
A metal–organic framework supported iridium catalyst for the gas phase hydrogenation of ethylene†
Ricardo A.
Perlata
a,
Michael T.
Huxley
a,
Zhaolin
Shi
b,
Yue-Biao
Zhang
 b,
Christopher J.
Sumby
*a and
Christian J.
Doonan
*a
b,
Christopher J.
Sumby
*a and
Christian J.
Doonan
*a
aDepartment of Chemistry and the Centre for Advanced Nanomaterials, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: christopher.sumby@adelaide.edu.au; christian.donan@adelaide.edu.au
bSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 201210, China
First published on 3rd November 2020
Abstract
The mutable structures of metal–organic frameworks (MOFs) allow their use as novel supports for transition metal catalysts. Herein we prepare an iridium bis(ethylene) catalyst bound to the neutral N-donors of a MOF structure and show that the compound is a stable gas phase ethylene hydrogenation catalyst. The data illustrate the need to carefully consider the inner sphere (support) and outer sphere (anion) chemistry.
Metal–organic frameworks (MOFs) are a class of porous, crystalline materials that are comprised of metal nodes interconnected by organic linkers to produce open networked structures.1,2 Due to their modular synthesis, crystallinity and high porosity, MOFs are attractive platform materials for exploring the gas phase activity of well-defined molecular catalysts in a heterogeneous environment.3 Two strategies that have been employed to successfully incorporate discrete organometallic catalysts within MOF pores are: (1) anchoring to the metal oxide nodes;4–6 and (2) binding to donor sites built into the organic linkers.3,7–12 Examples of the former have been reported by the groups of Gates, Gagliardi and Farha who showed that nature of the support has a marked influence on the chemistry of Ir and Ni ethylene hydrogenation catalysts bound to the metal nodes of Zr and Hf-based MOFs.4–6 Recently, we showed that a Rh(I) organometallic catalyst, 1·[Rh(C2H4)2]X (X = Cl, BF4), bound to the organic backbone of a manganese-based MOF (1, [Mn3L3] (L = bis-(4-carboxyphenyl-3,5-dimethyl-pyrazol-1-yl)methane)) via bispyrazole moieties was also active for the gas phase hydrogenation of ethylene.3 In contrast to the aforementioned Ir and Ni systems, the Rh cation is supported by neutral nitrogen donors rather than charged oxygen atoms and thus a counter anion is required for charge balance. Interestingly, we found that the activity of the catalyst was highly anion dependent; 1·[Rh(C2H4)2]BF4 showed much greater activity than the 1·[Rh(C2H4)2]Cl sample. Additionally, we observed that the Rh centres were prone to deactivation via the formation of nanoparticles under an excess of hydrogen. To further explore gas phase hydrogenation chemistry in 1 and expand our fundamental knowledge of MOFs as supports for site-isolated molecular catalysts, we installed an Ir(I) bis-ethylene organometallic catalyst into the pores of 1 and examined its reactivity towards ethylene hydrogenation in the gas phase. We found that the Ir catalyst is more stable towards hydrogen gas than the corresponding Rh species. Furthermore, we again observed that the catalytic activity is anion dependent, however, for the Ir catalyst the chloride complex shows greater reactivity than the BF4 species.
Using a post-synthetic metalation (PSMet) process developed to prepare 1·[Rh(C2H4)2]X (X = [RhCl2(C2H4)2]−, Cl−, BF4−),3 we reacted MOF 1 with [Ir(C2H4)2Cl]2 in ethanol at 65 °C under an ethylene atmosphere for three days (Fig. 1 and Fig. S5.3, ESI†). This provided the iridium metalated MOF, 1·IrCl (Fig. 1). To determine the extent of metalation, the yellow crystals of 1·IrCl were examined by Energy Dispersive X-ray (EDX) analysis. The data indicated ca. 85% occupancy of the chelating sites in 1, and an Ir![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cl ratio of 1:1. Furthermore, the bulk crystallinity of 1·IrCl, post metalation, was confirmed by Powder X-ray Diffraction (PXRD) data (Fig S4.1, ESI†).
:Cl ratio of 1:1. Furthermore, the bulk crystallinity of 1·IrCl, post metalation, was confirmed by Powder X-ray Diffraction (PXRD) data (Fig S4.1, ESI†).
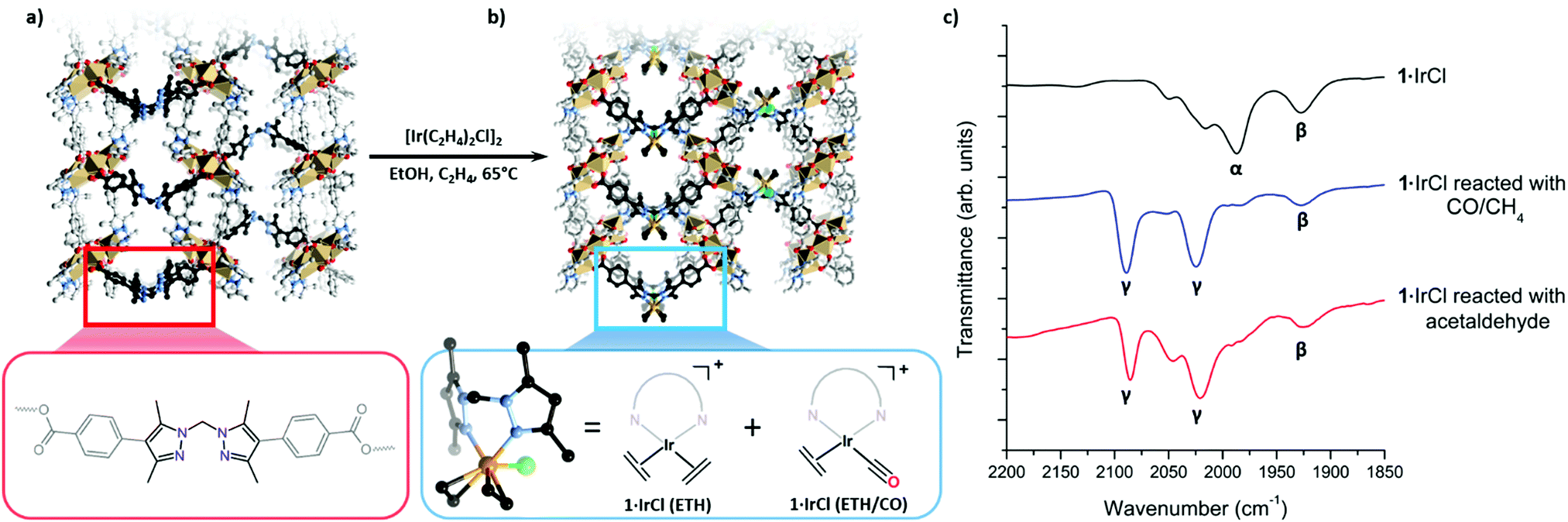 | ||
| Fig. 1 (a) A perspective view of the X-ray crystal structure of MOF 1 and a representation of the ligand moiety shown in the enlargement. (b) Post-synthetic metalation of 1 with [Ir(C2H4)2Cl]2 yields 1·IrCl, while the expanded panel shows that the chelated Ir centre can be modelled as a of bis-ethylene Ir(I) chloride complex (1·IrCl(ETH)) with an adjacent low occupancy Ir centre with an un-resolved coordination sphere, likely corresponding to the monocarbonyl ethylene Ir(I) (1·IrCl(ETH/CO)) complex observed via IR spectroscopy (Ir, orange; Cl, green; C, black/grey; N, blue; O, red; Mn, beige; H omitted for clarity). (c) IR spectra of 1·IrCl as synthesised, following exposure to CO/CH4 and following reaction with acetaldehyde in ethanol, displaying the changes in the CO stretching region. Bands attributed to Ir(I) monocarbonyl complexes are labelled α and β while the gem dicarbonyl stretches of 1·[Ir(CO)2] are labelled γ. | ||
The excellent condition of 1·IrCl single crystals prompted us to investigate the coordination sphere of the iridium centres using single crystal X-ray diffraction (SCXRD). However, refinement of the structure indicated considerable disorder of the metalation site (Fig. 1b). With the MOF backbone in place, a structural model was developed of the disordered metalation site by considering the electron density map and spectroscopic data for the sample. This reveals the main species is a bis(ethylene) complex of iridium, 1·[Ir(C2H2)2Cl] (referred to from this point as 1·IrCl(ETH)); the Ir3B atom is modelled with a site occupancy factor of 0.25, 50% occupancy of the metalation site due to the mirror plane. An ethylene ligand is bonded with Ir3B–C with bond lengths of 2.256(19) and 2.25(2) Å and slightly canted, as observed for the Rh(I) complex.3 A Cl atom was modelled with 0.25 occupancy bound to an axial coordination site of the iridium centre (Ir3B–Cl 2.646(13) Å). Unusually for this MOF, the linker where post-synthetic metalation occurs is identifiably disordered (B:A = 0.75:0.25) and the electron density map supports a second, low occupancy Ir position (Ir3A, 0.05, 10% occupancy) giving a total Ir occupancy of 0.6 that is slightly less than that determined by EDX analysis. Another Cl site, also refined at 0.05 to match the Ir3A occupancy, was located in the MOF pore in the pocket normally occupied by anions.3,13 The coordination environment of this second component could not be determined but partially occupied CO and ethylene ligands were included in the formula based on the spectroscopic evidence (see below). Together, these components suggest that the Ir centre in 1·IrCl is a mixture of the target bis-ethylene complex (1·IrCl(ETH)) and a potential carbonyl ethylene Ir(I) complex (1·IrCl(ETH/CO)) (Fig. 1b).
To probe the coordination sphere of the Ir sites, we activated samples of 1·IrCl and analysed the headspace above the sample with gas phase NMR following exposure to CO and H2, respectively. The enhanced binding strength of CO in comparison to ethylene will convert putative 1·IrCl(ETH) to 1·[Ir(CO)2] and allow direct measurement of bound ethylene in the reaction headspace. Furthermore, bound ethylene can be measured by the introduction of H2 which will reduce the ethylene ligand to ethane. These experiments confirmed the presence of slightly more than one molecule of ethylene per Ir site (S7.2, ESI†). Thus, these data are consistent with a significant proportion of the iridium in 1·IrCl being 1·IrCl(ETH) but also support the formation of the additional 1·IrCl(ETH/CO) species observed by SCXRD.
To investigate this second Ir species, IR spectroscopy measurements were conducted on the metalated MOF crystals. The presence of weak stretches between 1925–2080 cm−1 that could not be assigned to 1·IrCl(ETH) (Fig. 2a, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretches typically observed below 1600 cm−1)14–16 confirm an additional species is present. Although bis-ethylene Ir(I) complexes of substituted tris-pyrazolylborates and tris-pyrazolylmethanes are known to form Ir(III) alkyl hydride complexes upon heating,17–19 the Ir–H stretches (or the crotyl hydrides that subsequently form) appear at higher wavenumbers (2100 to 2200 cm−1)17 and are thus inconsistent with the data for 1·IrCl. As eluded to in the structural discussion above, we advance that the IR stretches observed in the spectrum of 1·IrCl correspond to C
C stretches typically observed below 1600 cm−1)14–16 confirm an additional species is present. Although bis-ethylene Ir(I) complexes of substituted tris-pyrazolylborates and tris-pyrazolylmethanes are known to form Ir(III) alkyl hydride complexes upon heating,17–19 the Ir–H stretches (or the crotyl hydrides that subsequently form) appear at higher wavenumbers (2100 to 2200 cm−1)17 and are thus inconsistent with the data for 1·IrCl. As eluded to in the structural discussion above, we advance that the IR stretches observed in the spectrum of 1·IrCl correspond to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretches from an Ir–CO moiety formed via the decarbonylation of ethanol during the PSMet of 1.15,20–22 We note that this chemistry has been observed for P,N,P-pincer complexes of Ir(I).20,22 Alcohol decarbonylation by simple Ir(I) complexes is thought to proceed via dehydrogenation of the alcohol to form the corresponding aldehyde which can subsequently undergo decarbonylation to form an Ir(III) monocarbonyl complex (Fig. 2b).20–22 The three day reaction time and relatively high temperature (65 °C) needed for metalation of 1 with [Ir(C2H4)2Cl]2 would provide suitable conditions for this reaction. To confirm that the CO originates from decarbonylation of ethanol, we performed two experiments: first, we metalated 1 with [Ir(C2H4)2Cl]2 in the presence of the expected aldehyde intermediate, acetaldehyde. After three days at 65 °C, the MOF crystals were analysed by IR spectroscopy which revealed a more pronounced monocarbonyl CO stretching band at 1980 cm−1 (Fig. S8.2, ESI†) and additional bands centred at 2088 and 2024 cm−1 which correspond to an Ir(I) dicarbonyl species (see Fig. 2b for proposed reaction mechanism). Secondly, we reacted 1·IrCl crystals with acetaldehyde at 65 °C in ethanol and observed a similar increase in the intensity of the CO stretches and formation of the dicarbonyl complex (Fig. 1c). The appearance of higher intensity CO stretching bands in the presence of acetaldehyde supports the hypothesis that ethanol decarbonylation is the origin of the unexpected carbonyl species present in 1·IrCl.
O stretches from an Ir–CO moiety formed via the decarbonylation of ethanol during the PSMet of 1.15,20–22 We note that this chemistry has been observed for P,N,P-pincer complexes of Ir(I).20,22 Alcohol decarbonylation by simple Ir(I) complexes is thought to proceed via dehydrogenation of the alcohol to form the corresponding aldehyde which can subsequently undergo decarbonylation to form an Ir(III) monocarbonyl complex (Fig. 2b).20–22 The three day reaction time and relatively high temperature (65 °C) needed for metalation of 1 with [Ir(C2H4)2Cl]2 would provide suitable conditions for this reaction. To confirm that the CO originates from decarbonylation of ethanol, we performed two experiments: first, we metalated 1 with [Ir(C2H4)2Cl]2 in the presence of the expected aldehyde intermediate, acetaldehyde. After three days at 65 °C, the MOF crystals were analysed by IR spectroscopy which revealed a more pronounced monocarbonyl CO stretching band at 1980 cm−1 (Fig. S8.2, ESI†) and additional bands centred at 2088 and 2024 cm−1 which correspond to an Ir(I) dicarbonyl species (see Fig. 2b for proposed reaction mechanism). Secondly, we reacted 1·IrCl crystals with acetaldehyde at 65 °C in ethanol and observed a similar increase in the intensity of the CO stretches and formation of the dicarbonyl complex (Fig. 1c). The appearance of higher intensity CO stretching bands in the presence of acetaldehyde supports the hypothesis that ethanol decarbonylation is the origin of the unexpected carbonyl species present in 1·IrCl.
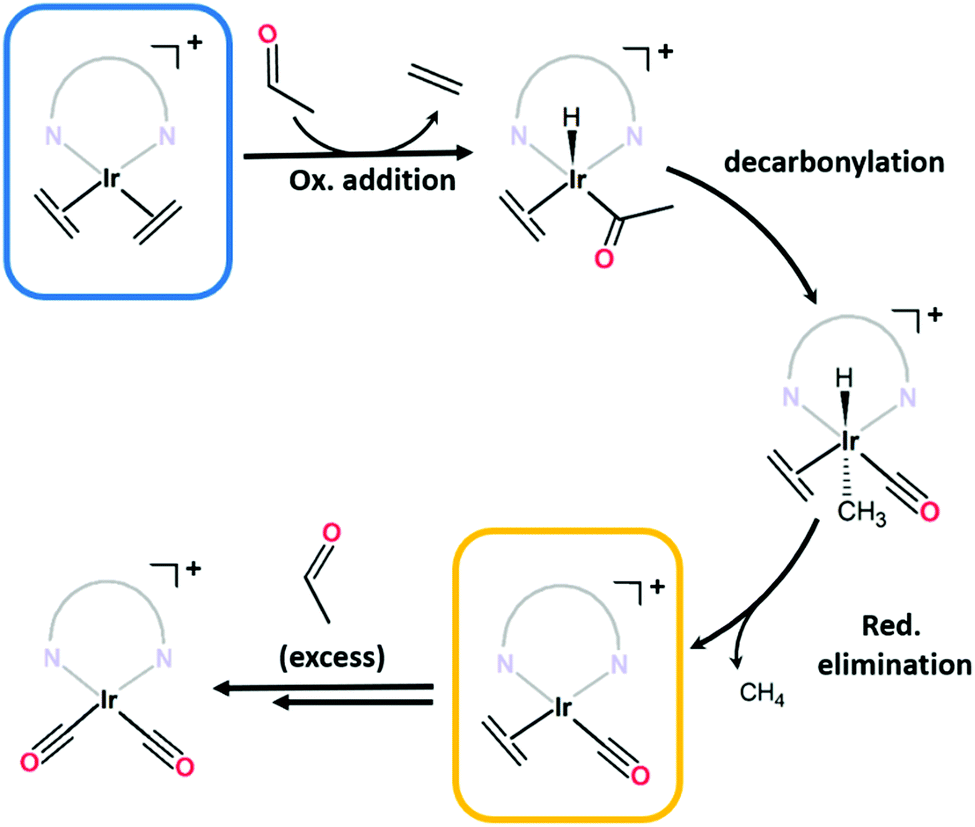 | ||
| Fig. 2 The proposed mechanism via which mono- and dicarbonyl complexes are generated from decarbonylation of acetaldehyde. The target bis(ethylene) complex 1·IrCl(ETH), and the 1·IrCl(ETH/CO) by product that is also observed, are shown in the blue and yellow boxes. Acetaldehyde can be formed by dehydrogenation of ethanol. | ||
The relatively low intensity of the ν(CO) stretches indicates that carbonyl complexes are a minor component of the Ir species in 1·IrCl. Thus, the combination of SCXRD, NMR and IR evidence suggests that 1·IrCl is mainly the targeted 1·IrCl(ETH) species (ca. 66%), which is anticipated to be a promising catalyst for gas-phase hydrogenation, and a mixture of Ir carbonyl complexes formed during synthesis, predominantly 1·IrCl(ETH/CO) (ca. 34%). Despite extensive attempts to form pure 1·[Ir(C2H4)2], metalation in solvents other than ethanol was unsuccessful either due to decomposition of the Ir(I) precursor or the unfavourable arrangement of the pyrazole sites of the MOF in these solvents.23
Given our previous observation that the coordinating tendency of the anion can significantly impact the catalytic activity of organometallic catalysts loaded into 1,3 we exchanged the chloride anion in 1·IrCl for tetrafluoroborate using facile anion exchange with NaBF4 in methanol under an ethylene atmosphere (S1.3, ESI†). EDX analysis confirmed complete removal of chloride from the sample (Table S1, ESI†). IR spectroscopy revealed minor changes in the position of the CO stretches (2020 cm−1) (Fig. S8.1, ESI†). The sample was determined to be ca. 58% 1·IrBF4(ETH) and 42% 1·IrBF4(ETH/CO) based on spectroscopic analysis, noting that this might be a slight over estimation of the latter due to facile loss of ethylene in 1·IrBF4.
With an enhanced understanding of the sample composition, we investigated the stability of 1·IrCl and 1·IrBF4 towards activation and hydrogen exposure. Activation from dry pentane at room temperature for 2 h yielded surface areas of 575 and 462 m2 g−1, for 1·IrCl and 1·IrBF4 respectively, which are consistent with that observed for other metalated samples of 1.24 Activated samples of 1·IrCl and 1·IrBF4 were exposed to pure hydrogen (1 bar), and unlike the analogous 1·[Rh(C2H4)2]X samples,3 the crystals did not become black (due to formation of Rh nanoparticles) but instead transformed from pale yellow to off-white. Furthermore, Transition Electron Microscopy (TEM) analysis of the sample showed no evidence of Ir nanoparticles (S3, ESI†). We then studied the gas-phase hydrogenation of ethylene using 1·IrCl and 1·IrBF4 in a 1:1 mixture of ethylene and hydrogen (2 bar) at 46 °C. Using gas-phase NMR to monitor the evolution of the reaction within a sealed high-pressure NMR tube, we observed the conversion of ethylene to ethane and consumption of hydrogen. Both catalysts are active with the 1·IrCl material retaining consistent activity over the course of four successive cycles (1·IrCl: initial TOF15% 44 h−1, average TOF15% over four cycles: 49 h−1) (Fig. 3b). The BF4 derivative is more active initially (TOF20% of 95 h−1) but loses some activity on cycling (average TOF15% over cycles 2–4: 24 h−1). This suggests that the chloride anion plays a role in stabilising the active catalyst, as has been observed in alcohol decarbonylation using molecular iridium catalysts.21
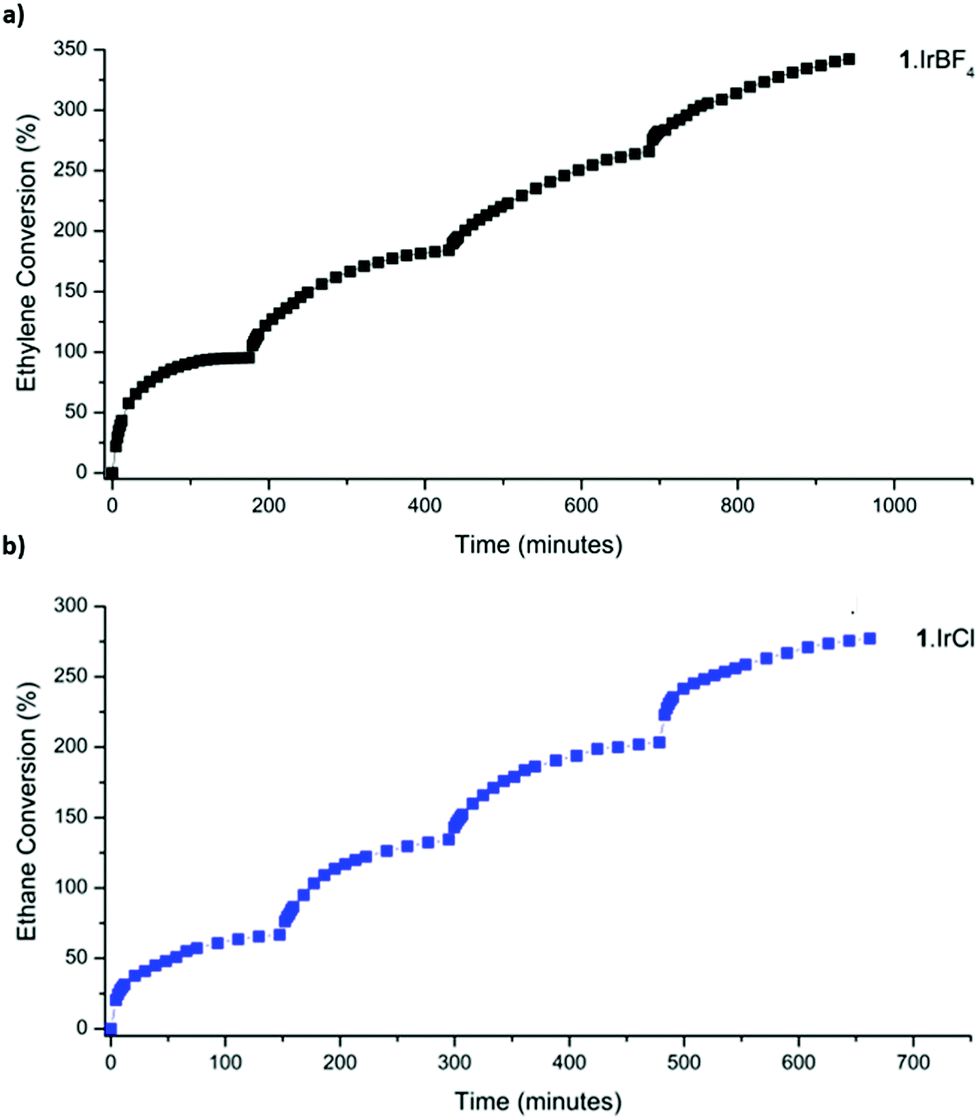 | ||
| Fig. 3 Catalysis data for the hydrogenation of ethylene at 46 °C (2 bar) using (a) 1·IrBF4 and (b) 1·IrCl. | ||
The average TOF15% over four cycles of 49 h−1 demonstrated by 1·IrCl is comparable to the activity reported for MOF-node supported [Ir(C2H4)2] species,4 although it most likely underestimates the activity of 1·IrCl(ETH) due to the presence of less active carbonyl species.25 Various studies have investigated the gas phase ethylene hydrogenation activity of [Ir(C2H4)2] complexes supported on the inorganic nodes of the MOFs UiO-66, UiO-67 and NU-1000, as well as zeolite and MgO supports.4,25–28 Under flow-reaction conditions operating at room temperature, these systems typically display TOFs up to 62 h−1 for the MOF supported systems and up to 2160 h−1 on HY Zeolite.26 Notably, these complexes are rendered charge neutral via the support, negating the need for the charge balancing anions employed in 1·IrCl and 1·IrBF4. Furthermore, the electron donating or withdrawing ability of the support plays a significant role in the catalytic activity of the supported Ir complex.25,26 The highly acidic Al sites in HY Zeolite generate electron deficient Ir centres which promote rapid hydrogenation (TOF 2160 h−1) due to their tendency to interact with and activate several reagent molecules simultaneously.25,29 Based on comparison of the ν(CO) stretching frequencies of zeolite-supported gem-dicarbonyl complexes (2109, 2038 cm−1)25 and our system (2088, 2025 cm−1), the Ir centre in 1·IrCl is more electron rich and therefore less active in hydrogenation (although it appears intermediate between Zeolite and MOF-node supported Ir centres,4,26 for example UiO-66 node-supported [Ir(CO)2]: ν(CO) 2074 cm−1, 1996 cm−1).
In conclusion, metalation of a porous MOF with [Ir(C2H4)2Cl]2 resulted in a solid-state catalyst active towards ethylene hydrogenation. A combination of SCXRD, IR and NMR spectroscopy, and EDX analysis elucidated that the MOF pores were decorated with non-homogeneous single-atom active sites including 1·IrCl(ETH) and carbonyl ligated by-products e.g.1·IrCl(ETH/CO). The Ir(I) ethylene complexes are stable catalysts (cycled 4 times with retention of activity) that compare favourably with MOF node supported variants in terms of activity. Furthermore, we found that the use of a neutral support allows for modulation of catalyst activity and stability via the counter anion. This work expands our knowledge of MOFs as supports for single site catalysts and shows how the chemistry can be tuned by both inner and outer-sphere effects.
CJS and CJD gratefully acknowledge the Australian Research Council for funding (DP190101402). This research was undertaken in part using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. RAP gratefully acknowledges Adelaide Scholarship International.
Conflicts of interest
There are no conflicts to declare.References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 974–985 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS.
- R. A. Peralta, M. T. Huxley, J. D. Evans, T. Fallon, H. Cao, M. He, X. S. Zhao, S. Agnoli, C. J. Sumby and C. J. Doonan, J. Am. Chem. Soc., 2020, 142, 13533–13543 CrossRef CAS.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS.
- X. Wang, X. Zhang, R. Pandharkar, J. Lyu, D. Ray, Y. Yang, S. Kato, J. Liu, M. C. Wasson, T. Islamoglu, Z. Li, J. T. Hupp, C. J. Cramer, L. Gagliardi and O. K. Farha, ACS Catal., 2020, 10, 8995–9005 CrossRef CAS.
- V. Bernales, D. Yang, J. Yu, G. Gümüşlü, C. J. Cramer, B. C. Gates and L. Gagliardi, ACS Appl. Mater. Interfaces, 2017, 9, 33511–33520 CrossRef CAS.
- X. Feng, Y. Song, Z. Li, M. Kaufmann, Y. Pi, J. S. Chen, Z. Xu, Z. Li, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 11196–11203 CrossRef CAS.
- T. Sawano, Z. Lin, D. Boures, B. An, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 9783–9786 CrossRef CAS.
- T. Sawano, P. Ji, A. R. McIsaac, Z. Lin, C. W. Abney and W. Lin, Chem. Sci., 2015, 6, 7163–7168 RSC.
- A. Burgun, C. J. Coghlan, D. M. Huang, W. Chen, S. Horike, S. Kitagawa, J. F. Alvino, G. F. Metha, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef.
- R. J. Young, M. T. Huxley, E. Pardo, N. R. Champness, C. J. Sumby and C. J. Doonan, Chem. Sci., 2020, 11, 4031–4050 RSC.
- M. I. Gonzalez, E. D. Bloch, J. A. Mason, S. J. Teat and J. R. Long, Inorg. Chem., 2015, 54, 2995–3005 CrossRef CAS.
- M. T. Huxley, R. J. Young, W. M. Bloch, N. R. Champness, C. J. Sumby and C. J. Doonan, Organometallics, 2019, 38, 3412–3418 CrossRef CAS.
- H. Huber, G. A. Ozin and W. J. Power, J. Am. Chem. Soc., 1976, 98, 6508–6511 CrossRef CAS.
- J. Hirsch, S. DeBeer George, E. I. Solomon, B. Hedman, K. O. Hodgson and J. N. Burstyn, Inorg. Chem., 2001, 40, 2439–2441 CrossRef CAS.
- S. B. Mohsin, M. Trenary and H. J. Robota, J. Phys. Chem., 1991, 95, 6657–6661 CrossRef CAS.
- I. I. Padilla-Martínez, M. L. Poveda, E. Carmona, M. A. Monge and C. Ruiz-Valero, Organometallics, 2002, 21, 93–104 CrossRef.
- C. K. Ghosh, J. K. Hoyano, R. Krentz and W. A. G. Graham, J. Am. Chem. Soc., 1989, 111, 5480–5481 CrossRef CAS.
- Y. Alvarado, O. Boutry, E. Gutiérrez, A. Monge, M. C. Nicasio, M. L. Poveda, P. J. Pérez, C. Ruíz, C. Bianchini and E. Carmona, Chem. – Eur. J., 1997, 3, 860–873 CrossRef CAS.
- J. G. Melnick, A. T. Radosevich, D. Villagran and D. G. Nocera, Chem. Commun., 2010, 46, 79–81 RSC.
- E. P. Olsen and R. Madsen, Chemistry, 2012, 18, 16023–16029 CrossRef CAS.
- S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Organometallics, 2006, 25, 3007–3011 CrossRef CAS.
- R. Peralta, M. Huxley, R. Young, O. M. Linder-Patton, J. D. Evans, C. J. Doonan and C. J. Sumby, Faraday Discuss., 2020 10.1039/D0FD00012D.
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS.
- J. Lu, P. Serna and B. C. Gates, ACS Catal., 2011, 1, 1549–1561 CrossRef CAS.
- D. Yang, S. O. Odoh, J. Borycz, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, ACS Catal., 2016, 6, 235–247 CrossRef CAS.
- A. Uzun and B. C. Gates, J. Am. Chem. Soc., 2009, 131, 15887–15894 CrossRef CAS.
- A. Uzun, V. A. Bhirud, P. W. Kletnieks, J. F. Haw and B. C. Gates, J. Phys. Chem. C, 2007, 111, 15064–15073 CrossRef CAS.
- J. Lu, P. Serna, C. Aydin, N. D. Browning and B. C. Gates, J. Am. Chem. Soc., 2011, 133, 16186–16195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details of the metalated MOFs, microscopy, powder X-ray diffraction, isotherm, and spectroscopy data, and methods for the catalysis experiments. CCDC 2025173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc06058e |
| This journal is © The Royal Society of Chemistry 2020 |