
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[Re(η6-arene)2]+ as a highly stable ferrocene-like scaffold for ligands and complexes†
Daniel
Hernández-Valdés
a,
Frédéric
Avignon
ab,
Peter
Müller
a,
Giuseppe
Meola
a,
Benjamin
Probst
 a,
Thomas
Fox
a,
Bernhard
Spingler
a and
Roger
Alberto
*a
a,
Thomas
Fox
a,
Bernhard
Spingler
a and
Roger
Alberto
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstr. 190, CH-8057 Zurich, Switzerland. E-mail: ariel@chem.uzh.ch
bDépartement de Chimie, École Normale Supérieure, PSL Research University, 75005 Paris, France
First published on 2nd April 2020
Abstract
Ferrocenes are versatile ligand scaffolds, complexes of which have found numerous applications in catalysis. Structurally similar but of higher redox stabilites are sandwich complexes of the [Re(η6-arene)2]+ type. We report herein routes for conjugating potential ligands to a single or to both arenes in this scaffold. Since the arene rings can freely rotate, the [Re(η6-arene)2]+ has a high degree of structural flexibility. Polypyridyl ligands were successfully introduced. The coordination of Co(II) to such a model tetrapyridyl-Re(I)-bis-benzene complex produced a bimetallic Re(I)–Co(II) complex. To show the stability of the resulting architecture, a selected complex was subjected to photocatalytic reactions. It showed good activity in proton reduction over a long time and did not decompose, corroborating its extraordinary stability even under light irradiation. Its activity compares well with the parent catalyst in turn over numbers and frequencies. The supply of electrons limits catalytic turnover frequency at concentrations below ∼10 μM. We also show that other ligands can be introduced along these strategies. The great diversity offered by [Re(η6-arene)2]+ sandwich complexes from a synthetic point allows this concept to be extended to other catalytic processes, comparable to ferrocenes.
Introduction
Over the past decades, organometallic chemistry has found its way into many fields of science. Among the plethora of organometallic compounds, sandwich complexes have been studied in great depth in particular. Metallocenes are a focus (ferrocene), but arene sandwiches are less common and as such less studied. The first bis-arene sandwich, [Cr(η6-C6H6)2], was reported by Fischer and Hafner et al. in 1955 by reducing CrCl3 with Al/AlCl3 in benzene.1 Syntheses of bis-arene sandwich complexes are less straight than the ones of their cyclopentadienyl counterparts and the arene ligands are less stably bound to the metal centres due to their uncharged nature. Moreover, in many cases arene sandwich complexes are prone to oxidation and/or ligand loss.2 Little attention was payed to [Re(η6-C6R6)2]+ type complexes in particular. Although known since the Fifties,3 little respective chemistry was developed over the past fifty years. Progress was recently made by Kudinov et al. and in our group by developing a high-yield, one step procedures from [ReO4]− and Zn/AlCl3 as activation/reduction agents.4–6 Direct syntheses with functionalized arenes were reported as well, however, the high reactivity of AlCl3 limits the compatibility to some aniline-type arenes.7 Therefore, post-synthetic modifications are often required to introduce functionalities into the basic [Re(η6-C6H6)2]+ framework, a comparable procedure as done with ferrocene. Because of its stability and great versatility in terms of derivatization, ferrocene has become landmark in organometallic chemistry and has played a fundamental role in many areas of chemistry. Numerous examples of applications in asymmetric catalysis, sensors, electroactive materials and anti-cancer agents abound in literature.8–15 Developing similar concepts based on the [Re(η6-C6H6)2]+ scaffold opens up a whole range of opportunities that we can explore.In contrast to its neighbouring elements, we found that [Re(η6-C6H6)2]+ type complexes are remarkably stable.4,5,16 For instance, [Re(η6-C6H6)2]+ does not decompose or lose benzene rings even when heated to 160 °C or when irradiated with UV light. [Re(η6-C6R6)2]+ offers structural flexibilities in three dimensions as depicted in Scheme 1. Rotation around the rhenium axis, as well as tilting of pendent functionalities is possible without a notable energy barrier. The symmetry of all signals observed in the NMR spectra (see ESI†) supports the notion of free rotation at room temperature.5–7,16,17
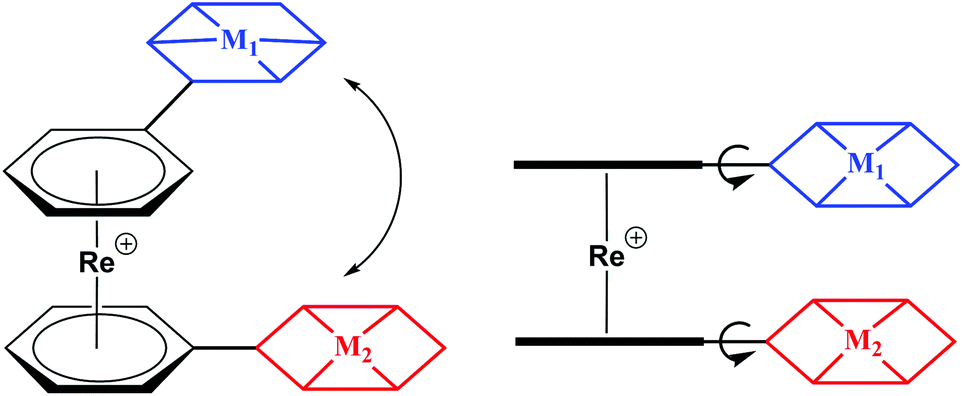 | ||
| Scheme 1 Different degrees of freedom for the basic arrangement of [Re(η6-C6H6)2]+ type complexes with two pendent coordinating modalities. | ||
Electrochemistry showed that Re(I) is reversibly oxidized at E°1/2 > +1.3 V (ReI/ReII). Irreversible reductions take place at low potentials <−2.0 V, both vs. Ag/AgCl.5,7 Compared to ferrocene, Re(I) bis-arene derivatives are much more difficult to oxidize and are generally stable against atmospheric oxygen (Fig. S1 and S2†).18–20 These properties make [Re(η6-C6H6)2]+ inert in several chemically interesting redox processes. A representative example is the reduction of H+ to H2 or CO2 to CO, both of which require substantial, negative potentials.21–26 Protons and/or CO2 can be reduced by molecular catalysts in systems that commonly involve multiple components; photosensitizers, electron donors and proton shuttles. The [Re(η6-C6H6)2]+ building block is an elegant way to combine two or more subunits in one molecule. Those subunits would be separated by a ring-ring distance of about 3 Å (Scheme 1), making communication between these two entities conjugated to the arene rings possible. This report shows that [Re(η6-C6H6)2]+ is suitable for conjugating different ligands to the arenes by common synthetic arene modification strategies. To show retention of catalytic activity and non-interference of the [Re(η6-C6H6)2]+ scaffold, the photocatalytic activity of a Re(I)–Co(II) heterobimetallic complex towards proton reduction is shown in detail and compared to the catalyst in its native form.
Results and discussion
Precursor syntheses
[Re(η6-C6H6)2](OTf) (1) was mono or bis-lithiated with LiN(i-Pr)2 (LDA) to form [Re(η6-C6H5Li)2]+. Reactions with electrophiles lead to e.g. the respective bromides and carboxylates.5 We extended the electrophiles to C2Cl6 or dimethylformamide (DMF) and prepared the mono-derivatized complexes [Re(η6-C6H6)(η6-C6H5Cl)](PF6) (2a+) and [Re(η6-C6H6)(η6-C6H5COH)](TFA) (3a+) or their bis-functionalized analogues [Re(η6-C6H5Cl)2](PF6) (2b+) and [Re(η6-C6H5COH)2](PF6) (3b+) (Scheme 2, “a” always denominates mono-functionalization). The ratio of mono- and bis-functionalization can be controlled by varying the amounts of LDA, but the selective formation of only one product could not be achieved. The reaction with DMF produces a mixture 3a+ and 3b+ as main products but also alcohols and carboxylic acids groups on the arene rings due to the high reactivity of these aldehydes. Since the solubilities of all products are very similar, clean separation of mono- and bis-aldehyde is challenging (full synthetic procedures and characterizations are given in the ESI†).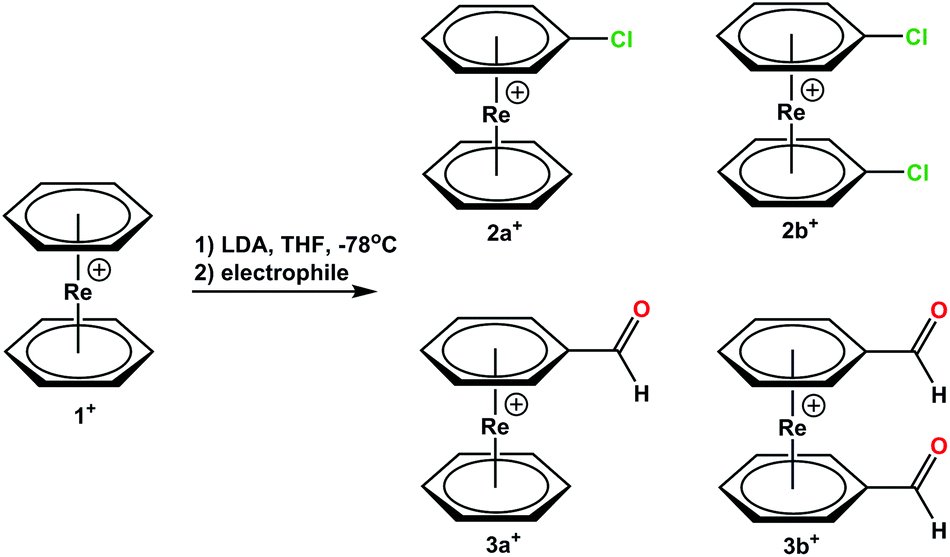 | ||
| Scheme 2 Reaction scheme for the functionalization of [Re(η6-C6H6)2](OTf). Reaction conditions: (1) Lithiation/deprotonation 1.5–2.5 eq. of LDA, 1.5 h, −78 °C, THF; (2) in situ electrophilic attack 5 h, −78 °C, THF; electrophiles: C2Cl6 and DMF. Combined yields: (63.1% for 2a+, 2b+ and 2c+; 43.2% for 3a+ and 3b+). | ||
Preparative HPLC with an isocratic gradient: 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95 CH3CN:H2O/TFA (0.1%) in 45 min, 40 mL min−1 leads to clean separation but UPLC-MS analysis evidenced that each fraction contained, besides 3a+ and 3b+, also the corresponding hydrate, supporting the high reactivity of the aldehyde groups attached to the electron poor arene rings. Obviously, there is an equilibrium established between these two species, which is more favourable for the aldehyde form.
:95 CH3CN:H2O/TFA (0.1%) in 45 min, 40 mL min−1 leads to clean separation but UPLC-MS analysis evidenced that each fraction contained, besides 3a+ and 3b+, also the corresponding hydrate, supporting the high reactivity of the aldehyde groups attached to the electron poor arene rings. Obviously, there is an equilibrium established between these two species, which is more favourable for the aldehyde form.
Cyclovoltammetry (CV) of 3a+ and 3b+ was performed to study electrochemical properties (Fig. 1). Cyclovoltammograms for complexes 3a+ and 3b+ show irreversible oxidations at +1.63 V and +1.7 V (vs. Ag/AgCl), respectively. These oxidations are anodically shifted between +0.3 and +0.4 V relative to 1+. The irreversible nature of the two oxidation processes in 3a+ suggests a ligand- and a metal-based oxidation, as observed for other sandwich complexes with functionalities bound to the arene rings.5,7 In 3b+ with two electron-withdrawing groups on the arenes, the metal-based oxidation is probably beyond the solvent window and only the oxidation of the aldehydes occurs.
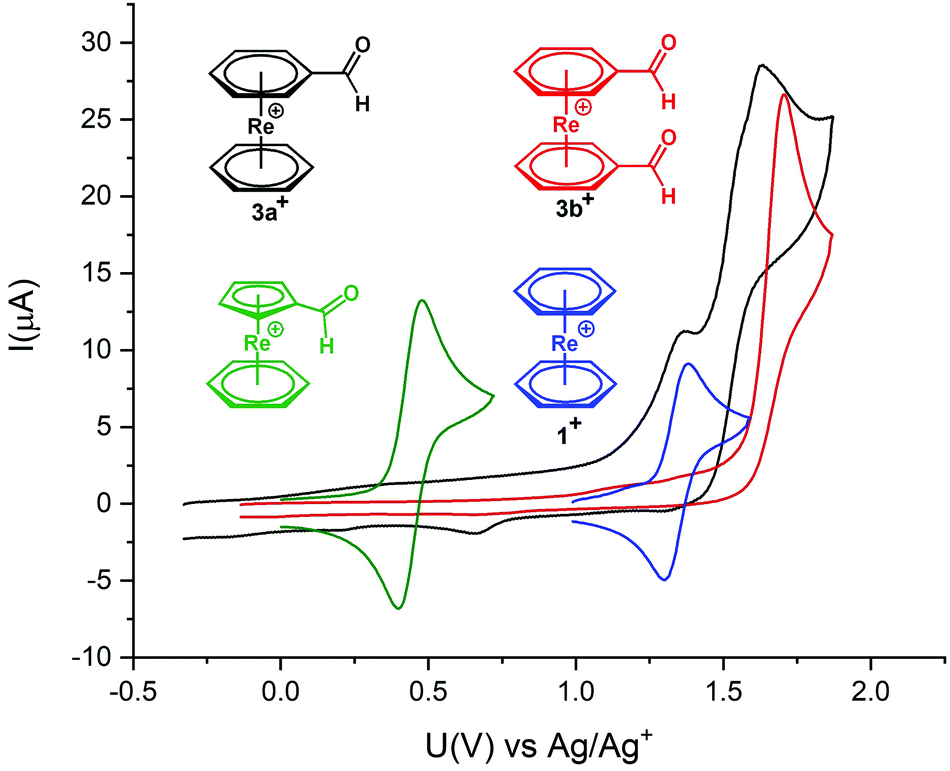 | ||
| Fig. 1 Cyclovoltammogram of complexes 1+ (blue), 3a+ (black), 3b+ (red) and mixed sandwich arene-Cp aldehyde (green); acetonitrile, 0.1 M [TBA][PF6] (electrolyte), glassy carbon working electrode (i.d. = 3 mm), Pt auxiliary electrode, and Ag/AgCl reference electrode, analyte concentrations 1 mM, voltage step: 6 mV, sweep rate: 0.1 V s−1. | ||
Meola et al. reported the mixed-ring complex [Re(η6-C6H6)(η5-C5H4COH)] which also comprises an aldehyde.17 It is interesting to compare the electrochemistry between this mixed-ring sandwich complex and 3a+, both containing an aldehyde function. For [Re(η6-C6H6)(η5-C5H4COH)], the reversible and thus metal-centred ReI/ReII oxidation takes place at +0.44 V (vs. Ag/AgCl) (Fig. 1). For 3a+ the oxidation is irreversible and the first oxidation is ligand- and not metal-centred and shifted by about 1 V.
Conjugation of ligands to complexes 2a+–3b+
The chloride and the aldehyde bearing arenes 2a+–3b+ are highly susceptible for nucleophilic attack due to the electron-withdrawing effect of the cationic rhenium centre. To exemplify the conjugation of additional ligands to the [Re(η6-arene)2]+ frameworks, appropriate aromatic heterocycles such as pyrrole or pyrazole were chosen for forming Carene–Nligand bonds to complex 1+. Pyrrole was deprotonated with NaH in dry THF.Nucleophilic substitution of Cl− in 2a+ gave [Re(η6-C6H6)(η6-C6H5-pyrrol)](PF6) (4a+) (Scheme 3) as the main product. The same procedure was applied to pyrazole, obtaining complex 5a+ [Re(η6-C6H6)(η6-C6H5-pyz)](PF6) in a 98% yield.
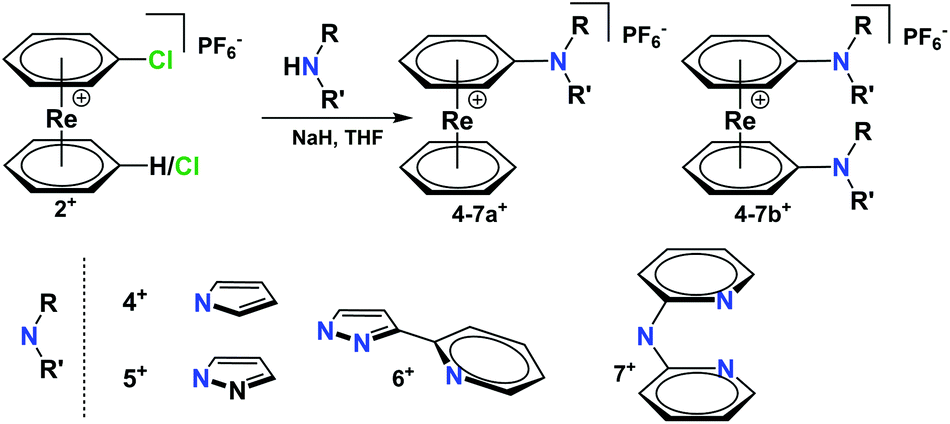 | ||
| Scheme 3 Reaction scheme for conjugation of five and six-membered N-heterocyclic rings to [Re(η6-C6H6)(η6-C6H5Cl)](PF6) and [Re(η6-C6H5Cl)2](PF6). Reaction conditions: (1) Deprotonation of the N-heterocyclic rings or 2,2′-dipyridylamine with NaH, 2 h, 60 °C, THF; (2) nucleophilic substitution 3 h, 60 °C, THF. | ||
Complex 5a+ carries a single sp2-N as a potential coordination site. To extend the synthetic strategy towards bi- and multidentate ligands, the reaction of 2b+ with 2-(1H-pyrazol-3-yl)pyridine (Hpyz-py) gave complex [Re(η6-C6H5-pyzpy)2](PF6) (6b+) in almost quantitative yield. Complex 6b+ has four nitrogen atoms for coordination to further metals centres. The crystal structure of 6b+ shows that the pyzpy units are coplanar (Fig. 2).
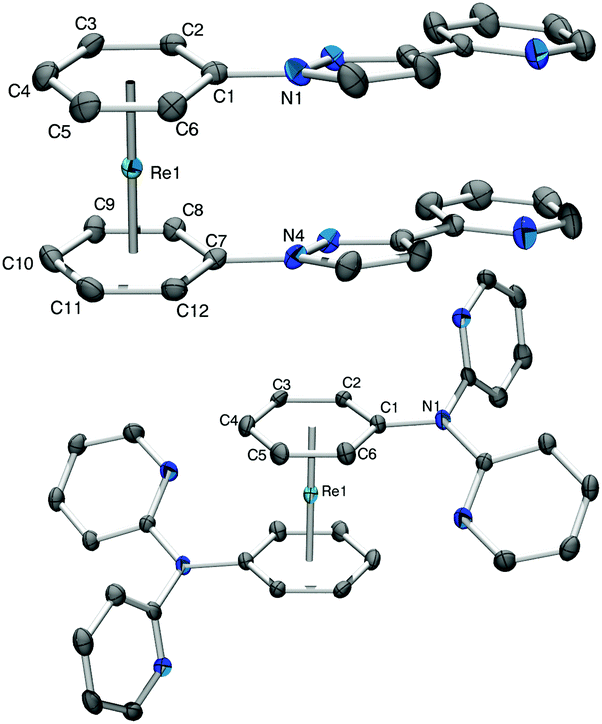 | ||
| Fig. 2 ORTEP representation of cations 6b+ (top) and 7b+ (bottom). Hydrogen atoms and anions have been omitted for clarity; thermal ellipsoids represent 50% probability. Selected bond lengths (Å) for 6b+: Re1–C1 2.276(2), Re1–C2 2.263(2), Re1–C3 2.246(2), Re1–C4 2.238(2), Re1–C5 2.231(2), R1–C6 2.237(2), Re1–C7 2.265(2), Re1–C8 2.260(2), Re1–C9 2.254(2), Re1–C10 2.237(2), Re1–C11 2.229(3), Re1–C12 2.240(2), C1–N1 1.400(3), C7–N4 1.405(3). For 7b+: Re1–C1 2.310(3), Re1–C2 2.237(3), Re1–C3 2.231(3), Re1–C4 2.241(3), Re1–C5 2.252(3), R1–C6 2.275(3), C1–N1 1.409(3). | ||
As obvious from the symmetry of the NMR signals, both rings in complex 6b+ rotate in solution, therefore, the attached ligands can coordinate either individually to two metal centres or via both pyzpy units to a single metal centre. The symmetry of all NMR spectra and therefore the free rotation around the rhenium axis is found for all [Re(arene)2]+ synthesized by our group so far.5–7,16,17,27
Pyridines are prototypical ligands in many catalytic processes, e.g. for proton reduction in photocatalysis.21,25 To extend the concept towards such polypyridyl ligands bound to the [Re(η6-C6H6)2]+ scaffold, 2,2′-dipyridylamine (Hdipyam) was deprotonated with NaH in dry THF and reacted with 2a+ and 2b+ respectively. The two complexes [Re(η6-C6H6)(η6-C6H5-dipyam)](PF6) (7a+) and [Re(η6-C6H5-dipyam)2](PF6) (7b+) were obtained as PF6− salt in 74% and 23.7% yield, respectively after separation by preparative HPLC and precipitation with NH4PF6. Single X-ray crystal structures of both complexes confirm their authenticities (see Fig. 2 and ESI†).
Compounds 7a+ and 7b+ are stable but light sensitive and decompose in acetonitrile solution by ligand loss. After exposing acetonitrile solutions of complexes 7a+ and 7b+ to light, UPLC-MS evidenced a new peak with a m/z of 248.11, corresponding to the protonated form of phenyl-di-pyridylamine (see ESI†).
A chemically different synthetic route to form ligands directly on the arene rings in [Re(η6-C6H6)2]+ starts from 3a+. Aldehydes are convenient functionalities for a number of reactions, yielding e.g. terpyridine in a Kröhnke reaction in methanol at r.t.28 Applying these conditions to the organometallic aldehyde 3a+, the complex [Re(η6-C6H5-terpy)(η6-C6H6)](PF6) (8a+) formed directly in the presence of acetylpyridine (Scheme 4). Precipitation with NH4PF6 gave complex 8a+ in good yields.
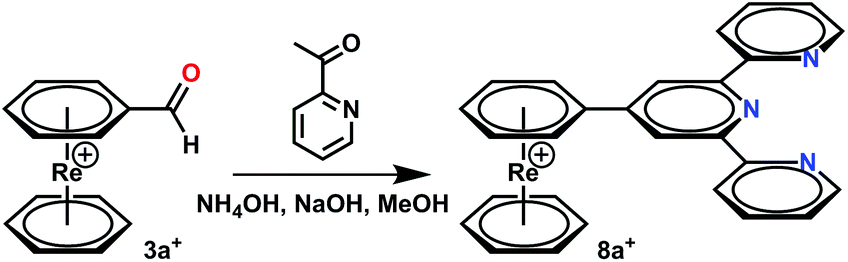 | ||
| Scheme 4 Reaction scheme for conjugation of terpyridine to [Re(η6-C6H5COH)(η6-C6H6)](TFA). Reaction conditions: Condensation reaction NaOH, 30 eq. acetylpyridine, 3 h, r.t., NH4OH solution (25% in mass). | ||
Ligand systems can also be directly conjugated to 1+ after lithiation as described before. For instance, mono- and bis-lithiated 1+ undergoes nucleophilic additions to carbonyl groups (Scheme 5). Several different keto-pyridines were employed in this way as electrophiles and products containing pendent ligands on one or both rings were isolated. Along this route, the mono and bis-substituted compounds 9a+/b+, 10a+/b+ and 11a+/b+ could be obtained and were fully characterized (see ESI†).
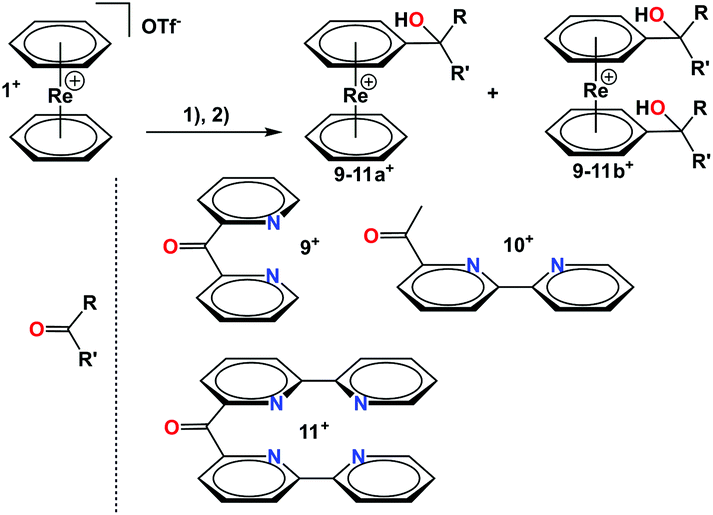 | ||
| Scheme 5 Reaction scheme for conjugation of polypyridyl ligands to [Re(η6-C6H6)2](OTf). Reaction conditions: (1) Lithiation/deprotonation 3.0 eq. of LDA, 1.5 h, −78 °C, THF; (2) addition to carbonyl 5 h, −78 °C, THF. | ||
In comparison with complexes 7a+ and 7b+, these new bis-arene based “ligands” are remarkably more stable; no decomposition was observed in the presence of light or coordinating solvents and they are all slightly water-soluble.
Synthesis of a bimetallic Re–Co catalyst
Compounds 11a+ and 11b+ are of special interest since Co(II) complexes of these tetra-pyridyl units represent a highly active class of catalysts for photocatalytic proton reduction.29–33 To support the hypothesis that the [Re(η6-C6H6)2]+ is a suitable scaffold, a Co(II) complex with 11a+ was prepared by reaction of 20 eq. CoBr2 in MeOH with the metal-free precursor. Complete complexation was attained after a few minutes according to UPLC-MS (see Fig. S5 and S6†). Washing the product multiple times with Et2O gave the pure product [12a]Br (Scheme 6).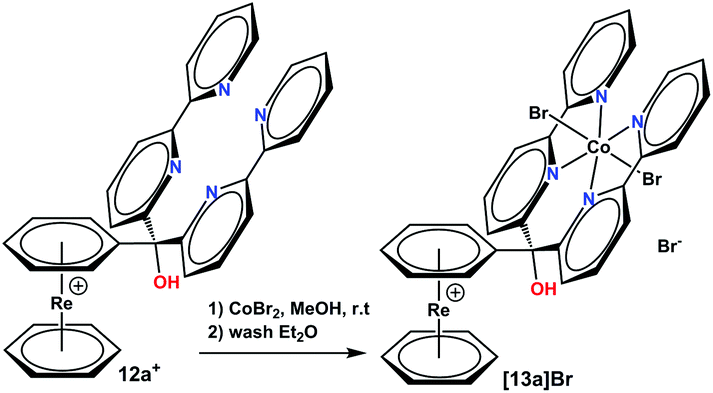 | ||
| Scheme 6 Synthesis of complex [12a]Br. Reaction conditions: (1) 20 eq. CoBr2, 1 h, r. t., in MeOH, (2) several washes with Et2O. | ||
The 1H NMR spectrum in deuterated methanol showed broad signals between −10 ppm and +80 ppm, in agreement with a paramagnetic nature of the complex. A single crystal X-ray structure analysis of 12a+ as trifluoromethanesulfonate (OTf) salt confirmed the formation of the Co(II) complex [12a]OTf (Fig. 3, full characterization in the ESI†).
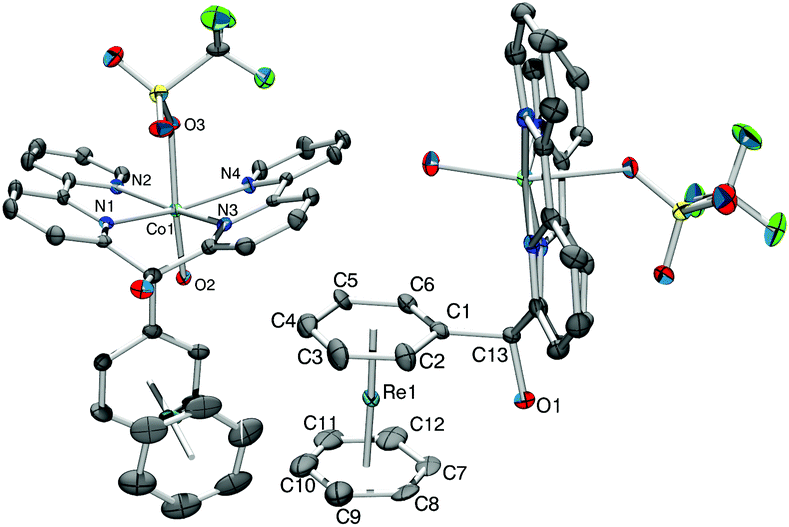 | ||
| Fig. 3 ORTEP representation of the crystal structure of [12a]OTf along two different views. Hydrogen atoms and two OTf-anions have been omitted for clarity; thermal ellipsoids represent 40% probability. Selected bond lengths (Å): Re1–C1 2.252(8), Re1–C2 2.235(9), Re1–C3 2.218(10), Re1–C4 2.244(10), Re1–C5 2.259(10), Re1–C6 2.228(9), Re1–C7 2.228(10), Re1–C8 2.198(10), Re1–C9 2.206(13), Re1–C10 2.206(13), Re1–C11 2.238(11), Re1–C12 2.226(12), C1–C13 1.550(11), N1–C01 2.100(7), N–C01 2.108(7), N3–C01 2.103(7), N4–C01 2.109(7), Co1–O2 2.064(6), Co1–O3 2.279(6). | ||
The crystal structure of complex [12a]OTf shows a strong Jahn–Teller distortion at the cobalt centre. The distance Co–O(H2O) and Co–O(OTf) are 2.064(6) Å and 2.279(6) Å, respectively and the angle O–Co–O is 165°. Additionally, the two bipyridyl subunits are bent in a book-shaped fashion and out of the equatorial coordination plane by around 22°. The tetrapyridyl Co(II) moiety orients in an almost perpendicular fashion to the bis-benzene subunit.
Complex [12a]Br is photostable. Irradiation of a 20 μM solution in water with LED light at 453 nm (photon flux of 0.35 ± 0.02 μE s−1) did not show any change in the UV spectrum after 8 h. After 24 h, minimal decomposition probably related to Co(II) decomplexation was observed (Fig. S20†). In addition, the UV/vis spectrum of complex [12a]Br did not change down to pH = 1.97 (Fig. S21†). Continuous additions of NaOH to a 20 μM solution lead though to irreversible changes in the UV/vis spectrum, indicating decomposition under strongly alkaline conditions (Fig. S22†).
Photocatalysis
Since electronically decoupled, the pendent [Re(η6-arene)2]+ unit in 12a+ should not influence the activity of the catalyst. Accordingly, 12a+ was subjected to photocatalysis with [Ru(bpy)3]2+ as photosensitizer (PS) and ascorbate as the sacrificial electron donor (SED) (Scheme S1†). To regenerate dehydroascorbic acid (DHA), the oxidized form of ascorbate, tris(2-carboxyethyl)phosphine (TCEP) was added.29–31,34 A comparison with the parent catalyst [Co(Phbpy2)Br2] (13), i.e.12a+ without [Re(η6-arene)2]+, was performed for assessing its innocence (Fig. 4 and Fig. S23†).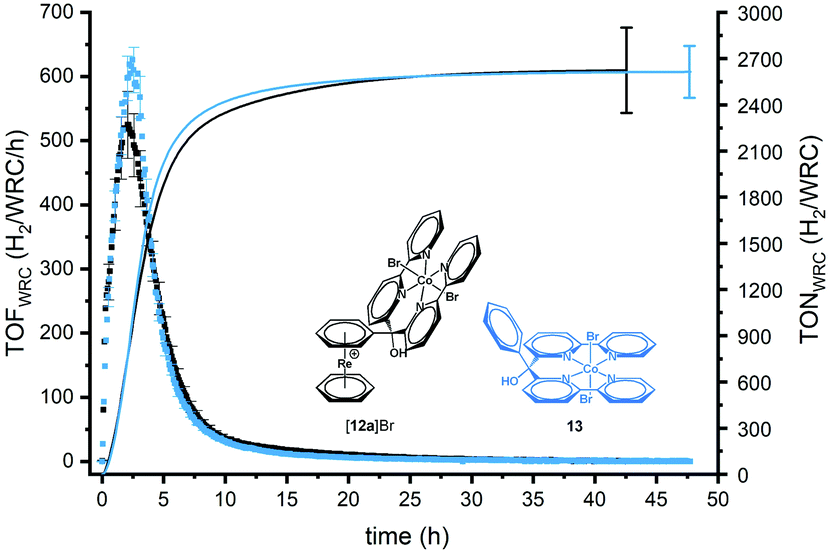 | ||
| Fig. 4 Photocatalysis experiment with [12a]Br in black and the parent catalyst 13 in blue. TOF (dots) (H2/Co/h) and TON (lines) (H2/Co) formed as a function of time for 5 μM catalyst. Experimental conditions: 10 mL solution, 0.5 mM [Ru(bpy)3]Cl2, 0.1 M NaAsc, 0.1 M TCEP, pH 5.0, 453 nm LED irradiation. | ||
Accordingly, photocatalytic proton reduction experiments were performed with [12a]Br in water at pH 5, 0.1 M TCEP, 0.1 M NaAsc and 0.5 mM PS at five different concentrations of catalyst (1 μM, 5 μM, 10 μM, 20 μM, 100 μM). H2 evolution was continuously monitored by in-line gas chromatography. Blank experiments in the absence of catalyst did not show significant amounts of H2 formation in comparison to experiments with catalyst. Mercury poisoning experiments excluded the formation of nanoparticles as catalytically active species (Fig. S24†).
Fig. 4 shows a typical H2 production profile as a function of time for 5 μM catalyst (for all concentrations, see Fig. S25†). The rate of H2 evolution increases rapidly with time, reaching a maximum after around 2 h (for 5 μM of catalyst). Then, the H2 formation rate decreases and catalysis ceases after 10 h. Depending on the WRC concentration, the periods of catalytic activities as well as the time needed for reaching the maximum turnover frequency (TOF) change. At higher initial concentrations of WRC, a delay until maximum TOF as well as longer catalysis time were observed, in line with previous studies by Joliat-Wick et al.29
Varying the concentrations of the WRC or the light intensity under otherwise identical condition are useful experiments to determine rate-limiting factors. As shown in Fig. 5a, the maximum TOF of H2 formation stays about constant at around 520 h−1 for concentrations <10 μM, indicating either electron transfer to or turnover of complex [12a]Br as rate limiting.
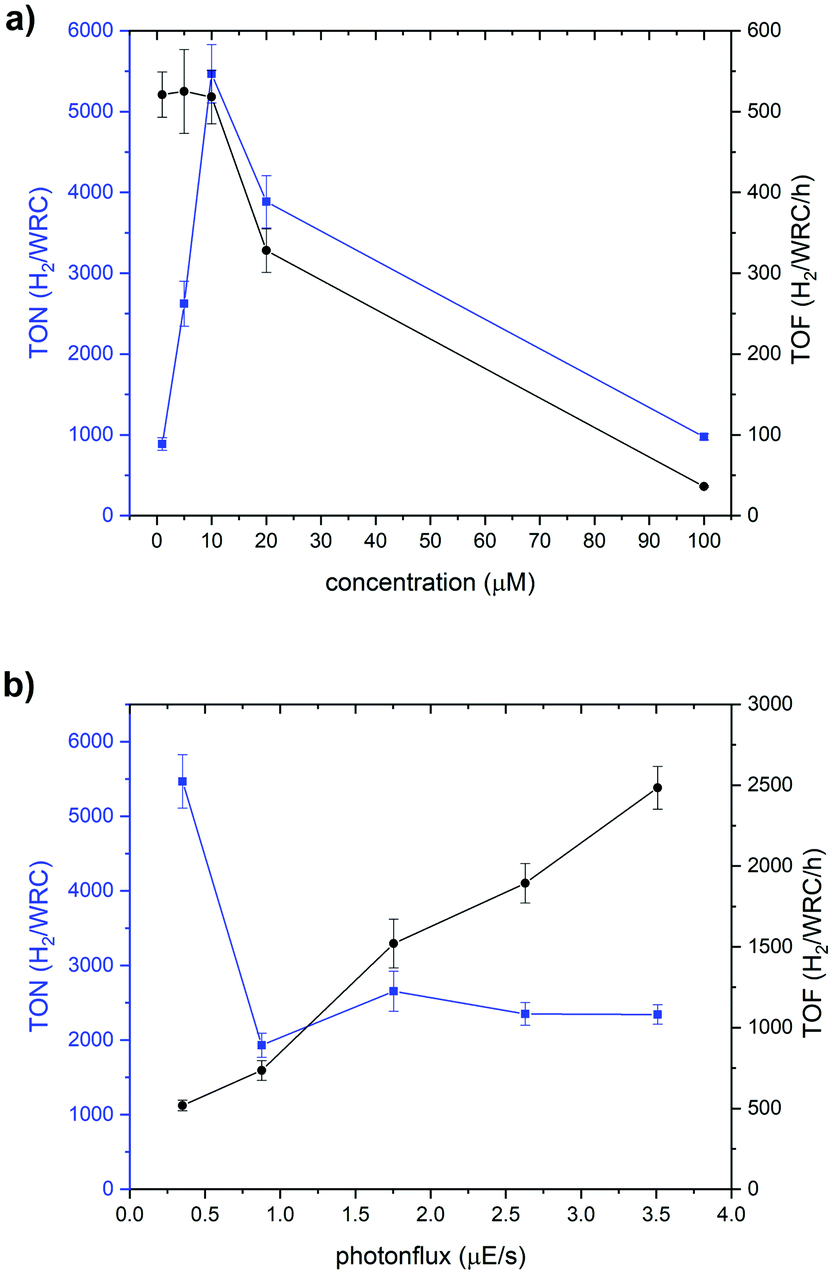 | ||
| Fig. 5 TON and maximum TOF for H2 evolution vs. (a) WRC concentration at a constant photon flux of 0.35 ± 0.02 μE s−1. (b) photonflux with 10 μM of WRC. Experimental conditions: 10 mL of solution, 0.500 mM [Ru(bpy)3]Cl2, 0.1 M NaAscO, 0.1 M TCEP, pH 5.0, 453 nm LED irradiation. Black circles denote the maximum TOF of H2 production and blue squares the turnover numbers. | ||
For concentrations above 10 μM, the TOFs decrease with increasing concentrations of WRC. At 20 μM and 100 μM, the maximum TOFs drop to 320 and 36 h−1 respectively. This effect is related to a limitation in the photocycle. At concentrations higher than 20 μM, the SED is almost completely consumed and for 100 μM in WRC, catalysis stops sharply after 70 h (Fig. S25b†) since no SED is available anymore. The limitation of the photocycle, i.e. the number of photons per time, can be shown by increasing the photon flux while keeping WRC concentrations constant. As obvious from Fig. 5b, TOFs increase linearly with increasing light intensity at 10 μM in WRC. A TOF of around 2500 h−1 is obtained upon irradiation of the sample with 3.5 μE s−1, corroborating the limitations by the photocycle. Increased light intensities result in shorter but faster catalyses, overall with reduced TONs since higher photon fluxes favour the decomposition of the PS−. All together, these observations support the view that the supply of electrons limits catalytic turnover frequency at concentrations below 10 μM and that the total photon flux limits it at higher concentrations. No negative interference with the [Re(η6-arene)2]+ scaffold on the catalytic activity is observed as similar performances have been obtained for catalysts purely based on cobalt polypyridyl complexes.29–31 Quantum yields were calculated from the maximum rate of H2 formation and the number of absorbed photons. Two photons are required for one H2 molecule. The quantum yields are between 1 and 10%, reaching the maximum at 20 μM in catalyst and 0.35 μE s−1 photonflux. Although catalyst 12a+ is relatively slow at low concentrations, the TONs and TOFs at higher concentrations are comparable with those of other systems with tetrapyridyl ligands.30
Conclusions
The sandwich complex [Re(η6-arene)2]+ represents a structural architecture to which additional functionalities such as ligands are conjugated in straight synthetic approaches. Additional reactive functions such as chlorides or aldehydes are introduced first on the arene rings, followed by ligand syntheses with these functions or, optionally, by direct coupling of intact ligands to the arenes. Thus, the respective opportunities provided by ferrocene are extended to [Re(η6-arene)2]+ scaffolds. The sandwich complex [Re(η6-arene)2]+ is cationic, which supports water solubility, and highly stable against redox reactions. Photocatalysis for H2 formation corroborates the light stability of the entity and comparison with the parent catalyst, i.e. catalyst without the [Re(η6-arene)2]+ unit, confirms its purely structural role. We emphasize that the options offered by the [Re(η6-C6H6)2]+ scaffold will go beyond the photocatalysis presented in this work. The high flexibility of the ligands to be conjugated can be equally exploited for other applications. Synergistic effects and communications between different modalities conjugated to the [Re(η6-C6H6)2]+ scaffold is also anticipated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the University of Zurich, University Research Priority Project “LightChEC”, and the Joint South Africa Switzerland Research Project IZLSZ2_170856 from the Swiss National Science Foundation.Notes and references
- E. O. Fischer and W. Hafner, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1955, 10, 665–668 Search PubMed.
- G. Pampaloni, Coord. Chem. Rev., 2010, 254, 402–419 CrossRef CAS.
- E. O. Fischer and A. Wirzmüller, Chem. Ber., 1957, 90, 1725–1730 CrossRef CAS.
- E. A. Trifonova, D. S. Perekalin, K. A. Lyssenko and A. R. Kudinov, J. Organomet. Chem., 2013, 727, 60–63 CrossRef CAS.
- G. Meola, H. Braband, P. Schmutz, M. Benz, B. Spingler and R. Alberto, Inorg. Chem., 2016, 55, 11131–11139 CrossRef CAS PubMed.
- M. Benz, H. Braband, P. Schmutz, J. Halter and R. Alberto, Chem. Sci., 2015, 6, 165–169 RSC.
- D. Hernández-Valdés, G. Meola, H. Braband, B. Spingler and R. Alberto, Organometallics, 2018, 37, 2910–2916 CrossRef.
- A. Togni, Angew. Chem., Int. Ed., 1996, 35, 1475–1477 CrossRef CAS.
- F. Rebière, O. Riant, L. Ricard and H. B. Kagan, Angew. Chem., Int. Ed., 1993, 32, 568–570 CrossRef.
- T. J. Colacot, Chem. Rev., 2003, 103, 3101–3118 CrossRef CAS PubMed.
- C. Ornelas, New J. Chem., 2011, 35, 1973–1985 RSC.
- R. Djeda, A. Rapakousiou, L. Liang, N. Guidolin, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2010, 49, 8152–8156 CrossRef CAS PubMed.
- H. Yang, Z. Zhou, K. Huang, M. Yu, F. Li, T. Yi and C. Huang, Org. Lett., 2007, 9, 4729–4732 CrossRef CAS PubMed.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 2017, 6–29 CrossRef CAS.
- D. R. Van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5986 CrossRef CAS PubMed.
- G. Meola, H. Braband, S. Jordi, T. Fox, O. Blacque, B. Spingler and R. Alberto, Dalton Trans., 2017, 46, 14631–14637 RSC.
- G. Meola, H. Braband, D. Hernández-Valdés, C. Gotzmann, T. Fox, B. Spingler and R. Alberto, Inorg. Chem., 2017, 56, 6297–6301 CrossRef CAS PubMed.
- S. M. Batterjee, M. Marzouk, M. Aazab and M. El-Hashash, Appl. Organomet. Chem., 2003, 17, 291–297 CrossRef CAS.
- M. Emilia, N. Silva, A. J. Pombeiro, J. J. F. da Silva, R. Herrmann, N. Deus, T. J. Castilho and M. F. C. Silva, J. Organomet. Chem., 1991, 421, 75–90 CrossRef.
- M. E. N. Silva, A. J. Pombeiro, J. J. F. da Silva, R. Herrmann, N. Deus and R. E. Bozak, J. Organomet. Chem., 1994, 480, 81–90 CrossRef CAS.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3–19 CrossRef CAS PubMed.
- C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- Q. Nadeem, G. Meola, H. Braband, R. Bolliger, O. Blacque, D. Hernández-Valdés and R. Alberto, Angew. Chem., Int. Ed., 2020, 59, 1197–1200 CrossRef CAS PubMed.
- F. Kröhnke, W. Zecher, J. Curtze, D. Drechsler, K. Pfleghar, K. E. Schnalke and W. Weis, Angew. Chem., Int. Ed., 1962, 1, 626–632 CrossRef.
- E. Joliat-Wick, N. Weder, D. Klose, C. Bachmann, B. Spingler, B. Probst and R. Alberto, Inorg. Chem., 2018, 57, 1651–1655 CrossRef CAS PubMed.
- S. Schnidrig, C. Bachmann, P. Müller, N. Weder, B. Spingler, E. Joliat-Wick, M. Mosberger, J. Windisch, R. Alberto and B. Probst, ChemSusChem, 2017, 10, 4570–4580 CrossRef CAS PubMed.
- E. Joliat, S. Schnidrig, B. Probst, C. Bachmann, B. Spingler, K. K. Baldridge, F. von Rohr, A. Schilling and R. Alberto, Dalton Trans., 2016, 45, 1737–1745 RSC.
- M. Guttentag, A. Rodenberg, C. Bachmann, A. Senn, P. Hamm and R. Alberto, Dalton Trans., 2013, 42, 334–337 RSC.
- C. Bachmann, M. Guttentag, B. Spingler and R. Alberto, Inorg. Chem., 2013, 52, 6055–6061 CrossRef CAS PubMed.
- C. Bachmann, B. Probst, M. Guttentag and R. Alberto, Chem. Commun., 2014, 50, 6737–6739 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1957333–1957341. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt00731e |
| This journal is © The Royal Society of Chemistry 2020 |