
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate substitution effects in the Fries rearrangement of aryl esters over zeolite catalysts†
Ronghe
Lin
a,
Sharon
Mitchell
 *a,
Thomas
Netscher
b,
Jonathan
Medlock
b,
René T.
Stemmler
b,
Werner
Bonrath
b,
Ulla
Létinois
b and
Javier
Pérez-Ramírez
*a
*a,
Thomas
Netscher
b,
Jonathan
Medlock
b,
René T.
Stemmler
b,
Werner
Bonrath
b,
Ulla
Létinois
b and
Javier
Pérez-Ramírez
*a
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, Zurich 8093, Switzerland. E-mail: msharon@chem.ethz.ch; jpr@chem.ethz.ch
bDSM Nutritional Products, Research and Development, PO Box 2676, Basel 4002, Switzerland
First published on 3rd June 2020
Abstract
The catalytic transformation of aryl esters to hydroxyacetophenones via Fries rearrangement over solid acids is of interest to avoid the use of corrosive and toxic Lewis and Brønsted acids traditionally applied. Microporous zeolites are known to catalyze the reaction of simple substrates such as phenyl acetate, but their application to substituted derivatives has received limited attention. To refine structure–activity relationships, here we examine the impact of various parameters including the solvent polarity, water content, acidic properties, and framework type on the reaction scheme in the Fries rearrangement of p-tolyl acetate over common solid acids. The results confirm the importance of providing a high concentration of accessible Brønsted acid sites, with beta zeolites exhibiting the best performance. Extension of the substrate scope by substituting methyl groups in multiple positions identifies a framework-dependent effect on the rearrangement chemistry and highlights the potential for the transformation of dimethylphenyl acetates. Kinetic studies show that the major competitive path of cleavage of the ester C–O bond usually occurs in parallel to the Fries rearrangement. The possibility of sequentially acylating the resulting phenol depends on the substrate and reaction conditions.
Introduction
The Fries rearrangement of aryl esters is a robust method for the synthesis of hydroxyaryl ketones, which are important building blocks and intermediates in the manufacture of fine chemicals and pharmaceuticals.1–4 This reaction is traditionally thermally-driven with the use of Lewis acids such as AlCl3, TiCl4, or BF3, metal triflates (e.g., Bi(OTf)3) or strong protic acids (HF or MeSO3H). In addition to being corrosive, the homogeneous catalytic systems are often consumed in a stoichiometric non-recyclable manner, generating substantial amounts of waste. To identify more sustainable alternatives, various heterogeneous catalysts have been actively explored in this reaction.5–19Zeolites have long attracted interest for selective organic syntheses because of their characteristic acidity, potential for confinement effects, and good thermal stability.20–25 Under appropriate conditions, they have been shown to present a viable alternative for conventional catalysts in the Fries rearrangement of simple substrates,5–11 with most previous studies focusing on phenyl acetate. In general, liquid-phase tests result in improved selectivity over gas phase reactions, and are typically conducted at 373–463 K with batch operation preferred over continuous.8 ZSM-5 (MFI) and beta (BEA) zeolites have been most widely studied. MFI is known to exhibit higher selectivity to para-hydroxyacetophenone.5–8 Comparatively, large-pore BEA was suggested to be more active in the rearrangement of bulkier phenyl benzoate and p-tolyl acetate.8,10 The dependence on the acidic properties is less clear. A linear correlation was reported between the conversion of phenyl acetate and the degree of ion exchange of protonic sites with alkali metals.11 In contrast, other studies found that the conversion was independent of the Si/Al ratio of BEA zeolites.8,26
The large number of products formed in the transformation of phenyl acetate over solid acids poses a major challenge for understanding the influence of distinct catalyst properties on the reaction scheme. Comparatively, the rearrangement of p-tolyl acetate is less complex (Scheme 1) and thus, can be valuable for gaining insight into the kinetics of the distinct reaction paths. In addition to the intramolecular Fries rearrangement, hydroxyacetophenones can form via an intermolecular reaction of the aryl ester with phenols. The latter often comprise a major side product of the reaction and are formed by the competitive cleavage of the ester bond. Previous studies of the rearrangement of phenyl acetate identified various factors that minimize the selectivity to phenols. The use of MFI zeolites was shown to yield lower amounts of phenol than BEA zeolites.8 The selectivity to rearrangement products over NU-2 zeolites was enhanced when the catalyst was dehydrated before the reaction, pointing to a link between phenol formation and the adventitious presence of water.6 The choice of solvent also had a pronounced effect on the product distribution, with polar solvents promoting the intermolecular reaction on BEA zeolites.7,9 In all of the above interconversions, appreciable rates of catalyst deactivation have been reported due to coking or inhibition by phenolic compounds.7,8,27 The formation of these polymeric deposits stems from the presence of highly reactive ketenes, formed by the dissociation of acetylium species from protonated phenyl acetate and their subsequent decomposition.8,27
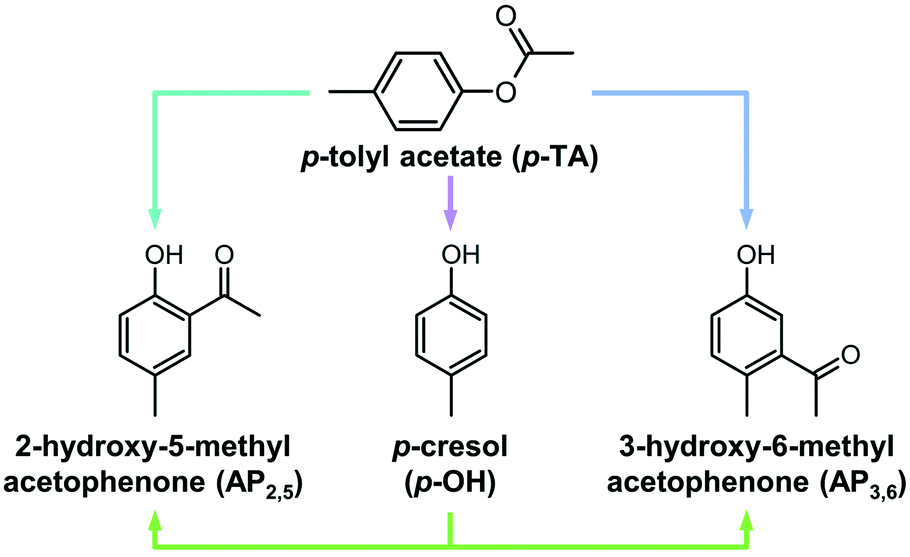 | ||
| Scheme 1 Possible routes to form 2-hydroxy-5-methylacetophenone (AP2,5) or 3-hydroxyl-6-methylacetophenone (AP3,6) from p-tolyl acetate (p-TA); the direct Fries rearrangement (blue arrows) or the sequential cleavage of the ester C–O bond (purple arrow) to p-cresol (p-OH) followed by C-acylation (green arrows). | ||
In contrast to other acid-catalysed reactions over zeolite catalysts, in which precise structure–activity relationships have been established, for example with the concentrations and strengths of distinct acid site and the porous properties,28–34 understanding of the Fries rearrangement remains limited. Currently, it is unclear where the reaction occurs on acid sites located within the micropores or on the external (non-microporous) surface of the zeolite crystal. Furthermore, the scarce exploration of substituted aryl esters relevant for the elaboration of complex intermediates in fine-chemical syntheses prohibits the generalization of the trends evidenced for phenyl acetate. In this study, we compare the scope of zeolite catalysts of distinct framework type for the Fries rearrangement of a broad set of methyl-substituted derivatives. To improve understanding, the influence of the reaction conditions (temperature, solvent), type of solid acid, and the associated concentration and strength of Brønsted acid sites and micropore diameter is examined in the transformation of p-tolyl acetate. A detailed analysis of the reaction scheme is undertaken over the best-performing beta zeolites to identify properties that influence acetophenone and p-cresol formation as well as catalyst deactivation. Comparison of the substrate scope of the distinct zeolite frameworks demonstrates the potential of beta zeolites in the Fries rearrangement of dimethylphenylacetates. The effects of substitution of the aryl esters are discussed with respect to the accessibility of the active sites within the zeolite micropores.
Materials
Physicochemical properties of the commercial zeolites and mesoporous materials studied in this work are listed in Table 1 and the XRD patterns and gas sorption isotherms are provided in Fig. S1 and S2.† Throughout the manuscript, zeolite samples are denoted by their framework code and nominal Si/Al ratio. All zeolites were calcined in static air at 823 K (heating rate = 5 K min−1) for 5 h prior to catalytic tests. Mesoporous MCM-41 was evaluated as received. Several other solid acids were synthesized for reference using ZrO2 as a carrier, including boron trioxide (B2O3/ZrO2), tungsten oxide (WO3/ZrO2) and phosphotungstic acid (HPA/ZrO2). The synthesis protocols of these reference materials are provided in Method S1.†| Catalyst | Commercial namea | V total (cm3 g−1) | V micro (cm3 g−1) | S BET (m2 g−1) | S meso (m2 g−1) | c Brønsted,Pyr (μmol g−1) | c Lewis,Pyr (μmol g−1) | T C3H6 (K) | c Brønsted,TPSR (μmolC3H6 g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a The cationic form of the as-received zeolites is indicated in parenthesis. b Determined from the amount of N2 adsorbed at p/p0 = 0.97. c BET method. d T-Plot method. e Concentrations of Brønsted and Lewis acid sites derived from pyridine-IR. f Temperature of the first desorption peak of propene and. g Concentration of Brønsted acid sites derived by TPSR of n-propylamine. | |||||||||
| FER(10) | Zeol CP914C (NH4+) | 0.18 | 0.14 | 295 | 24 | — | — | 661 | 1447 |
| MFI(15) | Zeolyst CBV3024E (NH4+) | 0.24 | 0.13 | 345 | 93 | 296 | 42 | 652 | 578 |
| BEA(12) | Zeolyst CP814E (NH4+) | 0.73 | 0.18 | 518 | 176 | 196 | 98 | 684 | 865 |
| BEA(15) | Clariant CZB30 (H+) | 0.36 | 0.22 | 546 | 118 | 150 | 85 | 653 | 459 |
| BEA(90) | Clariant CZB150 (H+) | 0.49 | 0.20 | 516 | 126 | 66 | 8 | 701 | 321 |
| BEA(220) | Tosoh HSZ-980HOA (H+) | 0.32 | 0.19 | 486 | 125 | 22 | 4 | 695 | 173 |
| MOR(10) | Zeolyst CBV21A (NH4+) | 0.25 | 0.18 | 381 | 35 | 152 | 27 | 678 | 390 |
| FAU(15) | Zeolyst CBV720 (H+) | 0.49 | 0.25 | 670 | 186 | 220 | 69 | 650 | 403 |
| MCM-41 | Sigma-Aldrich | 1.10 | 0.00 | 990 | 861 | — | — | 672 | 149 |
| WO3/ZrO2 | Self-prepared | 0.25 | 0.00 | 93 | 99 | — | — | 630 | 99 |
| HPA/ZrO2 | Self-prepared | 0.12 | 0.00 | 38 | 34 | — | — | 613 | 43 |
| B2O3/ZrO2 | Self-prepared | 0.15 | 0.00 | 141 | 130 | — | — | 618 | 160 |
Catalyst characterisation
Powder X-ray diffraction (XRD) patterns were recorded using a PANalytical X'Pert PRO-MPD diffractometer equipped with Ni-filtered Cu Kα radiation (λ = 0.1541 nm). Acquisition took place in the 2θ range of 5–70° with an angular step size of 0.05° and counting time of 1.4 s per step. Thermogravimetric analysis (TGA) was carried out on a Linseis DSC PT1600, equipped with a mass spectrometer (MS). The program involved dehydrating the samples at 373 K in flowing air followed by ramping the temperature from 373–1073 K at a rate of 10 K min−1 all under flowing air (100 cm3 min−1). The fragments with m/z of 18, 28 and 44 were followed by MS indicative of H2O, CO and CO2, respectively. The carbon content accumulated in the used solids was determined from the weight loss evidenced between 373–873 K. Nitrogen and argon isotherms were recorded using a Micromeritics TriStar analyser and a Micrometrics 3Flex instrument. Prior to analysis, samples were degassed overnight at 573 K. The temperature-programmed surface reaction (TPSR) of n-propylamine to propene and ammonia was measured using a Micromeritics Autochem II chemisorption analyser. Following degassing in a He flow (50 cm3 min−1) at 773 K for 2 h, the samples were saturated with n-propylamine at 473 K, purged with He at the same temperature to remove physisorbed amine and n-propylamine decomposition was monitored in the range of 473–773 K (10 K min−1 heating rate). Propene injections were used to develop response factors and the signal of propene during TPSR was followed by MS. This signal was also integrated to quantify the number of Brønsted acid sites. Fourier transform infrared spectroscopy (FTIR) of pyridine was performed in a Bruker IFS 66 spectrometer (65–4000 cm−1, 2 cm−1 optical resolution, 32 scans). Self-supporting wafers of samples were degassed under vacuum (10−3 mbar) at 693 K for 4 h, prior to adsorbing pyridine. Gaseous and weakly adsorbed molecules were subsequently removed by evacuation at 473 K for 30 min. The concentrations of Brønsted (cBrønsted,Pyr) and Lewis (cLewis,Pyr) acid sites were calculated from the band areas of adsorbed pyridine at 1545 and 1454 cm−1 using extinction coefficients of εBrønsted = 1.67 cm μmol−1 and εLewis = 2.94 cm μmol−1, respectively. Adsorbed species in the used catalysts were studied by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) on a Bruker Vertex 400 spectrometer equipped with liquid N2 cooled MCT detector and a Harrick diffuse reflection accessory (4000–700 cm−1, 4 cm−1 resolution, 32 scans). Prior to analysis, samples were degassed in Ar (20 cm3 min−1) for 1 h.Catalytic tests
p-Tolyl acetate was obtained from Sigma Aldrich, other acetates and reference compounds were prepared as described in Method S2.† Fries rearrangement and acylation reactions were studied in pressure tube reactors (V = 21 cm3, Ace Glass) equipped with a magnetic stirrer and heated using an oil bath. In a typical experiment, the pre-dried catalyst (340 mg, 573 K, 0.1 mbar, 12 h), and a solution of the substrate (2.5 mmol) in n-decane (10 cm3, Acros, ≥99%) unless otherwise indicated were added to the reactor. After replacing air with Ar three times (PanGas, purity 5.0), the reactor was heated to T = 393 or 423 K at autogenous pressure for 6 h. The reaction mixture was quenched with ice-water, and 0.01 cm3 of the liquid was added to 1 cm3 acetonitrile (99.9%, AcroSeal) or 40 ppm ascorbic acid (>99.9%, Acros) in dioxane (>99.9%, Acros, only for trimethylhydroquinone diacetate) for further analysis. Samples were analyzed using an Agilent 1260 Infinity HPLC equipped with an Agilent Zorbax C18 column and both DAD and RID detectors (150 × 3.0 mm, 3 μm, 298 K). The substrate and product concentrations were calibrated with reference to pure standards. The conversion of aryl ester and the product selectivity were calculated as follows: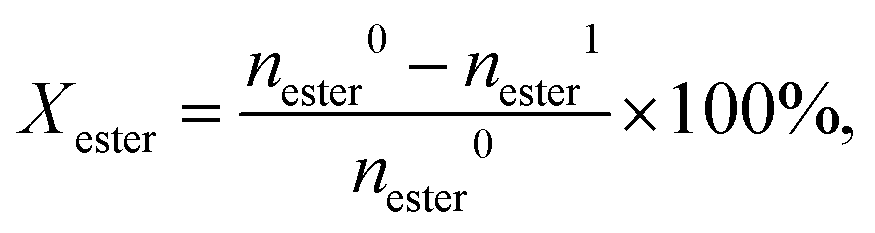 | (1) |
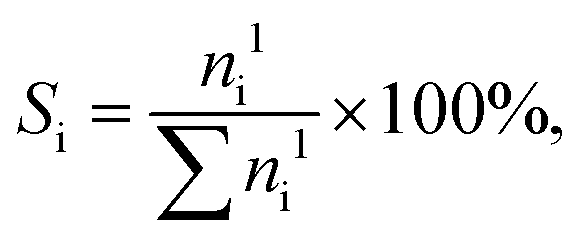 | (2) |
In exemplary cases, product assignments were ensured by additional GC-MS, HPLC-MS, 1H and 13C NMR measurements with reference to pure compounds (Method S2, Data S1†).
Results and discussion
Impact of the solvent and water content
Previous studies identified a potentially strong influence of the solvent on phenyl acetate rearrangement.7,9 To examine the impact on the Fries rearrangement of p-TA, the reaction was performed in solvents of distinct polarity; n-decane < toluene < nitrobenzene < N-methyl-2-pyrrolidone (NMP) < dimethylsulfoxide (DMSO) < water, using BEA(15) as a representative system (Fig. 1a). The product distribution shows a strong dependence on the solvent polarity, with the highest selectivity to the desired products observed in the least polar n-decane. It is noteworthy that AP2,5 was the main rearrangement product evidenced in all cases, and no AP3,6 was detected in n-decane, thus the solvent was selected for further studies. The only by-product detected was p-cresol, which was predominantly formed when the reaction was conducted in water.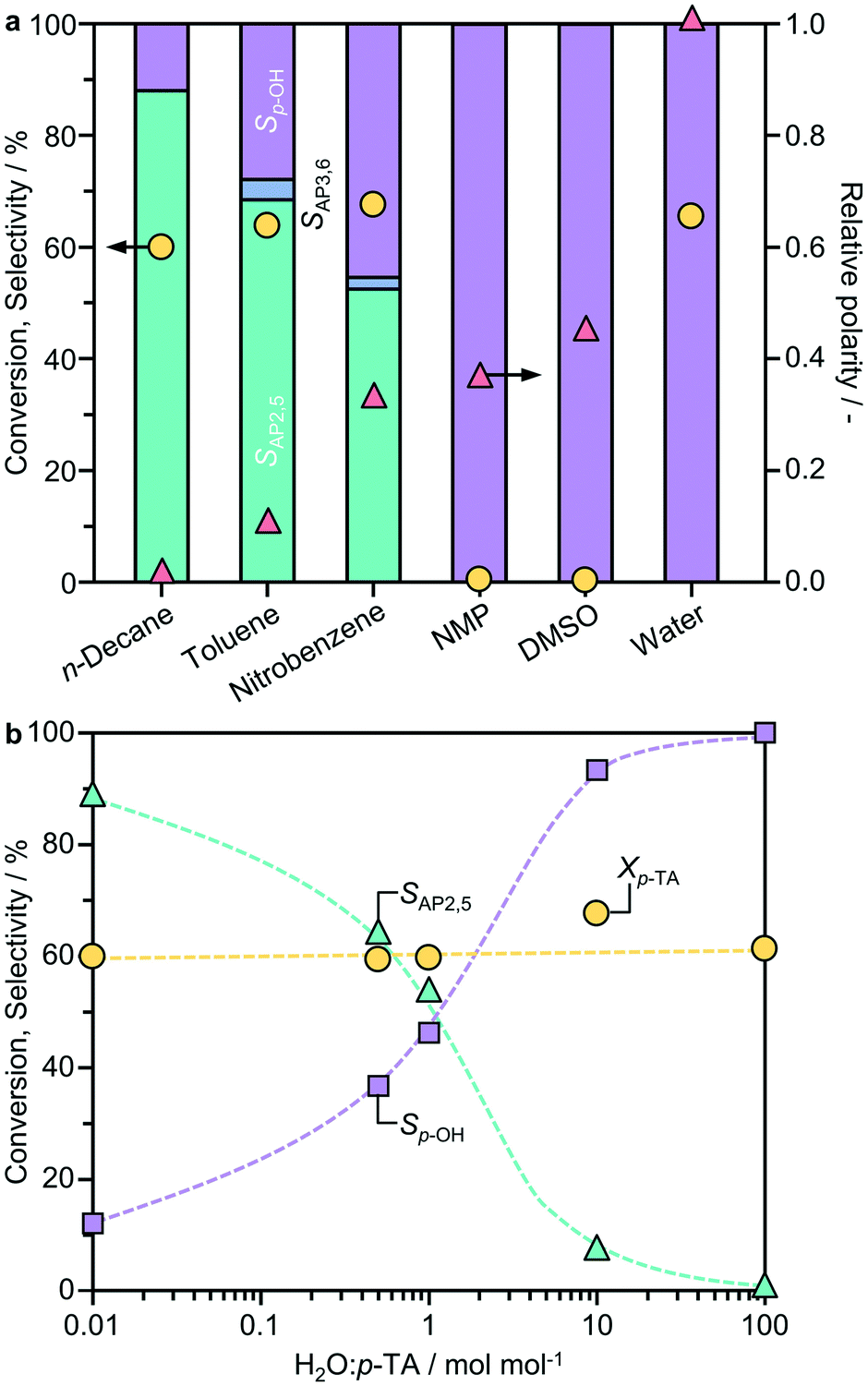 | ||
| Fig. 1 Influence of a) the solvent and b) the water content on the performance of BEA(15) in the Fries rearrangement of p-TA. The indicated polarity of the solvents is referenced to water. Conditions: 2.5 mmol p-TA, catalyst 340 mg, 10 cm3 solvent (n-decane was used to evaluate the effect of H2O), T = 423 K, t = 6 h. | ||
The influence of the water content on the reaction was studied in more detail using n-decane as the solvent (Fig. 1b). To eliminate the potential effect of the degree of catalyst hydration, the BEA(15) zeolite was pre-dried at 573 K under vacuum (0.1 mbar) overnight before introducing controlled amounts of water. Interestingly, the p-TA conversion remained virtually constant upon varying the H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-decane ratio. In contrast, the product selectivity changed radically with the highest selectivity of AP2,5 (88%) obtained over the pre-dried catalyst and more pronounced formation of p-cresol with increasing water content. The results are in agreement with previous studies on phenyl acetate rearrangement that revealed a decreased selectivity to phenol over catalysts dehydrated in situ.6 Accordingly, all catalysts were pre-dried under such conditions before testing unless otherwise specified.
:n-decane ratio. In contrast, the product selectivity changed radically with the highest selectivity of AP2,5 (88%) obtained over the pre-dried catalyst and more pronounced formation of p-cresol with increasing water content. The results are in agreement with previous studies on phenyl acetate rearrangement that revealed a decreased selectivity to phenol over catalysts dehydrated in situ.6 Accordingly, all catalysts were pre-dried under such conditions before testing unless otherwise specified.
Performance of zeolites and other solid acids
To examine the influence of important properties, the performance of a range of zeolites of different framework type (FER, MFI, BEA, MOR, and FAU) and other solid acids (MCM-41, B2O3/ZrO2, WO3/ZrO2, and HPA/ZrO2) was compared in the Fries rearrangement of p-TA in n-decane at 423 K (Fig. 2). BEA outperformed all the other catalysts, with a p-TA conversion of 60%, followed by FAU and MOR with conversion values of 22% and 12%, respectively, while the other catalysts exhibited much lower activity (<6% conversion). Interestingly, the selectivity to AP2,5 over the zeolite catalyst correlated with p-TA conversion in the order BEA > FAU > MOR > MFI > FER.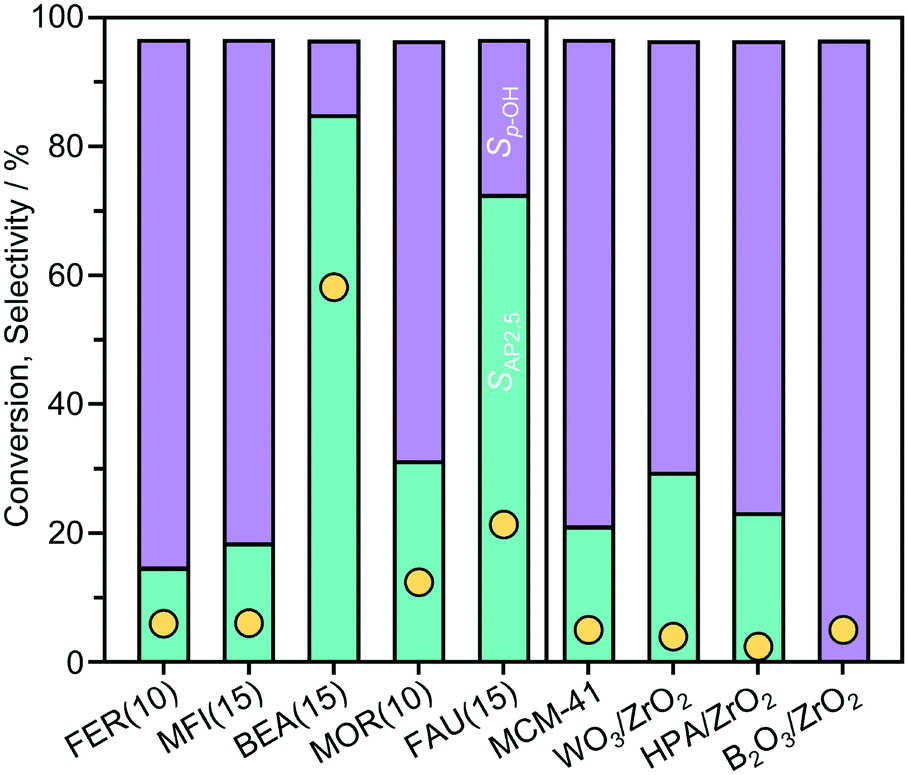 | ||
| Fig. 2 Conversion (yellow circles) and selectivity (turquoise and purple bars) observed in the Fries rearrangement of p-tolyl acetate over zeolites (left side, ordered by the maximum diameter of a sphere that can diffuse through the micropore channels) and other solid acids (right side). Conditions: 2.5 mmol p-TA, catalyst 340 mg, 10 cm3n-decane, T = 423 K, t = 6 h. | ||
To understand the performance of the distinct catalysts, the impact of the acidic and porous properties was examined in detail for the BEA and FAU-type zeolites (Table 1). The zeolite catalysts of similar composition (Si:Al ≤ 15) exhibit high concentrations of Brønsted acid sites in protonic form (cBrønsted,Pyr > 150 mol g−1). Thus, the variation in activity observed over these catalysts must be related to another property of the catalyst. The general correlation with the micropore diameter, classified with respect to the maximum diameter of a sphere that can diffuse through the micropore channels, suggests that a shape selective effect limits the activity of medium-pore FER(10) and MFI(15) zeolites, but does not directly explain the lower activity of MOR(10) compared to BEA(15) and FAU(15).
To gain further insight into the acidic properties, the catalysts were characterized by the temperature programmed surface reaction (TPSR) of n-propylamine (Fig. 3 and S3†).35–37 In comparison with the analysis of adsorbed pyridine, similar trends in the concentrations of Brønsted acid sites (cBrønsted,TPSR) derived by TPSR were observed with the composition of zeolites of a particular framework type (Table 1). However, the values were much higher which could relate to the smaller size of n-propylamine and thus the ability to probe additional sites. Analysis of the temperature of the first propene evolution peak, TC3H6, provides an indication of the acidic strength, and reveals that FAU(15), BEA(15), and MFI(15) have almost equivalent properties, while the production of propene is not observed until higher temperatures over MOR(15), indicative of a weaker character of the Brønsted acid sites. This observation could explain the limited activity exhibited by this catalyst.
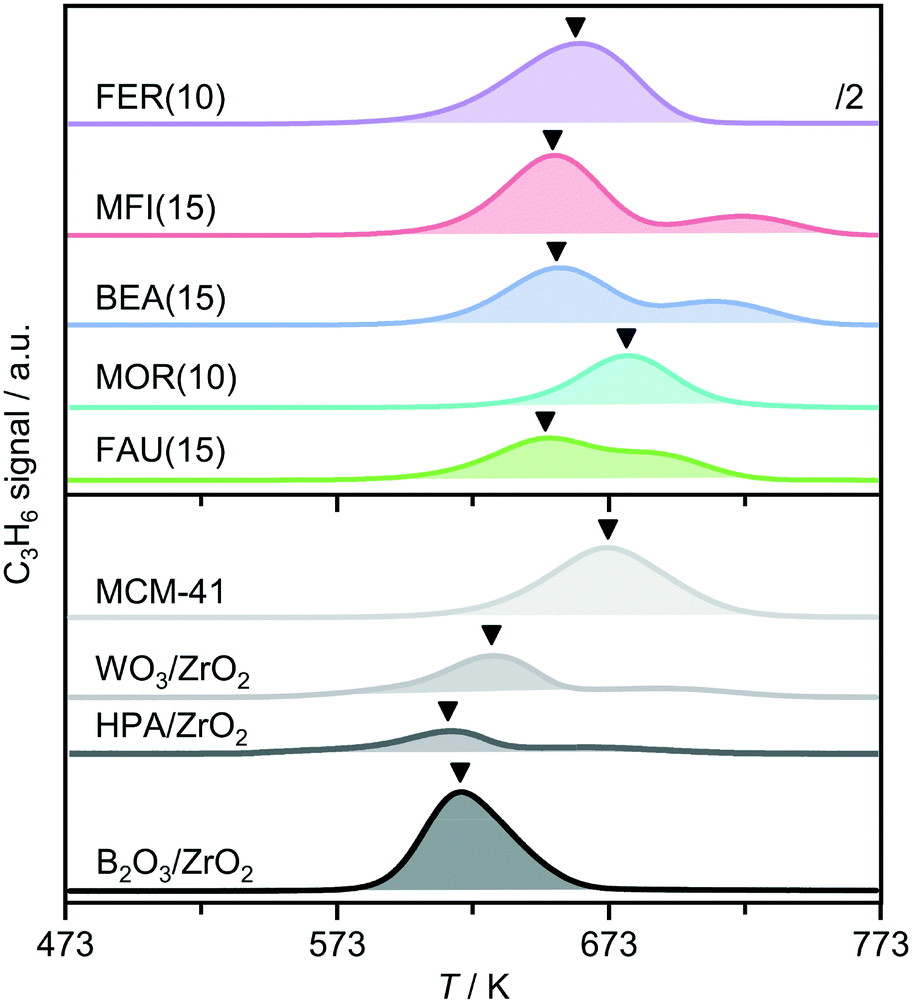 | ||
| Fig. 3 Propene evolution during the TPSR of n-propylamine over zeolites (top) and other solid acids (bottom, multiplied by a factor of 4 for improved visualization). The arrows indicate the position of the temperature of maximum propene evolution (TC3H6) while the shaded areas were used to derive the concentration of Brønsted acid sites. | ||
Compared to the zeolites, the other solid catalysts generally showed much lower concentrations of Brønsted acid sites (Table 1). In contrast, the lower TC3H6 values observed evidence a higher strength of acid sites in the ZrO2-supported materials. These results suggest that the lower activity seen over the latter catalysts (Fig. 2) primarily derives from the reduced density of active sites, especially considering that the surface area is mainly mesoporous and therefore accessibility constraints are not expected. This was confirmed by the increasing the amount of the WO2/ZrO2 catalyst, which exhibited the highest selectivity towards AP2,5 of the non-zeolitic solids. Tripling the quantity led to an approximately three times higher conversion (10%) without changing the selectivity.
The influence of the acid site density was further studied over the BEA zeolites by varying the Si/Al ratio (Table 1 and Fig. 4). The conversion of p-TA increased with the corresponding concentration of Brønsted acid sites. Comparatively, the selectivity to AP2,5 was constant (>85%) over a wide range, but dropped in the BEA(220) sample which exhibited the lowest acidity. Correspondingly, the selectivity to p-cresol showed the opposite trend. This finding confirms the role of Brønsted acidity for the efficient rearrangement of p-TA to acetophenone. Determination of the turnover frequencies per Brønsted acid site (BEA(11) = 17 h−1; BEA(15) = 25 h−1; BEA(90) = 37 h−1; BEA(220) = 18 h−1) evidences a maximum activity at a Si/Al ratio of 90. The close magnitudes of the mesopore surface area (Smeso, Table 1) in these materials indicates that this is not likely to be an effect of distinctions in the crystal size. It is possible that the increased hydrophobicity enhances diffusion of the substrate and products, but we cannot exclude other effects e.g. lower accessibility of Brønsted acid sites due to differences in their location, or their inefficient utilization due to steric effects related to the closer proximity of active sites.
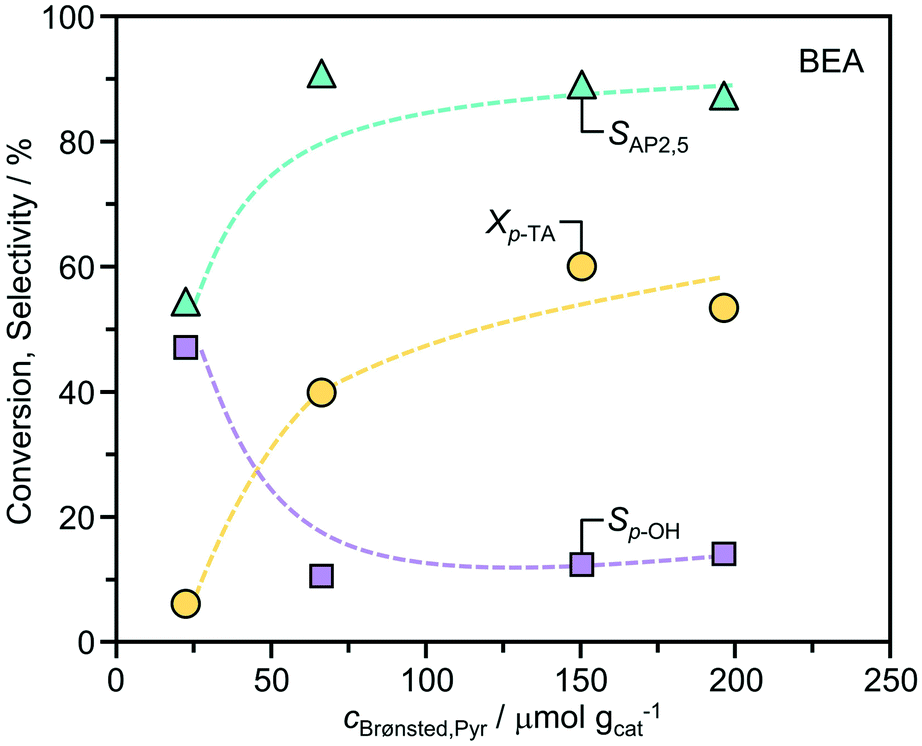 | ||
| Fig. 4 Correlation between the conversion of p-TA, the selectivity to distinct products, and the concentrations of Brønsted acid sites over distinct BEA zeolites. The reaction conditions were as reported in Fig. 2. | ||
Considering the similarity of the structures to p- and o-xylene, the molecular surface diameter of p-TA and AP2,5 are expected to be in the range of 5.85–6.80 Å, falling between the characteristic channel dimensions of the large-pore zeolites (6.3 and 7.4 Å for BEA and FAU, respectively) but exceeding the size of medium-pore MFI (ca. 5.0–5.5 Å). Overall, these results suggest that the rearrangement reaction can occur within the channels of the large-pore zeolites, but likely proceeds at the pore mouths of the other framework types.
Reaction kinetics and catalyst stability
Previous studies of the Fries rearrangement of phenyl acetate have highlighted the complexity of possible reaction networks over solid acids, which can form multiple products through distinct pathways. Owing to the simplicity of product distribution in the rearrangement of p-TA, additional tests were conducted over the best-performing BEA(15) catalyst to gain an insight into the mechanism. Fig. 5 presents the dependence of the product selectivity as a function of the conversion of p-TA in the presence of both polar and non-polar solvents. In n-decane the formation of AP2,5, the only rearrangement product observed, closely mirrored the consumption of p-TA suggesting a purely intramolecular reaction. The concentration continued to increase steadily until around 6 h, after which it remained relatively constant. The formation of small amounts of p-cresol occurred simultaneously in the first stages of the reaction, but then reached a plateau after 15 min, showing no further variation with time. A similar picture emerged when the reaction was conducted in nitrobenzene, but the formation of p-cresol reached a maximum more rapidly. The detrimental effect of p-cresol formation on the Fries rearrangement selectivity at low conversion can be clearly seen by correlating these values (Fig. 6a). Note that, due to the faster kinetics of the reaction in this solvent, the conversion already reached 40% after 15 min (the first sampling point) of reaction when studied at 423 K. Irrespective of the solvent, p-TA rearrangement shows typical first-order kinetics at the initial stages of reaction, as evidenced by the linear dependence of ln(cp-TA) with the time (Fig. 6b).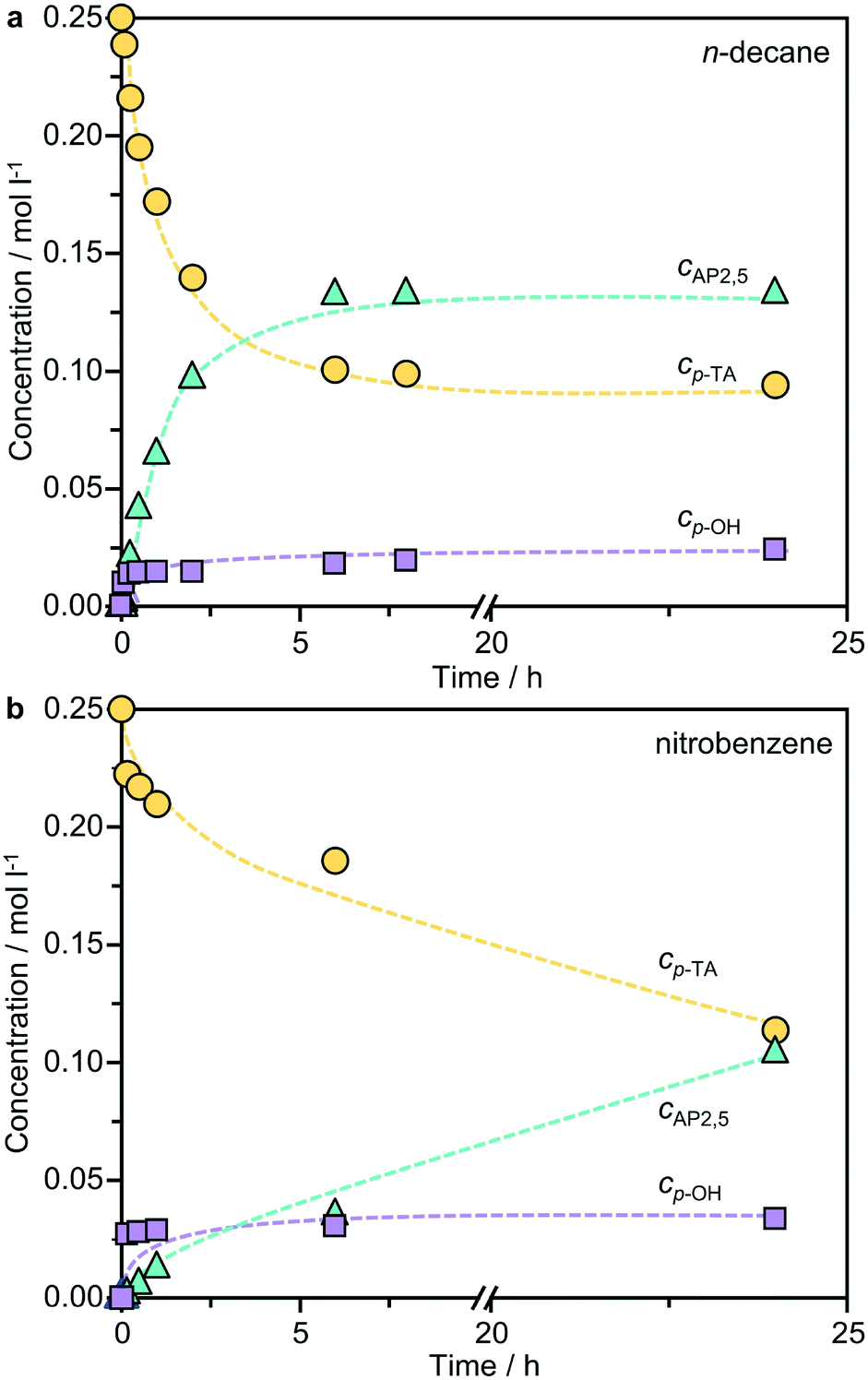 | ||
| Fig. 5 Evolution of the concentrations of the substrate and products with time in the Fries rearrangement of p-tolyl acetate over BEA(15) zeolite in a) n-decane and b) nitrobenzene. Conditions: p-TA 2.5 mmol, catalyst 340 mg, solvent 10 cm3, T = 423 K (for n-decane) or 393 K (for nitrobenzene). | ||
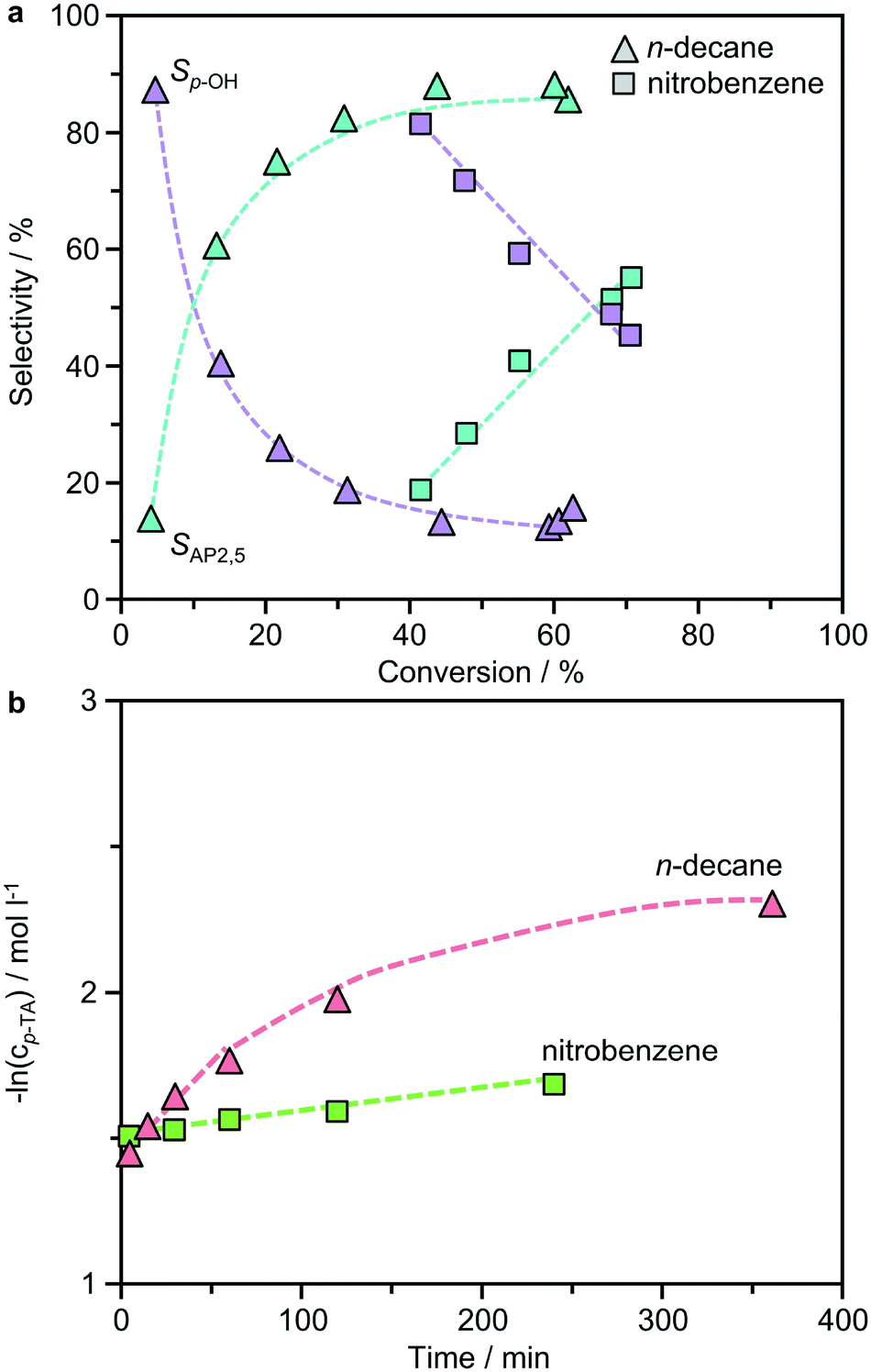 | ||
| Fig. 6 a) The correlation between selectivity and conversion and b) negative logarithm of the concentration of p-tolyl acetate versus time assuming first order reaction kinetics for the same solvents. Conditions: p-TA 2.5 mmol, catalyst 340 mg, solvent 10 cm3, T = 423 K or 393 K (for nitrobenzene in part b). | ||
The results suggest that the formation of the by-product likely occurs via a parallel reaction to the Fries rearrangement. To support this conclusion, two additional experiments were performed. In the first, p-cresol was added into the typical reaction mixture with a p-cresol:p-TA molar ratio of 1:5 in n-decane (Fig. 7a). In this case, a significant inhibition effect was observed as the p-TA conversion was largely reduced from 44% to 6%. However, the selectivity towards p-cresol (60%) was decreased compared to that observed without p-cresol addition at a similar conversion (>80%). The second experiment tested the possibility of catalyzing an acylation reaction between p-cresol and acetic anhydride over BEA(15) in the presence of n-decane (Fig. 7b). Acetic anhydride was selected as the acylating agent as it was detected by HPLC as a side-product in p-TA rearrangement. Already at room temperature the acylation reaction proceeds efficiently, resulting in 84% conversion of p-cresol with full selectivity towards p-TA within 5 min. Increasing the temperature to 423 K, which is a typical temperature for Fries rearrangement reactions, further increased the conversion to 96% with only minor formation of AP2,5 (<1%). These results support the hypothesis that the formation of AP3,6via the intermolecular acylation of p-cresol and acetic anhydride is unfavorable under Fries rearrangement conditions, and both are likely primary reaction products of p-TA. With prolonged reaction time, catalyst deactivation is apparent as evidenced from the deviation in the linearity of ln(cp-TA) (Fig. 6b).
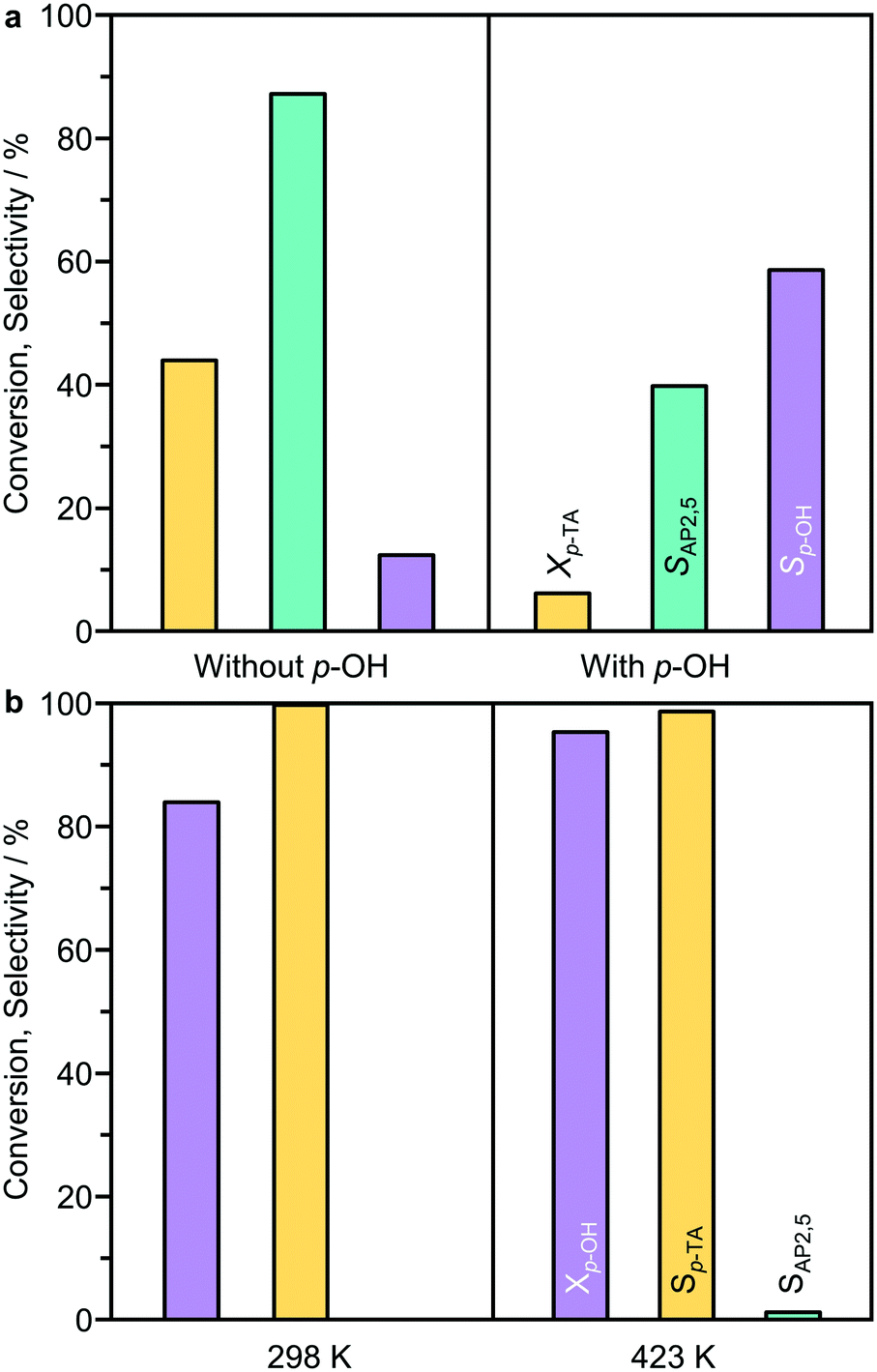 | ||
| Fig. 7 a) The effect of p-cresol addition on the Fries rearrangement of p-tolyl acetate. Conditions: p-TA 2.5 mmol, catalyst 340 mg, n-decane 10 cm3, T = 423 K. b) The acylation of p-cresol with acetic anhydride over BEA(15). Conditions: p-cresol 2.5 mmol, acetic anhydride 2.5 mmol, catalyst 340 mg, n-decane 10 cm3, t = 5 min (298 K) or 6 h (423 K). | ||
To gain insight into the cause of the deactivation, we conducted thermogravimetric analysis coupled with mass spectrometry (TGA-MS) in flowing air of the catalyst isolated and dried after use for different reaction times. Two distinct weight losses are evident (Fig. 8a and S4†), the first (of approximately 5 wt%) occurs at ca. 400–573 K and the second at ca. 600–873 K. The magnitude of the latter varies depending on the length of use. Both steps are accompanied by significant evolution of H2O and CO2. Comparison of the relative intensity of the two fragments reveals that the CO2:H2O ratio associated with the second stage weight change is much higher, indicative of the formation of condensed carbonaceous species as the precursors of coke. Comparison with time shows that the weight gain associated with coke was already quite pronounced at the beginning of the reaction (Fig. 8b), suggesting the fast and strong adsorption of organic species on the zeolite. To probe the possible impact of adsorbed species on the active sites, diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy was conducted (Fig. 8d). Comparison on the DRIFT spectra of the fresh and used catalysts shows significantly reduced intensities of bands at 3660 and 3740 cm−1 associated with bridging (Brønsted acid sites) and terminal silanol groups, respectively, as well as the appearance of multiple bands in the C–H region of the spectra (reflected in the broad envelop at 3100–2800 cm−1) at the beginning of the reaction, in line with the TGA-MS results. Taking into account the above observations, the deactivation of the zeolite catalyst can be attributed to the poisoning of the active sites due to the interaction with reaction intermediates and subsequent coke deposition. To confirm the catalyst reusability, the used catalyst was then first separated by centrifugation from the reaction mixture, followed by re-dispersing and washing with n-decane. After separation with centrifugation, the recovered solid (denoted as washed catalyst) was tested under typical rearrangement conditions by recharging with 10 cm3n-decane. Although the selectivity patterns were similar to those of the fresh catalyst, the washed catalyst showed much lower activity (39% versus 60%, Fig. 8c), indicating that washing is not sufficient to regenerate all the active sites. In contrast, oxidative regeneration performed at 823 K in flowing air led to complete removal of the organic species and the performance was fully restored.
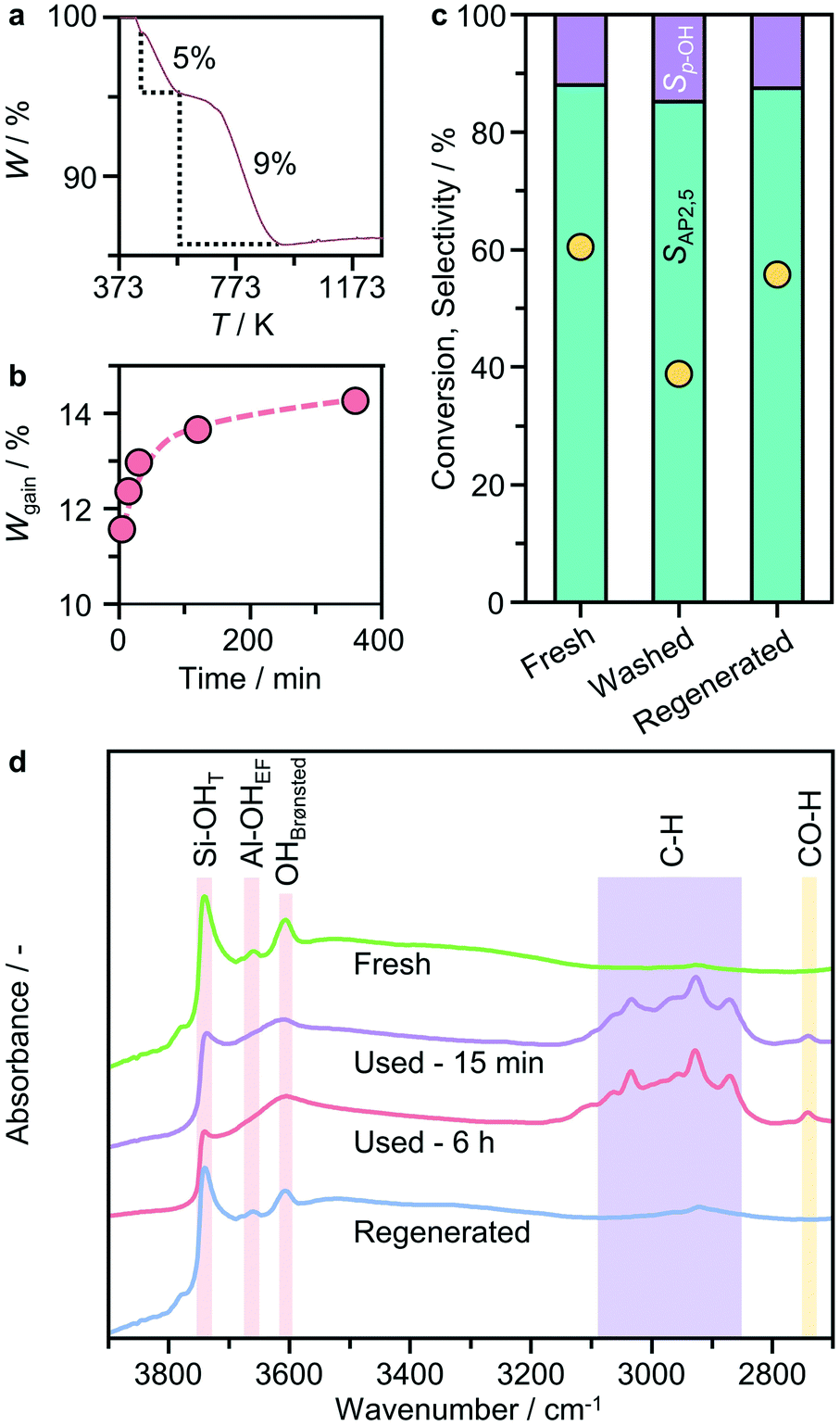 | ||
| Fig. 8 a) Representative TGA profile of BEA(15) after used in p-TA Fries rearrangement and b) the catalyst weight gain as a function of the reaction time estimated from the second weight loss step. c) Reusability of the catalysts after washing with n-decane (10 cm3) or regeneration in air at 823 K for 5 h. d) DRIFT spectra of the fresh, used and regenerated catalysts. The reaction conditions were as reported in Fig. 2. | ||
Other aryl acetates
To explore the scope of zeolites in Fries rearrangement, the reaction of a range of aryl esters from phenyl acetates to trimethylhydroquinone diacetate,38 was evaluated over the MFI(15), BEA(15), and FAU(15) zeolites (Fig. 9a and S5†). For this purpose, the reactions were conducted in anhydrous toluene instead of n-decane, as all of the substrates were soluble in this solvent. Low substrate concentrations were applied during the catalytic tests and no signs of immiscibility were detected. For p-tolyl acetate, the use of toluene led to slightly higher conversions than those observed in n-decane (Fig. 2) and in the case of FAU(15) an enhanced selectivity to acetophenones similar to that of BEA(15) was observed.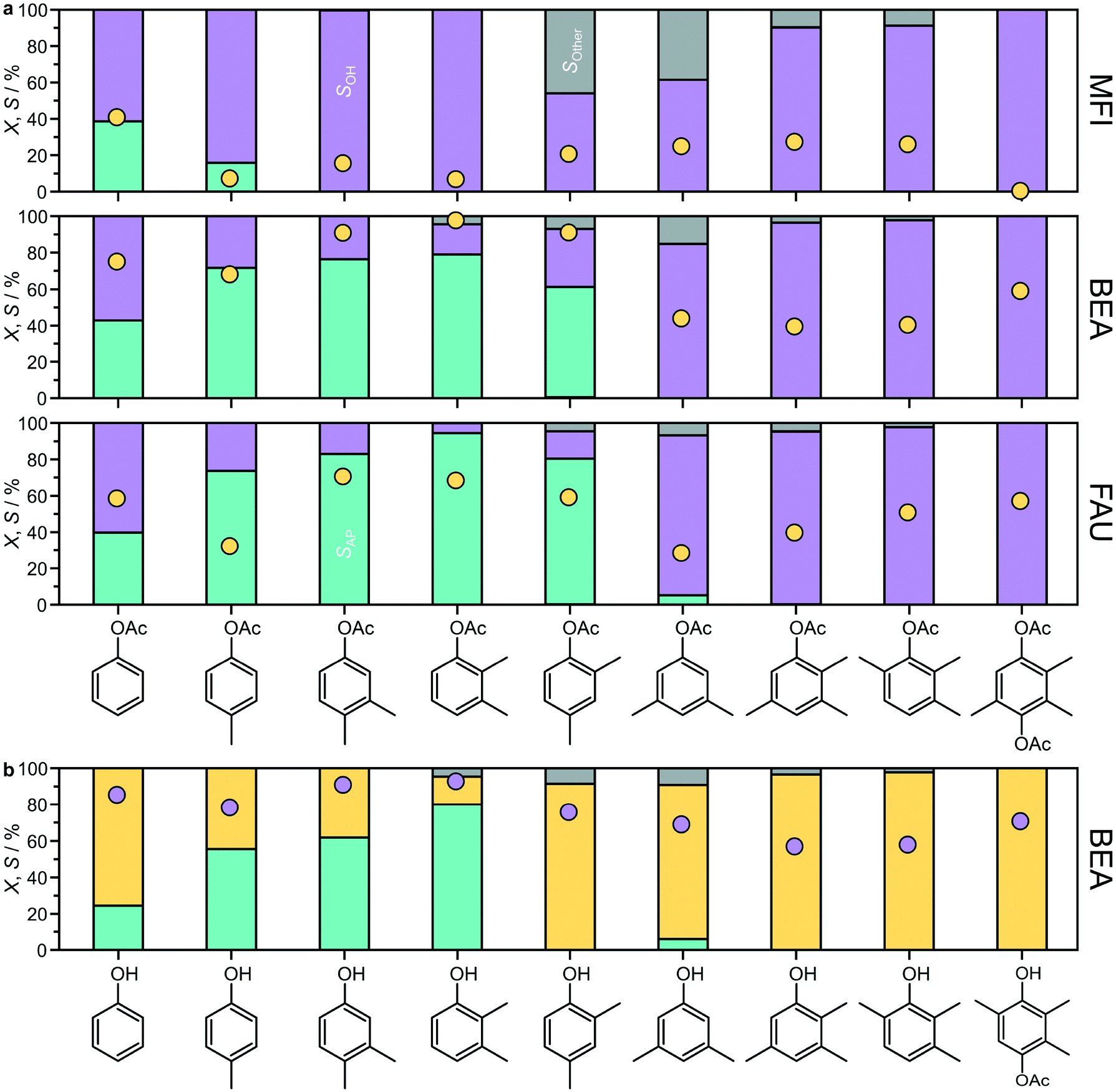 | ||
| Fig. 9 a) Conversion (yellow circles) and selectivity (bars) in the Fries rearrangement of distinct aryl esters over MFI, BEA or FAU zeolites. Conditions: substrate 2.5 mmol, catalyst 340 mg, toluene 10 cm3, T = 423 K, and t = 6 h. Bars: acetophenones (turquoise), phenols (purple), and other (grey). b) Conversion (purple circles) and selectivity (bars) observed over BEA(15) in the acylation of the indicated phenol by treatment with acetic anhydride. Conditions: substrate 2.5 mmol, acetic anhydride 2.5 mmol, catalyst 340 mg, toluene 10 cm3, T = 423 K, and t = 6 h. Bars: hydroxyacetophenones (turquoise), aryl esters (yellow), and other (grey). The specific product distributions are presented in Fig. S5.† | ||
The zeolite framework type strongly influenced the substrate scope of the catalyst. For substrates where Fries rearrangement was observed, the conversion of the aryl ester followed the general trend BEA(15) > FAU(15) > MFI(15). For MFI(15), Fries rearrangement products were only observed for phenyl acetate and p-tolyl acetate. This zeolite showed higher p-:o-selectivity in the conversion of phenyl acetate. It also yielded the highest AP3,6:AP2,5 ratio in the conversion of p-tolyl acetate. In addition to Fries rearrangement products and phenol, p-acetoxyacetophenone also formed in appreciable amounts during the reaction of phenyl acetates over MFI(15) and to a limited extent over BEA(15) and FAU(15). This product has been previously reported to result from the intermolecular reaction between two phenyl acetate molecules,15 which also originates stoichiometric amounts of phenol. Autoacylation was not observed for any of the other substrates studied, which could be due to a combination of steric and electronic effects of the methyl substituents on the phenyl ring.
Extending the substrate scope, large-pore BEA(15) could also catalyze the conversion of 3,4-, 2,3-, and 2,4-dimethylphenyl acetates. In these cases, the only rearrangement products were the o-substituted hydroxyacetophenones, which formed with high selectivity (57–79%). FAU(15) could also catalyze the Fries rearrangement of the dimethyl substituted phenyl acetates, in some cases with superior selectivity to BEA(15), and in addition catalyzed the Fries rearrangement of 3,5-dimethylphenylacetate to a very limited extent (<5%), also forming the o-substituted hydroxyacetophenone. However, neither of these zeolites could catalyze the Fries rearrangement of trimethylphenyl acetates or trimethylhydroquinone diacetate, instead primarily promoting the conversion to the corresponding phenols via cleavage of the C–O ester bond.
Considering the high selectivity often observed, the possibility of forming acetophenones via acylation of the corresponding substituted phenols with acetic anhydride was also studied over BEA(15) (Fig. 9b). High conversions were observed for all substrates, yielding the desired hydroxyacetophenones with a similar trend as observed in the Fries rearrangement. In this case, it was not possible to obtain 2-hydroxy-3,5-dimethylacetophenone from 2,4-dimethylphenol. Other products were also detected in the conversion of some of the di- and trimethyl substituted phenylacetates, sometimes in appreciable amounts (Fig. S5†). These included tolylacetophenone(s) originating from the solvent toluene as a co-reactant and arylated products containing an ester linkage.
Overall, these results provide a strong indication that the Fries rearrangement reaction requires some degree of confinement within the micropore channels of the zeolites. To evaluate the dependence of the substrate accessibility on the framework structure, the diameters of the molecular surface of selected phenyl acetates were estimated after optimization of their structures (Method S3,†Fig. 10a). Quantification of the effect of introducing methyl substituents in the meta-position to the acetate groups shows that each addition leads to an increase of ca. 1 Å, ranging from 6.3–8.5 Å. These values are very close to the maximal channel sizes of the zeolites, i.e., 5.8, 6.7, and 7.4 Å, respectively, for MFI, BEA and FAU. Superposition of the selected molecules on the pore entrances of the MFI-, BEA-, and FAU-type framework illustrates the bulky nature of the substrates (Fig. 10b). Even the diffusion of p-tolyl acetate into the MFI zeolite appears as if it would be strongly constrained and although the dimethyl substituted analogues have a better fit with the larger pore diameters of BEA and FAU zeolites, the reaction is likely to be largely catalysed by strong Brønsted acid sites located in the vicinity of the micropore entrances.
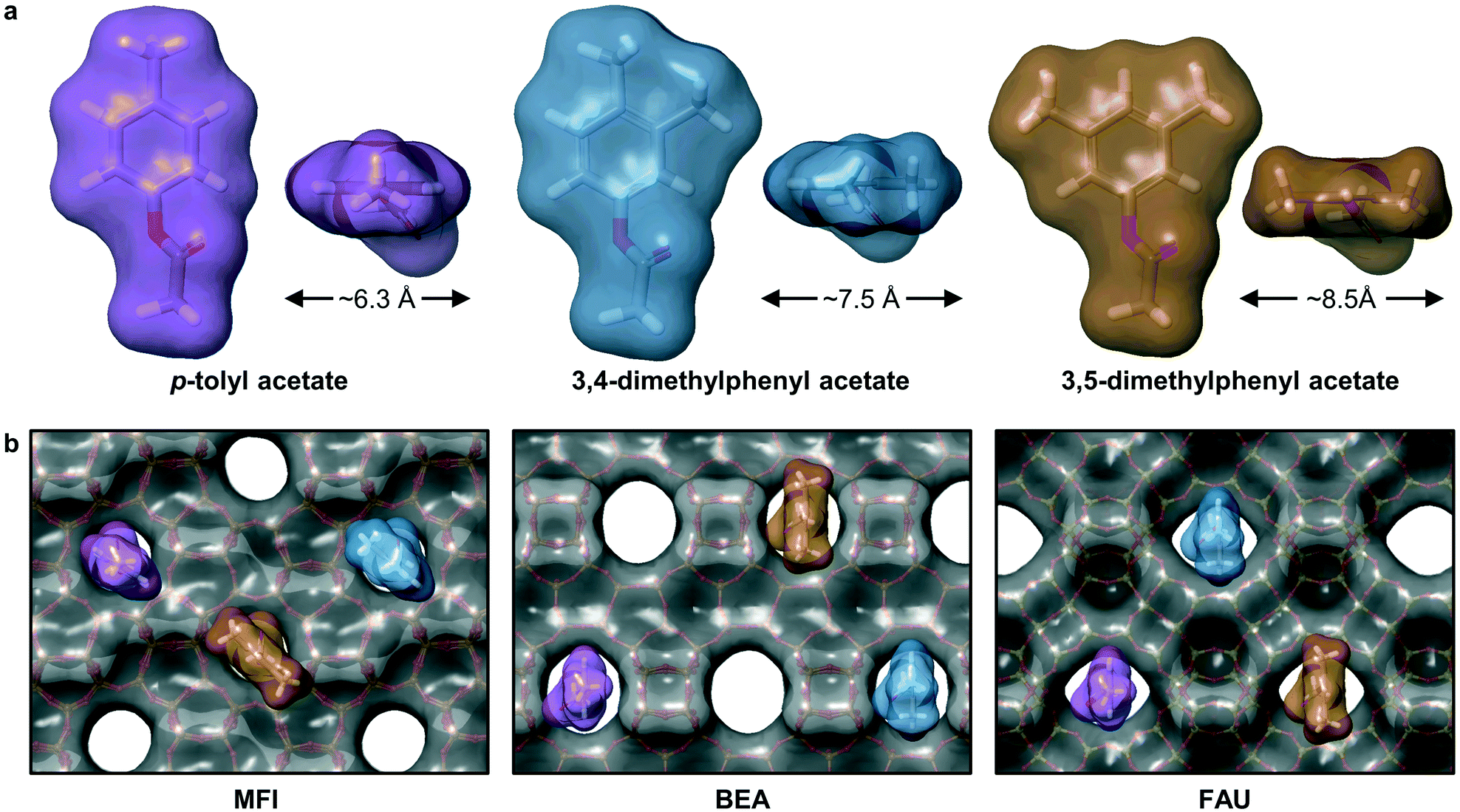 | ||
| Fig. 10 a) Computed molecular surface diameters of representative phenyl acetates illustrating the effect of adding a methyl group in meta-positions to the acetate functionality and b) superposition of the molecules on the pore entrances of the distinct zeolite frameworks studied. | ||
Conclusions
A broad family of different solid acid catalysts have been evaluated in the Fries rearrangement of p-tolyl acetate to assess the impacts of key conditions and properties on the performance. The use of non-polar solvents and the elimination of water was critical to maximize the activity and selectivity to acetophenones. Under the optimised conditions, high activity and selectivity to the rearrangement products are gained over large-pore zeolites (BEA and FAU), whereas small and medium pore zeolites displayed limited conversion and predominantly catalysed cleavage of the ester bond to form p-cresol, demonstrating a clear effect of the pore size. The low activity of ZrO2-supported solid acids was linked to the lower density of Brønsted acid sites exhibited by these materials. Detailed kinetic studies on the BEA zeolite suggest that the rearrangement and ester cleavage are likely parallel reactions. p-Cresol is readily converted to p-tolyl acetate upon the addition of acetic anhydride, suggesting that the Friedel-Crafts acylation of p-cresol to hydroxyacetophenone is not favoured under the reaction conditions. Furthermore, the accumulation of condensed organic species is identified as the main cause of catalyst deactivation, while the performance can be fully restored by oxidative thermal regeneration. Expanding the substrate scope to other substituted phenyl acetates uncovered the potential of BEA and FAU zeolites for the Fries rearrangement of dimethylphenyl acetates. Computation of the molecular structures highlighted the close fit of the molecular surface diameters with the channel dimensions of these frameworks, pointing towards a scenario dominated by pore-mouth catalysis in these transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Mr. S. Bischof and Mr. L. Bucher for analytical measurements and structure determination, and Mr. S. Ackermann, Mr. S. Läuger, Mr. S. Häss, Mr. M. Gerber, Mr. L. Niess for the preparation of substrates and product reference compounds, and Dr. E. Canet Martinez for support in NMR spectroscopy.References
- K. Fries and G. Finck, Ber. Dtsch. Chem. Ges., 1909, 41, 4271 CrossRef CAS.
- R. Martin, Org. Prep. Proced. Int., 1992, 24, 369 CrossRef CAS.
- G. Bringmann, D. Menche, J. Kraus, J. Mühlbacher, K. Peters, E. M. Peters, R. Brun, M. Bezabih and B. M. Abegaz, J. Org. Chem., 2002, 67, 5595 CrossRef CAS PubMed.
- M. Korb and H. Lang, Chem. Soc. Rev., 2019, 48, 2829 RSC.
- Y. Pouilloux, S. N. Gnep, P. Magnoux and G. Perot, J. Mol. Catal., 1987, 40, 231 CrossRef CAS.
- S. C. S. Cundy, R. Higgins, S. A. M. Kibby, B. M. Lowe and R. M. Paton, Tetrahedron Lett., 1989, 30, 2281 CrossRef.
- H. van Bekkum, A. J. Hoefnagel, M. A. van Koten, E. A. Gunnewegh, A. H. G. Vogt and H. W. Kouwenhoven, Stud. Surf. Sci. Catal., 1994, 83, 379 CrossRef CAS.
- A. Vogt, H. W. Kouwenhoven and R. Prins, Appl. Catal., A, 1995, 123, 37 CrossRef CAS.
- F. Jayat, M. J. S. Picot and M. Guisnet, Catal. Lett., 1996, 41, 181 CrossRef CAS.
- A. Heidekum, M. A. Harmer and W. F. Hölderich, J. Catal., 1998, 176, 260 CrossRef CAS.
- P. Adryan, R. A. Ismail and F. Roessner, Kinet. Catal., 2008, 49, 587 CrossRef CAS.
- E. F. Kozhevnikova, E. G. Derouane and I. V. Kozhevnikov, Chem. Commun., 2002, 1178 RSC.
- E. F. Kozhevnikova, J. Quartararo and I. V. Kozhevnikov, Appl. Catal., A, 2003, 245, 69 CrossRef CAS.
- J. H. Clark, M. G. Dekamin and F. M. Moghaddam, Green Chem., 2002, 4, 366 RSC.
- R. van Grieken, J. A. Melero and G. Morales, Appl. Catal., A, 2005, 289, 143 CrossRef CAS.
- P. T. Patil, K. M. Malshe, P. Kumar, M. K. Dongare and E. Kemnitz, Catal. Commun., 2002, 3, 411 CrossRef CAS.
- R. Sakthivel, H. Prescott and E. Kemnitz, J. Mol. Catal. A: Chem., 2004, 223, 137 CrossRef CAS.
- B. Bachiller-Baeza and J. A. Anderson, J. Catal., 2004, 228, 225 CrossRef CAS.
- M. Hechelski, A. Ghinet, B. Louvel, P. Dufrenoy, B. Rigo, A. Daich and C. Waterlot, ChemSusChem, 2018, 11, 1249 CrossRef CAS PubMed.
- W. Hölderich, M. Hesse and F. Näumann, Angew. Chem., Int. Ed. Engl., 1988, 27, 226 CrossRef.
- A. Corma and H. García, Chem. Rev., 2003, 103, 4307 CrossRef CAS PubMed.
- G. Sartori and R. Maggi, Chem. Rev., 2006, 106, 1077 CrossRef CAS PubMed.
- G. Perot and M. Guisnet, J. Mol. Catal., 1990, 61, 173 CrossRef CAS.
- M. Bejblová, D. Procházková and J. Čejka, ChemSusChem, 2009, 2, 486 CrossRef PubMed.
- S. Abate, K. Barbera, G. Centi, P. Lanzafame and S. Perathoner, Catal. Sci. Technol., 2016, 6, 2485 RSC.
- U. Freese, F. Heinrich and F. Roessner, Catal. Today, 1999, 49, 237 CrossRef CAS.
- E. Heitling, F. Roessner and E. van Steen, J. Mol. Catal. A: Chem., 2004, 216, 61 CrossRef CAS.
- W. W. Kaeding, C. Chu, L. B. Young, B. Weinstein and S. A. Butter, J. Catal., 1981, 67, 159 CrossRef CAS.
- M. Milina, S. Mitchell, N.-L. Michels, J. Kenvin and J. Pérez-Ramírez, J. Catal., 2013, 308, 398 CrossRef CAS.
- M. Milina, S. Mitchell, Z. Domínguez Trinidad, D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2012, 2, 759 RSC.
- M. Koehle, Z. Zhang, K. A. Goulas, S. Caratzoulas, D. G. Vlachos and R. F. Lobo, Appl. Catal., A, 2018, 564, 90 CrossRef CAS.
- T. C. Keller, J. Arras, S. Wershofen and J. Pérez-Ramírez, ACS Catal., 2014, 5, 734 CrossRef.
- M. Koehle, E. Saraçi, P. Dauenhauer and R. F. Lobo, ChemSusChem, 2017, 10, 91 CrossRef CAS PubMed.
- S. C. Oh, T. Nguyendo, Y. He, A. Filie, Y. Wu, D. T. Tran, I. C. Lee and D. Liu, Catal. Sci. Technol., 2017, 7, 1153 RSC.
- W. E. Farneth and R. J. Gorte, Chem. Rev., 1995, 95, 615 CrossRef CAS.
- C. Pereira and R. J. Gorte, Appl. Catal., A, 1992, 90, 145 CrossRef CAS.
- R. J. Gorte, Catal. Lett., 1999, 62, 1 CrossRef CAS.
- R. Hinze, M. C. Laufer, W. F. Hölderich, W. Bonrath and T. Netscher, Catal. Today, 2009, 140, 105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy00590h |
| This journal is © The Royal Society of Chemistry 2020 |