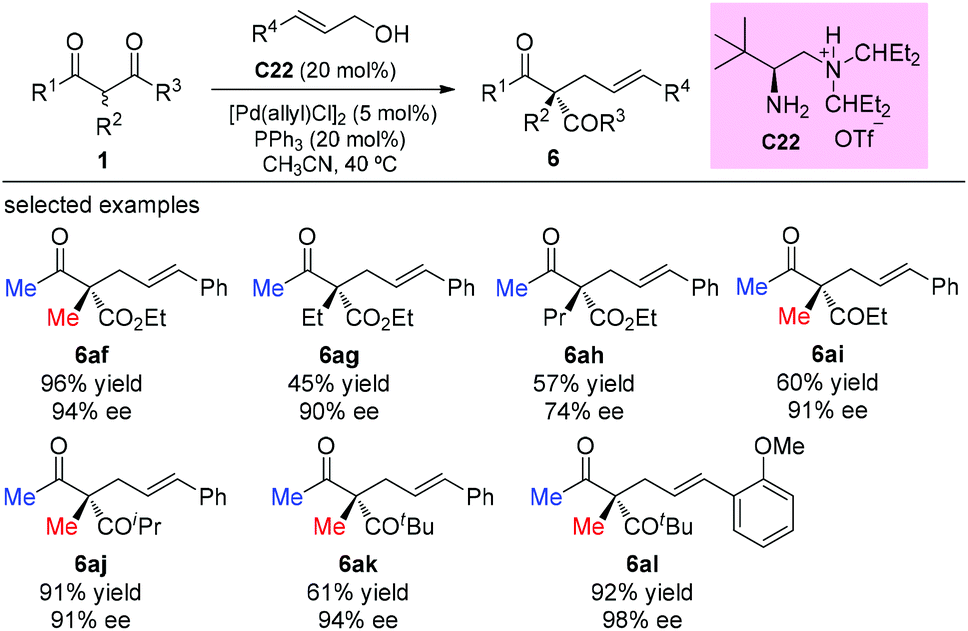
One year later, the same group developed an asymmetric allylic alkylation of acyclic β-ketocarbonyl compounds with free allylic alcohols under mild conditions. The reaction was enabled by a synergistic combination of a chiral primary amine and a palladium catalyst, and featured the enantioselective construction of quaternary stereocenters. As to the scope of the β-ketocarbonyl compounds, the methyl ketone unit and/or α-methyl substituent are still necessary to achieve good yields (Scheme 37).42
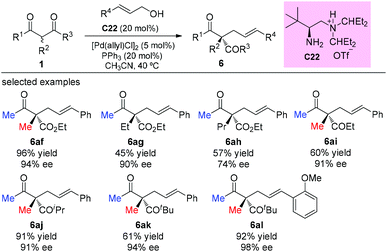 |
| | Scheme 37 Asymmetric allylation of β-ketocarbonyls via Pd-amine co-catalysis. | |
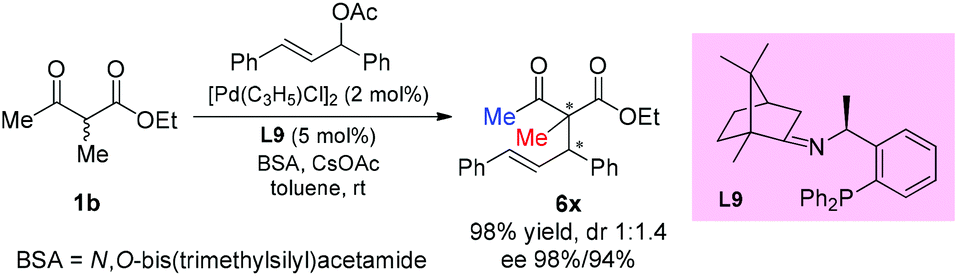
In 2016, the Xu group reported the asymmetric allylation reaction of methylene compounds using a chiral C-camphor-derived phosphine ligand L9. The reaction afforded the products in excellent yields with excellent ee values, and only one methyl-ketoester was tested (Scheme 38).43
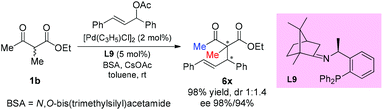 |
| | Scheme 38 Palladium-catalyzed asymmetric allylation using L9 as the ligand. | |
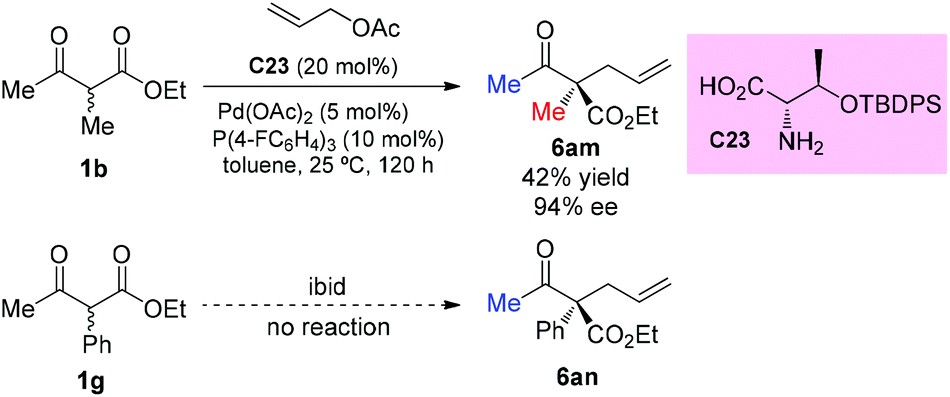
The strategy was later used by Yoshida and co-workers to achieve the highly enantioselective α-allylations of α-substituted β-ketoesters, particularly 2-oxocycloalkanecarboxylates. As shown in Scheme 39, the ethyl 2-methyl-3-oxobutanoate 1b as the acyclic α-substituted β-ketoester can also participate in the reaction, delivering 6am in 42% yield with 94% ee. However, α-phenyl substituted 1g cannot work under the standard conditions.44
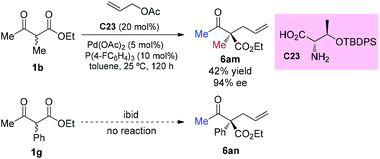 |
| | Scheme 39 Asymmetric α-allylation of acyclic β-ketoesters under Yoshida's conditions. | |
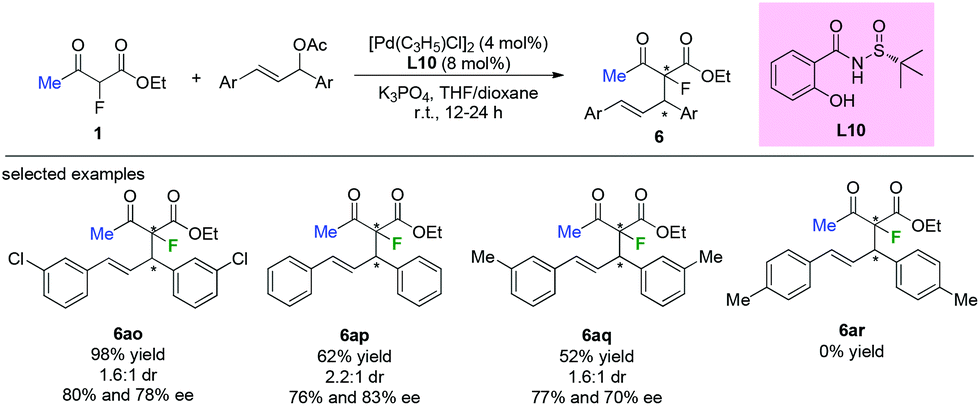
Chiral sulfinamide ligand L10 was used by the Zhao group in the Pd-catalyzed asymmetric allylation of ethyl 2-fluoroacetoacetate with a variety of allylating agents. Although most of the transformations could proceed in acceptable to satisfactory yields and with moderate enantioselectivities, low diastereoselectivities were achieved by this approach (Scheme 40). Furthermore, the substituent pattern on the phenyl ring of the allylic acetates led to significant limitation of the functional group tolerance.45
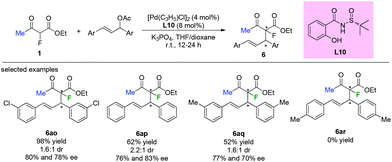 |
| | Scheme 40 Applications of a chiral sulfinamide ligand in Pd-catalyzed allylation. | |
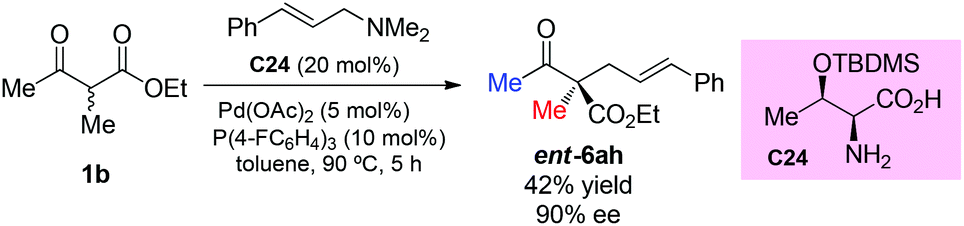
Recently, Tian et al. reported the use of allylic amines as the allylic reagent in the chiral α-amino acid/palladium-catalyzed asymmetric allylation of α-branched β-ketoesters. The reaction shows high enantioselectivity for cyclic ketoesters, and one example using a methyl ketone substrate was tested, affording ent-6ah in moderate yield with high ee (Scheme 41).46
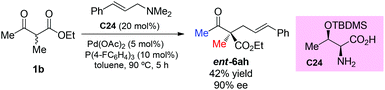 |
| | Scheme 41 Chiral α-amino acid/Pd-catalyzed asymmetric allylation with allylic amine. | |
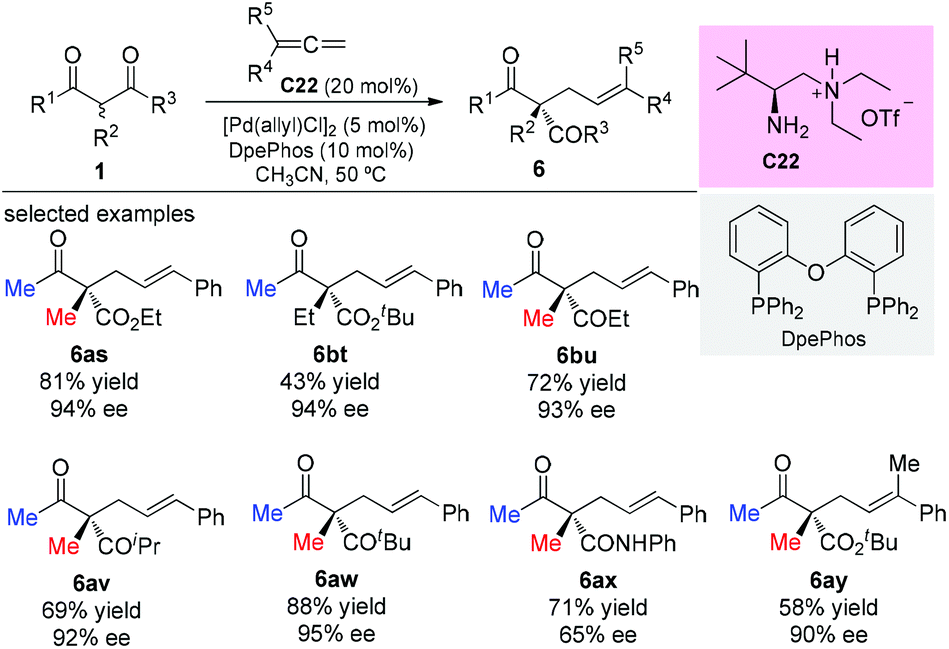
In 2017, the Luo group described a synergistic chiral primary amine/achiral palladium catalyzed enantioselective terminal addition to allenes with α-branched β-ketocarbonyls and aldehydes. The reaction afforded allylic adducts with all-carbon quaternary stereocenters with high regio- and enantioselectivities (Scheme 42). Again most of the β-ketocarbonyls contain two methyl substituents, showing the generality of the Methyl Rule.47a Later in 2019, Luo and co-workers developed a double-layered Sterimol model to account for the steric effect on enantioselectivity in dual primary amine/palladium-catalyzed asymmetric allylic alkylation reactions.47b
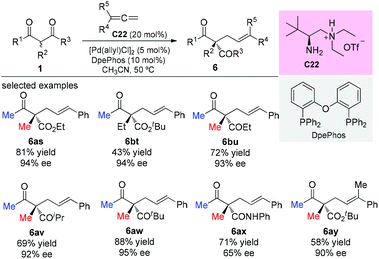 |
| | Scheme 42 Pd-Amine co-catalyzed asymmetric addition of β-ketocarbonyls to allenes. | |
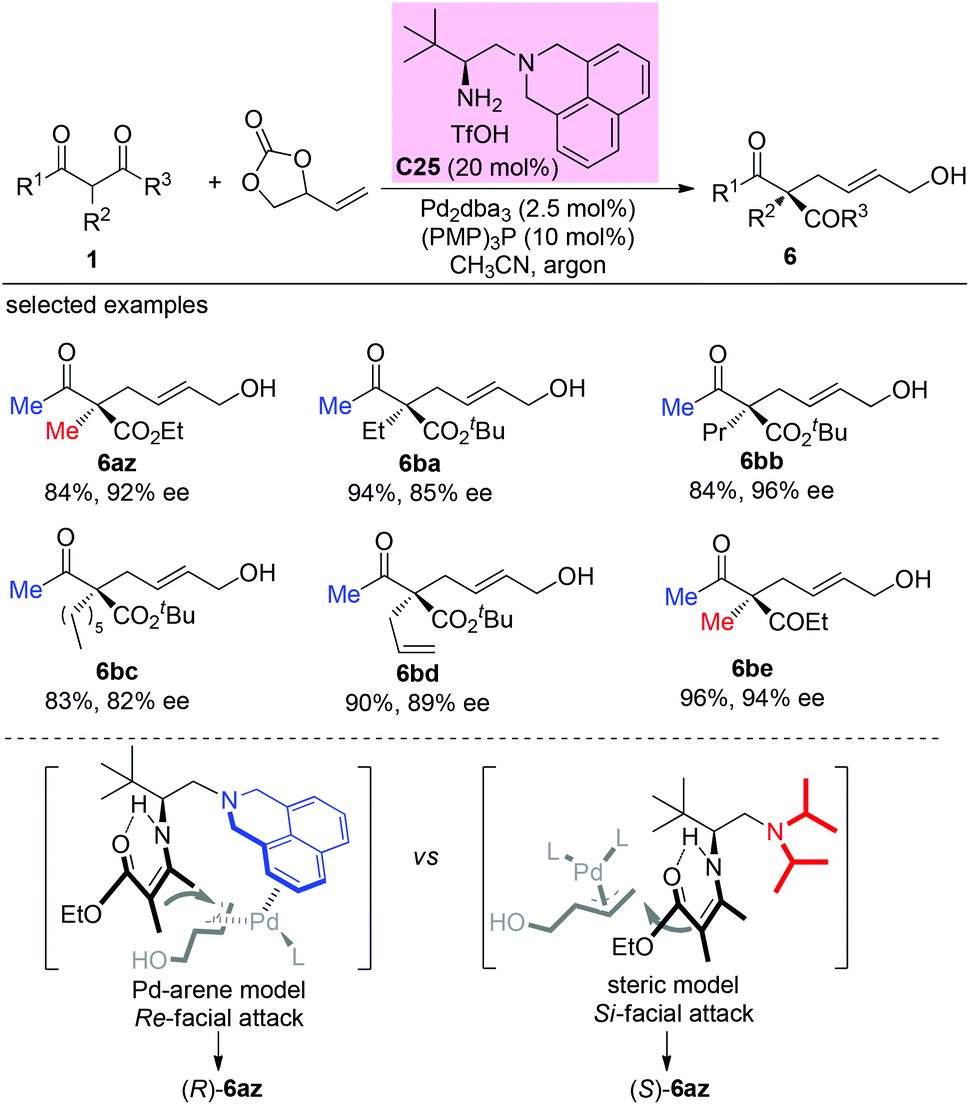
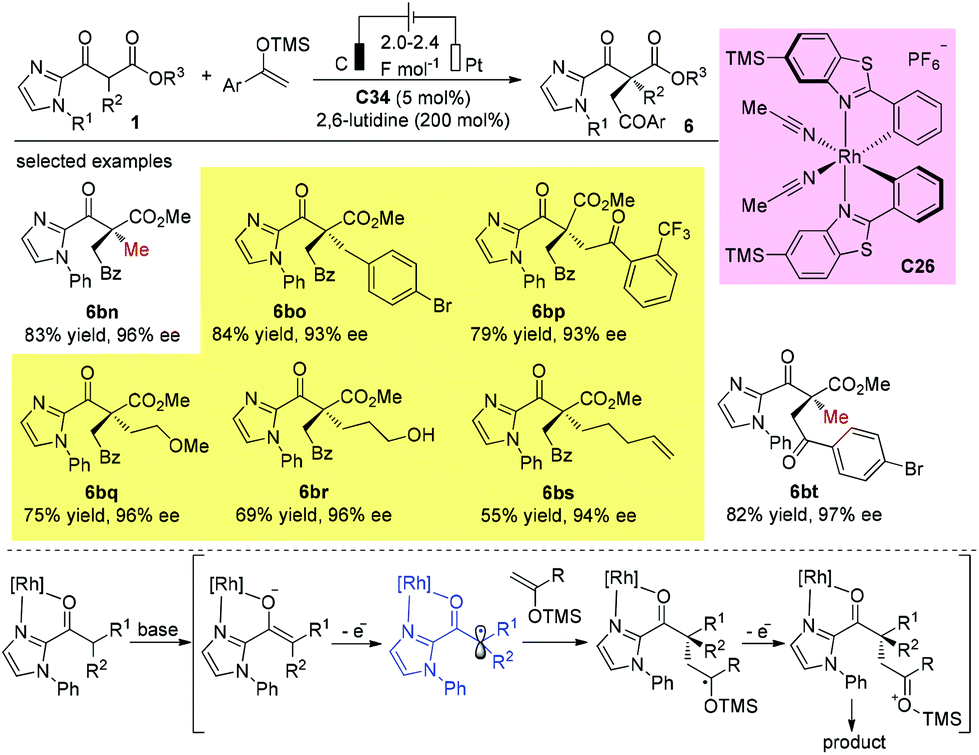
In 2020, Luo and co-workers reported a π-coordinating aminocatalyst/palladium synergistic catalysis for asymmetric allylic alkylation of α-branched β-ketocarbonyls using an arene-containing chiral primary amine as a dual aminocatalyst and ligand. A coordination between palladium and the arene group of C25 is proposed to release (R)-products. In contrast, a non-coordinating catalyst leads to the (S)-configuration of the products. Using catalyst C25, both cyclic and acyclic β-ketocarbonyls gave the allylic adducts with excellent regio-, stereo-, and enantioselectivities, and only methyl ketone-derived acyclic β-ketocarbonyls were tested (Scheme 43).48
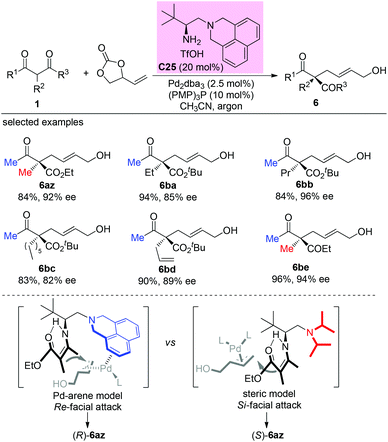 |
| | Scheme 43 Asymmetric allylation using Pd-primary amine co-catalysis. | |
A dual activation strategy merging primary amine catalysis and Lewis base activation was developed by Luo and co-workers in 2017, which achieved the asymmetric α-benzylation of α-branched β-ketocarbonyls. Enamines derived from β-ketocarbonyls could react effectively with in situ generated ortho-quinone methides under Lewis base activation, affording products with acyclic all-carbon quaternary stereocenters with excellent ee values (Scheme 44).49
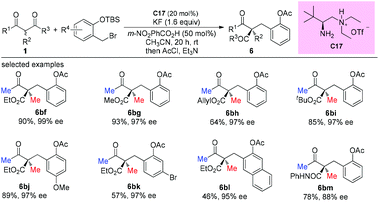 |
| | Scheme 44 Asymmetric benzylation via primary amine catalysis. | |
In 2018, Meggers and co-workers developed an elegant electricity-driven chiral Lewis acid catalysis for the oxidative cross-coupling of 2-acyl imidazoles with silyl enol ethers, which include products that bear all-carbon quaternary stereocenters. A chiral-at-metal rhodium catalyst C26 activates substrates towards anodic oxidation to form radical intermediates, which then undergo addition to enol ethers with high chemo- and enantioselectivities. The protocol successfully affords a series of quaternary β-ketoesters with two bulky α-substituents (Scheme 45).50
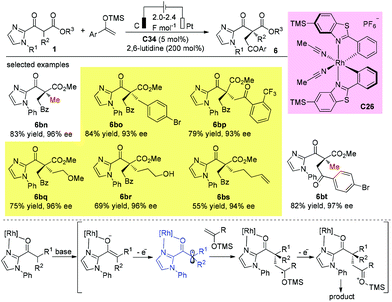 |
| | Scheme 45 Asymmetric oxidative cross-coupling of 2-acyl imidazoles with silyl enol ether. | |
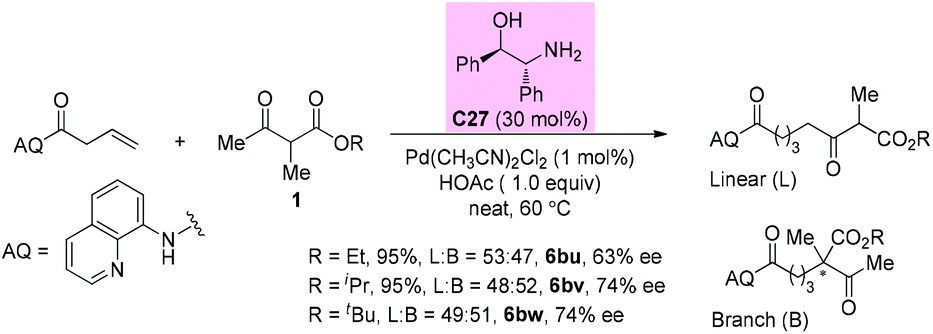
In 2019, the Shi group reported a synergistic palladium/enamine catalyzed asymmetric addition of ketones to non-activated alkenes under mild conditions. Primary amine C27 was identified as an effective catalyst in promoting the condensation of β-ketoester with an olefin. Unfortunately, in all cases, linear and branched products were formed in a similar ratio. For the branched products, moderate 63–74% ee values were detected (Scheme 46).51a
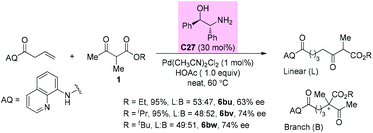 |
| | Scheme 46 Asymmetric alkylation via Pd-primary amine co-catalysis. | |
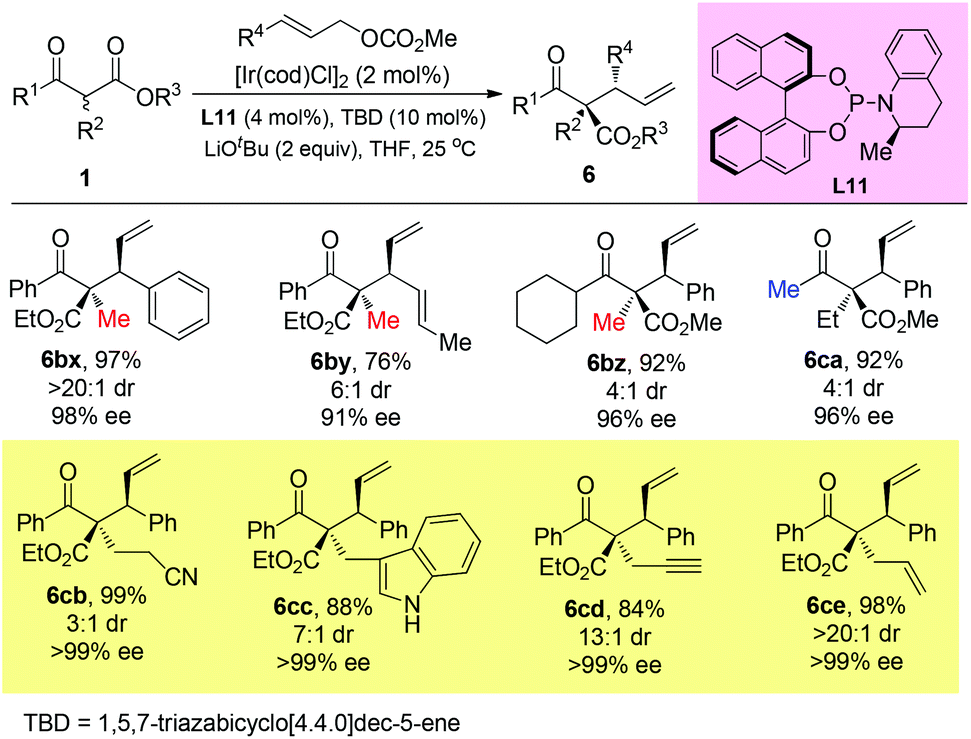
Stoltz and co-workers developed an elegant iridium-catalyzed asymmetric allylic alkylation of α-branched β-ketoesters using L11 as the ligand (Scheme 47).51b A strong base LiOtBu (2 equiv.) was added, and the system tolerated bulky substituents and afforded 6cb–6ce that beyond the Methyl Rule.
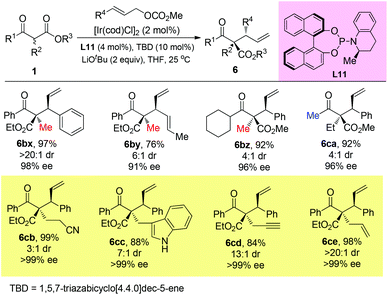 |
| | Scheme 47 Iridium-catalyzed asymmetric allylic alkylation. | |
7. α-Alkynylation of β-ketocarbonyls
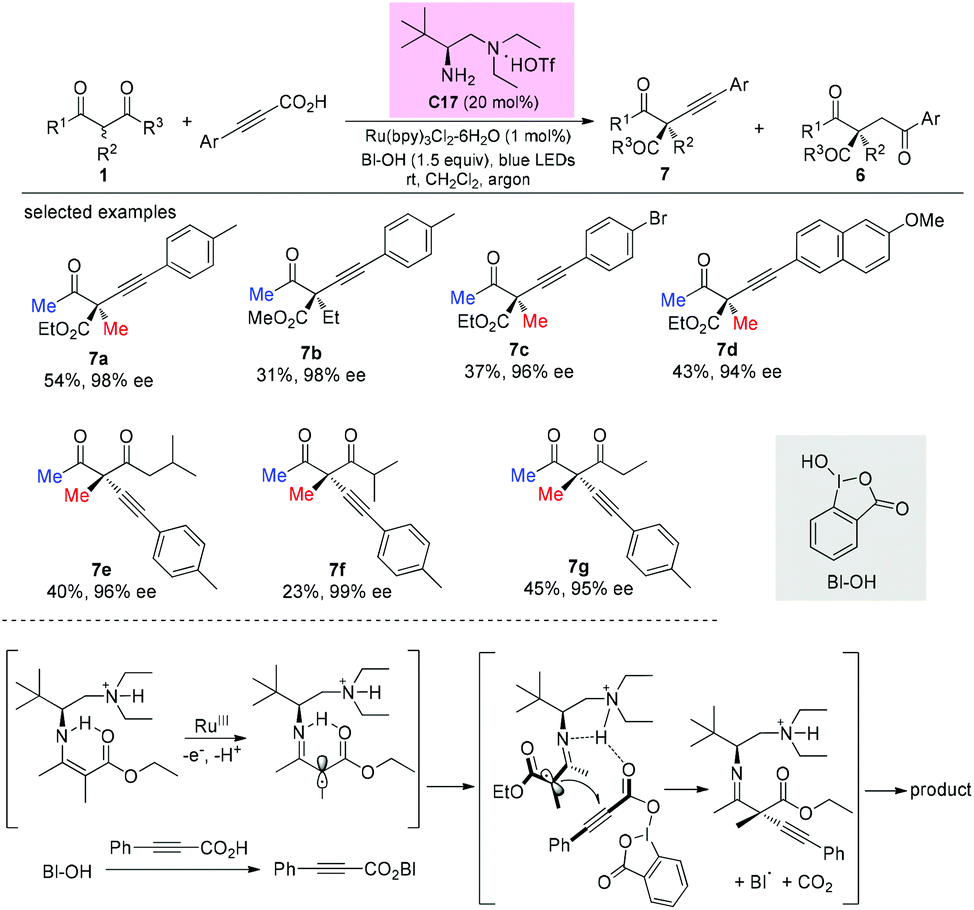
In 2017, an enantioselective decarboxylative coupling of propiolic acid and β-ketocarbonyls using a combination of chiral primary amine catalysis and visible-light photoredox catalysis was described by the Luo group. The reaction afforded alkynylation adducts with high enantioselectivity. An α-amino radical addition mechanism was proposed. In all cases, methyl ketone-derived ketoesters were used, and the alkylation side products were also formed (Scheme 48).52
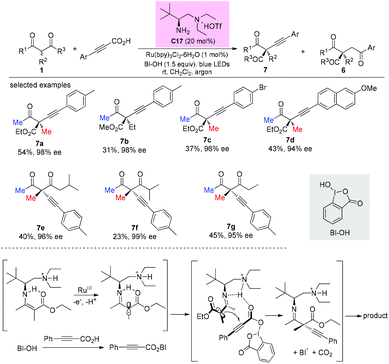 |
| | Scheme 48 Asymmetric alkynylation via primary amine-photocatalysis. | |
8. α-Oxygenation of β-ketocarbonyls
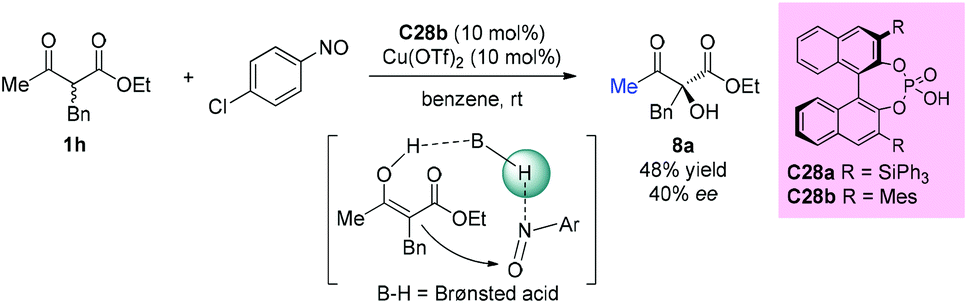
The first chiral phosphoric acid catalyzed α-hydroxylation of β-dicarbonyl compounds through a tandem aminoxylation/N–O bond heterolysis sequence using nitroso compounds as the oxygen source was reported by the Zhong group in 2009. With this protocol, a variety of substituted cyclic α-hydroxybenzo-β-ketoesters were obtained in the presence of 1 mol% catalyst C28a in good yields with high enantioselectivities. However, when an acyclic substrate was tested, low yield (48%) and ee value (40%) were obtained, even by employing increased loading of the modified catalyst C28b (10 mol%) and additional Cu(OTf)2 (10 mol%) as a co-catalyst (Scheme 49).53
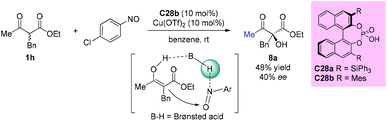 |
| | Scheme 49 α-Hydroxylation of β-ketoester catalyzed by chiral phosphoric acid. | |
The direct asymmetric α-benzoyloxylation of β-ketocarbonyls catalyzed by a chiral primary amine was revealed by Luo and co-workers in 2015. The protocol allows convenient access to highly enantioenriched α-hydroxy-β-ketocarbonyls. Methyl ketone-derived β-ketocarbonyls were still a suitable choice that could guarantee smooth reactions (Scheme 50).54
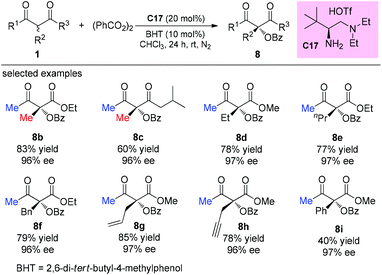 |
| | Scheme 50 Asymmetric α-benzoyloxylations of β-ketocarbonyls. | |
Subsequently, the same group disclosed a highly enantioselective primary amine catalyzed α,α-bis-functionalization of β-ketocarbonyls and cyclohexanones. The reaction used N-hydroxycarbamates as both nitrogen and oxygen sources under aerobic oxidative conditions to furnish chiral N,O-ketals with high yields and enantioselectivities. Four examples that are beyond the Methyl Rule were shown in the work, albeit with less satisfactory results. Mechanistically, a key highly activated imine intermediate is responsible for overcoming the substrate inertness (Scheme 51).55
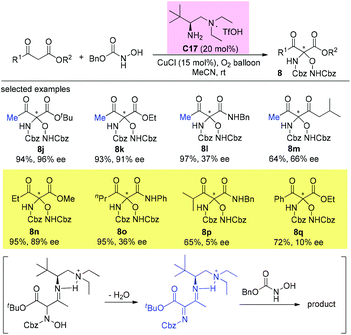 |
| | Scheme 51 Asymmetric α,α-bis-functionalization of β-ketocarbonyls and cyclohexanones. | |
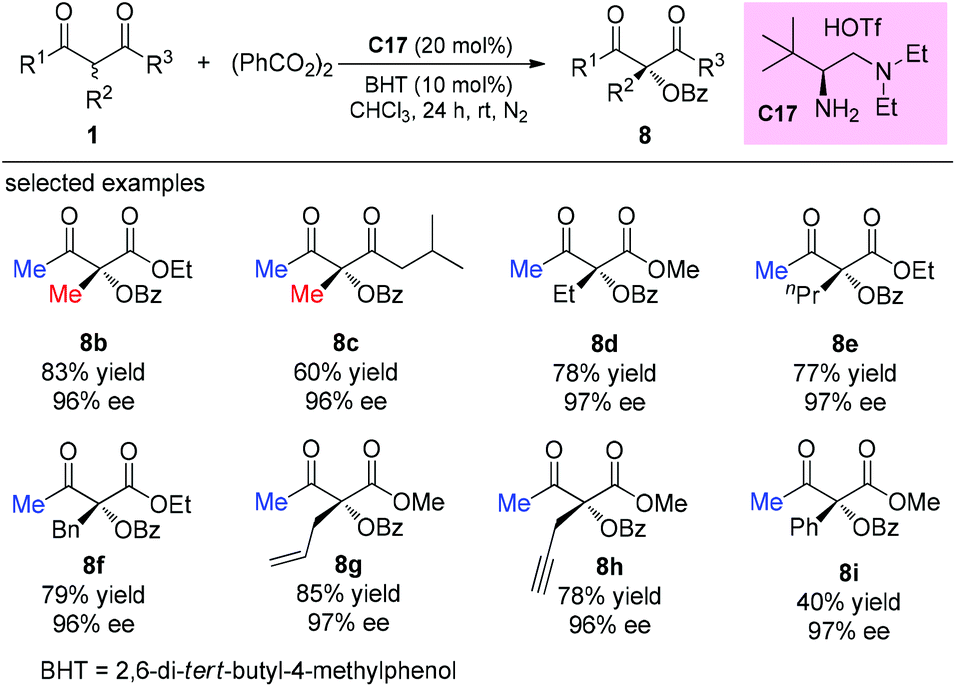
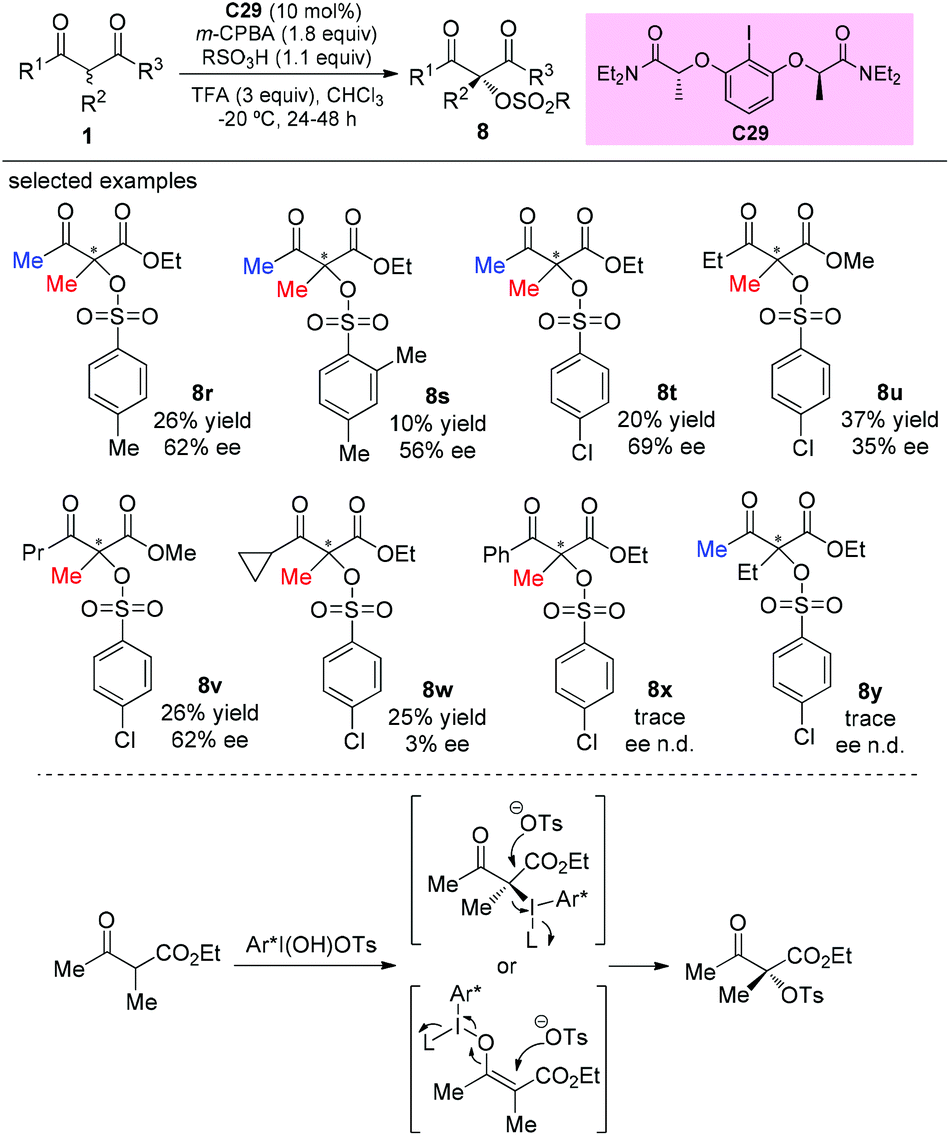
The catalytic asymmetric oxidation of α-substituted-β-ketoesters promoted by C2-symmetric iodoarene (C29) as a chiral agent and m-CPBA as the oxidant was reported by Xiong, Coeffard, and co-workers in 2016. In this work, the Methyl Rule is still followed, and it can be seen that phenyl ketone and α-ethyl derived substrates are inert to the reaction (8x–8y). Two possible pathways are involved in the production of the products (Scheme 52).56
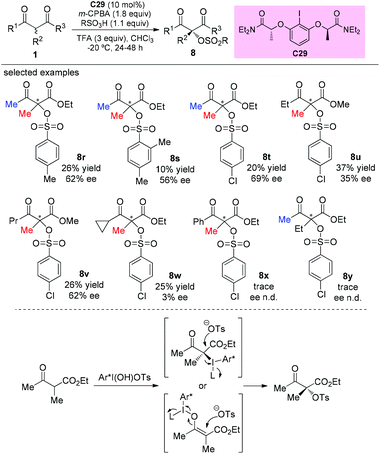 |
| | Scheme 52 Asymmetric oxidation of α-substituted β-ketoesters promoted by C2-symmetric iodoarene. | |
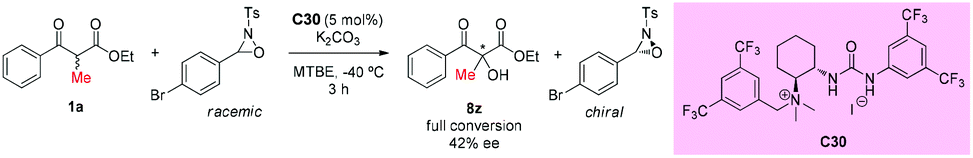
In the same year, an asymmetric α-hydroxylation reaction of β-ketoesters using oxaziridines as the [O+]-transfer reagents catalyzed by chiral bifunctional urea-containing ammonium salt (C30) was developed by the Waser group. This process contained a simultaneous kinetic resolution of the oxaziridine, and the match–mismatch scenario between the catalyst and the oxaziridine played a crucial role in the resolution. In the work, only one example of an acyclic substrate was surveyed. In contrast to the cyclic ketoesters, the transformation of the acyclic α-methyl ketoester substrate could be carried out in the presence of an additional base (e.g. K2CO3), but giving low enantioselectivity of the product (Scheme 53).57
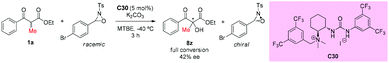 |
| | Scheme 53 Asymmetric α-hydroxylation reaction of β-ketoesters catalyzed by C30. | |
9. Mannich reaction of α-branched β-ketocarbonyls
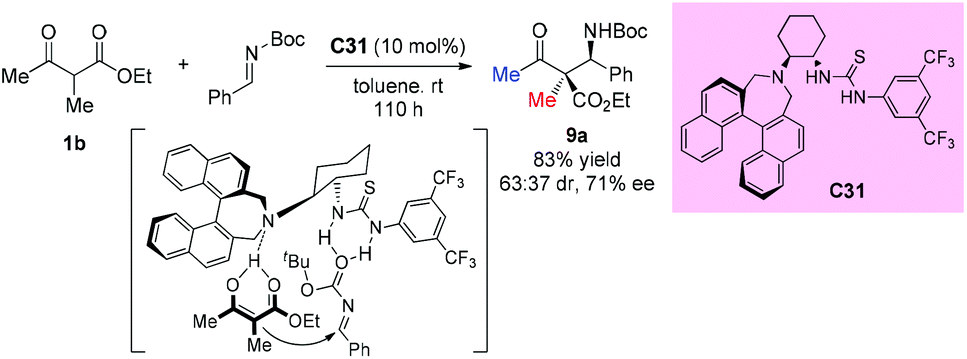
Enantioselective Mannich reactions of β-ketoesters with aldimines are efficient and powerful methods to prepare chiral β-amino carbonyl derivatives. In 2009, Guin et al. described the application of chiral bifunctional organocatalyst C31 in such type of reaction. However, acyclic substrates do not seem to be suitable for this protocol, giving 83% yield and moderate stereoselectivity (63:37 dr, 71% ee) even with prolonged reaction time (Scheme 54).58 A stereochemical model wherein the catalyst activates both the ketoester and the imine was proposed by the authors.
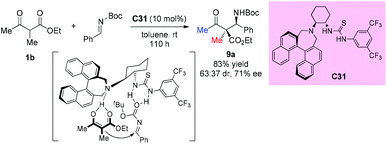 |
| | Scheme 54 Asymmetric Mannich reaction of β-ketoesters catalyzed by C31. | |
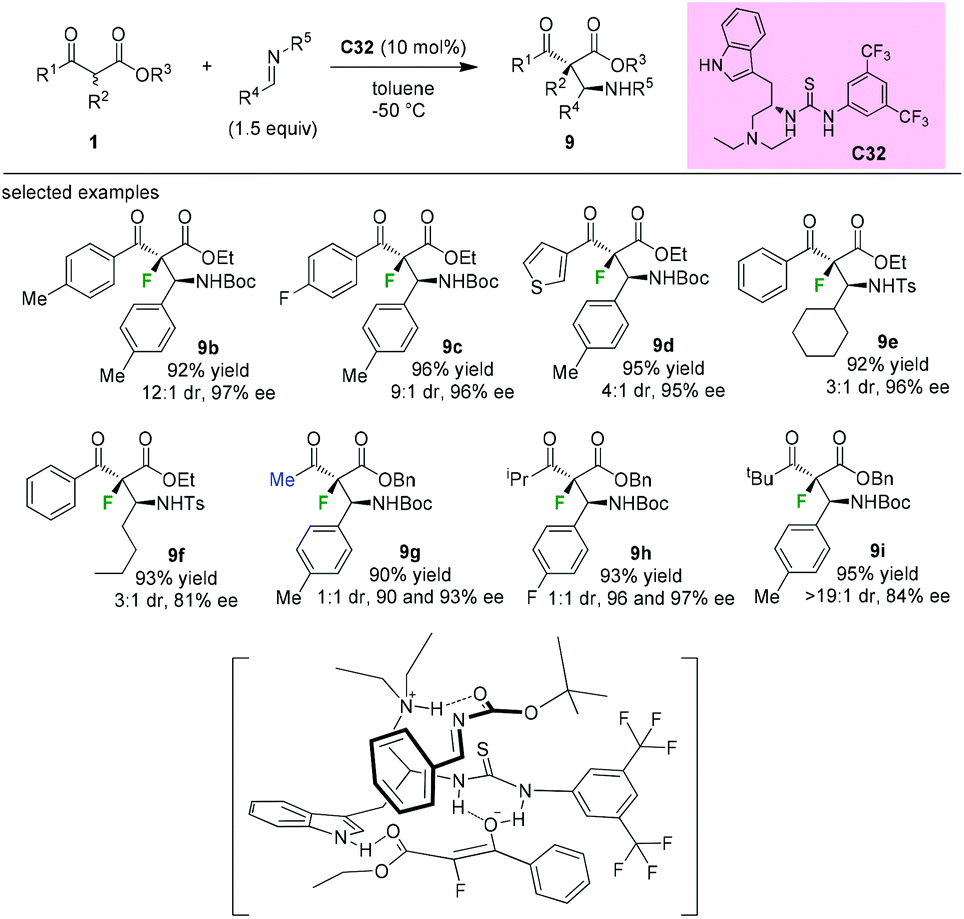
In 2009, Lu and co-workers introduced the synthesis of a novel tryptophan-based bifunctional thiourea catalyst C32, and its applications in the Mannich reaction of α-fluoro-β-ketoesters to afford fluorinated chiral molecules containing vicinal tetrasubstituted and tertiary stereogenic centers. Good diastereoselectivities and high enantioselectivities could be achieved by employing aromatic α-fluorinated substrates. The methyl and isopropyl ketoesters afforded 1:1 mixtures of diastereomers (9g and 9h), but with high enantioselectivities. Further study indicated that the tert-butyl α-fluorinated ketoester could be converted to the desired product 9i with a good dr value and a high ee value. DFT calculations support that the indole could assist the thiourea moiety in binding the ketoenolate through an additional hydrogen bonding, and the ammonium group binds the imine to bring it in proximity to the enolate (Scheme 55).59
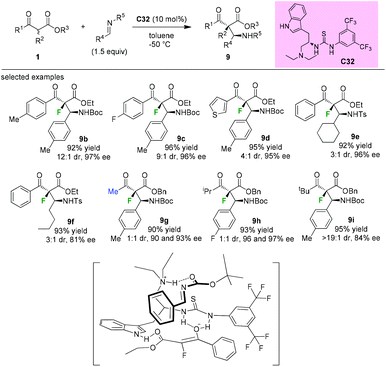 |
| | Scheme 55 Asymmetric Mannich reaction catalyzed by C32. | |
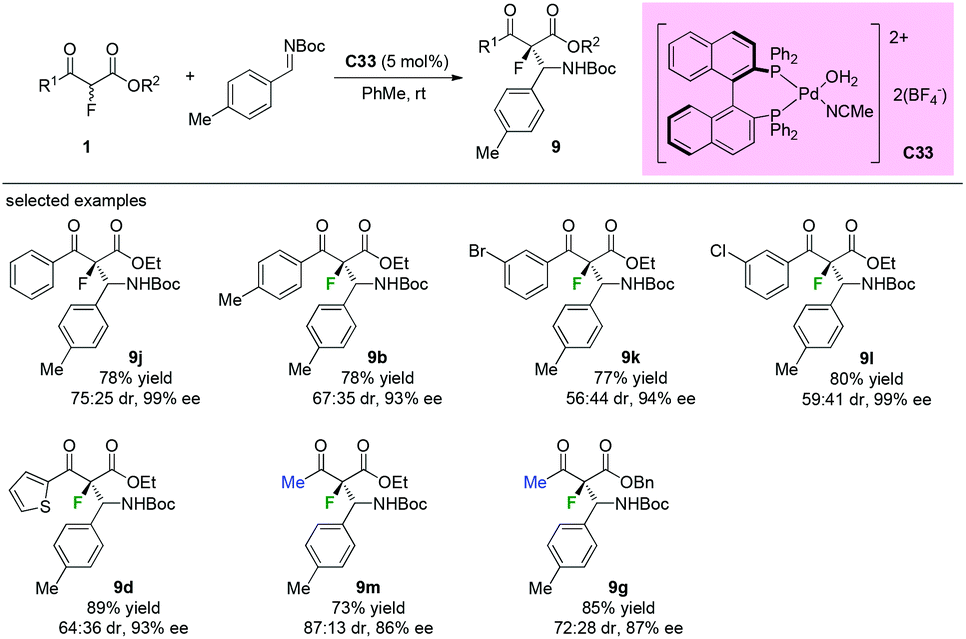
Similar reaction catalyzed by chiral palladium complex C33 was developed by Kim and co-workers in 2011. As shown in Scheme 56, both aromatic and methyl ketone-containing substrates could be well tolerated in this protocol, and the desired β-aminated products were obtained in good to high yields (73–89%) with high enantioselectivities (86–99% ee) and diverse diastereoselectivity (56:44–87:13) (Scheme 56).60
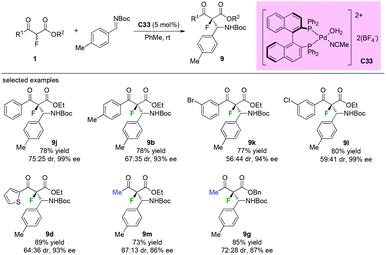 |
| | Scheme 56 Pd-Complex catalyzed Mannich reactions of the fluorinated β-ketoesters. | |
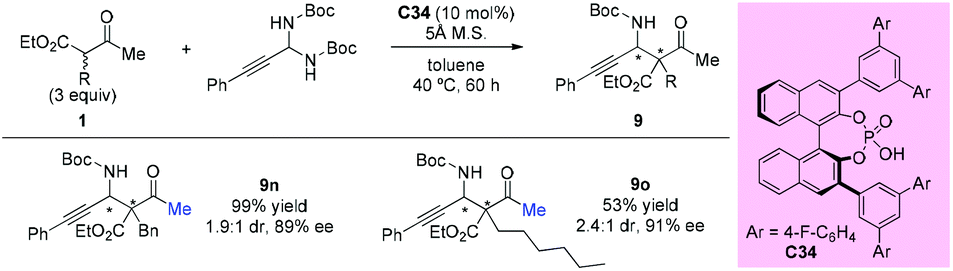
In 2013, the Maruoka group reported the chiral phosphoric acid catalyzed stereoselective Mannich-type reactions of in situ generated C-alkynyl imines with β-ketoesters. Among the dicarbonyls examined, two acyclic β-ketoesters were investigated and the afforded the adducted 9n and 9o with good enantioselectivities, albeit with low diastereoselectivities (Scheme 57).61
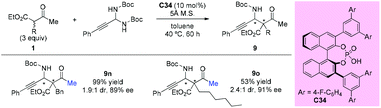 |
| | Scheme 57 Asymmetric Mannich-type reactions catalyzed by chiral phosphoric acid. | |
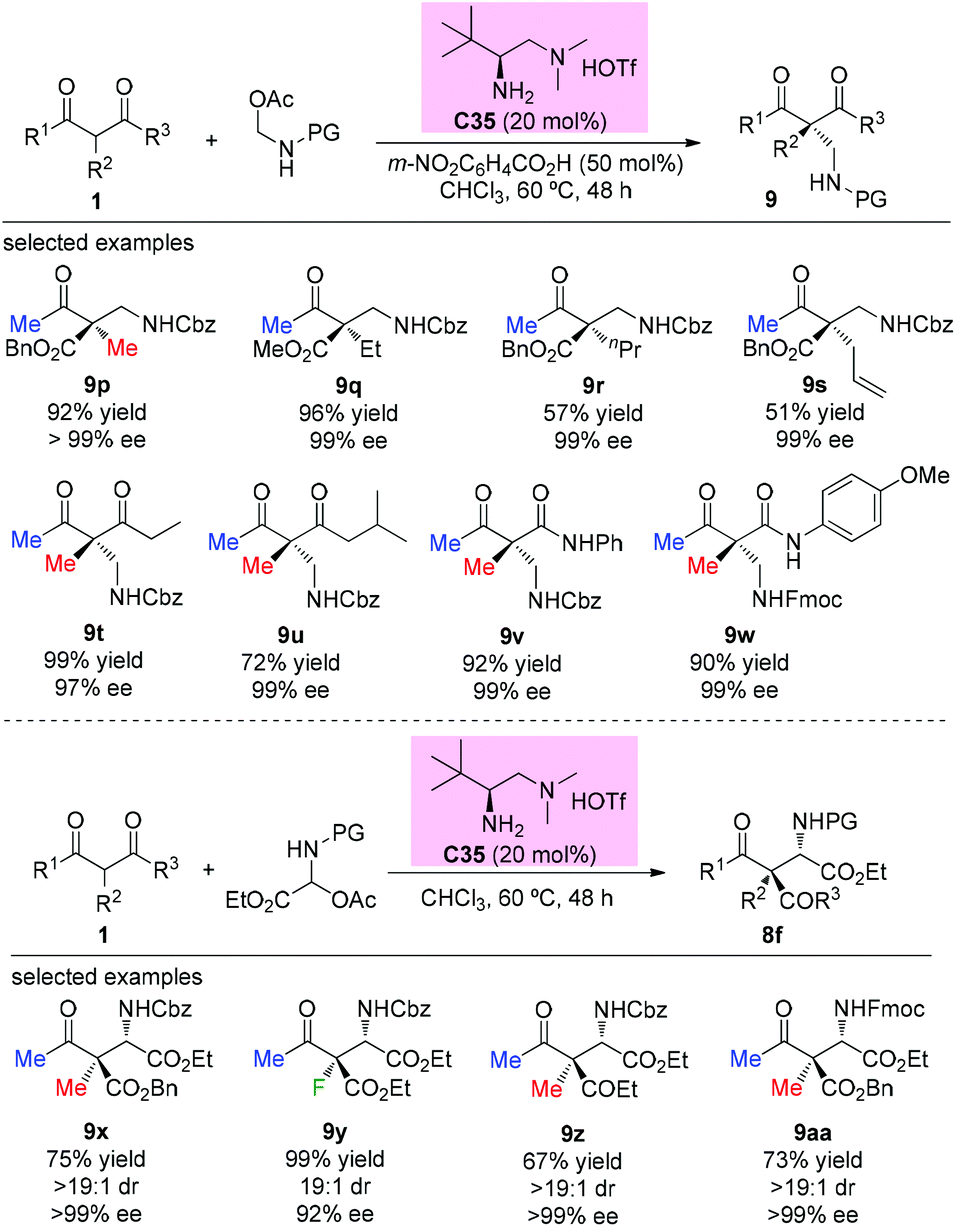
The first catalytic asymmetric Mannich reaction with α-branched ketones using enamine catalysis was achieved by the Luo group in 2017. A series of N,O-acetals were developed as bench stable surrogates for imines in the reaction with acyclic and cyclic β-ketocarbonyls. The primary amine catalyzed Mannich reactions occurred with high yields and excellent stereoselectivity, providing a straightforward approach for the synthesis of α- or β-amino carbonyls bearing quaternary centers. In the cases of acyclic α-branched β-ketocarbonyls, only methyl ketone derivatives were used (Scheme 58), further demonstrating the practicality of the Methyl Rule. It is worth mentioning that product 9y, with an α-fluoro group, was obtained in 99% yield with 92% ee.62a A similar catalytic reaction was also reported by the same group using tri/difluoro- or trichloroacetaldimine precursors, affording chiral CF3-, CF2H,- or CCl3-substituted amines in excellent yields and high enantioselectivity.62b Again the Methyl Rule was followed when quaternary stereocenters were constructed in the protocol.
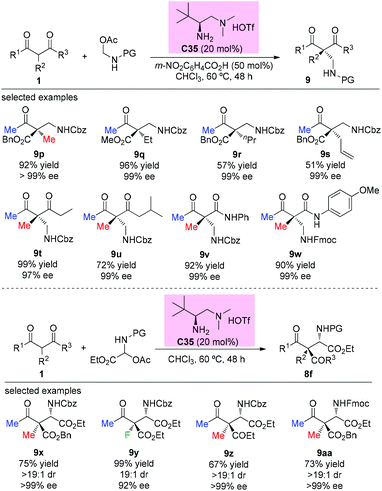 |
| | Scheme 58 Asymmetric Mannich reaction of β-ketocarbonyls with enamine catalysis. | |
10. α-Sulfenylation of β-ketocarbonyls
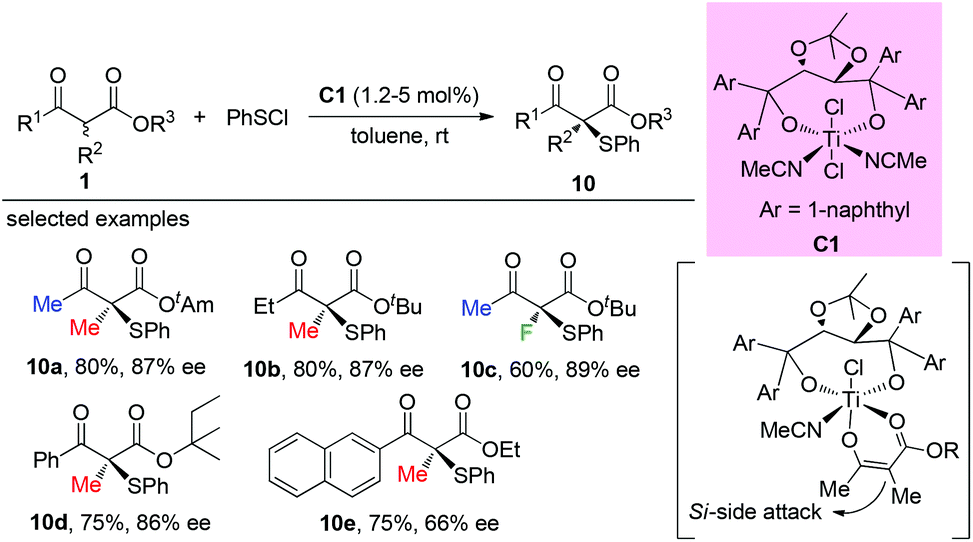
The Togni group developed for the first time the asymmetric α-sulfenylation of β-ketoesters using Ti-complex C1. In all cases, moderate to good enantioselectivities were observed, and the Methyl Rule was followed by the reactions (Scheme 59).63
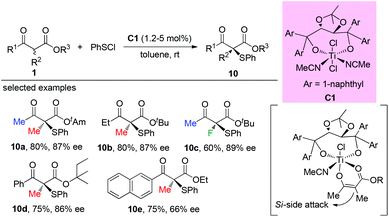 |
| | Scheme 59 Ti-Complex catalysed asymmetric α-sulfenylation. | |
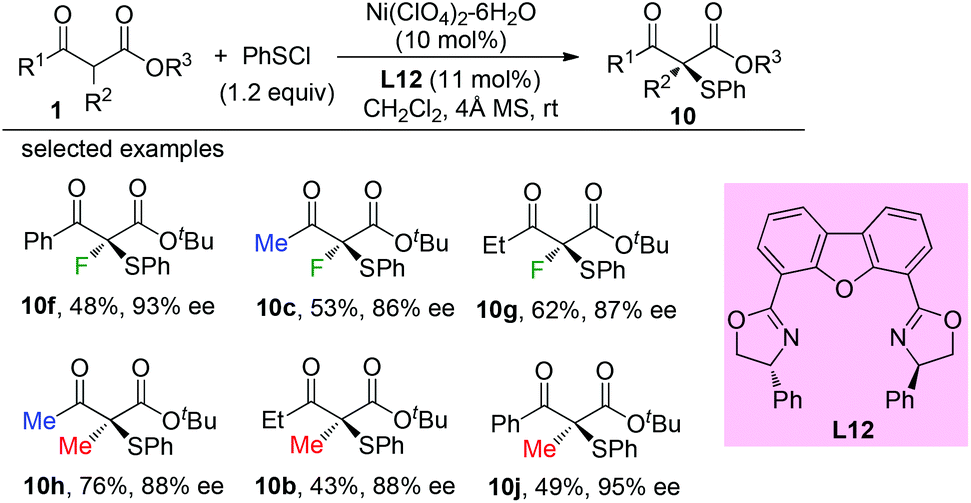
Later, a Ni-catalyzed asymmetric α-sulfenylation of β-ketoesters was developed by Shibata and co-workers. Ligand L12 was used to obtain the products with good to excellent ee. In all cases, α-methyl or fluoro-substituted β-ketoesters were used (Scheme 60).64
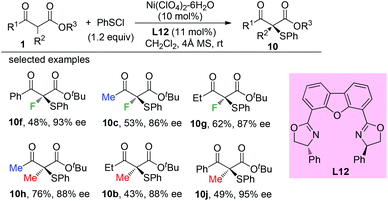 |
| | Scheme 60 Ni-Complex catalysed asymmetric α-sulfenylation. | |
In 2018, an organic primary amine catalysed direct α-sulfenylation of acyclic and cyclic β-ketocarbonyls was achieved by the Luo group. The corresponding products were obtained in good yields with excellent enantioselectivities. Besides cyclic ketoesters, only methyl ketones were tested, and the results are in accordance with the Methyl Rule (Scheme 61).65
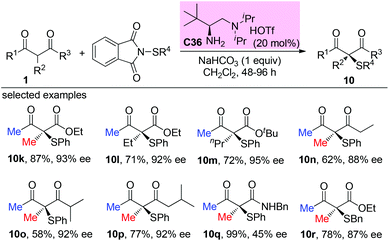 |
| | Scheme 61 Primary amine-catalzyed asymmetric α-sulfenylation. | |
11. Alternative approaches allowing access to quaternary β-ketocarbonyls with sterically more bulky substituents
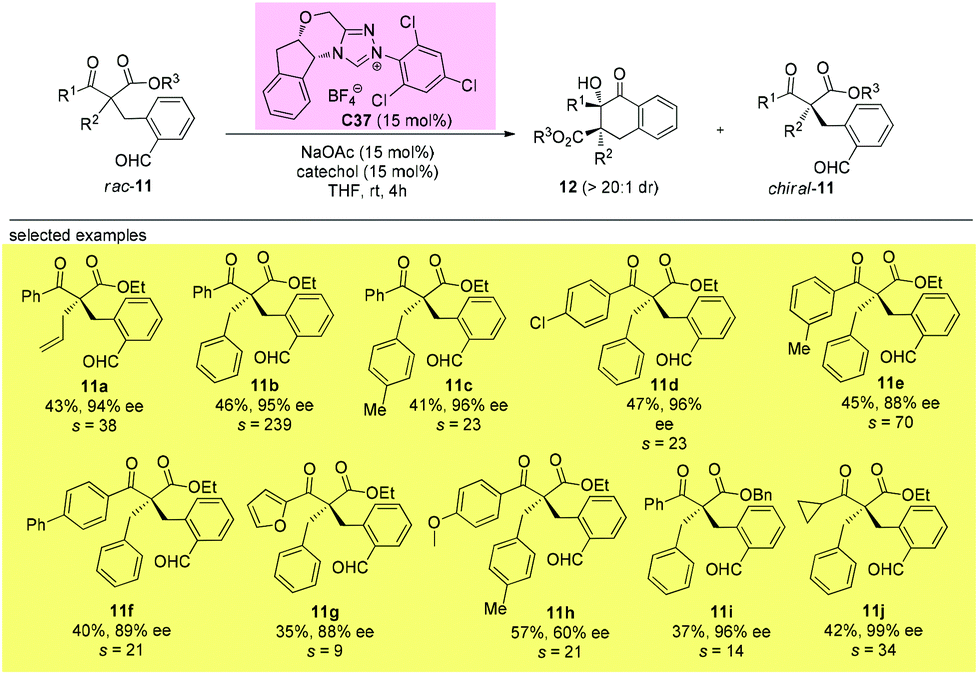
It can be seen from the above selected examples that the Methyl Rule actually widely exists in catalytic asymmetric transformations with α-branched β-ketocarbonyl participation. Among the fifty-nine reactions shown above, there are only three exceptions, and two of them use substrates bearing nitrogen or oxygen α-substituents, and the only successful reaction leading to products with two α-substituents that are all sterically bigger than Me employs a radical pathway. However, there are some alternative methods allowing access to enantioenriched α,α-disubstituted β-ketocarbonyls without limitations arising from the sterically bulky substituents, such as the kinetic resolution method.
In 2019, Fang and co-workers achieved the first catalytic kinetic resolution of α,α-disubstituted β-ketoesters using intramolecular benzoin reaction. The protocol tolerates two different α-benzyl type groups, and the ketone unit can be phenyl type groups. A series of previously unavailable β-ketoesters with all-carbon quaternary stereocenters can be obtained with good to excellent enantioselectivity. The employment of intramolecular reaction greatly overcame the low reactivity brought about by the bulky substituents, and a catechol additive proved important in enhancing the selectivity of the resolution (Scheme 62).66
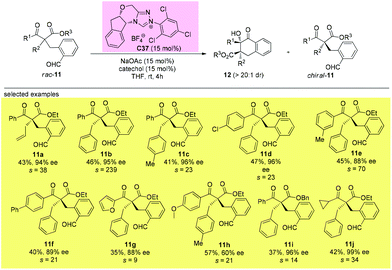 |
| | Scheme 62 Kinetic resolution of quaternary β-ketoesters via intramolecular benzoin reaction. | |
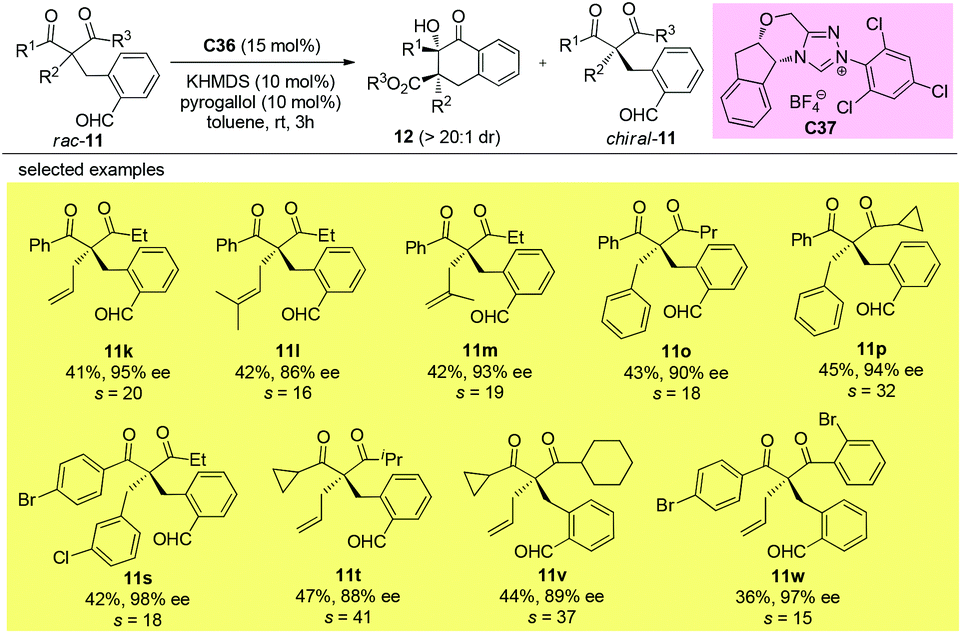
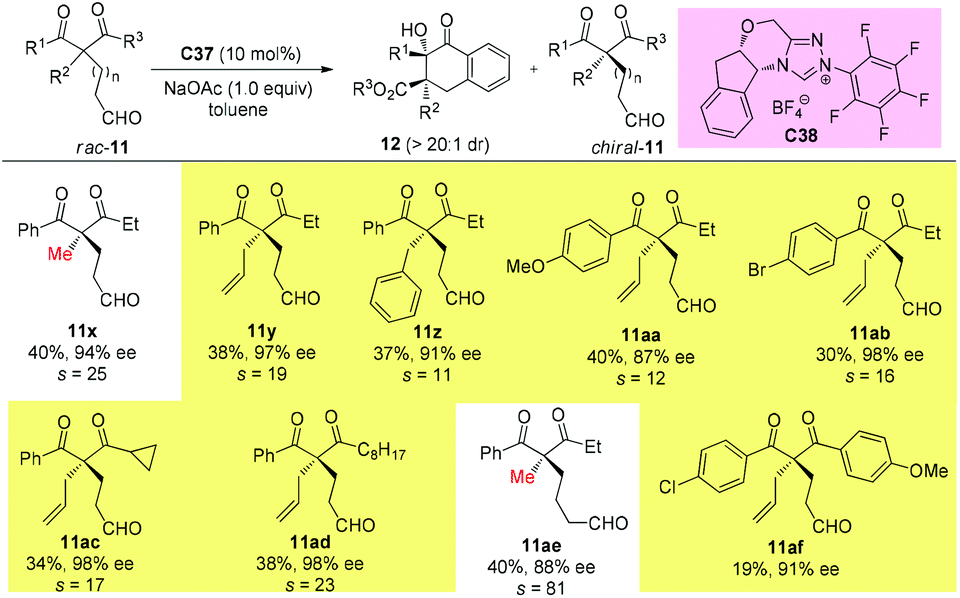
Using the same strategy, the Fang group also achieved the kinetic resolution of 2,2-disubstituted 1,3-diketones with more bulky substituents than the methyl group. The work is more challenging because the substrates contain two ketone moieties and a selective nucleophilic attack by the catalytically formed Breslow intermediate is crucial for the success of the work. Two basic kinetic resolution modes were disclosed by the report, and when the two ketone units showed comparable reactivities, divergent type resolution was applied; in contrast, when the two ketone moieties have apparently different reactivities, classical kinetic resolution takes place. Using this method, a large amount of enantioenriched 1,3-diketones with 2-substituents bulkier than Me can be obtained (Schemes 63 and 64).67
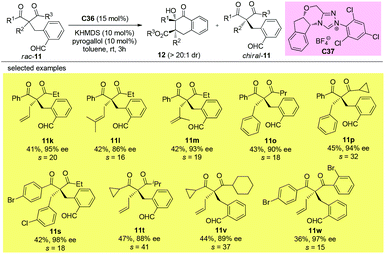 |
| | Scheme 63 Kinetic resolution of quaternary 1,3-diketones via intramolecular benzoin reaction. | |
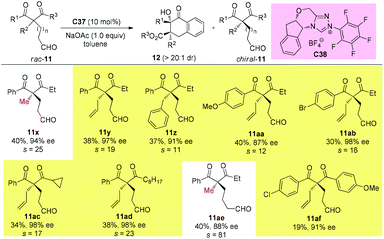 |
| | Scheme 64 Kinetic resolution of quaternary 1,3-diketones via intramolecular benzoin reaction. | |
12. Preliminary analysis of the origin of the Methyl Rule
The origin of the Methyl Rule can be analysed based on the different activation modes mentioned above. Basically, there are mainly three active intermediates derived from ketoesters involved in the transformations, which are enolate, enol, and enamine.
In the cases of enamine formation, the catalysts are usually primary amines, as witnessed by the Luo group in their elegant works. So first of all, the ketone unit must be able to react with primary amines to form enamines. Therefore, the reactivities of the ketone units play a crucial role in the success of the reaction. In the work shown in Schemes 63 and 64, Fang and co-workers have disclosed an empirical conclusion with respect to the keto reactivities using intramolecular benzoin reaction as the probe (Scheme 65).67 It can be seen that a methyl ketone actually shows competitive reactivity towards a cyclic ketone, and is more reactive than phenyl ketones and other aliphatic ketones.
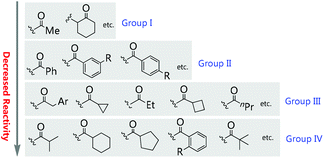 |
| | Scheme 65 Empirical conclusion using intramolecular benzoin reaction as the criterion. | |
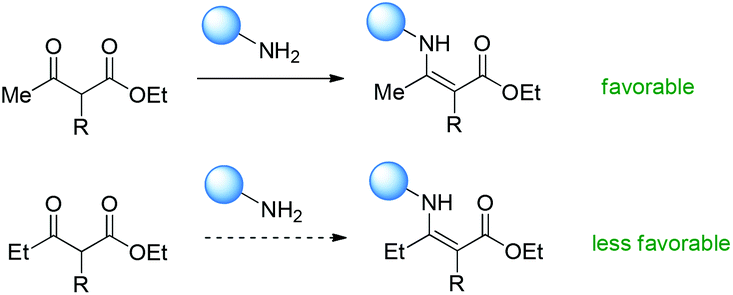
Therefore, Scheme 66 shows the major origin of the enamine intermediate-related transformations using β-ketoesters, that is, the primary amine catalyst can react smoothly with methyl ketones, but other ketones such as ethyl and phenyl ones are less favorable to react with primary amines, and result in low yields or no conversion. This can be clearly found from the results in Scheme 8.
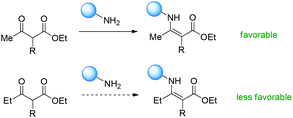 |
| | Scheme 66 Ketone reactivity as the major origin of the Methyl Rule using primary amines as the catalysts. | |
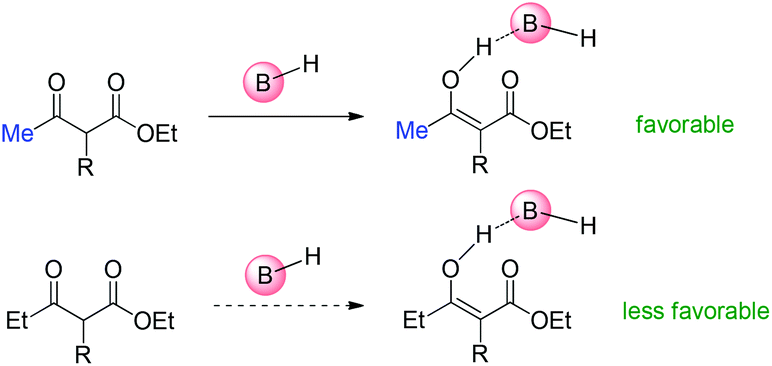
As to Brønsted acid-catalyzed reactions, the enol form is the active intermediate. In this case, the ability of β-ketocarbonyls to form the corresponding enols through tautomerization with the assistance of the catalysts is the key factor. Under the reaction conditions, it is considered that it is more easier for methyl ketone to form the corresponding enol than ethyl ketones (Scheme 67).68
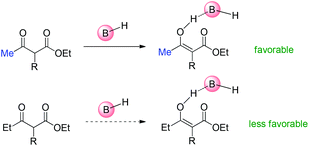 |
| | Scheme 67 Tautomerization ability as the major origin of the Methyl Rule using Brønsted acids as the catalysts. | |
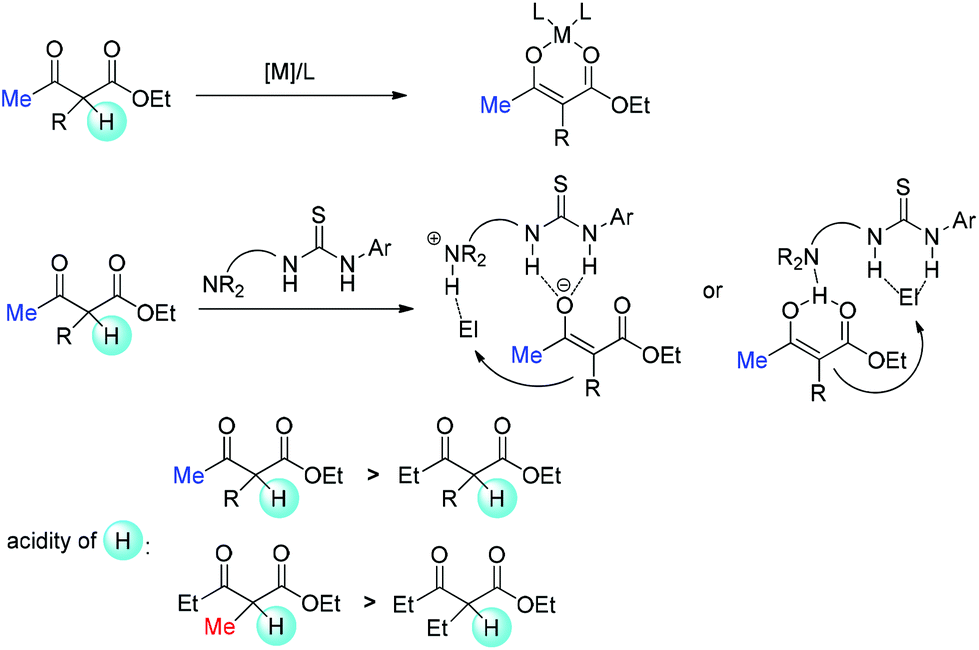
Relatively complicated situations will be encountered when enolate intermediates are applied in the reactions. To form enolates, deprotonation must occur. Therefore, the acidity of the α-proton in β-ketocarbonyls is one of the key parameters. When metal Lewis acid or bifunctional tertiary amine is used, relatively weak bases are present in the reaction. Hence, methyl ketones or methyl-substituted ketocarbonyls are easier to be deprotonated, and it is relatively more difficult for other substrates to form enolates (Scheme 68).69
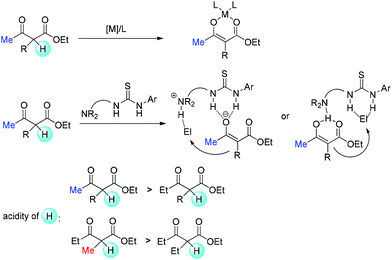 |
| | Scheme 68 Acidity of the α-proton as the origin of the Methyl Rule. | |
However, there are some cases where stoichiometric amounts of strong bases are used in the system, such as those that use phase transfer catalysts. In these cases, enolates can be definitely formed, and the difference is the reactivity of enolates with electrophiles at the following step. Now the steric bulkiness of the α-substituents and the nucleophilicity of the enolates may play crucial roles in determining the results (Scheme 69). Moreover, finding a catalytic system that can tolerate strong bases is also a challenging topic.
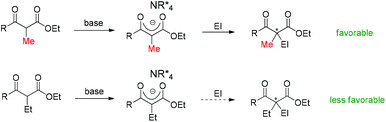 |
| | Scheme 69 Steric bulkiness of α-substituents and nucleophilicity of enolates as the origin of the Methyl Rule. | |
From the above analysis, it can be seen that methyl ketocarbonyls or α-methyl-substituted ketocarbonyls are actually relatively significant in asymmetric catalytic transformations. Furthermore, it should be noted that in many cases, two or more factors work together to affect the final outcomes. For instance, when an α-ethyl-substituted substrate can also form an enolate complex, then the steric bulkiness and the nucleophilicity start to affect the result. Anyway, all these reasons work together in most cases, but the final results make the Methyl Rule a relatively widely existing phenomenon.
13. Conclusions
This review summarizes the recent advances of diverse types of catalytic asymmetric reactions using acyclic α-branched β-ketocarbonyls, including asymmetric α-fluorination, Michael reaction, α-amination, aldol reaction, α-alkylation, α-oxygenation, Mannich reaction, and α-sulfenylation. Various catalytic systems have been developed and impressive progress has been achieved, providing a large number of protocols to construct various types of chiral quaternary or tetrasubstituted carbon stereocenters. As emphasized above, the α-substituents and ketone moieties within the β-ketocarbonyls are found to significantly affect the outcome of the reaction, especially the efficiency. Therefore, in this field, the Methyl Rule is a general principle that describes the feature of the reactivity of α-branched β-ketocarbonyls. However, complementary methods such as kinetic resolution have been developed to overcome the shortage brought about by the Methyl Rule. The possible origin of the Methyl Rule has also been analyzed according to the catalyst types. Nevertheless, the development of novel strategies and protocols which can break the restriction of the Methyl Rule and promote further the field is still highly needed.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank NSFC (21871260), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), Fujian Natural Science Foundation (2018J05035), and China Postdoctoral Science Foundation (2018M630734) for finacial support.
Notes and references
-
(a) K. Fuji, Asymmetric Creation of Quaternary Carbon Centers, Chem. Rev., 1993, 93, 2037–2066 CrossRef CAS;
(b) L. Zhang, N. Fu and S. Luo, Pushing the Limits of Aminocatalysis: Enantioselective Transformations of α-Branched β-Ketocarbonyls and Vinyl Ketones by Chiral Primary Amines, Acc. Chem. Res., 2015, 48, 986–997 CrossRef CAS;
(c) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS.
-
(a) L. Hintermann and A. Togni, Catalytic Enantioselective Fluorination of β-Ketoesters, Angew. Chem., Int. Ed., 2000, 39, 4359–4362 CrossRef CAS;
(b) S. Piana, I. Devillers, A. Togni and U. Rothlisberger, The Mechanism of Catalytic Enantioselective Fluorination: Computational and Experimental Studies, Angew. Chem., Int. Ed., 2002, 41, 979–982 CrossRef CAS.
- D. Y. Kim and E. J. Park, The Mechanism of Catalytic Enantioselective Fluorination: Computational and Experimental Studies, Org. Lett., 2002, 4, 545–547 CrossRef CAS.
- Y. Hamashima, K. Yagi, H. Takano, L. Tamás and M. Sodeoka, An Efficient Enantioselective Fluorination of Various β-Ketoesters Catalyzed by Chiral Palladium Complexes, J. Am. Chem. Soc., 2002, 124, 14530–14531 CrossRef CAS.
- Y. Hamashima, H. Takano, D. Hotta and M. Sodeka, Immobilization and Reuse of Pd Complexes in Ionic Liquid: Efficient Catalytic Asymmetric Fluorination and Michael Reactions with β-Ketoesters, Org. Lett., 2003, 5, 3225–3228 CrossRef CAS.
- Y. Hamashima, T. Suzuki, H. Takano, Y. Shimura, Y. Tsuchiya, K.-i. Moriya, T. Goto and M. Sodeoka, Highly Enantioselective Fluorination Reactions of β-Ketoesters and β-Ketophosphonates Catalyzed by Chiral Palladium Complexes, Tetrahedron, 2006, 62, 7168–7179 CrossRef CAS.
- K. Hayamizu, N. Terayama, D. Hashizume, K. Dodo and M. Sodeoka, Unique Features of Chiral Palladium Enolates Derived from β-Ketoamide: Structure and Catalytic Asymmetric Michael and Fluorination Reactions, Tetrahedron, 2015, 71, 6594–6601 CrossRef CAS.
- M. Frings and C. Bolm, Enantioselective Halogenation of β-Oxo Esters Catalyzed by a Chiral Sulfoximine-Copper Complex, Eur. J. Org. Chem., 2009, 4085–4090 CrossRef CAS.
-
(a) K. Shibatomi, A. Narayama, Y. Soga, T. Muto and S. Iwasa, Enantioselective gem-Chlorofluorination of Active Methylene Compounds Using a Chiral Spiro Oxazoline Ligand, Org. Lett., 2011, 13, 2944–2947 CrossRef CAS;
(b) K. Shibatomi, M. Kotozaki, N. Sasaki, I. Fujisawa and S. Iwasa, Williamson Ether Synthesis with Phenols at a Tertiary Stereogenic Carbon: Formal Enantioselective Phenoxylation of β-Keto Esters, Chem. – Eur. J., 2015, 21, 14095–14098 CrossRef CAS.
-
(a) K. Balaraman, R. Vasanthan and V. Kesavan, Enantioselective Fluorination of β-Ketoesters Using Tartrate Derived Bidentate Bioxazoline-Cu(II) Complexes, Tetrahedron: Asymmetry, 2013, 24, 919–924 CrossRef CAS;
(b) Y. Wang, H. Wang, Y. Jiang, C. Zhang, J. Shao and D. Xu, Fast, Solvent-Free and Highly Enantioselective Fluorination of β-Keto Esters Catalyzed by Chiral Copper Complexes in a Ball Mill, Green Chem., 2017, 19, 1674–1677 RSC.
- Y. You, L. Zhang and S. Luo, Reagent-Controlled Enantioselectivity Switch for the Asymmetric Fluorination of β-Ketocarbonyls by Chiral Primary Amine Catalysis, Chem. Sci., 2017, 8, 621–626 RSC.
- Y. Hamashima, D. Hotta and M. Sodeoka, Direct Generation of Nucleophilic Chiral Palladium Enolate from 1,3-Dicarbonyl Compounds: Catalytic Enantioselective Michael Reaction with Enones, J. Am. Chem. Soc., 2002, 124, 11240–11241 CrossRef CAS.
- Y. Hamashima, D. Hotta, N. Umebayashi, Y. Tsuchiya, T. Suzuki and M. Sodeoka, Catalytic Enantioselective Michael
Reaction of 1,3-Dicarbonyl Compounds via Formation of Chiral Palladium Enolate, Adv. Synth. Catal., 2005, 347, 1576–1586 CrossRef CAS.
- F. Wu, H. Li, R. Hong and L. Deng, Construction of Quaternary Stereocenters by Efficient and Practical Conjugate Additions to α,β-Unsaturated Ketones with a Chiral Organic Catalyst, Angew. Chem., Int. Ed., 2006, 45, 947–950 CrossRef CAS.
- M. Bella and K. A. Jørgensen, Organocatalytic Enantioselective Conjugate Addition to Alkynones, J. Am. Chem. Soc., 2004, 126, 5672–5673 CrossRef CAS.
- C. L. Rigby and D. J. Dixon, Enantioselective Organocatalytic Michael Additions to Acrylic Acid Derivatives: Generation of All-Carbon Quaternary Stereocentres, Chem. Commun., 2008, 3798–3800 RSC.
-
(a) Z. Chen, M. Furutachi, Y. Kato, S. Matsunaga and M. Shibasaki, A Stable Homodinuclear Biscobalt(III)–Schiff Base Complex for Catalytic Asymmetric 1,4-Addition Reactions of β-Keto Esters to Alkynones, Angew. Chem., Int. Ed., 2009, 48, 2218–2220 CrossRef CAS;
(b) M. Furutachi, Z. Chen, S. Matsunaga and M. Shibasaki, Catalytic Asymmetric 1,4-Additions of β-Keto Esters to Nitroalkene Promoted by a Bifunctional Homobimetallic Co2-Schiff Base Complex, Molecules, 2010, 15, 532–544 CrossRef CAS;
(c) H. Mitsunuma and S. Matsunaga, Dinuclear Ni2-Schiff Base Complex-Catalyzed Asymmetric 1,4-Addition of β-Keto Esters to Nitroethylene Toward γ2,2-Amino Acid Synthesis, Chem. Commun., 2011, 47, 469–471 RSC.
- J. R. Robinson, X. Fan, J. Yadav, P. J. Carroll, A. J. Wooten, M. A. Pericas, E. J. Schelter and P. J. Walsh, Air- and Water-Tolerant Rare Earth Guanidinium BINOLate Complexes as Practical Precatalysts in Multifunctional Asymmetric Catalysis, J. Am. Chem. Soc., 2014, 136, 8034–8041 CrossRef CAS.
- T. Tatsumi, T. Misaki and T. Sugimura, Organocatalytic 1,4-Addition Reaction of 2-Formyl(thio)esters to Vinylketones: An Efficient Access to Acyclic Chiral Building Blocks with a Quaternary Carbon Stereocenter, Chem. – Eur. J., 2015, 21, 18971–18974 CrossRef CAS.
- X. Han, J. Luo, C. Liu and Y. Lu, Asymmetric Generation of Fluorine-Containing Quaternary Carbons Adjacent to Tertiary Stereocenters: Uses of Fluorinated Methines as Nucleophiles, Chem. Commun., 2009, 2044–2046 RSC.
- Y. Lu, G. Zou and G. Zhao, Primary-Secondary Diamines Catalyzed Michael Reaction to Generate Chiral Fluorinated Quaternary Carbon Centers, Tetrahedron, 2015, 71, 4137–4144 CrossRef CAS.
- P. Vinayagam, M. Vishwanath and V. Kesavan, New Class of Bifunctional Thioureas from L-Proline: Highly Enantioselective Michael Addition of 1,3-Dicarbonyls to Nitroolefins, Tetrahedron: Asymmetry, 2014, 25, 568–577 CrossRef CAS.
- G. Chen, G. Liang, Y. Wang, P. Deng and H. Zhou, A Homodinuclear Cobalt Complex for the Catalytic Asymmetric Michael Reaction of β-Ketoesters to Nitroolefins, Org. Biomol. Chem., 2018, 16, 3841–3850 RSC.
- M. Marigo, K. Juhl and K. A. Jørgensen, Catalytic, Highly Enantioselective, Direct Amination of β-Ketoesters, Angew. Chem., Int. Ed., 2003, 42, 1367–1369 CrossRef CAS.
- C. Foltz, B. Stecker, G. Marconi, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Stereochemical Consequences of Threefold Symmetry in Asymmetric Catalysis: Distorting C3 Chiral 1,1,1-Tris(oxazolinyl)ethanes (“Trisox”) in CuII Lewis Acid Catalysts, Chem. – Eur. J., 2007, 13, 9912–9923 CrossRef CAS.
- A. Kumar, S. K. Ghosh and J. A. Gladysz, Tris(1,2-diphenylethylenediamine)cobalt(III) Complexes: Chiral Hydrogen Bond Donor Catalysts for Enantioselective α-Aminations of 1,3-Dicarbonyl Compounds, Org. Lett., 2016, 18, 760–763 CrossRef CAS.
- L. Benavent, F. Puccetti, A. Baeza and M. Gómez-Martínez, Readily Available Chiral Benzimidazoles-Derived Guanidines as Organocatalysts in the Asymmetric α-Amination of 1,3-Dicarbonyl Compounds, Molecules, 2017, 22, 1333 CrossRef.
- C. Xu, L. Zhang and S. Luo, Asymmetric Enamine Catalysis with β-Ketoesters by Chiral Primary Amine: Divergent Stereocontrol Modes, J. Org. Chem., 2014, 79, 11517–11526 CrossRef CAS.
- S. Santra, U. Maji and J. Guin, Enantioselective α-Amination of Acyclic 1,3-Dicarbonyls Catalyzed by N-Heterocyclic Carbene, Org. Lett., 2020, 22, 468–473 CrossRef CAS.
- C. Xu, L. Zhang and S. Luo, Merging Aerobic Oxidation and Enamine Catalysis in the Asymmetric α-Amination of β-Ketocarbonyls Using N-Hydroxycarbamates as Nitrogen Sources, Angew. Chem., Int. Ed., 2014, 53, 4149–4153 CrossRef CAS.
- D. Sandoval, C. P. Frazier, A. Bugarin and J. R. de Alaniz, Electrophilic α-Amination Reaction of β-Ketoesters Using N-Hydroxycarbamates: Merging Aerobic Oxidation and Lewis Acid Catalysis, J. Am. Chem. Soc., 2012, 134, 18948–18951 CrossRef CAS.
- W. Chen, Y. Wang, X. Mi and S. Luo, Enantioselective Oxidative Coupling of β-Ketocarbonyls and Anilines by Joint Chiral Primary Amine and Selenium Catalysis, Org. Lett., 2019, 21, 8178–8182 CrossRef CAS.
- N. Umebayashi, Y. Hamashima, D. Hashizume and M. Sodeoka, Catalytic Enantioselective Aldol-Type Reaction of β-Ketosters with Acetals, Angew. Chem., Int. Ed., 2008, 47, 4196–4199 CrossRef CAS.
- S. Mouri, Z. Chen, S. Matsunaga and M. Shibasaki, Direct Catalytic Asymmetric Aldol Reaction of β-Keto Esters with Formaldehyde Promoted by a Dinuclear Ni2-Schiff Base Complex, Chem. Commun., 2009, 5138–5140 RSC.
- K. Hiroi and Y. Suzuki, Chiral β-Amino Sulfoxides as Chiral Ligands in Palladium-Catalyzed Asymmetric Allylations, Heterocycles, 1997, 46, 77–81 CrossRef CAS.
- R. Kuwano and Y. Ito, Catalytic Asymmetric Allylation of Prochiral Nucleophiles, α-Acetamido-β-ketoesters, J. Am. Chem. Soc., 1999, 121, 3236–3237 CrossRef CAS.
- K. Nagata, D. Sano, Y. Shimizu, M. Miyazaki, T. Kanemitsu and T. Itoh, Catalytic Asymmetric Alkylation of α-Cyanocarboxylates and Acetoacetates Using a Phase-Transfer Catalyst, Tetrahedron: Asymmetry, 2009, 20, 2530–2536 CrossRef CAS.
- T. Hashimoto, K. Sasaki, K. Fukumoto, Y. Murase, N. Abe, T. Ooi and K. Maruoka, Phase-Transfer-Catalyzed Asymmetric Alkylation of α-Benzoyloxy-β-keto Esters: Stereoselective Construction of Congested 2,3-Dihydroxycarboxylic Acid Esters, Chem. - Asian J., 2010, 5, 562–570 CrossRef CAS.
- T. Kobayashi, R. Shioi, A. Ushie, H. Abe and H. Ito, Catalytic Asymmetric Total Synthesis of (+)-Artalbic Acid, Chem. Commun., 2016, 52, 9391–9393 RSC.
- H.-G. Cheng, B. Feng, L.-Y. Chen, W. Guo, X.-Y. Yu, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Rational Design of Sulfoxide-Phosphine Ligands for Pd-Catalyzed Enantioselective Allylic Alkylation Reactions, Chem. Commun., 2014, 50, 2873–2875 RSC.
-
(a) Y. Zhu, L. Zhang and S. Luo, Asymmetric α-Photoalkylation of β-Ketocarbonyls by Primary Amine Catalysis: Facile Access to Acyclic All-Carbon Quaternary Stereocenters, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS;
(b) W. Zhang, Y. Zhu, L. Zhang and S. Luo, Asymmetric α-Alkylation of β-Ketocarbonyls via Direct Phenacyl Bromide Photolysis by Chiral Primary Amine, Chin. J. Chem., 2018, 36, 716–722 CrossRef CAS.
- H. Zhou, L. Zhang, C. Xu and S. Luo, Chiral Primary Amine/Palladium Dual Catalysis for Asymmetric Allylic Alkylation of β-Ketocarbonyl Compounds with Allylic Alcohols, Angew. Chem., Int. Ed., 2015, 54, 12645–12648 CrossRef CAS.
- Q.-L. Liu, W. Chen, Q.-Y. Jiang, X.-F. Bai, Z. Li, Z. Xu and L.-W. Xu, A D-Camphor-Based Schiff Base as a Highly Efficient N,P Ligand for Enantioselective Palladium-Catalyzed Allylic Substitutions, ChemCatChem, 2016, 8, 1495–1499 CrossRef CAS.
- M. Yoshida, S. Yanob and S. Hara, Asymmetric Allylation of 2-Oxocycloalkanecarboxylates, Synthesis, 2017, 49, 1295–1300 CrossRef CAS.
- M. Zhao, Y. Tian and X. Zhao, Where Are They Now? Chiral Sulfinamide Ligands and Pd-Catalyzed Asymmetric Allylic Alkylations
of Ethyl 2-Fluoroacetoacetate, Synlett, 2017, 28, 1801–1806 CrossRef CAS.
- Y.-N. Xu, M.-Z. Zhu and S.-K. Tian, Chiral α-Amino Acid/Palladium-Catalyzed Asymmetric Allylation of α-Branched β-Ketoesters with Allylic Amines: Highly Enantioselective Construction of All-Carbon Quaternary Stereocenters, J. Org. Chem., 2019, 84, 14936–14942 CrossRef CAS.
-
(a) H. Zhou, Y. Wang, L. Zhang, M. Cai and S. Luo, Enantioselective Terminal Addition to Allenes by Dual Chiral Primary Amine/Palladium Catalysis, J. Am. Chem. Soc., 2017, 139, 3631–3634 CrossRef CAS;
(b) Y. Wang, H. Zhou, K. Yang, C. You, L. Zhang and S. Luo, Steric Effect of Protonated Tertiary Amine in Primary-Tertiary Diamine Catalysis: A Double-Layered Sterimol Model, Org. Lett., 2019, 21, 407–411 CrossRef CAS.
- Y. Wang, J. Chai, C. You, J. Zhang, X. Mi, L. Zhang and S. Luo, π-Coordinating Chiral Primary Amine/Palladium Synergistic Catalysis for Asymmetric Allylic Alkylation, J. Am. Chem. Soc., 2020, 142, 3184–3195 CrossRef CAS.
- Y. Zhu, W.-Z. Zhang, L. Zhang and S. Luo, Chiral Primary Amine Catalyzed Asymmetric α-Benzylation with In Situ Generated ortho-Quinone Methides, Chem. – Eur. J., 2017, 23, 1253–1257 CrossRef CAS.
- X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Electricity-Driven Asymmetric Lewis Acid Catalysis, Nat. Catal., 2019, 2, 34–40 CrossRef CAS.
-
(a) C. Wei, X. Ye, Q. Xing, Y. Hu, Y. Xie and X. Shi, Synergistic Palladium/Enamine Catalysis for Asymmetric Hydrocarbon Functionalization of Unactivated Alkenes with Ketones, Org. Biomol. Chem., 2019, 17, 6607–6611 RSC;
(b) W.-B. Liu, C. M. Reeves and B. M. Stoltz, Enantio-, Diastereo-, and Regioselective Iridium-Catalyzed Asymmetric Allylic Alkylation of Acyclic β-Ketoesters, J. Am. Chem. Soc., 2013, 135, 17298–17301 CrossRef CAS.
- D. Wang, L. Zhang and S. Luo, Enantioselective Decarboxylative α-Alkynylation of β-Ketocarbonyls via a Catalytic α-Imino Radical Intermediate, Org. Lett., 2017, 19, 4924–4927 CrossRef CAS.
- M. Lu, D. Zhu, Y. Lu, X. Zeng, B. Tan, Z. Xu and G. Zhong, Chiral Brønsted Acid-Catalyzed Enantioselective α-Hydroxylation of β-Dicarbonyl Compounds, J. Am. Chem. Soc., 2009, 131, 4562–4563 CrossRef CAS.
- D. Wang, C. Xu, L. Zhang and S. Luo, Asymmetric α-Benzoyloxylation of β-Ketocarbonyls by a Chiral Primary Amine Catalyst, Org. Lett., 2015, 17, 576–579 CrossRef CAS.
- C. Xu, L. Zhang and S. Luo, Catalytic Asymmetric Oxidative α-C−H N,O-Ketalization of Ketones by Chiral Primary Amine, Org. Lett., 2015, 17, 4392–4395 CrossRef CAS.
- Y. Feng, R. Huang, L. Hu, Y. Xiong and V. Coeffard, Chiral C2-Symmetric Iodoarene-Catalyzed Asymmetric α-Oxidation of β-Keto Esters, Synthesis, 2016, 48, 2637–2644 CrossRef CAS.
- J. Novacek, J. A. Izzo, M. J. Vetticatt and M. Waser, Bifunctional Ammonium Salt Catalyzed Asymmetric α-Hydroxylation of β-Ketoesters by Simultaneous Resolution of Oxaziridines, Chem. – Eur. J., 2016, 22, 17339–17344 CrossRef CAS.
- Y. K. Kang and D. Y. Kim, Organocatalytic Highly Enantio- and Diastereoselective Mannich Reaction of β-Ketoesters with N-Boc-aldimines, J. Org. Chem., 2009, 74, 5734–5737 CrossRef CAS.
- X. Han, J. Kwiatkowski, F. Xue, K.-W. Huang and Y. Lu, Asymmetric Mannich Reaction of Fluorinated Ketoesters with a Tryptophan-Derived Bifunctional Thiourea Catalyst, Angew. Chem., Int. Ed., 2009, 48, 7604–7607 CrossRef CAS.
- Y. K. Kang and D. Y. Kim, Catalytic Asymmetric Mannich-Type Reactions of Fluorinated Ketoesters with N-Boc Aldimines in the Presence of Chiral Palladium Complexes, Tetrahedron Lett., 2011, 52, 2356–2358 CrossRef CAS.
- T. Kano, T. Yurino and K. Maruoka, Organocatalytic Asymmetric Synthesis of Propargylamines with Two Adjacent Stereocenters: Mannich-Type Reactions of In Situ Generated C-Alkynyl Imines with β-Keto Esters, Angew. Chem., Int. Ed., 2013, 52, 11509–11512 CrossRef CAS.
-
(a) Y. You, L. Zhang, L. Cui, X. Mi and S. Luo, Catalytic Asymmetric Mannich Reaction with N-Carbamoyl Imine Surrogates of Formaldehyde and Glyoxylate, Angew. Chem., Int. Ed., 2017, 56, 13814–13818 CrossRef CAS;
(b) Y. You and S. Luo, Catalytic Asymmetric Mannich Type Reaction with Tri-/Difluoro- or Trichloroacetaldimine Precursors, Org. Lett., 2018, 20, 7137–7140 CrossRef CAS.
-
(a) M. Jereb and A. Togni, Titanium(IV)-Catalyzed Enantioselective Sulfenylation of β-Ketoesters, Org. Lett., 2005, 7, 4041–4043 CrossRef CAS;
(b) M. Jereb and A. Togni, TiIV-Catalyzed Asymmetric Sulfenylation of 1,3-Dicarbonyl Compounds, Chem. – Eur. J., 2007, 13, 9384–9392 CrossRef CAS.
- T. Ishimaru, S. Ogawa, E. Tokunaga, S. Nakamura and N. Shibata, Asymmetric Synthesis of α-Fluoro-α-fulfenyl-β-ketoesters Using DBFOX-Ph/Ni(II) Complex, J. Fluorine Chem., 2009, 130, 1049–1053 CrossRef CAS.
- L. Cui, Y. You, X. Mi and S. Luo, Catalytic Enantioselective α-Sulfenylation of β-Ketocarbonyls by Chiral Primary Amine, Org. Chem. Front., 2018, 5, 2313–2316 RSC.
- S. T. Zehra, G. Zhang, S. Yang and X. Fang, Kinetic Resolution of β-Ketoesters with Quaternary Stereocenters via a Carbene-Catalyzed Benzoin Reaction, Org. Biomol. Chem., 2019, 17, 2169–2173 RSC.
- W. Xu, Y. Li, R. Liu, S. Yang, J. Liu and X. Fang, Kinetic Resolution of 2,2-Disubstituted-1,3-diketones via Carbene Catalysis, Org. Chem. Front., 2019, 6, 290–298 RSC.
- For selected spectral evidence showing that it is more easier for acetyl acetate to form an enol form than propionyl acetate, see:
(a) E. Erbing, A. Vázquez-Romero, A. B. Gómez, A. E. Platero-Prats, F. Carson, X. Zou, P. Tolstoy and B. Martín-Matute, General, Simple, and Chemoselective Catalysts for the Isomerization of Allylic Alcohols: The Importance of the Halide Ligand, Chem. – Eur. J., 2016, 22, 15659–15663 CrossRef CAS;
(b) N. Nishiwaki, K. Kobiro, S. Hirao, J. Sawayama, K. Saigo, Y. Ise, M. Nishizawa and M. Ariga, One-step Synthesis of Differently Bis-functionalized Isoxazoles by Cycloaddition of Carbamoylnitrile Oxide with β-Keto Esters, Org. Biomol. Chem., 2012, 10, 1987–1991 RSC.
- For the working models of bifunctional tertiary amine catalyst–mediated reactions, see the following report and the references cited therein: P.-G. Ding, F. Zhou, Q.-H. Zhao, J.-S. Yu and J. Zhou, H-Bond Donor-Directed Switching of Diastereoselectivity in the Michael Addition of α-Azido Ketones to Nitroolefins, Chem. Sci., 2020, 11, 3852–3861 RSC.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*