
Rotation of a helical coordination polymer by mechanical grinding†
Bibhuti Bhusan
Rath
 a,
Goutam Kumar
Kole
b,
Samuel Alexander
Morris
*c and
Jagadese J.
Vittal
*a
a,
Goutam Kumar
Kole
b,
Samuel Alexander
Morris
*c and
Jagadese J.
Vittal
*a
aDepartment of Chemistry, National University of Singapore, 117543, Singapore. E-mail: chmjjv@nus.edu.sg; Fax: +65-6779-1691
bDepartment of Chemistry and Research Institute, SRM Institute of Science and Technology, Kattankulathur, Tamil Nadu 603203, India
cSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore. E-mail: smorris@ntu.edu.sg
First published on 7th May 2020
Abstract
Anisotropic cell volume expansion by mechanical grinding of the solid facilitates the concerted rotation of the photo-inert helical coordination polymer, which causes the misaligned arms containing olefin functional groups in the neighbouring strands to align to undergo [2+2] cycloaddition reaction in 83% yield.
Movements of molecules, especially rotational and vibrational motions are highly restricted in the solid state. However, free space around the functional groups and cooperativity among the molecules in crystals enable large motions of different types. Such molecular dynamics has been observed in molecular crystals as well as in coordination polymers (CPs) and metal–organic frameworks (MOFs) for their potential use in the fabrication of nanoscale devices.1–3 Realization of molecular motions resembling gears, gyroscopes and bearings are fascinating advancements in terms of molecular machines.4 Further bicycle-pedal or crankshaft motion is the most frequently observed in the solid state.5–14 Rotation of helical 1D column of phenylene rotators by the influence of temperature has been studied by Garcia-Garibay et al.15
On the contrary, solid state reactions facilitated by grinding, which is popularly called “mechanochemistry”, have been extensively explored.16–20 Kaupp examined various molecular dynamic processes occurring in the solid state by mechanical forces.21 Mechanical pressure upon the motor protein F1-ATPase, is known to induce unidirectional motions.22 Mechanical force can also cause the molecules to undergo bond stretching to a bond scission.23–25 In this manuscript, we describe the influence of mechanical grinding on the molecular rotation of a photostable helical coordination polymer, which causes the misaligned olefin groups in the neighbouring strands to align favourably to undergo solid state photochemical [2+2] cycloaddition reaction (Scheme 1).
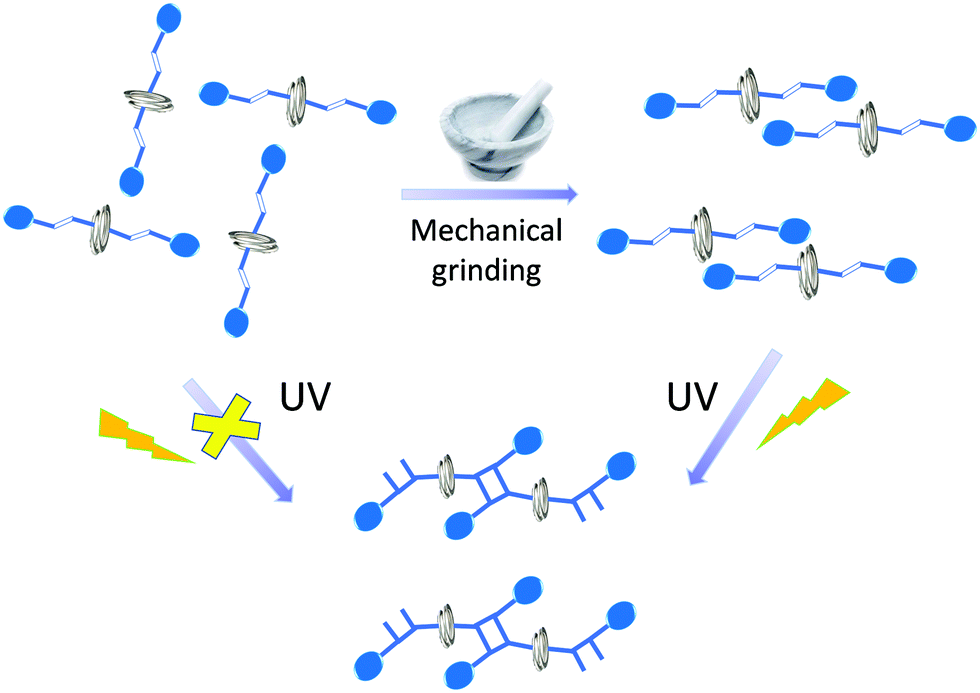 | ||
| Scheme 1 Rotational motion of helical coordination polymer facilitated by mechanical grinding and dimerization under UV irradiation. | ||
The helical coordination polymer, [(Ph3P)Ag(5-Spym)(CF3SO3)]·0.75MeOH, 1 was crystallized in the space group P21/n with Z = 8, from a methanolic solution containing AgCF3SO3, PPh3 and 5-styrylpyrimidine (5-Spym) by slow evaporation.‡ The asymmetric unit contains two [(Ph3P)Ag(5-Spym)(CF3SO3)] units and 1.5 MeOH. The crystal readily loses methanol from its lattice at room temperature and its single crystallinity. The two Ag(I) atoms have distorted tetrahedral geometry from the phosphorus atom of the PPh3 ligand, an oxygen atom of CF3SO3− anion, and two nitrogen atoms from two different 5-Spym ligands (Fig. 1a). The connectivity of the 5-Spym ligands to Ag(I) atoms generates helical chains which propagate along b-axis (Fig. 1b). The pitch of the [Ag(5-Spym)] helical chain is the length of the b-axis, 9.029 Å. Further, the 5-Spym arms of the two helical chains arising from two different Ag(I) atoms are orienting almost normal to each other forming a picket fence enclosing the PPh3 ligands (Fig. 1c).
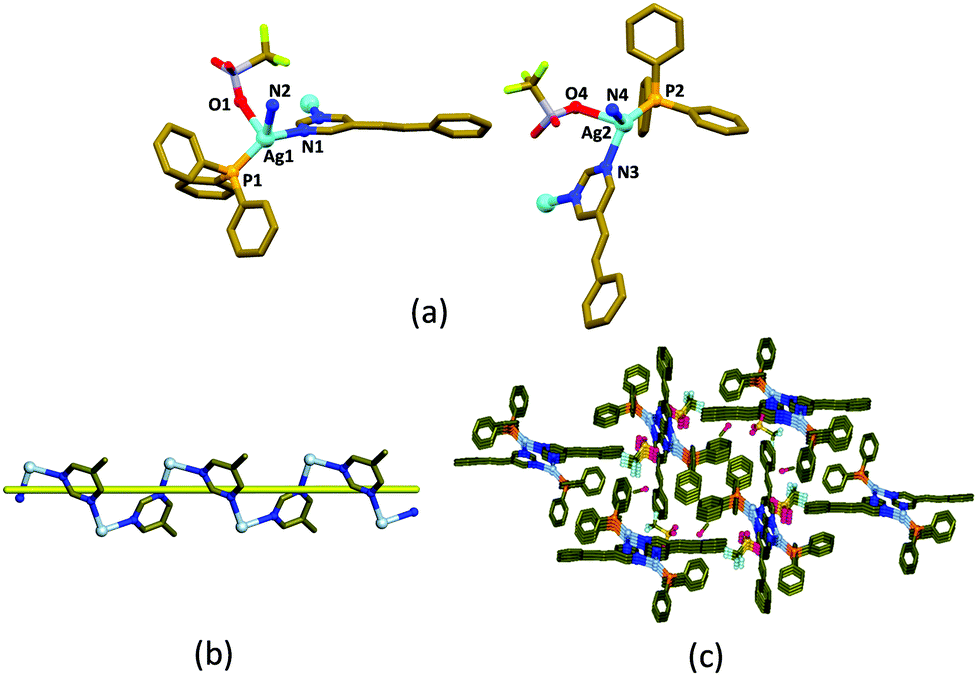 | ||
| Fig. 1 (a) Coordination geometry around Ag(I) atoms with selected labelling of atoms. (b) A helical polymer strand. (c) Packing of 1 showing different orientations of the 5-Spym arms in the helical one-dimensional CP when viewed from b-axis. The disorder and H-atoms are omitted for clarity. | ||
It is evident from the packing that there is no alignment of the olefin pairs between the helical chains satisfying the Schmidt's criteria for [2+2] cycloaddition reaction,26 and hence 1 is expected to be photo stable. This is also confirmed after exposing the crystals of 1 under UV light for a few hours. However, when the crystals were ground to powder using pestle and mortar and then exposed to UV light (λmax = 360 nm), the 1H-NMR spectrum of the irradiated powder in DMSO-d6, showed a multiplet peak centred at 4.76 ppm which indicate to the formation of cyclobutane photoproduct. It is surprising that the photo-inert crystals of 1 exhibit photoreactivity after grinding. Henceforth, the [2+2] cycloaddition reactivity was followed with different grinding times. After grinding, the powders were subjected to UV irradiation for 72 h and the 1H-NMR spectra were recorded.
Fig. 2a shows the 1H-NMR spectra at different time intervals of UV exposure for the 30 min ground sample. Gradual disappearance of the proton signals of the pyrimidine group at 9.1 ppm, and evolution of the pyrimidine proton peaks at 8.8 and 8.6 as well as the cyclobutane peaks at ca. 4.76 ppm with different UV exposure time shown in Fig. 2a indicate the progress of the photoreaction. The percentage formation of the cyclobutane photoproduct versus time of UV irradiation in Fig. 2b shows a maximum of 83% conversion of the 5-Spym ligand to rctt-1,3-bis(5′-pyrimidyl)-2,4-bis(phenyl)cyclobutane (rctt-bpcb)27 which could arise only from the head-to-tail alignment in the ground solid. This seems to be accompanied by some side reaction as inferred from the unidentified peaks in the 1H-NMR spectra. The dimerization is further complemented by the FT-IR spectral data (Fig. S3, ESI†). A comparison of the FT-IR spectra between the crystal of 1 and ground sample after 30 min grinding shows there is no change in the local coordination mode. For the 30 min ground sample irradiated under UV for 25 h, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at 1636 cm−1 and C–H out of plane bending at 965 cm−1 were completely disappeared suggesting the conversion of the olefin groups to cyclobutane group. In order to get further evidence for the head-to-tail dimerization in the photoproduct, rctt-bpcb was isolated after chemical treatment and crystallized in DMSO. The single crystal structure of the isolated product clearly validates the head-to-tail dimerization of the ligands.§
C stretching at 1636 cm−1 and C–H out of plane bending at 965 cm−1 were completely disappeared suggesting the conversion of the olefin groups to cyclobutane group. In order to get further evidence for the head-to-tail dimerization in the photoproduct, rctt-bpcb was isolated after chemical treatment and crystallized in DMSO. The single crystal structure of the isolated product clearly validates the head-to-tail dimerization of the ligands.§
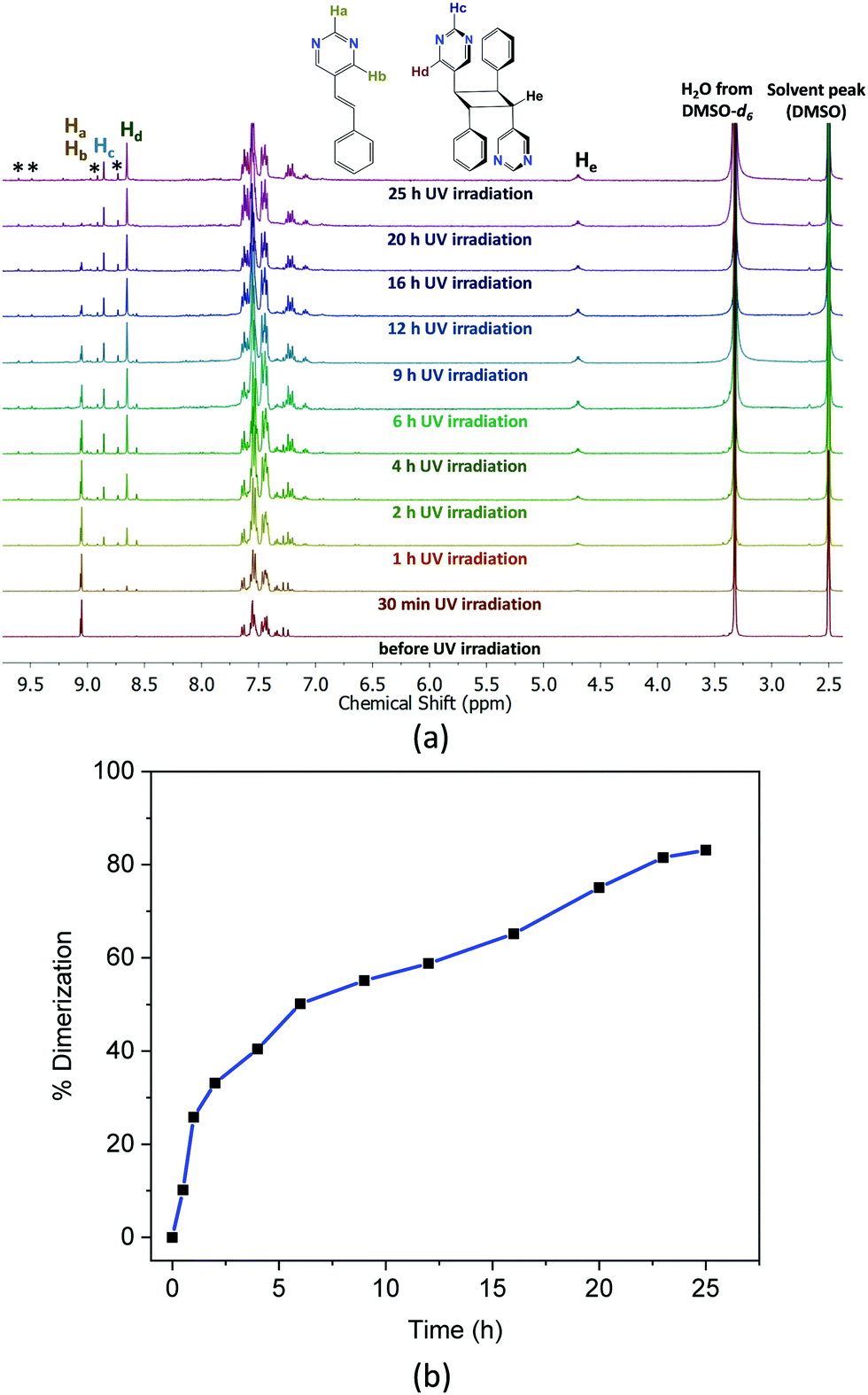 | ||
| Fig. 2 (a) 1H-NMR spectra of 1 (30 min grinding) in DMSO-d6 plotted at different times of UV irradiation. Complete absence of the Ha, Hb protons may indicate quantitative consumption of the 5-Spym ligand. However, some unidentified peaks indicate only 83% of rcct-bpcb photoproduct formed. (b) Time-dimerization plot for 30 min ground sample shows the progress of the photoreaction under UV light with time. | ||
The powder X-ray diffraction (PXRD) patterns in Fig. 3 show that there is a gradual change in crystallinity with increasing grinding time. Broader peaks suggest decrease in the crystallite size and crystallinity with increasing grinding time. The decrease in the crystallite size is also corroborated from the SEM images (Fig. S4, ESI†), where bigger particles transform to plate shaped microcrystals after 30 min grinding.
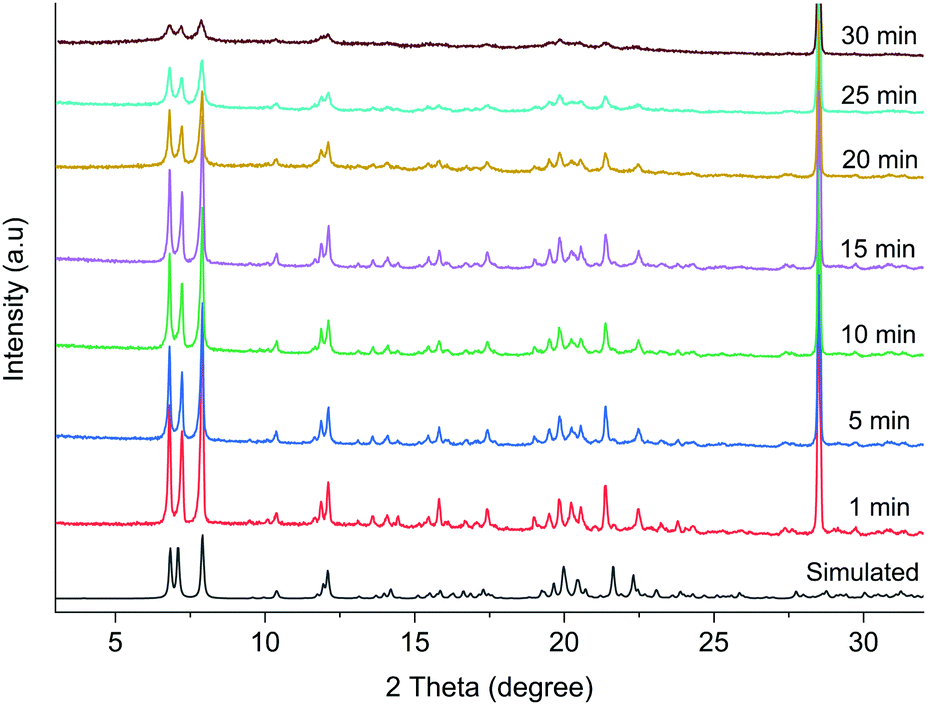 | ||
| Fig. 3 The PXRD patterns of 1 at different grinding time along with the simulated pattern from the single crystal data. | ||
Previously reported photoreactive helical CP chains with well-aligned olefin groups in the 5-Spym ligands,27 yielded 100% dimerized product upon UV irradiation. In this work, while powdered samples of 1 after 20 min and 25 min grinding resulted 40% and 59% dimerization respectively after 72 h of UV irradiation, a maximum dimerization of 83% was only achieved for 30 minute grinding sample in 25 h of UV irradiation. Further, the single crystals heated to 150 °C for 30 min were found to be photo-inert under UV light. Thus, the photoreactivity is not due to heat generated by grinding.
Orderly alignments of the olefin pairs are required for the solid-state photochemical [2+2] cycloaddition reaction and this can be achieved only in the solids with long-range order. Usually, grinding of a solid is expected to reduce the particle size. Hence, the amount of photoproduct will increase due to increase in the surface area as UV light can penetrate better. In this current study, there is no perfect alignment of the olefin pairs and the long-range order seems to be reduced due to increased amorphization of the 30 min ground sample as evident from the broadening of the peaks in the powder X-ray diffraction in Fig. 3. Further analysis by high resolution transmission electron microscopy (HRTEM) was done to examine the crystalline nature of the sample. The HRTEM images (Fig. S5, ESI†) clearly show lattice fringes and crystallinity, inferring that the reduced size in the crystallite size did not destroy the crystallinity completely. In order to investigate the volume expansion, if any, by grinding, the densities of the powders at different time of grinding were measured by neutral buoyancy method and the results are plotted in Fig. 4. The trend highlights that grinding decreases the density of the sample and hence, increase the cell volume.
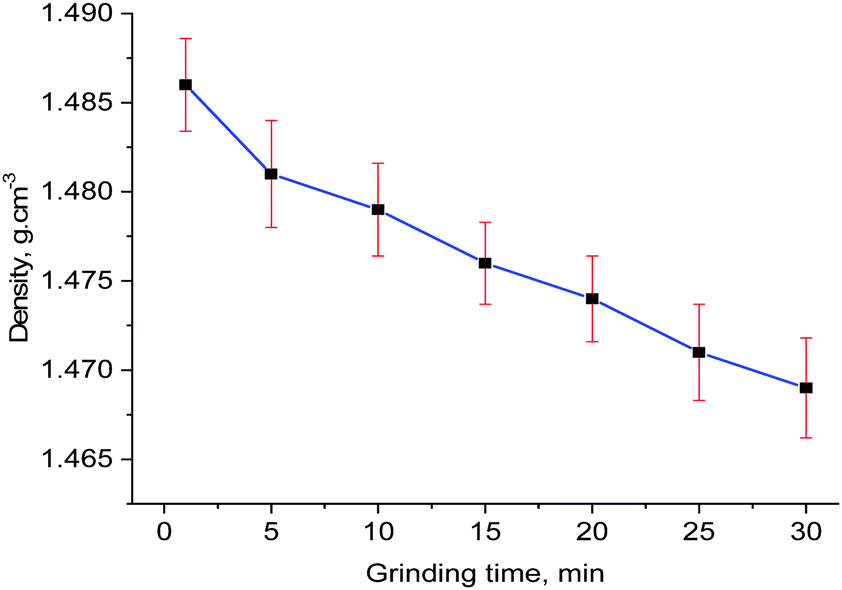 | ||
| Fig. 4 A plot of measured densities of the ground samples with grinding time. | ||
In order to get further insights into the nature of volume expansion, Pawley refinements were undertaken on all samples using the TOPAS program.28 The results are displayed in Table 1 which corroborates the results from the density measurements. The grinding time increases the lengths of a-axis while the helical b-axis remains relatively unaltered. The increase in a-axis lengths can be explained by the rotation of the helical chains along b-axis which brings the styryl arms closer together to have head-to-tail alignment, which in turn, satisfy the Schmidt's conditions for the photoreactivity.26 Apart from this, increased amorphization due to mechanical grinding could facilitate more rotational motion and the amorphous phase present could still give rise to head-to-tail alignment of the ligands. Also, the rotational motion and consequent photoreaction upon UV irradiation may occur on thin surfaces of the particles. This explains the enhanced photoreactivity of 30 min grinding sample with particle size around 11 nm.
| Sample | LP – a, Å | LP – b, Å | LP – c, Å | LP – β, ° | V, Å3 | ρ, g cm−3 (experimental) | Crystallite size (nm) |
|---|---|---|---|---|---|---|---|
| Complete details about the TOPAS28 results are given in ESI (Table S1). | |||||||
| 1 min grinding | 26.4759(5) | 9.1166(1) | 28.0833(5) | 111.524(1) | 6306.2(2) | 1.486(2) | 87.9(8) |
| 10 min grinding | 26.5179(6) | 9.11383 | 28.0879(6) | 111.527(2) | 6314.7(3) | 1.480(2) | 66.4(6) |
| 20 min grinding | 26.5310(7) | 9.1115(2) | 28.0967(6) | 111.565(2) | 6316.6(3) | 1.474(2) | 50.6(5) |
| 30 min grinding | 26.540(1) | 9.112(3) | 28.096(9) | 111.50(3) | 6321.8(4) | 1.469(3) | 11.5(2) |
In summary, a helical CP was found to be photo-inert in the solid state as the olefin groups in the neighbouring styryl arms were not suitably aligned to under [2+2] cycloaddition reaction. However, the ground samples showed increased photoreaction with increasing grinding time, and quantitative consumption of olefin bonds were observed in 30 min of ground sample after 25 h of UV irradiation. The photoproduct is expected to have a two-dimensional structure. Movements of the functional groups have been observed in CPs in the solid state.15–21 Further, mechanical grinding has been known to promote pedal motion of crisscrossed olefin pairs,8–14 movements of molecules,29–31 and change in chemical composition.32 Large molecular movement of ladders in the crystal lattice by mechanical grinding was reported to facilitate quantitative photodimerization.33 In a rare case, here we observed that simple mechanical grinding induced cooperative rotational movements of the neighbouring helices in 1, which facilitates the alignment of olefin bonds as confirmed by the solid state [2+2] cycloaddition reaction under UV light. Unusually, grinding causes anisotropic volume expansion as evident from the density measurements and cell data.
This work was financially supported by the Ministry of Education, Singapore (Grant No. Tier 1 WBS R-143-000-A12-114 and R-143-000-B13-114). We acknowledge the help of Ms Geok Kheng for collecting the single crystal X-ray intensity data.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- J. D. Dunitz, E. F. Maverick and K. N. Trueblood, Angew. Chem., Int. Ed. Engl., 1988, 27, 880–895 CrossRef.
- C. S. Vogelsberg and M. A. Garcia-Garibay, Chem. Soc. Rev., 2012, 41, 1892–1910 RSC.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9(8), 7746–7768 CrossRef CAS PubMed.
- M. K. Sharma and P. K. Bharadwaj, Inorg. Chem., 2011, 50, 1889–1897 CrossRef CAS PubMed.
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244–2252 RSC.
- T.-A. V. Khuong, G. Zepeda, C. N. Sanrame, H. Dang, M. D. Bartberger, K. N. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2004, 126, 14778–14786 CrossRef CAS PubMed.
- J. Harada and K. Ogawa, J. Am. Chem. Soc., 2004, 126, 3539–3544 CrossRef CAS PubMed.
- J. Harada, M. Harakawa and K. Ogawa, CrystEngComm, 2008, 1777–1781 RSC.
- S. Galli, P. Mercandelli and A. Sironi, J. Am. Chem. Soc., 1999, 121, 3767–3772 CrossRef CAS.
- B. R. Bhogala, B. Captain, A. Parthasarathy and V. Ramamurthy, J. Am. Chem. Soc., 2010, 132, 13434–13442 CrossRef CAS PubMed.
- A. M. P. Peedikakkal and J. J. Vittal, Chem. – Eur. J., 2008, 14, 5329–5334 CrossRef CAS PubMed.
- Q. Chu, D. C. Swenson and L. R. MacGillivray, Angew. Chem., Int. Ed., 2005, 44, 3569–3572 CrossRef CAS PubMed.
- A. Chanthapally, W. T. Oh and J. J. Vittal, Chem. Commun., 2014, 50, 451–453 RSC.
- S. Pérez-Estrada, B. Rodríguez-Molina, E. F. Maverick, S. I. Khan and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2019, 141, 2413–2420 CrossRef PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002–4011 CrossRef CAS PubMed.
- A. V. Trask and W. Jones, Top. Curr. Chem., 2005, 254, 41–70 CrossRef CAS.
- A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC.
- C. Mottillo and T. Friščić, Molecules, 2017, 22, 144 CrossRef PubMed.
- G. Kaupp, Curr. Opin. Solid State Mater. Sci., 2002, 6, 131–138 CrossRef CAS.
- H. Noji, D. Okuno and T. Ikeda, Chem. Sci., 2011, 2, 2086–2093 RSC.
- K. M. Wiggins, J. N. Brantley and C. W. Bielawski, Chem. Soc. Rev., 2013, 42, 7130–7147 RSC.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- G. De Bo, Chem. Sci., 2018, 9, 15–21 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS.
- B. B. Rath, G. K. Kole and J. J. Vittal, Cryst. Growth Des., 2018, 18(10), 6221–6226 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- M. Nagarathinam and J. J. Vittal, Angew. Chem., Int. Ed., 2006, 45, 4337–4341 CrossRef CAS PubMed.
- M. Nagarathinam and J. J. Vittal, Chem. Commun., 2008, 438–440 RSC.
- A. Chanthapally, W. T. Oh and J. J. Vittal, CrystEngComm, 2013, 15, 9324–9327 RSC.
- G. K. Kole, G. K. Tan and J. J. Vittal, Org. Lett., 2010, 12, 128–131 CrossRef CAS PubMed.
- H. A. Al-Mohsin, A. AlMousa, S. A. Oladepo, A. S. Jalilov, M. Fettouhi and A. M. P. Peedikakkal, Inorg. Chem., 2019, 58, 10167–10173 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1989895 and 1990101. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc02158j |
‡ Crystal data for 1 at 100(2) K (CCDC 1989895): C31.75H28AgF3N2O3.75PS, M = 725.46; monoclinic, P21/n; a = 26.6378(6), b = 9.0287(2), c = 27.6480(6) Å; β = 110.8540(10); V = 6213.9(2) Å3; Z = 8; ρcalc = 1.551 g cm−3; μ = 0.824 mm−1; GOF = 1.032; final R1 = 0.0532; wR2 = 0.0898 [for 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 716 data I > 2σ(I)]. 716 data I > 2σ(I)]. |
| § Crystal data for rctt-bpcb at 100(2) K (CCDC 1990101): C24H20N4, M = 364.44; monoclinic, P21/c; a = 10.4634(5), b = 8.3919(4), c = 10.5935(6) Å; β = 98.999(2); V = 918.74(8) Å3; Z = 2; ρcalc = 1.317 g cm−3; μ = 0.080 mm−1; GOF = 1.051; final R1 = 0.0505; wR2 = 0.1336 [for 2267 data I > 2σ(I)]. |
| This journal is © The Royal Society of Chemistry 2020 |