
Recent developments in biomolecule-based nanoencapsulation systems for antimicrobial delivery and biofilm disruption
Mark Louis P.
Vidallon
* and
Boon Mian
Teo
 *
*
School of Chemistry, Faculty of Science, Monash University, Clayton, VIC 3800, Australia. E-mail: boonmteo@gmail.com; mark.vidallon@monash.edu
First published on 19th October 2020
Abstract
Biomolecules are very attractive nanomaterial components, generally, due to their biocompatibility, biodegradability, abundance, renewability, and sustainability, as compared to other resources for nanoparticle-based delivery systems. Biomolecule-based nanoencapsulation and nanodelivery systems can be designed and engineered for antimicrobial cargos in order to surmount classical and current challenges, including the emergence of multi-drug resistant strains of microorganisms, the low effectiveness and limitations in the applicability of the present antimicrobials, and biofilm formation. This feature article highlights the recent applications and capabilities of biomacromolecule-based nanomaterials for the delivery and activity enhancement of antimicrobials, and disruption of biofilms. Unique properties of some nanomaterials, arising from specific biomacromolecules, were also emphasized. We expect that this review will be helpful to researchers in engineering new types of antimicrobial nanocarriers, hybrid particles and colloidal systems with tailored properties.
1. Biofilm production as a manifestation of antimicrobial resistance
Antimicrobial therapy has been one of the most successful advances of modern medicine, laying the foundations for complex and innovative medical interventions that have allowed lengthening of the expected human life span worldwide. However, existing antibiotics are losing efficacy over time due to the emergence of drug resistance in pathogenic microorganisms.1 From an evolutionary perspective, bacteria use two major genetic strategies to adapt to the effects of antibiotics, namely: mutations in genes, often associated with the mechanism of action of the compound; and acquisition of foreign DNA coding for resistance determinants through horizontal gene transfer.1,2 These genes encode for a variety of antimicrobial resistance mechanisms, which allow microorganism to: (1) produce substances that can destroy or modify the antimicrobial molecule, thus rendering it inactive; (2) generate molecules, structures and internal machineries that either prevent the antibiotic target from penetrating the cytoplasmic membrane or actively extrude the antimicrobial compound; (3) protect the target sites either by biosynthesizing compounds that compete with the antimicrobial molecule or by directly introducing alterations to the target site structures; and (4) develop resistant phenotypes as a result of global cell adaptive response. Antibiotic overuse and misuse has been strongly linked to the unceasing emergence and propagation of resistant strains.1Biofilm formation is strong evidence of this evolutionary adaptation, which also perpetuates antimicrobial resistance. Biofilms are sessile microbial communities attached to a living surface and embedded within a matrix of extracellular polymeric substances (EPS), including exopolysaccharides, proteins and nucleic acids, that they produce.3–5 Biofilms appear to be the predominant and natural state of some microorganisms compared to their planktonic state, despite the high growth and reproduction rate of planktonic cells. Biofilm operates as a favourable environment for microbial growth. It can act as a filter and trap nutrients from the surroundings and distribute these through water channels between microcolonies, which can also act as an expulsion or excretory system of harmful metabolites.4–6 Biofilms promote bacterial tolerance towards unfavourable environmental conditions, including water or blood flood flow, by attaching to biotic or abiotic surfaces. The EPS matrix also functions as a defensive, impenetrable layer (Fig. 1), which minimizes the exposure of cells to antimicrobial agents and protects the cells from other changes in environmental conditions. Microcolonies embedded within the biofilm matrix also have limited mobility, but are in high density, which favours transfer of plasmids or extracellular DNA, some of which contain antimicrobial resistance genes.3 Recent studies have shown that the efficacy of antibiotics against biofilms is also affected by various mechanisms, including the slow growth of biofilm organisms, spatial heterogeneity and different penetration pathways (voids and dense cell clusters) in the biofilm structure, and the presence of drug-resistant or drug-tolerant microbial physiology.7,8 Penetration of antimicrobials through the biofilm is also dependent on the antimicrobials used – while some molecules diffuse quickly and completely though some biofilms, others cannot.9
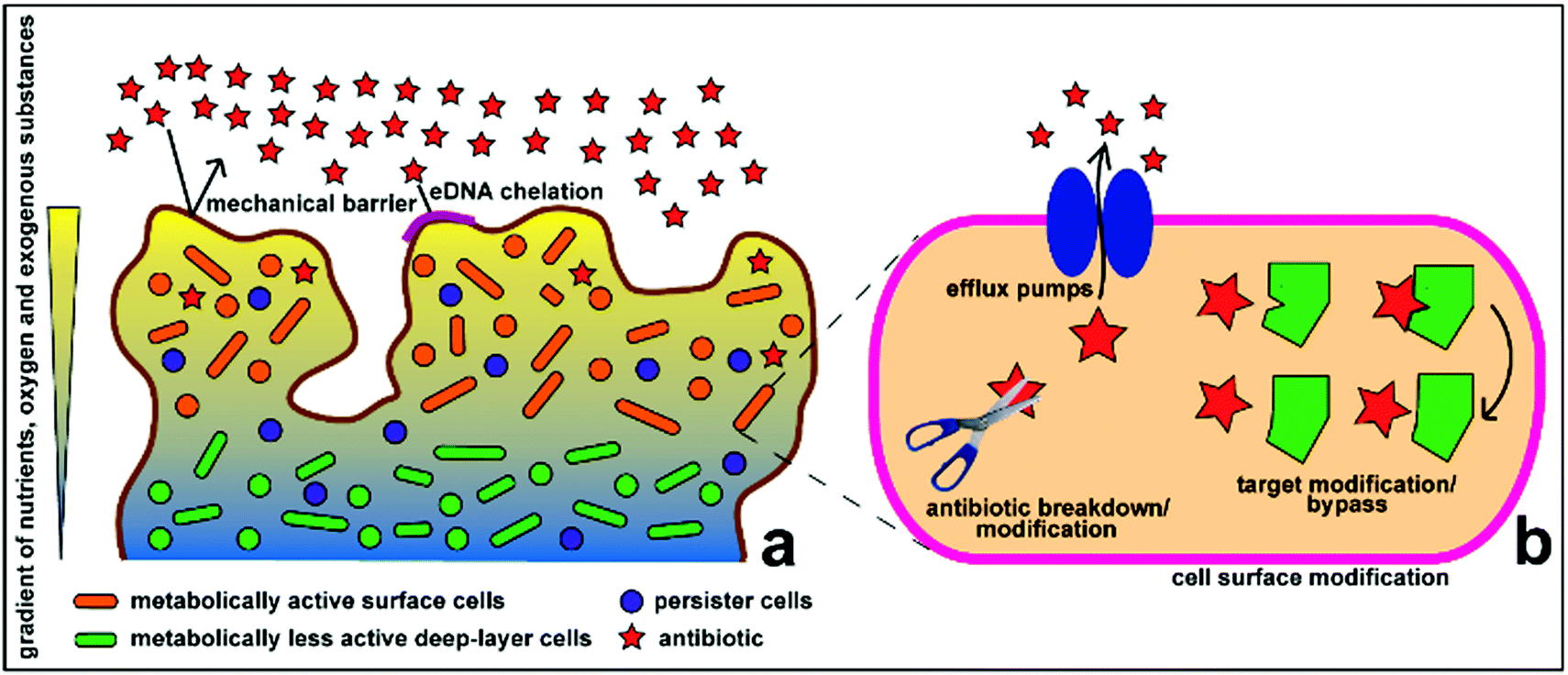 | ||
| Fig. 1 Schematic representation of the antimicrobial resistance mechanism of biofilms at the (a) community and (b) cellular levels. Reproduced with permission.10 Copyright 2015, MDPI. | ||
Biofilm formation has been a major challenge in medical sciences, especially in some disease, such as in cystic fibrosis, in wound infections, and in biomedical device-associated treatments, surgeries, implants and prostheses.5,11 Biofilms in indwelling medical devices are difficult to treat and are much more problematic to eradicate, compared to planktonic cells.6,12–15 Conventional intervention strategies for biofilm-associated infections include: (1) prevention of initial device contamination; (2) minimization of initial microbial cell attachment; (3) use of agents such as high-dose antibiotics, antibiotic combinations or antibiofilm agents in a catheter lock solution to penetrate the biofilm matrix and kill the embedded microorganisms; and (4) removal of the infected indwelling medical devices.16–18 Infections in prostheses are much more complicated and may necessitate expensive implant replacement and often times cause chronic and/or relapsing disease.12 Some of the promising, non-conventional approaches against antibiotic-resistant biofilms, include nanoparticle systems, natural bioactive compounds, anti-quorum sensing signalling molecules, matrix-degrading enzymes, photodynamic therapy, and CRISPR-CAS (gene editing technique).19 The succeeding sections of this work discuss a small portion of antimicrobial nanoparticle systems, which is focussed on biomacromolecule encapsulation materials for the delivery of antimicrobials and some examples of nanostructures that target biofilm disruption.
2. Encapsulation technology for antimicrobial therapy
Extensive research in nanotechnology, chemistry and materials science has demonstrated promising approaches, which may have high potential to circumvent previous challenges in designing antimicrobial carriers, specific for their practical use. Nanoencapsulation is a method where one or more substance (core material) is immobilized in some form of matrix or wall, termed as the shell, encapsulant, and wall or carrier material. The wall materials act as a physical barrier that gives rise to important physicochemical and biological properties of the resulting encapsulated product, such as protection or limited interaction of the core from its environment, thereby effecting improved stability during storage or preservation of bioactivity when applied in biological systems. By reducing the particle size, the delivery properties, solubility, and bioavailability of the nutraceutical can be improved, depending on the increased surface area per unit volume.20 These general properties of nanoparticle-based encapsulation and delivery systems are summarised in Fig. 2. Based on composition, these carriers can be classified as carbon-based (fullerenes), metallic, ceramic (inorganic non-metals), lipid-based, and polymeric nanoparticles. Even within the same class, each type of nanoparticle/nanoencapsulation system possesses unique properties, depending on their respective size, geometry, and structure.21,22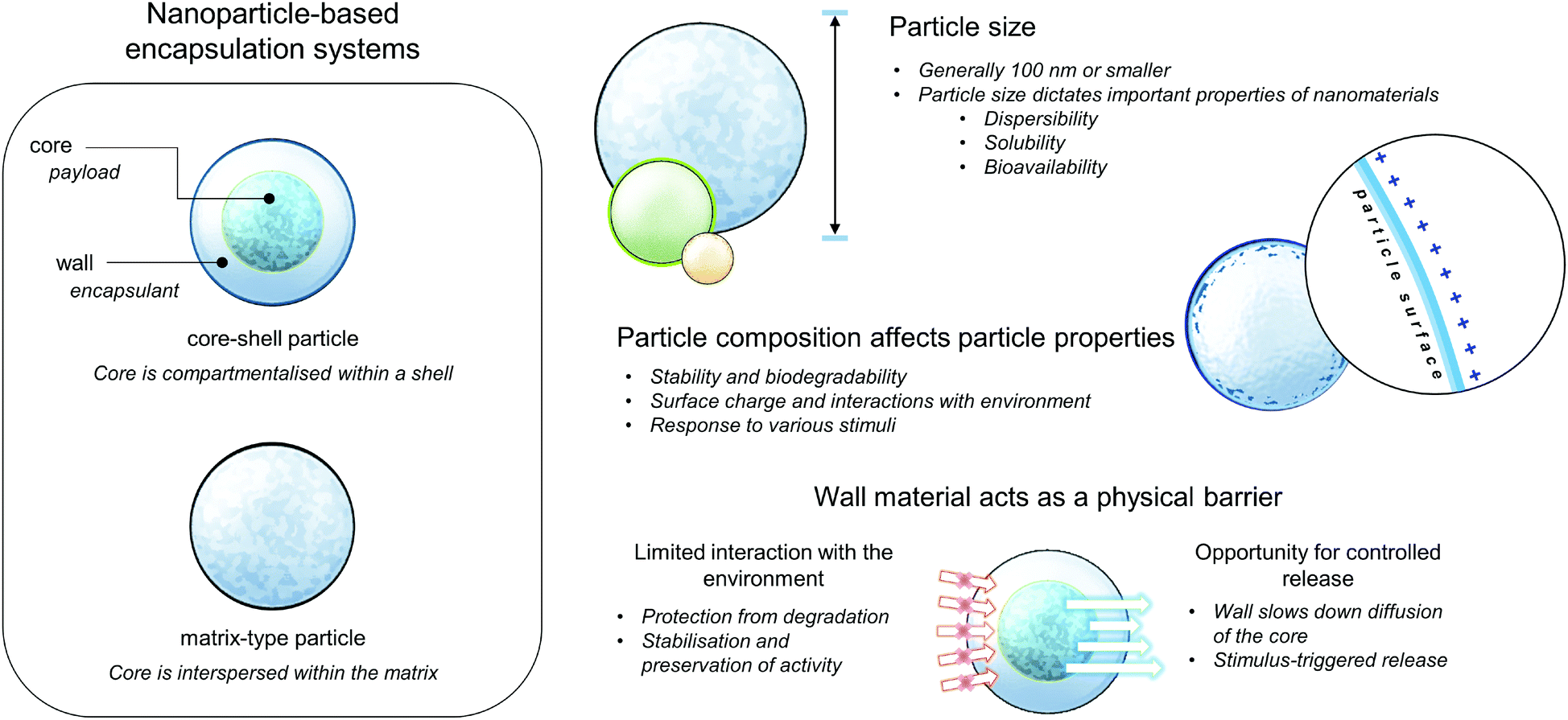 | ||
| Fig. 2 Cartoon depicting the general structures and properties of nanoparticle-based encapsulation systems. | ||
Utilisation of biomolecules in fabricating nanoencapsulation and delivery systems is being promoted due to their high abundance and relatively lower costs, in addition to their stability under various process conditions and biological conditions (during application) including high temperature and pH changes. Furthermore, these molecules are generally biodegradable and biocompatible unlike other materials used in the fabrication of drug delivery systems. Aside from these intrinsic properties that make them suitable as drug carriers, various biopolymers have a distinct set of characteristics that influences the final physiochemical and functional properties of the drug delivery system. Some of these properties can be improved or modified by creating composites and assemblies with nano-inorganics, such as gold and silver nanoparticles.23 This section enumerates and discusses some of the advantageous properties of various biomolecule-based carriers and products in recent literature, which can be used for antimicrobial delivery and activity enhancement. Structures and some of the important properties of these biomolecules are summarised in Fig. 3.
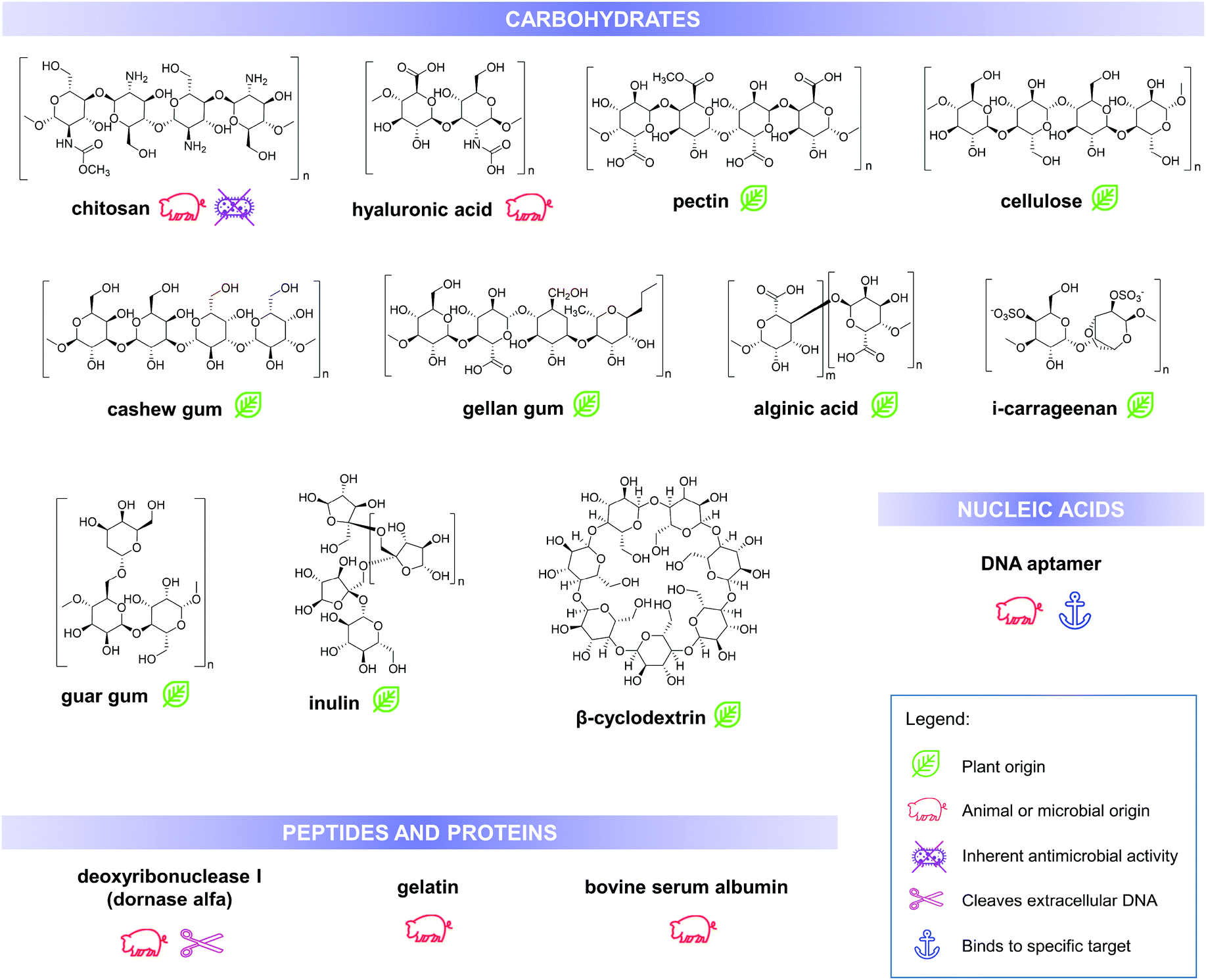 | ||
| Fig. 3 Chemical structures, biological source/origin and some of the important properties of the biomolecules, discussed in this work. | ||
2.1. Improved solubility, and stability during storage and in biological systems
 | ||
| Fig. 4 A schematic diagram showing some of the possible mechanisms of drug release from polymeric particle-based delivery systems, including passive diffusion, swelling, and polymer degradation. Reproduced with permission.71 | ||
This typical solubility problem of drugs, specifically antimicrobials, is frequently addressed using cyclodextrin inclusion complexes. A recent example of this system that is applied to antimicrobials is the oral carbapenem analog, tebipenem pivoxil (TP), in a β-cyclodextrin (TP-β-CD) complex.30 This inclusion complex enhanced the aqueous solubility of TP by two folds and effected faster solution rate at around the intestinal pH (7.2), compared with that at simulated gastric pH (1.2). Similarly, cefuroxime axetil (CA) in the β-cyclodextrin (CA-HPβCD) complex showed significantly improved dissolution profile, reaching a higher plateau on the solubility curve (80% after 60 minutes), compared to pure CA (72% after 165 minutes).31 In effect, a CA-HPβCD inclusion complex showed up to four-fold increase in antimicrobial activity against clinical isolates of Klebsiella pneumoniae and Pseudomonas aeruginosa.
A more complex system that applied the solubility-enhancing property of cyclodextrins is described in the study of Costa-Gouveia et al.32 Methods such as nanoemulsification have been utilised to fabricate delivery systems for ethionamide (ETH), an anti-tubercular with limited solubility and high toxicity, when administered at high doses. However, ETH, even as emulsions, was observed to undergo crystallisation in aqueous media and in biological fluids, which limits the efficacy of this delivery system. In the work described, ETH was co-encapsulated with ETH booster (BDM41906) using epichlorohydrin-crosslinked polymeric β-cyclodextrin, intended for combination therapy for tuberculosis. Encapsulated ETH did not crystallise over the one-month storage stability study, which might be due to the accommodation of the drug and the booster in the hydrophobic cyclodextrin cavities, as well as in the confined micro-domains in the crosslinked polymeric cyclodextrin nanoparticles. In addition to the prevention of crystallisation, the apparent solubility of ETH was also significantly increased, especially when the concentration of polymeric cyclodextrins used in encapsulation was increased. This would explain the observed, lower M. tuberculosis H37Rv-GFP CFU count in treated mice lungs by Microsprayer® as compared with those in lungs in the control group and ETH-treated group.
Itraconazole (ITZ) is another water-insoluble drug, which is being applied as an anti-fungal to counteract opportunistic infections in HIV patients. Burapapadh et al. reported that the encapsulation of this drug in Miglyol 812 (capric triglyceride)-pectin nanoemulsions.33 All physical mixtures of the components used in the fabrication of the nanomaterial showed a melting endothermic peak associated with the occurrence of crystalline ITZ. In the case of the nanoparticles, no melting peak of ITZ was observed, indicating that the drug was molecularly dispersed in the polymer. Furthermore, in a two-hour dissolution test conducted, dissolution of ITZ which was around 3–5% was significantly increased to 60%, 66% and 80%, upon encapsulation in high-methoxy pectin (HMP), low-methoxy pectin (LMP) and amidated low-methoxy pectin (ALMP), respectively. These observations can be related to the wall strength of the particles; unlike LMP and HMP particles, ALMP particles tend to rupture despite their excellent gel-forming properties, indicating the possibility of using this material for fast or immediate drug release formulations.
2.2. Antimicrobial encapsulants and additives enhance antimicrobial activity
Chitosan is one of the most extensively studied polymers as a shell material, matrix component, and additive to delivery systems due to its inherent antimicrobial activity. Table 1 shows some examples of chitosan-based antimicrobial encapsulation and delivery systems from recent reports in the literature. A widely accepted explanation for the antimicrobial activity of chitosan is the electrostatic interactions between the amino groups of the chitosan molecule and the negatively charged bacterial surface molecules, including lipopolysaccharides, which induces change in the penetrability of the bacterial membrane.34–36 Jeon et al. proposed an alternative explanation, after studying the molecular mechanisms of the antimicrobial activity of chitosan microparticles via in vivo and in vitro tests with Escherichia coli O157:H7 EDL933 (ATCC48935), intrauterine pathogenic Escherichia coli, Salmonella enterica CDC3041-1, and Klebsiella pneumonia.34 In their work, the outer membrane protein A (OmpA), an integral bacterial outer membrane protein embedded as a β-barrel structure that contributes to the structural integrity of the bacterial cell surface, is proposed to be a direct target of chitosan binding. The hydrogen bond interactions between the chitosan and this membrane protein inhibit OmpA function, which results in membrane disruption leading to apoptosis. Chitosan nanoparticles were also demonstrated to exhibit biofilm-disrupting capabilities, mainly due to the particle size that allows higher penetration rate compared to micro-sized antimicrobial agents.37 Furthermore, cationic chitosan interacts with the negative charge of the biofilm components, including the EPS matrix and extracellular DNA, and the microbial membrane, leading to the inhibition of surface colonisation and biofilm formation.38 This effect of chitosan nanoparticles was also shown to be effective even against dual-species biofilms of Streptococcus mutans and Candida albicans, a pair that is known to exhibit faster EPS formation and enhanced antimicrobial drug tolerance compared to their single-species biofilms.37| Active material | Encapsulation or delivery system | Test microorganism | Observed bioactivity |
|---|---|---|---|
| *MIC – minimum inhibitory concentration; MBC – minimum bactericidal concentration; MFC – minimum fungicidal concentration. | |||
| Ethionamide39 | Chitosan/alginate nanoparticles, stabilised by carrageenan | Mycobacterium tuberculosis H37RA | MIC: 0.61 μg mL−1 (NPs) vs. 0.43 μg mL−1 (free ethionamide) |
| Levofloxacin40 | TPP-crosslinked chitosan nanoparticles | Staphylococcus aureus | Three- to four-fold increase in antimicrobial activity |
| Escherichia coli | |||
| Mentha piperita essential oil41 | Chitosan-cinnamic acid nanogel | Aspergillus flavus | No growth from 1000 ppm and higher concentrations. |
| Ciprofloxacin42 | Alginate/high methoxy and low methoxy pectin beads with Guar gum alkyl amine-stabilised silver nanoparticles | Pseudomonas aeruginosa | Percentage inhibition after 8 h: |
| Staphylococcus aureus | 90% (P. aeruginosa) | ||
| Escherichia coli | 78% (B. cereus) | ||
| Bacillus cereus | 74% (S. aureus) | ||
| Rosemary essential oil (REO)43 | Chitosan-benzoic acid (CS-BA) nanogel | Staphylococcus aureus | MIC: 80 μg mL−1 (CS-BA); 40 μg mL−1 (encapsulated REO) |
| Nisin44 | Alginate-chitosan nanoparticles | Listeria monocytogenes | MIC: 500 IU mL−1 |
| Eugenol45 | Chitosan nanoparticles | Staphylococcus aureus | MIC (S. aureus): 195 μg mL−1 (NPs) vs. 363 μg mL−1 (free eugenol) |
| Escherichia coli O157:H7 | MIC (E. coli): 98 μg mL−1 (NPs) vs. 726 μg mL−1 (free eugenol) | ||
| Pseudomonas aeruginosa | MIC (P. aeruginosa): 195 μg mL−1 (NPs) vs. 726 μg mL−1 (free eugenol) | ||
| Salmonella sp. | MIC (Salmonella): 391 μg mL−1 (NPs) vs. 726 μg mL−1 (free eugenol) | ||
| Vancomycin46 | Layer-by-layer gelatin/chitosan coated nanoparticles | Staphylococcus aureus MRSA 134/94 | Significant inhibition of S. aureus growth with increased osteoblast proliferation on NP-coated titanium surfaces |
| Temporin B47 | TPP-crosslinked chitosan nanoparticles | Staphylococcus epidermidis | Reduction of temporin B cytotoxicity on mammalian cells; sustained antimicrobial action vs. S. epidermidis for at least four days |
| Nisin Z48 | Chitosan-coated liposomes | Staphylococcus aureus | MIC (S. aureus): 10 μg mL−1 (NPs) vs. 10 μg mL−1 (free nisin) |
| Listeria monocytogenes | MIC (L. monocytogenes): poor activity (NPs) vs. 50 μg mL−1 (free nisin) | ||
| Enterobacter faecalis | MIC (E. faecalis): 200 μg mL−1 (NPs) vs. 726 μg mL−1 (free nisin) | ||
| Cymbopogon citratus essential oil49 | Chitosan-oleic acid nanoemulsion | Escherichia coli (ATCC 25922 and ATCC 35218) Staphylococcus aureus (ATCC 29213 and ATCC 43300) | MBC/MFC values for nanoemulsions (with 1000 times lower oil content) were comparable or only a few times higher than those of the pure oil. |
| Gardnerella vaginalis | |||
| Pseudomonas aeruginosa | |||
| Chlamydia trachomatis | |||
| Lysozyme50 | TPP-crosslinked chitosan nanoparticles | Escherichia coli | MIC (E. coli): 0.156 μg mL−1 (NPs + lysozyme) vs. 0.625 μg mL−1 (NPs) |
| Bacillus subtilis | MIC (B. subtilis): 0.156 μg mL−1 (NPs + lysozyme) vs. 0.313 μg mL−1 (NPs) | ||
| Antimicrobial motif (Pep-H) of human neutrophil peptide-1 (HNP-1)51 | TPP-crosslinked chitosan nanoparticles | Mycobacterium tuberculosis H37Rv | NPs showed significant reduction in CFU (>90%) at 5–10 times lower concentrations than that observed for free Pep-H. |
| Homalomena pineodora essential oil52 | TPP-crosslinked chitosan nanoparticles | Diabetic wound pathogens: | Broad spectrum antimicrobial activity with 60-80% microbial growth reduction in 3D collagen wound models. |
| Bacillus cereus | |||
| Bacillus subtilis | |||
| Staphylococcus aureus Methicilin-resistant Staphylococcus aureus (MRSA) | |||
| Escherichia coli | |||
| Proteus mirabilis | |||
| Yersinia sp. | |||
| Klebsiella pneumonia | |||
| Shigella boydii | |||
| Salmonella typhimurium Acinetobacter anitratus Pseudomonas aeruginosa | |||
| Candida albicans | |||
| Candida utilis | |||
| Alginate lyase53 | TPP-crosslinked chitosan (low molecular weight) nanoparticles | Pseudomonas aeruginosa biofilm | Biofilm thickness: |
| 88 μm (no treatment) | |||
| 22 μm (NPs + alginate lyase) | |||
| 50 μm (free alginate lyase) | |||
| Increased biofilm sensitivity towards piperacillin, ceftazidime, and amikacin. | |||
The study of Breser et al. suggests that chitosan may have important bactericidal properties, against both the bacteria in preformed biofilms and the planktonic cultures of coagulase-negative Staphylococcus (CNS) isolates from chronic bovine mastitis.54 Remarkably, a combined treatment using chitosan and cloxacillin was not only able to inhibit bacterial biofilm establishment and increase preformed biofilm eradication, but it also reduced intracellular bacterial viability and induced a slight increase in interleukin-6 (IL-6) in infected MAC-T cells, indicating the possible contribution of chitosan to the epithelial cell response against CNS.
Another example of an antimicrobial polymer is aminocellulose. In the study of Ivanova et al., nanoparticles were fabricated via the layer-by-layer (LbL) technique using alternating antibacterial aminocellulose and hyaluronic acid (the stabilizer) on a biologically inert nanoparticle template.55 This biopolymer nanoparticle system seems to exhibit a mechanism of antimicrobial action similar to chitosan, which affects both the planktonic cells of E. coli and S. aureus and the total biofilm growth. Moreover, the functionalized NPs demonstrated enhanced capacity of the nano-layered aminocellulose to interact with and disrupt the negatively charged bacterial cell membranes at lower concentration in comparison to its bulk solution. On the other hand, the LbL functionalized NPs were able to inhibit the formation of biofilms by S. aureus and impede the biofilm growth by E. coli, without affecting the morphology of human fibroblast cells.
In addition to antimicrobial polymers, metal nanoparticles such as gold (AuNPs) and silver nanoparticles (AgNPs) possess intrinsic antimicrobial activity56–58 and in combination with various antimicrobials (Table 2), the bioactivity of the resulting assemblies and hybrids sometimes exceeds the capacity of the materials alone through synergism. AgNPs and AuNPs exert antimicrobial activity by binding to the microbial cell membrane, resulting in the disturbance of normal cell wall permeability and cellular respiration by dispelling the chemiosmotic gradient, leading to cell death. Furthermore, several studies revealed that AuNP- and AgNP-treated microorganisms have elevated reactive oxygen species (ROS) levels, a condition which may lead to cell inactivation. AuNP and AgNP that can penetrate the cell membrane can cause cell damage by interacting with intracellular proteins and DNA.42,59–62
| Nanometal-based carrier or nanometal-antimicrobial combination | Test microorganism | Observed bioactivity |
|---|---|---|
| *MIC – minimum inhibitory concentration. | ||
| Amphotericin B-conjugated biogenic silver nanoparticles62 | Candida albicans | MIC (C. albicans): 5 μg mL−1 (conjugated AgNPs) vs. 125 μg mL−1 (AgNPs) |
| Candida tropicalis | MIC (C. tropicalis): 1.5 μg mL−1 (conjugated AgNPs) vs. 62.5 μg mL−1 (AgNPs) | |
| Penicillin G-capped silver nanoconjugates63 | β-lactamase resistant Staphylococcus aureus and Escherichia coli | AgNP nanoconjugates showed six to eight times increase in the zones of inhibition in the test microorganisms compared to AgNPs. |
| Cationic dipeptide capped gold/silver nanohybrids64 | Escherichia coli Staphylococcus aureus | Nanohybrids exhibited 2–10 fold reduction in nano formulation dosage against tested microorganisms; |
| Salmonella typhimurium | ||
| Candida albicans | nanohybrid materials displayed non-cytotoxic behaviour. | |
| Candida glabrata | ||
| Gold-silver (Au-Ag) nanoparticle combination with doxycycline65 | Pseudomonas aeruginosa | Significant increase in the zones of inhibition in the test microorganisms compared to Au-Ag bimetallic NPs and doxycycline alone; |
| Staphylococcus aureus | ||
| Escherichia coli | Generation of reactive oxygen species by Au-Ag bimetallic NPs | |
| Micrococcus luteus | ||
| DNA aptamer-functionalised gold nanostructures66 | Methicillin-resistant Staphylococcus aureus | DNA aptamer-functionalised gold nanorods effectively inactivated 95% MRSA cells through hyperthermia via photothermal conversion of near-infrared radiation. |
| Nisin-gold assembly67 | Multidrug resistant (MDR) and non-resistant Enterobacter faecalis and Staphylococcus aureus | MIC (multidrug reistant E. faecalis and S. aureus): 156–313 U mL−1 (nisin-AuNPs) vs. >2500 U mL−1 (nisin only) |
| Bovine serum albumin-capped gold nanoparticles, functionalised with various antibiotics: streptomycin sulphate, neomycin sulphate, gentamicin sulphate and kanamycin sulphate68 | Escherichia coli ATCC 25922 | Antibiotic-conjugated AuNPs showed 1.4 to 3.6 times higher antimicrobial activity, compared to the pure antibiotics. |
| Pseudomonas aeruginosa ATCC 27853 | ||
| Staphylococcus aureus ATCC 25923 | ||
| Gold nanoparticle-sushi peptide and gold nanoparticle-polymyxin B conjugates69 | Salmonella typhi | MIC (S. typhi): ∼700 nM AuNP-sushi peptide conjugates |
| Gold nanoparticles with antimicrobial motif (Pep-H) of human neutrophil peptide-1 (HNP-1)50 | Mycobacterium tuberculosis H37Rv | Inhibition of M. tuberculosis growth: |
| 91–95% (1–5 μg mL−1 Pep-H AuNPs), | ||
| 59% (AuNPs) | ||
| 45% (1 μg mL−1 Pep-H) | ||
| 90% (5 μg mL−1 Pep-H) | ||
| Antimicrobial peptide (AMP) HPA3PHis loaded onto gold nanoparticle- DNA aptamer conjugate70 | Vibrio vulnificus | 90% reduction in intracellular V. vulnificus in HeLa cells, treated with 0.5 μM HPA3Phis and 1 nM AuNP-DNA aptamer conjugate |
| Silver nanoparticles physically combined with cefazolin, mupirocin, gentamycin, neomycin, tetracycline and vancomycin58 | Staphylococcus aureus ATCC 25922 | Synergism of AgNPs with various topical antibiotics against the test microorganisms; |
| Escherichia coli ATCC 10536 | AgNPs + topical antibiotics showed increased ROS level, membrane damage following protein release, K+ leakage and biofilm inhibition. | |
| Pseudomonas aeruginosa ATCC 25619 | ||
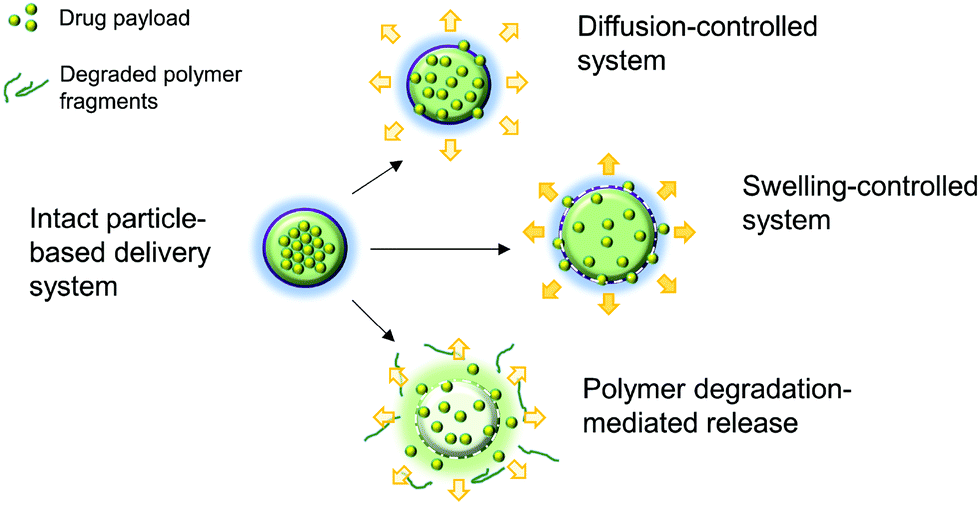
2.3. Controlled release and delivery to target sites of action
Aside from being a barrier between the core material and the external environment, wall materials can regulate the mode and rate of release in different media. Delivery systems can be engineered in such a way that the release of drugs from biopolymeric particles generally occurs via: passive diffusion; enhanced diffusion by swelling; polymer degradation and erosion; or a combination of these modes,72–74 as depicted in Fig. 5. In most of these systems, core materials are retained within the assembly until being exposed to a specific environment. Swelling-controlled release systems undergo swelling in an aqueous environment, allowing the entrapped core material to diffuse through the swollen network into the external environment. Diffusion-controlled systems are stable in the target environment and do not change in size through swelling or degradation; the drug diffuses through the pores or macromolecular structure of the polymer upon introduction of the delivery system to the biological environment, without inducing any change in the polymer itself.72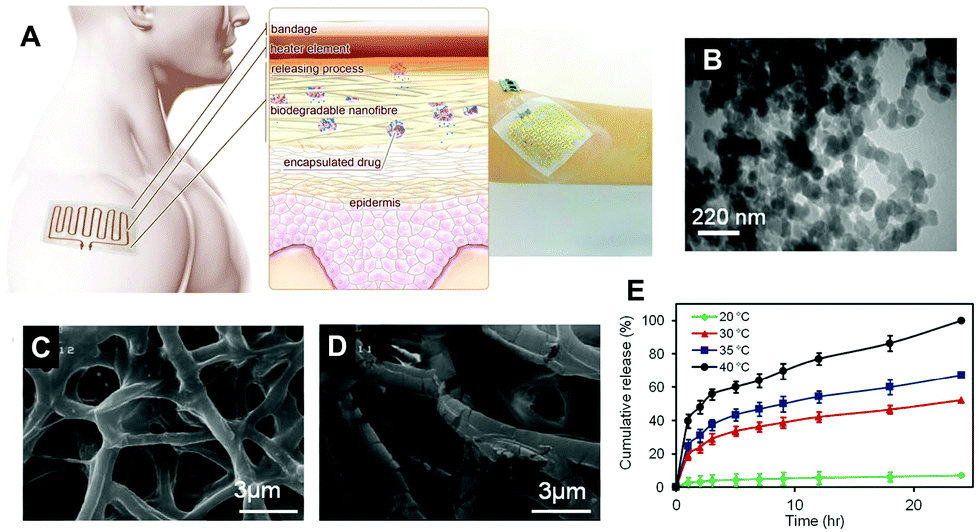 | ||
| Fig. 5 A schematic diagram showing the principle of the thermoresponsive drug delivery system, combined with a typical fabricated bandage with integrated heater and electronics (A). Transmission electron micrograph of thermoresponsive PEGylated chitosan nanocarriers (B). Scanning electron micrograph of a nanofibrous substrate, containing the thermoresponsive nanocarriers, heated to 38 °C (C) and 50 °C for one hour. Plots showing the temperature dependence of cefazolin release from the nanofibrous drug delivery system (E). Copyright, licensed under creative commons.86 | ||
Lima et al. demonstrated that increasing the degree of acetylation of cashew gum decreases the rate of release of hydrophobic drug amphotericin B.76 This was also observed in the other, where hydrophobic non-antimicrobial drugs, mostly chemotherapeutics, are encapsulated with hydrophobized polymers, such as the following systems: clonazepam in acetylated pullulan particles;77 paclitaxel in caproyl and oleyl hyaluronan micelles;78 and epirubicin in pullulan acetate nanoparticles.79
The effect of extended drug release from nanocarriers is demonstrated commonly observed as delayed effects or prolonged activity. In the study of Kumar et al., ketoconazole-loaded chitosan-gellan gum nanocomplexes showed improved inhibitory activity (>80%) against Aspergillus niger compared with the bare drug (40%) and the empty nanocomplexes (10%).80 The results suggest that the empty complexes have antifungal activity which may be attributed to chitosan and the higher activity of the ketoconazole-loaded nanocomplexes may be attributed to the synergistic effects of chitosan and ketoconazole. Significantly higher antimicrobial activity of the ketoconazole-loaded nanocomplexes only after a seven-day incubation period, compared to that after the three-day incubation period may suggest that encapsulation can prolong the antimicrobial effects of ketoconazole through controlled, sustained release.
De Matteis et al. showed interesting findings in the release properties of chitosan nanocarriers, containing bedaquiline, a diarylquinoline anti-mycobacterial drug, with and without conjugation with polyethylene glycol (PEGylation), in various release media. Both chitosan and PEGylated chitosan nanocarriers released similar amounts of bedaquiline in water and in Middlebrook 7H9 in a seven-day drug release study.81 Meanwhile, substantial bedaquiline release was only favoured from PEGylated chitosan nanocarriers in 0.9% NaCl and in RPMI, but not from chitosan nanocarriers. This would suggest that PEGylation changes the interactions of the nanoparticles with the surrounding PEGylation and alters the release properties of bedaquiline in various media. This suggests that the chitosan nanocarrier system can be modified by PEGylation to control or improve release properties, depending on its expected applications. Middlebrook 7H9 and RPMI media were used as Mycobacterium tuberculosis and infected macrophage culture media, respectively. The presence of proteins and salts in the release medium would influence the release of bedaquiline from chitosan and PEGylated chitosan nanocarriers. Release in 0.9% NaCl demonstrated the suitability of the PEGylated chitosan nanocarriers in nebulisation formulations.
Deacon et al. studied the effects of encapsulation of tobramycin in alginate/chitosan nanoparticles, prepared via ionic gelation.82 The encapsulated antibiotic showed a biphasic release pattern: 45% released after 90 min and up to 80% after 48 hours. A Galleria mellonella (greater wax moth) infection model was utilised to determine the inhibition of Pseudomonas aeruginosa infection-induced mortality. When the samples were administered 30 min after infection, no significant differences in survival rate were observed between the free and encapsulated tobramycin. However, in another set up, where free and encapsulated tobramycin were administered 96 hours prior to infection, tobramycin nanoparticle-treated larva had 80% survival while free tobramycin-treated moth larva only had 40% survival. These results suggest that the controlled release nature of the nanoparticles would provide an extended therapeutic window for the antimicrobial formulation.
Abdelkader et al. demonstrated that tripolyphosphate-crosslinked chitosan nanoparticles loaded with meropenem have two-fold lower minimum inhibitory concentrations against methicillin-resistant Staphylococcus aureus, meropenem-sensitive Escherichia coli, and meropenem-sensitive and meropenem-resistant Klebsiella pneumoniae compared with free meropenem.83 A rat systemic infection (sepsis) model also showed that meropenem-loaded nanoparticles can improved survival rates of rats with exceptional bacterial clearance, as compared to the animals treated with the free drug. These improvements in the antimicrobial activity of meropenem were due to two properties of the loaded nanoparticles: (1) positive surface charges of the nanoparticles, which enhanced the interactions between the drug-loaded nanoparticles and the negatively-charged bacterial cell walls, consequently allowing higher drug penetration into the bacterial cell; and (2) chitosan-to-TPP ratio-dependent slow release of meropenem from the nanoparticles over time, and limiting the degradation of the drug before exerting its antibacterial activity.
In another recent work of Patel et al.,84 DNase I-functionalised ciprofloxacin-loaded chitosan nanoparticles (CIP-CHNPs) were shown to have the enormous potential to inhibit Pseudomonas aeruginosa biofilm development and break the previously established biofilm extracellular matrix, without triggering severe lung toxicity. DNase-CIP-CHNPs significantly reduced the microbial count, thickness, and biomass, and showed the highest biofilm dispersal, compared to ciprofloxacin alone and CIP-CHNPs. These observed antimicrobial and anti-biofilm properties are attributed to the extracellular DNA-hydrolysing capability of DNase I, which improves both the nanoparticle penetration and drug diffusion in the biofilm matrix. In addition, chitosan nanoparticles effected the high biofilm penetration, due to their small particle size, and controlled ciprofloxacin release, which extended the antimicrobial efficiency of the delivery system over the 72 hour anti-biofilm studies.
Similarly, Amphotericin B, encapsulated in carboxymethyl-iota-carrageenan/gelatin nanoparticles also showed enhanced cellular uptake by Raw 264.7 cells in the study of Aparma et al.86 This result explains why the intracellular Candida glabrata survival was relatively low in cells treated with Amp B-loaded carboxymethyl-iota-carrageenan/gelatin nanoparticles, compared with the survival rates of those treated with non-encapsulated Amp B and Amp B-loaded gelatin nanoparticles.
A good representative of an enzyme-triggered release system is a cyclodextrin inclusion complex, as the cyclodextrins undergo degradation into glucose, in the presence of α-amylase and α-glucosidase, which are both found in the small intestines.88–90 Current data also show that cyclodextrins are easily degraded in the blood, and then excreted through urine. In the study of Kaneo et al., encapsulation of amphotericin B in hydrophobized cyclodextrin, specifically cholesterol-modified cyclodextrin nanoparticles, resulted in high plasma concentration of amphotericin B, compared with the Fungizone (commercially available amphotericin B formulation), with no haemolysis, which is commonly caused by amphotericin B.88 This suggests the possibility of using this delivery system for the intravenous administration of amphotericin B.
3. Conclusions and perspectives
Nature itself offers the essential components to combat this evolving problem of antimicrobial resistance and it is up to our human knowledge and creativity to utilise these components in the form of biomacromolecules and biologically derived materials to create effective, innovative and safe solutions. We hope that this review provides a bird's eye view of the capabilities of some of the recently reported biomolecule-based encapsulation and delivery systems and some insights into the possibilities in the development of these systems and technologies. Various biomolecules, especially carbohydrate biopolymers, proteins and peptides, have been successfully utilised as potential antimicrobial delivery vehicles or encapsulant modifiers with additional or altered physicochemical and biological properties, including: controlled and targeted release to sites of action; selective interactions with target microorganism/s; enhanced physical and chemical stability under biological conditions; increased solubility and uptake of the antimicrobial; enhanced antimicrobial activity; and specific targeting of microbial cell components and biofilms or processes leading to biofilm formation. With the abundance of biomolecules, we have an unimaginable number of combinations that can be tested and developed as carriers for the currently existing antimicrobials. In addition to these biomolecules, silver and gold nanoparticles and many other nanometals and nanoinogranics can also used as additives to alter the mode of action of the resulting nanoassemblies.Biomolecules are very attractive as nanomaterial components, generally, due to their biocompatibility, biodegradability, abundance, renewability, and sustainability, compared to other resources for nanoparticle-based delivery systems. The main challenges that we perceive in the clinical translation of these delivery systems are the compatibility and accumulation of these materials with the human body, as other properties arise when biomolecules are in their nanoparticle form. Most of the studies cited in this communication have reported successes in in vitro experiments and in vivo animal models. The human body's immune response to these delivery systems needs to be studied further. Storage stability testing, including physicochemical evaluation of the drug delivery system and monitoring of the chemical stability of the antimicrobial molecules over time, would also be a crucial part of any future work to guarantee the furtherance of new formulations into workable materials for industry-scale product development and their viability as new treatment options.
Increasing the complexity of the drug delivery system by using various biomacromolecules, nanometals and other additives, and modifying the drug payload may offer multiple modes of antimicrobial action, which is advantageous in improving antimicrobial efficacy and delaying the emergence of resistant strains. However, this increase in the complexity of the delivery system might also act as a double-edged sword for various reasons. Increased formulation complexity will lead to questions regarding the biocompatibility, potential bioaccumulation and side effects of the resulting hybrid material, as the property of the delivery system would most likely be different from the individual biocompatible components. In our view, all these encapsulation and delivery systems can be considered as works-in-progress, continuously being disassembled, reformulated and re-assembled, in our attempts to compete with the emergence of resistant microbial strains. While the clinical translation of these technologies might be slow and challenging, we predict that there will still be an increase in available information about these types of encapsulation systems in the coming years. While the search and syntheses of elusive “all-in-one” antimicrobial agents or antimicrobial delivery systems appears to be a long and difficult path, designing delivery systems for particular applications and treatments by changing the biopolymer or biomolecule compositions of the encapsulants seem to be a more practical option, particularly in this race against antimicrobial resistance. Tuning the drug delivery system components also seems to be a very attractive approach to create opportunities for on-demand localisation and/or release via external triggers or stimuli (heat,91–93 light,94–96 magnetic field,97–99 ultrasound,100–103etc.). The growing knowledge on these biomacromolecules and other nanomaterials is an opportunity that can be extended to other types of colloidal delivery systems, such as emulsion droplets, micro- and nanobubbles, and composite materials that can be fabricated with these colloids, including antimicrobial surfaces and coatings, fibres, gels, and implant-type biomedical devices.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4(2) CrossRef CAS , VMBF-0016-2015.
- C. M. Thomas and K. M. Nielsen, Nat. Rev. Microbiol., 2005, 3, 711–721 CrossRef CAS.
- N. Rabin, Y. Zheng, C. Opoku-Temeng, Y. Du, E. Bonsu and H. O. Sintim, Future Med. Chem., 2015, 7, 493–512 CrossRef CAS.
- S. Marić and J. Vranes, Period. Biol., 2007, 109, 115–121 Search PubMed.
- R. M. Donlan, Clin. Infect. Dis., 2001, 33, 1058–4838 CrossRef.
- A. B. Zoubos, S. P. Galanakos and P. N. Soucacos, Med. Sci. Monit., 2012, 18, RA89–RA96 CrossRef CAS.
- R. Singh, P. Ray, A. Das and M. Sharma, J. Antimicrob. Chemother., 2010, 65, 1955–1958 CrossRef CAS.
- E. Teirlinck, S. K. Samal, T. Coenye and K. Braeckmans, in Functionalized Nanomaterials for the Management of Microbial Infection, ed. R. Boukherroub, S. Szunerits and D. Drider, Elsevier, Boston, 2017, pp. 49–76 DOI:10.1016/B978-0-323-41625-2.00003-X.
- T. Song, M. Duperthuy and S. N. Wai, Antibiotics, 2016, 5, 23 CrossRef.
- A. Penesyan, M. Gillings and I. T. Paulsen, Molecules, 2015, 20, 5286–5298 CrossRef CAS.
- A. Algburi, N. Comito, D. Kashtanov, L. M. T. Dicks and M. L. Chikindas, Appl. Environ. Microbiol., 2017, 83, e02508–e02516 Search PubMed.
- C. R. Arciola, D. Campoccia and L. Montanaro, Nat. Rev. Microbiol., 2018, 16, 397–409 CrossRef CAS.
- A. Connaughton, A. Childs, S. Dylewski and V. J. Sabesan, Front. Med., 2014, 1, 22 Search PubMed.
- M. Clauss, A. Trampuz, O. Borens, M. Bohner and T. Ilchmann, Acta Biomater., 2010, 6, 3791–3797 CrossRef CAS.
- A. D. Verderosa, M. Totsika and K. E. Fairfull-Smith, Front. Chem., 2019, 7, 824 CrossRef CAS.
- S. Aslam, Am. J. Infect. Control, 2008, 36, S175.e179–S175.e111 CrossRef.
- H. Wu, C. Moser, H.-Z. Wang, N. Høiby and Z.-J. Song, Int. J. Oral Sci., 2015, 7, 1–7 CrossRef CAS.
- O. Ciofu, E. Rojo-Molinero, M. D. Macià and A. Oliver, APMIS, 2017, 125, 304–319 CrossRef.
- D. Sharma, L. Misba and A. U. Khan, Antimicrob. Resist. Infect. Control, 2019, 8, 76 CrossRef.
- O. Bayraktar, İ. Erdoğan, M. D. Köse and G. Kalmaz, in Nanostructures for Antimicrobial Therapy, ed. A. Ficai and A. M. Grumezescu, Elsevier, 2017, pp. 395–412 DOI:10.1016/B978-0-323-46152-8.00017-2.
- I. Khan, K. Saeed and I. Khan, Arab. J. Chem., 2019, 12, 908–931 CrossRef CAS.
- J. M. V. Makabenta, A. Nabawy, C.-H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2020 DOI:10.1038/s41579-020-0420-1.
- M. Rai, A. P. Ingle, I. Gupta and A. Brandelli, Int. J. Pharm., 2015, 496, 159–172 CrossRef CAS.
- R. Gruskiene, S. Jolanta and K. Tatjana, LWT, 2017, 82, 283–286 CrossRef CAS.
- T. Krivorotova, A. Cirkovas, S. Maciulyte, R. Staneviciene, S. Budriene, E. Serviene and J. Sereikaite, Food Hydrocolloids, 2016, 54, 56 CrossRef.
- J. Hadgraft and M. E. Lane, Expert Opin. Drug Delivery, 2016, 13, 817–830 CrossRef CAS.
- P. Khadka, J. Ro, H. Kim, I. Kim, J. T. Kim, H. Kim, J. M. Cho, G. Yun and J. Lee, Asian J. Pharm. Sci., 2014, 9, 304–316 CrossRef.
- X. Xia, K. Pethe, R. Kim, L. Ballell, D. Barros, J. Cechetto, H. Jeon, K. Kim and A. E. Garcia-Bennett, Nanomaterials, 2014, 4, 813–826 CrossRef.
- J. Leleux and R. O. Williams, III, Drug Dev. Ind. Pharm., 2014, 40, 289–300 CrossRef CAS.
- M. Paczkowska, D. Szymanowska-Powałowska, M. Mizera, D. Siąkowska, W. Błaszczak, H. Piotrowska-Kempisty and J. Cielecka-Piontek, PLoS One, 2019, 14, e0210694–e0210694 CrossRef CAS.
- M. Mizera, D. Szymanowska, A. Stasiłowicz, D. Siąkowska, K. Lewandowska, A. Miklaszewski, T. Plech, E. Tykarska and J. Cielecka-Piontek, Biomolecules, 2019, 10, 24 CrossRef.
- J. Costa-Gouveia, E. Pancani, S. Jouny, A. Machelart, V. Delorme, G. Salzano, R. Iantomasi, C. Piveteau, C. J. Queval, O.-R. Song, M. Flipo, B. Deprez, J.-P. Saint-André, J. Hureaux, L. Majlessi, N. Willand, A. Baulard, P. Brodin and R. Gref, Sci. Rep., 2017, 7, 5390 CrossRef.
- K. Burapapadh, H. Takeuchi and P. Sriamornsak, Asian J. Pharm. Sci., 2016, 11, 365–375 CrossRef.
- S. J. Jeon, M. Oh, W.-S. Yeo, K. N. Galvão and K. C. Jeong, PLoS One, 2014, 9, e92723 CrossRef.
- M. Kong, X. G. Chen, K. Xing and H. J. Park, Int. J. Food Microbiol., 2010, 144, 51–63 CrossRef CAS.
- V. N. Davydova, S. Y. Bratskaya, V. I. Gorbach, T. F. Solov'eva, W. Kaca and I. M. Yermak, Biophys. Chem., 2008, 136, 1–6 CrossRef CAS.
- R. Ikono, A. Vibriani, I. Wibowo, K. E. Saputro, W. Muliawan, B. M. Bachtiar, E. Mardliyati, E. W. Bachtiar, N. T. Rochman, H. Kagami, L. Xianqi, T. Nagamura-Inoue and A. Tojo, BMC Res. Notes, 2019, 12, 383 CrossRef.
- Y. Tan, S. Ma, M. Leonhard, D. Moser, G. M. Haselmann, J. Wang, D. Eder and B. Schneider-Stickler, Carbohydr. Polym., 2018, 200, 35–42 CrossRef CAS.
- S. Abdelghany, M. Alkhawaldeh and H. S. AlKhatib, J. Drug Delivery Sci. Technol., 2017, 39, 442–449 CrossRef CAS.
- Ameeduzzafar, S. S. Imam, S. N. Abbas Bukhari, J. Ahmad and A. Ali, Int. J. Biol. Macromol., 2018, 108, 650–659 CrossRef CAS.
- M. Beyki, S. Zhaveh, S. T. Khalili, T. Rahmani-Cherati, A. Abollahi, M. Bayat, M. Tabatabaei and A. Mohsenifar, Ind. Crops Prod., 2014, 54, 310–319 CrossRef CAS.
- G. A. Islan, A. Mukherjee and G. R. Castro, Int. J. Biol. Macromol., 2015, 72, 740–750 CrossRef CAS.
- N. Mohsenabadi, A. Rajaei, M. Tabatabaei and A. Mohsenifar, Int. J. Biol. Macromol., 2018, 112, 148–155 CrossRef CAS.
- P. Zimet, W. M. Álvaro, R. Caterina, B. Giannina, P. Helena, M. Iris and F. Ricardo, LWT, 2018, 91, 107–116 CrossRef CAS.
- Y. Shao, C. Wu, T. Wu, Y. Li, S. Chen, C. Yuan and Y. Hu, Carbohydr. Polym., 2018, 193, 144–152 CrossRef CAS.
- T. Sangfai, F. Dong, V. T. Tantishaiyakul, K. Jandt, C. Lüdecke, O. Boonrat and N. Hirun, eXPRESS Polym. Lett., 2017, 11, 73–82 CrossRef CAS.
- A. M. Piras, G. Maisetta, S. Sandreschi, M. Gazzarri, C. Bartoli, L. Grassi, S. Esin, F. Chiellini and G. Batoni, Front. Microbiol., 2015, 6, 372 Search PubMed.
- T. Niaz, S. Shabbir, T. Noor, A. Rahman, H. Bokhari and M. Imran, LWT, 2018, 96, 98–110 CrossRef CAS.
- M. C. Bonferoni, G. Sandri, S. Rossi, D. Usai, I. Liakos, A. Garzoni, M. Fiamma, S. Zanetti, A. Athanassiou, C. Caramella and F. Ferrari, Colloids Surf., B, 2017, 152, 385–392 CrossRef CAS.
- T. Wu, C. Wu, S. Fu, L. Wang, C. Yuan, S. Chen and Y. Hu, Carbohydr. Polym., 2017, 155, 192–200 CrossRef CAS.
- R. Sharma, R. Raghav, K. Priyanka, P. Rishi, S. Sharma, S. Srivastava and I. Verma, Sci. Rep., 2019, 9, 7866 CrossRef.
- N. A. S. Rozman, W. Y. Tong, C. R. Leong, M. R. Anuar, S. Karim, S. K. Ong, F. A. M. Yusof, W.-N. Tan, B. Sulaiman, M. L. Ooi and K. C. Lee, Sci. Rep., 2020, 10, 3307 CrossRef CAS.
- S. Li, Y. Wang, X. Li, B. S. Lee, S. Jung and M.-S. Lee, Int. J. Mol. Sci., 2019, 20, 4565 CrossRef CAS.
- M. L. Breser, V. Felipe, L. P. Bohl, M. S. Orellano, P. Isaac, A. Conesa, V. E. Rivero, S. G. Correa, I. D. Bianco and C. Porporatto, Sci. Rep., 2018, 8, 5081 CrossRef.
- A. Ivanova, K. Ivanova, J. Hoyo, T. Heinze, S. Sanchez-Gomez and T. Tzanov, ACS Appl. Mater. Interfaces, 2018, 10, 3314–3323 CrossRef CAS.
- A. Elbehiry, M. Al-Dubaib, E. Marzouk and I. Moussa, MicrobiologyOpen, 2019, 8, e00698 CrossRef.
- S. Onitsuka, T. Hamada and H. Okamura, Colloids Surf., B, 2019, 173, 242–248 CrossRef CAS.
- R. Geethalakshmi and D. V. L. Sarada, Ind. Crops Prod., 2013, 51, 107–115 CrossRef.
- G. Thirumurugan, J. V. L. N. Seshagiri Rao and M. D. Dhanaraju, Sci. Rep., 2016, 6, 29982 CrossRef CAS.
- B. Le Ouay and F. Stellacci, Nano Today, 2015, 10, 339–354 CrossRef CAS.
- Y. Cui, Y. Zhao, Y. Tian, W. Zhang, X. Lü and X. Jiang, Biomaterials, 2012, 33, 2327–2333 CrossRef CAS.
- A. Rai, A. Prabhune and C. C. Perry, J. Mater. Chem., 2010, 20, 6789–6798 RSC.
- A. Ahmad, Y. Wei, F. Syed, K. Tahir, R. Taj, A. U. Khan, M. U. Hameed and Q. Yuan, Microb. Pathog., 2016, 99, 271–281 CrossRef CAS.
- V. Ahmed, J. Kumar, M. Kumar, M. B. Chauhan, M. Vij, M. Ganguli and N. S. Chauhan, J. Biotechnol., 2013, 163, 419–424 CrossRef CAS.
- M. Bajaj, S. K. Pandey, T. Nain, S. K. Brar, P. Singh, S. Singh, N. Wangoo and R. K. Sharma, Colloids Surf., B, 2017, 158, 397–407 CrossRef CAS.
- A. Fakhri, S. Tahami and M. Naji, J. Photochem. Photobiol., B, 2017, 169, 21–26 CrossRef CAS.
- I. Ocsoy, S. Yusufbeyoglu, V. Yılmaz, E. S. McLamore, N. Ildız and A. Ülgen, Colloids Surf., B, 2017, 159, 16–22 CrossRef CAS.
- Pradeepa, K. Udaya Bhat and S. M. Vidya, J. Drug Delivery Sci. Technol., 2017, 37, 20–27 CrossRef CAS.
- L. Rastogi, A. J. Kora and J. Arunachalam, Mater. Sci. Eng., C, 2012, 32, 1571–1577 CrossRef CAS.
- R. Singh, S. Patil, N. Singh and S. Gupta, Sci. Rep., 2017, 7, 5792 CrossRef.
- B. Lee, J. Park, M. Ryu, S. Kim, M. Joo, J.-H. Yeom, S. Kim, Y. Park, K. Lee and J. Bae, Sci. Rep., 2017, 7, 13572 CrossRef.
- E. M. Martín del Valle, M. A. Galán and R. G. Carbonell, Ind. Eng. Chem. Res., 2009, 48, 2475–2486 CrossRef.
- A. K. Bajpai and J. Choubey, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 42, 253–275 CrossRef.
- J. Shi, Z. Zhang, W. Qi and S. Cao, Int. J. Biol. Macromol., 2012, 50, 747–753 CrossRef CAS.
- N. A. O. Pitombeira, J. G. Veras Neto, D. A. Silva, J. P. A. Feitosa, H. C. B. Paula and R. C. M. de Paula, Carbohydr. Polym., 2015, 117, 610–615 CrossRef CAS.
- M. R. Lima, H. C. B. Paula, F. O. M. S. Abreu, R. B. C. da Silva, F. M. Sombra and R. C. M. de Paula, Int. J. Biol. Macromol., 2018, 108, 523–530 CrossRef CAS.
- S.-W. Jung, Y.-I. Jeong and S.-H. Kim, Int. J. Pharm., 2003, 254, 109–121 CrossRef CAS.
- D. Smejkalová, K. Nešporová, M. Hermannová, G. Huerta-Angeles, D. Cožíková, L. Vištejnová, B. Safránková, J. Novotný, J. Kučerík and V. Velebný, Int. J. Pharm., 2014, 466, 147–155 CrossRef.
- H.-z. Zhang, F.-p. Gao, L.-r. Liu, X.-m. Li, Z.-m. Zhou, X.-d. Yang and Q.-q. Zhang, Colloids Surf., B, 2009, 71, 19–26 CrossRef CAS.
- S. Kumar, P. Kaur, M. Bernela, R. Rani and R. Thakur, Int. J. Biol. Macromol., 2016, 93, 988–994 CrossRef CAS.
- L. De Matteis, D. Jary, A. Lucía, S. García-Embid, I. Serrano-Sevilla, D. Pérez, J. A. Ainsa, F. P. Navarro and J. M. de la Fuente, Chem. Eng. J., 2018, 340, 181–191 CrossRef CAS.
- J. Deacon, S. M. Abdelghany, D. J. Quinn, D. Schmid, J. Megaw, R. F. Donnelly, D. S. Jones, A. Kissenpfennig, J. S. Elborn, B. F. Gilmore, C. C. Taggart and C. J. Scott, J. Controlled Release, 2015, 198, 55–61 CrossRef CAS.
- A. Abdelkader, M. A. El-Mokhtar, O. Abdelkader, M. A. Hamad, M. Elsabahy and O. N. El-Gazayerly, Carbohydr. Polym., 2017, 174, 1041–1050 CrossRef CAS.
- K. K. Patel, A. K. Agrawal, M. M. Anjum, M. Tripathi, N. Pandey, S. Bhattacharya, R. Tilak and S. Singh, Appl. Nanosci., 2020, 10, 563–575 CrossRef CAS.
- M. Sandhya, V. Aparna, S. Maneesha K, B. Raja, R. Jayakumar and S. Sathianarayanan, Int. J. Biol. Macromol., 2018, 110, 133–139 CrossRef CAS.
- V. Aparna, A. R. Melge, V. K. Rajan, R. Biswas, R. Jayakumar and C. Gopi Mohan, Int. J. Biol. Macromol., 2018, 110, 140–149 CrossRef CAS.
- A. Tamayol, A. Hassani Najafabadi, P. Mostafalu, A. K. Yetisen, M. Commotto, M. Aldhahri, M. S. Abdel-wahab, Z. I. Najafabadi, S. Latifi, M. Akbari, N. Annabi, S. H. Yun, A. Memic, M. R. Dokmeci and A. Khademhosseini, Sci. Rep., 2017, 7, 9220 CrossRef.
- Y. Kaneo, K. Taguchi, T. Tanaka and S. Yamamoto, J. Drug Delivery Sci. Technol., 2014, 24, 344–351 CrossRef CAS.
- L. R. Lumholdt, R. Holm, E. B. Jørgensen and K. L. Larsen, Carbohydr. Res., 2012, 362, 56–61 CrossRef CAS.
- H. Kondo, H. Nakatani and K. Hiromi, Carbohydr. Res., 1990, 204, 207–213 CrossRef CAS.
- M. G. Arafa, R. F. El-Kased and M. M. Elmazar, Sci. Rep., 2018, 8, 13674 CrossRef.
- G. Qing, X. Zhao, N. Gong, J. Chen, X. Li, Y. Gan, Y. Wang, Z. Zhang, Y. Zhang, W. Guo, Y. Luo and X.-J. Liang, Nat. Commun., 2019, 10, 4336 CrossRef.
- R. Canaparo, F. Foglietta, F. Giuntini, C. Della Pepa, F. Dosio and L. Serpe, Molecules, 2019, 24, 1991 CrossRef CAS.
- Z. Lu, A. J. Quek, S. P. Meaney, R. F. Tabor, B. Follink and B. M. Teo, ACS Appl. Bio Mater., 2020, 3, 5880–5886 CrossRef CAS.
- Z. Lu, Z. Zhang and Y. Tang, ACS Appl. Bio Mater., 2019, 2, 4485–4492 CrossRef CAS.
- Z. Yang, Z. Sun, Y. Ren, X. Chen, W. Zhang, X. Zhu, Z. Mao, J. Shen and S. Nie, Mol. Med. Rep., 2019, 20, 5–15 CAS.
- G. R. Rodrigues, C. López-Abarrategui, I. de la Serna Gómez, S. C. Dias, A. J. Otero-González and O. L. Franco, Int. J. Pharm., 2019, 555, 356–367 CrossRef CAS.
- J. Owen, C. Crake, J. Y. Lee, D. Carugo, E. Beguin, A. A. Khrapitchev, R. J. Browning, N. Sibson and E. Stride, Drug. Delivery Transl. Res., 2018, 8, 342–356 CrossRef CAS.
- P. M. Price, W. E. Mahmoud, A. A. Al-Ghamdi and L. M. Bronstein, Front. Chem., 2018, 6, 619 CrossRef CAS.
- G. LuTheryn, P. Glynne-Jones, J. S. Webb and D. Carugo, Microb. Biotechnol., 2020, 13, 613–628 CrossRef CAS.
- J. Y. Lee, C. Crake, B. Teo, D. Carugo, M. de Saint Victor, A. Seth and E. Stride, Adv. Healthcare Mater., 2017, 6, 1601246 CrossRef.
- Y.-Y. Fu, L. Zhang, Y. Yang, C.-W. Liu, Y.-N. He, P. Li and X. Yu, Int. J. Nanomed., 2019, 14, 1805–1815 CrossRef CAS.
- H. Horsley, J. Owen, R. Browning, D. Carugo, J. Malone-Lee, E. Stride and J. L. Rohn, J. Controlled Release, 2019, 301, 166–175 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |