
Characterization of heterogeneous aryl–Pd(II)–oxo clusters as active species for C–H arylation†
Taeil
Shin‡
a,
Minjun
Kim‡
a,
Younjae
Jung
a,
Sung June
Cho
 *b,
Hyunwoo
Kim
*a and
Hyunjoon
Song
*a
*b,
Hyunwoo
Kim
*a and
Hyunjoon
Song
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: hwkim@kaist.edu; hsong@kaist.ac.kr
bDepartment of Chemical Engineering, Chonnam National University, Gwangju 61186, Republic of Korea. E-mail: sjcho@chonnam.ac.kr
First published on 28th October 2020
Abstract
C–H arylation with heterogeneous palladium was investigated. The surface oxidation of Pd nanoparticles with a hypervalent iodine reagent, [Ph2I]BF4, resulted in the generation of Pd(II)–aryl–oxo clusters, which were characterized as the crucial intermediate.
There is a long-standing debate for actual catalytic mechanisms through homogeneous and heterogeneous pathways in various organic transformations, while even using homogeneous precursors.1–3 The formation of small Pd nanoparticles (NPs) has been reported during the Pd-catalyzed cross-coupling reactions including Suzuki–Miyaura, Sonogashira, and Mizoroki–Heck reactions.4–10 The Pd NPs could be yielded by the aggregation of homogeneous Pd precursors, such as Pd(OAc)2 (OAc = acetate) and Pd(dba)2 (dba = dibenzylideneacetone). Recently, in situ formation of Pd NPs has been identified after the catalytic C–H bond functionalization.11–14 It has been proposed that the heterogeneous surface potentially catalyzed the reactions on the basis of multiple control experiments. For instance, Glorius and co-workers reported the regioselective arylation of thiophenes and polyaromatic hydrocarbons using Pd/C in the presence of hypervalent iodine(III) reagents.15,16 The reaction mechanism was proposed to use heterogeneous Pd(0) and Pd(II) species similar to the homogeneous counterpart.15–17 However, “Pd(II) NPs” is not a proper term for the intermediates considering the strong electrostatic repulsion between Pd(II) species. It is still rarely explored what the actual catalytic species are in the heterogeneous reactions.
The metal NPs have been commonly used for low-valent transition metal catalysis due to their zero-valent nature. Recently, Toste and Somorjai expanded the heterogeneous catalysis into high-valent metal chemistry that the homogeneous counterparts usually carried out.18–21 We have also characterized high valent nanostructures such as Pd(II), Pd(IV), and Rh(III), and investigated various organic transformations, hydroalkoxylation, coupling of epoxides and CO2, and C–H halogenation. Moreover, Pd nanocatalysts were used for a tandem C–H halogenation and cross-coupling reaction, mediated by heterogeneous Pd(0) and Pd(IV) catalysts.22,23
In the present study, we revisited the Pd NPs-catalyzed C–H arylation reaction of indoles and other heterocycles, and characterized the “heterogeneous” surface intermediates. By various spectroscopic analyses, the active species were characterized to be aryl–Pd(II)–oxo clusters bearing Pd(II)–C bonds on their surface, which were used to demonstrate the plausible heterogeneous Pd(0)/Pd(II) reaction mechanisms.
To identify the heterogeneous species in the Pd NPs-catalyzed C–H arylation, we have synthesized uniform Pd NPs by thermal decomposition of Pd–oleylamine complex according to the literature.24 The transmission electron microscopy (TEM) image shows that the particles are uniformly spherical and highly monodisperse with an average diameter of 4.2 ± 0.4 nm (Fig. 1a). The X-ray diffraction (XRD) shows a single broad peak at 40°, corresponding to the (111) reflection of Pd face-centered-cubic structure (JCPDS No. 46-1043) (Fig. S1, ESI†). The average domain size was estimated to be 4.7 nm from the FWHM of the (111) peak by using the Debye–Scherrer equation.25
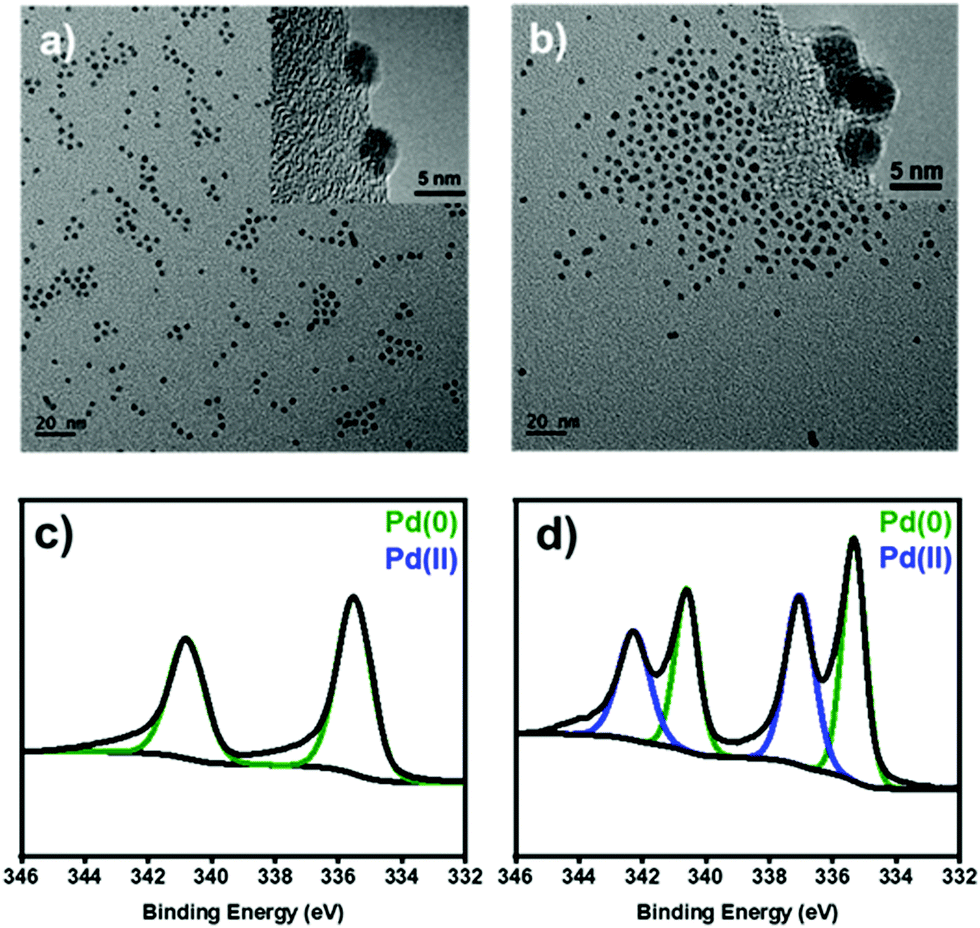 | ||
| Fig. 1 (a and b) TEM images and (c and d) XPS spectra at the Pd 3d level of (a and c) the pristine and (b and d) oxidized Pd NPs after the treatment with [Ph2I]BF4. | ||
Before the reaction study, proper solvents were selected using the Pd NPs treated with diphenyliodonium salt. In ethanol, the Pd NPs were leached out and agglomerated due to high solvent polarity (Fig. S2, ESI†). In contrast, the Pd NPs were stable in THF, maintaining its original colloidal morphology (Fig. 1b).
The reaction conditions of the C–H arylation of indole were screened in the presence of the Pd NPs and [Ph2I]BF4 (Table 1). Under the standard condition, the arylation selectively occurred at the C2-position of indole (1b) with 91% and 81% yields in ethanol and THF, respectively (Table 1, entries 1 and 2).
|
|
||||||
|---|---|---|---|---|---|---|
| No.a | Catalyst | Solvent | Yield (%) | Selectivityb (%) | ||
| 1b | 1c | 1d | ||||
| a Conditions: indole (0.2 mmol) [Ph2I]BF4 (0.26 mmol) and solvent (2.0 mL) without exclusion of air and moisture. b Determined by gas chromatography-mass spectrometry (GC-MS) analysis. Mesitylene was used as an internal standard. | ||||||
| 1 | Pd NPs | EtOH | 91 | 99 | — | — |
| 2 | Pd NPs | THF | 80 | 99 | — | — |
| 3 | PdCl2 | THF | 42 | 80 | 10 | 10 |
| 4 | Pd(TFA)2 | THF | 47 | 81 | 18 | 1 |
| 5 | Pd(OAc)2 | THF | 65 | 76 | 12 | 12 |
| 6 | Pd(OAc)2 | EtOH | 72 | 76 | 7 | 17 |
Interestingly, homogeneous Pd(II) reagents, PdCl2, Pd(TFA)2, and Pd(OAc)2 showed superior reactivity to the Pd NPs (Table S1, ESI†), but provided the products with moderate yields (42–72%). However, significant amounts of the arylation also occurred at the C3 position (1c) and biarylation at the C2 and C3 positions (1d) (Table 1, entries 3–6). The homogeneous Pd catalysts showed a large difference in reactivity and selectivity compared to the heterogeneous Pd NPs. However, the two reaction pathways are not clearly distinguished because the heterogeneous Pd was formed after the reaction with Pd(TFA)2 or Pd(OAc)2 (Fig. S3, ESI†).26
We next investigated the reaction scope of the Pd NPs-catalyzed C–H arylation of heterocyclic compounds (Scheme 1). The heterocyclic substrate was heated at 60 °C for 24 h with 1.3 equiv. of [Ph2I]BF4 and 5 mol% of Pd NPs in THF. Under the reaction condition, N-heterocyclic compounds including indole (1a), 1,5,6,7-tetrahydro-4H-indol-4-one (2a), methyl 2-carboxylate pyrrole (3a), methyl 1H-indole-5-carboxylate (4a) 5-methoxy-1H-indole (5a) and 5-methyl-1H-indole (6a) transformed into the corresponding phenylated products in 80%, 84%, 81%, 94%, 85% and 89% yields, respectively. Remarkably, the C2 arylation products were exclusively generated over the C3 arylation ones in excellent regioselectivities of >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all reactions. O-Heterocyclic compounds such as benzofuran (7a), 3-methyl benzofuran (8a), 2-furaldehyde (9a), and 2-pentylfuran (10a) also provided the C2 arylation products (7b–10b) with excellent regioselectivity in 77%, 84%, 84%, and 52% yields, respectively. In the case of C–H arylation of sulfur-containing heterocycles, benzo[b]thiophene (11a) was not effective (25% yield), but 3-methylthiophene (12a) provided the C3-arylated product (12c) with good yield of 71%. Both sulfur-containing heterocycles showed the regioselectivity at the C3 position (>99:1) in accordance with the literature.16
:1 in all reactions. O-Heterocyclic compounds such as benzofuran (7a), 3-methyl benzofuran (8a), 2-furaldehyde (9a), and 2-pentylfuran (10a) also provided the C2 arylation products (7b–10b) with excellent regioselectivity in 77%, 84%, 84%, and 52% yields, respectively. In the case of C–H arylation of sulfur-containing heterocycles, benzo[b]thiophene (11a) was not effective (25% yield), but 3-methylthiophene (12a) provided the C3-arylated product (12c) with good yield of 71%. Both sulfur-containing heterocycles showed the regioselectivity at the C3 position (>99:1) in accordance with the literature.16
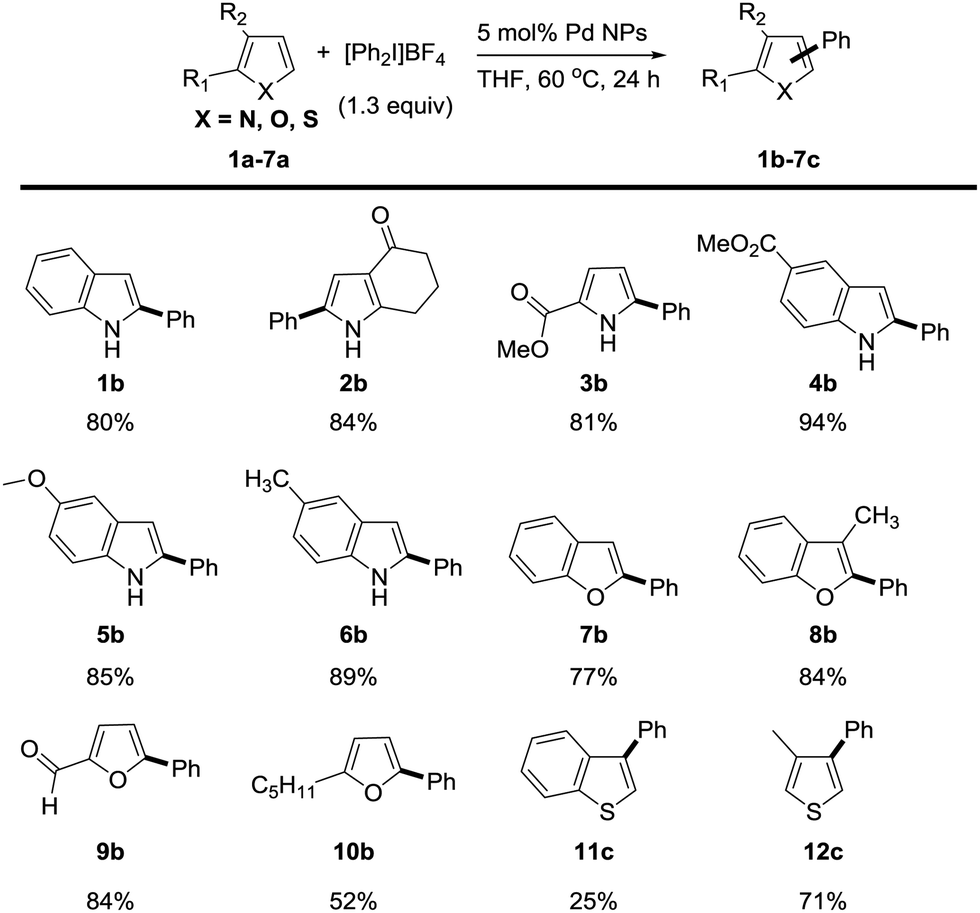 | ||
| Scheme 1 Substrate scope of indoles, furans, and thiophenes. aConditions: indole (0.2 mmol), [Ph2I]BF4 (0.26 mmol) and solvent (2.0 mL) without exclusion of air and moisture. Isolated yields are given. | ||
To confirm the involvement of the heterogeneous catalytic species in the Pd NPs-catalyzed C–H arylation, we performed hot filtration and mercury poisoning tests.27 The reaction mixture at 60 °C was filtered, and the filtrate did not contain any trace of Pd species over the detection limit (<0.01 ppm) analyzed by inductively coupled plasma-optical emission spectrometry (ICP-OES) (Fig. S5, ESI†). The addition of 2a and [Ph2I]BF4 into the filtrate did not proceed the reaction at all at 60 °C for 6 h, also supporting the absence of the catalytic Pd species in the filtrate. For the mercury poisoning test, a drop of mercury to the reaction mixture of 2b completely stopped the reaction (Fig. S6, ESI†). These control experiments indicate that the C–H arylation mainly occurred by the heterogeneous catalytic species of the Pd NPs.15,16
Generally, the heterogeneous catalyst has a high level of reusability compared to the homogeneous catalyst. After the reaction, the Pd NPs were recovered and reused five more times. The recycle experiments provided the arylated product within the narrow range of 80–86% conversions under 6 cycles (Fig. S7, ESI†). After the recycling experiments, the morphology of the catalyst was preserved as the original spherical structure well. Because only the surface was oxidized to form active species, the original hardy Pd(0) frameworks of the catalysts may lead to a high degree of reliability (see below).
Under the present oxidative reaction conditions, Pd(0) species are easily oxidizable to Pd(II) or Pd(IV) species. To clarify the oxidation states of Pd NPs during the reaction, the Pd NPs were treated with [Ph2I]BF4 in THF at 60 °C without the substrates. The X-ray photoelectron spectroscopy (XPS) shows that the pristine Pd NPs have the major two peaks at 340.2 and 335.8 eV in the Pd 3d region, corresponding to Pd(0) species (Fig. 1c, green).28 After the treatment, two new peaks distinctively emerge at 341.8 and 336.7 eV (Fig. 1d, blue), indicating the formation of Pd(II) species by surface oxidation.29 The infrared spectroscopy of the oxidized NPs showed that a new peak appeared at 730 cm−1, correlated with the typical out of plane bending peak of monosubstituted rings (Fig. S8, ESI†).30 These measurements reveal the existence of Ph–Pd(II) species under the reaction conditions.
X-ray absorption spectroscopy provides direct information of the oxidation state and coordination environment at the metal centres.31 In X-ray absorption near-edge structure (XANES), the characteristic white line at 24370 eV with the subsequent pattern at 24388 eV for oxidized Pd NPs (red) is close to that of the pristine Pd NPs (green), rather than the intense peak at 24371 eV of PdO (black). It indicates that the major fraction of the oxidized Pd NPs still involves Pd(0) species (Fig. 2a). The Fourier transform of k2-weighted Pd K-edge extended X-ray absorption fine structure (EXAFS), however, shows significantly distinct features from Pd(0) (Fig. 2b and Fig. S9, ESI†). The pristine Pd NPs show a strong single peak (green) identical to that of Pd foils, which corresponds to the Pd–Pd bond length at 2.7 Å. The coordination number of the Pd–Pd bond was simulated to be 5.0 due to the less-coordinated surface sites exposed outward (Fig. 2c). On the other hand, the oxidized Pd NPs (red) have distinct signals at 1.6–2.2 Å, corresponding to the overlaps of Pd–C and Pd–O bonds, prior to the strong peak for the Pd–Pd bond. Using the average Pd–C and Pd–O bond lengths observed in organometallic compounds,32,33 the optimal fitting provided the coordination numbers of Pd–Pd, Pd–C, and Pd–O to be 4.8, 1.2, and 1.1, respectively (Fig. 2c). Based on these spectroscopic measurements, it is reasonable to identify that the oxidized NPs are composed of polymeric aryl–Pd(II)–oxo clusters, –(Pd(II)(Ph)–O)–, bearing both Pd–Ph and Pd–O bonds. The charge of Pd(II) was stabilized by the coordination of aryl and oxo ligands. The large fraction of the Pd species is still Pd(0), indicating that the pristine Pd NPs are partially oxidized by [Ph2I]BF4. The oxidized domains are potentially located at the surface, in consideration of the coordination number (4.8) of Pd–Pd bonds nearly identical to that of the pristine NPs and large Pd(II) peaks in the XPS spectrum. These aryl–Pd(II)–oxo clusters are robust in aprotic solvents such as THF and retain their nanoscale morphology.
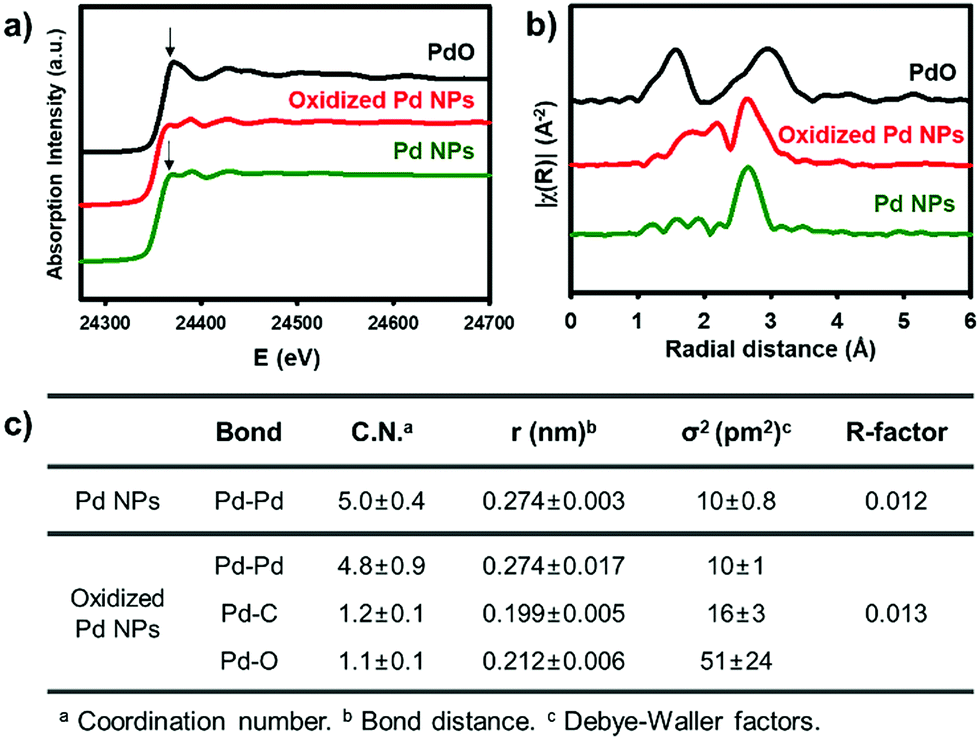 | ||
| Fig. 2 (a) Pd K-edge XANES and (b) FT of k2-weighted Pd K-edge EXAFS spectra. (c) Structural parameters obtained from the EXAFS refinement. | ||
To propose the catalytic mechanism of C–H arylation, a model reaction, arylation of indole (1a), was selected. To demonstrate the arylation reactions that occurred on the surface of Pd(II) with phenyl, we performed three control experiments. First, the catalytic arylation was carried out with excess indole (5 equiv.) in the presence of [Ph2I]BF4 (1 equiv.). The arylated product (1c) was isolated in 15% yield. Then the surface oxidation states of Pd NPs were characterized by XPS. The spectrum at the Pd 3d level after the arylation showed the large decrease of Pd(II) (Fig. 3a and b). It evidenced that coordinated phenyl groups on the Pd(II) surface were used as arylation sources. Second, the mixture of Pd NPs and [Ph2I]BF4 were heated to 60 °C for 24 h in THF. After the reaction, biphenyl was solely yielded without the original substrates (Fig. S10, ESI†). In comparison, the biphenyl compounds were not detected without Pd NPs. These results confirmed the formation of Pd(II)–aryl species during the reaction.34 Third, the addition of H2O increased the product yields (Fig. S11, ESI†) and the fraction of Pd(II) species in the XPS spectrum (Fig. S12, ESI†). However, the addition of O2 did not affect the yield. Thus, we could rationally propose that H2O participates in the catalyst generation and the C–H arylation. As a result, we propose a plausible catalytic cycle for the C–H arylation on the heterogeneous Pd (Fig. 3c). The surface of Pd NPs is oxidized to generate –(Pd(II)(Ph)O)– clusters (II), which mediate C–H activation to form the intermediate (III). The subsequent reductive elimination provides the intermediate (IV), which is further oxidized to the –(Pd(II)(Ph)O)– clusters (II) by the action of [Ph2I]BF4 and H2O.
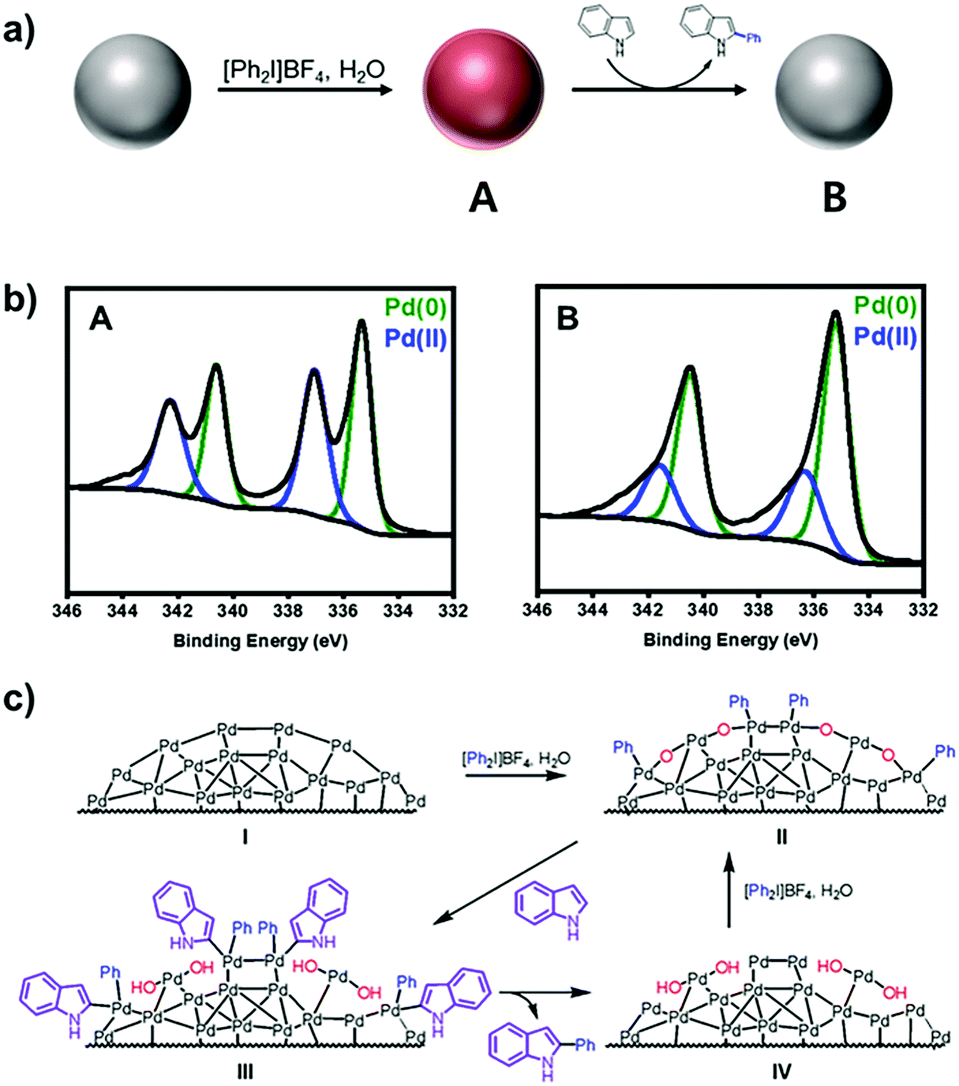 | ||
| Fig. 3 (a) Reaction scheme and (b) XPS spectra at the Pd 3d level of (A) oxidized Pd NPs after the treatment with [Ph2I]BF4 and (B) after C–H arylation in air. (c) Proposed catalytic cycle. | ||
In conclusion, we investigated the heterogeneous catalytic system promoting C–H arylation reactions of heterocycle compounds in high yield and regioselectivity. Multiple spectroscopic analyses revealed that the oxidized Pd(II) intermediates were –(Pd(II)(Ph)O)– clusters generated on the Pd(0) surface. Such heterogeneous Pd(II) species are expected to additional features superior to homogeneous catalysis, including easy access to regioselectively demanded substrates. These heterogeneous catalytic systems with homogeneous substrates would offer synergistic characteristics merging the advantages of both systems, such as enhanced catalytic performances with separation and recycle abilities.
This work was supported by Saudi Aramco-KAIST CO2 management center. The authors thank for the support by the National Research Foundation of Korea (NRF) funded by the Korea Government (MSIT) (2018R1A2B3004096, 2018R1A5A1025208, and 2020R1A2C2013420). SJC thanks the Global Frontier Center for Hybrid Interface Materials (GFHIM, grant no. NRF-2015M3A6B106526633). The authors acknowledge the Pohang Accelerator Laboratory (PAL) for beamline use. EXAFS experiments at PLS were supported by MSIP and POSTECH.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- A. Biffis, P. Centomo, A. D. Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS.
- L. X. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS.
- Y. Zhao, L. Du, H. Li, W. Xie and J. Chen, J. Phys. Chem. Lett., 2019, 10, 1286–1291 CrossRef CAS.
- S. Shabbir, S. Lee, M. Lim, H. Lee, H. Ko, Y. Lee and H. Rhee, J. Organomet. Chem., 2017, 846, 296–304 CrossRef CAS.
- S. Santoro, S. I. Kozhushkov, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 3471–3493 RSC.
- A. J. Reay and L. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289–16307 RSC.
- L. Djakovitch and F.-X. Felpin, ChemCatChem, 2014, 6, 2175–2187 CrossRef CAS.
- D. Pla and M. Gómez, ACS Catal., 2016, 6, 3537–3552 CrossRef CAS.
- L. J. S. Fairlamb, A. R. Kapdi, A. F. Lee, G. Sanchez, G. Lopez, J. L. Serrano, L. Garcia, J. Perez and E. Perez, Dalton Trans., 2004, 3970–3981 RSC.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderckx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC.
- J. G. de Vries, Dalton Trans., 2006, 421–429 RSC.
- T. E. Storr, C. G. Baumann, R. J. Thatcher, S. D. Ornellas, A. C. Whitwood and L. J. S. Fairlamb, J. Org. Chem., 2009, 74, 5810–5821 CrossRef CAS.
- K. D. Collins, R. Honeker, S. Vásquez-Céspedes, D.-T. D. Tang and F. Glorius, Chem. Sci., 2015, 6, 1816–1824 RSC.
- D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809–1813 CrossRef CAS.
- D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS.
- R. Ye, A. V. Zhukhovitskiy, C. V. Deraedt, F. D. Toste and G. A. Somorjai, Acc. Chem. Res., 2017, 50, 1894–1901 CrossRef CAS.
- C. Deraedt, R. Ye, W. T. Ralston, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2017, 139, 18084–18092 CrossRef CAS.
- A. J. Brown, D. Pinkowicz, M. R. Saber and K. R. Dunbar, Angew. Chem., Int. Ed., 2015, 52, 5864–5868 CrossRef.
- Y. Li, J. H.-C. Liu, C. A. Witham, W. Huang, M. A. Marcus, S. C. Fakra, P. Alayoglu, Z. Zhu, C. M. Thompson, A. Arjun, K. Lee, E. Gross, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2011, 133, 13527–13533 CrossRef CAS.
- K. Kim, Y. Jung, S. Lee, M. Kim, D. Shin, H. Byun, S. J. Cho, H. Song and H. Kim, Angew. Chem., Int. Ed., 2017, 56, 6952–6956 CrossRef CAS.
- M. Kim, S. Lee, K. Kim, D. Shin, H. Kim and H. Song, Chem. Commun., 2014, 50, 14938–14941 RSC.
- M. Kim, J. C. Park, A. Kim, K. H. Park and H. Song, Langmuir, 2012, 28, 6441–6447 CrossRef CAS.
- B. D. Hall, D. Zanchet and D. Ugarte, J. Appl. Crystallogr., 2000, 33, 1335–1341 CrossRef CAS.
- V. Arun, P. O. V. Reddy, M. Pilania and D. Kumar, Eur. J. Org. Chem., 2016, 2096–2100 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS.
- X. Fan, F. Wang, T. Zhu and H. He, J. Environ. Sci., 2012, 24(3), 507–511 CrossRef CAS.
- L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, J. Phys. Chem. C, 2012, 116, 19342–19348 CrossRef CAS.
- M. Margoshes and V. A. Fassel, Spectrochim. Acta, 1955, 7, 14–24 CAS.
- K. R. Priolkar, P. Bera, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro and N. P. Lalla, Chem. Mater., 2002, 14, 2120–2128 CrossRef CAS.
- P. L. Alsters, J. Boersma, W. J. J. Smeets, A. L. Spek and G. van Koten, Organometallics, 1993, 12, 1639–1647 CrossRef CAS.
- J. M. Keith, W. A. Goddard and J. Oxgaard, J. Am. Chem. Soc., 2007, 129, 10361–10369 CrossRef CAS.
- Rearrangement of Pd(II)–Ph species could provide Ph–Pd(II)–Ph species, resulting in Pd(0) and biphenyl.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc06716d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |