DOI:
10.1039/C9QM00546C
(Review Article)
Mater. Chem. Front., 2020,
4, 12-28
pH-Controlled motions in mechanically interlocked molecules
Received
28th August 2019
, Accepted 30th September 2019
First published on 30th September 2019
Abstract
Mechanically interlocked molecules (MIMs), especially rotaxanes and catenanes, allow large-amplitude movement at the molecular level, making them the perfect prototypes for artificial molecular switches and machines. By applying external stimuli, the noncovalent interactions between their subcomponents can be interrupted and formed reversibly, leading to mechanical motions. Among various stimuli, pH stimulation is one of the most powerful and commonly used means of controlling motions by acids and bases. In this review, we summarize the pH-controlled mechanical motions including translocation in rotaxanes, circumrotation in catenanes and other kinds of motion in more sophisticated mechanomolecules, and discuss their operating mechanisms. In addition, we present several more recent developments of alternative stimuli for pH responsive motions.

He-Ye Zhou
| He-Ye Zhou received his BS degree from Xiangtan University. In 2016, he joined the laboratory of Prof. Chuan-Feng Chen at Institute of Chemistry, Chinese Academy of Sciences (ICCAS) to pursue his PhD in chemistry. His current research interest is the construction and applications of mechanically interlocked molecules based on triptycene-derived macrocyclic hosts. |

Ying Han
| Ying Han received her BS from Capital Normal University (CNU) in 2007, MS degree from CNU in 2010 under the guidance of Prof. Sheng-Li Cao, and PhD in 2013 from ICCAS under the guidance of Prof. Chuan-Feng Chen. She is now working as an associate professor at ICCAS. Her current research interests are supramolecular chemistry based on new synthetic hosts and molecular machines. |

Chuan-Feng Chen
| Chuan-Feng Chen has been working as a full professor of organic chemistry at Institute of Chemistry, Chinese Academy of Sciences since 2001. His current research interests include supramolecular chemistry based on synthetic macrocyclic hosts, chiral organic light-emitting materials and helicene chemistry. |
1. Introduction
Mechanically interlocked molecules (MIMs),1 typically rotaxanes, catenanes and knots, have attracted widespread attention, not only because of their charming topological structures,2 but also as a consequence of their ability to undergo controllable motion between their subcomponents.3 At first, introducing a mechanical bond into a molecule makes it possible to change the co-conformations of MIMs under external stimuli. Then, the interlocked structures can constrain the magnitude of the subcomponents’ movement by an entanglement in three-dimensional space. As a result, the MIMs which contain two or more different recognition sites can provide the perfect prototypes for the design and synthesis of artificial molecular switches and machines.4 This research field was highlighted by the Nobel prize in 2016 for Chemistry to Stoddart and Sauvage, alongside Feringa.5 Rotaxanes6 are described as compounds that comprise a dumbbell-shaped guest threaded through the cavity of a ring. If it is properly designed, translational motion of the ring along its associated dumbbell can be triggered under proper stimulation (Fig. 1a). Catenanes,7 which comprise two or more interlocking ring-shaped component parts, can interconvert between co-conformations by circumrotational motion of a small ring with respect to a larger one (Fig. 1b). Taking advantage of the interconversion between different co-conformations of MIMs, more diverse and sophisticated kinds of motion have been successfully fulfilled at the molecular level.8
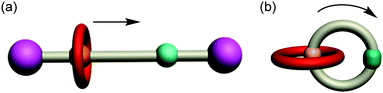 |
| | Fig. 1 Cartoon illustrations of mechanical motions: (a) translocation in typical [2]rotaxanes; (b) circumrotation in typical [2]catenanes. | |
The external stimuli, including chemical,9 photochemical,10 and electrochemical11 stimuli, are considered as an energy input to regulate the co-conformations of the MIMs, generating controllable motions. Among various external stimuli, pH stimulation is considered as one of the most convenient and efficient methods to regulate the subcomponents’ motion by readily available acids and bases, which belongs to chemical stimulation.12 Until recently, various types of macrocyclic hosts (crown ethers, cucurbiturils, tetracationic cyclophanes, amide-based macrocycles, calixarenes, etc.) have been incorporated in MIMs that perform pH-triggered motions. These pH-responsive systems have two obvious features: (i) MIMs (host or guest) usually contain reactive donor atoms (e.g. nitrogen and oxygen atoms), which are easily protonated and deprotonated with acids and bases; (ii) non-covalent interactions, particularly, hydrogen bonding, hydrophobic effects, and electrostatic interactions are very sensitive to pH variation. Therefore, acid/base has been one of the most useful stimuli for controllable motions in MIMs.
We herein present an overall review of pH-responsive motions in MIMs. In the first section, we focus on operating translational motions in rotaxane architectures based on different recognition motifs. Then, the circumrotational motions in typical [2]catenanes are discussed. Next, we turn our attention to the motions beyond linear translation and circumrotation in more sophisticated mechanomolecules. Finally, we describe the recent progress made on actuating pH-responsive motions with alternative stimuli instead of acids and bases. This review will not discuss pH-switchable association and disassociation between host and guest.
2. Operating translocation in rotaxanes
Rotaxanes, the most studied interlocked molecules, have been a versatile platform to construct stimuli-responsive molecular shuttles. Within rotaxane architectures, the associated thread usually contains two or more recognition sites with different binding affinities for the rings. By weakening or increasing the affinity of the binding sites under external stimuli, the macrocycle can be driven to shuttle from one site to another site. When there exists a protonated ammonium, pyridinium or phenolic unit in MIMs, the association and disassociation process is readily controlled by acids and bases. The addition or removal of protons in a ring or thread might change the existing hydrogen-bonding, electrostatic interactions or hydrophobic effect dramatically, resulting in mechanical movement. In this section, we focus on the acid/base-controlled translocation in rotaxanes. Different recognition motifs based on various macrocyclic hosts and their operating mechanism will be discussed.
2.1 pH-Controlled translocation based on cucurbiturils
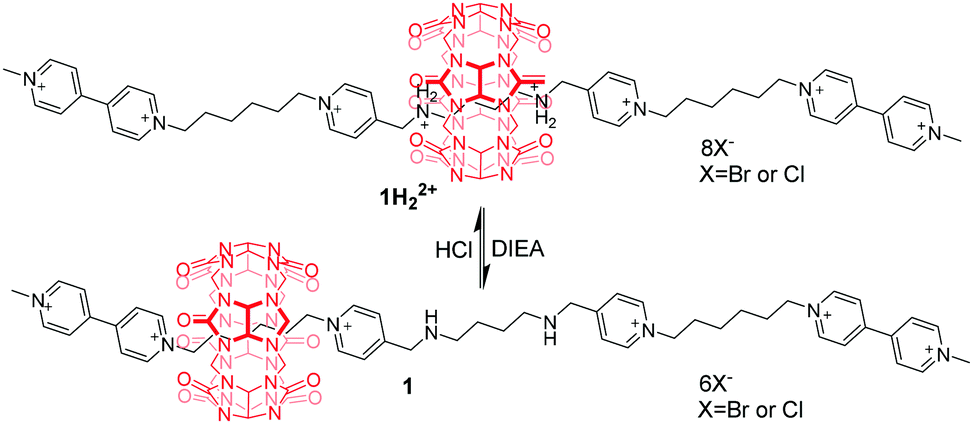
Cucurbit[n]urils (CB[n]),13 prepared by condensation of glycoluril with formaldehyde in acidic medium, are an important family of macrocyclic compounds in supramolecular and mechanically bonded systems. They are well known for their extraordinary affinity towards alkanediammoniums in water driven by the hydrophobic effect and ion–dipole interactions between the ammoniums and the polar carbonyl groups at the portals of CB[n]. Accordingly, host–guest binding between CBs and ammoniums is strongly pH-dependent, which paves the way for constructing pH-controllable molecular motions. In this respect, pioneering works of pH-driven shuttling were performed in 1990 by Mock14 and Kim.15 A bistable [2]rotaxane 1H22+ with electrostatic bipyridinium (BIPY2+) stoppers was designed and synthesized.16 The switchable translational motion of the CB[6] was driven by pH variation (Fig. 2). Under the acidic condition, the CB[6] resides exclusively on the central protonated diaminobutane unit promoted by hydrophobic effects and hydrogen-bonding interactions. Upon deprotonation of the butanediammonium in 1H22+ by diisopropylethylamine (DIEA), the CB[6] ring moves to one of the peripheral bis(pyridinium) hexane units in 1. But the reverse process requires hydrochloric acid plus thermal activation.
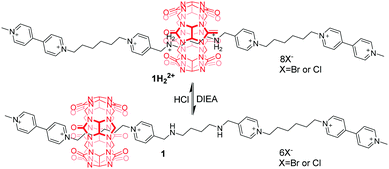 |
| | Fig. 2 pH-Controlled CB[6]-based [2]rotaxane 1 with electrostatic BIPY2+ stoppers. | |
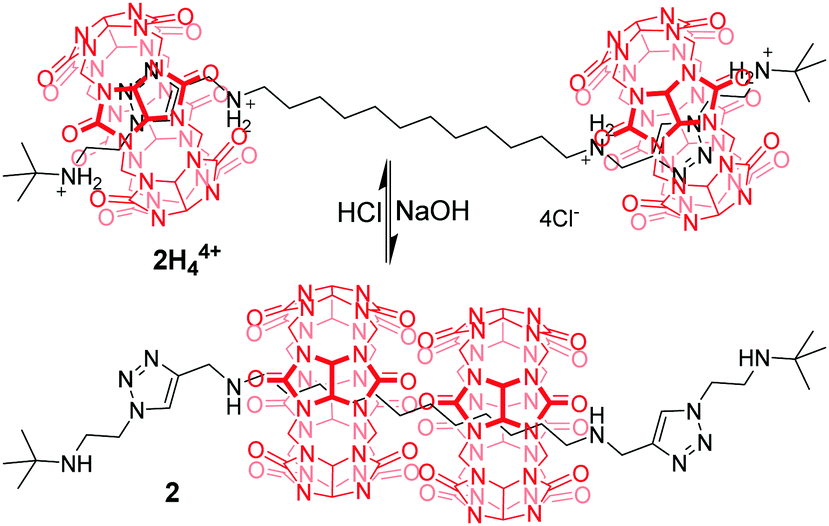
A few examples of pH-dependent [3]rotaxanes based on CB[6] have been reported by Tuncel.17 [3]rotaxane 2H44+ (Fig. 3) was prepared by CB[6]-catalyzed 1,3-dipolar cycloaddition reaction, which is composed of two CB[6] rings, each encircling triazole-bis(ammonium) units near the termini of the tert-butyl group. When NaOH was used to neutralize the ammonium groups, two CB[6] rings moved to the more hydrophobic dodecamethylene spacer. Conversely, the rings return to the triazole units in 2 after the amines have been reprotonated with HCl. Similar motifs were also incorporated into a tetrapodal [5]rotaxane to give a pH-driven radial translocation.18 Based on the similar working mechanism, CB[7] was also used for the construction of pH-controlled motions.19
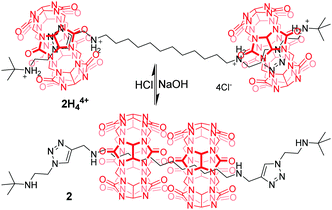 |
| | Fig. 3 Tuncel's acid/base switchable palindromic [3]rotaxane 2. | |
2.2 pH-Controlled translocation based on tetracationic cyclophanes
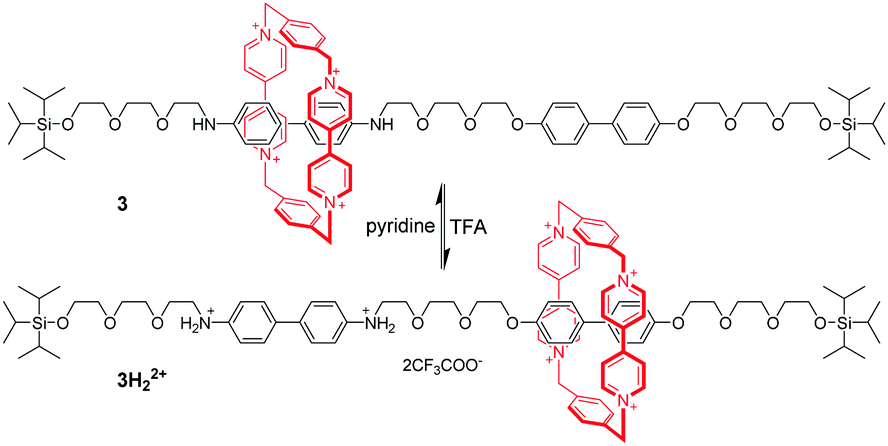
Tetracationic cyclophanes,20 which consist of two π-electron-deficient 4,4′-bipyridinium units, are capable of strongly binding with π-electron-rich guests. One of the most famous cyclophanes is cyclobis(paraquat-p-phenylene) (CBPQT4+), developed by the group of Stoddart,21 which is known as the “blue box”. Taking advantage of the CBPQT4+, the molecular shuttle was achieved based on π-associated donor–acceptor interactions.22 The [2]rotaxane 3 contains the CBPQT4+ and a linear molecule axle with benzidine and biphenol recognition sites (Fig. 4). Under neutral pH and at 229 K, the macrocycle mainly located at the benzidine position in the dumbbell, because the binding affinity of CBPQT4+ for the benzidine derivative was about 10 times greater than that for the corresponding biphenol derivative. Addition of excess trifluoroacetic acid (TFA) to pronate benzidine resulted in macrocyclic translocation to the biphenol station owing to electrostatic repulsion. Subsequently TFA-induced shuttling motion could be fully reversed by neutralization of TFA with pyridine. Another important feature was that this translational motion could also be triggered by electrochemical means. Since Stoddart's invention of the switchable molecular shuttle, chemists have used molecular switching to perform a variety of “on”/“off” tasks with synthetic MIMs.
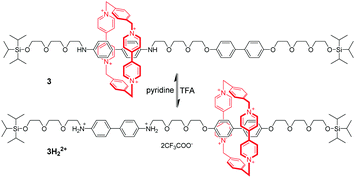 |
| | Fig. 4 pH-Switchable molecular shuttle 3 driven by π-associated donor–acceptor interactions. | |
2.3 pH-Controlled translocation based on crown ethers
Crown ethers and their derivatives have been continuingly popular for the fabrication of pH-responsive MIMs. They are of versatile binding ability towards an enormous variety of organic guests, especially dialkylammonium ions, based on hydrogen-bond interactions and electrostatic interaction. These interactions can be destroyed simply by removing the hydrogen linked to the nitrogen in the guest molecules with appropriate base. Hence, lots of pH-responsive crown ether-based MIMs have been reported in the last few decades.
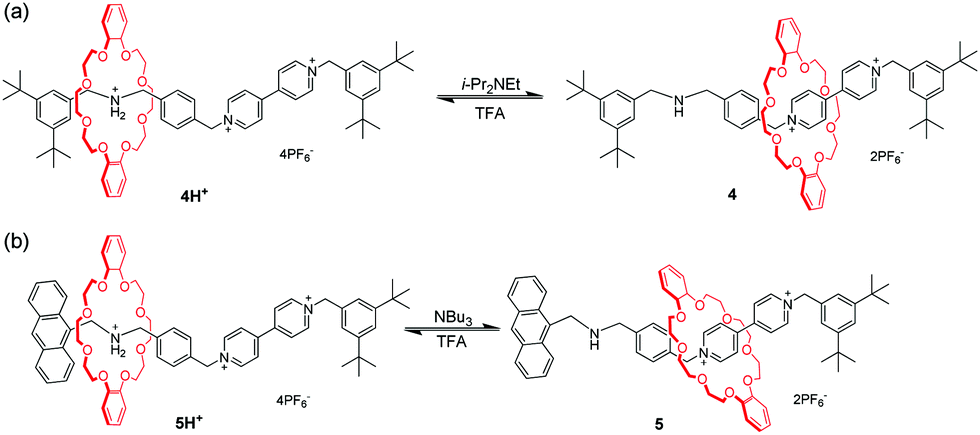
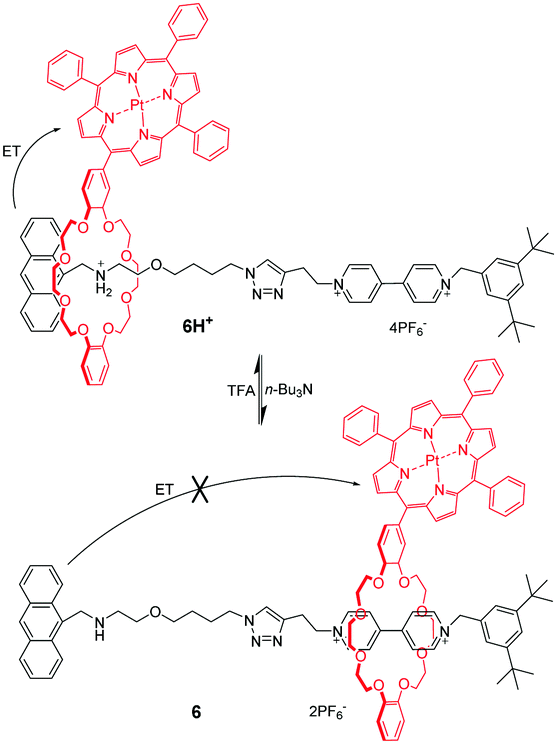
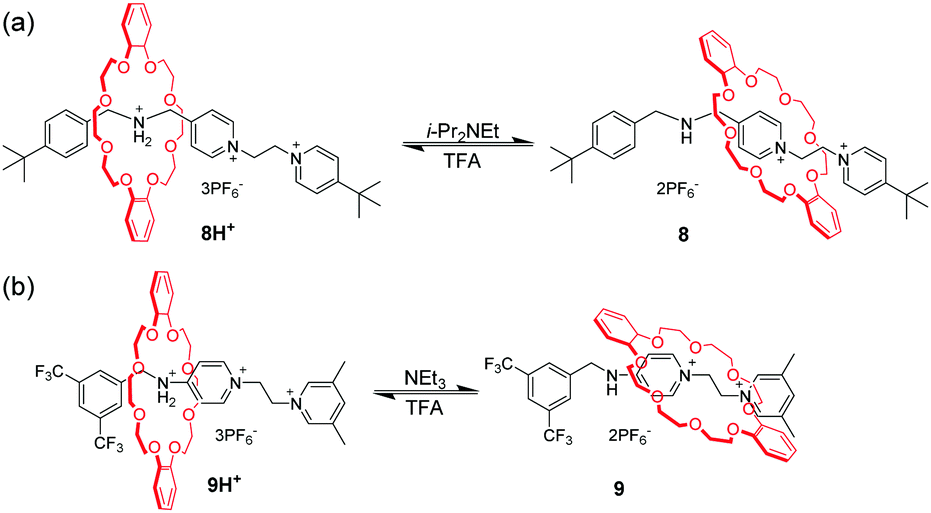
Among the crown ethers and their derivatives, dibenzo-24-crown-8 (DB24C8) is the most commonly designed and synthesized pH-responsive molecular switch and machine. Different recognition sites for DB24C8 have been explored to construct pH-switchable rotaxanes. Dialkylammomium is usually employed as a reversible control unit and another permanent cation as a second recognition site. This strategy has generated various examples owing to the discovery of novel recognition stations for DB24C8. Early research was founded by Stoddart23 that [2]rotaxane containing a dibenzylammonium unit (R2NH2+) and bipyridinium unit (BIPY2+) can be switched by acid/base. The key to this design lies in the fact that DB24C8 binds R2NH2+ more strongly than BIPY2+. Thus, the DB24C8 ring encircles the R2NH2+ site almost exclusively in 4H+ (Fig. 5a), whereas conversion to the corresponding amine 4 with i-Pr2NEt causes the ring to shuttle to the BIPY2+ unit. The rotaxane 4H+ can be easily regenerated by simply reprotonating the amine with TFA. Subsequently, a pH-controlled molecular switch 5H+ (Fig. 5b) was developed referring to the mechanism behind the operation of 4H+.24 The stopper adjacent to the R2NH2+ center was replaced with anthracene as a fluorophore. This modification made it possible to investigate the detailed kinetics of the shuttling process by using the stopped-flow spectroscopic technique.25 In 2016, Ma and co-workers26 introduced this operating mechanism into a molecular shuttle whose shuttling was encoded by room temperature phosphorescence (RTP) signals. As shown in Fig. 6, [2]rotaxane 6 comprises a Pt(II) porphyrin-containing DB24C8 threaded onto two different recognition sites and the anthracene moiety as a one terminal stopper. In the protonated state, the macrocycle was located on the R2NH2+ site close to the anthracene unit, and the porphyrin unit exhibited strong RTP emission due to the distance-dependent energy transfer between the anthracene and the porphyrin. However, deprotonation of the R2NH2+ unit could engender the displacement of the macrocycle to the BIPY2+ station, and its RTP emission dramatically decreased.
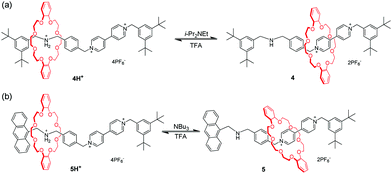 |
| | Fig. 5 Acid/base-driven molecular shuttles 4 and 5 containing a dibenzylammonium unit and bipyridinium unit. | |
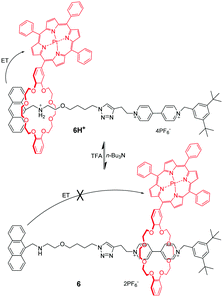 |
| | Fig. 6 [2]Rotaxane 6 with pH-switchable room temperature phosphorescence. | |
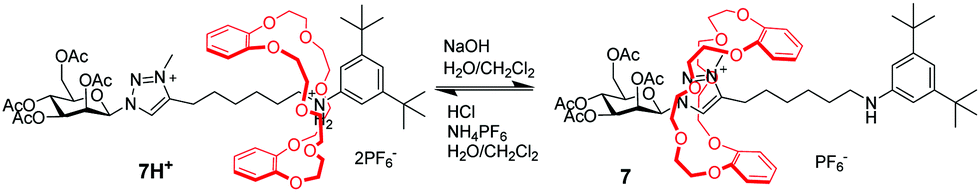
Since Coutrot's report in 2008,27N-methyltriazolium has become another popular molecular station for the crown ether, which is easily generated via methylation of the triazole formed during a copper(I)-catalyzed azide-alkene cycloaddition (CuAAC) ‘click’ stoppering reaction in a high yield. The triazolium ion has a much poorer affinity for the DB24C8 than the ammonium, which makes triazoliums an ideal second molecular station. As a consequence, in the bistable [2]rotaxane 7H+, the DB24C8 ring initially resides around the anilinium site (Fig. 7). However, deprotonation of the ammonium station triggers the shuttling motion of the crown ether toward the triazolium station. This process can be willingly reset by addition of acid. From then on, a number of MIMs have adopted the trizolium motif to develop pH-sensitive molecular machines.28 For example, functionalized [2]rotaxanes with pH-switchable fluorescent outputs have been achieved.29 Besides, triazolium-containing [3]rotaxane, [4]rotaxane and [5]rotaxane molecular machines were reported by several researchers, including Liu,30 Li,31 Tian,32 Qu33 and so on.34 In recent years, Leigh et al. creatively used a crown ether and triazolium motif to prepare an appealing rotaxane-based switchable organocatalyst.35
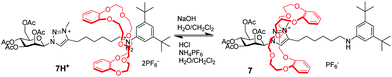 |
| | Fig. 7 pH-Switchable [2]rotaxane adopting a triazolium moiety. | |
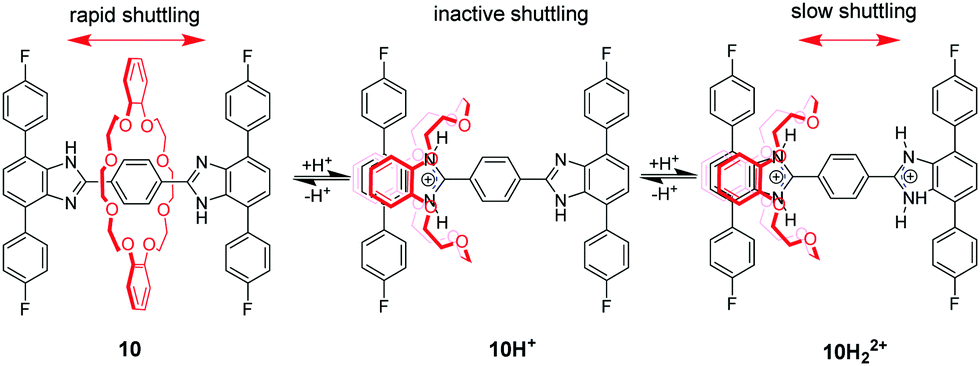
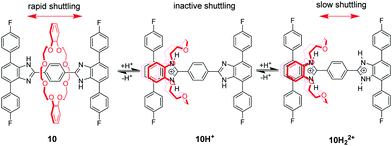
Loeb et al.36 found that the 1,2-bis(pyridinium)ethane dication (BPE2+) could also serve as a binding site for DB24C8. Subsequently, by utilizing BPE2+ as a second recognition site, acid/base-controllable molecular shuttles containing DB24C8 were constructed by Stoddart and Loeb (Fig. 8).37 The benzimidazolium ion is another common recognition template for developing MIMs with crown ethers due to their rigid core and pH responsiveness. However, the interaction of DB24C8 with either the imidazolium or phenylbenzimidazolium cation is quite weak. Hence, Loeb and co-workers developed a new T-shaped benzimidazolium cation which could severe as an efficient template for the formation of [2]pseudorotaxanes with crown ethers.38 This templating motif was incorporated into acid–base controllable molecular shuttles. [2]Rotaxane 10H22+ containing two benzimidazole recognition sites on a single axle but only a single macrocyclic wheel could function as a molecular shuttle by acid–base chemistry (Fig. 9).39 The neutral form of the molecular shuttle 10 shows an extremely rapid shuttling due to lower barrier. Addition of one equivalent of acid to give 10H+ arrests the shuttling due to a protonated site which is complexed and a neutral site which is uncomplexed. Diprotonation produces 10H22+ containing two recognition sites and the shuttling is resumed but at a much lower rate due to an increased barrier to translational motion. More interestingly, this molecular shuttle for the first time was embedded into a metal–organic framework (MOF) material allowing rapid shuttling motions in its skeletal framework.40 In addition, Loeb's group reported acid–base switchable [2]- and [3]rotaxane molecular shuttles by combining 1,2-bis(pyridinium)ethane and benzimidazolium recognition motifs.41
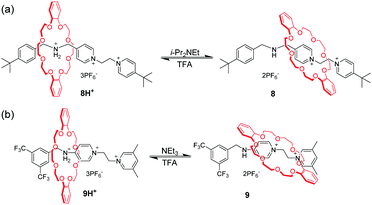 |
| | Fig. 8 pH-Switchable [2]rotaxane utilizing BPE2+ as a second recognition site. | |
 |
| | Fig. 9 Rigid H-shaped [2]rotaxane shuttle 10. | |
2.4 pH-Controlled translocation based on benzylic amide macrocycles
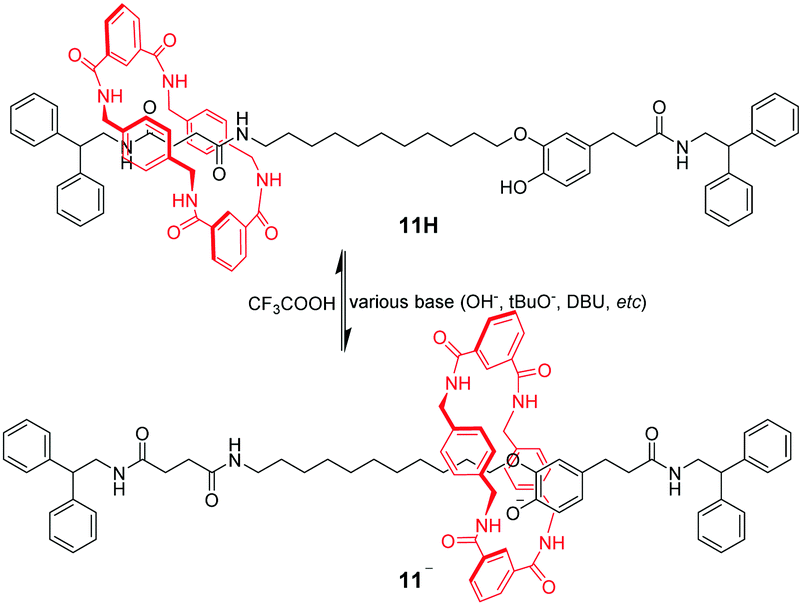
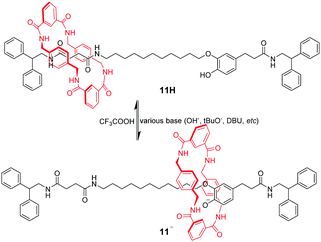
Amide-based macrocycles also bind neutral guests through hydrogen bonding, which have been effectively used for pH-triggered molecular shuttles. The first example of pH-switchable shuttling involved anion recognition of a benzylic amide macrocycle by Leigh's group.42 [2]Rotaxane 11H contains a benzylic amide ring threaded onto an axle incorporating two potential hydrogen bonding stations (Fig. 10). One of them is a coumaric amide chromophore with a phenolic group that can be deprotonated by various bases to afford an oxyanion. In the neutral form, the macrocycle resides preferentially on the succinamide station in DMF. Under basic conditions, the macrocycle shuttles away to participate in strong hydrogen bonding interactions with the corresponding phenolic oxyanion of 11−. Subsequent addition of TFA reprotonates the phenolic group and returns the ring to the succinamide location.
 |
| | Fig. 10 pH-Switchable shuttling involved anion recognition. | |
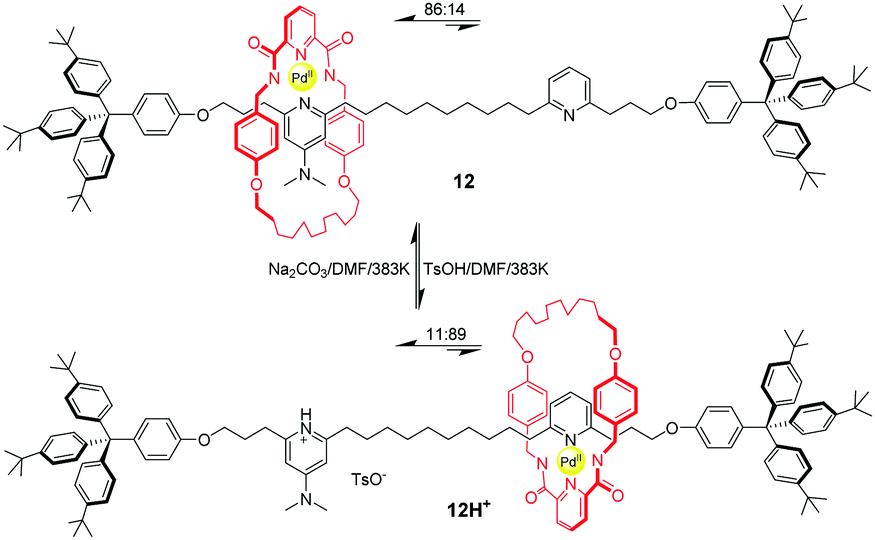
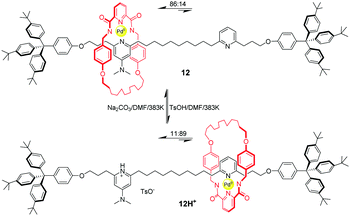
Another of Leigh's example of pH-triggered shuttling motion was based on a Pd(II)-complexed benzylic amide macrocycle.43 Pyridine (PY) and dimethylaminopyridine (DMAP) were chosen as recognition sites to form a Pd-complex with an amide macrocycle. And DMAP is a stronger base which coordinates preferentially to Pd(II) ion ensconced in the macrocycle in 12 (Fig. 11). The addition of an equivalent of p-toluenesulfonic acid (TsOH) protonates the DMAP unit selectively, thus leading to migration of the macrocycle to the PY site. As a consequence, the ratios of translational isomer 12 and 12H+ could be actuated from 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 to 11:89 by acid–base chemistry. Later, they developed a second generation of this molecular shuttle, which shows significantly increased rates of shuttling and improved co-conformational bias.44
:14 to 11:89 by acid–base chemistry. Later, they developed a second generation of this molecular shuttle, which shows significantly increased rates of shuttling and improved co-conformational bias.44
 |
| | Fig. 11 pH-Switchable palladium-complexed molecular shuttle 12. | |
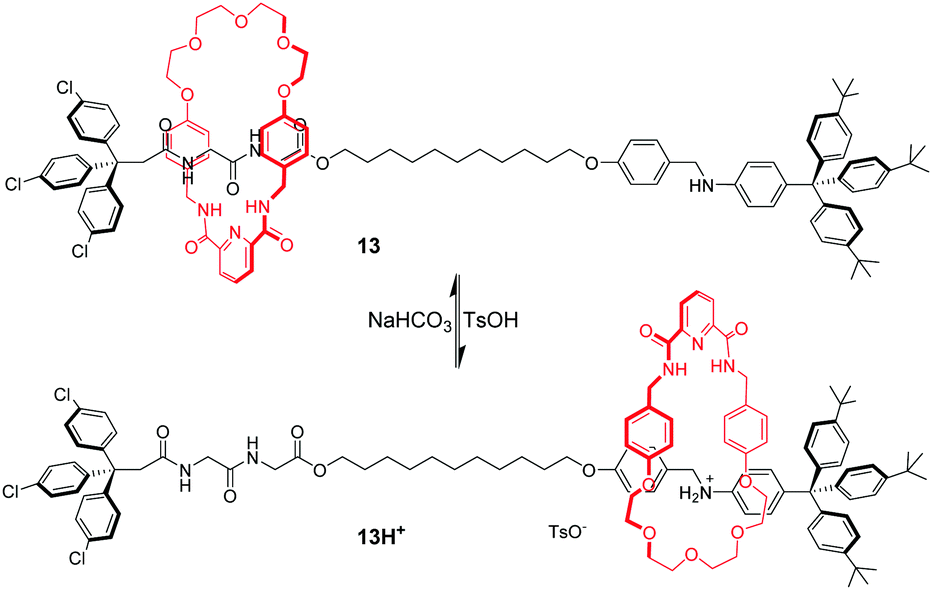
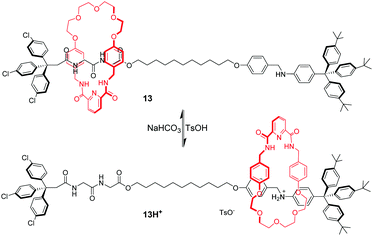
By integrating amide–amide hydrogen bonding and crown ether–ammonium cation interactions, a pH-switchable molecular shuttle with dual binding mode was achieved.45 As illustrated in Fig. 12, the thread component of rotaxane 13 contains glycylglycine (GlyGly) and anilinium as recognition sites and an isophthalamide group and polyether chain in the macrocycle. The ring encircles a dipeptide unit stabilized by amide–amide hydrogen bonds in the neutral form. When aniline is protonated with TsOH, polyether–ammonium cation interactions dominate and the macrocycle migrates to the anilinium station in 13H+. By using the same principle, several pH-controlled molecular shuttles based on this hybrid polyether/amide macrocycle were also developed by Li,46 Chiu47 and Wang.48
 |
| | Fig. 12 pH-Controlled molecular shuttle 13 based on a hybrid polyether/amide macrocycle. | |
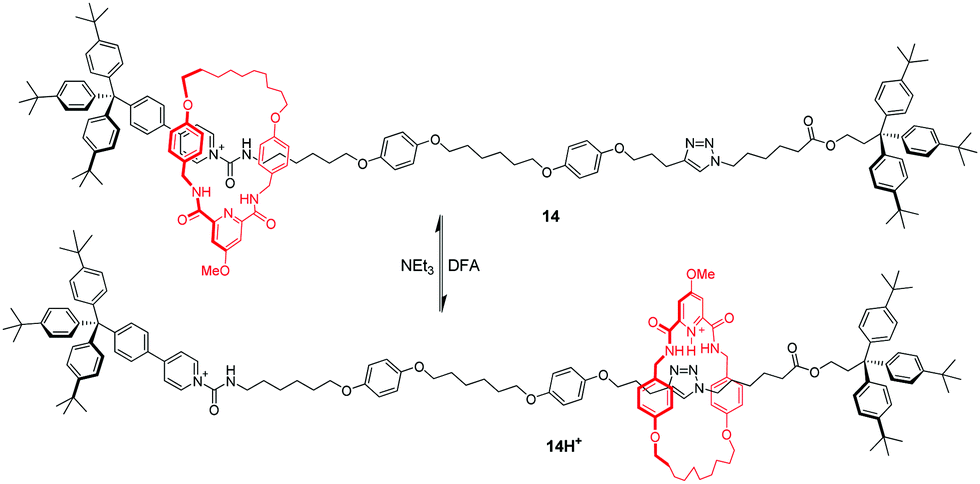
Lüning and co-workers49 also reported a pH-induced molecular shuttle using pyridine-modified benzylic amide macrocycle. The shuttling process can be operated by protonation/deprotonation of the ring. In [2]rotaxane 14 in Fig. 13, the benzylic amide ring initially resides at one end of the dumbbell where two supramolecular interactions favor this orientation: hydrogen bonds and donor–acceptor interactions with amide and pyridinium units adjacent to the stopper. After the protonation of ring's pyridyl group with difluoroacetic acid (DFA), the macrocycle shuttles to the triazole station as a result of electrostatic repulsion and an increased propensity to interact with the triazole units.
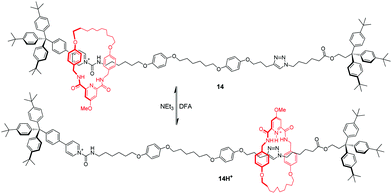 |
| | Fig. 13 pH-Induced molecular shuttle 14 using a pyridine-modified benzylic amide macrocycle. | |
2.5 pH-Controlled rotaxanes based on calix[6]arene
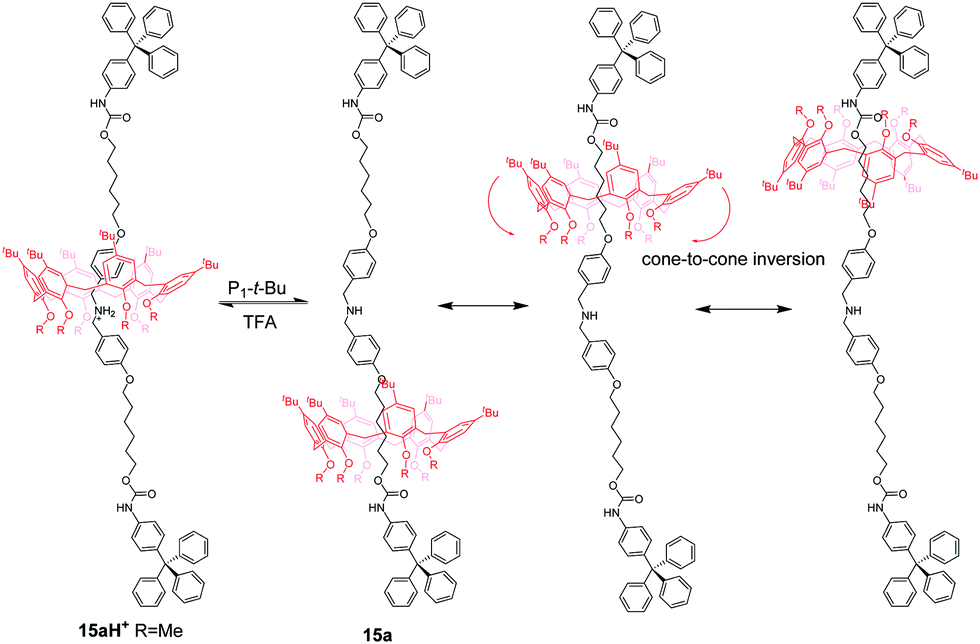
Calix[6]arene (CA6), with an electron-rich cavity, tends to bind protonated secondary ammonium or tertiary ammonium, which can be efficiently controlled by acid/base. Neri and co-workers50 demonstrated the first example of calix[6]arene-based molecular shuttles. As shown in Fig. 14, a dibenzylammonium-containing axle molecule thread into the cavity of a calix[6]arene derivative in the [2]rotaxanes 15aH+ and 15bH+, which differ only in their methoxyl and n-hexyloxyl group appended at the calix[6]arene lower rim. And the macrocycle surrounds exclusively the dibenzylammonium site in a cone conformation for the formation of hydrogen bonds between its OR groups and ammonium. Upon deprotonation of the ammonium with phosphazene base P1tBu (N-tert-butyl-N′,N′,N′′,N′′,N′′′,N′′′-hexamethylphosphorimidic triamide), the biased shuttling motion in neutral [2]rotaxane was observed to give two translational isomers, 15L and 15U, in which the alkoxy moieties at the lower rim or the tert-butyl groups at the upper rim are facing the trityl stopper, respectively. And the rate of the two isomers of 15a is much more biased (K = 32) than that (K = 1.5) of 15b, which may be ascribed to the formation of stronger hydrogen-bonding interactions in 15a with the urethane group linked to the stoppers. More interestingly, the CA6 ring of 15a can undergo a cone-to-cone inversion of the macrocycle via through-the-annulus passage of the small OMe group (Fig. 15).
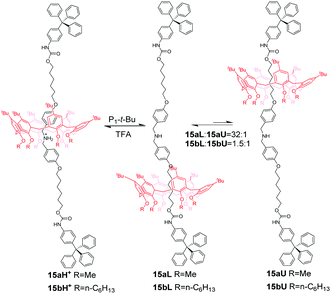 |
| | Fig. 14 pH-Induced molecular shuttle 15 based on calix[6]arene. | |
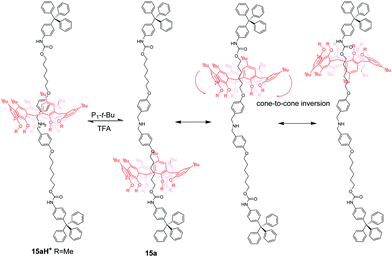 |
| | Fig. 15 Combined translocation/inversion motion of the calix[6]-wheel in [2]rotaxane 15a. | |
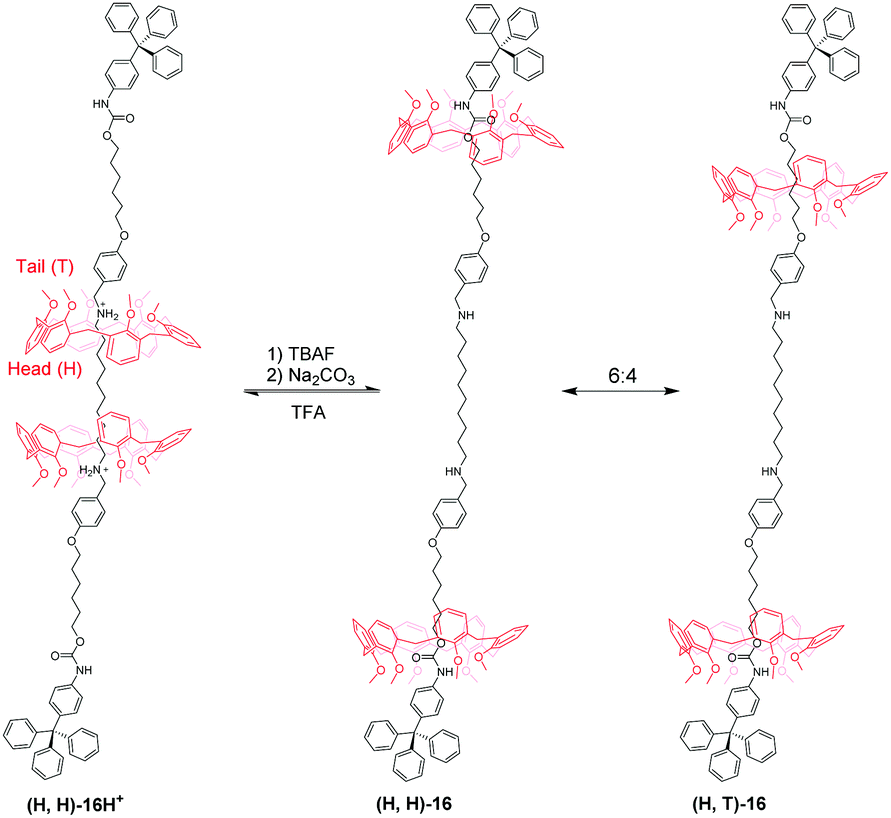
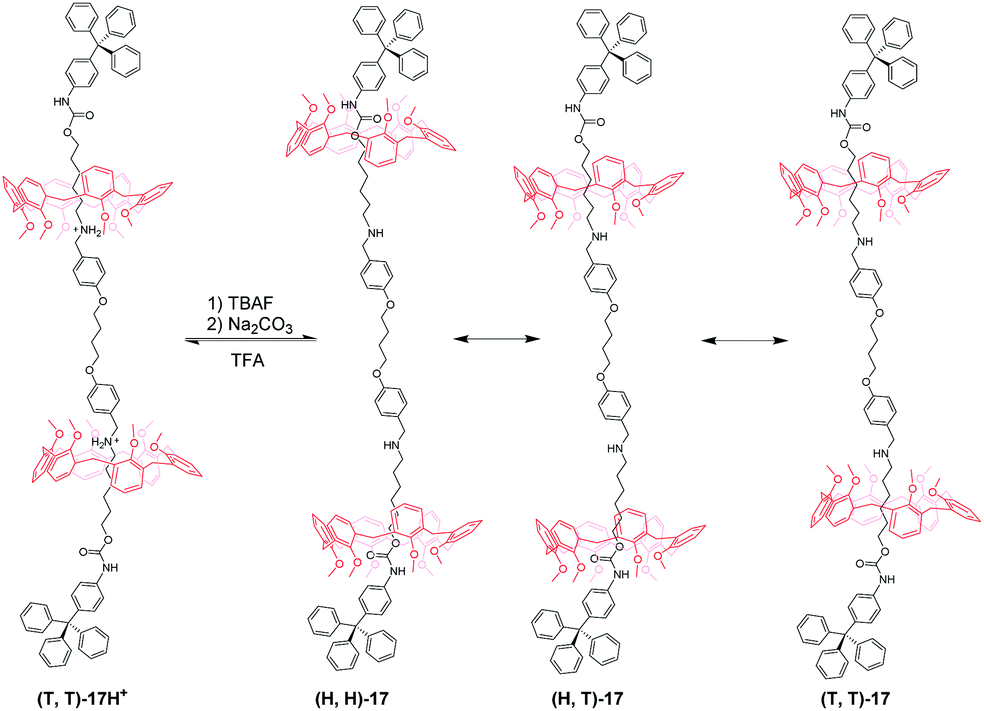
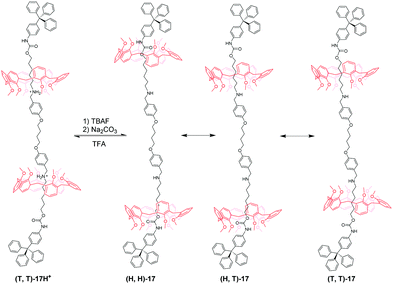
Based on an analogous motif, Neri's group51 also reported a pH-controllable [3]rotaxane to investigate their shuttling behaviour. They prepared methoxyl-containing [3]rotaxane (H,H)-16H+ (Fig. 16) with head-to-head stereosequence of the two calix[6]arenes and tail-to-tail [3]rotaxane (T,T)-17H+ (Fig. 17). When neutralizing the (H,H)-16H+ and (T,T)-17H+, the two macrocycles move from alkylbenzylammonium stations to new positions where aliphatic chains are inside the cavity of CA6. Simultaneously, the CA6 rings can experience cone-to-cone inversion to give three possible translational isomers, which feature as head-to-head (H,H), head-to-tail (H,T), and tail-to-tail (T,T). As a result, only two isomers were observed in 16, including (H,H)-16 and (H,T)-16 with a ratio of 6:4. But in 17, there exist (H,H)-17, (H,T)-17, and (T,T)-17 in a 55:25:20 ratio. Finally, the acidic treatment of neutral rotaxane fully restored the original state.
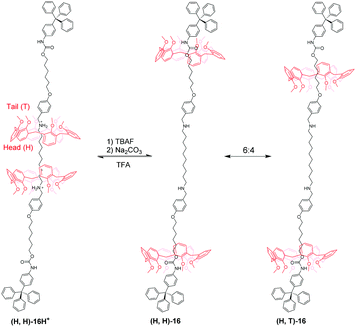 |
| | Fig. 16 pH-Controlled calix[6]arene-based [3]rotaxane 16. | |
 |
| | Fig. 17 pH-Controlled calix[6]arene-based [3]rotaxane 17. | |
2.6 Directional shuttling in naphthotube-based rotaxane
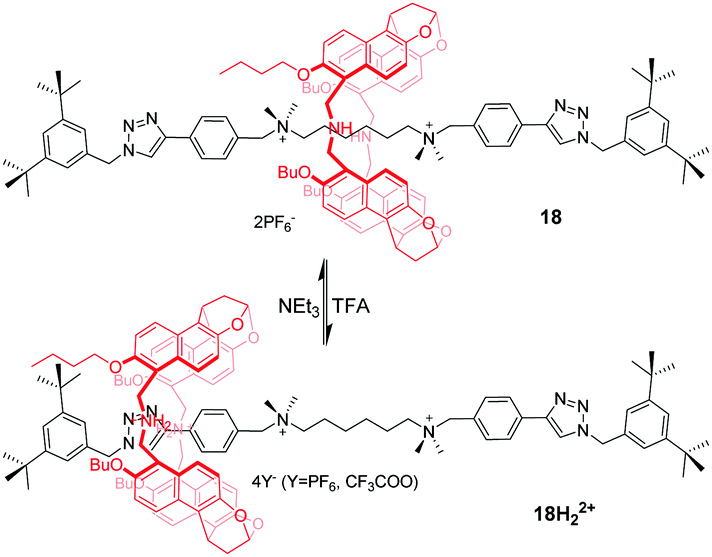
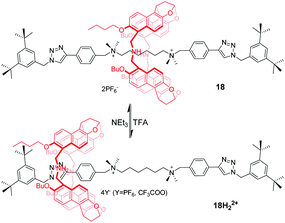
Jiang et al.52 reported naphthol-based macrocyclic receptors, namely naphthotubes, with two secondary amine groups in their backbones. Thus, two different binding properties of this macrocycle can be expected as its electron-rich cavity can be switched to positively charged by adding acid. In this way, they designed and synthesized a molecular shuttle 18 in which the macrocycle locates around the centre di(quaternary ammonium) station (Fig. 18). Once 18 is protonated, the macrocycle moves away from the centre station due to weakened binding and charge repulsion. Unexpectedly, the macrocycle only stays at one phenyl triazole station that is closer to the butyl groups of the macrocycle, generating a directional shuttling. This can be explained by multiple weak interactions in 18H22+ synergistically supporting this energy-minimized structure. The subsequent addition of NEt3 causes rotaxane 18H22+ to regain its original state.
 |
| | Fig. 18 Directional shuttling in naphthotube-based rotaxane. | |
3. Actuating circumrotation in catenanes
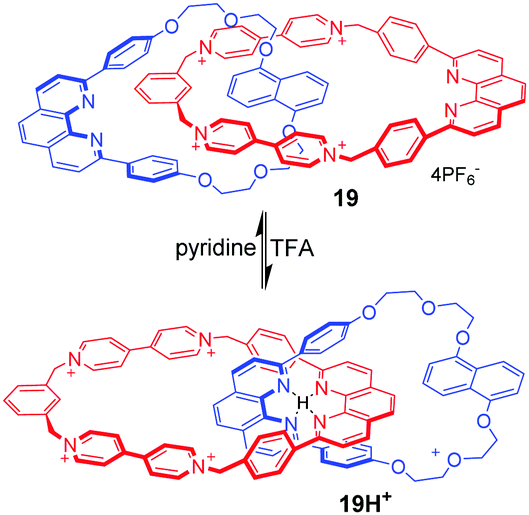
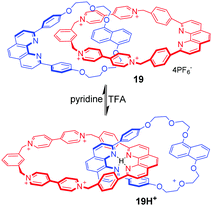
Circumrotation in catenanes is another typical mechanical motion in MIMs. In principle, all above-mentioned operating mechanisms in rotaxanes can be used in catenane architectures, giving rise to rotational motions. However, only a few examples have been successfully achieved for this goal, mainly due to the larger synthetic challenge of catenanes than rotaxanes. At the early stage, Stoddart and Sauvage53 constructed a hybrid [2]catenane that performed circumrotation motion of one ring within the other triggered by adding acid–base. As shown in Fig. 19, the [2]catenane 19 contains a core of two dpp-type ligands (dpp = 2,9-diphenyl-1,10-phenanthroline), π-electron rich aromatic rings (1,5-dioxynaphthalene) and a π-electron deficient component (tetracationic cyclophane). Under acidic conditions, the two dpp fragments in 19H+ are entwined around the proton. Under basic conditions, the 19 adopts another entwined topography for the formation of a π–π donor–acceptor complex between 1,5-dioxynaphthalene and bipyridinium.
 |
| | Fig. 19 pH-Responsive hybrid [2]catenane 19. | |
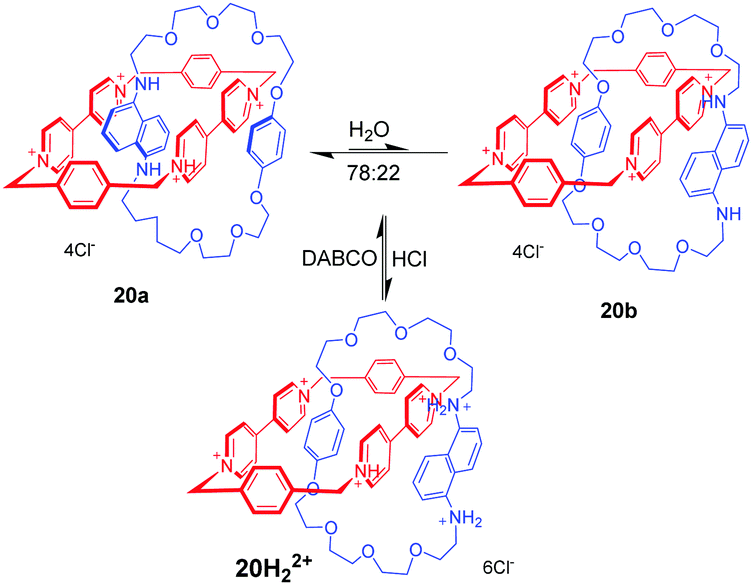
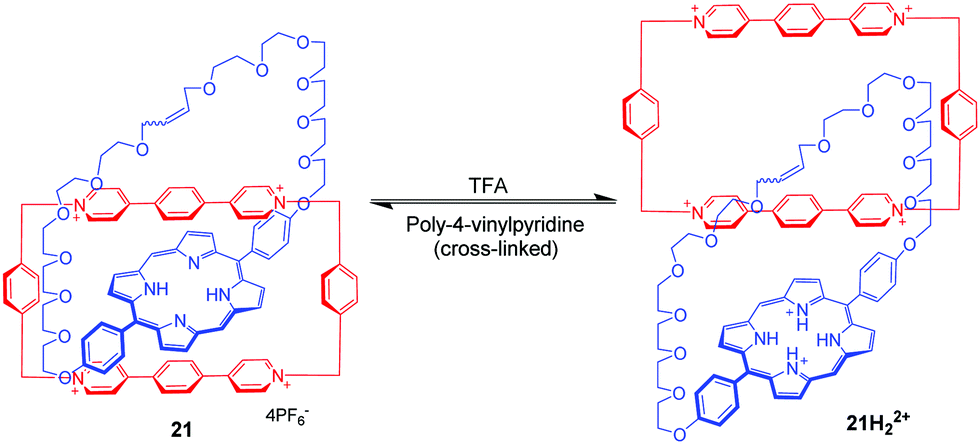
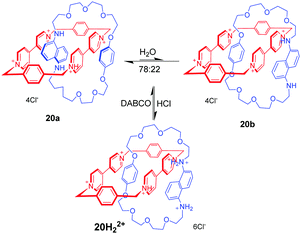
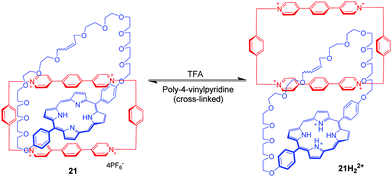
Donor–acceptor [2]catenanes could also perform as a pH-triggered molecular switch, which was demonstrated by Stoddart. For example, [2]catenane 20 consists of a tetracationic cyclophane (CBPQT4+) and a crown ether containing a hydroquinone(HQ) unit and a 1,5-diaminonaphthalene (DAN) unit (Fig. 20).54 And the DAN and HQ units compete for the cavity of the CBPQT4+ ring in a ratio of 78:22 in aqueous solution, giving two circumrotational isomers, 20a and 20b. After protonation of the DAN unit with HCl, the HQ units exclusively reside in the CBPQT4+ ring's cavity on account of coulombic repulsion. And this switching process can be reset by adding DABCO (1,4-diazabicyclo[2.2.2]octane). This switchable rotation can be repeated more than five times by alternately adding acid and base in water. Another pH-controllable donor–acceptor [2]catenane 21 involves an extended tetracationic cyclophane (ExBox4+) and porphyrin-containing polyether (Fig. 21).55 The acid–base actuation depends on the protonation and deprotonation of the nitrogen atoms of the porphyrin unit. The neutral porphyrin unit resides inside the cavity of the cyclophane on account of π-associated charge-transfer interactions. Addition of TFA protonates the porphyrin ring which drives the cyclophane away from the porphyrin unit to minimize the repulsive interactions. The switch can be reversibly restored to their original structure by the addition of base. Taking advantage of the pH-responsiveness of the porphyrin unit, Rowan and Nolte also reported a series of acid–base switchable MIMs from porphyrin–glycoluril cage compounds.56
 |
| | Fig. 20 A donor–acceptor [2]catenane 20 which is operated with acid–base in water. | |
 |
| | Fig. 21 pH-Controllable [2]catenane 21. | |
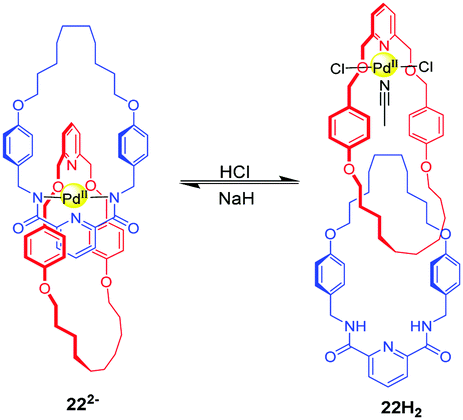
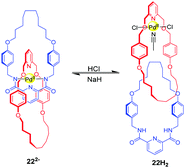
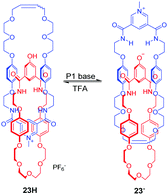
Other pH-induced circumrotational motion involves the benzylic amide macrocycle which was reported by several groups. For example, the [2]catenane 22H2 is composed of two pyridyl rings. The mechanism of rotational motion in this catenane relies on the manipulation of palladium (II) coordination modes via acid–base chemistry (Fig. 22).57 When the amide groups of the ‘blue’ macrocycle in 22H2 are deprotonated with a strong base (NaH), both rings participate in the square planar coordination in 222−. After neutralization with HCl, only a ‘red’ ring is coordinated to the palladium metal ion, regenerating 22H2. These different coordination modes cause a rotational motion in the catenane. Beer and co-workers also reported a pH-switchable [2]catenane 23H driven by anion recognition.58 This catenane comprises a pyridinium-based isophthalamide ring and a phenolic containing ring (Fig. 23). Initially, two rings intertwine in 23H with the pyridinium ring motif between the electron rich hydroquinone groups stabilized by aromatic donor–acceptor interactions and hydrogen bonds. Upon the addition of base, the phenol group is deprotonated to form a phenolate anion, and the rings undergo a circumrotation so that the phenolate anion can associate with pyridinium bis-amide via hydrogen bonding. The rotation process can be reversed with the addition of TFA. Lin and co-workers reported the pyridyl amide [2]catenane containing two pyridyl macrocycles and one of them was modified with tetraphenylethene (TPE).59 The acid–base controllable rotation endows it with enabled aggregation-induced emission.
 |
| | Fig. 22 Rotational motion in [2]catenane 22H2 relies on the manipulation of palladium(II) coordination modes by acid–base. | |
 |
| | Fig. 23 pH-Switchable [2]catenane 23H driven by anion recognition. | |
4. pH-Controlled motions beyond translocation and circumrotation
To date, most of the design motifs involved in synthesized molecular machines are based on relatively simple MIMs that execute a single movement of subcomponents. Actually, the artificial molecular machines are expected to perform more sophisticated tasks like natural machines. Although it is impossible to attain by the current generation of molecular machines, scientists have been trying to explore some of the advances by applying the basic principles in rotaxanes and catenanes.60 Several specific topological architectures constricted by mechanical bonds are adopted to carry out the sophisticated tasks inspired by nature. Representative examples include molecular elevators,61 molecular muscles,62 molecular ratchets63 and so on. pH-Stimulation is proved to be the most efficient means to fulfil this challenge. In this section, we will present how to operate several sophisticated motions in MIMs by acid–base chemistry.
4.1 Molecular elevators
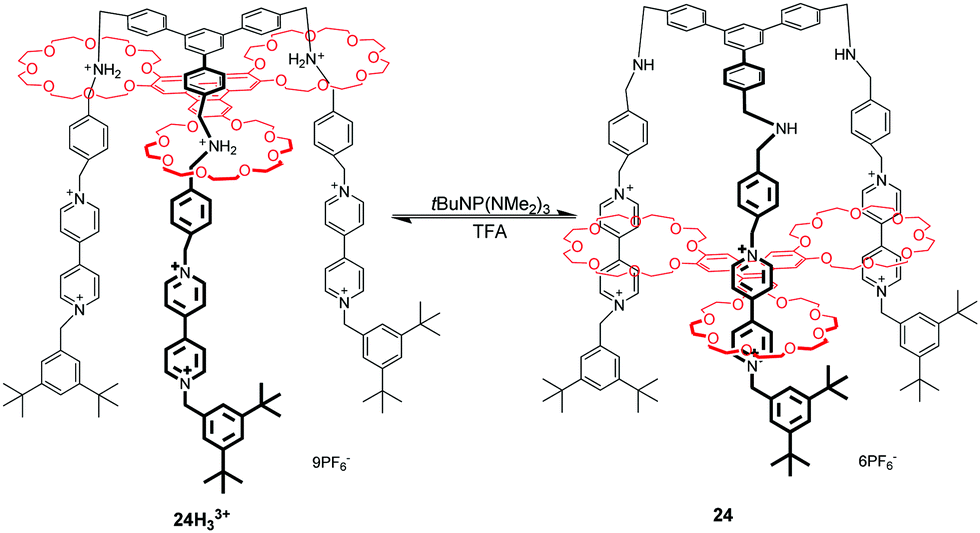
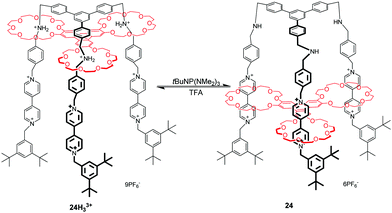
The advent of molecular elevators is a landmark of synthesized molecular machines, which was firstly reported by Stoddart's group in 2004.61 Molecular elevators are a class of rotaxanes which express the up-and-down motion of their platform component interlocked with a rig-like component. As shown in Fig. 24, the triply interlocked rotaxane 24H33+ is made up of a multivalent platform of rings containing three crown ether macrocycles and the trifurcated rig-like component possesses dibenzylammonium and BIPY2+ recognition sites. The movement of the elevator depends on the protonation and deprotonation of the dibenzylammonium ions with acid and base. It has been estimated from a thermodynamic analysis that the elevator movement from the upper to lower level could generate a force of up to 200 pN. When investigating how the rig and platform components move with respect to each other, they found that the translational motions of each ring operate in a stepwise manner, rather than by a concerted motion of the entire platform, owing to the conformational flexibility of the rig.64 This well-defined mechanical movement also reveal that the multivalent compounds can be harnessed to perform nontrivial mechanical movements.65
 |
| | Fig. 24 Stoddart's molecular elevator. | |
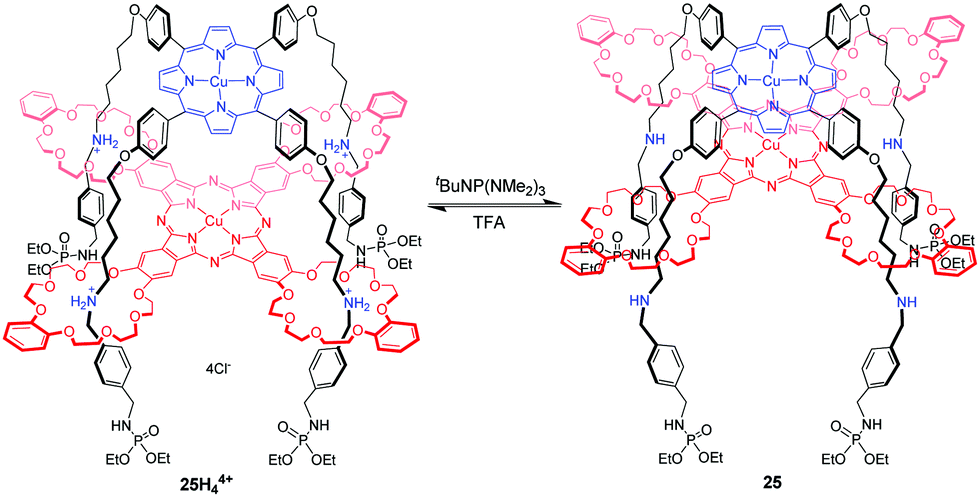
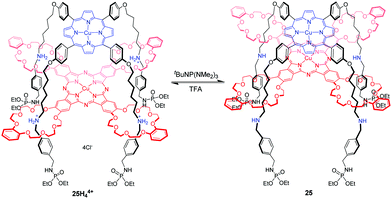
Another example of a molecular elevator was introduced by Tanaka66 in the form of four-fold rotaxane 25H44+, in which the Cu(II) porphyrin rig with four alkylammonium chains is mechanically linked to crown ethers of a Cu(II) phthalocyanine tetramacrocycle by a facial stacking (Fig. 25). The spin–spin interactions between the Cu2+ centers are sensitively influenced by their distance and relative spatial configuration. This expectation was confirmed by electron paramagnetic resonance (EPR) spectroscopy, which revealed that the spin-exchange interactions occur only in compressed 25 and not in 25H44+. By acid–base stimuli, the switchable spin–spin communication between mechanically interlocked metal complexes was well demonstrated. This concept would encourage us to prepare the supramolecular architectures with switchable functions related to nanomagnetism, conductivity, and photonic properties.
 |
| | Fig. 25 Tanaka's molecular elevator. | |
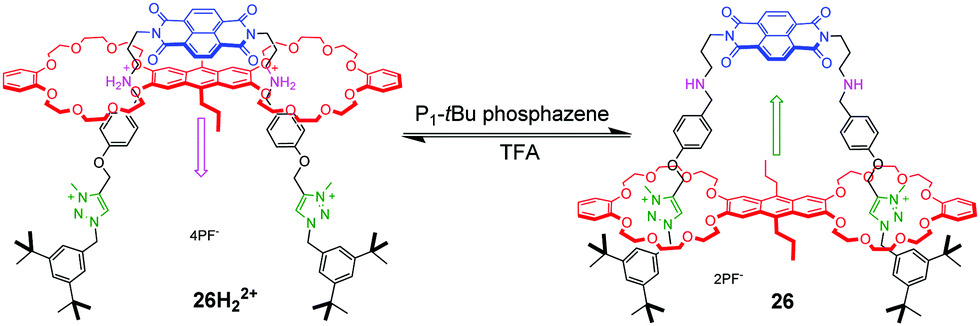
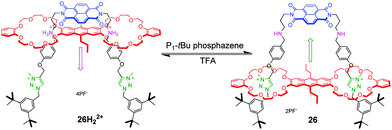
pH-Controlled palindromic motions also have been exploited to tune the interaction between naphthalene diimide (NDI) and anthracene chromophores in a double-leg donor–acceptor molecular elevator 26H22+.67 As shown in Fig. 26, a bis-24C8 macrocycle with an electron-rich anthracene moiety in the middle was chosen for the platform component, and the rig component contains a NDI unit with dialkylammonium and methyl triazolium stations on its legs. The distance between the two different platforms can be adjusted by acid and base. The similar operation of a bipodal molecular elevator with a ferrocene platform was also demonstrated by Qu.68
 |
| | Fig. 26 Double-leg donor–acceptor molecular elevator 26. | |
4.2 Contracting and stretching motion in [c2]daisy chains
Molecular muscles, which exhibit expansion and contraction on the nanoscale, are another typical mode of motion by mimicking biology. Rotaxane architectures, especially supramolecular daisy chains,69 have been a promising candidate for the design and synthesize of artificial molecular muscles.70 A daisy chain is a special case of MIMs created by interlocked monomers that consist of both a linear thread (guest) and a ring (host). In these systems, shuttling of the macrocycle along the thread results in a relative motion that either contracts or expands the entire molecule under the external stimuli.
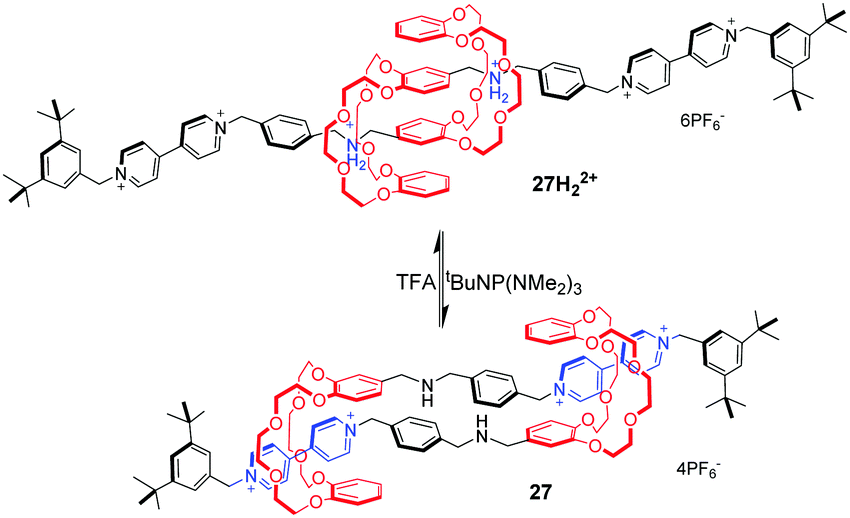
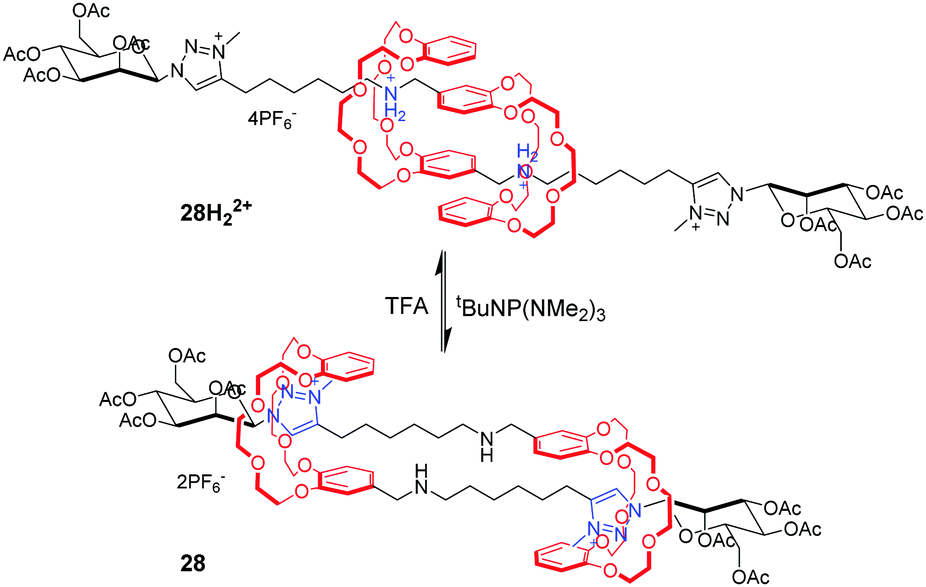
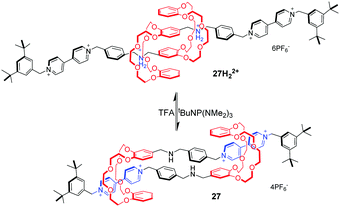
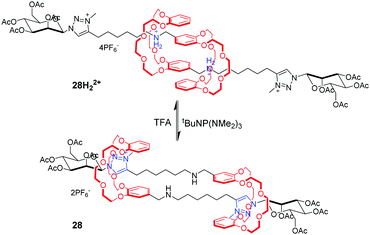
[c2]Daisy chains, with double-threaded architecture of the rotaxane dimer, are an ideal module for the construction of artificial molecular muscles by means of antiparallel sliding among its two interlocked rings. A large proportion of [c2]daisy chains are based on crown ethers and dialkylammonium ions, where contracting and stretching motions are readily actuated by acid/base. Stoddart and co-workers71 for the first time reported the pH-controllable [c2]daisy chain, in which the two DB24C8 rings switch between two different recognition sites, dibenzylammonium and bipyridinium. As a result, the 27H22+ adopted an extended state under acidic conditions, or a contracted state in 27 under basic conditions, in a similar manner to human muscles (Fig. 27). The simulation results show that the longitudinal molecular length of deprotonated 27 became approximately 29% (0.9 nm) shorter than the original extended length in 27H22+. Analogous to this operating mechanism, Coutrot and co-workers72 prepared the pH-switchable [c2]daisy chain 28H22+ by substitution of bipyridinium units with their triazolium units (Fig. 28).
 |
| | Fig. 27 pH-Actuated [c2]daisy chain 27. | |
 |
| | Fig. 28 Coutrot's type of [c2]daisy chain. | |
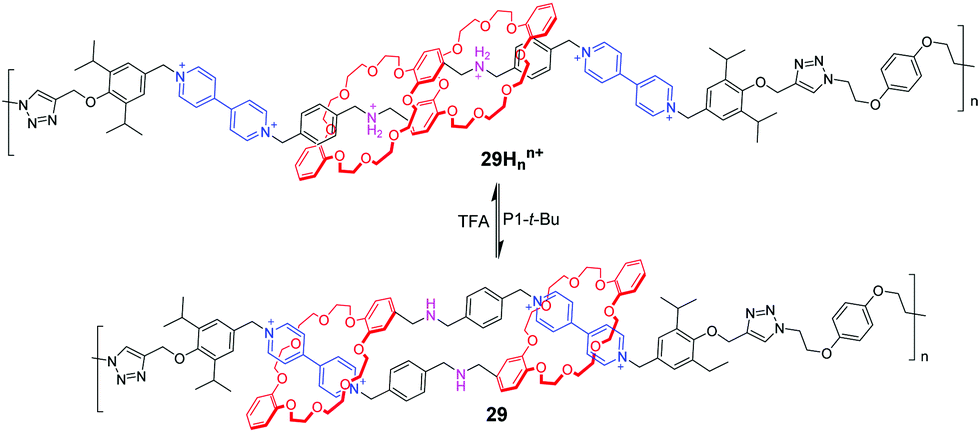
In order to amplify the molecular level movements into nanoscale or microscale motions, these [c2]daisy chain topologies have been incorporated into polymer scaffolds so as to generate mechanically interlocked macromolecules. Stoddart and co-workers73 synthesized a bistable poly[c2]daisy chain 29Hnn+ using click chemistry (Fig. 29). This mechanically interlocked polymer had 11 repeating [c2]daisy chain monomers on average. The experiments confirmed that a concerted contractile motion is indeed achievable along a polymer backbone by acid and base. They also found that the extension/contraction movements of the polymeric [c2]daisy chain are actually faster than those of the corresponding monomer. These observations demonstrate important information that the complexity of MIMs will promote their performance in materials.
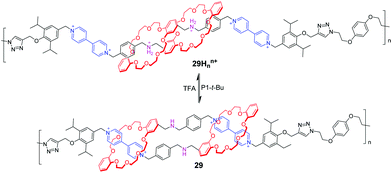 |
| | Fig. 29 Acid–base actuation of [c2]daisy chains 29. | |
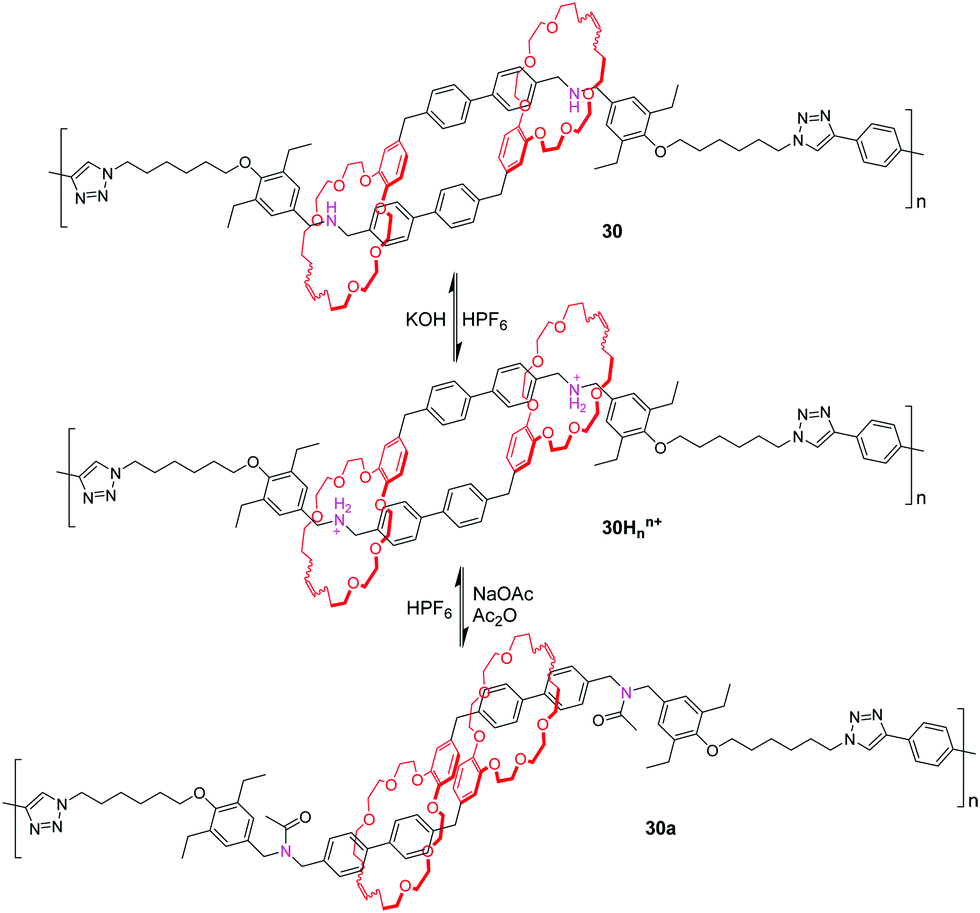
By utilizing olefin metathesis, Grubbs and co-workers74 synthesized a [c2]daisy chain dimer with the ammonium binding site. With addition of potassium hydroxide, the ammonium is deprotonated to give the unbound analogue due to the absence of a secondary binding site. Then, the system can be reset to the contracted, bound conformation by hexafluorophosphoric acid, enhancing its utility as a synthetic molecular actuator. Subsequently, the click polymerization of this [c2]daisy chain gives a linear polymer 30Hnn+, which possesses about 22 repeating units. Like the monomeric dimer, the polymer could also be readily deprotonated and reprotonated by alternately adding base and acid (Fig. 30). The multiangle laser light scattering (MALLS) detection analysis showed that the polymer had a radius of gyration (Rg) of 14.8 nm in the contracted conformation. Deprotonation of 30Hnn+ to generate 30 did not induce lengthening (Rg = 13.5 nm), likely due to poor solubility and π–π slipped-stacking interactions, but acylated polymer 30a showed a 48% increase in size (Rg = 21.4 nm).
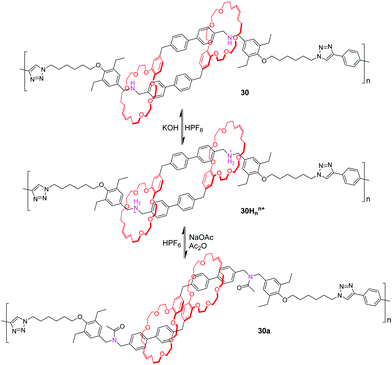 |
| | Fig. 30 pH-Controllable [c2]daisy chain dimer polymer. | |
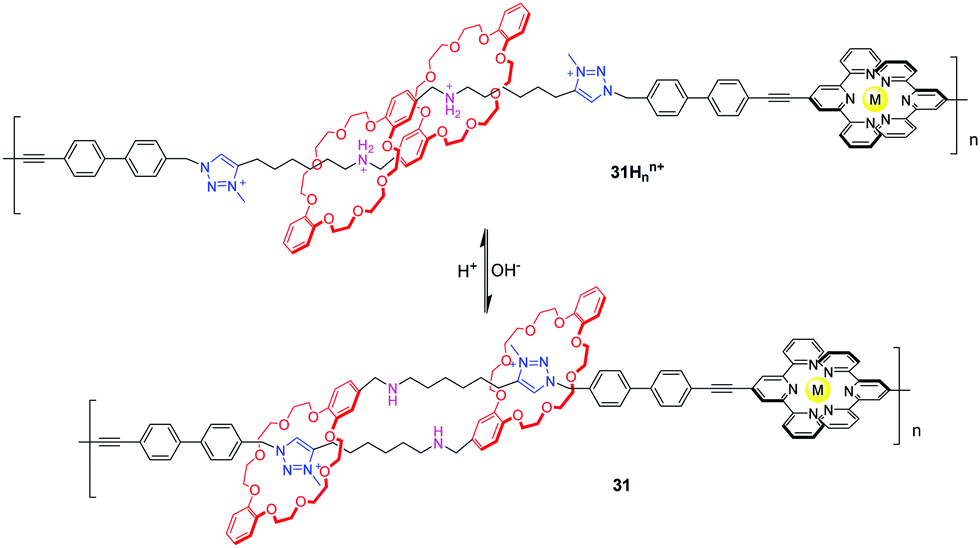
In the cases of Stoddart and Grubbs, the low molecular weight problem impeded the performance of the poly[c2]daisy chain in actuating contractile motions on mesoscopic scales. Giuseppone et al.75 solved this problem by taking metallosupramolecular polymerization of the [c2]daisy chain monomers. They synthesized a [c2]daisy chain analogous to Coutrot's 28, which featured terpyridines as stoppers. The addition of Zn(II) or Fe(II) triggered the supramolecular polymerizations of the [c2]daisy chain dimer for the formation of the bisterpyridine complexes. As a result, more than 3000 repeating units were embedded into a single-strand polymer chain. Thus, the poly[c2]daisy chains can amplify the tiny motions of rotaxane-based molecular muscles by four orders of magnitude. The X-ray scattering experiments revealed that the contour length of the protonated polymer 31Hnn+ (Fig. 31) is longer (15.9 μm) compared to the deprotonated polymer (9.4 μm). The same group also demonstrated that [c2]daisy chains of this type can be modified with alternative stoppering groups to induce the fabrication of other supramolecular polymers, and that the pH-actuated contractile motions influence the morphologies of the corresponding aggregates.76
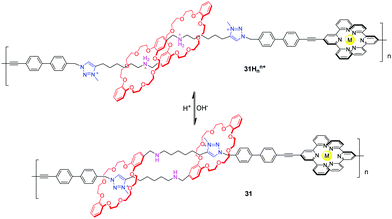 |
| | Fig. 31 pH-Controllable metallosupramolecular poly[c2]daisy chain. | |
4.3 Tightening and loosening in lasso rotaxanes
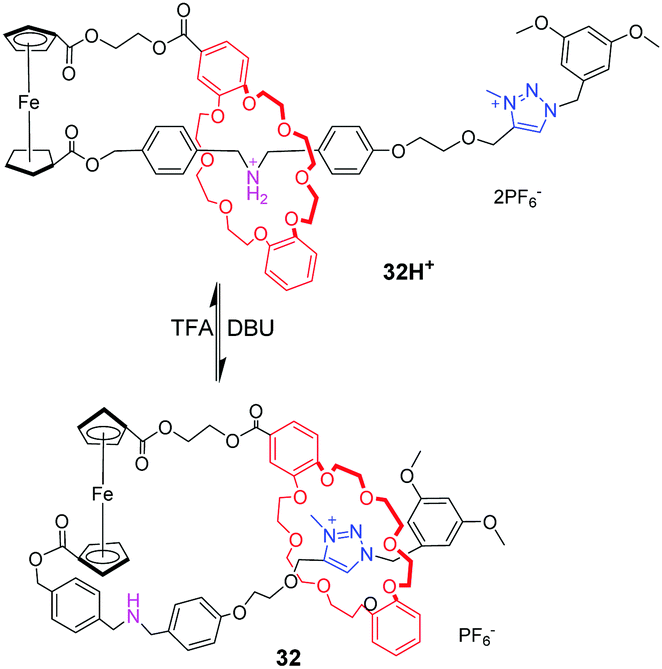
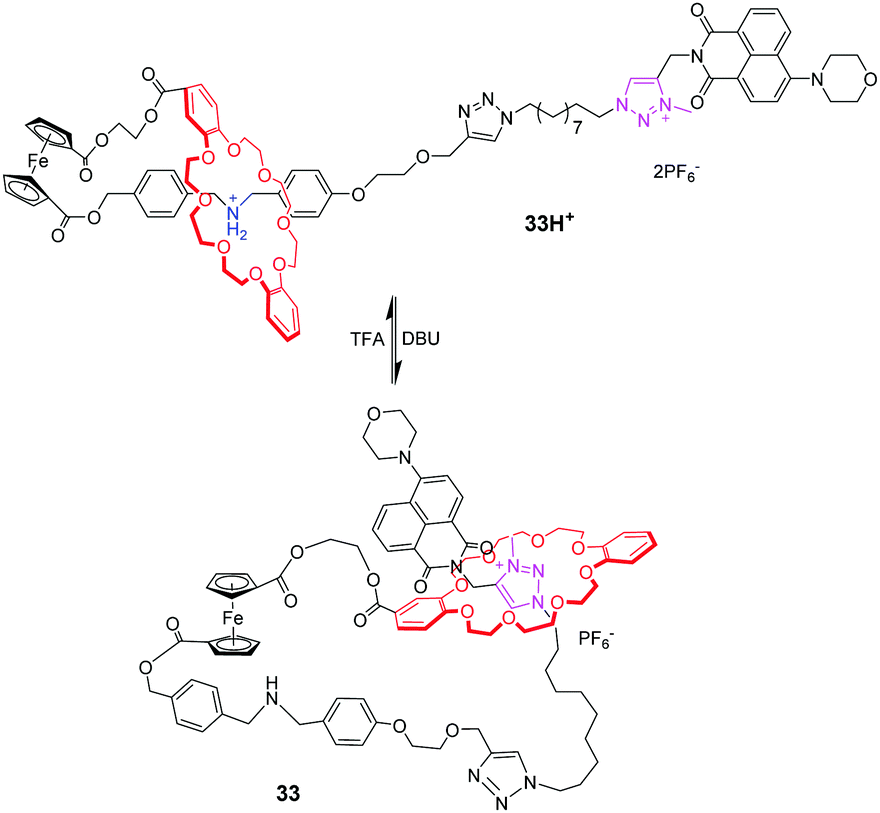
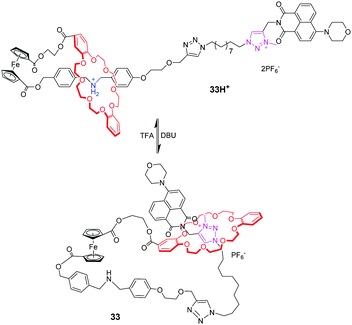
[c1]Daisy chains, also known as lasso, are the simplest rotaxanes in the family of daisy chains. Due to their unique self-entangled structure, the tightening and loosening motions can be achieved by gliding of the ring between different stations. Several examples of pH-responsive [c1] daisy chains are based on dialkylammonium and crown ether recognition motifs. An early example was reported by Qu and co-workers,77 in which a ferrocene unit as an electrochemically active centre is covalently linked to the DB24C8 macrocycle and a thread with a dibenzylammonium site (DBA) and a N-methyltriazolium (MTA) site (Fig. 32). In the protonated state, the DB24C8 encircles the DBA station in 32H+, which makes the ferrocene unit show a reversible oxidation–reduction property. Upon deprotonating the ammonium with base, the DB24C8 moves to the MTA site in 32, resulting in the electrochemically irreversible state. This switchable motion was also designed for application in fluorescence output.78 As shown in Fig. 33, the [1]rotaxane 33H+ comprises a ferrocenyl (Fc) unit covalently connected with a DB24C8 macrocycle and a thread with different stations. The 4-morpholinnaphthalimide (MA) was chosen as a terminated stopper, whose fluorescence could be adjusted on and off by a distance-dependent photoinduced electron transfer (PET) process between an electron-rich Fc unit and an electron-deficient MA fluorophore. Under acidic conditions, the DB24C8 resides in the ammonium site forcing the lasso to adopt a tightened co-conformation. At this point, the ferrocenyl unit remains too far away from the morpholin–naphthalimide to quench its fluorescence, the latter being monitored by the naked eye. Deprotonation of the ammonium results in the shuttling of the DB24C8 around the triazolium, hence loosening the lasso. In this specific co-conformation, the ferrocenyl unit is much closer to the tail, now authorizing a strong PET from the electron-rich ferrocene to the electron-deficient naphthalimide. Furthermore, the fluorescence output signal was also redox-dependent by oxidizing/reducing the ferrocene unit.
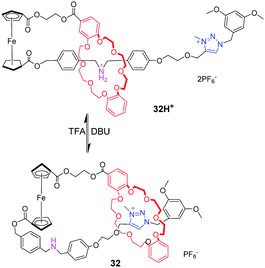 |
| | Fig. 32 pH-Switchable lasso rotaxane 32. | |
 |
| | Fig. 33 Lasso rotaxane 33 with switchable fluorescence output. | |
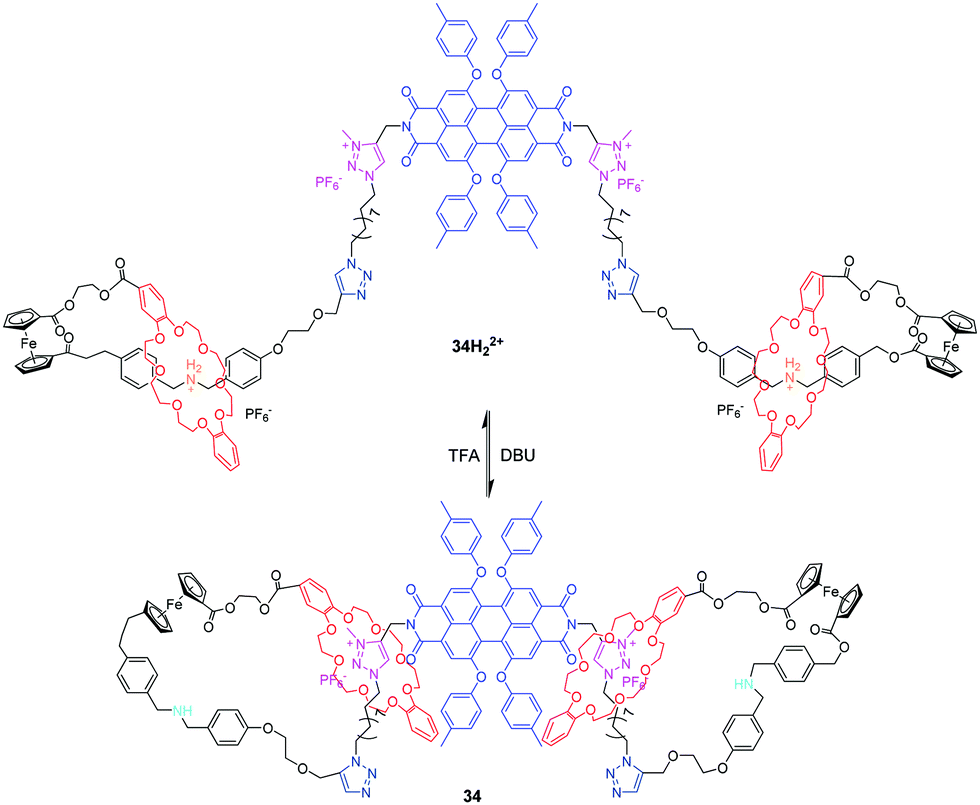
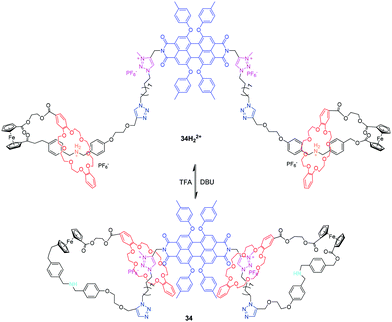
Qu et al. has also investigated the pH-controlled molecular motions of a multifunctional bis-branched [1]rotaxane with reversible fluorescence changes and tunable molecular aggregations.79 As shown in Fig. 34, bis[1]rotaxane 34H22+ comprises a perylenebisimide (PBI) core and two bistable [1]rotaxane arms containing two different stations and terminal ferrocene units. The translational movement of the DB24C8 between ammonium and triazolium units can be controlled by acid/base, giving rise to a change in molecular length of about 1.7 nm. Interestingly, the molecular dynamics simulations indicated that these extension and contraction movements were accompanied by a simultaneous rotational motion of the ferrocene units, thus generating a dual-mode movement type. More importantly, the movement of DB24C8 rings can adjust the system fluorescence through the PET process and nano-structural morphologies induced by PBI aggregations with Zn2+. The same group later adapted this ferrocene-linked [1]rotaxane into a more complex star-shaped [1](n)rotaxane, including tris[1]rotaxane and tetrakis[1]rotaxane, which expand and contract radially in response to pH changes.80
 |
| | Fig. 34 pH-Controllable bis[1]rotaxane 34. | |
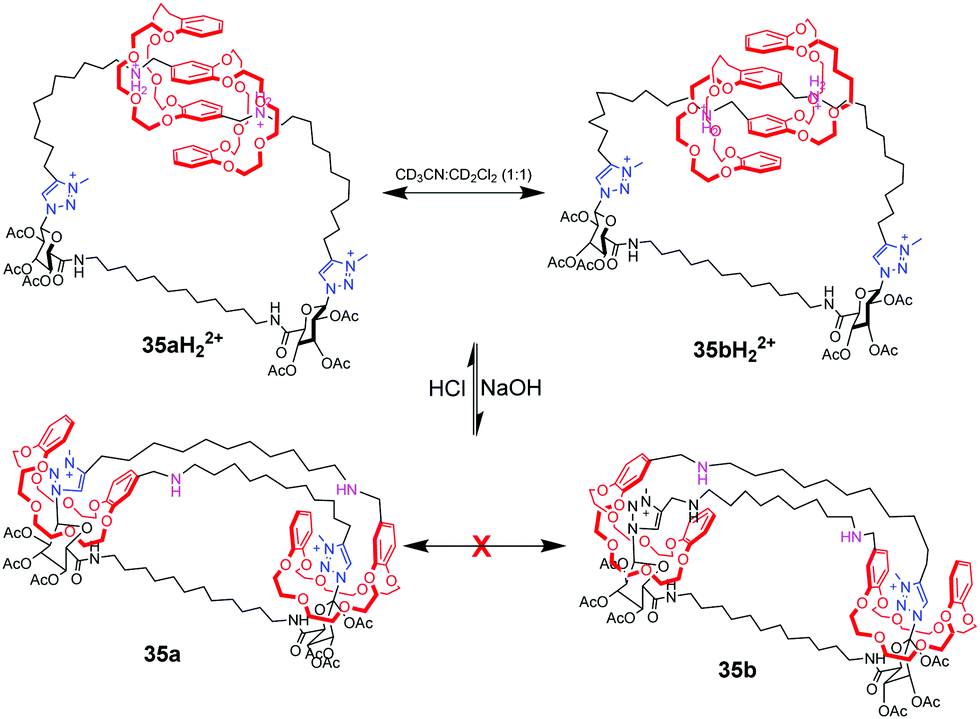
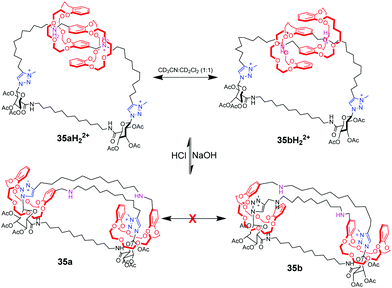
Coutrot et al. also have stimulated remarkable progress toward pH-controlled motions in lasso rotaxanes.81 They ingeniously described the constriction and dilation of a double-lasso architecture (Fig. 35), which derives from an end-activated [c2]daisy chain. The two DB24C8 rings can slide along the ultramacrocyclic loop upon variation of pH. At low pH, the double lasso adopts a relatively loose conformation, allowing fast exchange between isomers 35aH22+ and 35bH22+, thus behaving like a ‘‘jump rope’’. The addition of base triggered the shuttling motion of the DB24C8 around the triazolium stations, generating a tightened conformation. In this conformation, the jump rope movement was inhibited.
 |
| | Fig. 35 pH-Triggered motion in double-lasso architecture. | |
4.4 Wing-flapping motion in a [2](2)rotaxane
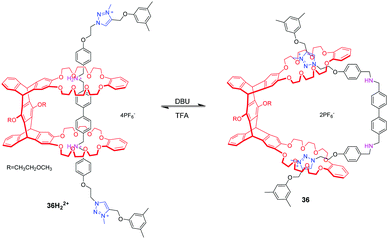
By imitating bionic machines, Chen and co-workers82 constructed a novel [2](2)rotaxane based on a pentiptycene-derived bis(crown ether) host. It was found that the shuttle process of two DB24C8 rings between the dibenzylammonium and N-methyltriazolium stations can be operated by acid/base stimulus. In the protonated state, two DB24C8 encircle the ammonium station without any torsion of the host. Deprotonation of the ammonium ion with DBU causes the movement of DB24C8 to triazolium stations, just like a butterfly spreading its wings (Fig. 36).
 |
| | Fig. 36 pH-Driven wing-flapping motion in a [2](2)rotaxane 36. | |
4.5 Molecular pulley
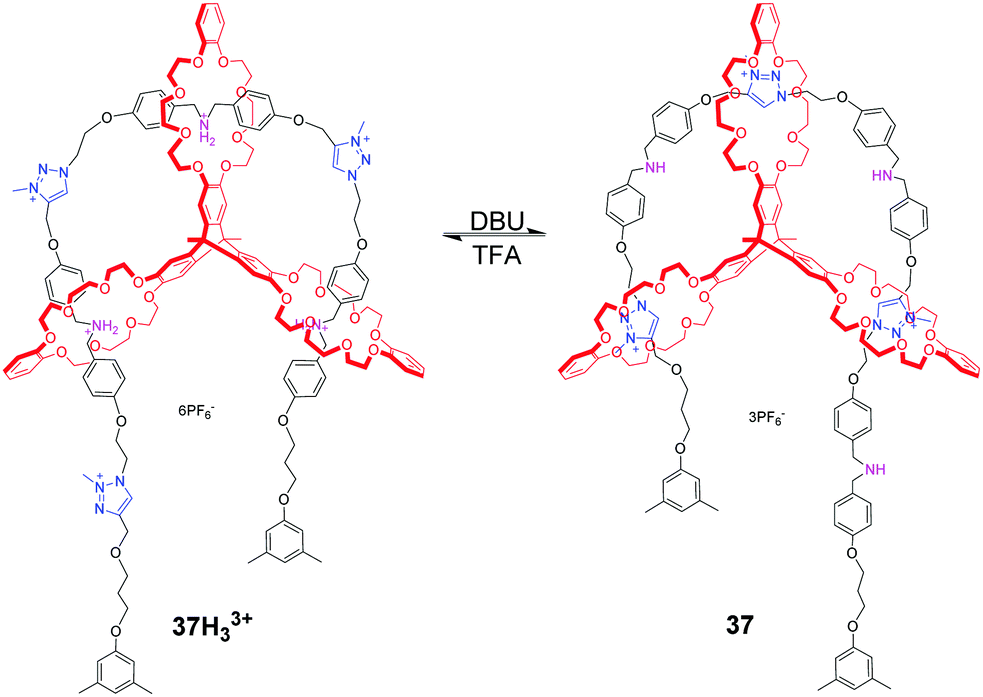
Chen et al. reported a pulley-like motion of the triply interlocked [2]rotaxane, which combines the features of plain rotary motion and linear translocation.83 In 37H33+ as shown in Fig. 37, the triptycene-derived tris(crown ether) host (TC), with three DB24C8 rings positioned in three orientations, threads onto a linear molecular axle which incorporates three dibenzylammonium and three N-methyltriazolium sites. And the three DB24C8 rings all encircle ammonium salt sites. Upon deprotonating these sites with DBU, each DB24C8 shuttles to the triazolium station, generating the direction-biased motion. The sliding back of the dibenzylammonium sites into the DB24C8 rings is possible through reprotonation of the amines using TFA. As a result, the back and forth motion in an arc-shaped trajectory was found in this [2]rotaxane, mimicking the function of the fundamental pulley device. This work presents a new mode of motion that combines translocation and circumrotation in a typical [2]rotaxane and [2]catenane.
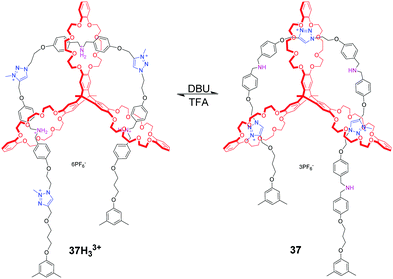 |
| | Fig. 37 pH-Controlled molecular pulley based on a triply interlocked [2]rotaxane 37. | |
4.6 Stepwise motion in a multivalent [2](3)catenane
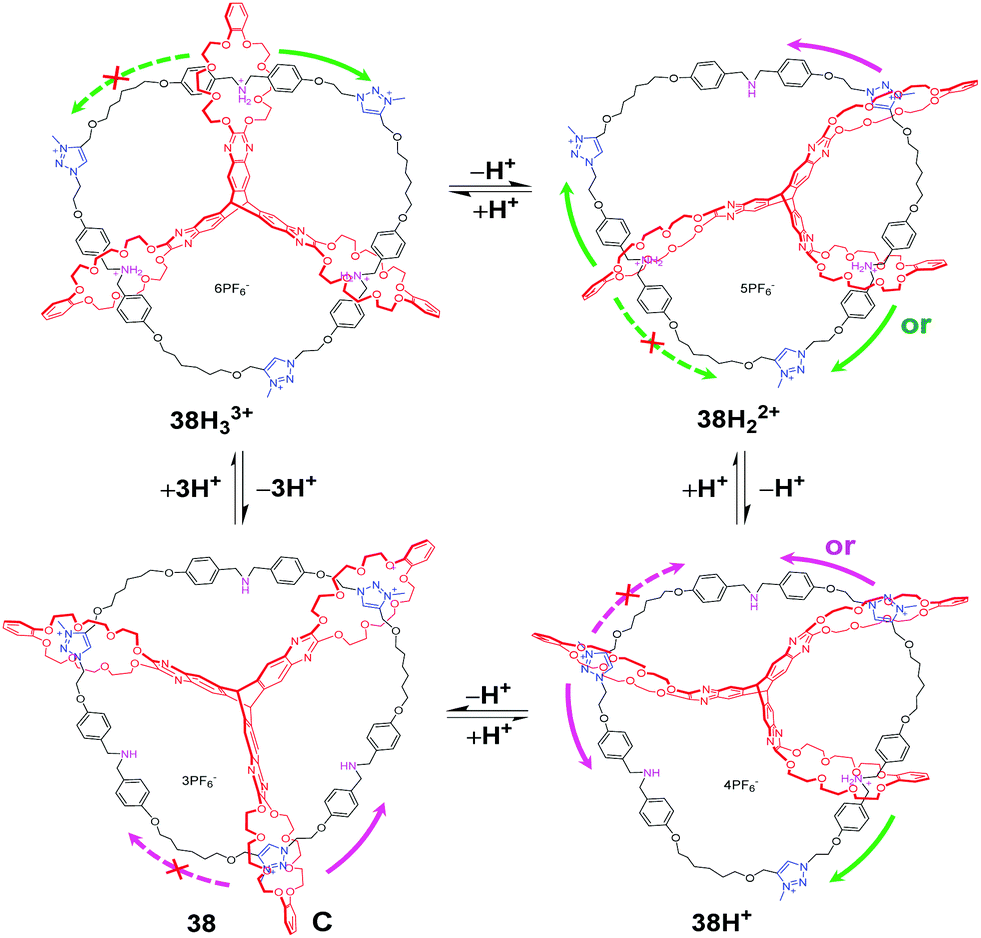
Based on a pyrazine-extended triptycene-derived tris-(crown ether) host (eTC), another unique topologically linked interlocked molecule, [2](3)catenane 38H33+ (Fig. 38), was also reported by Chen's group.84 The three crown ether of eTC successively threads onto a circular ditopic guest with three dibenzyl ammonium (DBA) sites and three N-methyltriazolium (MTA) sites. Taking advantage of the multivalent nature featuring multiple and sequential binding, the acid–base triggered motion can be actuated in a stepwise manner. As the DBU is added to deprotonate the DBA sites, one of three crown ether rings in 38H33+ begins to move to the MTA site, then two, and finally all three rings encircle the MTA sites. Thus, the four protonation states of the [2](3)catenane can be detected and identified by 1H NMR titration experiments, which confirms the multistep process. And the different thermodynamic data of dissociation equilibria, corresponding to pKa values was also determined by an indicator method. Moreover, an uncontemplated selectivity in the stepwise motion was observed where the B24C8 ring exclusively moved to nearer the MTA sites in 38H33+ owing to the special interlocked topology of the [2](3)catenane.
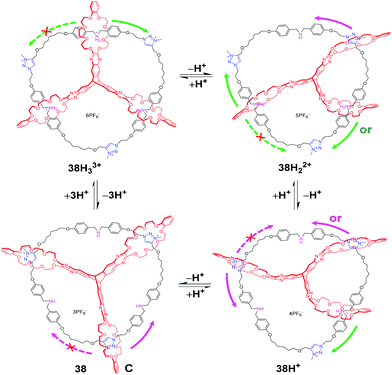 |
| | Fig. 38 Stepwise motion in [2](3)catenane 38 actuated by acid–base. | |
5. Controlling pH-responsive motions by alternative stimuli
Although acids and bases have been an efficient and commonly used stimuli for pH-responsive motions, the energy supply strategy is relatively simple. Besides, the waste products generated from acid–base neutralization reaction accumulate and will eventually affect the operation of the systems. Hence, the utilization of alternative stimuli instead of acids and bases will improve the performance of molecular machines. In this regard, a decarboxylation of strategy was used to power acid–base operated molecular machines. Photo-induced proton-transfer strategy was incorporated into pH-responsive molecular switches. Recently, a step-by-step reaction with pH oscillating properties has been found to power translational motion triggered by a chemical fuel pulse. In the following section, we will give a detailed introduction about this progress.
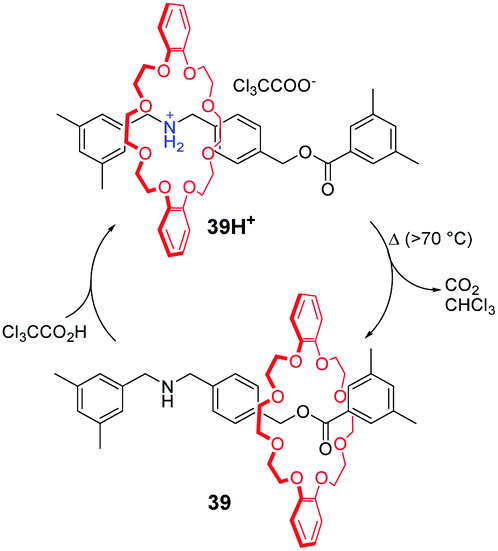
Taking advantage of pH-responsiveness of ammonium/crown ether-type MIMs, Takata and co-workers85 constructed a thermo-responsive rotaxane shuttling system. As shown in Fig. 39, the [2]rotaxane 39H+ is equipped with a trichloroacetate counterion (CCl3COO−), which is the key feature. Under the condition of heat, the CCl3COO− extracts a proton from the dibenzylammonium group and undergoes thermal decomposition to CO2 gas and volatile CHCl3. In the meantime, the macrocycle moves away from the nitrogen atom to generate the neutral rotaxane 39. The 39H+ is reproduced by the addition of trichloroacetic acid (CCl3COOH), providing a novel switching system. More importantly, this shuttling motion was well-repeated without accumulated residues of the salts compared to previous acid–base stimuli. Later, they embedded this thermoresponsive rotaxane in the side chains of poly[2]rotaxanes to control the helical pitch of a polyacetylene backbone.86
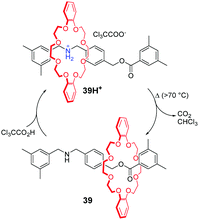 |
| | Fig. 39 Decarboxylation of trichloroacetate counteranions driving the shuttling motion. | |
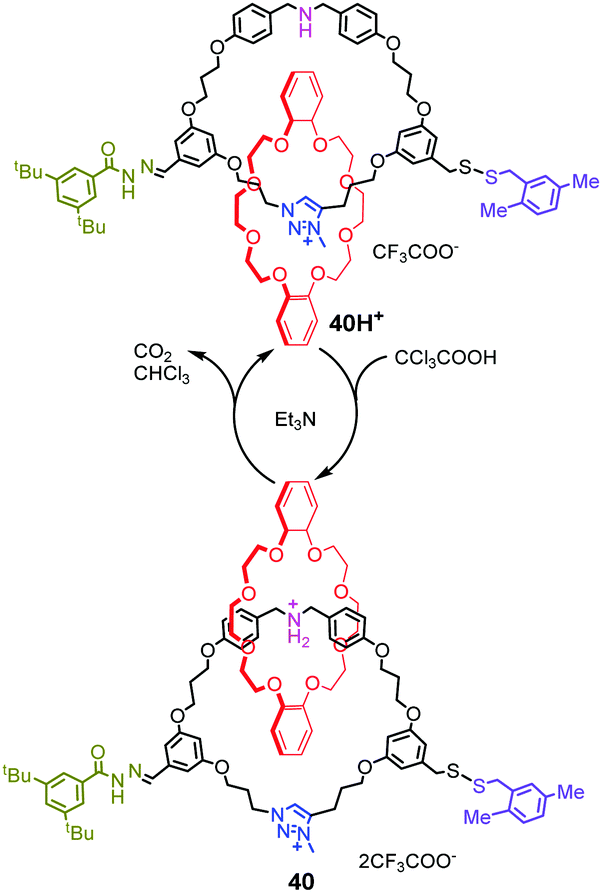
A similar decarboxylation strategy was also introduced into rotary and linear molecular motors by Leigh's group87 They designed an energy ratchet mechanism based on acid–base oscillations that power molecular motors. The acid–base oscillations are operated by base-promoted decarboxylation of CCl3COOH. As shown in Fig. 40, [2]catenane 40H+ comprises two interlocked rings: a crown ether ring and a larger ring with a dibenzylamine unit, triazolium site and two potential blocking groups. Upon addition of excess CCl3COOH, the dibenzylamine site is protonated and the hydrazone undergoes exchange via a transient aldehyde, leading to 180° directional rotation. In the meantime, triethylamine catalyzes decarboxylation of the trichloroacetic acid, generating CO2 and CHCl3. And the reaction solution becomes sufficiently basic to deprotonate the dibenzylammonium group and promote disulfide exchange, which resulted to another 180° directional rotation of the motor. As a whole, the crown ether completes one 360° directional rotation after one acid–base cycle. Based on this operating mechanism, [3]catenane rotary molecular motors and a linear molecular pump were also realized. Recently, they reported that a rotaxane catalyst can be switched using CCl3COOH as fuel pulses.88
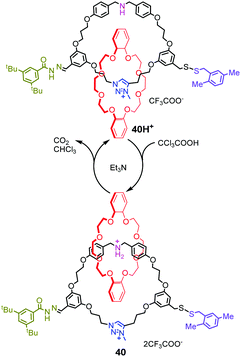 |
| | Fig. 40 Operation of a rotary motor using base-promoted decarboxylation. | |
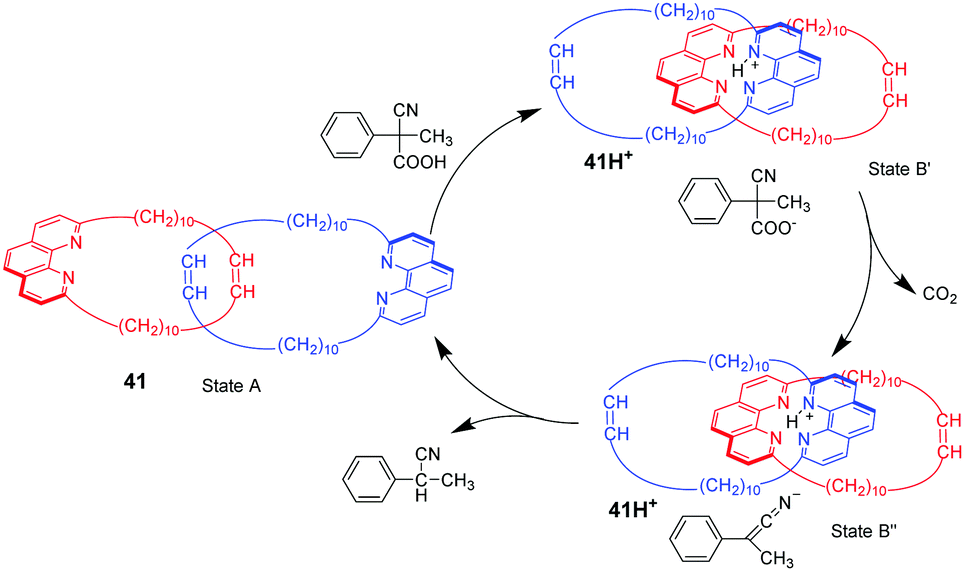
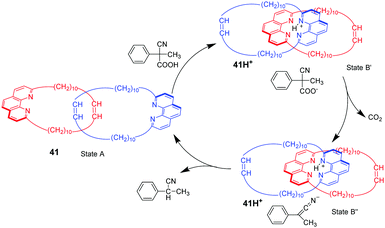
2-Cyano-2-phenylpropanoic acid is another choice of substrate for acid–base responsive molecular switching by the strategy of decarboxylation reaction, which has been reported by Stefano and co-workers.89 As shown in Fig. 41, [2]catenane 41 consists of two identical interlocked macrocycles featuring 1,10-phenanthroline units in their backbone without any precisely defined co-conformation (state A). The addition of 2-cyano-2-phenylpropanoic acid can provide protons to stabilize the ternary complex (state B′) by hydrogen bonding, producing the proton catenate 41H+. Then, 2-cyano-2-phenylpropanoic acid can undergo smooth decarboxylation in the presence of NEt3, leading to state B′′. Finally, rate-determining proton transfer from 41H+ to the anion, restores the catenane to its neutral form 41. This process demonstrated an autonomous back and forth motion by tactful coupling of the decarboxylation of 2-cyano-2-phenylpropanoic acid to an acid–base operated [2]catenane. Later, Schmittel and co-workers employed 2-cyano-2-phenylpropanoic acid as a chemical fuel to switch a [2]rotaxane shuttle with a feature of oscillating emission.90
 |
| | Fig. 41 Switching motions of catenane 41 fueled by 2-cyano-2-phenylpropanoic acid. | |
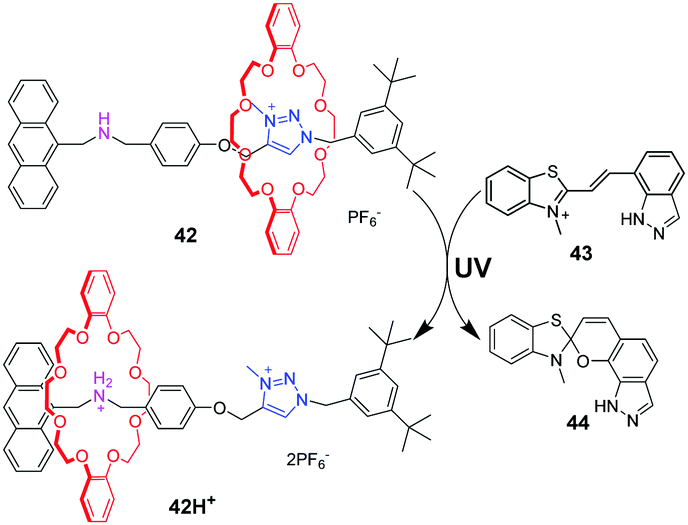
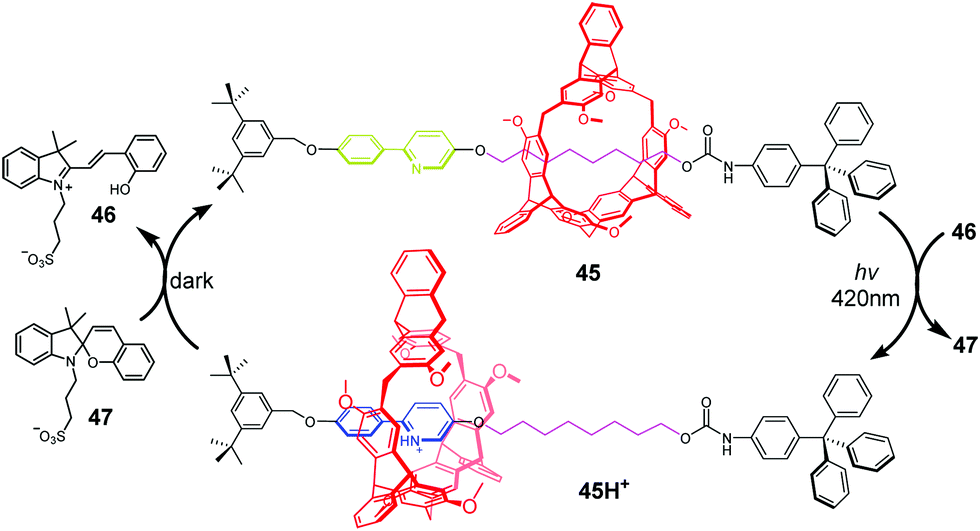
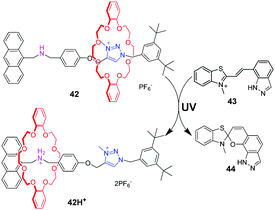
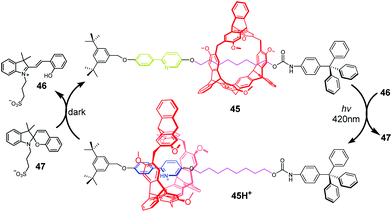
Photoacids or photobases, which exhibit an increase in their acidity or basicity upon photoexcitation, provide a new method to drive acid–base switchable molecular machines through a photo-induced proton-transfer (PIPT) strategy. Jiang and co-workers91 firstly reported a non-photoresponsive molecular shuttle coupled to a photoacid. In the [2]rotaxane 42 (Fig. 42), the DB24C8 resides at the triazolium station. The photoacid 43 can protonate the dibenzylamine after irradiation by a 365 nm ultraviolet lamp, generating 42H+. However, they failed to trigger the deprotonation step in the dark to reset the system to its original state. In order to find a more compatible system, Chen and coworkers developed an acid/base responsive complexation between helic[6]arene and pyridinium, which can be efficiently controlled by coupling to a photoacid.92 Subsequently, they designed and synthesized the [2]rotaxane 45 featuring the helic[6]arene/pyridinium recognition motif. It was found that photoacid 46 could protonate the pyridine part of 45 after 420 nm light or sunlight irradiation, which induces the macrocycle to move to the pyridium site from the alkyl group site. Moreover, the systems can be reversibly restored to their original structures under dark conditions. This acid/base-controlled palindromic motion could be switched repeatedly more than 50 times with excellent reproducibility (Fig. 43).
 |
| | Fig. 42 Light-controlled switching of the pH-responsive molecular shuttle. | |
 |
| | Fig. 43 Reversible shuttling motion controlled by photoacid. | |
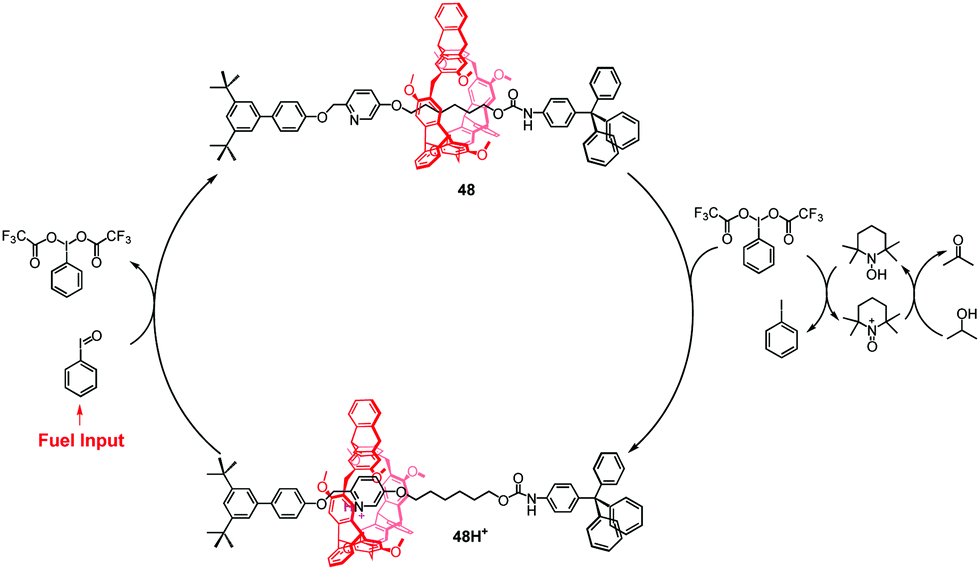
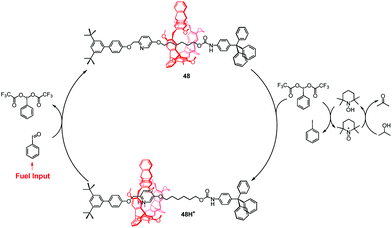
Recently, Chen et al. reported a new step-by-step reaction system with a pH oscillation feature that powers the acid–base controllable molecular shuttle.93 The rotaxane 48H+ (Fig. 44) is composed of a helic[6]arene interlocked onto a thread, which has a protonated pyridinium site and the alkyl chain site. The pH oscillation reaction-powered motion of the rotaxane was triggered by PhIO, which was considered as a fuel input. The PhIO can absorb the TFA from 48H+ to produce [bis(trifluoroacetoxy)iodo]benzene (BTAIB), leading to the movement of the macrocycle to the alkyl group in 48. Then, the newly produced BTAIB could be utilized to participate in the TEMPO-catalyzed oxidation of isopropanol to acetone. In the meantime, TFA could be gradually released from BTAIB, which resulted in the formation of 48H+ again. This work represents the new pH oscillating chemical fuel powered mechanical movement of a molecular shuttle.
 |
| | Fig. 44 Switchable motion of the [2]rotaxane powered by step-by-step reaction. | |
6. Summary and outlook
In this review, we summarized the pH-controlled motions that arise as a result of a mechanical bond. These motions involve non-covalent interactions, including hydrogen bonding, hydrophobic effects, electrostatic interactions and others, and are usually sensitive to pH variation, which makes them one of the most powerful and commonly used means of controlling molecular motions. By adopting different host–guest recognition motifs, two types of motions including translocations in rotaxanes and circumrotations in catenanes could be efficiently controlled by acid–base chemistry. By integrating these simple movements, more diverse and sophisticated motions at the molecular level have been realized as well. These highly complex molecular machines would stimulate remarkable improvement toward understanding physics and biology. Finally, we also highlighted the pH-dependent motions controlled by alternative stimuli instead of acids and bases, which could overcome the disadvantages of acid–base stimuli and generate novel modes of motion.
We believe that pH-stimulation will continue to play a key role in artificial molecular machines, particularly in multivalent and multicomponent systems. By integrating the operating mechanisms that appeared in this review, we might realize more complex molecular-level motion like biological molecular machines. Moreover, the introduction of various functional groups into MIMs is also helpful to construct pH-responsive functional materials.94 Last but not least, pH gradients are widespread in biological and physiological systems, which can provide the perfect environment for us to power molecular motions in living organisms. Taking advantage of pH gradients, we might operate molecular shuttles in lipid bilayers for transmembrane transport,95 and adopt [2]rotaxane as a nanovalve to selectively modulate drug release.96 We can foresee that this combination might promote the application of artificial molecular machines in biology and medicine.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (21772205, 21521002) for financial support.
Notes and references
-
(a) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802 RSC;
(b) L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard Iii and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17 RSC.
- S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Angew. Chem., Int. Ed., 2017, 56, 11166 CrossRef CAS PubMed.
-
C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, 2016 Search PubMed.
-
(a) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS;
(b) J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 11102 CrossRef CAS;
(c) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS.
-
(a) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094 CrossRef CAS;
(b) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080 CrossRef CAS;
(c) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060 CrossRef CAS.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398 CrossRef CAS.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110 CrossRef.
-
(a) C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186 CrossRef CAS;
(b) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592 RSC.
-
(a) A. Carlone, S. M. Goldup, N. Lebrasseur, D. A. Leigh and A. Wilson, J. Am. Chem. Soc., 2012, 134, 8321 CrossRef CAS;
(b) Z. Meng, J.-F. Xiang and C.-F. Chen, Chem. Sci., 2014, 5, 1520 RSC;
(c) M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235 CrossRef CAS.
-
(a) M. Baroncini, S. Silvi, M. Venturi and A. Credi, Angew. Chem., Int. Ed., 2012, 51, 4223 CrossRef CAS;
(b) G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70 CrossRef CAS;
(c) M. Baroncini, M. Canton, L. Casimiro, S. Corra, J. Groppi, M. La Rosa, S. Silvi and A. Credi, Eur. J. Inorg. Chem., 2018, 4589 CrossRef CAS PubMed;
(d) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543 CrossRef CAS.
-
(a) C. Pezzato, M. T. Nguyen, D. J. Kim, O. Anamimoghadam, L. Mosca and J. F. Stoddart, Angew. Chem., Int. Ed., 2018, 57, 9325 CrossRef CAS;
(b) Y. Wang, T. Cheng, J. Sun, Z. Liu, M. Frasconi, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13827 CrossRef CAS;
(c) H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 3496 CrossRef.
-
(a) K. C.-F. Leung, C.-P. Chak, C.-M. Lo, W.-Y. Wong, S. Xuan and C. H. K. Cheng, Chem. – Asian J., 2009, 4, 364 CrossRef CAS;
(b) G.-H. Weng, B. Zhu, Y. Ye and S. Li, Chin. J. Org. Chem., 2015, 35, 309 CrossRef CAS.
-
(a) J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS;
(b) L. Isaacs, Acc. Chem. Res., 2014, 47, 2052 CrossRef CAS.
- W. L. Mock and J. Pierpont, J. Chem. Soc., Chem. Commun., 1990, 1509 RSC.
- S. Im Jun, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Tetrahedron Lett., 2000, 41, 471 CrossRef.
- J. W. Lee, K. Kim and K. Kim, Chem. Commun., 2001, 1042 Search PubMed.
-
(a) D. Tuncel, Ö. Özsar, H. B. Tiftik and B. Salih, Chem. Commun., 2007, 1369 RSC;
(b) D. Tuncel and M. Katterle, Chem. – Eur. J., 2008, 14, 4110 CrossRef CAS.
- D. Tuncel, N. Cindir and Ü. Koldemir, J. Incl. Phenom. Macrocycl. Chem., 2006, 55, 373 CrossRef CAS.
-
(a) V. Sindelar, S. Silvi and A. E. Kaifer, Chem. Commun., 2006, 2185 RSC;
(b) W. Wu, S. Song, X. Cui, T. Sun, J.-X. Zhang and X.-L. Ni, Chin. Chem. Lett., 2018, 29, 95 CrossRef CAS.
- E. J. Dale, N. A. Vermeulen, M. Juríček, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Acc. Chem. Res., 2016, 49, 262 CrossRef CAS.
- B. Odell, M. V. Reddington, A. M. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547–1550 CrossRef.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133 CrossRef CAS.
- M. Martínez-Díaz, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1997, 36, 1904 CrossRef.
- P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. Fyfe, M. T. Gandolfi, M. Gómez-López, M. Martínez-Díaz and A. Piersanti, J. Am. Chem. Soc., 1998, 120, 11932 CrossRef CAS.
- S. Garaudée, S. Silvi, M. Venturi, A. Credi, A. H. Flood and J. F. Stoddart, ChemPhysChem, 2005, 6, 2145 CrossRef.
- X. Ma, J. Zhang, J. Cao, X. Yao, T. Cao, Y. Gong, C. Zhao and H. Tian, Chem. Sci., 2016, 7, 4582 RSC.
- F. Coutrot and E. Busseron, Chem. – Eur. J., 2008, 14, 4784 CrossRef CAS.
-
(a) F. Coutrot, ChemistryOpen, 2015, 4, 556 CrossRef CAS;
(b) P. Waelès, K. Fournel-Marotte and F. Coutrot, Chem. – Eur. J., 2017, 23, 11529 CrossRef;
(c) S. Chao, C. Romuald, K. Fournel-Marotte, C. Clavel and F. Coutrot, Angew. Chem., Int. Ed., 2014, 53, 6914 CrossRef CAS;
(d) M. Berg, S. Nozinovic, M. Engeser and A. Lützen, Eur. J. Org. Chem., 2015, 5966 CrossRef CAS;
(e) G. Ragazzon, A. Credi and B. Colasson, Chem. – Eur. J., 2017, 23, 2149 CrossRef CAS;
(f) Z. Meng, J.-F. Xiang and C.-F. Chen, J. Am. Chem. Soc., 2016, 138, 5652 CrossRef CAS.
-
(a) R. Arumugaperumal, V. Srinivasadesikan, M. V. Ramakrishnam Raju, M.-C. Lin, T. Shukla, R. Singh and H.-C. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 26491 CrossRef CAS PubMed;
(b) W. Zhou, H. Zhang, H. Li, Y. Zhang, Q.-C. Wang and D.-H. Qu, Tetrahedron, 2013, 69, 5319 CrossRef CAS;
(c) W. Yang, Y. Li, J. Zhang, Y. Yu, T. Liu, H. Liu and Y. Li, Org. Biomol. Chem., 2011, 9, 6022 RSC.
- Q. Jiang, H.-Y. Zhang, M. Han, Z.-J. Ding and Y. Liu, Org. Lett., 2010, 12, 1728 CrossRef CAS.
- W. Yang, Y. Li, J. Zhang, N. Chen, S. Chen, H. Liu and Y. Li, Small, 2012, 8, 2602 CrossRef CAS.
-
(a) Z.-Q. Cao, H. Li, J. Yao, L. Zou, D.-H. Qu and H. Tian, Asian J. Org. Chem., 2015, 4, 212 CrossRef CAS;
(b) J.-N. Zhang, H. Li, W. Zhou, S.-L. Yu, D.-H. Qu and H. Tian, Chem. – Eur. J., 2013, 19, 17192 CrossRef CAS.
- H. Zhang, Q. Liu, J. Li and D.-H. Qu, Org. Lett., 2013, 15, 338 CrossRef CAS.
- C. M. Álvarez, H. Barbero and D. Miguel, Eur. J. Org. Chem., 2015, 6631 CrossRef.
-
(a) V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, J. Am. Chem. Soc., 2014, 136, 4905 CrossRef CAS;
(b) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775 CrossRef CAS;
(c) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC;
(d) K. Eichstaedt, J. Jaramillo-Garcia, D. A. Leigh, V. Marcos, S. Pisano and T. A. Singleton, J. Am. Chem. Soc., 2017, 139, 9376 CrossRef CAS.
- S. J. Loeb and J. A. Wisner, Angew. Chem., Int. Ed., 1998, 37, 2838 CrossRef CAS.
-
(a) A. M. Elizarov, S.-H. Chiu and J. F. Stoddart, J. Org. Chem., 2002, 67, 9175 CrossRef CAS;
(b) S. J. Vella, J. Tiburcio and S. J. Loeb, Chem. Commun., 2007, 4752 RSC.
- N. Noujeim, K. Zhu, V. N. Vukotic and S. J. Loeb, Org. Lett., 2012, 14, 2484 CrossRef CAS.
- K. Zhu, V. N. Vukotic and S. J. Loeb, Angew. Chem., Int. Ed., 2012, 51, 2168 CrossRef CAS.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514 CrossRef CAS.
- K. Zhu, V. N. Vukotic and S. J. Loeb, Chem. – Asian J., 2016, 11, 3258 CrossRef CAS.
- C. M. Keaveney and D. A. Leigh, Angew. Chem., Int. Ed., 2004, 43, 1222 CrossRef CAS.
- J. D. Crowley, D. A. Leigh, P. J. Lusby, R. T. McBurney, L.-E. Perret-Aebi, C. Petzold, A. M. Z. Slawin and M. D. Symes, J. Am. Chem. Soc., 2007, 129, 15085 CrossRef CAS PubMed.
- D. A. Leigh, P. J. Lusby, R. T. McBurney and M. D. Symes, Chem. Commun., 2010, 46, 2382 RSC.
- D. A. Leigh and A. R. Thomson, Org. Lett., 2006, 8, 5377 CrossRef CAS.
-
(a) H. Zheng, W. Zhou, J. Lv, X. Yin, Y. Li, H. Liu and Y. Li, Chem. – Eur. J., 2009, 15, 13253 CrossRef CAS;
(b) Y. Zhao, Y. Li, S.-W. Lai, J. Yang, C. Liu, H. Liu, C.-M. Che and Y. Li, Org. Biomol. Chem., 2011, 9, 7500 RSC.
- S.-Y. Hsueh, C.-T. Kuo, T.-W. Lu, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-h. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9170 CrossRef CAS.
- L. Liu, Y. Liu, P. Liu, J. Wu, Y. Guan, X. Hu, C. Lin, Y. Yang, X. Sun, J. Ma and L. Wang, Chem. Sci., 2013, 4, 1701 RSC.
- B. Hesseler, M. Zindler, R. Herges and U. Lüning, Eur. J. Org. Chem., 2014, 3885 CrossRef CAS.
- T. Pierro, C. Gaeta, C. Talotta, A. Casapullo and P. Neri, Org. Lett., 2011, 13, 2650 CrossRef CAS.
- C. Talotta, C. Gaeta and P. Neri, Org. Lett., 2012, 14, 3104 CrossRef CAS.
- J.-S. Cui, Q.-K. Ba, H. Ke, A. Valkonen, K. Rissanen and W. Jiang, Angew. Chem., Int. Ed., 2018, 57, 7809 CrossRef CAS.
- D. B. Amabilino, C. O. Dietrich-Buchecker, A. Livoreil, L. Pérez-García, J.-P. Sauvage and J. F. Stoddart, J. Am. Chem. Soc., 1996, 118, 3905 CrossRef CAS.
- S. Grunder, P. L. McGrier, A. C. Whalley, M. M. Boyle, C. Stern and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 17691 CrossRef CAS PubMed.
- M. Juríček, J. C. Barnes, N. L. Strutt, N. A. Vermeulen, K. C. Ghooray, E. J. Dale, P. R. McGonigal, A. K. Blackburn, A.-J. Avestro and J. F. Stoddart, Chem. Sci., 2014, 5, 2724 RSC.
- R. G. Coumans, J. A. Elemans, A. E. Rowan and R. J. Nolte, Chem. – Eur. J., 2013, 19, 7758 CrossRef CAS.
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Chem. Commun., 2005, 4919 RSC.
- K.-Y. Ng, V. Felix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281 RSC.
- M. V. Ramakrishnam Raju and H.-C. Lin, Org. Lett., 2014, 16, 5564 CrossRef CAS.
- L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9397 CrossRef CAS.
- J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef.
- M. C. Jiménez, C. D. Buchecker and J. P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284 CrossRef.
-
(a) M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058 CrossRef CAS;
(b) V. Serreli, C. F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523 CrossRef CAS.
- J. D. Badjic, C. M. Ronconi, J. F. Stoddart, V. Balzani, S. Silvi and A. Credi, J. Am. Chem. Soc., 2006, 128, 1489 CrossRef CAS.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472 CrossRef CAS.
- Y. Yamada, M. Okamoto, K. Furukawa, T. Kato and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 709 CrossRef CAS.
- Z.-J. Zhang, M. Han, H.-Y. Zhang and Y. Liu, Org. Lett., 2013, 15, 1698 CrossRef CAS.
- H. Li, X. Li, Y. Wu, H. Ågren and D.-H. Qu, J. Org. Chem., 2014, 79, 6996 CrossRef CAS.
-
(a) P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. Fyfe, P. T. Glink, J. F. Stoddart, A. J. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294 CrossRef CAS;
(b) M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284 CrossRef;
(c) J. Rotzler and M. Mayor, Chem. Soc. Rev., 2013, 42, 44 RSC.
-
(a) A. Goujon, E. Moulin, G. Fuks and N. Giuseppone, CCS Chem., 2019, 1, 83 CAS;
(b) R. Tao, Q. Zhang, S. Rao, X. Zheng, M. Li and D. Qu, Sci. China: Chem., 2019, 62, 245 CrossRef CAS.
- J. Wu, K. C.-F. Leung, D. Benítez, J.-Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470 CrossRef CAS.
-
(a) F. Coutrot, C. Romuald and E. Busseron, Org. Lett., 2008, 10, 3741 CrossRef CAS;
(b) C. Romuald, E. Busseron and F. Coutrot, J. Org. Chem., 2010, 75, 6516 CrossRef CAS PubMed;
(c) P. Waelès, B. Riss-Yaw and F. Coutrot, Chem. – Eur. J., 2016, 22, 6837 CrossRef PubMed.
-
(a) L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y.-W. Yang, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126 CrossRef CAS;
(b) M. Hmadeh, L. Fang, A. Trabolsi, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Mater. Chem., 2010, 20, 3422 RSC.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631 CrossRef CAS.
- G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504 CrossRef CAS.
-
(a) A. Wolf, E. Moulin, J.-J. Cid, A. Goujon, G. Du, E. Busseron, G. Fuks and N. Giuseppone, Chem. Commun., 2015, 51, 4212 RSC;
(b) A. Goujon, G. Du, E. Moulin, G. Fuks, M. Maaloum, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2016, 55, 703 CrossRef CAS;
(c) A. Goujon, G. Mariani, T. Lang, E. Moulin, M. Rawiso, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 4923 CrossRef CAS;
(d) A. Goujon, T. Lang, G. Mariani, E. Moulin, G. Fuks, J. Raya, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 14825 CrossRef CAS.
- H. Li, H. Zhang, Q. Zhang, Q.-W. Zhang and D.-H. Qu, Org. Lett., 2012, 14, 5900 CrossRef CAS.
- H. Li, J.-N. Zhang, W. Zhou, H. Zhang, Q. Zhang, D.-H. Qu and H. Tian, Org. Lett., 2013, 15, 3070 CrossRef CAS.
- H. Li, X. Li, Z.-Q. Cao, D.-H. Qu, H. Ågren and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 18921 CrossRef CAS.
- H. Li, X. Li, H. Ågren and D.-H. Qu, Org. Lett., 2014, 16, 4940 CrossRef CAS.
-
(a) C. Romuald, A. Ardá, C. Clavel, J. Jiménez-Barbero and F. Coutrot, Chem. Sci., 2012, 3, 1851 RSC;
(b) C. Clavel, C. Romuald, E. Brabet and F. Coutrot, Chem. – Eur. J., 2013, 19, 2982 CrossRef CAS.
- Y.-X. Ma, Z. Meng and C.-F. Chen, Org. Lett., 2014, 16, 1860 CrossRef CAS.
- Z. Meng and C.-F. Chen, Chem. Commun., 2015, 51, 8241 RSC.
- Z. Meng, Y. Han, L.-N. Wang, J.-F. Xiang, S.-G. He and C.-F. Chen, J. Am. Chem. Soc., 2015, 137, 9739 CrossRef CAS.
- Y. Abe, H. Okamura, K. Nakazono, Y. Koyama, S. Uchida and T. Takata, Org. Lett., 2012, 14, 4122 CrossRef CAS.
- N. Zhu, K. Nakazono and T. Takata, Chem. Commun., 2016, 52, 3647 RSC.
- S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340 CrossRef CAS.
- C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876 CrossRef CAS.
-
(a) J. A. Berrocal, C. Biagini, L. Mandolini and S. Di Stefano, Angew. Chem., Int. Ed., 2016, 55, 6997 CrossRef CAS;
(b) C. Biagini, F. Di Pietri, L. Mandolini, O. Lanzalunga and S. Di Stefano, Chem. – Eur. J., 2018, 24, 10122 CrossRef CAS;
(c) C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. Di Stefano, Chem. Sci., 2018, 9, 181 RSC.
- A. Ghosh, I. Paul, M. Adlung, C. Wickleder and M. Schmittel, Org. Lett., 2018, 20, 1046 CrossRef CAS.
- L.-P. Yang, F. Jia, J.-S. Cui, S.-B. Lu and W. Jiang, Org. Lett., 2017, 19, 2945 CrossRef CAS.
- Q. Shi and C.-F. Chen, Org. Lett., 2017, 19, 3175 CrossRef CAS.
- Q. Shi and C.-F. Chen, Chem. Sci., 2019, 10, 2529 RSC.
-
(a) Z.-Q. Cao, Q. Miao, Q. Zhang, H. Li, D.-H. Qu and H. Tian, Chem. Commun., 2015, 51, 4973 RSC;
(b) Q. Zhang, S.-J. Rao, T. Xie, X. Li, T.-Y. Xu, D.-W. Li, D.-H. Qu, Y.-T. Long and H. Tian, Chem, 2018, 4, 2670 CrossRef CAS.
- S. Chen, Y. Wang, T. Nie, C. Bao, C. Wang, T. Xu, Q. Lin, D.-H. Qu, X. Gong, Y. Yang, L. Zhu and H. Tian, J. Am. Chem. Soc., 2018, 140, 17992 CrossRef CAS.
-
(a) H. Meng, M. Xue, T. Xia, Y.-L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690 CrossRef CAS;
(b) Q. Duan, Y. Cao, Y. Li, X. Hu, T. Xiao, C. Lin, Y. Pan and L. Wang, J. Am. Chem. Soc., 2013, 135, 10542 CrossRef CAS.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab