
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV light promoted ‘Metal’/‘Additive’-free oxidation of alcohols: investigating the role of alcohols as electron donors†
Preet Kamal Walia,
Manik Sharma,
Manoj Kumar and
Vandana Bhalla *
*
Department of Chemistry, UGC Sponsored Centre for Advanced Studies-II, Guru Nanak Dev University, Amritsar-143005, Punjab, India. E-mail: vanmanan@yahoo.co.in
First published on 6th November 2019
Abstract
UV light promoted selective oxidation of primary and secondary alcohols has been demonstrated under ‘metal-free’ and ‘additive-free’ conditions. Under the optimized conditions, a variety of aromatic, heteroaromatic, and alicyclic alcohols have been examined for their transformations to the corresponding carbonyl compounds. The mechanistic studies emphasize the important role of substrate (alcohol) and solvent (DMSO) in the generation of superoxide radical which is a vital intermediate for the transformation. This study also highlights the role of air as the oxidant in the oxidation process. Further, the practical application of the strategy has also been demonstrated for the oxidation of the alcoholic moiety in cholesterol.
Introduction
Aldehydes and ketones are ubiquitous building blocks for the synthesis of various natural/unnatural materials, biologically active molecules, pharmaceuticals, agrochemicals, and fine chemicals.1a–d The requirement of these derivatives as starting precursors for the preparation of important materials provide sufficient impetus to develop new approaches for the facile synthesis of these derivatives providing a demand and supply balance. Among various approaches, transition metal catalyzed controlled oxidation of alcohols is one of the best approaches for the preparation of aldehydes/ketones (Fig. 1).2a–i Though the use of transition metals enables the reaction to proceed rapidly the requirement for precious metals as catalytic centres, addition of toxic additives, expensive ligands, external oxidants (such as osmium oxide, permanganate, MnO2, hypochlorite, and O2 balloons) in stoichiometric amounts and high temperature conditions make these transformations a costly and eco-unfriendly affair.3a–g To overcome the shortcomings associated with transition metal based catalytic systems, ‘metal-free’ approaches for the oxidation of alcohols using photosensitizers have also been explored. These systems are relatively economical and non-toxic4a–d but the reaction conditions require the presence of large excesses of promoters, prolonged reaction time and high thermal conditions for achieving the final product in high yield.5a–i Recently, organic photosensitizers have been explored for carrying out oxidative organic transformations (Fig. 1).6a–g In these photocatalytic systems under irradiation, reactive oxygen species (ROS) are generated which catalyze the reaction. Very recently, the application of 9-fluorenone has been demonstrated as a photocatalyst for the aerobic oxidation of alcohols under visible light.6b Aerobic oxidations are deemed benign from the ‘green’ chemistry point of view as they employ inexpensive and abundant air as a ‘green oxidant’, thus, avoiding the requirement of hazardous and toxic oxidants and byproducts. Though the reported strategy is good but the use of organic materials as photosensitizers leave enough scope for further improvement for making the reaction more ‘green’.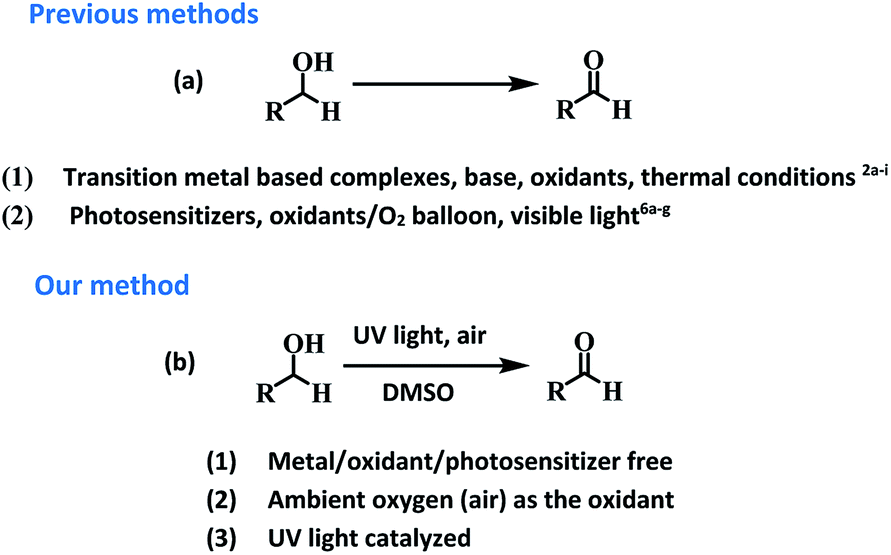 | ||
| Fig. 1 (a) Previous methods for the oxidation of alcohols to aldehydes. (b) Photosensitizer free method for synthesizing aldehydes using ambient oxygen (air) as the oxidant in the presence of UV light. | ||
Long back, Hergenrother et al.7 reported aerobic oxidation of alcohols to aldehydes by passing a stream of air at 190 °C for 4–48 h using DMSO as a solvent. The study highlighted the important role of DMSO in the oxidation process. The strategy did not work well for the preparation of secondary alcohols and the hard reaction conditions such as use of stream of air, prolonged heating at 190 °C (4–48 h) restricted the real time application of the approach. Both the above studies motivated us to examine the possibility of photopromoted, ROS catalyzed oxidation of alcohols under metal/additive/photosensitizer free conditions. To examine the validity of the idea, we chose benzyl alcohol as the model reactant and examined its photopromoted air oxidation in DMSO. Gratifyingly, the reaction worked well and after 66 h, the final product was obtained in 66% yield. Interestingly, photopromoted air oxidation also worked well in case of secondary alcohols. The mechanistic studies clearly indicate the important role of substrate (alcohol) in generation of superoxide radical anion. To the best of our knowledge, there is no report in literature regarding oxidative transformation of alcohols to carbonyl compounds without using any metal based catalytic system or photosensitizer. The work being presented in this manuscript highlights the importance of UV light as irradiation source, air as oxidant, DMSO as solvent and potential of substrate (primary/secondary alcohols) to act as electron donor for air oxidation of a variety of primary and secondary alcohols.
Results and discussion
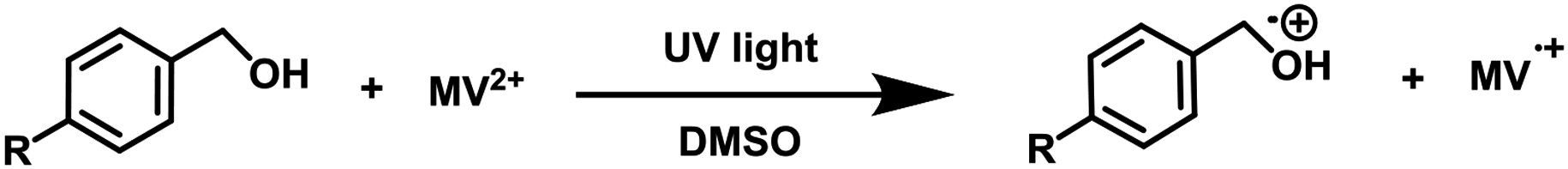
To examine the photopromoted oxidation of alcohols, we chose oxidation of benzyl alcohol as the model reaction. First of all, we determined the potential of benzyl alcohol to oxidize air by carrying out cyclic voltammetric studies. Reduction potential of benzyl alcohol in DMSO was found to be −0.78 V (Fig. S1, ESI†) vs. SCE which is sufficient for the reduction of molecular oxygen to its superoxide radical form (O2/˙O2−).6a,8a,b Next, we examined the capacity of benzyl alcohol to serve as electron donor in DMSO using methyl viologen dichloride as electron acceptor.9a,b We irradiated benzyl alcohol under UV lamp in presence of methyl viologen (MV2+) using DMSO as solvent under inert conditions. After 14 h, the color of the reaction mixture was changed from colorless to dark blue. The absorption spectrum of the blue colored reaction mixture showed the presence of absorption bands at 395 and 603 nm (Fig. 2) corresponding to the reduced viologen species (MV˙+) (Scheme 1).10a,b Thus, the absorption studies of benzyl alcohol in presence of methyl viologen confirmed the potential of benzyl alcohol to act as electron donor.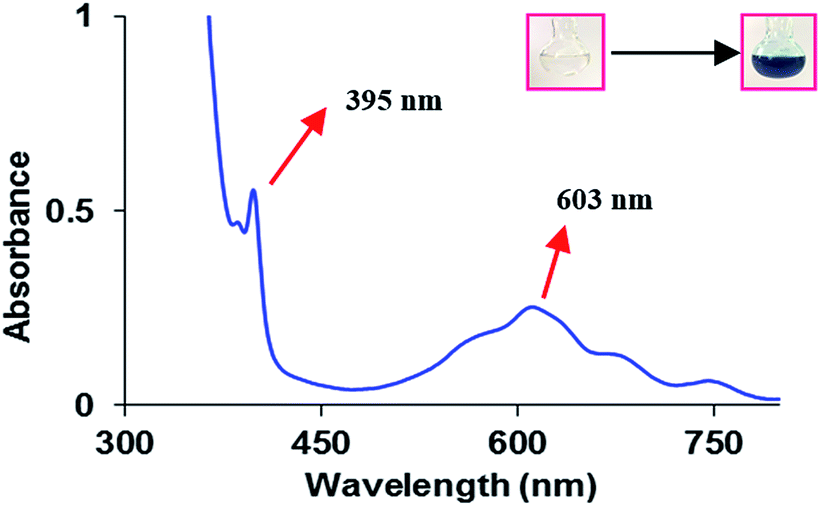 | ||
| Fig. 2 UV-Vis Spectra of reduced form of methyl viologen (MV˙+) generated by the reaction of benzyl alcohol (0.01 mM) and methyl viologen dichloride (MV2+) (0.2 mM) in DMSO under UV light irradiation. Inset: showing the change in color of reaction mixture after 14 h. | ||
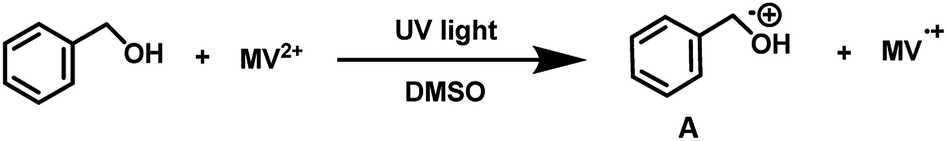 | ||
| Scheme 1 Reduction of methyl viologen (MV2+) to its reduced species (MV˙+) by benzyl alcohol. | ||
Encouraged by these results, we examined the oxidation of benzyl alcohol 1a under aerial conditions using UV light as source of irradiation in DMSO as solvent. To our pleasure, the target product 2a was attained in good yield (66%) (Table 1, entry 1). Notably, ambient air was used as the oxidant and oxygen source under mild conditions in this UV light promoted reaction. We also monitored the reaction for another 10 h (i.e. 76 h) but no significant change in the reaction was observed (TLC). On switching the solvent to DMF, ACN, H2O and toluene, yield of the product was drastically affected (Table 1, entries 2–5). Additionally, the reaction did not proceed under nitrogen atmosphere which clearly suggest the importance of aerial conditions (Table 1, entry 6 and Scheme 2). Furthermore, no transformation was observed under visible light irradiation and under dark conditions (Table 1, entries 7 and 8 and Scheme 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Light source | Solventb | Atmosphere | Time | Yield (%) |
| a Reaction conditions: benzyl alcohol (1 mmol), solvent (4 mL), air, 30 °C.b DMSO = dimethyl sulfoxide; H2O = water; ACN = acetonitrile; and DMF = N,N-dimethylformamide. | |||||
| 1 | UV light | DMSO | Ambient oxygen | 66 h | 66% |
| 2 | UV light | H2O | Ambient oxygen | 66 h | <10% |
| 3 | UV light | ACN | Ambient oxygen | 66 h | Negligible |
| 4 | UV light | DMF | Ambient oxygen | 66 h | Negligible |
| 5 | UV light | Toluene | Ambient oxygen | 66 h | Negligible |
| 6 | UV light | DMSO | Nitrogen | 66 h | No reaction |
| 7 | Visible light | DMSO | Ambient oxygen | 66 h | No reaction |
| 8 | No UV light | DMSO | Ambient oxygen | 66 h | No reaction |
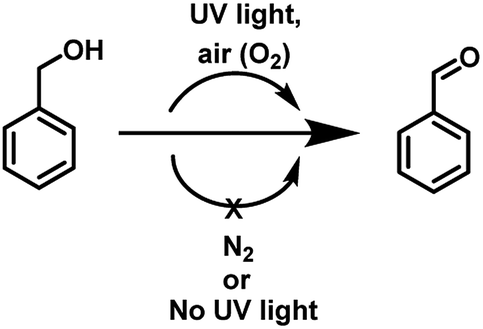 | ||
| Scheme 2 Conversion of benzyl alcohol to benzaldehyde under different conditions. | ||
With optimized reaction conditions in hand, the substrate scope for aerobic oxidation of alcohols in DMSO was surveyed. Substituted benzyl alcohols bearing electron-donating groups (Scheme 3, entries 2b–2d) showed better activity in comparison with those containing electron-withdrawing groups (Scheme 3, entries 2e and 2f). The reaction efficiency was not affected by the presence of bulky group at para-position (Scheme 3, entry 2i). Several alcohols including aromatic, alicyclic and heteroaromatic rings were smoothly oxidized and furnished the final compounds in good to moderate yields (Scheme 3, entries 2h, 2o and 2q). However, the alcohols having strong electron-withdrawing group (4-nitrobenzyl alcohol), heteroaromatic (furfuryl alcohol) and aliphatic (5-hexen-1-ol) alcohol failed to give the desired product (Scheme 3, entries 2g, 2m and 2n).
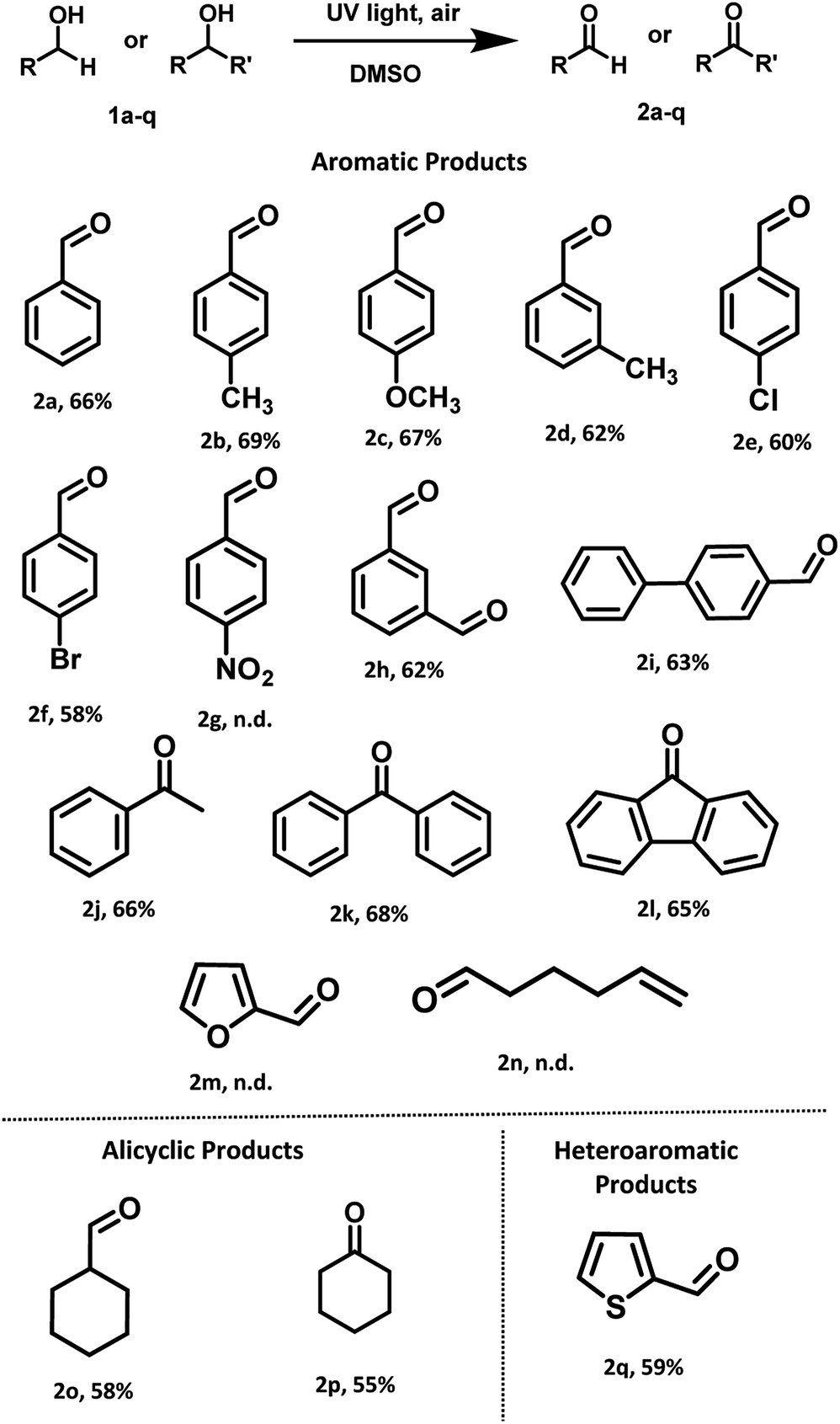 | ||
| Scheme 3 General substrate scope for the oxidations of primary and secondary alcohols under UV light (254 nm) irradiation. | ||
To get deep insight into the mechanism, we checked the electron transport abilities of 4-methylbenzyl alcohol, 4-methoxybenzyl alcohol, 4-bromobenzyl alcohol, 4-chlorobenzyl alcohol and 4-nitrobenzyl alcohol (see Table 2). We carried out the reaction of respective alcohol in presence of methyl viologen (MV2+) in DMSO under UV light irradiation under inert conditions. The color of the reaction mixture in case of these alcohols except for the 4-nitrobenzyl alcohol changed from colorless to dark blue in 0.5–16 h, thus, indicating the formation of (MV˙+) (Table 2, entries 1–6). The reactions were also monitored by absorption studies which confirmed the reduction of methyl viologen (MV2+) to MV˙+ (Fig. S2–S5, ESI†). These studies suggest the low potential of 4-nitrobenzyl alcohol to transport electrons which may be attributed to the presence of stronger electron withdrawing (nitro) group. To rule out the role of DMSO as electron donor, we also carried out the blank experiment in which methyl viologen (MV2+) was added to DMSO and the solution was kept under UV light irradiation under inert conditions. Even after 16 h, there was no color change in the reaction mixture (Table 2, entry 7), which supports our assumption that DMSO is not an electron donor in this case. On the basis of these studies, we propose that, inability of the electron deficient alcohols to transfer electrons is the reason behind their unsuccessful oxidation to carbonyl compounds.
|
||||
|---|---|---|---|---|
| Entry | Alcohol | Solvent | Time taken to reduce MV2+ to MV˙+ | Color change |
| 1 | Benzyl alcohol (R = H) | DMSO | 14 h | Yes |
| 2 | 4-Methylbenzyl alcohol (R = CH3) | DMSO | 0.5 h | Yes |
| 3 | 4-Methoxybenzyl alcohol (R = OCH3) | DMSO | 12 h | Yes |
| 4 | 4-Bromobenzyl alcohol (R = Br) | DMSO | 16 h | Yes |
| 5 | 4-Chlorobenzyl alcohol (R = Cl) | DMSO | 16 h | Yes |
| 6 | 4-Nitrobenzyl alcohol (R = NO2) | DMSO | 24 h | No |
| 7 | None | DMSO | 16 h | No |
After successful investigation of oxidation of different primary alcohols, we examined the UV light promoted oxidation of unactivated secondary alcohols. To our delight, different secondary alcohols showed excellent reactivity under the optimized reaction conditions. In fact, the desired oxidized products were conveniently isolated for both the internal and terminal secondary alcohols, in moderate yields (Scheme 3, entries 2j–2l). Notably, unactivated cyclic alcohol was also oxidized to the corresponding ketone in moderate yield (Scheme 3, entry 2p).
Furthermore, we also examined the UV light promoted oxidation of more complex steroid i.e. (cholesterol). In case of cholesterol, to our delight, the formation of the desired ketone was observed in 64% yield (72 h) (Scheme 4). To the best of our knowledge, currently there is no ‘metal-free’ approach known for the oxidation of cholesterol using air as the oxidant.
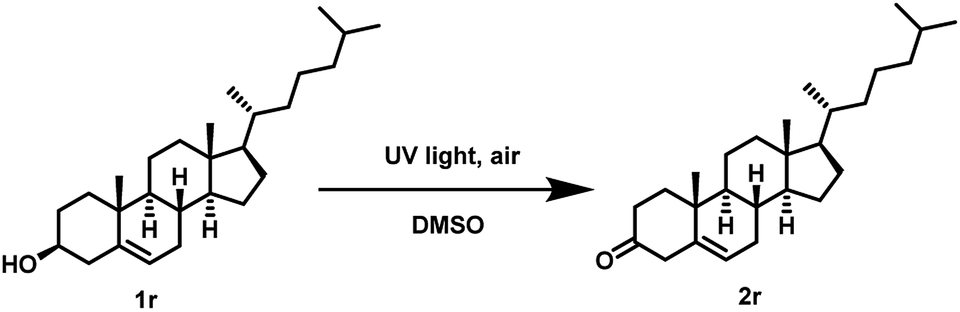 | ||
| Scheme 4 Oxidation of alcoholic moiety in cholesterol. | ||
After the evaluation of substrate scope, we carried out several control experiments to understand the mechanism of the reaction (see Table 3). First of all, we carried out the model reaction in the presence of TEMPO, however, the reaction did not progress indicating the radical mechanism (entries 1 and 2, Table 3). We were also successful in isolating the para-adduct between 4-methylbenzylalcohol and TEMPO whose structure was confirmed from ESI-MS studies (Fig. S22, ESI†). Similarly, addition of BHT afforded the desired product in negligible yield (entries 3 and 4, Table 3). The model reaction showed a slight decrease in the yield in the presence of tertiary butanol, ruling out the presence of hydroxide radical as reactive species (entry 5, Table 3). Further addition of CuCl2 in the reaction exhibited lower yield that clearly suggested the involvement of a single electron in the reaction process (entry 6, Table 3). However, using benzoquinone as quencher, presence of superoxide radical anion in the reaction system was revealed (entry 7, Table 3). We propose that under UV light irradiation, substrate itself reduces the air to generate the superoxide radical anion which is responsible for the formation of target product. Further the presence of H2O2 in the reaction mixture was detected by adding KI/CH3COOH to the reaction mixture. The color of the reaction mixture was changed from colorless to brown (Fig. S6, ESI†).11a,b To elaborate the mechanism, we examined the model reaction in which H2O2 is introduced externally, however, no significant change in reaction in terms of rate/yield of final product was observed. This study ruled out the participation of H2O2 in the oxidative transformation.
|
||||
|---|---|---|---|---|
| Entry | Quenchera | Equivalent | Yield | Notes |
| a TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl; BHT = butylated hydroxytoluene. | ||||
| 1 | TEMPO | 0.5 | 11 | Radical scav. |
| 2 | TEMPO | 1.0 | <5% | Radical scav. |
| 3 | BHT | 0.5 | 12 | Radical scav. |
| 4 | BHT | 1.0 | <5% | Radical scav. |
| 5 | tert-Butanol | 1.0 | 54% | Hydroxide radical scav. |
| 6 | CuCl2 | 1.0 | <5% | Electron scav. |
| 7 | Benzoquinone | 1.0 | <5% | Superoxide radical anion scav. |

Taking into account all the experimental evidences, we propose that under UV light irradiation, benzyl alcohol in the presence of DMSO is transformed to the corresponding radical cation (A) (vide supra) with simultaneous reduction of O2 (from air) to generate oxidant superoxide radical anion. The radical cation (A) then reacted with the superoxide anion to generate peroxide radical which abstract hydrogen atom from intermediate B to furnish the desired product (Scheme 5). We believe that DMSO2 (ref. 6b) is generated as byproduct in the reaction sequence which is generated after the oxidation of DMSO by the in situ-generated H2O2. The byproduct DMSO2 is removed during the aqueous workup of the reaction mixture (Fig. S23, ESI†).
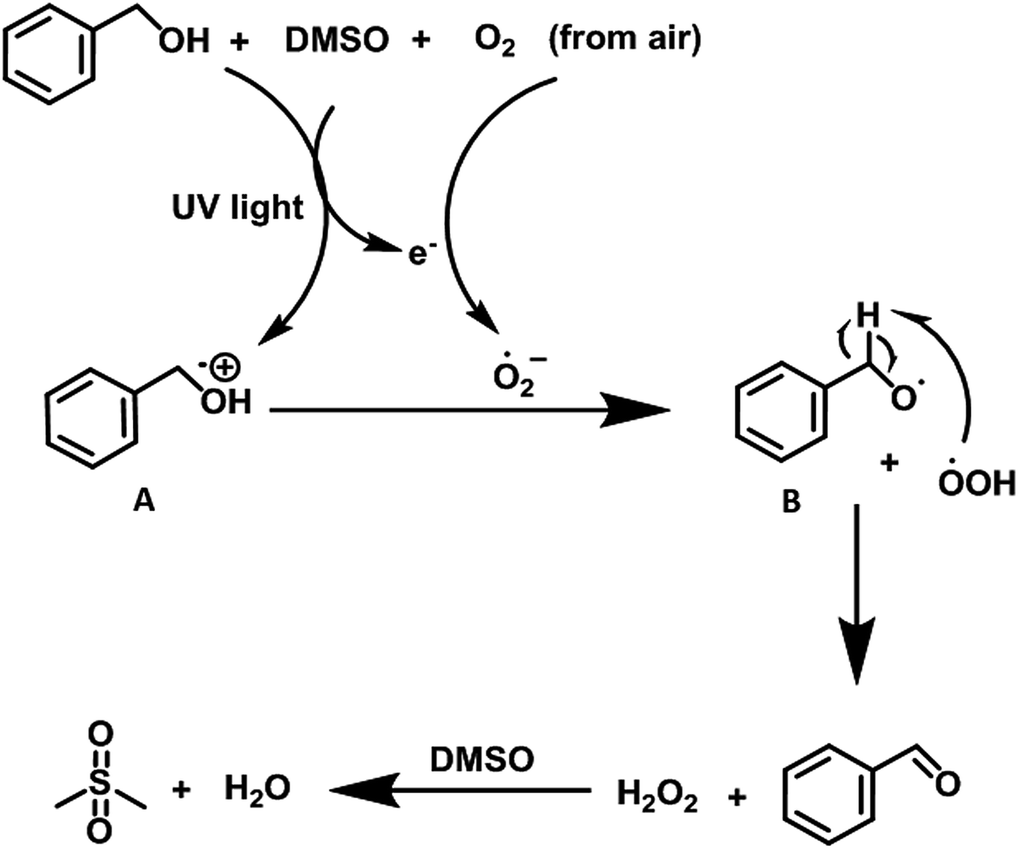 | ||
| Scheme 5 Proposed reaction mechanism for the oxidations of alcohols. | ||
Conclusions
In conclusion, the present study demonstrates the important role of alcohols themselves as electron donors for their selective oxidative transformations to the corresponding carbonyl compounds in absence of any metal/oxidant and external photosensitizer. The photopromoted oxidation exhibited wide substrate scope and especially high reactivity toward aromatic, alicyclic, and heteroaromatic alcohols. The practical application of the approach has been demonstrated by carrying out selective oxidation of steroid (cholesterol), which is a key component for many pharmaceuticals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. B. and M. K. are thankful to Science and Engineering Research Board (SERB), New Delhi (ref. no. CRG/2018/001274) and (ref no. EMR/2016/003473) for financial support respectively. P. K. W. is thankful to UGC for Senior Research Fellowship (SRF). We are also thankful to UGC (New Delhi, India) for the “University with Potential for Excellence” (UPE) project.Notes and references
- (a) J. E. Backvall, Modern Oxidation Methods, Wiley−VCH, New York, 2004 CrossRef; (b) J. Kochi and R. Sheldon, Metal Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed; (c) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS; (d) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS.
- (a) R. Ray, S. Chandra, D. Maiti and G. K. Lahiri, Chem.–Eur. J., 2016, 22, 8814–8822 CrossRef CAS; (b) J. N. Jaworski, S. D. McCann, I. A. Guzei and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56, 3605–3610 CrossRef CAS; (c) H. Su, K. X. Zhang, B. Zhang, H. H. Wang, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, J. Am. Chem. Soc., 2017, 139, 811–818 CrossRef CAS; (d) B. Xu, J. P. Lumb and B. A. Arndtsen, Angew. Chem., Int. Ed., 2015, 54, 4208–4211 CrossRef CAS; (e) S. Zavahir, Q. Xiao, S. Sarina, J. Zhao, S. Bottle, M. Wellard, J. Jia, L. Jing, Y. Huang, J. P. Blinco, H. Wu and H. Y. Zhu, ACS Catal., 2016, 6, 3580–3588 CrossRef CAS; (f) C. J. Weiss, P. Das, D. L. Miller, M. L. Helm and A. M. Appel, ACS Catal., 2014, 4, 2951–2958 CrossRef CAS; (g) Y. Yuan, N. Yan and P. J. Dyson, Inorg. Chem., 2011, 50, 11069–11074 CrossRef CAS; (h) S. Gowrisankar, H. Neumann, D. Goerdes, K. Thurow, H. Jiao and M. Beller, Chem.–Eur. J., 2013, 19, 15979–15984 CrossRef CAS; (i) Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- (a) G. Tojo and M. I. Fernández, Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer Science, NewYork, 2006 Search PubMed; (b) H. B. Friedrich, Platinum Met. Rev., 1999, 43, 94–102 CAS; (c) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC; (d) K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef CAS; (e) K. E. Pfitzner and J. G. Moffatt, J. Am. Chem. Soc., 1963, 85, 3027–3028 Search PubMed; (f) J. R. Parikh and W. V. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS; (g) J. D. Albright and L. Goldman, J. Am. Chem. Soc., 1965, 87, 4214–4216 CrossRef CAS.
- (a) W. Adam, C. R. Saha-Moller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499–3548 CrossRef CAS; (b) B. Karimi, A. Biglari, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210–7213 CrossRef CAS; (c) X. Zhang, X. Fu, Y. Zhang, Y. Zhu and J. Yang, Catal. Lett., 2016, 146, 945–950 CrossRef CAS; (d) H. Watanabe, S. Asano, S. Fujita, H. Yoshida and M. Arai, ACS Catal., 2015, 5, 2886–2894 CrossRef CAS.
- (a) R. Liu, X. Liang, C. Dong and X. Hu, J. Am. Chem. Soc., 2004, 126, 4112–4113 CrossRef CAS; (b) B. Karimi, A. Biglari, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210–7213 CrossRef CAS PubMed; (c) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS; (d) M. S. Laeini and A. Shaabani, ChemistrySelect, 2017, 2, 9084–9087 CrossRef CAS; (e) S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC; (f) Y. Yan, X. Tong, K. Wang and X. Bai, Catal. Commun., 2014, 43, 112–115 CrossRef CAS; (g) J. Zhu, X. J. Zhao, P. C. Wang and M. Lu, Chem. Lett., 2013, 42, 1505–1507 CrossRef CAS; (h) X. Wang, R. Liu, Y. Jin and X. Liang, Chem.–Eur. J., 2008, 14, 2679–2685 CrossRef CAS; (i) X. He, Z. Shen, W. Mo, N. Sun, B. Hu and X. Hu, Adv. Synth. Catal., 2009, 351, 89–92 CrossRef CAS.
- (a) W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS; (b) W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, ACS Catal., 2018, 8, 5425–5430 CrossRef CAS; (c) F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS; (d) K. Walsh, H. F. Sneddon and C. J. Moody, Org. Lett., 2014, 16, 5224–5227 CrossRef CAS; (e) Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS; (f) Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC; (g) W. Zhang, A. Bariotaki, I. Smonou and F. Hollmann, Green Chem., 2017, 19, 2096–2100 RSC.
- V. J. Traynelis and W. L. Hergenrother, J. Am. Chem. Soc., 1964, 86, 298–299 CrossRef CAS.
- (a) N. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (b) S. Ghosh, N. A. Kouame, L. Ramos, S. Remita, A. Dazzi, A. D. Besseau, P. Beaunier, F. Goubard, P. H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS.
- (a) L. A. Vermeulen and M. E. Thompson, Nature, 1992, 358, 656–658 CrossRef CAS; (b) B. Limburg, E. Bouwman and S. Bonnet, J. Phys. Chem. B, 2016, 120, 6969–6975 CrossRef CAS.
- (a) W. W. Yong, H. Lu, H. Li, S. Wang and M. T. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 10828–10834 CrossRef CAS; (b) B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS.
- (a) X. F. Wang, S. Sh. Yu, C. Wang, D. Xue and J. Xiao, Org. Biomol. Chem., 2016, 14, 7028–7037 RSC; (b) L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06490g |
| This journal is © The Royal Society of Chemistry 2019 |