
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlecstatin-1 induces an immunogenic cell death signature in colorectal tumour spheroids†
Debora
Wernitznig
 *a,
Samuel M.
Meier-Menches
b,
Klaudia
Cseh
a,
Sarah
Theiner
b,
Dominik
Wenisch
a,
Andreas
Schweikert
ab,
Michael A.
Jakupec
ac,
Gunda
Koellensperger
b,
Andreas
Wernitznig
d,
Wolfgang
Sommergruber
de and
Bernhard K.
Keppler
ac
*a,
Samuel M.
Meier-Menches
b,
Klaudia
Cseh
a,
Sarah
Theiner
b,
Dominik
Wenisch
a,
Andreas
Schweikert
ab,
Michael A.
Jakupec
ac,
Gunda
Koellensperger
b,
Andreas
Wernitznig
d,
Wolfgang
Sommergruber
de and
Bernhard K.
Keppler
ac
aInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Währinger Straße 42, 1090, Vienna, Austria. E-mail: debora.wernitznig@univie.ac.at
bDepartment of Analytical Chemistry, University of Vienna, Währinger Straße 38, 1090, Vienna, Austria
cResearch Platform ‘Translational Cancer Therapy Research’, University of Vienna, Währinger Straße 42, 1090, Vienna, Austria
dBoehringer Ingelheim RCV GmbH & Co KG, 1121, Vienna, Austria
eUniversity of Applied Sciences, Dept. Biotechnology, Helmut Qualtinger Gasse 2, 1030, Vienna, Austria
First published on 9th December 2020
Abstract
Organometallic metal(arene) anticancer agents were believed to confer low selectivity for potential cellular targets. However, the ruthenium(arene) pyridinecarbothioamide (plecstatin-1) showed target selectivity for plectin, a scaffold protein and cytolinker. We employed a three-dimensional cancer spheroid model and showed that plecstatin-1 limited spheroid growth, induced changes in the morphology and in the architecture of tumour spheroids by disrupting the cytoskeletal organization. Additionally, we demonstrated that plecstatin-1 induced oxidative stress, followed by the induction of an immunogenic cell death signature through phosphorylation of eIF2α, exposure of calreticulin, HSP90 and HSP70 on the cell membrane and secretion of ATP followed by release of high mobility group box-1.
Significance to metallomicsOur work highlights the anticancer effects of plecstatin-1, an organoruthenium drug candidate that selectively targets plectin, in tumour spheroids. The treatment with plecstatin-1 resulted in the expected disruption of the cytoskeleton and phosphorylation of the stress marker eIF2α. This was accompanied by the induction of an immunogenic cell death signature, including calreticulin, high mobility group protein B and extracellular ATP. Thus, together with oxaliplatin and KP1339, plecstatin-1 is among the few metallodrugs that induces an immunogenic cell death signature in vitro and add additional evidence of the promise of metallodrugs in modulating immunogenic mechanisms as an anticancer strategy. |
Introduction
In the past few years, the interest in the therapeutic potential of metal based anticancer agents has substantially increased.1,2 Their wide-spectrum-effect against different types of cancer as well as their ease of chemical modification open up promising options in the treatment of cancer.1,2 The success of platinum complexes such as cisplatin, carboplatin and oxaliplatin, which are used as first-line anticancer drugs, also contributed to the expansion on the number of studies in this field.3,4 However, the adverse effects and the development of drug resistance limit the application of Pt–drugs in the clinics,3–5 thus spurring interest in other metal centres. Among several new classes of non-platinum metal-based complexes, ruthenium-based compounds have emerged as an interesting alternative to Pt-anticancer agents.7 The ruthenium(III) coordination complexes NAMI-A and KP1019/NKP-1339 reached the stage of clinical evaluation.5,6 Especially, the phase I study of NKP-1339 (BOLD-100, Scheme 1) suggested excellent tolerability also at elevated doses and showed a prolonged disease stabilization in some cases.8,9 Many derivatives of this class have been synthesized and studied for their antineoplastic properties. Besides the ruthenium(III) coordination complexes, more recent research efforts focussed on organometallic ruthenium(II) derivatives.10 The elucidation of their modes of action turned out as a considerable challenge.10–14 For many years, non-platinum drug candidates were believed to have a wide-spectrum mode of action, mainly relying on DNA-targeting.3,4 However, there is now substantial evidence that some of these next-generation metal-based compounds have non-conventional modes of action, which excludes DNA-targeting.8,11–15 For example, NKP-1339 was found to be a first-in-class GRP78 modulator.5 Furthermore, we were recently able to validate plectin-targeting for [chlorido(η6-p-cymene)(N-fluorophenyl-2-pyridinecarbothioamide)ruthenium(II)] chloride, termed plecstatin-1 (Scheme 1).16 Plectin is a scaffold protein and cytolinker that connects cellular membranes and cytoskeletal components such as intermediate filaments, actin microfilaments and microtubules.17–20 Mutations in plectin were shown to cause a number of diseases.19 Moreover, many studies have shown an association between plectin expression with cancer progression and metastasis in vitro and in vivo. Loss or mutations in plectin result in tissue degeneration and loss of cellular structural stability. Plectin depletion reduces the speed, directionality, and persistence of migration, as well as centrosome orientation.18,20 In analogy, plecstatin-1 was shown to exert anti-migratory effects in cancer cell culture models.10 Moreover, treating HCT116 colon carcinoma cells in monolayers with plecstatin-1 led to the phosphorylation of eukaryotic initiation factor 2-alpha (eIF2α) and down-stream activation of the cytoprotective integrated stress response (ISR), characterized by mitochondrial stress.21 EIF2α is also involved in the mode of action of NKP-13396,8 and a cyclometalated gold(III) N-heterocyclic carbene anticancer drug.16 Interestingly, eIF2α is a master regulator of cellular stress management. It is not only implicated in ISR,22 but also in endoplasmic reticulum (ER) stress responses, such as the unfolded protein response (UPR).23 It acts as a molecular switch and may dictate cell survival or death under stress. Of note, phosphorylation of eIF2α is a direct biomarker of immunogenic cell death (ICD).24 Indeed, a small number of metal-based anticancer agents is able to induce an immune response towards cancer cells, which represents an emerging strategy in the treatment of neoplastic cells.25,26 Classic cytotoxic chemotherapy has been considered immunosuppressive, but recent studies demonstrated that it may also interact positively with immunotherapy.27–29 Oxaliplatin (Scheme 1) is a prominent example to induce cell death accompanied by the release of damage-associated-molecular-patterns (DAMPs), rendering cancer cells detectable by the immune system.25 These events may lead to long term responses in immunocompetent hosts, which characterizes immunogenic cell death (ICD).31,32 Interestingly, cisplatin or carboplatin do not induce such effects.29 | ||
| Scheme 1 Representative metal-based anticancer agents that induce hallmarks of immunogenic cell death (ICD). | ||
We have recently shown that NKP-1339 is the first ruthenium(III) compound to trigger a sustained endoplasmic reticulum (ER) stress response and to induce the hallmarks of ICD,26e.g. calreticulin (CRT) exposure on the cell membrane, release of ATP and high mobility group box-1 (HMGB-1) from dying cancer cells in vitro. These hallmarks are essential for the development of an immune response.29 CRT surface exposure induces dendritic cell (DC) maturation and uptake of tumour antigens. Secretion of ATP from dying cells stimulates purinergic P2RX7 receptors on the surface of DCs leading to the activation of the NLRP3 inflammasome.24 Additionally, ATP stimulates IFN-γ production by tumour-specific cytotoxic T cells and functions as a potent chemoattractant for several cell types involved in immune responses. Finally, HMGB-1 binds to toll-like receptor 4 (TLR-4) on the surface of DCs to optimize the presentation of antigens from dying tumour cells. Besides the ‘classical’ ICD signature, heat shock proteins (HSPs) are implicated in activation of ICD as well. HSPs are intracellular chaperones induced by cellular stress including oxidative stress, chemotherapeutic drugs and irradiation. Specifically, heat shock protein 70 (HSP70) and heat shock protein 90 (HSP 90) may amplify the innate immune response once they reach the extra-cellular space. Together with other DAMPs, they support the cross-presentation of tumour-related antigens by DCs to CD4+ and CD8+ T cells resulting in specific immunity against the tumour.24–32
In this study, it is aimed to validate the mode of action of plecstatin-1 with respect to plectin-targeting and the impact on cell architecture in 3D tumour spheroids of the colon carcinoma cell lines HCT116, HCT-15 and HT-29 including uptake and distribution studies.30 Additionally, the involvement of eIF2α phosphorylation was confirmed in tumour spheroids and unexpectedly connected to the induction of an ICD signature.
Results and discussion
Plecstatin-1 treatment affects growth, morphology and colony formation
The resazurin cytotoxicity assay was employed to quantitatively measure proliferation and relative cytotoxicity of plecstatin-1 against three different colon adenocarcinoma cell lines (HCT-116, HCT-15 and HT-29) grown as monolayers (2D) and as multicellular tumour spheroids (3D) (Fig. ST1, ESI†). The complex was more active in all cell lines grown as monolayers in comparison to cells grown as spheroids, where up to a 10-fold lower potency was observed. Resazurin sodium salt provides accurate measurements of the number of dead cells in a population at a specific time, however, it is difficult to distinguish between dying cells and those that are marked for future death. Therefore, the long-term proliferative capacity of plecstatin-1 treated cells was investigated as an indirect measurement of cell death.33,34 In a first step using cell monolayers, the colony formation assay (CFA) was combined with a resazurin cytotoxicity test. Plecstatin-1 treatment significantly decreased the number of colonies at IC50 concentration compared to the control in HT-29 and HCT-116 cells. Almost no colonies were observed after 7 days of incubation with the compound (Fig. SF1, ESI†).Subsequently, the effects of plecstatin-1 were investigated in 3D multicellular tumour spheroids. These in vitro models better recapitulate the tumour architecture and biology found in vivo in comparison to cells grown in monolayers.36–38 Tumour spheroids represent a suitable model for mode of action studies of plecstatin-1 because it was shown to exert strong effects on non-mitotic microtubules by plectin modulation.10 In a first series of experiments, multicellular spheroids of HCT-116, HCT-15 and HT-29 were incubated with plecstatin-1 (200 μM) for 11 days. The effect of plecstatin-1 was cell line dependent (Fig. 1). Plecstatin-1-treated HCT-15 and HCT-116 multicellular spheroids were smaller and displayed a qualitative loss of compactness compared to untreated cells (Fig. 1A). However, in the more resistant HT-29 spheroids no pronounced reduction in size was observed compared to the untreated control, even though cells located in the periphery of the spheroids displayed an apoptotic morphology. Similar results could be observed in cisplatin treated spheroids. However, cisplatin was unable to induce loss of compactness and apoptotic morphology of HCT-116 spheroids as observed for plecstatin-1 and oxaliplatin treated cells. Furthermore, oxaliplatin greatly reduced the size of the spheroids, including the HT-29 cell line. HT-29 and HCT-15 spheroids were incubated for a shorter period of 6 days with plecstatin-1 at increasing concentrations. The diameter of the spheroids was measured before treatment, at day 3 and at day 6 post-treatment. In HT-29 spheroids the apoptotic morphology could be observed despite reduced exposure time to the compound (Fig. SF2, ESI†). In contrast, the cytostatic effect of the compound was already observed at sub-IC50 concentrations in HCT-15 spheroids and a significant difference in size in comparison to untreated cells was observed in spheroids exposed to plecstatin-1 for 6 days (Fig. SF2, ESI†). Furthermore, the proliferation status of cells in the tumour spheroids was assessed. Plecstatin-1 treated spheroids (200 μM) were cryo-sectioned followed by immunofluorescence-labelling, using the KI67 marker. In plecstatin-1 treated HCT116 spheroids, there is a ca. 3-fold decrease in the number of KI67+ cells (Fig. SF3, ESI†), indicating reduced proliferative capacity of the cells compared to the control, which further supports the findings of the cytotoxicity test and growth/morphology evaluation (Fig. 1B).
 | ||
| Fig. 1 (A) Representative pictures of HCT-15, HCT-116 and HT-29 multicellular spheroids treated with plecstatin-1 (200 μM), oxaliplatin (20 μM) and cisplatin (20 μM) for 11 days. Changes in the morphology can be observed in all cell lines upon treatment with plecstatin-1. (B) Representative immunofluorescence analysis (confocal microscopy) of paraformaldehyde (PFA)-fixed HCT-116 spheroids. A 3-fold reduction in the number of KI67+ cells is observed after 72 h treatment with plecstatin-1 (200 μM) in comparison to the untreated control. Scale Bar 50 μm. | ||
In order to elucidate whether the reduced proliferation of tumour spheroids would be due to cytostatic or cytotoxic properties of plecstatin-1 treatment, we decided to evaluate whether plecstatin-1 is able to induce apoptosis. The induction of apoptosis was accessed by flow cytometry. Spheroids were treated for 72 h with plecstatin-1 (200 μM), dissociated and incubated with an annexin-V-FITC-conjugated antibody and propidium iodide (PI). Plecstatin-1 treatment induced a relatively minor apoptotic response in HCT-15 and HCT-116 spheroids, with a slightly stronger effect on HCT-15, where cell death levels reached ca. 30% (Fig. 2). In contrast, the HT-29 spheroids were not affected by treatment with plecstatin-1 with no significant change in the number of apoptotic cells (10% of cell death) compared to the untreated control (Fig. 2).
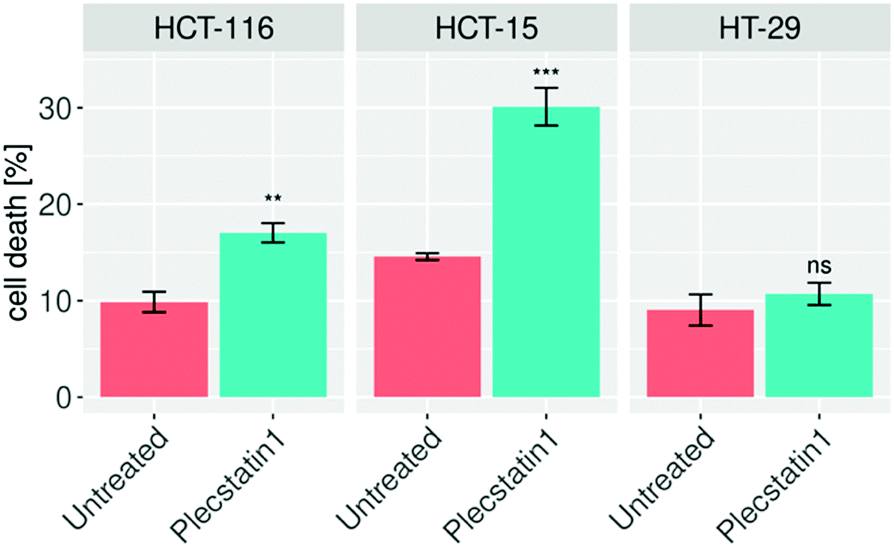 | ||
| Fig. 2 Tumour spheroids were treated for 72 h with plecstatin-1, stained with annexin-V/propidium iodide and analysed by flow cytometry. Apoptosis could be observed in HCT-15 and HCT-116 treated spheroids, while HT-29 were not significantly affected by the drug treatment. Mean + STD (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Tukey's range test), not significant (ns). | ||
Apoptosis is regulated by cytochrome-c release from mitochondria,61,62 which eventually triggers the activation of caspases.31–33 Indeed, cytochrome-c was found to be released from the mitochondria into the cytosol of HCT-116 tumour spheroids upon treatment for 48 h (Fig. 3 and Fig. SF4A, ESI†). Caspase-3 is responsible for the majority of proteolysis during apoptosis, and detection of cleaved caspase-3 is considered a reliable marker for dying or dead cells by apoptosis.33,39 Therefore, treated HCT-116 spheroids were immuno-labelled for cleaved caspase-3. Plecstatin-1 treatment induced cleavage of caspase-3 (Fig. SF4B, ESI†), confirming our findings regarding the induction of apoptosis obtained by flow cytometry. These effects were less pronounced in the less responsive HT-29 spheroids (Fig. SF5 and SF6, ESI†).
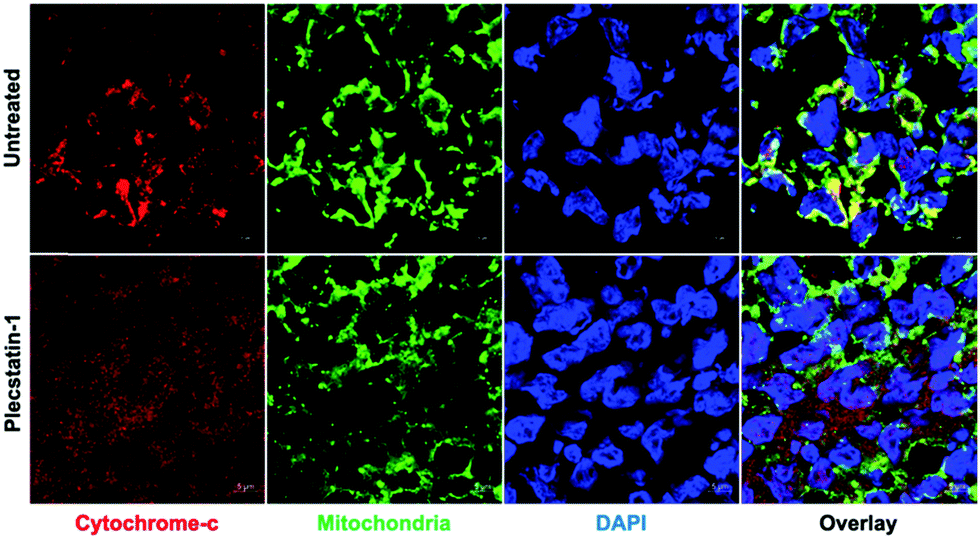 | ||
| Fig. 3 Representative immunofluorescence analysis (confocal microscopy) of HCT-116 spheroids. Mitochondria integrity is shown in untreated spheroids, while cytochrome c release from mitochondria is observed upon treatment with plecstatin-1 (200 μM) for 72 h. Note that the cytochrome-c immunostaining overlapped, as indicated with yellow immunofluorescence, with the mitochondrial marker in the cytoplasm of control cells. However, in plecstatin-1 treated cells cytochrome-c leaked from mitochondria and was distributed distinctly from the mitochondrial marker. | ||
Plecstatin-1 only moderately induces single-strand DNA breaks
In order to evaluate DNA damage as a possible cause of apoptosis-induction, the interaction of plecstatin-1 with DNA in a cell-free environment was tested by an electrophoretic dsDNA plasmid assay. The intact pUC19 plasmid (2.7 kbp) holds the most negative supercoiled (sc) form and upon drug interaction is gradually converted to the open circular (oc) form. The investigated compound (50 μM) was incubated with pUC19 at 37 °C and allowed to interact for 6 h. Plecstatin-1 did not strongly interact with plasmid DNA (Fig. SF7, ESI†). It moderately induced single-strand DNA breaks at elevated concentrations and partially converted the plasmid to its oc form only to a low degree of 5% after 30 minutes. The interaction between plasmid DNA and plecstatin-1 increased over time and after 6 h incubation it reached 14% of the oc form. Due to this very mild interaction with DNA, this mechanism possibly does not play a relevant role in the mode of action of plecstatin-1 in cells.102Ru is mainly distributed to the periphery of tumour spheroids as revealed by LA-ICP-MS
High-resolution laser ablation (LA) in combination with inductively coupled plasma time-of-flight mass spectrometry (ICP-TOF-MS) was used to determine the multi-element distribution in colon cancer HT-29 and HCT-116 tumour spheroids after treatment with plecstatin-1. Tumour spheroids were treated with 400 μM of plecstatin-1 for 5 days. Highest ruthenium accumulation was observed in the proliferating cells at the outer rim of the spheroid sections of both colon cancer cell lines (Fig. 4) in accordance with the immunofluorescence analysis of Ki67 (Fig. 1B), where a 3-fold reduction in the number of Ki67+ cells could be observed mainly in the outer rim of the spheroids. Significantly lower levels of ruthenium (about 1–2 orders of magnitude) were detected in the areas of quiescent cells of the spheroids indicating increased uptake of plecstatin-1 into proliferating cells. This is in accordance with previous LA-ICP-MS studies on platinum compounds in tumour spheroids, where highest platinum levels corresponded to proliferating cells, whereas quiescent cells displayed low platinum concentrations.40,41 Furthermore, plecstatin-1 was previously found to accumulate preferentially in the outer rims of a CT-26 tumour in vivo.42 For the HT-29 tumour spheroid sections, higher ruthenium accumulation was also observed for the compound in areas of deeper layers of the spheroids. Interestingly, plecstatin-1 was less potent in HT-29 spheroids probably indicating a protective transport mechanism from the proliferating area into the necrotic core. | ||
| Fig. 4 Signal intensity maps of 102Ru+ obtained by LA-ICP-TOF-MS of HT-29 (A) and HCT-116 (B) tumour spheroids after treatment with 400 μM of plecstatin-1 and for 5 days. High-resolution laser ablation images were obtained with a pixel size of 2.5 μm was performed at a repletion rate of 200 Hz. | ||
In addition to ruthenium, several elements of biological relevance were quantified in the spheroid sections by LA-ICP-TOF-MS (Fig. 4, 5 and Fig. SF8–SF11, ESI†). The phosphorus distribution followed the same pattern as ruthenium with higher levels in proliferating cells and lower levels in quiescent cells. This is also in accordance with a previous LA-ICP-MS study on HCT-116 tumour spheroids, where it was shown that phosphorus can be used as a marker for cell density and tissue thickness.40,41 Proliferating cells displayed 4 to 5 times higher levels of magnesium and calcium, in comparison to quiescent cells. The qualitative distribution pattern is in accordance to the ruthenium distribution.
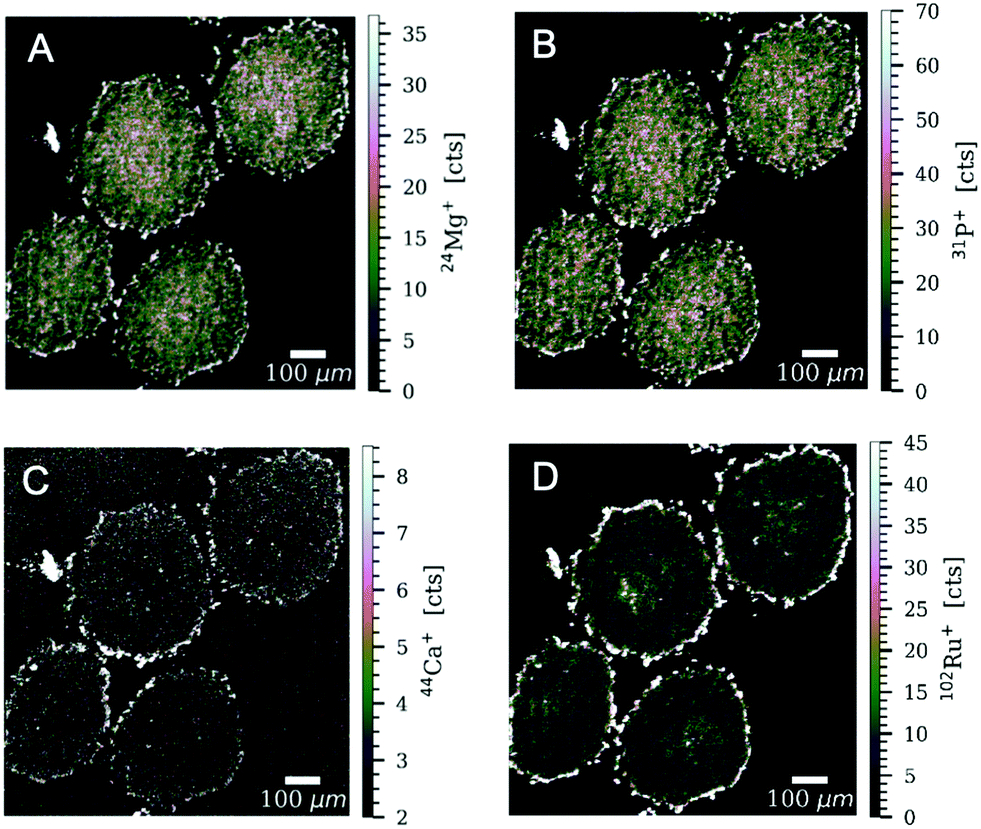 | ||
| Fig. 5 Signal intensity maps of (A) 24Mg+, (B) 31P+, (C) 44Ca+ and (D) 102Ru+ obtained by LA-ICP-TOF-MS of HT-29 tumour spheroids after treatment with 400 μM of plecstatin-1 for 5 days. High-resolution laser ablation images were obtained with a pixel size of 2.5 μm and a repetition rate of 200 Hz. | ||
Plecstatin-1 treatment leads to rearrangement of plectin and a-tubulin but not to induction of EMT in tumour spheroids of colon carcinoma
Plectin is a large intermediate filament associated protein. It functions as a cytoskeletal crosslinker and signalling scaffold, greatly influencing the cytoskeleton.43–46It has a multidomain structure which enables interactions with several different proteins, significantly affecting their function. It also binds to transmembrane receptors, proteins of the sub-plasma membrane protein skeleton, components of the nuclear envelope, and several kinases with known roles in migration, proliferation, and energy metabolism of cells. Multiple studies have shown plectin expression to be associated with cancer progression and metastasis.18,47 We examined subcellular localization of plectin by treating spheroids for 72 h with plecstatin-1 (200 μM) followed by the immune-staining against pan-plectin. Strikingly, a collapse of the punctuated plectin-network into larger aggregates was observed in plecstatin-1 treated HCT-116 spheroids (Fig. 6A) in addition to a reduction of the mean fluorescence signal (MFI) indicating loss of plectin (Fig. SF12, ESI†). We further examined cytoskeletal components upon treatment with plecstatin-1, namely α-tubulin and F-actin. Being a major component of microtubules and tightly associated with plectin,43,44 α-tubulin was also severely affected by the treatment with plecstatin-1. In untreated spheroids, α-tubulin structures were intact, showing an extensive network of microtubules (Fig. 6B and Fig. SF13, ESI†). In plectstatin-1 treated HCT-116 spheroids, a collapse of these structures was observed, in analogy to plectin (Fig. 6 and Fig. SF12, ESI†) and confirmed by a 5-fold reduction of the MFI of this protein in comparison to untreated spheroids (Fig. 6B). This effect was reduced in the less responsive HT-29 spheroid (Fig. SF14, ESI†). In immortalised keratinocytes of a plectin knock-out model, it was previously found that plecstatin-1 had a pronounced effect on α-tubulin, while the intracellular distribution of plectin remained unchanged.10 We also probed for F-actin integrity in tumour spheroids. Plecstatin-1 treated spheroids displayed a disrupted F-actin cytoskeleton, characterized by the formation of F-actin aggregates and general reduction of the fluorescence signal while untreated cells did not show such changes (Fig. SF15 and F16, ESI†). Again, actin disruption was more pronounced in the HCT-116 compared to the HT-29 spheroids.
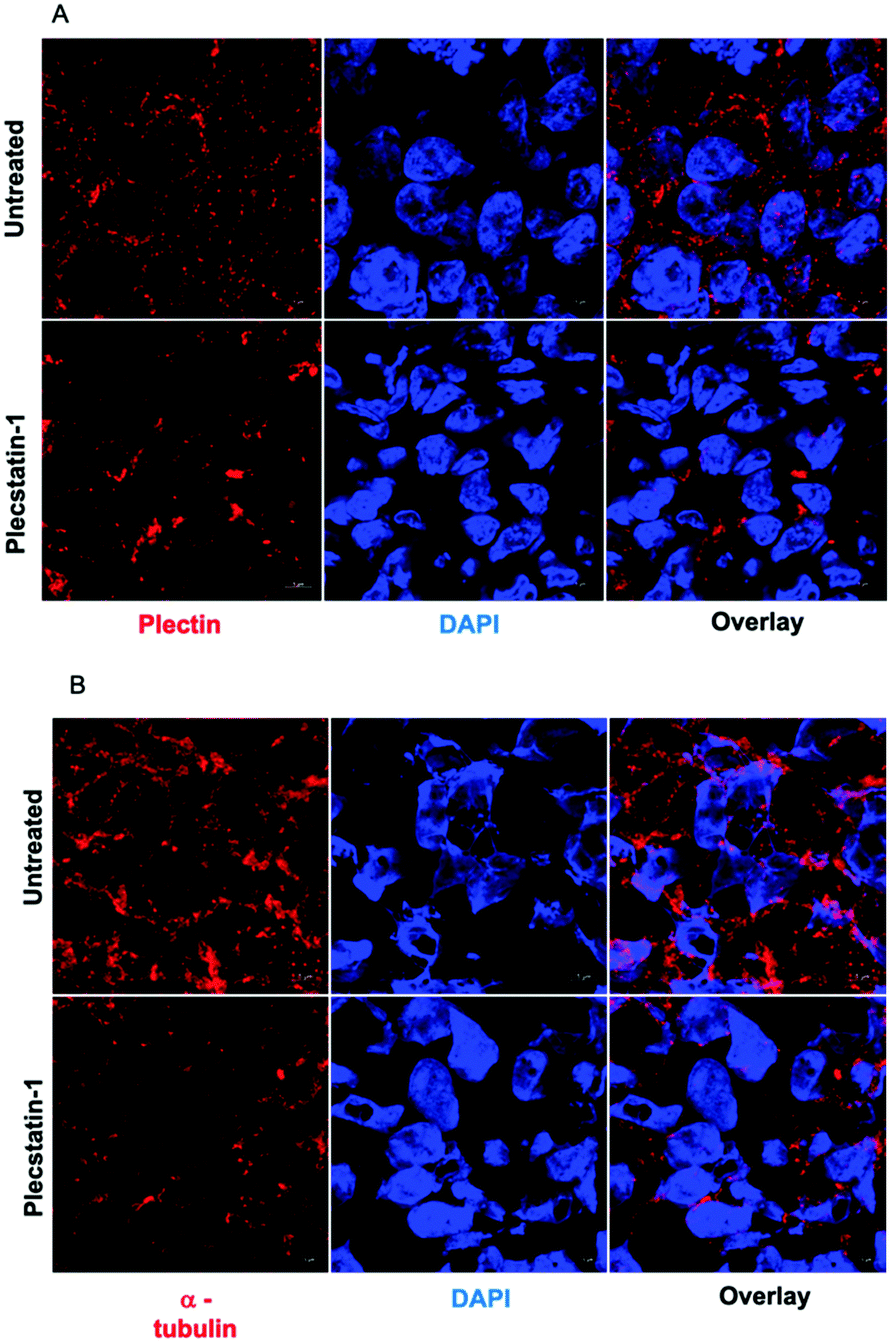 | ||
| Fig. 6 (A) Representative immunofluorescence analysis (confocal microscopy) of PFA fixed HCT-116 spheroids. Spheroids were labelled against a plectin antibody. Plecstatin-1 treatment (200 μM) led to a collapse of the plectin-network. (B) Representative immunofluorescence analysis (confocal microscopy) of PFA fixed HCT-116 spheroids. Spheroids were labelled against an α-tubulin antibody. Treatment with plecstatin-1 (200 μM) led to a major collapse of the α-tubulin network. | ||
Since plectin actively regulates cytoskeletal organization and migratory properties of cells,18,47 we decided to investigate whether modulation of plectin, as a consequence of plecstatin-1 treatment, would be able to influence the epithelial to mesenchymal transition (EMT). EMT is a process in which epithelial cells acquire a mesenchymal phenotype.49–51 Moreover, epithelial cells form layers where they are in close contact with each other, while mesenchymal cells are loosely organized in a three-dimensional extracellular matrix. Tumour cells undergoing EMT are characterized by increased motility and invasiveness, which favours their dissemination to other sites and the formation of metastasis.63 Furthermore, they become resistant to apoptosis and anticancer drugs, support immunosuppression and behave as cancer stem-like cells.52 During this process, cancer cells undergo tight junction dissolution, disruption of apical-basal polarity, and reorganization of the cytoskeletal architecture, which enable cells to develop an invasive phenotype.52–57
This process is characterized by a decrease of epithelial cell junction proteins (e.g. E-cadherin) and an increase of vimentin, fibronectin and N-cadherin.53–57 In this experimental setup, plecstatin-1 treatment did not induce the loss of E-cadherin (Fig. SF17 and SF18, ESI†) and neither concomitant increase of vimentin and n-cadherin in HCT-116 or HT-29 spheroids (Fig. SF19–SF22, ESI†). To support these findings, we selected the highly invasive HT1080 fibrosarcoma cell line to perform a spheroid invasion assay. Briefly, HT1080 spheroids were embedded in Matrigel and exposed to the compound (40 μM) for 72 h (Fig. SF23, ESI†). Untreated HT1080 spheroids were larger in size and displayed an invasive morphology, while HT1080 spheroids treated with plecstatin-1 remained smaller and did not invade the matrix. This trend was observed also after longer incubation times (>144 h). Collectively our data demonstrates that plecstatin-1 treatment greatly impacts the tumour architecture and invasiveness in multicellular spheroids, which supports the observations obtained by previous findings,10 where plecstatin-1 treatment hindered the invasion capacity of HCT-116 grown as spheroids. Thus, the plectin-targeting property of plecstatin-1 seems to prevail in 3D tumour spheroid models and emphasizes the strong impact of plecstatin-1 on the cellular architecture by a rearrangement of the α-tubulin network. In addition, plecstatin-1 treatment did not seem to induce EMT. These findings further highlight the potential of plecstatin-1 to reduce tumour invasiveness as well as its potential as an anti-metastatic drug.
Plecstatin-1 induces oxidative stress
To evaluate the generation of ROS upon treatment with plecstatin-1 (200 μM), spheroids were treated for 24 h. Spheroids were dissociated, incubated with Cellrox® and PI and then analysed by flow cytometry. The number of ROS+ cells was increased in all cell lines tested upon treatment, with the strongest effect, ca. 9-fold compared to the untreated controls on HCT-116 spheroids and the least effect on HT-29 cells, where a ca. 2-fold increase could be observed (Fig. 7A). ROS induction seems to correlate with sensitivity. To get a better overview of the distribution of ROS within the spheroids, plecstatin-1-treated and untreated spheroids were incubated with Cellrox® and PI and analysed by confocal microscopy. Three-dimensional reconstructions of optical sections displayed a large number of ROS+/PI− (viable cells) as well as ROS+/PI+ (non-viable cells) populations in plecstatin-1 treated HCT-116 spheroids (Fig. 7B and Fig. SF24, ESI†). In untreated spheroids, only few ROS+/PI− as well as ROS+/PI+ cells were observed. In accordance with the ruthenium accumulation evidenced by the LA-ICP-TOF-MS experiments, ROS were also generated mainly in the outer rims of the tumour spheroid. Thus, changes in cytoarchitecture are also accompanied by the induction of ROS and the co-localization of ruthenium in sensitive tumour spheroids. | ||
| Fig. 7 (A) Multicellular spheroids were treated for 24 h with plecstatin-1 (200 μM) and the induction of ROS was assessed by flow cytometry. Plecstatin-1 induced ROS in all cell lines, with a pronounced effect in HCT-116 spheroids. (B) Representative three-dimensional reconstruction of HCT-116 spheroids double-labelled with Cellrox® and PI. Confocal microscope images were obtained from a stack of optical sections from the spheroids. Green puncta correspond to ROS+ cells, red puncta to PI+ and yellow puncta ROS+/PI+ (colocalization). The untreated sample (on the left) displays few ROS+ cells. There is an increase in ROS+ cells in plecstatin-1 samples (on the right) treated for 24 h. | ||
Generation of ROS was previously observed for plecstatin-1 in cell culture monolayers.10,21 Together with a mitochondrial stress signature related to the cytoprotective integrated stress response (ISR), these processes culminated in the phosphorylation of eIF2α.21 Importantly, eIF2α is a master regulator of several stress response pathways, also including endoplasmic reticulum (ER) stress upon increased ROS, production as part of the unfolded protein response (UPR).64,65 ISR is an integral part of cellular responses to diverse stresses including those affecting the mitochondria.60 One of the main effects of eIF2α phosphorylation at Ser51 is the general inhibition of protein synthesis. This process allows cells to either recover from stress or be eliminated if the damage is beyond repair.22,66,67 Although the ISR is primarily a pro-survival homeostatic program, aiming to optimize the adaptive cellular response to stress, exposure to severe stress, either in intensity or duration, will overwhelm the capacity of this adaptive response and drive signalling toward cell death.22
Treatment of HCT-116 tumour spheroids with plecstatin-1 (200 μM) for 24 h clearly induced the phosphorylation of eIF2α (Fig. 8). Altogether, these results show that plecstatin-1 induced an oxidative stress response and phosphorylated the master regulator eIF2α of the cellular stress response management.
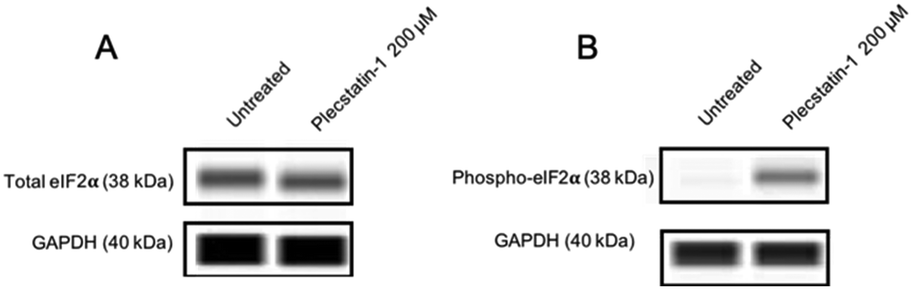 | ||
| Fig. 8 Representative immunoblot images from a capillary western blot system (WES). (A) The total load of eIF2α and GAPDH for HCT-116 spheroids is shown. (B) Phosphorylation is observed upon treatment with plecstatin-1 for 24 h. | ||
Plecstatin-1 induces an ICD signature in vitro
Phosphorylation of eIF2α serves a key role during stress responses such as ISR and UPR, but is also an essential mediator for ICD.22,67ICD is followed by the translocation of CRT from the ER lumen to the plasma membrane, which facilitates the transfer of tumour associated antigens to DCs.68–70 Kroemer and colleagues determined using a machine-learning approach that the main ER stress-related mechanism essential for starting an ICD-response is the phosphorylation of eIF2α together with its downstream consequences, e.g. CRT exposure, stress granule formation and autophagy induction, meaning that eIF2α-phosphorylation is pathognomonic for ICD.24 Additionally, eIF2a phosphorylation has a clinical importance, since it has been recently shown to serve as a biomarker that correlates with improved immunosurveillance a baseline as well as therapeutic responses to a variety of anticancer drugs.27,29,32 Having established that plecstatin-1 is able to induce the phosphorylation of eIF2α in HCT-116 tumour spheroids, we investigated whether the compound would be able to trigger an ICD signature. For this, spheroids were treated for 24 h with plecstatin-1 (200 μM). Qualitative analysis by immunofluorescence double-labelling with an anti-CRT antibody and the membrane marker, wheat germ agglutinin (WGA) showed an induction of CRT expression upon plecstatin-1 treatment of HCT-116 spheroids and a favoured distribution to membranes, indicated by the colocalization (yellow regions) of CRT and WGA (Fig. 9A and Fig. SF25, ESI†). This finding was supported by quantitative analysis of CRT+ cells by flow cytometry (Fig. 9B). Again, the percentage of CRT+ cells (20%) was highest in HCT-116 tumour spheroids treated with plecstatin-1, while HT-29 spheroids showed lowest responsiveness. In all cases, plecstatin-1 led to an increase (up to 4-fold) translocation of CRT to the plasma membrane in comparison to the untreated control.
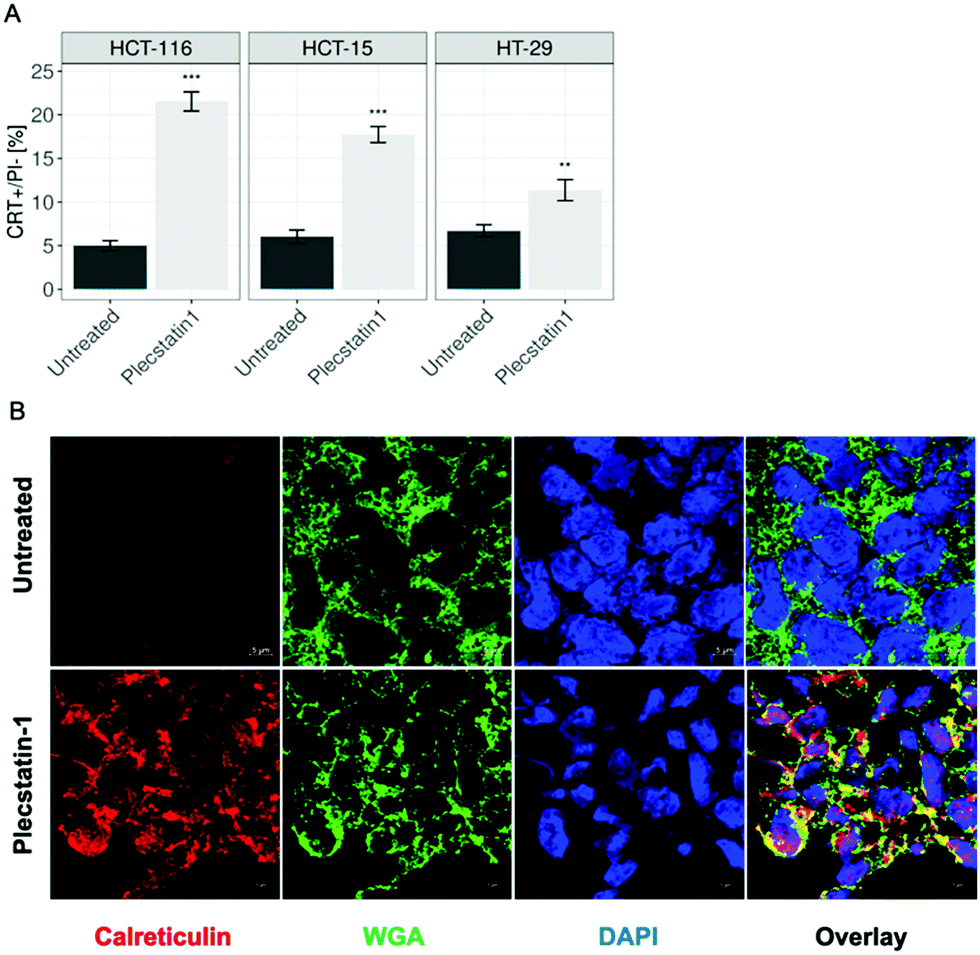 | ||
| Fig. 9 (A) Analysis of CRT membrane exposure of treated spheroids after plecstatin-1 (200 μM) exposure for 24 h obtained by flow cytometry. Mean + STD (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Tukey's range test). (B) Representative pictures of HCT-116 spheroids. Spheroids were labelled against a calreticulin antibody, as well as WGA for membrane staining. CRT translocation can be observed upon treatment with plecstatin-1 after 24 h (colocalization yellow regions). | ||
HMGB-1 represents a further marker for ICD induction. The protein first transits from the nucleus into the cytoplasm and then is secreted or passively released through the permeabilized plasma membrane.69 The extracellular levels of HMGB-1 were analysed by ELISA by collecting supernatants of plecstatin-1 treated spheroids after 72 h incubation with the compound. Indeed, HMGB-1 levels in the supernatant increased ca. 5-fold in plecstatin-1 treated spheroids compared to untreated controls, with a stronger effect on HCT-116, cells compared to HT-29 cells where a 3-fold increase was observed (Fig. 10A).
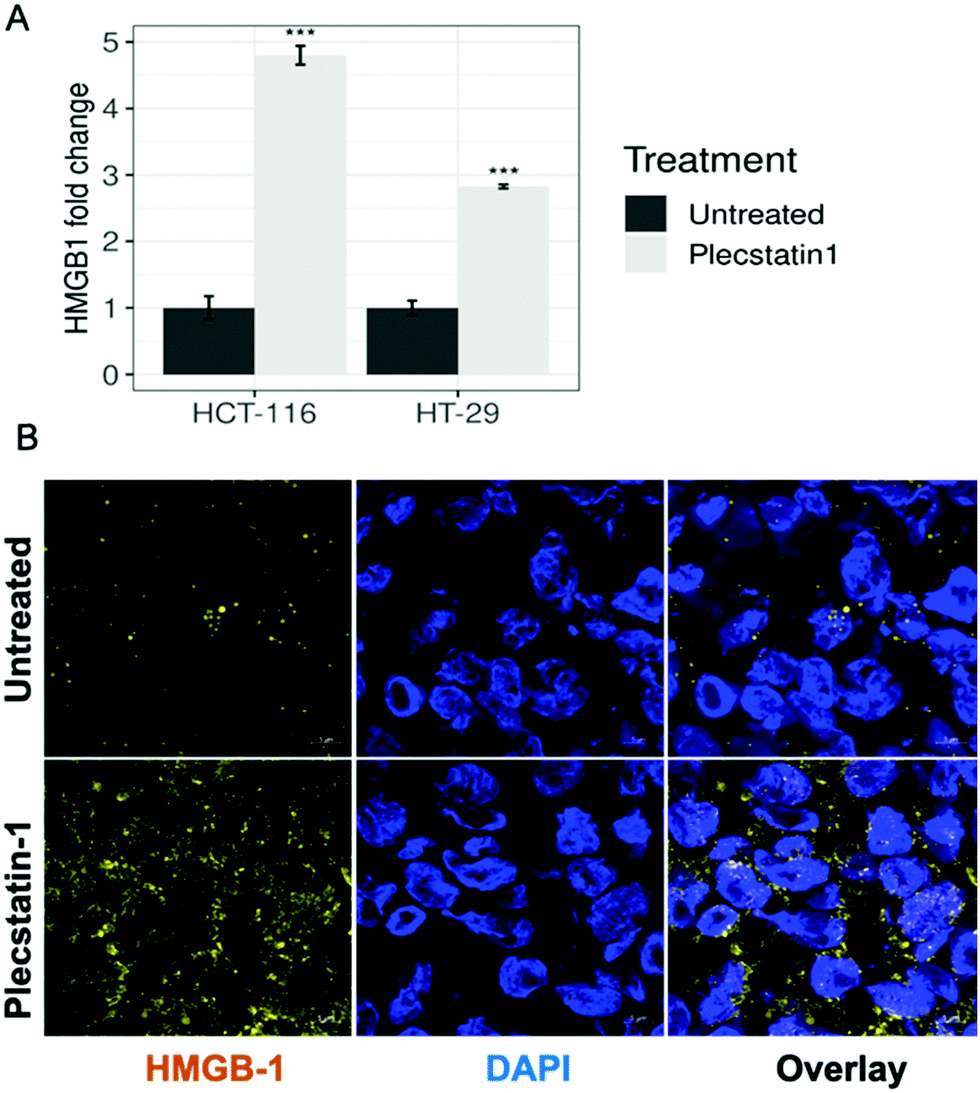 | ||
| Fig. 10 (A) HMGB-1 release from tumour spheroids upon treatment with plecstatin-1 (200 μM) for 72 h. Supernatants were collected and HMGB-1 levels were measured by ELISA. Mean ± STD (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Tukey's range test). (B) Representative immunofluorescence analysis (confocal microscopy) HCT-116 spheroids. Spheroids were labelled against a HMGB-1 antibody. HMGB-1 translocation to and enrichment in the cytoplasm can be observed upon treatment with plecstatin-1 after 72 h. | ||
Additionally, the release of HMGB-1 was qualitatively assessed by means of immunofluorescence analysis in HCT-116 (Fig. 10B and Fig. SF27, ESI†) and HT-29 spheroids (Fig. SF28, ESI†). In untreated spheroids, HMGB-1 is mainly located within the nuclei, whereas upon plecstatin-1 treatment, HMGB-1 can be found and is enriched in the extracellular space.
In both cases, HMGB-1 was induced upon treatment with plecstatin-1 and observed within the cytoplasmic compartment. Again, the effect was stronger in HCT-116 compared to HT-29 spheroids. HMGB-1 is a protein that is normally localized in the nucleus. The translocation of HMGB-1 constitutes a major cellular danger signal enabling the stimulation of pro-inflammatory and immunostimulatory pathways, more specifically through binding to pattern recognition receptors such as toll-like receptor-2 (TLR2) and TLR4.29,69
Extracellular levels of ATP constitute another important marker of ICD on the metabolite level.70 Once purinergic receptors (especially of the P2X7 type) are activated by increasing levels of extracellular ATP (eATP), several downstream effects on the immune system are observed. The main events consist of the activation of DCs, T cells, stimulation of CD4+/CD8+ effector T cells and NKT cells, induction of Treg apoptosis and generation of Th17 lymphocytes, together with high levels of IL-1β, IL-6, IL-17 in the tumour microenvironment.71 This process is essential for inducing an ICD response, stimulating host-specific immune responses against the tumour.71,72Prae-mortem release of ATP relies on an autophagy-dependent process involving the caspase-3 mediated proteolytic activation of pannexin-1 channels.70,73 To assess ATP release, supernatants from plecstatin-1 treated and untreated spheroids were collected and analysed by means of a luminescence assay. The results showed that plecstatin-1 treated spheroids released considerable amounts of ATP into the supernatant (Fig. 11), featuring a stronger ATP release (of about 10 times more compared to untreated controls) in HCT116 tumour spheroids in comparison to HT-29 spheroids. The emerging evidence that purinergic signalling influences cell proliferation and migration, tumour angiogenesis as well as invasiveness and metastatic diffusion in different types of cancer has substantially increased the number of studies in the field.71–74 It has been recently shown that P2Y2 receptors mediate the phosphorylation of EGFR, thus promoting the proliferation and tumorigenesis of head and neck squamous cell carcinoma cell lines.74 Furthermore, blocking P2X7 receptors has been shown to be an efficient way to prevent tumour growth in cancers where this receptor is overexpressed.71 The administration of P2X7 inhibitors and antagonists decreased cancer cell growth and spreading in animal models of various cancers.71,72 In the other hand, eATP plays an immunostimulatory role in the host, by increasing chemotaxis and activation of DCs, macrophages, neutrophils among other immune cell types, which lead to increased cell death, apoptosis and reduced growth of tumours. These effects seem to be cancer-type specific and have been reported on nasopharyngeal carcinoma and colon carcinoma, prostate cancer and melanoma. The complex interaction of eATP and its hydrolysis product, adenosine in the tumour microenvironment with cancer cells and immune cells highlight the ambiguous effects that the activation of purinergic receptors has on the tumour side and on the host side.71,72 Therefore, further studies will be required to identify the best strategy (stimulation or inhibition) to target the purinergic receptor network, hopefully identifying the best approach to boost anti-tumour immune response, and limit tumour growth.71,72
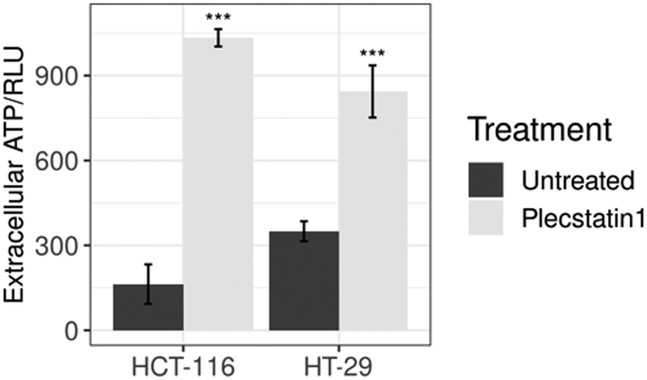 | ||
| Fig. 11 Analysis of ATP levels in supernatants from treated spheroids after drug exposure (200 μM) for 24 h expressed in relative light unit (RLU). Mean ± STD (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Tukey's range test). | ||
Our data demonstrates that plecstatin-1 treatment successfully induced an ICD signature in vitro, characterized by the translocation of CRT to the plasma membrane, release of ATP and HMGB-1, which represent key mechanisms taking place in the course of ICD.24
Finally, plecstatin-1 treatment of tumour spheroids led also to an increase in the fluorescence signal of the two chaperones HSP70 and HSP90 and a preferred co-localization to plasma membrane (yellow regions) was observed in comparison to untreated controls (Fig. SF26, ESI†). HSP70 and HSP90 are ER-resident chaperones58,59 that mediate protein transport, ensure proper protein folding, and protect newly synthesized polypeptide chains against denaturation.35,75 These chaperones accumulate within cells in response to different cellular insults including anticancer chemotherapy, allowing cells to overcome unfavourable conditions. HSP70 is specifically induced upon treatment with divalent metal compounds.76,77 This cytoprotective effect is due to their ability to inhibit apoptosis and in the case of HSP70, even autophagy.75–77 However, once in the extracellular space, HSP70 and HSP90 have an immunogenic role and have been shown to bind to high affinity receptors on the surface of antigen presenting cells (APCs), which leads to the activation and maturation of DCs, a process that enables the cross presentation to CD8+ cytotoxic T cells.76
Conclusions
Previous studies showed the target selectivity of plecstatin-1 towards plectin.10,21 Therefore, we aimed at better understanding whether and how the treatment with plecstatin-1 would impact biological processes in cancer cells. Our work employed multicellular spheroids to further characterize the response of cancer cells upon treatment with plecstatin-1. We observed that plecstatin-1 treatment reduced spheroid growth and decreased the colony-forming ability of colon adenocarcinoma cells. Furthermore, our data revealed that plecstatin-1 induced apoptosis to a minor degree via the intrinsic pathway, causing cytochrome-c release and caspase-3 activation. Additionally, plecstatin-1 generated an oxidative stress response. Furthermore, our approach revealed that the activation of the eIF2α axis induced an ICD signature consisting of: translocation of CRT, HSP90 and HSP70 to the plasma membrane, as well as the release of HMGB-1 and secretion of ATP. Plecstatin-1 did not strongly interact with DNA, emphasizing proteins as the relevant binding partners. Furthermore, we observed that plectin and other important structural proteins, namely F-actin and α-tubulin are highly impacted by the treatment with plecstatin-1. These structural proteins are important players in invasiveness and metastatic processes.46–51 Further studies are required to understand and fully characterize the intriguing mechanisms that shape and influence the cytoskeletal homeostasis of cancer cells induced by plecstatin-1. Finally, our work highlights another ruthenium compound that is able to induce an ICD signature in vitro with potential to be used in future in vivo experiments. It is of utmost urgency to precisely understand how plecstatin-1 and other metallodrugs may activate the immune system as an anticancer strategy, especially in times where immunotherapies regimens are increasingly applies in the treatment of different cancer types.Experimental
Cell culture
Human colon carcinoma cell lines HCT-116 and HT-29 were kindly provided by Brigitte Marian, Institute of Cancer Research, Department of Medicine I, Medical University of Vienna, Austria and HCT-15 was obtained from ATCC® (CCL-247™). HCT-116 and HT-29 cells were maintained in McCoy's 5a medium supplemented with 10% fetal calf serum (FCS) and L-glutamine. HCT-15 cells were grown in RPMI 1640 medium supplemented with 10% FCS and L-glutamine. All cells were cultured as adherent monolayers in 75 cm2 flasks and kept in a humidified incubator at 37 °C with 5% CO2. All cell culture media and reagents were obtained from Sigma-Aldrich (Vienna, Austria), and all plastic dishes, plates and flasks were from StarLab (Germany) unless stated otherwise.Spheroid formation
For spheroid generation, HCT-116, HT-29 and HCT-15 cells were harvested from culture flasks by trypsinization, resuspended in their respective supplemented medium and seeded in ultra-low attachment round-bottom 96-well plates (Nunclon SpheraTM, Thermo Fisher Scientific) at a density of 10.000 viable cells per well. Plates were incubated at 37 °C with 5% CO2 for 96 h to allow spheroid formation and then used for the experiments.Compounds
Plecstatin-1, oxaliplatin and cisplatin were synthesized at the Faculty of Chemistry, University of Vienna, as previously described.18,30–32Cytotoxicity test
Plectstatin-1 was first dissolved in DMSO, and stock solutions (800 μM) were prepared in the appropriate medium according to the cell line and diluted stepwise to obtain a serial dilution. 100 μL of the respective dilutions were added to each well, and the plates were incubated for 96 h at 37 °C with 5% CO2. All compound-solutions were freshly prepared before use. A 440 μM resazurin sodium salt (Sigma-Aldrich, Austria) solution in PBS solution was prepared and 20 μL were added to each well. The plates were incubated for 16 h at 37 °C with 5% CO2. Fluorescence was measured with a Synergy HT reader (BioTek).Treatment
Spheroids were incubated with the compounds for the respective number of hours indicated for each experiment at 37 °C with 5% CO2 and then processed for further experiments. In all experiments performed within this work, plecstatin-1 was given at a concentration of 200 μM unless stated otherwise. The concentrations were carefully selected based on the 50% (inhibitory concentrations IC50) obtained by the cytotoxicity test after 96 h of drug exposure.Colony formation assay
HCT-116 and HT-29 cells were harvested from culture flasks by trypsinization, resuspended in their respective supplemented medium and seeded in 6 well plates at a density of 3.000 viable cells per well. Plates were incubated at 37 °C with 5% CO2 for 24 h to allow proper cell attachment to the wells. Cells were treated and incubated with plecstatin-1 for 7 days. Plates were washed with PBS and fixed with methanol for 30 minutes at 4 °C. Cells were washed twice with PBS and stained with a crystal violet solution (Sigma Aldrich) for 5 minutes. Plates were washed with tap water to remove excess dye and air dried.Spheroid growth
HCT-116, HCT-15 and HT-29 spheroids were treated with plecstatin-1, oxaliplatin and cisplatin for 11 days. Pictures were taken before treatment and after treatment to compare growth among the different treatment groups. In the other experimental setup, the diameter of spheroids was measured before and after treatment (6 days) by using a CKX41 inverted microscope and Cell^F software (Olympus).Flow cytometry analysis of apoptosis
After treatment, spheroids were collected and pooled. Samples were washed with PBS followed by dissociation with Tryple Express (Gibco, Austria) for 15 minutes at 37 °C. 500 μL medium containing 10% FCS were added to each tube to stop trypsinization. Samples were centrifuged at 2200 g for 3 minutes and were incubated with anti-annexin-V FITC conjugated antibody (eBioscience) for 15 minutes followed by nuclei staining using propidium iodide (Sigma Aldrich) in annexin-V binding buffer solution. Samples were analysed immediately by using a Guava EasyCyte flow cytometer (Merck/Millipore). The results were analysed using the FlowJo software.Immunofluorescence staining
Spheroids were collected, pooled and embedded in TissueTek (Sakura, Japan). Samples were frozen at −80 °C until further processing. Samples were cut in 5 μm thick sections by using a Cryostat (Leica, Germany) and placed onto Superfrost slides (Thermo Fisher Scientific, Austria). For immunofluorescence staining, slides were brought to room temperature and fixed with a 0.25% PFA solution for 15 minutes followed by three washing steps with PBS. Samples were then permeabilized with a 0.1% Triton-X solution for 10 minutes for the following target/staining: Ki-67 (#9129, Cell Signaling Technology), cleaved caspase-3 (#9661, Cell Signaling Technology), cytochrome-c (#ab13575 Abcam), mitochondria (#ab92824, Abcam), plectin (#12254, Cell Signaling Technology), α-tubulin (#3873, Cell Signaling Technology), E-cadherin (#ab1416 Abcam), vimentin (#ab92547, Abcam), N-cadherin (#ab76057 Abcam), ZO-1 (#ab190085 Abcam), HMGB-1 (#3935, Cell Signaling Technology). In the case of Calreticulin (#ab2907 Abcam), HSP70 (#4873 Cell Signaling Technology), and HSP90 (#4877 Cell Signaling Technology), samples were not permeabilized. After additional washing steps, samples were incubated with the respective primary antibody at 4 °C overnight. Samples were washed with PBS and incubated with the secondary antibody: goat anti-rabbit Alexa Fluor 594 (#ab150080, Abcam), goat anti-mouse Alexa Fluor 488 (#ab150117, Abcam), goat anti-mouse Alexa Fluor 594 (#8890 Cell Signaling Technology), goat anti-rat Alexa Fluor 555 (#4417 Cell Signaling Technology) for 45 minutes at room temperature. For F-actin staining, slides were incubated with phalloidin Alexa Fluor 488 (#A12379, Molecular Probes). For calreticulin, HSP70 and HSP90, slides were incubated with a wheat germ agglutinin solution (Invitrogen) according to the manufacturer's instructions for 20 minutes and washed three times with PBS. Slides were mounted with ProlonGold Antifade Reagent with DAPI (Molecular Probes) and analysed with a confocal laser scanning microscope Zeiss LSM780. Images were processed with the Zen software (Zeiss).Detection of reactive oxygen species (ROS)
After treatment, spheroids were collected and pooled. Samples were washed with PBS followed by dissociation with Tryple Express (Gibco, Austria) for 15 minutes at 37 °C. 500 μL medium containing 10% FCS were added to each tube to stop trypsinization. Samples were centrifuged at 2200 g for 3 minutes and were incubated with Cellrox® (#C10492, Molecular Probes) dye according to manufacturer's instructions. Samples were analysed immediately by a Guava EasyCyte flow cytometer (Merck/Millipore). The results were analysed using the FlowJo software (Tree star). For the confocal imaging approach, spheroids were treated for 24 h with plecstatin-1 and labelled with Cellrox® (#C10444, Molecular Probes) and propidium iodide (PI) for 1 h. Spheroids were analysed with a confocal laser scanning microscope Zeiss LSM780. Images were processed with the Zen software Zeiss.Electrophoretic dsDNA plasmid assay
300 ng of the pUC19 plasmid (2686 bp), were incubated with the test compound (50 μM), for different time periods (15 min up to 6 h), with continuous shaking at 37 °C. Upon completion of the reaction. 4 μL of a 6× DNA loading dye were added to the 20 μL reaction solution. The DNA samples were loaded onto a 1% agarose gel (1% w/v in 1 × TBE) and subjected to electrophoresis, initially at 60 V for 5 min and subsequently at 120 V for 90 min, in 1 × TBE buffer. DNA visualisation was achieved by ethidium bromide (EtBr) staining of the agarose gel in 1 × TBE (0.75 μg mL−1) for 20 min. After electrophoresis, images were taken with the GelDoc-It Imaging System Fusion Fx7 (Vilber Lourmat, Germany).Western blotting
Spheroids were collected, pooled and washed three times with cold PBS and placed on ice. Cells were then lysed for 3 h with lysis buffer consisting of Cell Lytic M (Sigma-Aldrich), phosphatase inhibitor cocktail (Roche, Switzerland) and cOmplete (Roche, Switzerland). Lysates were centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 minutes at 4 °C, and supernatants were transferred to a fresh microfuge tube. For protein concentration measurements, 5 μL of sample were mixed with 1 ml Bradford solution (Biorad), and wave lengths were measured with a spectrophotometer at 590 nm. A WES Protein Simple System was used for protein detection according to manufacturer's instructions. The following antibodies were used: phospho-eIF-2alpha (#9721), eIF-2alpha (#5324), both from Cell Signaling Technologies, and GAPDH (#ab181602, Abcam) as loading control.
000g for 30 minutes at 4 °C, and supernatants were transferred to a fresh microfuge tube. For protein concentration measurements, 5 μL of sample were mixed with 1 ml Bradford solution (Biorad), and wave lengths were measured with a spectrophotometer at 590 nm. A WES Protein Simple System was used for protein detection according to manufacturer's instructions. The following antibodies were used: phospho-eIF-2alpha (#9721), eIF-2alpha (#5324), both from Cell Signaling Technologies, and GAPDH (#ab181602, Abcam) as loading control.
ATP measurements
Spheroids were treated for 24 h, and supernatants were collected and centrifuged at 5000g for 5 minutes. Supernatants were transferred into a white opaque flat-bottom 96-well culture plate (PerkinElmer), and 100 μL of CellTiter-Glo solution (Promega) were added to each well. Samples were incubated for 15 minutes at room temperature, and luminescence was recorded by a Synergy HT reader (BioTek).ELISA HMGB-1
Supernatants originated from spheroid cultures were collected after 72 h of drug-treatment centrifuged and stored at −20 °C until the measurements. Supernatants were brought to room temperature and were processed according to the manufacturer's instructions (Human HMGB1 ELISA Kit, FineTest). OD absorbance values were measured Synergy HT reader (BioTek) at 450 nm.LA-ICP-TOF-MS analysis
An Analyte Excite 193 nm ArF* excimer-based laser ablation system (Teledyne Photon Machines, Bozeman, MT, USA) was coupled to an icpTOF 2R (TOFWERK AG, Thun, Switzerland) TOF-based ICPMS instrument. The laser ablation system is equipped with the tube-type COBALT ablation cell and the aerosol rapid introduction system (ARIS), a low dispersion mixing bulb (developed at Ghent University and commercially available via Teledyne Photon Machines). The ARIS was used to introduce an Ar make-up gas flow (∼1.10 L min−1) into an optimized He carrier gas flow of 0.50 L min−1 before entering the plasma. The LA and ICP-TOFMS settings were optimized at the start of each experiment while ablating NIST SRM612 glass certified reference material (National Institute for Standards and Technology, Gaithersburg, MD, USA). Optimization was based on high intensities for 24Mg+, 89Y+, 115In+ and 238U+, low oxide formation based on the 238U16O+/238U+ ratio (<2%) and low laser-induced elemental fractionation based on the 238U+/232Th+ ratio (∼1). Laser ablation sampling was performed in fixed dosage mode 2, at a repetition rate of 100 Hz and using a square spot size with 5 μm in diameter. The line scans overlapped one another by 2.5 μm. Tumour spheroids were removed quantitatively using a fluence of 1.5 J cm−2. The icpTOF 2R ICP-TOFMS instrument has a specified mass resolution (R = m/Δm) of 6000 (FWHM definition) and allows the analysis of ions from m/z = 14–256. The integration and read-out rate match the laser ablation repetition rate. The instrument was equipped with a torch injector of 2.5 mm inner diameter and nickel sample and skimmer cones with a skimmer cone insert of 2.8 mm in diameter. A radio frequency power of 1400 W, an auxiliary Ar gas flow rate of ∼0.90 L min−1 and a plasma Ar gas flow rate of 15 L min−1 was used. Data were recorded using TofPilot 1.3.4.0 (TOFWERK AG, Thun, Switzerland). ICP-TOFMS data were saved in the open-source hierarchical data format (HDF5, http://www.hdfgroup.org). Post-acquisition data processing was performed with Tofware v3.1.0, which is a TOFWERK data analysis package and used as add-on on IgorPro (Wavemetric Inc., Oregon, USA). The data processing comprised the following steps: (1) drift correction of the mass peak position in the spectra over time via time-dependent mass calibration (2) determining the peak shape (3) fitting and subtracting the mass spectral base-line and (4) calculating high-resolution peak fits for peak deconvolution. LA-ICP-TOFMS data were further processed with HDIP version 1.2.6. beta11 (Teledyne Photon Machines, Bozeman, MT, USA).Invasion assay
Cells were grown for 4 days in MEM medium in 96-well plates (Nunclon Sphera, Thermo Fisher) and their size was measured using an inverted microscope (Olympus CKX41, 4× objective) and the Cell^F software. Spheroids of well comparable size (diameter: ca. 500 μm; matched pairs with differences in max. diameter <25 μm within each pair) were selected for the spheroid invasion assay. Growth factor reduced Matrigel (Corning) was diluted to a final concentration of 300 μg mL−1 with or without plecstatin-1/MEM. One hundred fifty microliter of these solutions were added to the wells containing spheroids in 50 μL medium. The plate was incubated in a humidified atmosphere with 5%CO2 at 37 °C. Both untreated and treated spheroids were monitored for 144 h. Diameters were measure before treatment (0 h) and at 72 h after treatment and are expressed in μm.Statistics
All the results originate from at least three technical and biological replicates. Tukey's range test was applied to analyse the apoptosis, ROS, HMGB-1 and ATP data.Conflicts of interest
There are no conflicts to declare.References
- J. Iida, E. T. Bell-Loncella, M. L. Purazo, Y. Lu, J. Dorchak, R. Clancy, J. Slavik, M. L. Cutler and C. Shriver, J. Transl. Med., 2016, 14, 48 CrossRef.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 17, 599–616 CrossRef.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS.
- C. Riccardi, D. Musumeci, M. Trifuoggi, C. Iraci, L. Paduano and D. Montesarchi, Pharmaceuticals, 2019, 12, 146 CrossRef CAS.
- L. S. Flocke, R. Trondl, M. A. Jakupec and B. K. Keppler, Invest. New Drugs, 2016, 34, 261–268 CrossRef CAS.
- E. Alessio and L. Messori, Molecules, 2019, 24, 10 CrossRef.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- H. A. Burris, S. Bakewell, J. C. Bendell, J. Infante, S. F. Jones, D. R. Spigel, G. J. Weiss, R. K. Ramanthan, A. Ogden and D. Von Hoff, ESMO Open, 2017, 1(6), 1–8 CrossRef.
- S. J. Bakewell, D. F. Rangel, D. P. Ha, J. Sethuraman, R. Crouse, E. Hadley, T. L. Costich, X. Zhou, P. Nichols and A. S. Lee, Europe PMC, 2018, 9(51), 29698–29714 Search PubMed.
- S. M. Meier, D. Kreutz, L. Winter, M. H. M. Klose, K. Cseh, T. Weiss, A. Bileck, B. Alte, J. C. Mader, S. Jana, A. Chatterjee, A. Chattacharyya, M. Hejl, M. A. Jakupec, P. Heffeter, W. Berger, C. H. Hartinger, B. K. Keppler, G. Wiche and C. Gerner, Angew. Chem., 2017, 56(28), 8267–8271 CrossRef CAS.
- Y. Wang, W. Qin, H. Shi, H. Chen, X. Chai, J. Liu, P. Zhang, Z. Li and Q. Zhang, Dalton Trans., 2020, 49, 972–976 RSC.
- D. Kreutz, C. Gerner and S. Meier-Menches, Metal-based Anticancer Agents, 2019, 246–270 CAS.
- J. Liu, H. Lai, Z. Xiong, B. Chen and T. Chen, Chem. Commun., 2019, 55, 9904–9914 RSC.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119(2), 1058–1137 CrossRef CAS.
- S. Jia, R. Wang, K. Wu, H. Jiang and Z. Du, Molecules, 2019, 24(3), 581 CrossRef.
- S. Carboni, A. Zucca, S. Stoccoro, L. Maiore, M. Arca, F. Ortu, C. Artner, B. Keppler, S. M. Meier-Menches, A. Casini and M. A. Cinellu, Inorg. Chem., 2018, 57(23), 14852–14865 CrossRef CAS.
- C. de Pascalis, C. Pérez-Gonzáles, S. Seetharaman, B. Boeda, B. Vianay, M. Burute, C. Leduc, N. Borghi, X. Trepat and S. Etienne-Manneville., J. Cell Biol., 2018, 217(9), 3031–3044 CrossRef CAS.
- T. C. Burch, M. T. Watson and J. O. Nyalwidhe, PLoS One, 2013, 8(5), 1–13 CrossRef.
- A. C. Raymond, B. Gao, L. Girard, J. D. Minna and D. Udugamasooriya, Sci. Rep., 2019, 9, 14954 CrossRef.
- G. Wiche and L. Winter, Bioarchitecture, 2011, 1(1), 14–20 CrossRef.
- S. M. Meier-Menches, K. Zappe, A. Bileck, D. Kreutz, A. Tahir, M. Cichna-Markl and C. Gerner, Metallomics, 2019, 11, 118–127 RSC.
- K. Pakos-Zebrucka, I. Koryga, K. Mnich, M. Ljujic, A. Samali and A. M. Gorman, EMBO Rep., 2016, 17, 1374–1395 CrossRef CAS.
- C. N. Spaan, W. L. Smith, J. F. van Lidth de Jeude, B. J. Meijer, V. Muncan, G. R. van den Brink and J. Heijmans, Cell Death Dis., 2019, 10, 490 CrossRef.
- L. Bezu, A. Sauvat, J. Humeau, L. C. Gomes-da-Silva, K. Iribarren, S. Forveille, P. Garcia, L. Zhao, P. Liu, L. Zitvogel, L. Senovilla, O. Kepp and G. Kroemer, Cell Death Differ., 2018, 8, 1375–1393 CrossRef.
- A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L. Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J.-P. Pignon, V. Jooste, P. van Endert, M. Ducreux, L. Zitvogel, F. Piard and G. Kroemer, Oncogene, 2010, 29(4), 482–491 CrossRef CAS.
- D. Wernitznig, K. Kiakos, G. Del Favero, N. Harrer, H. Machat, A. Osswald, M. A. Jakupec, A. Wernitznig, W. Sommegruber and B. K. Keppler, Metallomics, 2019, 11(6), 1044–1048 RSC.
- Q. Wang, X. Ju, J. Wang, Y. Fan, M. Ren and H. Zhang, Cancer Lett., 2018, 438, 17–23 CrossRef CAS.
- G. Kroemer, L. Galluzzi, O. Kepp and L. Zitvogel, Annu. Rev. Immunol., 2013, 31, 51–72 CrossRef CAS.
- L. Galluzzi, J. Humeau, A. Buqué, L. Zitvogel and G. Kroemer, Nat. Rev. Clin. Oncol., 2020 DOI:10.1038/s41571-020-0413-z.
- R. F. S. Lee, S. Theiner, A. Meibom, G. Koellensperger, B. K. Keppler and P. J. Dyson, Metallomics, 2017, 9, 365–381 RSC.
- I. Martins, O. Kepp, F. Schlemmer, S. Adjemian, M. Tailler, S. Shen, M. Michaud, L. Menger, A. Gdoura, N. Tajeddine, A. Tesniere, L. Zitvogel and G. Kroemer, Oncogene, 2010, 30, 1147–1158 CrossRef.
- L. Galluzzi, et al. , Immunother. Cancer, 2020, 8(1) DOI:10.1136/jitc-2019-000337.
- L. C. Crowley and N. J. Waterhouse, Cold Spring Harbor Protoc., 2016, 11 DOI:10.1101/pdb.prot087312.
- L. C. Crowley, M. E. Christensen and N. J. Waterhouse, Cold Spring Harbor Protoc., 2016, 8 DOI:10.1101/pdb.prot087171.
- G. Andocs, N. Meggyeshazi, L. Balogh, S. Spisak, M. E. Maros, P. Balla, G. Kiszner, I. Teleki, C. Kovago and T. Krenacs, Cell Stress Chaperones, 2015, 1, 37–46 CrossRef.
- K. Stock, M. F. Estrada, S. Vidic, K. Gjerde, A. Rudisch, V. E. Santo, M. Barbier, S. Blom, S. C. Arundkar, I. Selvam, A. Osswald, Y. Stein, S. Gruenewald, C. Brito, W. van Weerden, V. Rotter, E. Boghaert, W. Sommergruber, Y. Chong, R. de Hoogt and R. Graeser, Sci. Rep., 2016, 1(6), 28951 CrossRef.
- S. P. Rebelo, C. Pinto, T. R. Martins, N. Harrer, M. F. Estrada, P. Loza-Alvarez, J. Cabecadas, P. M. Alves, E. J. Gualda, W. Sommergruber and C. Brito, Biomaterials, 2018, 163, 185–197 CrossRef CAS.
- R. L. F. Amaral, M. Miranda, P. D. Marcato and K. Swiech, Front. Physiol., 2017, 8, 605 CrossRef.
- E. A. Slee, C. Adrain and S. J. Martin, J. Biol. Chem., 2001, 276, 7320–7326 CrossRef CAS.
- E. Schreiber-Brynzak, V. Pichler, P. Heffeter, B. Hanson, S. Theiner, I. Lichtscheidel-Schultz, C. Kornauth, L. Bamonti, V. Dhery, D. Groza, D. Berry, W. Berger, M. Galanski, M. A. Jakupec and B. K. Keppler, Metallomics, 2016, 8(4), 422–433 RSC.
- S. Theiner, E. Schreiber-Brynzak, M. A. Jakupec, M. Galanski, G. Koellensperger and B. K. Keppler, Metallomics, 2016, 8(4), 398–402 RSC.
- M. H. M. Klose, S. Theiner, C. Kornauth, S. M. Meier-Menches, P. Heffeter, W. Berger, G. Koellensperger and B. K. Keppler, Metallomics, 2018, 57(5), 2851–2864 Search PubMed.
- G. Wiche and L. Winter, Bioarchitecture, 2011, 1(1), 14–20 CrossRef.
- K. Andrä, B. Nikolic, M. Stöcher, D. Drenckhahn and G. Wiche, Genes Dev., 1998, 12(21), 3442–3451 CrossRef.
- A. C. Raymond, B. Gao, L. Girard, J. D. Minna and D. G. Udugamasooriya, Sci. Rep., 2019, 9, 14954 CrossRef.
- Y. Ni, X. Wang, X. Yin, X. Liu, H. Wang, X. Liu, J. Zhang, H. Gao, B. Shi and S. Zhao, J. Cell. Mol. Med., 2018, 22(11), 5450–5467 CrossRef CAS.
- M. Sutoh Yoneyama, S. Hatakeyama, T. Habuchi, T. Inoue, T. Nakamura, T. Funyu, G. Wiche, C. Ohyama and S. Tsuboi, Eur. J. Cell Biol., 2014, 93(4), 157–169 CrossRef CAS.
- I. Pastushenko and C. Blanpain, Trends Cell Biol., 2019, 29(3), 212–226 CrossRef CAS.
- X. Li, J. Strietz, A. Bleilevens, E. Stickeler and J. Maurer, Int. J. Mol. Sci., 2020, 21(2), 404 CrossRef CAS.
- Y. Zhang and R. A. Weinberg, Front. Med., 2018, 12(4), 361–373 CrossRef.
- F. Marcucci, G. Stassi and R. De Maria, Nat. Rev. Drug Discovery, 2016, 15(5), 311–325 CrossRef CAS.
- A. Dongre, M. Rashidian, F. Reinhardt, A. Bagnato, Z. Keckesova, H. L. Ploegh and R. A. Weinberg, Cancer Res., 2017, 77(15), 3982–3989 CrossRef CAS.
- C. Loh, J. Y. Chai, T. F. Tang, W. F. Wong, G. Sethi, M. K. Shanmugam, P. P. Chong and C. Y. Looi, Cells, 2019, 8(10), 1118 CrossRef CAS.
- E. Forte, I. Chimenti, P. Rosa, F. Angelini, F. Pagano, A. Calogero, A. Giacomelio and E. Messina, Cancers, 2017, 9(8), 98 CrossRef.
- M. P. Serres, M. Samwer, B. A. Truong Quang, G. Lavoie, U. Perera, D. Görlich, G. Charras, M. Petronczki, P. P. Roux and E. K. Paluch, Dev. Cell, 2020, 52(2), 210–222 CrossRef CAS.
- K. Strouhalova, M. Prechová, A. Gandalovicová, J. Brábek, M. Gregor and D. Rosel, Cancers, 2020, 12(1), 184 CrossRef CAS.
- S. Osmanagic-Myers, S. Rus, M. Wolfram, D. Brunner, W. H. Goldman, N. Bonakdar, I. Fischer, S. Reipert, A. Zuzuarregui, G. Walko and G. Wiche, J. Cell Sci., 2015, 128(22), 4138–4150 CrossRef CAS.
- J. Radons, Cell Stress Chaperones, 2016, 21(3), 379–404 CrossRef CAS.
- C. Boudesco, S. Cause, G. Jego and C. Garrido, Methods Mol. Biol., 2018, 1709, 371–396 CrossRef CAS.
- W. Y. Zhang, Q. Y. Yi, Y. J. Wang, F. Du, M. He, B. Tang, D. Wan, Y. J. Liu and H. L. Huang, Eur. J. Med. Chem., 2019, 10(151), 568–584 Search PubMed.
- M. Seervi, J. Joseph, P. K. Sobhan, B. C. Bhavya and T. R. Santhoshkumar, Cell Death Dis., 2011, 2(9) DOI:10.1038/cddis.2011.90.
- T. Kamogashira, K. Hayashi, C. Fujimoto, S. Iwasaki and T. Yamasoba, npj Aging Mech. Dis., 2017, 3, 2 CrossRef.
- T. Y. Na, L. Schecterson, A. M. Mendonsa and B. M. Gumbiner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(11), 5931–5937 CrossRef CAS.
- K. Rajesh, J. Krishnamoorthy, U. Kazimierczak, C. Tenkerian, A. I. Papadakis, S. Wang, S. Huang and A. E. Koromilas, Cell Death Dis., 2015, 6, 1591 CrossRef.
- C. D. Ochoa, R. F. Wu and L. S. Terada, Mol. Aspects Med., 2018, 63, 18–29 CrossRef CAS.
- Y. Lin, M. Jiang, W. Chen, T. Zhao and Y. Wei, Biomed. Pharmacother., 2019, 118 DOI:10.1016/j.biopha.2019.109249.
- Y. Chern, J. C. T. Wong, G. S. W. Cheng, A. Yu, Y. Yin, D. F. Schaeffer, H. F. Kennecke, G. Morin and I. T. Tai, Cell Death Dis., 2019, 10, 504 CrossRef.
- J. Humeau, J. Bezu, O. Kepp and G. Kroemer, Mol. Cell. Oncol., 2020, 7(5) DOI:10.1080/23723556.2020.1776570.
- P. Liu, L. Zhao, F. Loos, K. Iribarren, S. Lachkar, H. Zhou, L. C. Gomes-da-Silva, G. Chen, L. Bezu, G. Boncompain, F. Perez, L. Zitvogel, O. Kepp and G. Kroemer, Sci. Rep., 2017, 7, 14915 CrossRef.
- I. Martins, Y. Wang, M. Michaud, Y. Ma, A. Q. Sukkurwala, S. Shen, O. Kepp, D. Métivier, L. Galluzzi, J.-L. Perfettini, L. Zitvogel and G. Kroemer, Cell Death Differ., 2014, 21, 79–91 CrossRef CAS.
- P. de Andrade Mello, R. Coutinho-Silva and L. E. Baggio Savio, Front. Immunol., 2017, 8, 1526 CrossRef . Purinergic receptors.
- L. Feng, Y. Cai, M. Zhu, L. Xing and X. Wang, Cancer Cell Int., 2020, 20, 110 CrossRef CAS.
- F. Pietrocola, J. M. Bravo-San Pedro, L. Galluzzi and G. Kroemer, Autophagy, 2017, 13(12), 2163–2170 CrossRef CAS.
- L. T. Woods, K. J. Jasmer, K. M. Forti, V. C. Shanbhag, J. M. Camden, L. Erb, M. J. Petris and G. A. Weisman, Oral Oncol., 2020, 109 DOI:10.1016/j.oraloncology.2020.104808.
- M. Murata, P. Gong, K. Suzuki and S. Koizumi, J. Cell. Physiol., 1999, 180, 105–113 CrossRef CAS.
- V. R. Wiersma, M. Michalak, T. M. Abdullah, E. Bremer and P. Eggleton, Front. Oncol., 2015, 5, 7 Search PubMed.
- R. Azhwar, L. Panneerselvam, K. Sundarraj and E. Perumal, Heat Shock Proteins and Stress, 2018, pp. 39–78 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mt00227e |
| This journal is © The Royal Society of Chemistry 2020 |