
C–F bond activation by pentamethylcyclopentadienyl-aluminium(I): a combined experimental/computational exercise†
Oleksandr
Kysliak
a,
Helmar
Görls
a and
Robert
Kretschmer
 *ab
*ab
aInstitute of Inorganic and Analytical Chemistry (IAAC), Friedrich Schiller University Jena, Humboldtstraße 8, 07743 Jena, Germany
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena Philosophenweg 7, 07743 Jena, Germany. E-mail: robert.kretschmer@uni-jena.de
First published on 3rd February 2020
Abstract
The reaction of (Cp*Al)4 with a series of fluoro(hetero)arenes has been investigated and C–F bond activation was observed with perfluorotoluene, pentafluoropyridine as well as 1,2,3,4-tetrafluoro-, pentafluoro- and hexafluorobenzene. The reaction mechanism has been probed by means of DFT calculations and the computational findings are in good agreement with the experimental observations.
Since the first report about the isolation of a monovalent aluminium compound, i.e., tetrameric (Cp*Al)41 (1) by the group of Schnöckel and the isolation of the aluminium(I) β-diketiminate 2 by Roesky and co-workers in 2000,2Fig. 1, subvalent aluminium compounds and studies on their reactivity have received considerable interest.3 Both compounds differ from each other not only with respect to their electronic structure but also in terms of their synthetic accessibility. Although the tetramer 1 is preferred over the respective monomer 1′ by about 150 kJ mol−1,4 the reactivity of dissolved (Cp*Al)4 is due to the presence of monomeric AlCp* possessing a lone pair and a vacant orbital at the aluminium atom. 2 exists as monomer in both the solid state and in solution, and is considered as an aluminium analogue of N-heterocyclic carbenes. While the synthesis of 1 has been improved from yields of 44%1 up to 93%5 and without the requirement of strong reducing agents, 2 is not as readily obtained.2,6 In distinct contrast, the reactivity of 23a,7 is much more explored compared to 1,8 for which reports have been faded after an initial period of intense research.
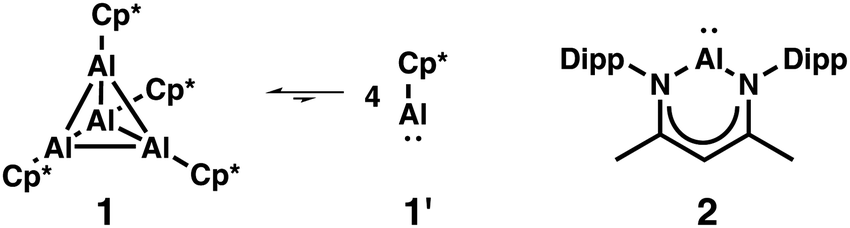 | ||
| Fig. 1 Monovalent aluminium compounds 1 and 2. Cp* = pentamethylcyclopentadienyl, Dipp = 2,6-Diisopropylphenyl. | ||
The oxidative addition of strong σ-bonds has been believed to be limited to transition metals, but the last decade has witnessed that main-group elements are also able to split strong σ-bonds.9 The activation of C–F bonds is particularly challenging, due to their high bond dissociation energies and only a few examples incorporating Al(I) and Mg(I) derivatives have been reported in the last five years.7a,b,10 For aluminium, the activation of both, aliphatic and aromatic C–F bonds has been reported, but all originate from 2, which lacks access in decent yields. As the readily available pentamethylcyclopentadienyl-aluminium(I) (1) has been shown to activate Si–F bonds,11 we wondered whether it also allows for the activation of aromatic and heteroaromatic C–F bonds and our findings are reported herein.
(Cp*Al)4 (1)1 was treated with an excess of fluorobenzene, 1,2-difluorobenzene, 1,3,5-trifluorobenzene, 1,2,3,4-tetrafluorobenzene, pentafluorobenzene, hexafluorobenzene, pentafluoropyridine, and perfluorotoluene, Scheme 1. While slow oxidative addition of pentafluoropyridine 3a to 1 is already observed at room temperature, as evidenced by 1H NMR spectroscopy, heating to 90 °C is desirable in order to achieve complete conversion. Here, exclusive and regioselective activation of the C–F bond in 4-position, i.e., para to the nitrogen moiety, takes place.
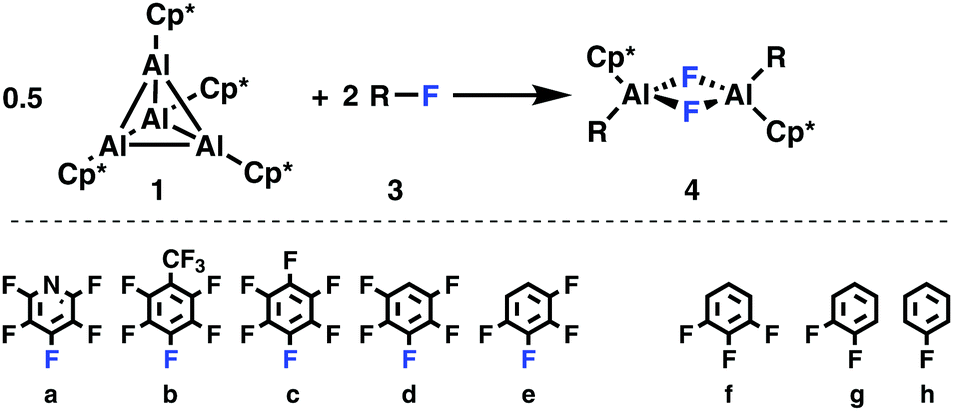 | ||
| Scheme 1 Oxidative addition of the fluoro(hetero)arenes 3a–e to 1 is observed experimentally, while 3f–h remained unreactive at 90 °C. | ||
Activation of perfluorotoluene (3b) necessitates heating to 90 °C for 15 minutes, while for penta- (3d) and hexafluorobenzene (3c) heating for 24 hours is required. To achieve complete conversion of 1,2,3,4-tetrafluorobenzene, the reaction time has to be increased to five days. The less fluorine substituted benzenes, i.e., 1,3,5-C6H3F3, 1,2-C6H4F2, and C6H5F, do not show any reactivity despite heating to 90 °C for several days. According to the 1H NMR spectra, the conversion of 1 is quantitative in cases of 3a–e and the respective aluminium(III) complexes are obtained in crystalline yields ranging from 23 to 57%. As observed for pentafluoropyridine, reaction of 1 with perfluorotoluene, penta- and tetrafluorobenzene occurs regioselectively by activation of the C–F bond in 4, 3, and 2 position, respectively. Such a regioselectivity has also been observed in case of the 2/C6H2F4 and 2/C6HF5 couples.10a,b
Single-crystals of the aluminium(III) complexes 4a–e were obtained and allowed for an X-ray diffraction analysis. The respective molecular structures in the solid state are illustrated in Fig. 2 and Fig. S1 (ESI†). Each Cp*AlF(R) fragment represents a part of a centrosymmetric dimer in which both distorted tetrahedral aluminium atoms are fluorine-bridged. The distances between the aluminium atom and the Cp* plane take values between 1.876 (4a) and 1.918 (4e) Å, thus being comparable with values obtained for other (Cp*AlXR)2 species as reported before.12 Notably, the chlorine analogue of 4c has been obtained by reacting Cp*2AlCl with B(C6F5)3 and possesses similar structural features.13 The aluminium–carbon and aluminium–fluorine bond lengths are in the range of 1.990(2) to 2.0049(14) Å and 1.8391(19) to 1.8513(19) Å, hence comparable to those found in the related β-diketiminate aluminium(III) complexes.10a,b
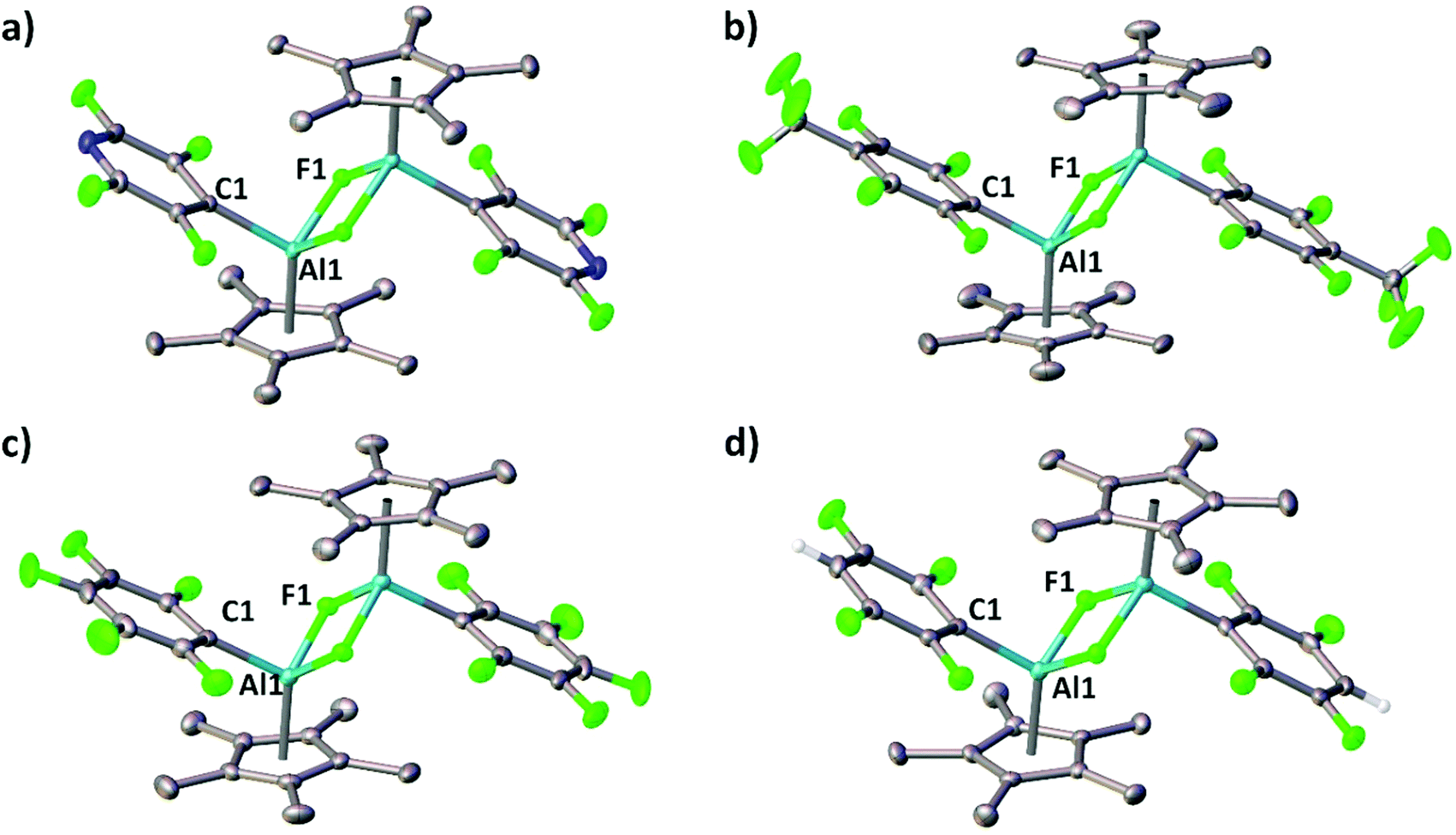 | ||
| Fig. 2 Solid-state structures of 4a–d (hydrogen atoms except the aromatic protons are omitted for the sake of clarity). Selected bond lengths [Å] and angles [°] with calculated values (M06-2X/6-31G(d)//M06-2X(SMD)/6-311+G(d,p)) in square brackets: (a) 4a Al1–C1 2.0049(14) [1.992], Al1–F1 1.8391(9) [1.849], C1–Al1–F1 99.01(5) [98.66]; (b) 4b Al1–C1 1.9987(16) [1.990], Al1–F1 1.8410(9) [1.843], C1–Al1–F1 99.68(5) [99.15]; (c) 4c Al1–C1 1.990(2) [1.984], Al1–F1 1.8403(12) [1.846], C1–Al1–F1 100.33(7) [99.79]; (d) 4d Al1–C1 1.994(4) [1.981], Al1–F1 1.8513(19) [1.850], C1–Al1–F1 101.82(11) [100.04]. | ||
The complexes 4a–e were fully characterised including 1H, 13C and 19F NMR as well as IR spectroscopy; 27Al-NMR resonances could not been detected despite extended numbers of scans. The 1H and 13C NMR spectra reveal one singlet for the Cp* methyl resonances in the range of 1.37–1.63 ppm and 9.4–9.7 ppm, respectively. The observed steady downfield shift in going from 4a to 4e accounts for the increased deshielding due to the increasing Al–Cp* separation. The 19F NMR spectra feature a broad singlet, due to J coupling to 27Al (I = 5/2) at about −109 ppm, which appears downfield shifted compared to the other yet reported values of dinuclear aluminium compounds with bridging fluorine groups but more electron rich ligands.14 Furthermore, the additional 19F resonances have the expected pattern characteristic for fluoro(hetero)arene substituents. Notably, the room temperature 1H and 19F NMR spectra of 4b, 4c, and 4d, respectively, reveal two sets of resonances in different proportions ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in case of 4b to 1:12 for 4c. As the effect is most pronounced for 4b, diffusion-ordered (DOSY) NMR experiments have been performed (Fig. S30, ESI†). Both 1H resonances (1.4 and 1.65 ppm) show a mono-exponential and comparable diffusion behaviour, which indicates a similar hydrodynamic radius and makes a conceivable monomer–dimer equilibrium unlikely. Variable-temperature (VT) 1H and 19F NMR experiments in the 233–333 K range were also performed and a temperature dependence of the chemical shifts and incipient coalescence at 333 K was observed. Hence, we speculated that besides the dimeric species 4 observed in the solid state, an isomeric form 4′ exists in solution, in which the two fluoroaryl substituents are located on the same instead of opposing sides of the Al2F2 plane. Based on the NMR data the conceivable formation of AlCp*R2/AlCp*F2 couples or the respective adducts by ligand redistribution is unlikely as one would not only expect two sets of 1H resonances but also more complex 19F spectra.
:4 in case of 4b to 1:12 for 4c. As the effect is most pronounced for 4b, diffusion-ordered (DOSY) NMR experiments have been performed (Fig. S30, ESI†). Both 1H resonances (1.4 and 1.65 ppm) show a mono-exponential and comparable diffusion behaviour, which indicates a similar hydrodynamic radius and makes a conceivable monomer–dimer equilibrium unlikely. Variable-temperature (VT) 1H and 19F NMR experiments in the 233–333 K range were also performed and a temperature dependence of the chemical shifts and incipient coalescence at 333 K was observed. Hence, we speculated that besides the dimeric species 4 observed in the solid state, an isomeric form 4′ exists in solution, in which the two fluoroaryl substituents are located on the same instead of opposing sides of the Al2F2 plane. Based on the NMR data the conceivable formation of AlCp*R2/AlCp*F2 couples or the respective adducts by ligand redistribution is unlikely as one would not only expect two sets of 1H resonances but also more complex 19F spectra.
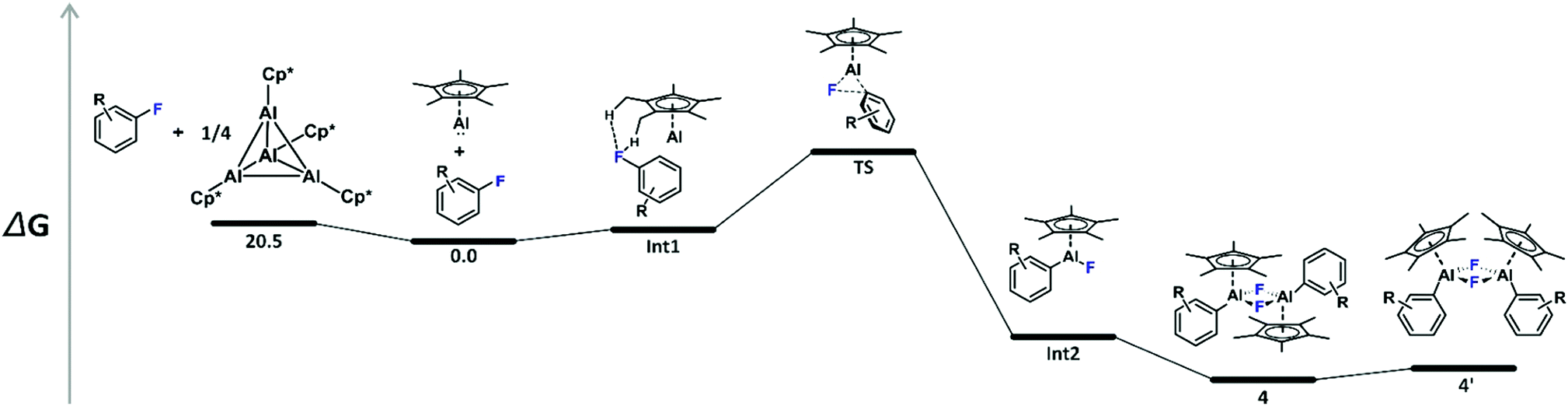
In order to rationalize the experimental findings and to reveal the origin of the second set of resonances in the 1H and 19F NMR spectra of 4b, 4c, and 4d, the oxidative addition reactions have been explored by means of density functional theory (DFT) calculations on the M06-2X/6-31G(d)//M06-2X(SMD)/6-311+G(d,p) level of theory.15 Notably, C–F bond activation by 2 has recently also been investigated computationally.10f,16 A schematic potential-energy surface is depicted in Table 1 along with the respective energies. The calculated tetramerization enthalpy of −159.8 kJ mol−1 is in good agreement with the experimental value (−150 kJ mol−1).17 However, based on the Gibbs free energies, tetramerization is in contrast to the experiment (−60.6 kJ mol−1 at 298 K) endergon by 20.5 kJ mol−1 due to the overestimation of entropic contributions to gas-phase calculations, which has been discussed before for various substituted aluminium cyclopentadienyls.18 In consequence, all energies given in Table 1 are referenced to the 1′/3 couples. Formation of the encounter complex Int1 is exothermic but endergonic for all substrates investigated. Int1 features weak noncovalent C–H⋯F–C interactions19 involving the fluorine atoms of the fluoro(hetero)arenes and the methyl groups of the Cp* unit, with H⋯F distances between 2.36 and 2.50 Å. Next, C–F bond activation occurs in a concerted manner, i.e., simultaneous C–F bond breaking and Al–C as well as Al–F bond making via the three-membered transition structure TS. The transition state involve alternating electron transfer from the aluminium lone pair to the antibonding σ* orbital of the C–F bond and from the fluorine lone pair to the vacant p-type orbital at aluminium. The calculated activation energies are in good agreement with the experimental parameters and explain then non-occurring C–F bond activation in case of the substrates 3f–h, Table 1. Please note that the transition state energies reported for 2 are by 33.3 to 67.4 kJ mol−1 lower,16c which reflects well the differences observed experimentally and attributes to the higher HOMO LUMO separation in case of 1′, Fig. S2 (ESI†). The thus formed Lewis-acidic tricoordinated aluminium(III) species Int2 dimerises to give 4 in which the aluminium atoms are stabilised by an electron octet. The stabilisation is expressed by a gain in free energy of 90.8 to 98.7 kJ mol−1. However, a second isomer 4′, with almost parallel oriented fluoro(hetero)arene and adjacent Cp* substituents, Fig. 3, is by only 8.0 to 14.7 kJ mol−1 higher in energy compared to 4, which explains the experimental observation of the second species. Please note that according to DFT calculations the 4′ isomer should be most pronounced in case of 4e but the small energy differences are within the uncertainty of the computational protocol. Furthermore, our attempts to locate a transition structure connecting 4 and 4′ on the potential-energy surfaces remained unsuccessful despite several efforts.
|
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | Int1 | Pos | TS | Int2 | 4 | 4′ | Substrate | Int1 | Pos | TS | Int2 | 4 | 4′ |
| C6F5N (3a) | 27.8 (−20.8) | 2 | 146.2 (94.2) | −249.7 (−298.4) | −338.5 (−464.9) | −325.8 (−458.0) | 1,2,3,4-C6H2F4 (3e) | 32.0 (−11.7) | 1 | 170.3 (119.2) | −262.4 (−314.2) | −358.6 (−478.5) | −353.9 (−472.8) |
| 32.4 (−19.4) | 3 | 136.8 (82.3) | −312.0 (−363.1) | −404.4 (−533.1) | −396.8 (−523.4) | 2 | 145.4 (91.6) | −293.9 (−346.8) | −388.7 (−513.9) | −380.7 (−506.4) | |||
| 4 | 102.8 (49.2) | −308.5 (−360.09) | −399.3 (−528.9) | −391.2 (−520.8) | |||||||||
| CF3C6F5 (3b) | 30.9 (−23.5) | 2 | 116.8 (61.8) | −313.6 (−368.8) | −359.5 (−501.7) | −370.6 (−510.7) | 1,3,5-C6H3F3 (3f) | 33.2 (−14.8) | 189.6 (137.5) | −228.8 (−274.2) | −316.5 (440.9) | −299.6 (−427.6) | |
| 3 | 132.9 (78.1) | −314.1 (−365.0) | −408.1 (−539.7) | −395.8 (−531.8) | |||||||||
| 4 | 108.5 (56.0) | −311.2 (−363.0) | −409.9 (−535.0) | −396.1 (−531.4) | |||||||||
| C6F6 (3c) | 30.4 (−21.5) | 132.8 (80.5) | −311.9 (−363.6) | −409.0 (−529.8) | −394.3 (−525.3) | o-C6H4F2 (3g) | 28.8 (−12.8) | 176.9 (126.9) | −249.2 (−300.5) | −337.4 (−459.1) | −324.2 (−444.7) | ||
| C6HF5 (3d) | 29.2 (−12.8) | 1 | 164.7 (109.3) | −272.3 (−322.89) | −363.9 (−491.3) | −359.9 (−481.6) | C6H5F (3h) | 26.1 (−12.2) | 202.4 (150.6) | −213.6 (−270.2) | −295.5 (−408.2) | −270.8 (−387.2) | |
| 33.6 (−19.3) | 2 | 146.8 (92.2) | −301.0 (−354.1) | −398.3 (−523.4) | −390.6 (−517.2) | ||||||||
| 3 | 132.2 (80.4) | −303.9 (−355.9) | −398.5 (−524.9) | −386.9 (−516.1) | |||||||||
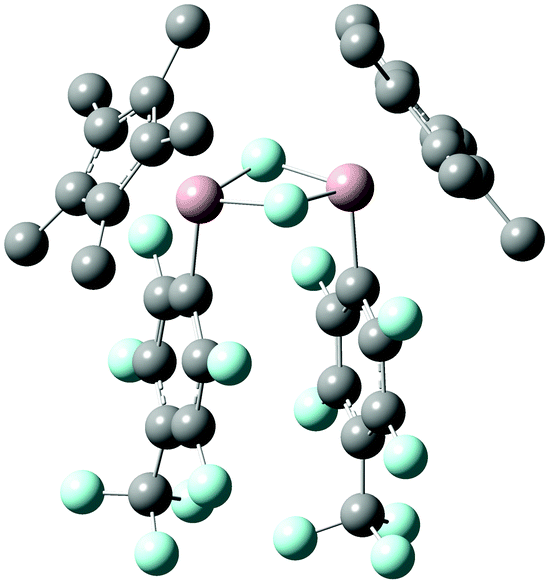 | ||
| Fig. 3 Molecular structure of 4b′ calculated at the M06-2X/6-31G(d) level of theory. Hydrogen atoms are omitted for the sake of clarity. Aluminium, carbon, and fluorine atoms are represented in rose, grey, and turquoise, respectively. | ||
The work is dedicated to John E. Bercaw on the occasion of his 75th birthday. The project was financially supported by the Deutsche Forschungsgemeinschaft (DFG, KR4782/3-1), and the Friedrich Schiller University Jena. We thank the Rechenzentrum of the Friedrich Schiller University Jena for the allocation of computer time and Rica Patzschke and Friederike Pielenz for helpful discussion and assistance with the NMR measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564 CrossRef
.
- C. M. Cui, H. W. Roesky, H. G. Schmidt, M. Noltemeyer, H. J. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274 CrossRef CAS PubMed
-
(a) Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387 CrossRef CAS
- J. Gauss, U. Schneider, R. Ahlrichs, C. Dohmeier and H. Schnoeckel, J. Am. Chem. Soc., 1993, 115, 2402 CrossRef CAS
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Chem. Commun., 2013, 49, 2858 RSC
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810 CrossRef CAS
-
(a) C. Bakewell, B. J. Ward, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 2348 RSC
-
(a) S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed. Engl., 1993, 32, 1729 CrossRef
-
(a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed
-
(a) M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994 RSC
- S. Schulz, T. Schoop, H. W. Roesky, L. Häming, A. Steiner and R. Herbst-Irmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 919 CrossRef CAS
- S. Schulz, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, J. Organomet. Chem., 1995, 493, 69 CrossRef CAS
- C. L. B. Macdonald, J. D. Gorden, A. Voigt, S. Filipponi and A. H. Cowley, Dalton Trans., 2008, 1161 RSC
-
(a) C. Schnitter, K. Klimek, H. W. Roesky, T. Albers, H.-G. Schmidt, C. Röpken and E. Parisini, Organometallics, 1998, 17, 2249 CrossRef CAS
-
(a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS
-
(a) Y. Kim, H. Cho and S. Hwang, Bull. Korean Chem. Soc., 2017, 38, 282 CrossRef CAS
- M. Huber and H. Schnöckel, Inorg. Chim. Acta, 2008, 361, 457 CrossRef CAS
- W. W. Tomlinson, D. H. Mayo, R. M. Wilson and J. P. Hooper, J. Phys. Chem. A, 2017, 121, 4678 CrossRef CAS PubMed
-
P. Panini and D. Chopra, in Hydrogen Bonded Supramolecular Structures, ed. Z.-T. Li and L.-Z. Wu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 37–67 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: For crystallographic, computational, and experimental details, and XYZ-coordinates. CCDC 1971668–1971672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc00003e |
| This journal is © The Royal Society of Chemistry 2020 |