
Multivalent metal ion hybrid capacitors: a review with a focus on zinc-ion hybrid capacitors
Liubing
Dong
 a,
Wang
Yang
a,
Wu
Yang
a,
Yang
Li
b,
Wenjian
Wu
c and
Guoxiu
Wang
*a
a,
Wang
Yang
a,
Wu
Yang
a,
Yang
Li
b,
Wenjian
Wu
c and
Guoxiu
Wang
*a
aCentre for Clean Energy Technology, University of Technology Sydney, NSW 2007, Australia. E-mail: guoxiu.wang@uts.edu.au; Fax: +62-2-95141460; Tel: +62-2-95141741
bSchool of Photovoltaic and Renewable Energy Engineering, University of New South Wales, NSW 2052, Australia
cDepartment of Materials Science and Engineering, Dongguan University of Technology, Dongguan 523808, China
First published on 16th April 2019
Abstract
Multivalent metal ion hybrid capacitors have been developed as novel electrochemical energy storage systems in recent years. They combine the advantages of multivalent metal ion batteries (e.g., zinc-ion batteries, magnesium-ion batteries, and aluminum-ion batteries) with those of supercapacitors, and are characterized by good rate capability, high energy density, high power output and ultralong cycle life. Herein, after a brief introduction to supercapacitors and multivalent metal ion batteries, we reviewed the recent progress in research on multivalent metal ion hybrid capacitors, with a focus on zinc-ion hybrid capacitors, from the perspectives of design concept, configuration, electrochemical behavior and energy storage mechanism. An outlook of the future research regarding multivalent metal ion hybrid capacitors was also presented. This review will be beneficial for researchers around the world to have a better understanding of multivalent metal ion hybrid capacitors and develop novel electrochemical energy storage systems to meet the demands of rapidly developing electric vehicles and wearable/portable electronic products.
 Liubing Dong | Liubing Dong was born in 1989. He received his PhD degree in materials science and engineering from Tsinghua University in 2018. He is now a postdoctoral research associate at the Center for Clean Energy Technology, University of Technology Sydney. His current research mainly focuses on supercapacitors (especially flexible supercapacitors), multivalent-ion hybrid capacitors and rechargeable aqueous zinc-ion batteries. |
 Guoxiu Wang | Guoxiu Wang is the Director of the Centre for Clean Energy Technology and a Distinguished Professor at the University of Technology Sydney (UTS), Australia. Professor Wang is an expert in materials chemistry, electrochemistry, energy storage and conversion, and battery technologies. His research interests include lithium ion batteries, lithium-air batteries, sodium-ion batteries, lithium–sulfur batteries, supercapacitors, hydrogen storage materials, fuel cells, 2D materials such as graphene and MXenes, and electrocatalysis for hydrogen production. |
1. Introduction
Supercapacitors are characterized by fast charge–discharge rate, high power density, a broad application temperature range and ultralong service life. Thus they have become an indispensable kind of electrochemical energy storage device in consumer electronics, the transportation field and electricity systems.1–3 Nevertheless, their low energy density severely limits their large-scale applications. By contrast, batteries (e.g., widely used lithium-ion batteries at present) exhibit relatively high energy density but low power output, poor rate performance and inferior cycling stability.2,4 To integrate these advantages of supercapacitors and batteries into one device, the concept of hybrid capacitors was developed. In a hybrid capacitor, one electrode is a battery-type faradaic electrode as an energy source and the other is a capacitive electrode as a power source.2 Similar to rechargeable batteries (e.g., lithium-ion batteries, sodium-ion batteries and potassium-ion batteries), hybrid capacitors are generally named based on their internal shuttling cations, such as lithium-ion hybrid capacitors, sodium-ion hybrid capacitors and potassium-ion hybrid capacitors. These univalent metal ion hybrid capacitors, especially the former two types, have been widely studied.5–13In recent years, portable electronics and electric vehicles have developed rapidly and set higher demands for energy density, power density, security and production cost of electrochemical energy storage devices.14–16 In the above-mentioned batteries/hybrid capacitors, the alkali metals Li, Na and K are extremely reactive and the introduced organic electrolytes are flammable, which brings enormous security risks. Besides these issues, a shortage of lithium resources and their uneven distribution in the earth have made costs higher for lithium-ion batteries/hybrid capacitors. Against that background, multivalent ion storage mechanisms have been proposed and applied to design novel electrochemical energy storage systems. Compared with the energy storage systems based on univalent metal ions (Li+, Na+, K+, etc.), the systems based on multivalent metal ions (e.g., Zn2+, Mg2+, Ca2+ and Al3+) may possess some distinct advantages, such as fast charge transfer dynamics and high capacity and energy density.17 On the basis of multivalent ion storage mechanisms, rechargeable zinc-ion batteries,18,19 magnesium-ion batteries,20–22 calcium-ion batteries,23–25 and aluminum-ion batteries26–29 have been developed and are being widely studied. Some reviews also have separately presented these multivalent metal ion batteries.30–34 However, multivalent metal ion hybrid capacitors, which are also designed based on multivalent ion storage mechanisms and exhibit many unique merits as novel energy storage devices, have not yet been systematically overviewed.
Herein, we reviewed the recent progress in research on multivalent metal ion hybrid capacitors. Considering that multivalent metal ion hybrid capacitors are constructed by integrating one capacitor-like electrode with one battery-like electrode (which refers to electrodes for multivalent metal ion batteries herein), the energy storage mechanisms and typical characteristics of separate supercapacitors and multivalent metal ion batteries were expounded first. The design concept, configuration, electrochemical behavior, energy storage mechanism and applicability of multivalent metal ion hybrid capacitors, especially aqueous zinc-ion hybrid capacitors, for micro-scale devices were then comprehensively elaborated. An outlook of the development directions for multivalent metal ion hybrid capacitors was also provided based on our overview of the published studies. We believe this overview can effectively collate the research systems for multivalent metal ion hybrid capacitors and thus provide guidance for designing, fabricating and exploiting novel high-performance electrochemical energy storage devices.
2. Introduction to supercapacitors and multivalent metal ion batteries
2.1 Supercapacitors
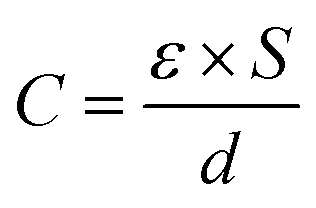 | (1) |
 | ||
| Fig. 1 Schematic illustrations of (a) parallel-plate capacitors, (b) carbon electrode based EDLCs, (c) Stern model of the electric double layer formed on carbon–electrolyte interfaces, and (d) pseudocapacitive behavior of a MnO2 electrode. | ||
Porous carbon materials with high surface areas were used earlier to construct supercapacitors.36–39 When carbon bulk is immersed in an electrolyte, the electrolyte will infiltrate open pores inside the carbon bulk, accompanied by the formation of a carbon–electrolyte interface. Each sufficiently small part of the interface can be regarded as a parallel-plate capacitor (Fig. 1b). It is noted from a microcosmic perspective that a capacitance of 3–40 μF cm−2 can be obtained on the carbon–electrolyte interface.40,41 Carbon morphology and electrolyte composition are decisive to the capacitance values. Considering the large specific surface area of porous carbon materials, the carbon–electrolyte interface contains a large multiplicity of “parallel-plate capacitors”, and thus is able to store a large amount of charges by electrostatic accumulation. Such a way of storing electricity is logically called electric double layer capacitors (EDLCs). Deeper research has shown that charge distribution on the carbon–electrolyte interface is not exactly the same as that in parallel-plate capacitors. Instead, on the electrolyte side, under the influence of electrostatic forces, ion diffusion and some other factors, charged ions are considered to form a Stern layer and diffuse layer,42,43 as illustrated in Fig. 1c. Despite this, the concept of an electric double layer is still a good approximation and remains widely used today.
Various carbon materials have been studied and used as EDLC electrodes, such as activated carbon particles (ACs),44–47 carbon nanotubes (CNTs),48–52 graphene nanosheets,53–57 carbon nanofibers (CNFs),58–61 activated carbon fibers (ACFs)62–67 and some other forms of carbon materials.68–71 To achieve a high capacitance, the specific surface area of these EDLC electrode materials is required to be high (e.g., 1000–4000 m2 g−1). On the other hand, specific surface area is not the sole factor determining the capacitance of carbon materials. The pore structures and electrical properties of carbon materials also have a crucial effect on their capacitance values.62,72–75 This is because the above parameters influence the transport behaviors of electrons and electrolyte ions in different ways, while charge storage on carbon–electrolyte interfaces in EDLCs needs the co-participation of electrons and ions. Pores which are too small do not allow the electrolyte to access, and relatively poor electrical conductivity impedes the electron transmission from current collectors to carbon–electrolyte interfaces. It is worth mentioning that besides the three factors of specific surface area, electrical resistance and pore structure, the organizational architecture of the carbon materials in the corresponding EDLC electrodes has to be taken into account. For instance, ACFs themselves are highly conductive and possess a high specific surface area and abundant pores on their surface, but ACF constructed felt electrodes may show inferior capacitive performance, associated with the high porosity and poor electrical conductivity of ACF felt electrodes.62,76,77 Similarly, densely stacked graphene electrodes hinder the infiltration of electrolyte ions onto the surface of each graphene nanosheet and thus lead to a low capacitance, regardless of the exceptional physical properties of the graphene nanosheets themselves.56,78,79 Therefore, constructing sufficient channels for fast transport of electrons and electrolyte ions inside electrodes is critical to achieve good electrochemical performance of carbon materials.
Typical pseudocapacitive materials include transition metal oxides/hydroxides (e.g., MnO2, RuO2, Fe2O3, TiO2, ZnO and V2O5),63,84–92 conductive polymers (e.g., polyaniline (PANI), polypyrrole (PPy) and polythiophene),64,65,93–99 heteroatom-doped carbon materials (e.g., N-doped graphene),100–106 and some other new nanomaterials (e.g., metal–organic-framework materials, MXene nanosheets, black-phosphorus nanoflakes and MoS2).107–122
MnO2 materials possess a high theoretical capacitance of 1370 F g−1 and a wide window of operating potential such as 0–0.9 V vs. Ag/AgCl in Na2SO4 aqueous electrolyte. Two mechanisms have been proposed to explain the pseudocapacitive behaviors of MnO2, as expressed by eqn (2) and (3),85,123,124
| MnO2 + C+ + e− ⇔ MnOOC | (2) |
| (MnO2)surface + C+ + e− ⇔ (MnO−2C+)surface | (3) |
Conductive polymers are widely used as pseudocapacitive materials in supercapacitors due to their high pseudocapacitance, superior chemical stability, good electrical conductivity (vs. transition metal oxides) and simple synthesis process. Taking PANI as an example, PANI electrode materials can be synthesized through chemical polymerization or electrochemical polymerization of aniline monomers.64,65,138–140 Note that these polymerization processes are typically carried out in acid solutions, because in this case, the synthesized PANI materials are doped with protons, which is helpful to optimize the electrical and electrochemical properties of PANI. When used as a supercapacitor electrode, PANI exhibits pseudocapacitive characteristics, attributed to reversible ion doping/de-doping during charge/discharge processes.141,142 Unfortunately, such an energy storage mode is accompanied by severe swelling and shrinkage of PANI chains. As a consequence, pure PANI electrode materials often exhibit poor cycling stability.64,143–145 PANI/CNTs, PANI/graphene and some other PANI/carbon composite electrode materials have been studied to prevent aggregation of PANI nanoparticles, facilitate electron transport in electrodes and alleviate the severe volume change of PANI.64,77,146–149
As mentioned above, good electrical properties, high specific surface areas and controllable pore structures endow carbon electrode materials with excellent rate performance and superior cycling performance, whereas the electrostatic charge–discharge mechanism limits carbon materials' capacitance. Heteroatom (e.g., N, O, B and P atoms or combinations of them) doping is an effective way to improve the charge storage ability of carbon materials.100–104,150–153 The introduced heteroatom functional groups on the carbon surface contribute a large pseudocapacitance in different ways. Oxygen functional groups are believed to conduct redox reactions between phenol or quinone and acidic or alkaline electrolytes.101 As summarized by Wang et al.,101 nitrogen functional groups can serve as electron donors to attract protons or improve the charge density of space charge layers,154–156 strengthen oxidation/reduction of quinones157 and introduce redox reactions of amine groups.158 P and/or B doping not only directly introduces heteroatom functional groups on the carbon surface but also brings additional surface oxygen groups, and these functional groups endow the carbon materials with pseudocapacitance through complex faradaic reactions with a wide range of potentials.100
In recent years, MXene nanosheets, black-phosphorus nanoflakes, MoS2 and some other two-dimensional (2D) nanomaterials have been synthesized and studied as high-performance electrodes for pseudocapacitors. We are giving a brief introduction to MXene materials as an example herein. MXenes possess superior pseudocapacitive performance such as good rate capability and high volumetric capacitance, benefiting from their high metallic conductivity, unique 2D structure and highly reversible surface redox reactions.113,116,159,160 MXenes are produced by etching out the A layer from Mn+1AXn phases (also denoted as MAX), in which M is an early transition metal or a solid solution of early transition metals, A is mainly a group IIIA or IVA element, X is C and/or N, and n = 1, 2 or 3. After etching the A layer, functional groups (OH, O, F, H, etc.) terminate the surface of each MXene layer. Also, the weak bonds between MXene layers allow for the intercalations of inorganic cations (such as H+, Li+, Na+ and K+), water molecules, organic compounds and even ionic liquids, which results in different interlayer distances of MXene sheets. All in all, various MAX phase precursors (e.g., Ti2AlC, V2AlC, (V0.5Cr0.5)3AlC2, Ti3AlCN, Ta4AlC3, and Nb4AlC3) with different surface functional terminations and adjustable interlayer distances make it possible to synthesize hundreds or more kinds of MXenes with particular physical–chemical features. Considering the ultrahigh volumetric capacitance of MXenes compared with the best all-carbon EDLC materials, the research on MXenes paves a new way for developing high-performance supercapacitors. Nevertheless, MXene materials are relatively easily oxidized by oxygen in air or at higher anodic potentials in electrochemical systems. This means that synthesized MXene materials must be carefully preserved in an inert atmosphere, and symmetric MXene supercapacitors are limited by low operating voltages in aqueous electrolytes (such as ∼0.6 V for titanium carbide MXene electrode-based symmetric supercapacitors), while low operating voltages will result in a modest energy density. Therefore, enhancing the stability of the MXenes will greatly extend the application of MXenes in electrochemical energy storage. In addition, MXene-based composite electrodes, such as MXene/carbon,114,161–164 MXene/conductive polymers,165–169 and MXene/transition metal oxides,170–172 have been reported for high-performance supercapacitors.
2.2 Multivalent metal ion batteries
| Zn | Mg | Ca | Al | Li | |
|---|---|---|---|---|---|
| Standard potential (V vs. standard hydrogen electrode) | −0.76 | −2.37 | −2.87 | −1.67 | −3.04 |
| Gravimetric capacity (mA h g−1) | 823 (Zn/Zn2+) | 2205 (Mg/Mg2+) | 1337 (Ca/Ca2+) | 2980 (Al/Al3+) | 3860 (Li/Li+) |
| Volumetric capacity (A h L−1) | 5845 (Zn/Zn2+) | 3833 (Mg/Mg2+) | 2073 (Ca/Ca2+) | 8046 (Al/Al3+) | 2046 (Li/Li+) |
| Ionic diameter (Å) | 0.75 (Zn2+) | 0.72 (Mg2+) | 1.00 (Ca2+) | 0.53 (Al3+) | 0.76 (Li+) |
| Element abundance in the Earth’s crust (%) | 0.0079 | 2.9 | 5.0 | 8.2 | 0.0017 |
2.2.1.1 Cathode materials for ZIBs.
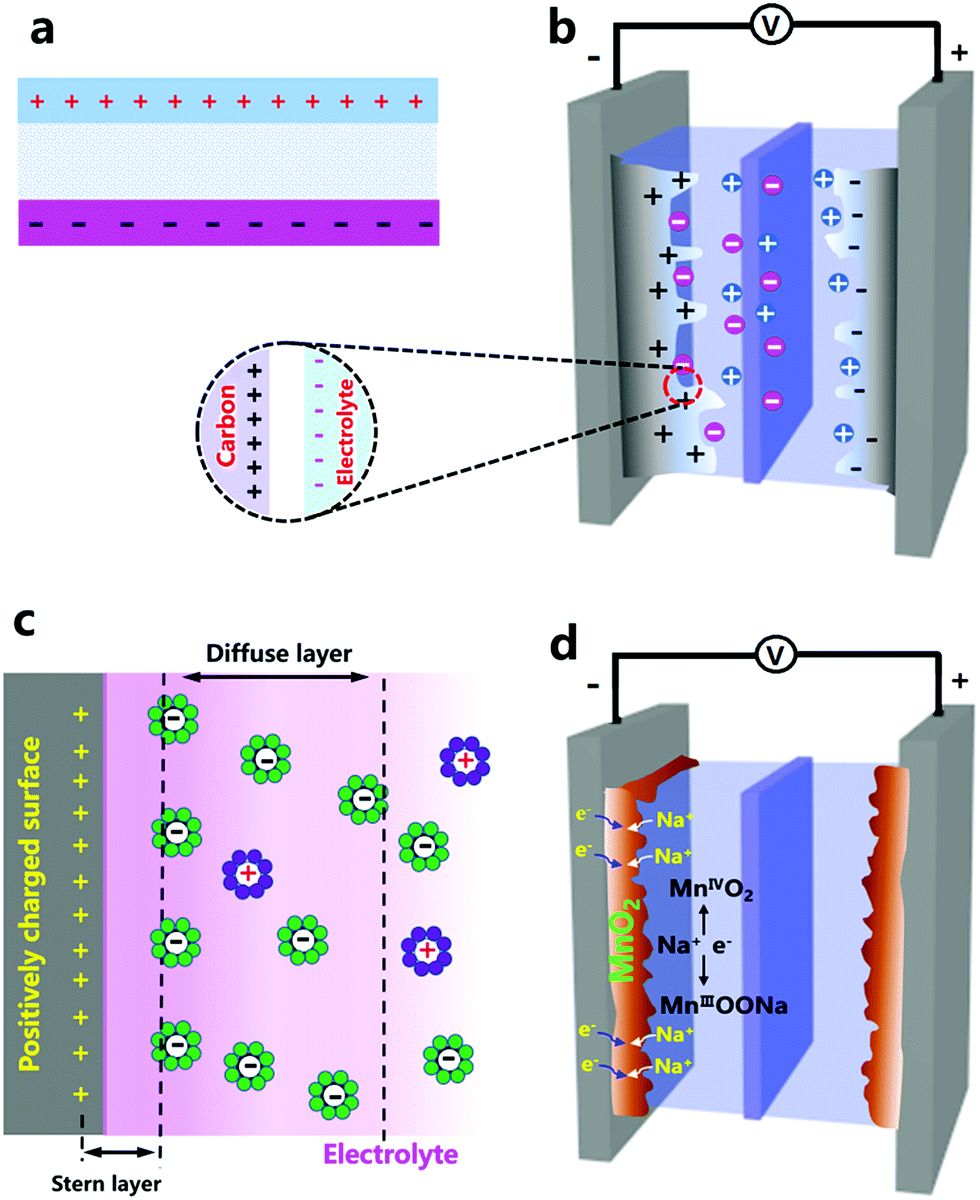
Manganese-based cathode materials for ZIBs. Tunnel structures endow MnO2 with good Zn2+-storage ability. Xu et al. reported α-MnO2-based ZIBs (Fig. 2a and b).18 They proved the reversible insertion/extraction of Zn2+ ions into/from α-MnO2: during discharging, the zinc anode rapidly dissolved as Zn2+ ions, and Zn2+ ions in the electrolyte intercalated into the α-MnO2 cathode; in the charging process, Zn2+ ions reversibly de-intercalated from the cathode, and deposited on the zinc anode. This process can be described by eqn (4).
| Zn2+ + 2e− + 2MnO2 ⇔ ZnMn2O4 | (4) |
 | ||
| Fig. 2 (a) Schematics of MnO2//Zn ZIBs and (b) corresponding CV curves of a Zn anode and MnO2 cathode. Adapted from ref. 18 with permission from Wiley-VCH. (c) Structure schematic, (d) rate performance and (e) energy storage mechanism of Zn0.25V2O5//Zn ZIBs. Adapted from ref. 19 with permission from Nature Publishing Group. | ||
The α-MnO2 cathode delivered a high capacity of 210 mA h g−1 at a charge/discharge rate of 0.5C, and maintained 68 mA h g−1 at a high rate of 126C. Subsequently, other MnO2 polymorphs (β-MnO2, γ-MnO2, and ε-MnO2) and other manganese oxides with a manganese valence state of less than +4 (e.g., Mn2O3 and Mn3O4) were studied for ZIBs.174–178 Their poor electrical conductivity causes a poor rate capability, and structural transformation and manganese dissolution during charge/discharge processes often result in poor cycling stability. Cation-deficient spinel ZnMn2O4 was then prepared and used in ZIBs. Its capacity could retain 94% over 500 charge/discharge cycles at 500 mA g−1.194
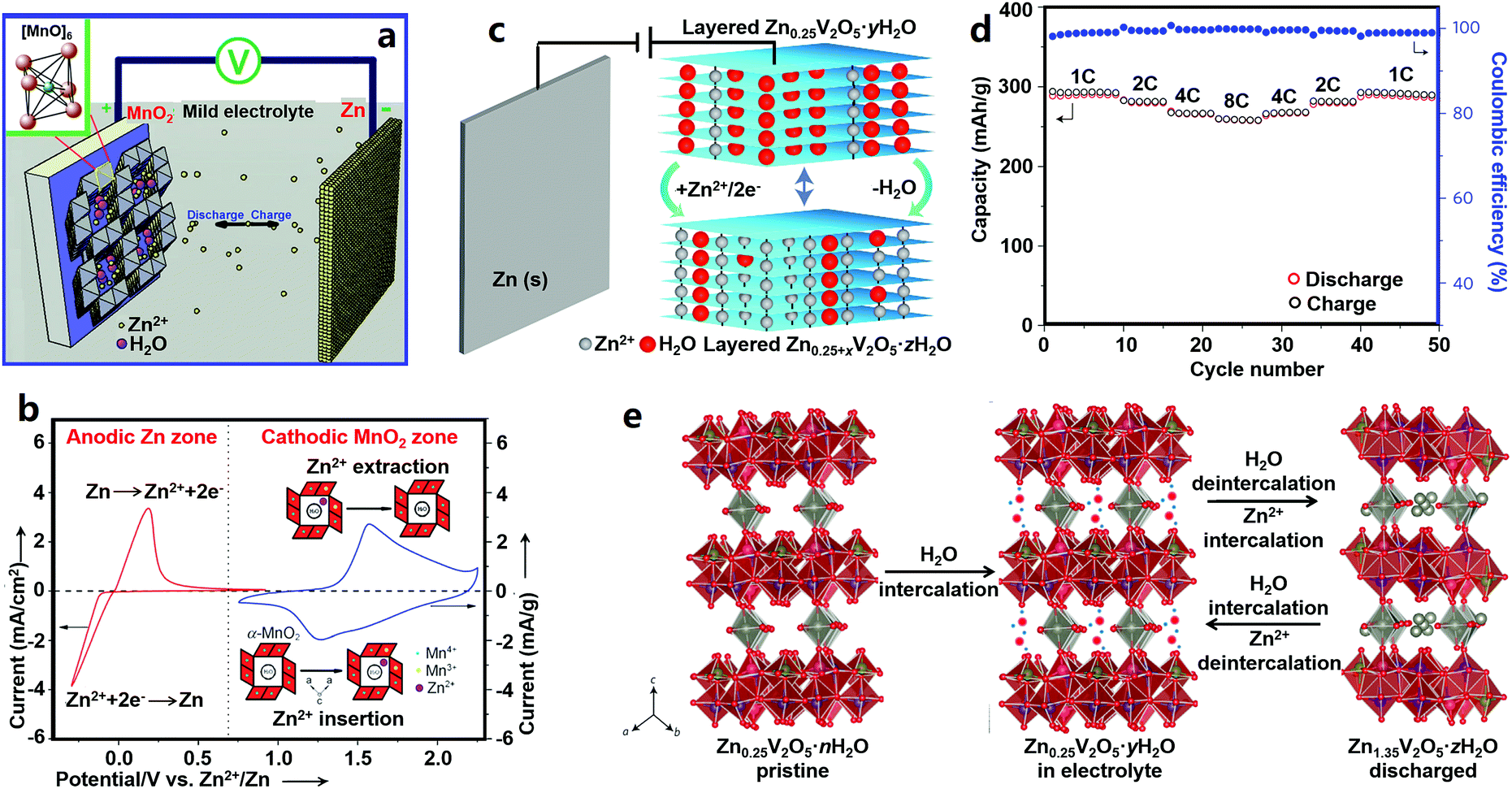
Vanadium-based cathode materials for ZIBs. Vanadium-based cathode materials were recently explored as high-performance ZIB cathodes. For instance, Zn0.25V2O5·nH2O composed of [V–O] layers and pillars of Zn2+ and H2O molecules was first applied in ZIBs by Nazar's group (Fig. 2c–e).19 The interlayer distance of ZnxV2O5·nH2O first increased after being immersed in an aqueous electrolyte as a result of water intercalation, and then in the discharge process, Zn2+ ions intercalated into while water molecules extracted from the ZnxV2O5·nH2O host, leading to a change of the interlayer distance. Zn2+ de-intercalation and water molecule re-insertion happened during recharging. Consequently, the Zn0.25V2O5·nH2O cathode showed good electrochemical performance with a specific energy of ∼250 W h kg−1, a capacity of 220 mA h g−1 at a 15C rate, and a capacity retention of about 80% after 1000 charge/discharge cycles. Other vanadium-based cathode materials such as Zn2(OH)VO4,195 Na1.1V3O7.9@rGO,196 and H2V3O8 (ref. 180) display good and stable Zn2+-ion storage behaviors. The NaV3O8-type layered structure (e.g., Na5V12O32 and HNaV6O16·4H2O) and β-Na0.33V2O5-type tunneled structure (e.g., Na0.76V6O15) have also been explored by Liang's group.197 Due to the higher charge density of Zn2+, some Na+ in Na5V12O32 are more likely to be displaced by Zn2+ to form a new phase of Zn4V2O9, which may result in an unstable crystal structure because the Na+ in Na0.76V6O15 can act as a strong “pillar” to stabilize the whole structure. Potassium vanadate oxides such as K2V8O21, K0.25V2O5, and K2V6O16·1.57H2O were also explored by the same group.198 Further, other vanadium oxides including V2O5·nH2O,179,199 VO2 nanofibers,200 and V5O12·6H2O nanobelts201 also possess good electrochemical properties as Zn2+ storage hosts. Although vanadium-based cathode materials have better cycling stability and higher capacity than manganese-based cathodes as a whole, it is a thorny issue that vanadium-based compounds can be toxic to humans and other living creatures.
Prussian blue analog cathode materials for ZIBs. Prussian blue analog cathodes generally exhibit good cycling performance, but their reversible capacities are low. ZnHCF cathodes studied by Liu et al. showed a discharge capacity of only 65.4 mA h g−1 at 1C.185 CuHCF cathodes delivered a maximum discharge capacity of 50–60 mA h g−1.186–188 And for FeHCF cathodes, their specific capacity was reported to be less than 50 mA h g−1.189
2.2.1.2 Zn anodes for ZIBs. Metallic Zn anodes are generally believed to be stable in neutral or slightly acidic aqueous electrolytes (e.g., ZnSO4 aqueous electrolyte) and will not form dendrites during repeated stripping/electroplating processes.18,19,173,202 But in practical experiments, we sometimes find that Zn anodes do not work normally after tens or hundreds of charge/discharge cycles of ZIBs. The following reasons may give rise to this problem. (i) The Zn electrode actually acts not only as an anode material but also as a current collector. Stripping of Zn inevitably causes morphological changes of the current collector (especially when the mass of the Zn electrode is not excessive), and this subsequently affects the electroplating process of Zn2+, forming an uneven Zn electrode surface (Fig. 3a).203 This effect accumulates in repeated stripping/electroplating processes and finally causes the failure of the Zn anode and the ZIB. (ii) Oxygen dissolved in aqueous electrolytes may corrode the Zn anode under the influence of electric field and form insulating zinc oxides on the Zn anode surface. The strategies of surface coating and three-dimensional (3D) architectural design have been proposed to modify Zn anodes to obtain long-term cycling stability (Fig. 3),203–206 which will also provide guidance to design ultralong-life and high-rate metal electrode-based hybrid capacitors.
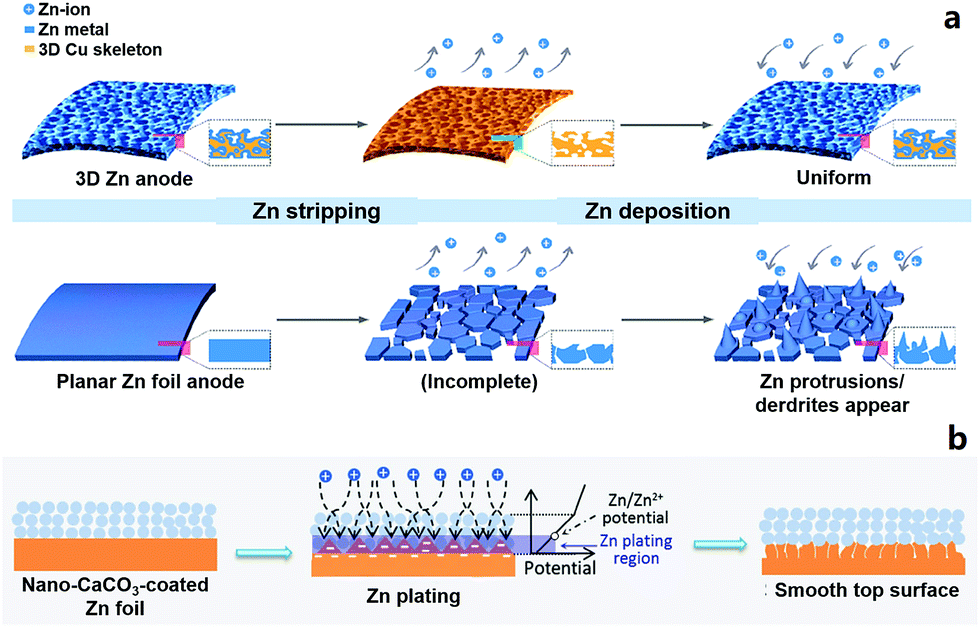 | ||
| Fig. 3 Schematic diagrams of Zn striping/electroplating on (a) a 3D Zn electrode (upper) and commercial Zn foil (lower). The 3D Zn electrode achieves a long-term cycling stability. Reproduced from ref. 203 with permission from American Chemical Society. (b) Schematic illustration of morphology evolution for a CaCO3 nanoparticle coated Zn electrode during the Zn striping/electroplating process. Reproduced from ref. 205 with permission from Wiley-VCH. | ||
2.2.2.1 Cathode materials for MIBs. Chevrel phases, namely ternary molybdenum chalcogenides (MxMo6T8, where M is a metal element and T is a chalcogen element), are the typical cathode materials for MIBs. They have unique structural features and mutable cation species in the crystal structure.217–230 Chevrel phases can be viewed as a stack of Mo6X8 blocks (Fig. 4a), and each Mo6X8 block consists of Mo6 octahedra inside a cubic cage with eight chalcogen atoms at the corners. Mg atoms occupy twelve interstitial sites formed in the channels between these Mo6X8 blocks. Insertion/extraction of Mg2+ in/from the Mo6S8 cathode can deliver a reversible capacity of 124.4 mA h g−1 (maximum theoretical value), corresponding to the reactions as given by eqn (5) and (6).218
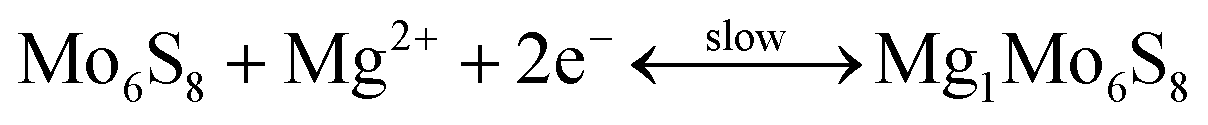 | (5) |
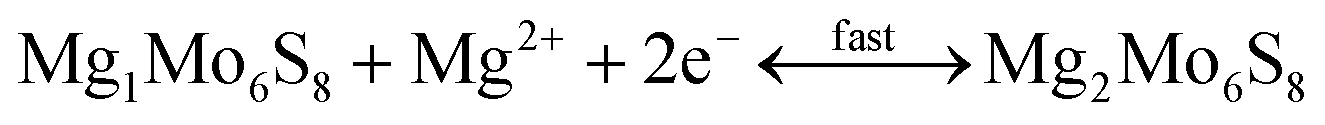 | (6) |
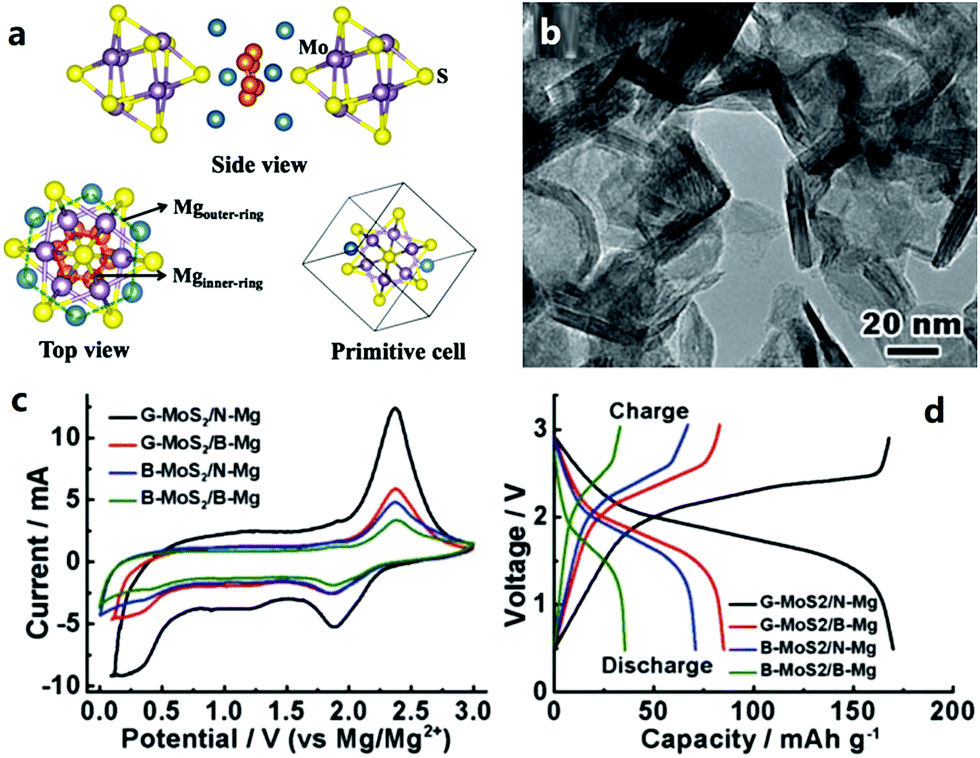 | ||
| Fig. 4 (a) Crystal structure of MoxMo6S8. Reproduced from ref. 219 with permission from Elsevier. (b) Transmission electron microscopy image of graphene-like MoS2 and (c) and (d) its electrochemical behaviors as a MIB cathode (G-MoS2, B-MoS2, N-Mg and B-Mg represent a graphene-like MoS2 cathode, bulk MoS2 cathode, ultrasmall Mg nanoparticle anode, and bulk Mg anode, respectively). Reproduced from ref. 221 with permission from Wiley-VCH. | ||
For these Chevrel phase materials, reduced particle size, incorporation of Cu and substitution of S with Se can further enhance their electrochemical properties.
Molybdenum disulfide (MoS2) has a 2D layered structure (each layer is composed of covalently bonded S–Mo–S trilayers and these layers are linked into a 3D structure through van der Waals forces). As a cathode material for MIBs, its theoretical capacity is 223.2 mA h g−1, according to the electrochemical reactions as expressed by eqn (7).221
| 6MoS2 + 4Mg2+ + 8e− ⇔ Mg4Mo6S12 | (7) |
Various MoS2 materials with different micro-morphologies have been investigated during last decades, such as graphene-like nanosheets (Fig. 4b–d), zigzag nanoribbons, nanotubes and so on.221–224 In addition to Chevrel phases and MoS2, some other transition metal sulfides have also been studied as cathode materials for MIBs, for instance, TiS2,207,225,226 VS2,207 ZrS2,207 CuS,227 NiS,227 NbS3,228 and CoS.229 These cathode materials presented high ionic mobility ascribed to anion polarizability, but their electrochemical performance was modest.
Vanadium pentoxide (V2O5) was studied as a cathode material for MIBs due to its low cost, high theoretical capacity (589 mA h g−1, for the reduction of V5+ to V3+), and high open-circuit voltage (∼2.66 V).212 However, intercalation of Mg2+ ions in V2O5 is a slow process and the electrical conductivity of V2O5 itself is low.230 To solve these problems, numerous research studies have been carried out to decrease the crystallite size of V2O5 and hybridize V2O5 with conductive materials.231–235 Manganese oxides have also been investigated as attractive cathode materials for MIBs owing to their low toxicity and natural abundance. It has been demonstrated that for the Mn2O3 cathode, an open-circuit voltage of 2.40 V and a capacity of 224 mA h g−1 could be obtained.207 Besides, MnO2 cathodes displayed a high capacity (308 mA h g−1 for the reduction of Mn4+ to Mn3+) and an open-circuit voltage of ∼2.9 V.236 Modification of MnO2 has also been investigated to optimize the transport behavior of Mg2+ in MnO2 cathodes.237–240 MoO3 is an orthorhombic layered material, which exhibited an open-circuit voltage of 2.28 V and an intercalation capacity of 143 mA h g−1 when used as a MIB cathode.207 In addition, Mg2+ storage behaviors of Co3O4, RuO2, WO3, U3O8, TiO2 and lead oxides have been investigated.207,241–244 Other classes of materials have been preliminarily studied as cathode materials for MIBs, including transition metal phosphates,245,246 transition metal silicates,247,248 organic materials,249,250 Prussian blue analogs,251,252 and metal chlorides.253 Further research is still needed.
2.2.2.2 Anode materials for MIBs. The passivation of Mg metal in most conventional electrolytes is an important issue needing to be considered in choosing anode materials for MIBs.254 Bi and Sn as anodes for MIBs have been investigated, because Bi can form intermetallic compounds with Mg and Sn has good compatibility with conventional battery electrolytes.255–258 In addition, 2D materials including defective graphene and black phosphorus have been studied as anodes for MIBs due to their open morphology and unique physical properties.259,260 Other materials such as nanoporous-carbon and Li4Ti5O12 are also worthy of attention.261,262
2.2.2.3 Electrolytes for MIBs. In recent decades, researchers have been developing suitable electrolytes which allow reversible insertion/extraction of Mg2+ into/from Mg anodes. Previously, electrolytes such as Mg(ClO4)2 or Mg(CF3SO3)2 dissolved in THF, acetonitrile (AN) or propylene carbonate were employed in MIBs. However, the high charge density of Mg2+ resulted in large de-solvation energies at cathodes.263 Afterwards, Mg(AlCl2BuEt)2 in THF has proven to be a suitable electrolyte for MIBs with cathodes of Chevrel phase materials.208
2.2.3.1 Cathode materials for CIBs. According to computational and experimental results, a 3D tunnel structure and/or a layered structure is beneficial for Ca2+ insertion/extraction. For example, CaMn2O4 was predicted to be a potential cathode material, attributed to its outstanding stability.267,268 V2O5 as a typical layered structure material can accommodate the intercalation of large Ca2+ and has an advantage from the perspective of energy density.269,270 Recently, Chevrel phase materials have also been investigated theoretically for CIBs. Indeed, Mo6X8 nanostructured materials have potential for Ca2+ insertion even at an average voltage of 1.25 V (vs. Ca/Ca2+).271,272 Unfortunately, no experimental results of Chevrel phases for CIBs have been reported to date. Prussian blue analogs have been investigated as insertion electrodes for CIBs.23,265,273 These materials have a cubic framework with the general formula of AxMFe(CN)6·yH2O (where A = Li, Na, Mg, Ca, etc. and M = Ba, Ti, Mn, Fe, Co or Ni). However, their capacities are unsatisfactory.
2.2.3.2 Anode materials for CIBs. Ca metal anodes are attractive due to their ultrahigh volumetric and gravimetric capacity. A solid electrolyte interphase (SEI) layer easily forms on the surface of Ca, but it is difficult for Ca2+ ions to diffuse through this SEI layer.24 Alloy materials such as Ca–Si alloy and Ca–Sn alloy can provide attractive electrochemical capacities while avoiding the issue of surface passivation; thus, they are possible alternative anodes for MIBs (Fig. 5).24,25,248
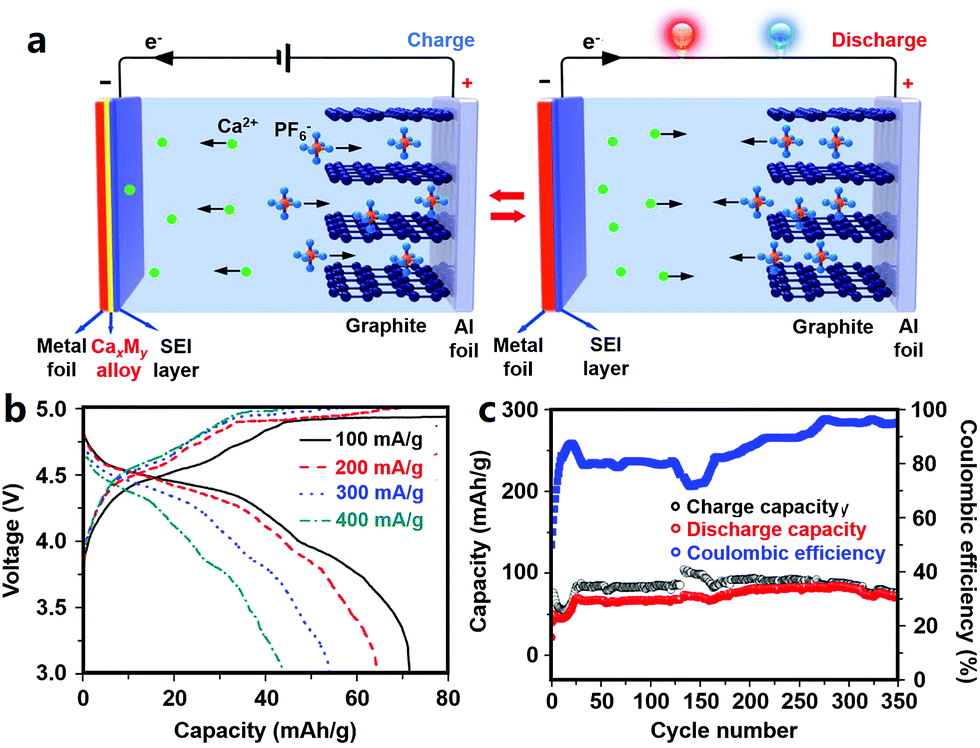 | ||
| Fig. 5 (a) Working schematics of a CIB with a graphite cathode and a metal foil anode that can form an alloy with Ca; (b) rate performance and (c) cycling behavior of the CIB. Adapted from ref. 25 with permission from Nature Publishing Group. | ||
2.2.3.3 Electrolytes for CIBs. Ponrouch et al.24 reported some organic electrolyte combinations from dissolving Ca(ClO4)2, Ca(BF4)2, and Ca(TFSI)2 salts in mixed carbonate solvents (propylene carbonate and ethylene carbonate), which offer a wide electrochemical window. However, these electrolytes require elevated temperatures and still exhibit poor reversibility of Ca2+ insertion/extraction. In another recent study, THF-based electrolytes with Ca(BF4)2 salts for CIBs were shown to operate at room temperature for Ca2+ insertion/extraction, although they showed a lower coulombic efficiency.274
2.2.4.1 Electrolytes for AIBs. Aqueous electrolytes based on AlCl3 or Al2(SO4)3 salts in water have been widely used for AIBs because of their low cost, simple operation and environmental friendliness.279–281 However, in aqueous electrolytes, a passive oxide layer (Al2O3) will form and the reversible deposition of Al3+ is hindered by a competing intrinsic hydrogen evolution reaction, leading to a low efficiency of the Al anode.282 To overcome these limitations, non-aqueous electrolytes have been applied in AIBs. Initially, molten salts such as the binary NaCl–AlCl3 system or the ternary KCl–NaCl–AlCl3 system were considered as possible electrolytes for AIBs.283,284 In these molten salt systems, bare Al3+ does not exist, only in the forms of AlCl4− and Al2Cl7−. The stripping/deposition process of Al follows eqn (8).283
| 2AlCl−4 ⇔ Al2Cl−7 + Cl− | (8) |
However, these molten salt electrolytes have a high melting point, which limits the practical application of AIBs. Therefore, ionic liquids with high ionic conductivity, low volatility and high electrochemical/chemical stability were then investigated as room-temperature electrolytes for AIB systems.285,286 The most widespread ionic liquid electrolytes for AIBs are AlCl3 in imidazolium based ionic liquids, in which imidazolium cations are typically 1-butyl-3-methylimidazolium, 1-ethyl-3-methylimidazolium (EMIM), etc.26,27,278,285–287
2.2.4.2 Cathode materials for AIBs. Graphite cathodes together with Al anodes can be used to construct a low-cost AIB system (Fig. 6). The redox reactions during charge–discharge processes inside Al/graphite AIBs (the operating voltage window is around 0.5–2.5 V) can be expressed by eqn (9) and (10).26
| Al + 7AlCl−4 ⇔ 4Al2Cl−7 + 3e− | (9) |
| Cn[AlCl4] + e− ⇔ Cn + AlCl−4 | (10) |
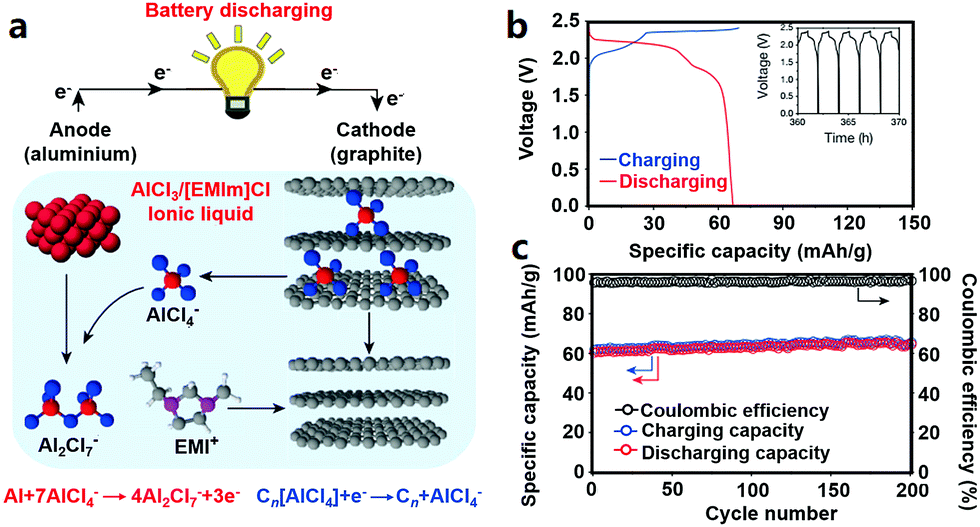 | ||
| Fig. 6 (a) Schematic of the graphite//Al AIB with an AlCl3/[EMIm]Cl ionic liquid electrolyte; (b) charge–discharge curves at a current density of 66 mA g−1 and (c) cycling behavior of the pyrolytic graphite//Al cell. Reproduced from ref. 26 with permission from Nature Publishing Group. | ||
Similarly, 3D graphitic foam and fluorinated natural graphite have been studied as cathode materials for AIBs; while the former showed modest capacity and the latter displayed low coulombic efficiency (about 75%).26,278,288 Furthermore, anatase TiO2, V2O5, CuHCF, Mo6S8, chloroaluminate-doped conducting polymers and FeS2 also allow reversible Al3+-storage in AIBs.280,287,289–292
Multivalent metal ion batteries including ZIBs, MIBs, CIBs and AIBs are overviewed above. Considering that multivalent metal ion batteries are often compared to monovalent metal ion batteries such as lithium-ion batteries (LIBs), sodium-ion batteries (SIBs) and potassium-ion batteries (KIBs), we thereby give a very brief introduction to monovalent metal ion batteries herein. The three types of monovalent metal ion batteries work based on a “rocking-chair”-type mechanism: during charge, Li+/Na+/K+ ions move from the cathode to the anode through the electrolyte, and then move back when the batteries are discharged.293–295 LIBs have been commercialized since 1991 and are widely applied in modern society. Up to now, LiCoO2, LiMn2O4, LiNi0.8Co0.15O2, LiNixMnyCo1−x−yO2 and LiFePO4 have been developed as cathode materials of LIBs, and the anode materials include carbon-based materials (e.g., graphite, which is the current dominant anode material in commercial LIBs), Li4Ti5O12 and Si/C composites.296–298 The electrolytes for LIBs are prepared by dissolving lithium salt (e.g., LiPF6, LiClO4 and LiAsF6) in organic solvents (e.g., dimethyl carbonate, diethyl carbonate, ethylene carbonate, propylene carbonate, ethyl methyl carbonate and a mixture of them).297 The growing demand of LIBs around the world and increasing shortage of lithium resources push up the price of LIBs. This promotes the research studies on SIBs and KIBs. Relatively, SIBs and KIBs possess the following advantages:299,300 (i) Na element and K element have abundant distribution on the Earth; (ii) standard electrode potentials of Na/Na+ and K/K+ are very low (−2.71 V for Na/Na+ and −2.93 V for K/K+, respectively, only 0.11–0.33 V higher than that of Li/Li+), which is beneficial for achieving a high energy density; (iii) Na+ and K+ have higher mobility than Li+ (in propylene carbonate solution), making it possible for SIBs and KIBs to realize a good high-rate performance. For SIBs,293 the cathode materials can be Na layered oxides (e.g., NaxMO2, where M = Co, V and Mo), 3D polyanion oxides and so on, and the anode materials contain carbonaceous materials, titanium oxides, some alloys and phosphorus. For KIBs, as summarized by Xu et al.294 the available cathode materials include layered metal oxides, metal hexacyanometalates, polyanionic compounds and organic compounds, and the anode materials are divided into carbon-based materials (e.g., graphite, soft carbon and hard carbon) and alloy-based materials (e.g., Sn-based anode, Bi-based anode and so on).
3. Multivalent metal ion hybrid capacitors
3.1 Zinc-ion hybrid capacitors (ZHCs)
3.1.1.1 ZHCs with an aqueous electrolyte. Based on the above advantages of Zn metal electrodes, we constructed extremely safe, high-rate and ultralong-life ZHCs in our previous work.202 In the ZHCs (Fig. 7a and b), high surface area AC was used as the cathode, metallic Zn foil was used as the anode and ZnSO4 aqueous solution was used as the electrolyte. The AC particles had a Brunauer–Emmett–Teller (BET) surface area of 1923 m2 g−1 and most of the pores on the AC surface had a size of 1.2–2.7 nm. The constructed AC//Zn ZHCs worked in a voltage range of 0.2–1.8 V. Different from the rectangular-shaped CV curves of EDLCs, redox peaks were observed in the CV curves of the ZHCs (Fig. 7c). Preservation of these redox peaks even at a large scan rate of 100–1000 mV s−1 demonstrated rapid kinetics of electrochemical reactions inside ZHCs. The ZHCs exhibited a high capacity of 121 mA h g−1 (calculated based on the mass of AC) at a charge/discharge current of 0.1 A g−1, as well as a good rate performance (could be charged/discharged within 15 s at a large current of 20 A g−1, corresponding to a discharge capacity of 41 mA h g−1) (Fig. 7d). High capacity and good rate performance endowed the ZHCs with a high energy density of 84 W h kg−1 and a high power output of 14.9 kW kg−1. In addition, the ZHCs presented an excellent cycling performance, with 91% capacity retention after 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge/discharge cycles at 1 A g−1 (Fig. 7e).
000 charge/discharge cycles at 1 A g−1 (Fig. 7e).
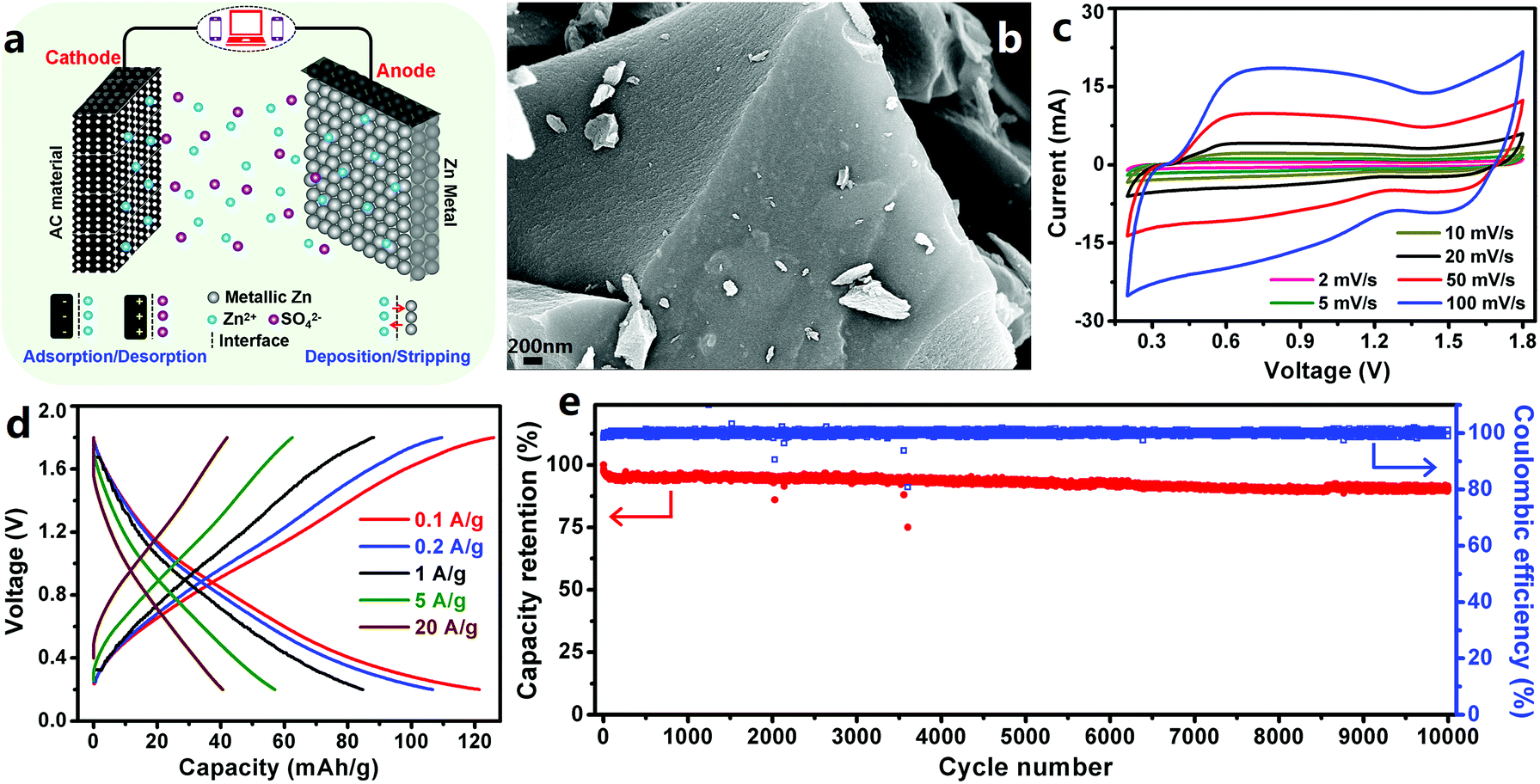 | ||
| Fig. 7 (a) Schematic of a ZHC with the AC cathode, Zn foil anode and ZnSO4 aqueous electrolyte; (b) scanning electron microscopy (SEM) image of the AC cathode; (c) CV curves, (d) charge–discharge profiles and (e) long-term cycling stability test of the AC//Zn ZHC. Reproduced from ref. 202 with permission from Elsevier. | ||
The energy storage mechanism for the ZHCs is summarized as follows. For Zn anodes, the primary electrode reaction is stripping/plating of Zn/Zn2+, as expressed by eqn (11).
| Zn ⇔ Zn2+ + 2e− | (11) |
For the AC cathode, due to its high specific surface area and appropriate pore structure, ion adsorption/desorption inevitably becomes one way for it to store energy, as confirmed by double layer capacitive behaviors of symmetric AC supercapacitors with the ZnSO4 electrolyte. Specifically, when the operating voltage is higher than the potential of zero charge (pzc), differential capacitance is minimum at the pzc. In Fig. 7c, we can see that the pzc of the AC material is around 1.3 V vs. Zn/Zn2+, ZSO42− anion adsorption/desorption on/from the AC surface is dominant, and when the operating voltage is lower than the pzc, it is mainly Zn2+ cation adsorption/desorption. Besides the aforementioned electrode reactions, a precipitation/dissolution reaction of Zn4SO4(OH)6·5H2O (also expressed as ZnSO4[Zn(OH)2]3·5H2O) on both electrodes also contributes capacity to ZHCs. The precipitation/dissolution of Zn4SO4(OH)6·5H2O, as described by eqn (12),
| 4Zn2+ + 6OH− + SO2−4 + 5H2O ⇔ Zn4SO4(OH)6·5H2O↓ | (12) |
| {C}⋯O + H+ + e− ⇔ {C}⋯OH | (13) |
| {C}⋯O + Zn2+ + 2e− ⇔ {C}⋯O⋯Zn | (14) |
We also utilized other AC materials to produce AC//Zn ZHCs and found that higher specific surface areas of the AC cathode were helpful to enhance the capacity of ZHC devices.202 Overall, metallic Zn electrode-based aqueous ZHCs have shown great potential in achieving a much higher energy density than aqueous symmetric supercapacitors and general asymmetric supercapacitors (e.g., MnO2//Na2SO4 (aq)//AC supercapacitors), benefiting from the following factors: (i) improved capacity due to the utilization of a high capacity Zn electrode and (ii) higher operating voltage (vs. aqueous symmetric supercapacitors and some asymmetric supercapacitors). Besides these factors, excellent cycling stability and fast kinetics of Zn2+ stripping/plating on Zn anode surfaces and ion adsorption/desorption on/from cathode surfaces endow metallic Zn electrode-based ZHCs with ultralong lifetime and superior rate performance.
Tian et al. applied CNTs as cathode materials to assemble CNT//ZnSO4 (aq)//Zn ZHCs and investigated the effect of CNT oxidation on ZHCs' electrochemical behaviors.306 Unoxidized CNTs showed simple electric double layer capacitive behavior. However, after oxidation treatment, an additional reversible faradaic process with participation of divalent Zn2+ and oxygen functional groups could be introduced to CNT cathodes according to eqn (6) and (7), thus leading to a notably enhanced capacity of CNT//Zn ZHCs. However, the low specific surface area (120–211 m2 g−1) of these CNT cathodes led to a relatively low capacity (about 27 mA h g−1 deduced according to related descriptions in the literature).
Besides carbon materials, conductive polymers of PANI and PPy have been employed as cathodes to prepare ZHCs by Han and Li et al.307,308 Note that the assembled conductive polymer//Zn devices were called zinc-ion batteries in some literature, but they are categorized as ZHCs herein (a detailed discussion is given in the following). Han et al. designed a graphene@PANI composite hydrogel with a 3D nano-architecture and they directly used this as a free-standing cathode for graphene@PANI//ZnSO4 (aq)//Zn ZHCs (Fig. 8a–e).307 Zn2+ ions were stored in the cathode through two pathways: electric double layer capacitance of graphene and pseudocapacitance of PANI (in fact, due to the nanoscale size and relatively high specific surface area of the deposited PANI particles, PANI also could contribute an electric double layer capacitance besides pseudocapacitance). The ZHCs operated in a voltage range of 0.4–1.6 V (Fig. 8c and d). Compared with the aforementioned AC//ZnSO4 (aq)//Zn ZHCs, the graphene@PANI//ZnSO4 (aq)//Zn ZHCs exhibited more obvious redox peaks in CV curves, associated with the redox transition of the leucoemeraldine/emeraldine and emeraldine/pernigraniline states of PANI (i.e., reversible ion doping/de-doping of PANI, which is considered as a pseudocapacitive behavior in traditional supercapacitors as discussed in Section 2.1.2. This is why we classify PANI//Zn as a ZHC.) The capacity of this cathode was 154 mA h g−1 (ca. 437 F g−1, calculated based on the dry weight of the graphene@PANI electrode) at a charge/discharge current density of 0.1 A g−1. The 3D porous structure and highly conductive graphene framework endowed the cathode with a superior rate performance: a capacity of 106 mA h g−1 was achieved at 5 A g−1. The ZHCs showed a maximum energy density and power density of 205 W h kg−1 and 2455 W kg−1, respectively. As mentioned above, conductive polymers often suffer from poor cycling stability as supercapacitor electrodes, because of their severe swelling-shrinkage during charge–discharge processes. A similar problem occurred in the graphene@PANI//Zn ZHC systems: 80.5% capacity retention was achieved after 6000 charge/discharge cycles (Fig. 8e), worse than that of AC//Zn ZHCs (Fig. 7e). Similarly, Li et al. recently reported an aqueous PPy//Zn ZHC (Fig. 8f–h).308 For this ZHC, the cathode was prepared through chemical polymerization of pyrrole on graphite nanosheets/polyvinyl alcohol aerogel, the anode was fabricated by electrodepositing Zn on a graphite paper and the electrolyte was a mixed aqueous solution of ZnCl2 and NH4Cl. CV curves at 5–100 mV s−1 for the ZHC presented quasi-rectangular shapes, quite different from the typical CV curves of zinc-ion batteries (clearly separated and intense oxidation peaks and reduction peaks always exist in the CV curves of zinc-ion batteries). In a voltage range of 0.6–1.6 V, the ZHC delivered a discharge capacity of ∼151.1 mA h g−1 at 0.5 A g−1, but the corresponding coulombic efficiency was only about 88%. In addition, this ZHC showed a good rate performance with a capacity value of 87.6 mA h g−1 at 16 A g−1, whereas its capacitance retention was only 76.7% after 1000 charge/discharge cycles, indicating unsatisfactory cycling stability.
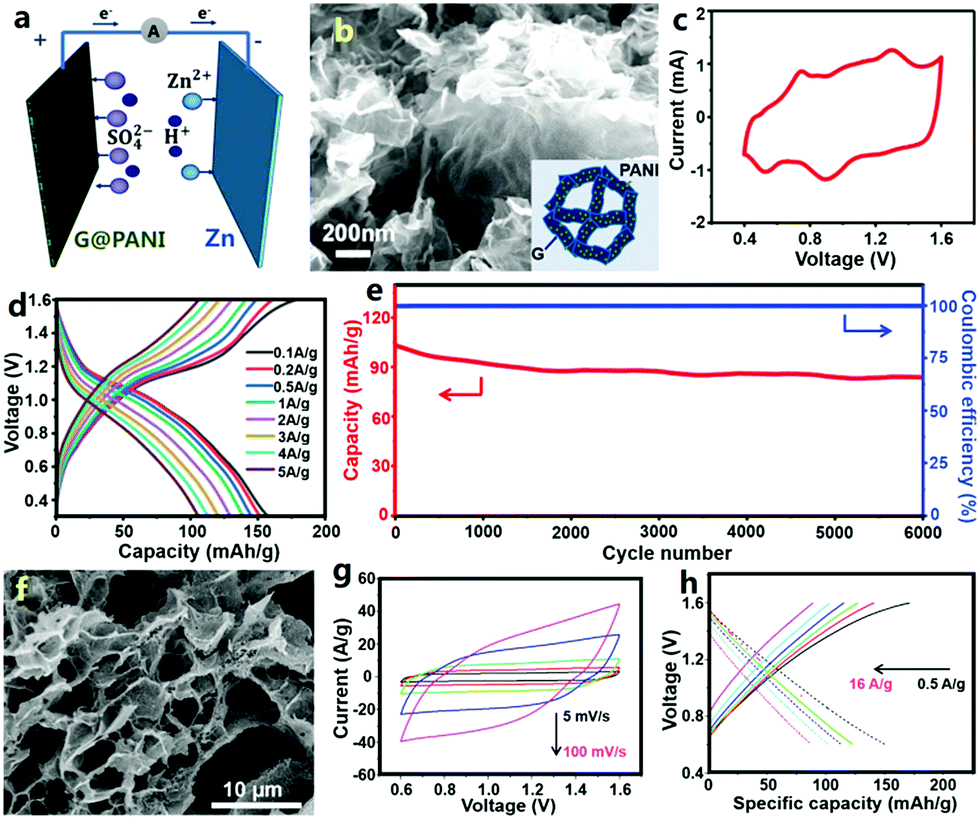 | ||
| Fig. 8 (a) Schematic of a graphene@PANI//Zn ZHC with the ZnSO4 aqueous electrolyte; (b) SEM image of the graphene@PANI composite hydrogel cathode (inset: structure illustration); (c) CV curves, (d) charge–discharge profiles and (e) cycling performance of the graphene@PANI//Zn ZHC. Adapted from ref. 307 with permission from The Royal Society of Chemistry. (f) SEM image of pyrrole/graphite nanosheets/polyvinyl alcohol aerogel and (g) and (h) its electrochemical behaviors as a cathode of a Zn metal based ZHC. Adapted from ref. 308 with permission from Wiley-VCH. | ||
3.1.1.2 ZHCs with an organic electrolyte. Organic carbon//Zn ZHCs were also studied. Wang et al. utilized coconut shells as raw materials to prepare bio-carbon with the assist of the carbonization-activation method (Fig. 9a).309 The obtained bio-carbon materials had a BET surface area of 2596–3554 m2 g−1. They constructed ZHCs based on the bio-carbon cathode, metallic Zn foil anode and organic Zn(CF3SO3)2 electrolyte (the organic solvent was AN or a mixture of dimethoxyethane and 1,3-dioxolane). The organic ZHCs worked in a voltage range of 0–1.8 V, delivering a discharge capacitance of 170 F g−1 (ca. 85 mA h g−1) at a charge/discharge current of 0.1 A g−1 and an energy density of 52.7 W h kg−1 at a power density of 1725 W kg−1 (Fig. 9b and c). The cycling stability of such organic ZHCs (91% capacity retention after 20
000 charge/discharge cycles at 2 A g−1) was comparable to that of our aforementioned aqueous ZHCs.202 Yoo et al. assembled organic ZHCs with an ACF cloth cathode, metallic Zn foil anode and zinc bis(trifluoromethylsulfonyl)imide (i.e., Zn[(CF3SO2)2N]2)/AN electrolyte.310 The ZHCs exhibited quasi box-shaped CV curves and linear charge (and discharge) curves without voltage plateaus in a voltage range of 0–1.8 V. Their capacity reached ∼55 mA h g−1 at a current of 5 mA cm−2. Above 98% of initial capacity was retained after 10000 charge/discharge cycles, implying a very good cycling performance.
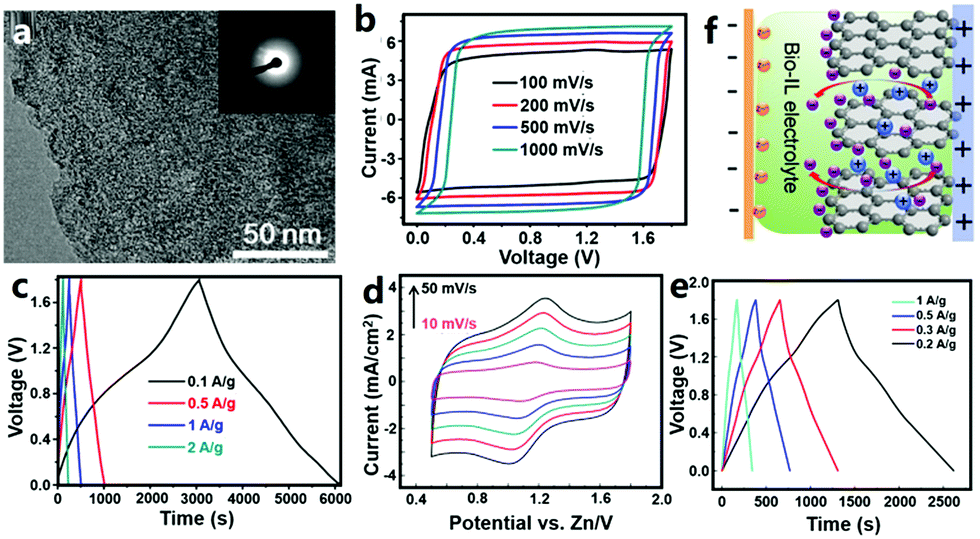 | ||
| Fig. 9 (a) Transmission electron microscopy image of a bio-carbon material and (b) and (c) its electrochemical behaviors as the cathode of a Zn metal based ZHC with a Zn(CF3SO3)2/AN electrolyte. Adapted from ref. 309 with permission from Elsevier. (d) CV curves (scan rate: 10–50 mV s−1), (e) charge–discharge profiles and (g) energy storage mechanism illustration of a ZHC with a sponge-like Zn anode, a natural graphite cathode and a biodegradable ionic liquid electrolyte. Adapted from ref. 311 with permission from Springer. Copyright, 2018. | ||
3.1.1.3 ZHCs with an ionic liquid electrolyte. Ionic liquids possess a series of unique characteristics including high ionic conductivity, negligible vapor pressure, excellent thermal and chemical stability, non-flammability, high safety and a wide electrochemical window; therefore, ionic liquid electrolytes have been widely studied in the fields of rechargeable batteries and supercapacitors. Liu et al. reported a ZHC composed of a sponge-like Zn anode, a natural graphite cathode and a biodegradable ionic liquid electrolyte (Fig. 9d–f).311 The electrolyte was prepared by dissolving Zn(OAc)2 in the mixture of choline acetate (70 wt%) and water (30 wt%). The constructed ZHC showed deviated rectangular shaped CV curves in a voltage range of 0.5–1.8 V, including a redox process at around 1.2 V (Fig. 9d). According to galvanostatic charge–discharge tests performed in a voltage range of 0–1.8 V, a discharge capacity of ∼73 mA h g−1 at 0.2 A g−1 and an energy density of 52.6 W h kg−1 at a power density of 145 W kg−1 were realized (Fig. 9e). Unfortunately, such a ZHC exhibited rapidly dropped capacity at large currents (e.g., 2 A g−1). The authors considered this to be caused by side reactions occurring at higher charge/discharge rates between the Zn electrode and the ionic liquid electrolyte.311,312 The energy storage mechanism of the ZHC was proposed as follows: during the charge process, Zn2+ ions were reduced on the Zn anode, while anions accumulated on and intercalated into the pseudocapacitive graphite cathode; reversed processes occurred during discharging (Fig. 9f).
Various ZHCs with aqueous electrolytes, organic electrolytes and ionic liquid electrolytes are summarized above. In comparison, aqueous ZHCs exhibit good rate performance benefiting from high ionic conductivity of aqueous electrolytes. Also, aqueous ZHCs are safe, low-cost and environmentally benign. In addition, it is worth noting that in conventional supercapacitors such as symmetric AC//AC supercapacitors and asymmetric MnO2//AC supercapacitors, application of organic electrolytes and ionic liquid electrolytes is beneficial for achieving a wider operating voltage window than with aqueous electrolytes, thus delivering a higher energy density.313–316 For example, Lewandowski et al. studied a symmetric carbon//carbon supercapacitor in different electrolytes and found that when aqueous, organic or ionic liquid electrolyte was applied, the supercapacitor could be charged up to 1, 2.5 and 3–3.5 V, respectively.314 In contrast, the reported Zn metal electrode-based ZHCs possess similar operating voltages in aqueous, organic and ionic liquid electrolytes. One important reason is that the dissolution/deposition potential of Zn/Zn2+ on Zn anodes is not strictly restricted by the hydrogen evolution of water. Despite this, by controlling the surface characteristics of cathode materials (e.g., changing the surface functional groups of carbon cathodes), a wider operating voltage window of ZHCs is still possible to be realized in organic and ionic liquid electrolytes. Furthermore, heteroatom-doped carbon materials and conductive metal–organic-framework materials exhibit good capacitive performance in conventional supercapacitors (e.g., with a H2SO4 electrolyte) and are worth studying as ZHC electrodes, while the operating voltage window and specific electrochemical behavior of these material-constructed ZHCs need to be investigated in future research studies.
In our previous research, we first studied MnO2//AC ZHCs in which MnO2 nanorods were used as the cathode material, high specific surface area AC particles were used as the anode and several types of aqueous solutions (including ZnSO4 solution, ZnSO4/MnSO4 mixture solution and Zn(CF3SO3)2 solution) were used as the electrolyte (Fig. 10).317 These ZHCs operated in a voltage range of 0–2.0 V. The corresponding CV curves were composed of a quasi-rectangular characteristic with redox peaks, and in galvanostatic charge–discharge profiles, the charge and discharge curves were not linear, which were indicative of an integration of battery behavior and capacitive behavior. MnO2 and AC materials possess different Zn2+-storage abilities; thus, the electrochemical properties of MnO2//ZnSO4 (aq)//AC ZHCs with different mass ratios of the MnO2 cathode to the AC anode should be investigated. When the mass ratio was 1:2.5, the assembled ZHC exhibited a maximum discharge capacity of 54.1 mA h g−1 and an energy density of 34.8 W h kg−1 (calculated based on the total weight of MnO2 and AC), much higher than the figures for asymmetric MnO2//AC supercapacitors with a Na2SO4 aqueous electrolyte. We would like to emphasize that even though MnO2//ZnSO4 (aq)//AC and MnO2//Na2SO4 (aq)//AC systems have the same cathode material (i.e., MnO2), anode material (i.e., AC) and operating voltage window (i.e., 0–2.0 V), they are essentially different in energy storage mechanisms and electrochemical performance. MnO2//Na2SO4 (aq)//AC forms an asymmetric supercapacitor. Its energy storage is realized by electrostatic accumulation of charges on the AC surface and pseudocapacitive behavior of MnO2 (fast and reversible redox reactions occur on the MnO2 surface, as illustrated in Fig. 1d). In contrast, MnO2//ZnSO4 (aq)//AC forms a hybrid capacitor. Its energy storage is mainly through electrostatic accumulation of charges on the AC surface and Zn2+-ion intercalation/de-intercalation into/from MnO2 channels. In addition, formation/dissolution of Zn4(OH)6SO4·nH2O on both the MnO2 cathode and the AC anode is another way to store electrochemical energy for MnO2//ZnSO4 (aq)//AC ZHCs, as pointed out in our previous research.317 Nevertheless, the formation/dissolution process of Zn4(OH)6SO4·nH2O is only partially reversible, which adversely affects the capacity and cycling performance of MnO2//ZnSO4 (aq)//AC ZHCs. Meanwhile, manganese dissolution from the MnO2 cathode also goes against ZHCs' cycling stability. As a result, up to 35% capacity decay was detected after 3000 charge/discharge cycles of MnO2//ZnSO4 (aq)//AC ZHCs.317 To minimize the above issues and optimize ZHCs' electrochemical performance, electrolyte composition was regulated, including (i) introducing Mn2+ in the ZnSO4 (aq) electrolyte to suppress manganese dissolution and (ii) replacing SO42− anions with CF3SO3− anions to inhibit generation of Zn4(OH)6SO4·nH2O. Overall, the former strategy improved the discharge capacity of ZHCs and the latter strategy significantly extended ZHCs' lifetime.
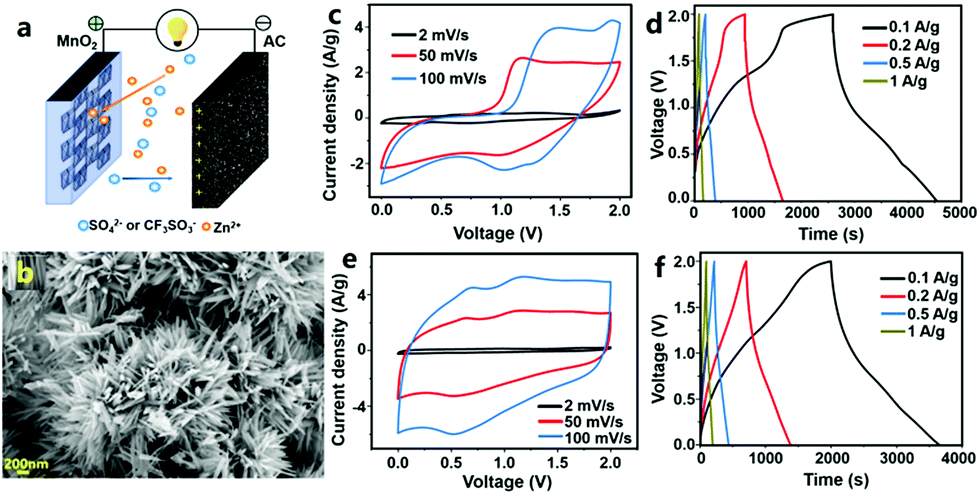 | ||
| Fig. 10 (a) Schematic of a MnO2//AC ZHC; (b) SEM image of a MnO2 nanorod cathode and its electrochemical properties inside the MnO2//AC ZHC with the aqueous electrolyte of (c) and (d) ZnSO4 and (e) and (f) Zn(CF3SO3)2. Adapted from ref. 317 with permission from Elsevier. | ||
As summarized in Section 2.2.1, besides MnO2, other types of manganese oxides and vanadium oxides are capable of storing Zn2+ ions in aqueous rechargeable zinc-ion batteries. In theory, these materials can be used to construct ZHCs with AC anodes. Even though the operating potentials of vanadium oxides (generally 0.2–1.6 V vs. Zn/Zn2+) are lower than that of manganese oxides (1.0–1.8 V vs. Zn/Zn2+), which will lead to a narrower voltage window of ZHCs, the higher Zn2+-storage capacity and good rate performance of vanadium oxide cathodes will be beneficial for ZHCs to achieve high energy density and power density. Similarly, besides AC powder, diverse kinds of capacitor-like electrode materials can be applied as anodes for ZHCs, such as CNTs, graphene, ACFs, CNFs, conductive polymers and so on.
Sun et al. designed a ZHmC by integrating an oxidized CNT (oCNT) micro-cathode, a Zn micro-anode and a gelatin/ZnSO4 electrolyte (Fig. 11a–e).327 The authors first pasted CNT paper onto polyimide tape, followed by laser etching to form a 6-finger interdigitated pattern and oxygen plasma treatment, and then, the Zn micro-anode was prepared through an in situ electroplating method in ZnSO4 aqueous solutions (during the electroplating process, one interdigitated oCNT micro-electrode acted as the working electrode and other ones acted as the reference and counter electrodes). The fabricated ZHmC operated in a voltage range of 0.2–1.8 V, and its CV curves could be regarded as a combination of broad redox peaks and fairly rectangular characteristic (Fig. 11b). The authors believed that the charge storage reactions inside the ZHmC were not diffusion-controlled but confined to the electrode surface, judged from the linear relationship between the peak current and scan rates (Fig. 11c). The detailed electrode reactions are the same as those inside macroscopic aqueous carbon//Zn ZHCs as reviewed in Section 3.1.1.1. The ZHmC displayed an areal capacitance of 83.2 mF cm−2 at a current of 1 mA cm−2 and a good rate performance with 78% capacitance retention when charge/discharge current was increased from 1 to 10 mA cm−2, leading to a high areal energy density of 29.6 μW h cm−2 (corresponding to a volumetric energy density of 0.015 W h cm−3) and a high power density of 8 mW cm−2 (corresponding to a volumetric power density of 4 W cm−3) (Fig. 11d). Besides, the capacity of the ZHmC retained 87.4% after 6000 charge/discharge cycles, and more importantly, the electrochemical performance of the ZHmC could recover (or become even higher than) its original level through further in situ electroplating of Zn again. The ZHmC displayed a certain flexibility; it could be bent and twisted (Fig. 11e). After 3000 bending cycles (one cycle: bending from 0° to 45° and then recovery to 0°), ZHmC capacitance retained 89.5% of its initial value. This preliminarily proves that ZHC systems are capable of being applied in the field of wearable/portable electronics.
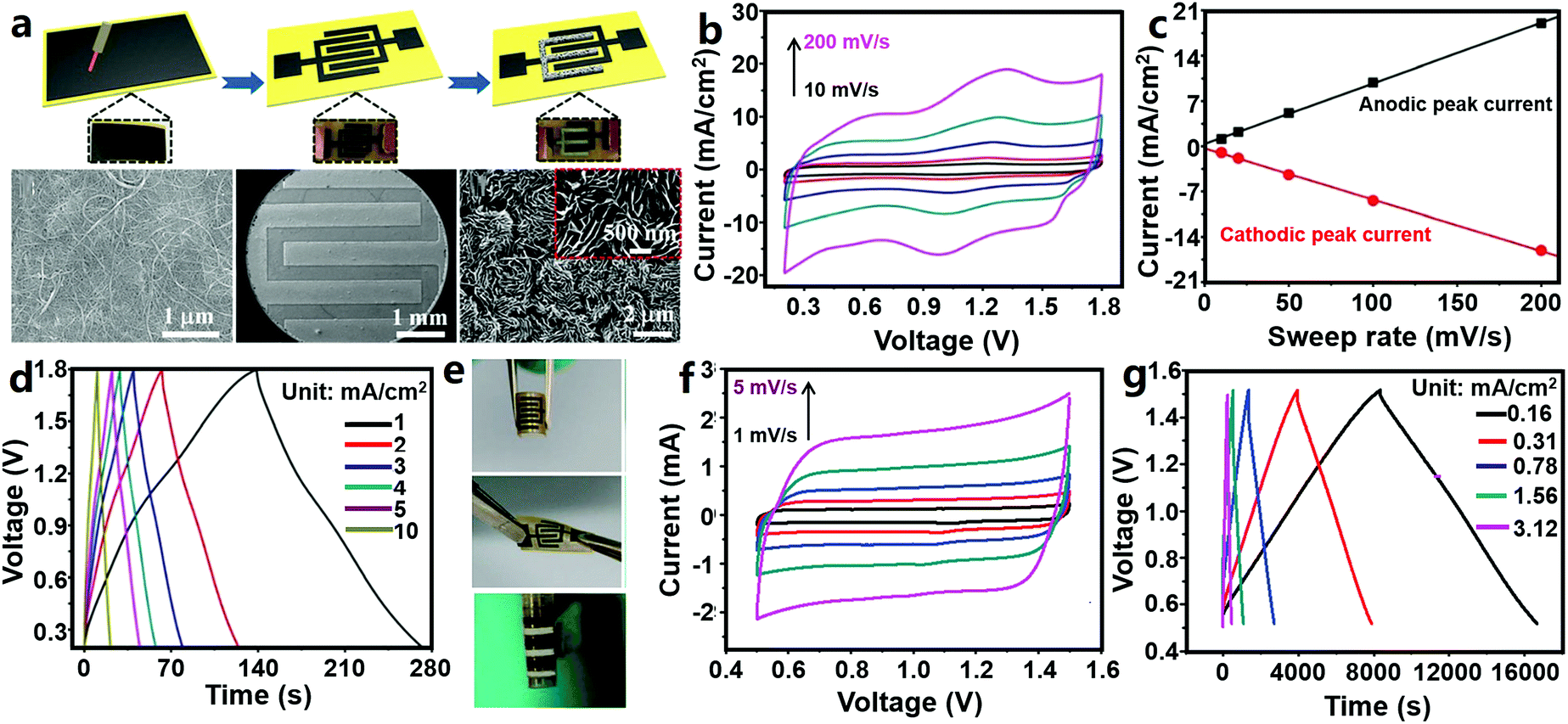 | ||
| Fig. 11 (a) Schematic illustration of the preparation process of an oCNT//Zn ZHmC and corresponding SEM images in each step (from left to right: pristine CNT paper, an interdigitated microelectrode and a Zn micro-anode); (b) CV curves at 10–200 mV s−1, (c) relation curve between the peak current and the scan rate, (d) charge–discharge profiles and (e) photographs in bent and twist states of the oCNT//Zn ZHmC. Adapted from ref. 327 with permission from The Royal Society of Chemistry. (f) CV (scan rate: 1–5 mV s−1) and (g) charge–discharge curves of an AC//Zn ZHmC. Adapted from ref. 328 with permission from Wiley-VCH. | ||
Similarly, Zhang et al. reported an AC//Zn ZHmC (Fig. 11f and g), in which an interdigitated Ti/Au pattern served as the current collector, electrochemically deposited Zn nanosheets served as the anode, a coated AC/acetylene black/sodium carboxymethylcellulose electrode served as the cathode, and ZnSO4 aqueous solution served as the electrolyte.328 The thickness and loading of Zn nanosheets were controlled by adjusting the electrodeposition time. According to the authors' research, during charge/discharge processes of the AC//Zn ZHmC in a voltage range of 0.5–1.5 V, Zn stripping/plating occurred on the Zn anode and only a physical adsorption/desorption process existed on the AC cathode, because the used AC particles almost did not contain oxygen functional groups, which means that the AC cathode could not contribute pseudocapacitance. Also, the AC//Zn ZHmC worked in a narrower operating voltage window compared with the AC//Zn ZHCs discussed in Fig. 7. Overall, there are some differences between the two devices in the energy storage mechanism. Consequently, CV curves of the ZHmC showed a nearly rectangular shape even at low scan rates of 1–5 mV s−1 (Fig. 11f). Despite this, capacitance of the ZHmC reached 1297 mF cm−2 at a current density of 0.16 mA cm−2 (equaling 259.4 F g−1 at a current density of 0.05 A g−1) and 600 mF cm−2 at 3.12 mA cm−2, implying a high areal capacitance and good rate performance (Fig. 11g). The maximum areal energy density and power density were 115.4 μW h cm−2 and 3.0 mW cm−2, respectively, significantly superior to those of many other previously reported micro-supercapacitors. In addition, for the ZHmC, an excellent cycling stability was also achieved with 100% capacitance retention over 10000 charge/discharge cycles.
As overviewed above and summarized in Table 2, various ZHCs with different configurations and energy storage mechanisms have been proposed. These ZHCs exhibit attractive electrochemical behaviors (such as the integration of high energy density and high power density). Benefiting from high security and superior electrochemical performance, the ZHCs are considered to have enormous application potential in wearable/portable energy storage, micro-scale devices and some other fields. By contrast, there are only several reports on hybrid capacitors based on multivalent Mg2+, Ca2+ and Al3+, as discussed in the following.
| Configuration | Electrochemical performance | Ref. | ||||
|---|---|---|---|---|---|---|
| Cathode | Anode | Electrolyte | Voltage (V) | Capacity | Capacity retention | |
| a Calculated based on the total mass of MnO2 and AC. | ||||||
| AC | Zn foil | ZnSO4 (aq) | 0.2–1.8 | 121 mA h g−1 (0.1 A g−1) | 91% after 10000 cycles |
202 |
| 41 mA h g−1 (20 A g−1) | ||||||
| Oxidized CNTs | Zn foil | ZnSO4 (aq); ZnSO4–PVA gel | 0–1.8 | ∼27 mA h g−1 (10 mV s−1) | — | 306 |
| Graphene@PANI | Zn foil | ZnSO4 (aq) | 0.4–1.6 | 154 mA h g−1 (0.1 A g−1) | 80.5% after 6000 cycles | 307 |
| 106 mA h g−1 (5 A g−1) | ||||||
| PPy | Zn/graphite paper | ZnCl2 + NH4Cl (aq) | 0.6–1.6 | 151.1 mA h g−1 (0.5 A g−1); 87.6 mA h g−1 (16 A g−1); | 76.7% after 1000 cycles | 308 |
| Bio-carbon | Zn foil | Zn(CF3SO3)2 in AN or the mixture of dimethoxyethane and 1,3-dioxolane | 0–1.8 | 85 mA h g−1 (0.1 A g−1) | 91% after 20000 cycles |
309 |
| ACF cloth | Zn foil | Zn[(CF3SO2)2N]2/AN | 0–1.8 | ∼55 mA h g−1 (5 mA cm−2) | 96% after 10000 cycles |
310 |
| Natural graphite | Sponge-like Zn | Zn(OAc)2/choline acetate/water | 0–1.8 | ∼73 mA h g−1 (0.2 A g−1) | ∼100% after 1000 cycles | 311 |
| MnO2 nanorods | AC | ZnSO4 (aq) | 0–2.0 | 54.1 mA h g−1 (0.1A g−1)a | 65.3% after 3000 cycles | 317 |
| MnO2 nanorods | AC | Zn(CF3SO3)2 (aq) | 0–2.0 | 46.4 mA h g−1 (0.1A g−1)a | 93.4% after 5000 cycles | 317 |
| Oxidized CNTs | Zn | ZnSO4–gelatin | 0.2–1.8 | 83.2 mF cm−2 (1 mA cm−2); 65 mF cm−2 (10 mA cm−2) | 87.4% after 6000 cycles | 327 |
| AC | Zn | ZnSO4 (aq) | 0.5–1.5 | 1297 mF cm−2 (0.16 mA cm−2); 600 mF cm−2 (3.12 mA cm−2) | 100% after 10000 cycles |
328 |
3.2 Other multivalent metal ion hybrid capacitors
Very recently, Wu et al. reported a CHC, in which high surface area AC was used as the cathode, Sn foil was the anode and an organic Ca(PF6)2 solution (solvent: a mixture of ethylene carbonate, propylene carbonate, dimethyl carbonate and ethyl methyl carbonate in a volume ratio of 1:1:1:1) was used as the electrolyte (Fig. 12).331 The CHC worked in a voltage range of 1.5–4.8 V, much higher than that of other types of multivalent metal ion hybrid capacitors. This is conducive to obtaining a high energy density. When the CHC was charged, PF6− anions adsorbed on the AC surface, and meanwhile, Ca2+ cations from the electrolyte moved onto the Sn foil to form a Ca7Sn6 alloy. During subsequent discharging, PF6− anions desorbed from the AC surface and a dealloying process (i.e., from the Ca7Sn6 alloy to Sn) occurred on the anode (Fig. 12a). Correspondingly, the CV curves of the CHC were nonideal rectangular and galvanostatic charge–discharge curves were nonideal straight (Fig. 12b). As a result, the CHC possessed a reversible capacity of 92 mA h g−1 at a charge/discharge current of 0.1 A g−1. Fast ion adsorption/desorption on/from the cathode and fast kinetics of the alloying/dealloying process on the anode endowed the CHC with a good rate capability: only 2 min was needed to fully charge or discharge the CHC. In addition, it was proved that the Sn anode was relatively stable during the alloying/dealloying process, which was beneficial for the CHC to achieve a superior cycling performance: 84% capacity could be retained after 1000 charge/discharge cycles (Fig. 12c). Even though such a cycling stability is much worse than that of many other kinds of multivalent metal ion hybrid capacitors, it is the highest to date for Ca2+ based electrochemical energy storage.
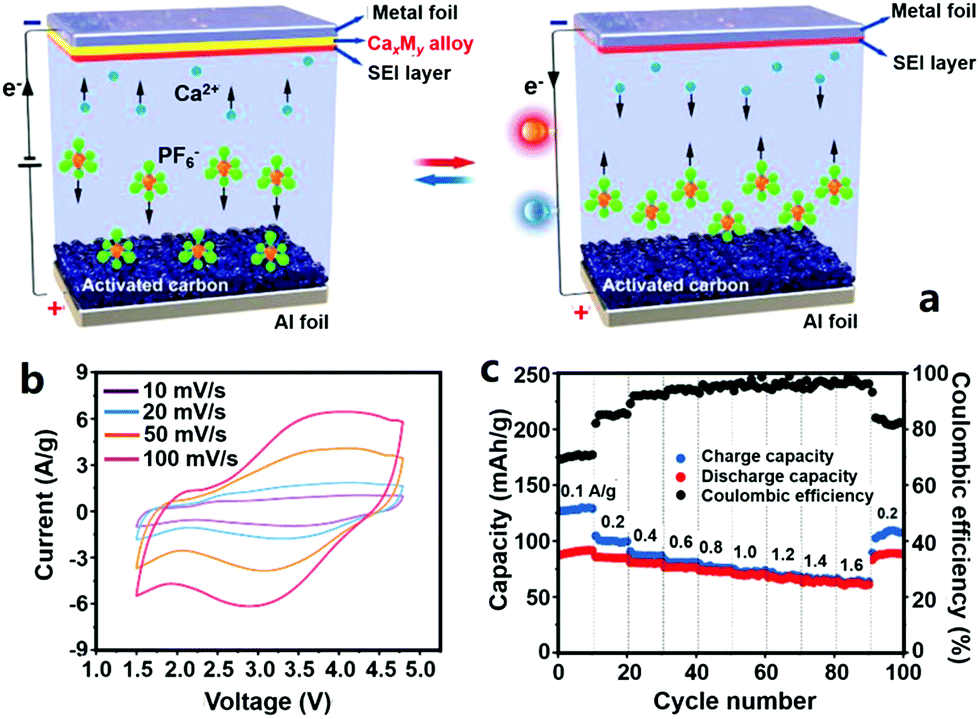 | ||
| Fig. 12 (a) Schematic illustration of a CHC system with an AC cathode, a metal foil anode (e.g., Sn foil) and an organic Ca(PF6)2 electrolyte; electrochemical performance of the CHC: (b) CV curves and (c) rate capability and coulombic efficiency. Reproduced from ref. 331 with permission from Wiley-VCH. | ||
Wang et al. first synthesized conductive PPy coated MoO3 nanotubes (MoO3@PPy) (Fig. 13a) and used them as an anode material for Al3+ storage.332 In 0.5 M Al2(SO4)3 aqueous electrolyte, the MoO3@PPy anode delivered a capacity of about 140 mA h g−1 at 1 A g−1 in an operation voltage window of −0.4 to 0.5 V (vs. saturated calomel electrode). Reversible intercalation/de-intercalation of Al3+ in MoO3@PPy was described as follows.
| MoO3 + xAl3+ + 3xe− ⇔ AlxMoO3 (0 ≤ x ≤ 0.27) | (15) |
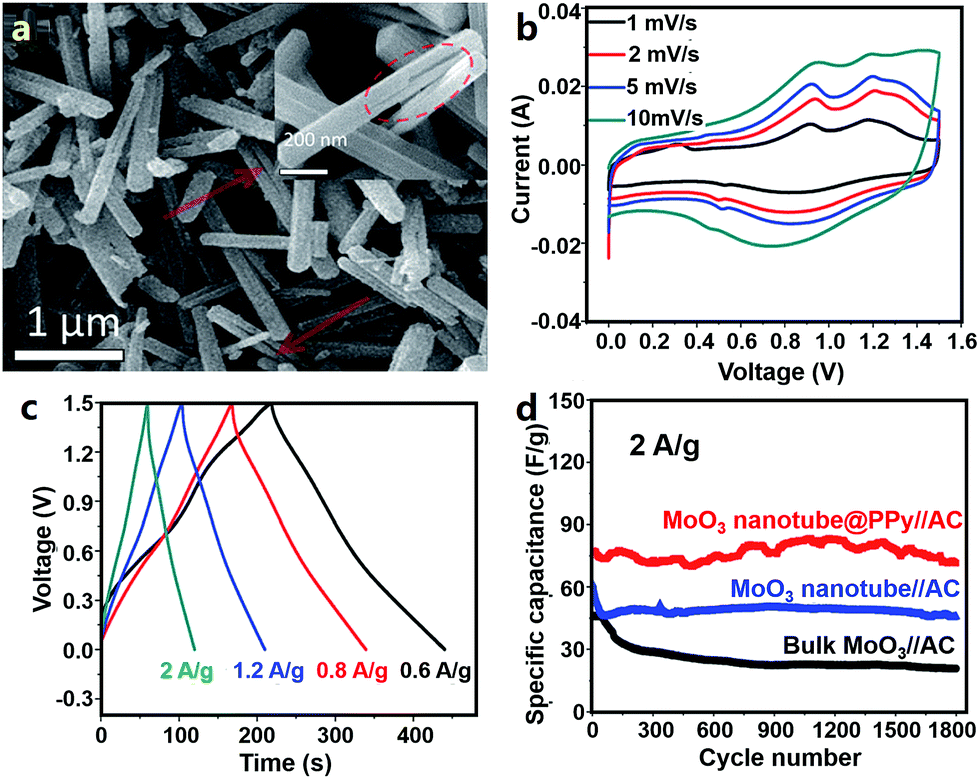 | ||
| Fig. 13 (a) SEM image of MoO3@PPy materials; (b) CV curves, (c) charge–discharge profiles and (d) cycling behavior of an AHC system comprising a MoO3@PPy anode, an AC cathode and an Al2(SO4)3 aqueous electrolyte. Adapted from ref. 332 with permission from The Royal Society of Chemistry. | ||
AC electrodes in an Al2(SO4)3 aqueous electrolyte exhibited typical electric double layer capacitive behavior in a voltage range of 0.5–1.1 V (vs. saturated calomel electrode). The authors then constructed an AHC comprising a MoO3@PPy anode, an AC cathode and an Al2(SO4)3 aqueous electrolyte (Fig. 13b–d). The AHC functioned from 0 to 1.5 V and its CV curves showed broad redox peaks associated with Al3+ intercalation/de-intercalation into/from MoO3@PPy. Furthermore, the AHC displayed a good rate performance. Its energy density was 28 and 20 W h kg−1 at a power density of 460 and 2840 W kg−1, respectively. After 1800 charge/discharge cycles, the capacity of the AHC retained ∼93%, demonstrating a relatively good cycling stability. The authors attributed the good electrochemical performance of the AHC to the following factors: (i) the small diameter but large charge number of Al3+ ions benefited the high capacity, (ii) PPy coating decreased the charge transfer resistance and effectively protected MoO3 from acid etching, and (iii) the nanotube structure of MoO3 made it easier for the electrolyte to infiltrate active materials and also buffered the volume change of active materials during Al3+ intercalation/de-intercalation. In addition, as pointed out by the authors, the capacity of the used AC cathode in the AHC was relatively low; thus, the energy density of the AHC device could be further enhanced by optimizing the cathode material. Afterwards, Li et al. studied an asymmetric Al3+-ion supercapacitor with a single-wall CNT (SWNT)/W18O49 nanowire negative electrode, an SWNT/PANI positive electrode and 1 M AlCl3 aqueous electrolyte.333 Even though such a supercapacitor is beyond the scope of hybrid capacitors, its SWNT/PANI electrode working in a potential range of 0–0.9 V (vs. Ag/AgCl) may be a potential candidate for AHC cathodes.
Based on the above discussions, we can see that various electrode materials and electrolytes are available for constructing ZHCs, whereas only a few configurations have been proved to be feasible for MHCs, CHCs, and AHCs. Besides, ZHCs are superior to the other types of hybrid multivalent metal ion capacitors in terms of rate capability and cycling stability, while CHCs and MHCs especially the former ones possess higher operating voltage, which is beneficial for achieving a higher energy density.
4. Conclusions and future outlook
Multivalent metal ion-based electrochemical energy storage is booming. Research studies on multivalent metal ion hybrid capacitors including ZHCs, MHCs, CHCs and AHCs are reviewed in this work. For each type of multivalent metal ion hybrid capacitor, its design concept, configuration, electrochemical behavior and energy storage mechanism are summarized. Overall, these multivalent metal ion hybrid capacitors integrate various advantages of conventional supercapacitors and multivalent metal ion batteries, exhibiting superior rate performance, high power density, high energy density and long cycle life. These merits give multivalent metal ion hybrid capacitors a promising future in electric vehicles and wearable/portable electronics. Furthermore, in our opinion, future research on multivalent metal ion hybrid capacitors will focus on the following aspects.(i) Synthesis of high-performance electrode materials for multivalent metal ion hybrid capacitors. Up to now, there have been only a few reports about MHCs, CHCs and AHCs, due to limited choices of battery-like electrodes for these systems. More types of electrode materials for MIBs, CIBs and AIBs should be developed and investigated in corresponding multivalent metal ion hybrid capacitors. Particularly, for practical applications of metal electrode-based hybrid capacitors such as Zn foil-based ZHCs, the energy density of the whole device (instead of the cathode material only) has to be taken into account. This requires the metal electrodes to possess a long-term stability and fast kinetics even though their mass in the hybrid capacitors is relatively low (relative to the mass of the counter electrode). In addition, capacitor-like electrodes beyond AC materials should be further studied for these hybrid capacitors. We hope our overview of electrode materials for supercapacitors (EDLCs and pseudocapacitors) and multivalent metal ion batteries in Section 2 can offer some inspiration.
(ii) Detailing of actual electrochemical processes inside multivalent metal ion hybrid capacitors. Multivalent ion storage mechanisms are a relatively novel concept. They have not been completely understood in battery systems due to the distinct characteristics of multivalent ions (being different from univalent ions in charge number, polarization strength and ionic diameter). Besides this, interactions between multivalent ions and supercapacitive materials such as carbons may be not pure adsorption/desorption. Therefore, comprehensive methods including electrochemical measurements and physical–chemical analysis should be applied to extrapolate the energy storage mechanism of multivalent metal ion hybrid capacitors. This will benefit science by enriching the understanding of multivalent ion storage mechanisms.
(iii) Development of various configurations of multivalent metal ion hybrid capacitors. For ZHCs, Zn electrode based ZHCs and metal oxide//carbon ZHCs have been proposed, while for MHCs, CHCs and AHCs, only metal electrode-based hybrid capacitors have been designed. Other kinds of configurations of multivalent metal ion hybrid capacitors need to be developed to suit diverse applications. The construction of various configurations of multivalent metal ion hybrid capacitors relies on research into high-performance electrode materials, electrolytes and multivalent ion storage mechanisms.
(iv) Study of multivalent metal ion hybrid capacitors for flexible and wearable energy storage. As discussed above, multivalent metal ion hybrid capacitors display a series of merits such as notably enhanced energy density compared to conventional supercapacitors and higher safety than Li-ion batteries. Therefore, multivalent metal ion hybrid capacitors with good deformability based on reasonable electrode/device structure designs have a wide possible range of applications for flexible/wearable energy storage. These flexible hybrid capacitors can also be combined with flexible solar cells, flexible sensors and other electronics for multifunctional smart applications in future society.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the Australian Research Council through the ARC Discovery projects (DP160104340 and DP170100436), the High-level Talents Project of Dongguan University of Technology (KCYKYQD2017016), and the Leading Talents of Innovation and Entrepreneurship of the Dongguan City D2017(16).Notes and references
- B. E. Conway and W. G. Pell, J. Solid State Electrochem., 2003, 7, 637 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- Z. Zhang, M. Li, Y. Gao, Z. Wei, M. Zhang, C. Wang, Y. Zeng, B. Zou, G. Chen and F. Du, Adv. Funct. Mater., 2018, 28, 1802684 CrossRef.
- J. Ding, W. Hu, E. Paek and D. Mitlin, Chem. Rev., 2018, 118, 6457 CrossRef CAS PubMed.
- Y. Ma, H. Chang, M. Zhang and Y. Chen, Adv. Mater., 2015, 27, 5296 CrossRef CAS PubMed.
- H. Li, L. Cheng and Y. Xia, Electrochem. Solid-State Lett., 2005, 8, A433 CrossRef CAS.
- Y. Wang and Y. Xia, Electrochem. Commun., 2005, 7, 1138 CrossRef CAS.
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093 CrossRef PubMed.
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Electrochem. Commun., 2015, 60, 172 CrossRef CAS.
- A. L. Comte, Y. Reynier, C. Vincens, C. Leys and P. Azaïs, J. Power Sources, 2017, 363, 34 CrossRef.
- S. Dong, Z. Li, Z. Xing, X. Wu, X. Ji and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 15542 CrossRef CAS PubMed.
- L. Dong, C. Xu, Y. Li, Z. H. Huang, F. Kang, Q. H. Yang and X. Zhao, J. Mater. Chem. A, 2016, 4, 4659 RSC.
- Z. Lv, W. Li, L. Yang, X. J. Loh and X. Chen, ACS Energy Lett., 2019, 4, 606 CrossRef CAS.
- W. Liu, M. S. Song, B. Kong and Y. Cui, Adv. Mater., 2017, 29, 1603436 CrossRef PubMed.
- C. Xu, Y. Chen, S. Shi, J. Li, F. Kang and D. Su, Sci. Rep., 2015, 5, 14120 CrossRef CAS PubMed.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- O. Chusid, Y. Gofer, H. Gizbar, Y. Vestfrid, E. Levi, D. Aurbach and I. Riech, Adv. Mater., 2003, 15, 627 CrossRef CAS.
- T. Ichitsubo, T. Adachi, S. Yagi and T. Doi, J. Mater. Chem., 2011, 21, 11764 RSC.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243 RSC.
- A. L. Lipson, B. Pan, S. H. Lapidus, C. Liao, J. T. Vaughey and B. J. Ingram, Chem. Mater., 2015, 27, 8442 CrossRef CAS.
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169 CrossRef CAS PubMed.
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H. M. Cheng, Nat. Chem., 2018, 10, 667 CrossRef CAS PubMed.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 324 CrossRef CAS PubMed.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610 RSC.
- H. Chen, F. Guo, Y. Liu, T. Huang, B. Zheng, N. Ananth, Z. Xu, W. Gao and C. Gao, Adv. Mater., 2017, 28, 1605958 CrossRef PubMed.
- L. D. Reed, S. N. Ortiz, M. Xiong and E. J. Menke, Chem. Commun., 2015, 51, 14397 RSC.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- R. J. Gummow, G. Vamvounis, M. B. Kannan and Y. He, Adv. Mater., 2018, 30, 1801702 CrossRef PubMed.
- S. K. Das, S. Mahapatra and H. Lahan, J. Mater. Chem. A, 2017, 5, 6347 RSC.
- V. Verma, S. Kumar, W. Manalastas Jr, R. Satish and M. Srinivasan, Adv. Sustainable Syst., 2019, 3, 1800111 CrossRef.
- Z. A. Zafar, S. Imtiaz, R. Razaq, S. Ji, T. Huang, Z. Zhang, Y. Huang and J. A. Anderson, J. Mater. Chem. A, 2017, 5, 5646 RSC.
- M. Jayalakshmi and K. Balasubramaniam, Int. J. Electrochem. Sci., 2008, 3, 1196 CAS.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483 CrossRef.
- H. I. Becker, U.S. Pat. 2800616 (to General Electric), 1957.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937 CrossRef CAS.
- C. Niu, E. K. Sichel, R. Hoch, D. Moy and H. Tennent, Appl. Phys. Lett., 1997, 70, 1680 CrossRef.
- K. Metenier, V. Bertagna and F. Béguin, Appl. Phys. Lett., 2000, 77, 2421 CrossRef.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11 CrossRef CAS.
- H. O. Stern, Z. Elektrochem. Angew. Phys. Chem., 1924, 30, 508 Search PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219 CrossRef PubMed.
- V. Subramanian, C. Luo, A. M. Stephan, K. S. Nahm, S. Thomas and B. Wei, J. Phys. Chem. C, 2007, 111, 7527 CrossRef CAS.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109 CrossRef CAS.
- Z. Z. Pan, L. Dong, W. Lv, D. Zheng, Z. Li, C. Luo, C. Zheng, Q. H. Yang and F. Kang, Chem.–Asian J., 2017, 12, 503 CrossRef CAS PubMed.
- X. He, Y. Geng, J. Qiu, M. Zheng, S. Long and X. Zhang, Carbon, 2010, 48, 1662 CrossRef CAS.
- L. Dong, C. Xu, Y. Li, Z. Pan, G. Liang, E. Zhou, F. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 9313 CrossRef CAS PubMed.
- D. Song, W. Guo, T. Zhang, P. Lu, A. Guo, F. Hou, X. Yan and J. Liang, Surf. Innovations, 2019, 7, 10 CrossRef.
- Z. Niu, W. Zhou, J. Chen, G. Feng, H. Li, W. Ma, J. Li, H. Dong, Y. Ren, D. Zhao and S. Xie, Energy Environ. Sci., 2011, 4, 1440 RSC.
- J. H. Chen, W. Z. Li, D. Z. Wang, S. X. Yang, J. G. Wen and Z. F. Ren, Carbon, 2002, 40, 1193 CrossRef CAS.
- K. H. An, W. S. Kim, Y. S. Park, Y. C. Choi, S. M. Lee, D. C. Chung, D. J. Bae, S. C. Lim and Y. H. Lee, Adv. Mater., 2001, 13, 497 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326 CrossRef CAS PubMed.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806 CrossRef CAS PubMed.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833 CrossRef CAS PubMed.
- X. He, N. Zhang, X. Shao, M. Wu, M. Yu and J. Qiu, Chem. Eng. J., 2016, 297, 121 CrossRef CAS.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366 CrossRef CAS.
- C. Kim and K. S. Yang, Appl. Phys. Lett., 2003, 83, 1216 CrossRef CAS.
- E. J. Ra, E. Raymundo-Piñero, Y. H. Lee and F. Béguin, Carbon, 2009, 47, 2984 CrossRef CAS.
- A. Chinnappan, C. Baskar, S. Baskar, G. Ratheesh and S. Ramakrishna, J. Mater. Chem. C, 2017, 5, 12657 RSC.
- L. Dong, C. Xu, Q. Yang, J. Fang, Y. Li and F. Kang, J. Mater. Chem. A, 2015, 3, 4729 RSC.
- L. Dong, C. Xu, Y. Li, C. Wu, B. Jiang, Q. Yang, E. Zhou, F. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 1675 CrossRef CAS PubMed.
- L. Dong, G. Liang, C. Xu, W. Liu, Z. Z. Pan, E. Zhou, F. Kang and Q. H. Yang, Nano Energy, 2017, 34, 242 CrossRef CAS.
- J. Wang, L. Dong, C. Xu, D. Ren, X. Ma and F. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 10851 CrossRef CAS PubMed.
- C. Kim, Y. O. Choi, W. J. Lee and K. S. Yang, Electrochim. Acta, 2004, 50, 883 CrossRef CAS.
- K. Babel and K. Jurewicz, J. Phys. Chem. Solids, 2004, 65, 275 CrossRef CAS.
- Z. Ling, Z. Wang, M. Zhang, C. Yu, G. Wang, Y. Dong, S. Liu, Y. Wang and J. Qiu, Adv. Funct. Mater., 2016, 26, 111 CrossRef CAS.
- X. He, N. Zhao, J. Qiu, N. Xiao, M. Yu, C. Yu, X. Zhang and M. Zheng, J. Mater. Chem. A, 2013, 1, 9440 RSC.
- X. He, H. Ma, J. Wang, Y. Xie, N. Xiao and J. Qiu, J. Power Sources, 2017, 357, 41 CrossRef CAS.
- S. Dong, X. He, H. Zhang, X. Xie, M. Yu, C. Yu, N. Xiao and J. Qie, J. Mater. Chem. A, 2018, 6, 15954 RSC.
- F. C. Wu, R. L. Tseng, C. C. Hu and C. C. Wang, J. Power Sources, 2005, 144, 302 CrossRef CAS.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Béguin, Carbon, 2005, 43, 1293 CrossRef CAS.
- J. Huang, B. G. Sumpter and V. Meunier, Angew. Chem., Int. Ed., 2008, 120, 530 CrossRef.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730 CrossRef CAS PubMed.
- Q. Yang, L. Dong, C. Xu and F. Kang, RSC Adv., 2016, 6, 12525 RSC.
- Y. Li and C. Chen, J. Mater. Sci., 2017, 52, 12348 CrossRef CAS.
- Y. Meng, Y. Zhao, C. Hu, H. Cheng, Y. Hu, Z. Zhang, G. Shi and L. Qu, Adv. Mater., 2013, 25, 2326 CrossRef CAS PubMed.
- L. Kou, T. Huang, B. Zheng, Y. Han, X. Zhao, K. Gopalsamy, H. Sun and C. Gao, Nat. Commun., 2014, 5, 3754 CrossRef CAS PubMed.
- X. Wang, G. Li, Z. Chen, V. Augustyn, X. Ma, G. Wang, B. Dunn and Y. Lu, Adv. Energy Mater., 2011, 1, 1089 CrossRef CAS.
- L. Kong, C. Zhang, J. Wang, W. Qiao, L. Ling and D. Long, ACS Nano, 2015, 9, 11200 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P. L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518 CrossRef CAS PubMed.
- Y. Gogotsi and R. M. Penner, ACS Nano, 2018, 12, 2081 CrossRef CAS PubMed.
- M. Huang, F. Li, F. Dong, Y. X. Zhang and L. L. Zhang, J. Mater. Chem. A, 2015, 3, 21380 RSC.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184 CrossRef CAS.
- Z. S. Wu, W. Ren, D. W. Wang, F. Li, B. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835 CrossRef CAS PubMed.
- W. Chen, R. B. Rakhi, L. Hu, X. Xie, Y. Cui and H. N. Alshareef, Nano Lett., 2011, 11, 5165 CrossRef CAS PubMed.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595 CrossRef CAS.
- G. Zhang, X. Xiao, B. Li, P. Gu, H. Xue and H. Pang, J. Mater. Chem. A, 2017, 5, 8155 RSC.
- C. Wu, F. Feng and Y. Xie, Chem. Soc. Rev., 2013, 42, 5157 RSC.
- X. Lu, G. Wang, T. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690 CrossRef CAS PubMed.
- J. G. Wang, F. Kang and B. Wei, Prog. Mater. Sci., 2015, 74, 51 CrossRef CAS.
- V. Gupta and N. Miura, Electrochim. Acta, 2006, 52, 1721 CrossRef CAS.
- A. Eftekhari, L. Li and Y. Yang, J. Power Sources, 2017, 347, 86 CrossRef CAS.
- Y. Huang, H. Li, Z. Wang, M. Zhu, Z. Pei, Q. Xue, Y. Huang and C. Zhi, Nano Energy, 2016, 22, 422 CrossRef CAS.
- R. B. Ambade, S. B. Ambade, R. R. Salunkhe, V. Malgras, S. H. Jin, Y. Yamauchi and S. H. Lee, J. Mater. Chem. A, 2016, 4, 7406 RSC.
- G. Cai, P. Darmawan, M. Cui, J. Wang, J. Chen, S. Magdassi and P. S. Lee, Adv. Energy Mater., 2016, 6, 1501882 CrossRef.
- B. Anothumakkool, B. Soni, S. N. Bhange and S. Kurungot, Energy Environ. Sci., 2015, 8, 1339 RSC.
- L. Dong, G. Liang, C. Xu, D. Ren, J. Wang, Z. Z. Pan, B. Li, F. Kang and Q. H. Yang, J. Mater. Chem. A, 2017, 5, 19934 RSC.
- X. Zhao, A. Wang, J. Yang, G. Sun, L. Sun and T. Zhang, Chem. Mater., 2010, 22, 5463 CrossRef CAS.
- D. W. Wang, F. Li, Z. G. Chen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2008, 20, 7195 CrossRef CAS.
- D. Hlicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438 CrossRef.
- L. F. Chen, X. D. Zhang, H. W. Wang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092 CrossRef CAS PubMed.
- D. Hlicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. D. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026 CrossRef PubMed.
- J. I. Paredes and J. M. Munuera, J. Mater. Chem. A, 2017, 5, 7228 RSC.
- N. Zhang, F. Liu, S. D. Xu, F. Y. Wang, Q. Yu and L. Liu, J. Mater. Chem. A, 2017, 5, 22631 RSC.
- J. Cheng, S. Chen, D. Chen, L. Dong, J. Wang, T. Zhang, T. Jiao, B. Liu, H. Wang, J. J. Kai, D. Zhang, G. Zheng, L. Zhi, F. Kang and W. Zhang, J. Mater. Chem. A, 2018, 6, 20254 RSC.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30 CrossRef CAS.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220 CrossRef CAS PubMed.
- W. H. Li, K. Ding, D. R. Tian, M. S. Yao, B. Nath, W. H. Deng, Y. Wang and G. Xu, Adv. Funct. Mater., 2017, 27, 1702067 CrossRef.
- C. Hao, B. Yang, F. Wen, J. Xiang, L. Li, W. Wang, Z. Ben, B. Xu, Z. Zhao, Z. Liu and Y. Tian, Adv. Mater., 2016, 28, 3194 CrossRef CAS PubMed.
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2018, 8, 1702093 CrossRef.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78 CAS.
- M. Q. Zhao, C. E. Ren, Z. Ling, M. R. Lukatskaya, C. Zhang, K. L. V. Aken, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2015, 27, 339 CrossRef CAS PubMed.
- Y. Wen, T. E. Rufford, X. Chen, N. Li, M. Lyu, L. Dai and L. Wang, Nano Energy, 2017, 38, 368 CrossRef CAS.
- Z. Ling, C. E. Ren, M. Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676 CrossRef CAS PubMed.
- H. Li, Y. Hou, F. Wang, M. R. Lohe, X. Zhuang, L. Niu and X. Feng, Adv. Energy Mater., 2017, 7, 1601847 CrossRef.
- W. Bao, X. Tang, X. Guo, S. Choi, C. Wang, Y. Gogotsi and G. Wang, Joule, 2018, 2, 778 CrossRef CAS.
- M. R. Lukatskaya, S. Kota, Z. Lin, M. Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P. L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- J. B. Cook, H. S. Kim, Y. Yan, J. S. Ko, S. Robbennolt, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2016, 6, 1501937 CrossRef.
- J. B. Cook, H. S. Kim, T. C. Lin, C. H. Lai, B. Dunn and S. H. Tolbert, Adv. Energy Mater., 2017, 7, 1601283 CrossRef.
- H. Y. Lee and J. B. Goodneough, J. Solid State Chem., 1999, 144, 220 CrossRef CAS.
- S. C. Pang, M. A. Anderson and T. W. Chapman, J. Electrochem. Soc., 2000, 147, 444 CrossRef CAS.
- Y. Hu, Y. Wu and J. Wang, Adv. Mater., 2018, 30, 1802569 CrossRef PubMed.
- A. Gambou-Bosca and D. Bélanger, Electrochim. Acta, 2016, 201, 20 CrossRef CAS.
- R. B. Li, L. L. Yu, S. Li, J. Fan, R. Luo and J. T. Zhao, Electrochim. Acta, 2018, 284, 52 CrossRef CAS.
- G. Yu, L. Hu, N. Liu, H. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. Bao, Nano Lett., 2011, 11, 4438 CrossRef CAS PubMed.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174 CrossRef CAS PubMed.
- Z. Li, J. Wang, S. Liu, X. Liu and S. Yang, J. Power Sources, 2011, 196, 8160 CrossRef CAS.
- T. Gu and B. Wei, J. Mater. Chem. A, 2016, 4, 12289 RSC.
- C. Choi, H. J. Sim, G. M. Spinks, X. Lepró, R. H. Baughman and S. J. Kim, Adv. Energy Mater., 2016, 6, 1502119 CrossRef.
- T. H. Lee, D. T. Pham, R. Sahoo, J. Seok, T. H. T. Luu and Y. H. Lee, Energy Storage Materials, 2018, 12, 223 CrossRef.
- T. Zhai, S. Xie, M. Yu, P. Fang, C. Liang, X. Lu and Y. Tong, Nano Energy, 2014, 8, 255 CrossRef CAS.
- Y. Fu, X. Gao, D. Zha, J. Zhu, X. Ouyang and X. Wang, J. Mater. Chem. A, 2018, 6, 1601 RSC.
- T. Zhai, X. Lu, F. Wang, H. Xia and Y. Tong, Nanoscale Horiz., 2016, 1, 109 RSC.
- Y. Huang, Y. Li, Z. Hu, G. Wei, J. Guo and J. Liu, J. Mater. Chem. A, 2013, 1, 9809 RSC.
- S. Bhadra, N. K. Singha and D. Khastgir, J. Appl. Polym. Sci., 2007, 104, 1900 CrossRef CAS.
- Y. Cao and T. E. Mallouk, Chem. Mater., 2008, 20, 5260 CrossRef CAS.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397 RSC.
- J. H. Park and O. O. Park, J. Power Sources, 2002, 111, 185 CrossRef CAS.
- X. Li, H. Zhang, G. Wang and Z. Jiang, J. Mater. Chem., 2010, 20, 10598 RSC.
- X. Wang, J. Deng, X. Duan, D. Liu, J. Guo and P. Liu, J. Mater. Chem. A, 2014, 2, 12323 RSC.
- K. Wang, L. Li, Y. Liu, C. Zhang and T. Liu, Adv. Mater. Interfaces, 2016, 3, 1600665 CrossRef.
- X. Liu, N. Wen, X. Wang and Y. Zheng, ACS Sustainable Chem. Eng., 2015, 3, 475 CrossRef CAS.
- F. Xiao, S. Yang, Z. Zhang, H. Liu, J. Xiao, L. Wan, J. Luo, S. Wang and Y. Liu, Sci. Rep., 2015, 5, 9359 CrossRef CAS PubMed.
- W. Fan, C. Zhang, W. W. Tjiu, K. P. Pramoda, C. He and T. Lu, ACS Appl. Mater. Interfaces, 2013, 5, 3382 CrossRef CAS PubMed.
- J. Li, W. Lu, Y. Yan and T. W. Chou, J. Mater. Chem. A, 2017, 5, 11271 RSC.
- E. H. Jo, H. D. Jang, H. Chang, S. K. Kim, J. H. Choi and C. M. Lee, ChemSusChem, 2017, 10, 2210 CrossRef CAS PubMed.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752 CrossRef CAS PubMed.
- S. Dai, Z. Liu, B. Zhao, J. Zeng, H. Hu, Q. Zhang, D. Chen, C. Qu, D. Dang and M. Liu, J. Power Sources, 2018, 387, 43 CrossRef CAS.
- Z. J. Han, C. Huang, S. S. Meysami, D. Piche, D. H. Seo, S. Pineda, A. T. Murdock, P. S. Bruce, P. S. Grant and N. Grobert, Carbon, 2018, 126, 305 CrossRef CAS.
- L. F. Chen, Y. Lu, L. Yu and X. W. Lou, Energy Environ. Sci., 2017, 10, 1777 RSC.
- D. Hulicova, J. Yamashita, Y. Soneda, H. Hatori and M. Kodama, Chem. Mater., 2005, 17, 1241 CrossRef CAS.
- D. Hulicova, M. Kodama and H. Hatori, Chem. Mater., 2006, 18, 2318 CrossRef CAS.
- G. Lota, K. Lota and E. Frackowiak, Electrochem. Commun., 2007, 9, 1828 CrossRef CAS.
- C. O. Ania, V. Khomenko, E. Raymundo-Piñero, J. B. Parra and F. Béguin, Adv. Funct. Mater., 2007, 17, 1828 CrossRef CAS.
- E. Frackowiak, G. Lota, J. Machnikowski, C. Vix-Guterl and F. Béguin, Electrochim. Acta, 2006, 51, 2209 CrossRef CAS.
- C. Zhang, B. Anasori, A. Seral-Ascaso, S. H. Park, N. McEvoy, A. Shmeliov, G. S. Duesberg, J. N. Coleman, Y. Gogotsi and V. Nicolosi, Adv. Mater., 2017, 29, 1702678 CrossRef PubMed.
- M. D. Levi, M. R. Lukatskaya, S. Sigalov, M. Beidaghi, N. Shpigel, L. Daikhin, D. Aurbach, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2015, 5, 1400815 CrossRef.
- X. Xie, M. Q. Zhao, B. Anasori, K. Maleski, C. E. Ren, J. Li, B. W. Byles, E. Pomerankseva, G. Wang and Y. Gogotsi, Nano Energy, 2016, 26, 513 CrossRef CAS.
- Y. Dall'Agnese, P. Rozier, P. L. Taberna, Y. Gogotsi and P. Simon, J. Power Sources, 2016, 306, 510 CrossRef.
- P. Yu, G. Cao, S. Yi, X. Zhang, C. Li, X. Sun, K. Wang and Y. Ma, Nanoscale, 2018, 10, 5906 RSC.
- J. Yan, C. E. Ren, K. Maleski, C. B. Hatter, B. Anasori, P. Urbankowski, A. Sarycheva and Y. Gogotsi, Adv. Funct. Mater., 2017, 27, 1701264 CrossRef.
- J. Fu, J. Yun, S. Wu, L. Li, L. Yu and K. H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 34212 CrossRef CAS PubMed.
- A. VahidMohammadi, J. Moncada, H. Chen, E. Kayali, J. Orangi, C. A. Carrero and M. Beidaghi, J. Mater. Chem. A, 2018, 6, 22123 RSC.
- X. Lu, J. Zhu, W. Wu and B. Zhang, Electrochim. Acta, 2017, 228, 282 CrossRef CAS.
- C. Chen, M. Boota, X. Xie, M. Zhao, B. Anasori, C. E. Ren, L. Miao, J. Jiang and Y. Gogotsi, J. Mater. Chem. A, 2017, 5, 5260 RSC.
- M. Zhu, Y. Huang, Q. Deng, J. Zhou, Z. Pei, Q. Xue, Y. Huang, Z. Wang, H. Li, Q. Huang and C. Zhi, Adv. Energy Mater., 2016, 6, 1600969 CrossRef.
- R. Zhao, M. Wang, D. Zhao, H. Li, C. Wang and L. Yin, ACS Energy Lett., 2018, 3, 132 CrossRef CAS.
- R. B. Rakhi, B. Ahmed, D. Anjum and H. N. Alshareef, ACS Appl. Mater. Interfaces, 2016, 8, 18806 CrossRef CAS PubMed.
- H. Jiang, Z. Wang, Q. Yang, M. Hanif, Z. Wang, L. Dong and M. Dong, Electrochim. Acta, 2018, 290, 695 CrossRef CAS.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, J. Jo, S. Kim, V. Mathew and J. Kim, J. Power Sources, 2015, 288, 320 CrossRef CAS.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121 CrossRef CAS.
- B. Lee, H. R. Lee, H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, Chem. Commun., 2015, 51, 9265 RSC.
- B. Jiang, C. Xu, C. Wu, L. Dong, J. Li and F. Kang, Electrochim. Acta, 2017, 229, 422 CrossRef CAS.
- J. Hao, J. Mou, J. Zhang, L. Dong, W. Liu, C. Xu and F. Kang, Electrochim. Acta, 2018, 259, 170 CrossRef CAS.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao, K. T. Mueller, L. Mai, J. Liu and J. Yang, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed.
- P. He, Y. Quan, X. Xu, M. Yan, W. Yang, Q. An, L. He and L. Mai, Small, 2017, 13, 1702551 CrossRef PubMed.
- J. H. Jo, Y. K. Sun and S. T. Myung, J. Mater. Chem. A, 2017, 5, 8367 RSC.
- H. Qin, Z. Yang, L. Chen, X. Chen and L. Wang, J. Mater. Chem. A, 2018, 6, 23757 RSC.
- X. Dai, F. Wan, L. Zhang, H. Cao and Z. Niu, Energy Storage Materials, 2019, 17, 143 CrossRef.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y. K. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 15530 RSC.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- R. Trócoli and F. L. Mantia, ChemSusChem, 2015, 8, 481 CrossRef PubMed.
- T. Gupta, A. Kim, S. Phadke, S. Biswas, T. Luong, B. J. Hertzberg, M. Chamoun, K. Evans-Lutterodt and D. A. Steingart, J. Power Sources, 2016, 305, 22 CrossRef CAS.
- Z. Jia, B. Wang and Y. Wang, Mater. Chem. Phys., 2015, 149–150, 601 CrossRef CAS.
- Z. Liu, P. Bertram and F. Endres, J. Solid State Electrochem., 2017, 21, 2021 CrossRef CAS.
- W. Xu, C. Sun, K. Zhao, X. Cheng, S. Rawal, Y. Xu and Y. Wang, Energy Storage Materials, 2019, 16, 527 CrossRef.
- Y. Zeng, Z. Lai, Y. Han, H. Zhang, S. Xie and X. Lu, Adv. Mater., 2018, 30, 1802396 CrossRef PubMed.
- F. Wang, E. Hu, W. Sun, T. Gao, X. Ji, X. Fan, F. Han, X. Q. Yang, K. Xu and C. Wang, Energy Environ. Sci., 2018, 11, 3168 RSC.
- W. Liu, J. Hao, C. Xu, J. Mou, L. Dong, F. Jiang, Z. Kang, J. Wu, B. Jiang and F. Kang, Chem. Commun., 2017, 53, 6872 RSC.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894 CrossRef CAS PubMed.
- D. Chao, C. Zhu, M. Song, P. Liang, X. Zhang, N. H. Tiep, H. Zhao, J. Wang, R. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1803181 CrossRef PubMed.
- Y. Cai, F. Liu, Z. Luo, G. Fang, J. Zhou, A. Pan and S. Liang, Energy Storage Materials, 2018, 13, 168 CrossRef.
- X. Guo, G. Fang, W. Zhang, J. Zhou, L. Shan, L. Wang, C. Wang, T. Lin, Y. Tang and S. Liang, Adv. Energy Mater., 2018, 8, 1801819 CrossRef.
- B. Tang, G. Fang, J. Zhou, L. Wang, Y. Lei, C. Wang, T. Lin, Y. Tang and S. Liang, Nano Energy, 2018, 51, 579 CrossRef CAS.
- J. Zhou, L. Shan, Z. Wu, X. Guo, G. Fang and S. Liang, Chem. Commun., 2018, 54, 4457 RSC.
- J. Ding, Z. Du, L. Gu, B. Li, L. Wang, S. Wang, Y. Gong and S. Yang, Adv. Mater., 2018, 30, 1800762 CrossRef PubMed.
- N. Zhang, M. Jia, Y. Dong, Y. Wang, J. Xu, Y. Liu, L. Jiao and F. Cheng, Adv. Funct. Mater., 2019, 29, 1807331 CrossRef.
- L. Dong, X. Ma, Y. Li, L. Zhao, W. Liu, J. Cheng, C. Xu, B. Li, Q. H. Yang and F. Kang, Energy Storage Materials, 2018, 13, 96 CrossRef.
- Z. Kang, C. Wu, L. Dong, W. Liu, J. Mou, J. Zhang, Z. Chang, B. Jiang, G. Wang, F. Kang and C. Xu, ACS Sustainable Chem. Eng., 2019, 7, 3364 CrossRef CAS.
- C. Shen, X. Li, N. Li, K. Xie, J. G. Wang, X. Liu and B. Wei, ACS Appl. Mater. Interfaces, 2018, 10, 25446 CrossRef CAS PubMed.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 1801090 CrossRef.
- G. H. An, S. Cha and J. I. Sohn, Appl. Surf. Sci., 2019, 467–468, 1157 CrossRef CAS.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724 CrossRef CAS PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265 RSC.
- D. Aurbach, Y. Cohen and M. Moshkovich, Electrochem. Solid-State Lett., 2001, 4, A113 CrossRef CAS.
- C. Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 2012, 76, 270 CrossRef CAS.
- M. M. Huie, D. C. Bock, E. S. Takeuchi, A. C. Marschilok and K. J. Takeuchi, Coord. Chem. Rev., 2015, 287, 15 CrossRef CAS.
- P. Novák, R. Imhof and O. Haas, Electrochim. Acta, 1999, 45, 351 CrossRef.
- D. Lv, T. Xu, P. Saha, M. K. Datta, M. L. Gordin, A. Manivannan, P. N. Kumta and D. Wang, J. Electrochem. Soc., 2013, 160, A351 CrossRef CAS.
- E. Levi, A. Mitelman, D. Aurbach and M. Brunelli, Chem. Mater., 2007, 19, 5131 CrossRef CAS.
- E. Levi, G. Gershinsky, D. Aurbach, O. Isnard and G. Ceder, Chem. Mater., 2009, 21, 1390 CrossRef CAS.
- E. Levi, E. Lancry, A. Mitelman, D. Aurbach, G. Ceder, D. Morgan and O. Isnard, Chem. Mater., 2006, 18, 5492 CrossRef CAS.
- M. D. Levi, E. Lancry, H. Gizbar, Z. Lu, E. Levi, Y. Gofer and D. Aurbach, J. Electrochem. Soc., 2004, 151, A1044 CrossRef CAS.
- T. Kaewmaraya, M. Ramzan, J. M. Osorio-Guillén and R. Ahuja, Solid State Ionics, 2014, 261, 17 CrossRef CAS.
- L. F. Wan, B. R. Perdue, C. A. Apblett and D. Prendergast, Chem. Mater., 2015, 27, 5932 CrossRef CAS.
- Y. Liang, R. Feng, S. Yang, H. Ma, J. Liang and J. Chen, Adv. Mater., 2011, 23, 640 CrossRef CAS PubMed.
- X.-L. Li and Y.-D. Li, J. Phys. Chem. B, 2004, 108, 13893 CrossRef CAS.
- S. Yang, D. Li, T. Zhang, Z. Tao and J. Chen, J. Phys. Chem. C, 2012, 116, 1307 CrossRef CAS.
- Y. Liu, L. Jiao, Q. Wu, Y. Zhao, K. Cao, H. Liu, Y. Wang and H. Yuan, Nanoscale, 2013, 5, 9562 RSC.
- Z. L. Tao, L. N. Xu, X. L. Gou, J. Chen and H. T. Yuan, Chem. Commun., 2004, 2080 RSC.
- X. Sun, P. Bonnick and L. F. Nazar, ACS Energy Lett., 2016, 1, 297 CrossRef CAS.
- N. Amir, Y. Vestfrid, O. Chusid, Y. Gofer and D. Aurbach, J. Power Sources, 2007, 174, 1234 CrossRef CAS.
- W. Yuan and J. R. Günter, Solid State Ionics, 1995, 76, 253 CrossRef CAS.
- D. He, D. Wu, J. Gao, X. Wu, X. Zeng and W. Ding, J. Power Sources, 2015, 294, 643 CrossRef CAS.
- L. Yu and X. Zhang, J. Colloid Interface Sci., 2004, 278, 160 CrossRef CAS PubMed.
- V. Shklover, T. Haibach, F. Ried, R. Nesper and P. Novák, J. Solid State Chem., 1996, 123, 317 CrossRef CAS.
- L. Jiao, H. Yuan, Y. Wang, J. Cao and Y. Wang, Electrochem. Commun., 2005, 7, 431 CrossRef CAS.
- L. F. Jiao, H.-T. Yuan, Y. C. Si, Y. J. Wang and Y. M. Wang, Electrochem. Commun., 2006, 8, 1041 CrossRef CAS.
- Y. Cheng, Y. Shao, V. Raju, X. Ji, B. L. Mehdi, K. S. Han, M. H. Engelhard, G. Li, N. D. Browning, K. T. Mueller and J. Liu, Adv. Funct. Mater., 2016, 26, 3446 CrossRef CAS.
- G. Sai Gautam, P. Canepa, W. D. Richards, R. Malik and G. Ceder, Nano Lett., 2016, 16, 2426 CrossRef CAS PubMed.
- R. Zhang, X. Yu, K. W. Nam, C. Ling, T. S. Arthur, W. Song, A. M. Knapp, S. N. Ehrlich, X. Q. Yang and M. Matsui, Electrochem. Commun., 2012, 23, 110 CrossRef CAS.
- N. Kumagai, S. Komaba, H. Sakai and N. Kumagai, J. Power Sources, 2001, 97–98, 515 CrossRef CAS.
- C. Ling, R. Zhang and F. Mizuno, ACS Appl. Mater. Interfaces, 2016, 8, 4508 CrossRef CAS PubMed.
- K. W. Nam, S. Kim, S. Lee, M. Salama, I. Shterenberg, Y. Gofer, J. S. Kim, E. Yang, C. S. Park, J. S. Kim, S. S. Lee, W. S. Chang, S. G. Doo, Y. N. Jo, Y. Jung, D. Aurbach and J. W. Choi, Nano Lett., 2015, 15, 4071 CrossRef CAS PubMed.
- J. Huang, A. S. Poyraz, K. J. Takeuchi, E. S. Takeuchi and A. C. Marschilok, Chem. Commun., 2016, 52, 4088 RSC.
- R. E. Dueber, J. M. Fleetwood and P. G. Dickens, Solid State Ionics, 1992, 50, 329 CrossRef CAS.
- T. E. Sutto and T. T. Duncan, Electrochim. Acta, 2012, 80, 413 CrossRef CAS.
- E. Sheha, Graphene, 2014, 3, 36 CrossRef.
- S. Su, Z. Huang, Y. NuLi, F. Tuerxun, J. Yang and J. Wang, Chem. Commun., 2015, 51, 2641 RSC.
- K. Makino, Y. Katayama, T. Miura and T. Kishi, J. Power Sources, 2001, 99, 66 CrossRef CAS.
- Z. D. Huang, T. Masese, Y. Orikasa, T. Mori, T. Minato, C. Tassel, Y. Kobayashi, H. Kageyama and Y. Uchimoto, J. Mater. Chem. A, 2014, 2, 11578 RSC.
- Z. Feng, J. Yang, Y. NuLi, J. Wang, X. Wang and Z. Wang, Electrochem. Commun., 2008, 10, 1291 CrossRef CAS.
- Y. Zheng, Y. NuLi, Q. Chen, Y. Wang, J. Yang and J. Wang, Electrochim. Acta, 2012, 66, 75 CrossRef CAS.
- Y. NuLi, Z. Guo, H. Liu and J. Yang, Electrochem. Commun., 2007, 9, 1913 CrossRef CAS.
- H. Sano, H. Senoh, M. Yao, H. Sakaebe and T. Kiyobayashi, Chem. Lett., 2012, 41, 1594 CrossRef CAS.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748 CrossRef CAS PubMed.
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, K. Oh-ishi, A. Okazawa, N. Kojima, R. Kurono, S. Nishimura and A. Yamada, J. Mater. Chem. A, 2013, 1, 13055 RSC.
- R. Zhang, C. Ling and F. Mizuno, Chem. Commun., 2015, 51, 1487 RSC.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, J. Electroanal. Chem., 1999, 466, 203 CrossRef CAS.
- Y. Shao, M. Gu, X. Li, Z. Nie, P. Zuo, G. Li, T. Liu, J. Xiao, Y. Cheng, C. Wang, J. G. Zhang and J. Liu, Nano Lett., 2014, 14, 255 CrossRef CAS PubMed.
- Z. Liu, J. Lee, G. Xiang, H. F. J. Glass, E. N. Keyzer, S. E. Dutton and C. P. Grey, Chem. Commun., 2017, 53, 743 RSC.
- N. Singh, T. S. Arthur, C. Ling, M. Matsui and F. Mizuno, Chem. Commun., 2013, 49, 149 RSC.
- W. Jin, Z. Li, Z. Wang and Y. Q. Fu, Mater. Chem. Phys., 2016, 182, 167 CrossRef CAS.
- D. Er, E. Detsi, H. Kumar and V. B. Shenoy, ACS Energy Lett., 2016, 1, 638 CrossRef CAS.
- W. Jin, Z. Wang and Y. Q. Fu, J. Mater. Sci., 2016, 51, 7355 CrossRef CAS.
- M. P. Siegal, W. G. Yelton, B. R. Perdue, D. F. S. Gallis and H. L. Schwarz, J. Electrochem. Soc., 2016, 163, A1030 CrossRef CAS.
- N. Wu, Y. C. Lyu, R. J. Xiao, X. Yu, Y. X. Yin, X. Q. Yang, H. Li, L. Gu and Y. G. Guo, NPG Asia Mater., 2014, 6, e120 CrossRef CAS.
- D. B. Le, S. Passerini, J. Guo, J. Ressler, B. B. Owens and W. H. Smyrl, J. Electrochem. Soc., 1996, 143, 2099 CrossRef CAS.
- D. Aurbach, R. Skaletsky and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3536 CrossRef CAS.
- P. Padigi, G. Goncher, D. Evans and R. Solanki, J. Power Sources, 2015, 273, 460 CrossRef CAS.
- T. Ouchi, H. Kim, B. L. Spatocco and D. R. Sadoway, Nat. Commun., 2016, 7, 10999 CrossRef CAS PubMed.
- M. E. A. Dompablo, C. Krich, J. Nava-Avendaño, N. Biškup, M. R. Palacín and F. Bardé, Chem. Mater., 2016, 28, 6886 CrossRef CAS.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964 RSC.
- A. Parija, D. Prendergast and S. Banerjee, ACS Appl. Mater. Interfaces, 2017, 9, 23756 CrossRef CAS PubMed.
- D. Wang, H. Liu, J. D. Elliott, L. M. Liu and W.-M. Lau, J. Mater. Chem. A, 2016, 4, 12516 RSC.
- M. Smeu, M. S. Hossain, Z. Wang, V. Timoshevskii, K. H. Bevan and K. Zaghib, J. Power Sources, 2016, 306, 431 CrossRef CAS.
- T. R. Juran and M. Smeu, Phys. Chem. Chem. Phys., 2017, 19, 20684 RSC.
- A. L. Lipson, S.-D. Han, S. Kim, B. Pan, N. Sa, C. Liao, T. T. Fister, A. K. Burrell, J. T. Vaughey and B. J. Ingram, J. Power Sources, 2016, 325, 646 CrossRef CAS.
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2017, 17, 16 CrossRef PubMed.
- Q. Li and N. J. Bjerrum, J. Power Sources, 2002, 110, 1 CrossRef CAS.
- G. L. Holleck, J. Electrochem. Soc., 1972, 119, 1158 CrossRef CAS.
- P. R. Gifford and J. B. Palmisano, J. Electrochem. Soc., 1988, 135, 650 CrossRef CAS.
- J. V. Rani, V. Kanakaiah, T. Dadmal, M. S. Rao and S. Bhavanarushi, J. Electrochem. Soc., 2013, 160, A1781 CrossRef CAS.
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743 RSC.
- F. Wang, F. Yu, X. Wang, Z. Chang, L. Fu, Y. Zhu, Z. Wen, Y. Wu and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 9022 CrossRef CAS PubMed.
- Y. Liu, S. Sang, Q. Wu, Z. Lu, K. Liu and H. Liu, Electrochim. Acta, 2014, 143, 340 CrossRef CAS.
- F. Ambroz, T. J. Macdonald and T. Nann, Adv. Energy Mater., 2017, 7, 1602093 CrossRef.
- Z. J. Karpinski and R. A. Osteryoung, Inorg. Chem., 1984, 23, 1491 CrossRef CAS.
- N. Takami and N. Koura, Electrochim. Acta, 1988, 33, 1137 CrossRef CAS.
- H. Sun, W. Wang, Z. Yu, Y. Yuan, S. Wang and S. Jiao, Chem. Commun., 2015, 51, 11892 RSC.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J. F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564 CrossRef CAS PubMed.
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203 CrossRef CAS.
- Y. Wu, M. Gong, M. C. Lin, C. Yuan, M. Angell, L. Huang, D. Y. Wang, X. Zhang, J. Yang, B. J. Hwang and H. Dai, Adv. Mater., 2016, 28, 9218 CrossRef CAS PubMed.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959 RSC.
- Y. J. He, J. F. Peng, W. Chu, Y. Z. Li and D. G. Tong, J. Mater. Chem. A, 2014, 2, 1721 RSC.
- L. Geng, G. Lv, X. Xing and J. Guo, Chem. Mater., 2015, 27, 4926 CrossRef CAS.
- H. Wang, Y. Bai, S. Chen, X. Luo, C. Wu, F. Wu, J. Lu and K. Amine, ACS Appl. Mater. Interfaces, 2015, 7, 80 CrossRef CAS PubMed.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- Y. S. Xu, S. Y. Duan, Y. G. Sun, D. S. Bin, X. S. Tao, D. Zhang, Y. Liu, A. M. Cao and L. J. Wan, J. Mater. Chem. A, 2019, 7, 4334 RSC.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Adv. Funct. Mater., 2018, 28, 1802938 CrossRef.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 RSC.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292 CrossRef.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Chem. Rec., 2018, 18, 459 CrossRef CAS PubMed.
- J. C. Pramudita, D. Sehrawat, D. Goonetilleke and N. Sharma, Adv. Energy Mater., 2017, 7, 1602911 CrossRef.
- F. R. McLarnon and E. J. Cairns, J. Electrochem. Soc., 1991, 138, 645 CrossRef CAS.
- A. A. Mohamad, J. Power Sources, 2006, 159, 752 CrossRef CAS.
- Y. Shen and K. Kordesch, J. Power Sources, 2000, 87, 162 CrossRef CAS.
- B. Lee, H. R. Seo, H. R. Lee, C. S. Yoon, J. H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, ChemSusChem, 2016, 9, 2948 CrossRef CAS PubMed.
- K. Jurewicz, E. Frackowiak and F. Béguin, Appl. Phys. A, 2004, 78, 981 CrossRef CAS.
- Y. Tian, R. Amal and D. W. Wang, Front. Energy Res., 2016, 4, 34 Search PubMed.
- J. Han, K. Wang, W. Liu, C. Li, X. Sun, X. Zhang, Y. An, S. Yi and Y. Ma, Nanosacle, 2018, 10, 13083 RSC.
- X. Li, X. Xie, R. Lv, B. Na, B. Wang and Y. He, Energy Technol., 2019, 1801092 CrossRef.
- H. Wang, M. Wang and Y. Tang, Energy Storage Materials, 2018, 13, 1 CrossRef CAS.
- H. D. Yoo, S. D. Han, R. D. Bayliss, A. A. Gewirth, B. Genorio, N. N. Rajput, K. A. Persson, A. K. Burrell and J. Cabana, ACS Appl. Mater. Interfaces, 2016, 8, 30853 CrossRef CAS PubMed.
- Z. Liu, G. Li, T. Cui, A. Borodin, C. Kuhl and F. Endres, J. Solid State Electrochem., 2018, 22, 91 CrossRef CAS.
- Z. Liu, G. Pulletikurthi and F. Endres, ACS Appl. Mater. Interfaces, 2016, 8, 12158 CrossRef CAS PubMed.
- R. R. Salunkhe, C. Young, J. Tang, T. Takei, Y. Ide, N. Kobayashi and Y. Yamauchi, Chem. Commun., 2016, 52, 4764 RSC.
- A. Lewandowski, A. Olejniczak, M. Galinski and I. Stepniak, J. Power Sources, 2010, 195, 5814 CrossRef CAS.
- Y. Liang, F. Liang, H. Zhong, Z. Li, R. Fu and D. Wu, J. Mater. Chem. A, 2013, 1, 7000 RSC.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872 CrossRef CAS PubMed.
- X. Ma, J. Cheng, L. Dong, W. Liu, J. Mou, L. Zhao, J. Wang, D. Ren, J. Wu, C. Xu and F. Kang, Energy Storage Materials, 2019 DOI:10.1016/j.ensm.2018.10.020.
- M. Beidaghi and Y. Gogotsi, Energy Environ. Sci., 2014, 7, 867 RSC.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480 CrossRef CAS PubMed.
- W. Gao, N. Singh, L. Song, Z. Liu, A. L. M. Reddy, L. Ci, R. Vajtai, Q. Zhang, B. Wei and P. M. Ajayan, Nat. Nanotechnol., 2011, 6, 496 CrossRef CAS PubMed.
- M. F. El-Kady and R. B. Kaner, Nat. Commun., 2013, 4, 1475 CrossRef PubMed.
- P. Huang, C. Lethien, S. Pinaud, K. Brousse, R. Laloo, V. Turq, M. Respaud, A. Demortière, B. Daffos, P. L. Taberna, B. Chaudret, Y. Gogotsi and P. Simon, Science, 2016, 351, 691 CrossRef CAS PubMed.
- Y. Da, J. Liu, L. Zhou, X. Zhu, X. Chen and L. Fu, Adv. Mater., 2019, 31, 1802793 CrossRef PubMed.
- H. Xiao, Z. S. Wu, F. Zhou, S. Zheng, D. Sui, Y. Chen and X. Bao, Energy Storage Materials, 2018, 13, 233 CrossRef.
- K. Guo, Y. Wan, N. Yu, L. Hu, T. Zhai and H. Li, Energy Storage Materials, 2018, 11, 144 CrossRef.
- Q. Li, H. Cheng, X. Wu, C. F. Wang, G. Wu and S. Chen, J. Mater. Chem. A, 2018, 6, 14112 RSC.
- G. Sun, H. Yang, G. Zhang, J. Guo, X. Jin, Y. Zhao, L. Jiang and L. Qu, Energy Environ. Sci., 2018, 11, 3367 RSC.
- P. Zhang, Y. Li, G. Wang, F. Wang, S. Yang, F. Zhu, X. Zhuang, O. G. Schmidt and X. Feng, Adv. Mater., 2018, 31, 1806005 CrossRef PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, R. E. Doe, C. C. Fischer, G. Ceder and D. Aurbach, J. Electrochem. Soc., 2014, 161, A410 CrossRef CAS.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- N. Wu, W. Yao, X. Song, G. Zhang, B. Chen, J. Yang and Y. Tang, Adv. Energy Mater., 2019, 1803865 CrossRef.
- F. Wang, Z. Liu, X. Wang, X. Yuan, X. Wu, Y. Zhu, L. Fu and Y. Wu, J. Mater. Chem. A, 2016, 4, 5115 RSC.
- K. Li, Y. Shao, S. Liu, Q. Zhang, H. Wang, Y. Li and R. B. Kaner, Small, 2017, 13, 1700380 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |