
Supramolecular polymerization and cyclization of dioxynaphthalene motif bridged bifunctional UPys: minor variations in the molecular skeleton and drastic differences in self-assembly†
Tangxin
Xiao
 *a,
Lixiang
Xu
a,
Julian
Götz
b,
Ming
Cheng
c,
Frank
Würthner
*b,
Jiande
Gu
a,
Xuejun
Feng
*a,
Zheng-Yi
Li
a,
Xiao-Qiang
Sun
a and
Leyong
Wang
ac
*a,
Lixiang
Xu
a,
Julian
Götz
b,
Ming
Cheng
c,
Frank
Würthner
*b,
Jiande
Gu
a,
Xuejun
Feng
*a,
Zheng-Yi
Li
a,
Xiao-Qiang
Sun
a and
Leyong
Wang
ac
aJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, 213164, China. E-mail: xiaotangxin@cczu.edu.cn; xuejun-f@163.com
bUniversität Würzburg, Institut für Organische Chemie & Center for Nanosystems Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
cSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China
First published on 21st October 2019
Abstract
The relationship between molecular structure and macroscopic characteristics is a fundamental issue for materials design. Some minor changes in molecular structure may have a great impact on the molecular self-assembly process. Therefore, it is of interest to design new isomeric building blocks for supramolecular self-assembly, offering inspiration for the development of smart materials by supramolecular engineering. In this study, two dioxynaphthalene (DNP) group bridged bifunctional ureidopyrimidinone (UPy) monomers (DNP1 and DNP2) were synthesized. The chemical structures of the two isomers are very similar and the only difference is that DNP1 is 2,6-substituted and DNP2 is 1,5-substituted on the DNP motif. Interestingly, these two isomers exhibit completely different supramolecular self-assembly behaviors. Highly viscous supramolecular polymers were obtained in a concentrated solution of DNP1, while crystals based on highly stable cyclic monomers were precipitated from a concentrated solution of DNP2. Since DNP is an electron rich group, the host–guest behaviors of DNP1 and DNP2 with a blue-box were further studied, which also showed different ring-threading capabilities.
Introduction
Scientists have long endeavored to comprehend the interplay between non-covalent and covalent bond formation, in order to study and understand how function is related to self-assembly.1–4 Moreover, minor changes in molecular structure may impact the inherent molecular properties and further transfer this difference to the final assemblies.5 Therefore, controlling the linking sequence of groups in one molecule is a potential alternative method to adjust the function of its corresponding supramolecular materials. Supramolecular polymers, in which low molecular weight monomers are assembled into polymeric structures by non-covalent interactions, attract much interest due to their highly tunable functional groups and dynamic nature.6–9 So far, supramolecular polymerization controlled by different means have been established, such as pathway control,10,11 orthogonal self-assembly,12–14 living supramolecular polymerization,15,16etc. Meanwhile, a large number of advanced supramolecular polymers with excellent properties have been developed by using various non-covalent interactions as a driving force, including macrocycle-based host–guest interactions,17,18 multiple hydrogen bonding,19,20 metal–ligand coordination,21,22 or integration of these forces.23–25 On the one hand, obtaining a supramolecular polymer with a high degree of polymerization is one of the goals of developing practical supramolecular materials. On the other hand, optimization of discrete aggregates with well-defined architectures in self-assembled systems is also one of the targets of supramolecular chemists.26,27 Therefore, it would be interesting for the final assemblies to shift between highly viscous supramolecular polymers and well-defined discrete cyclic oligomers via minor structural adjustments in the molecule.The ureidopyrimidinone (UPy) unit which can undergo quadruple hydrogen bonding is a promising supramolecular building block owing to its high dimerization constant (Kdim > 107 M−1 in chloroform), self-complementary nature, and ease of synthesis.28,29 In recent years, a large amount of supramolecular polymers30–35 and well-defined supramolecular assemblies based on UPy have been constructed.36,37
By connecting two UPy units to tetraphenylethene motif, Tang and co-workers found that the supramolecular polymerizabilities and fluorescence properties of the stereoisomers depend strongly on the configuration of the molecules.38 Additionally, the π–π stacking interaction also plays a key role in supramolecular self-assembly.39,40 It is noteworthy that the UPy motif can not only dimerize through quadruple hydrogen bonding but also exhibit π–π stacking capability with aromatic groups.41,42
Previously, Zhang and co-workers synthesized 1,5-substituted and 2,6-substituted dioxynaphthalene (DNP)-based amphiphiles, both of which could complex with electron-deficient naphthalene diimide group-based amphiphiles through charge-transfer interactions to form supra-amphiphiles, leading to the further formation of rod-like and sheet-shape nanostructures, respectively.43 Based on the above mentioned appealing examples and encouraged by our experiences on various non-covalent interactions,44–46 we attempted to combine UPy and DNP groups to fabricate supramolecular assemblies with various topological architectures, so as to deepen the understanding of the relationship between molecular structures and supramolecular self-assembly. As shown in Fig. 1, two kinds of DNP-bridged bifunctional UPy monomers (DNP1 and DNP2) were designed and synthesized. The chemical structures of the two monomers are similar and the only difference is that DNP1 is substituted in the 2,6-position, while DNP2 is substituted in the 1,5-position on the DNP moiety. According to the ring-chain equilibrium mechanism, there is a critical polymerization concentration (CPC) upon concentration increase in the ring-opening polymerization process of bifunctional UPys, suggesting that the cyclic monomer is predominantly below the CPC and linear polymers are the main species above the CPC in solution.47 This study shows that the self-assembly behaviors of DNP1 and DNP2 are totally different although they have similar molecular structure. Monomer DNP1 exhibited typical ring-opening supramolecular polymerization behavior and a highly viscous supramolecular polymer was obtained. In stark contrast, DNP2 could not open its ring and maintained an extremely stable cyclic monomer structure throughout the concentration range (0–650 mM), resulting in precipitation of crystals from its concentrated solution. Herein, the ring tension and the π–π interaction in their cyclic monomer play important roles in the self-assembly process. Moreover, mechanically interlocked molecules have drawn much interest in recent years.48–50 Since the DNP group is electron rich, the different self-assembly behaviors of DNP1 and DNP2 with electron-deficient cyclobis(paraquat-p-phenylene)cyclophane (the so-called “blue-box”) have also been explored.
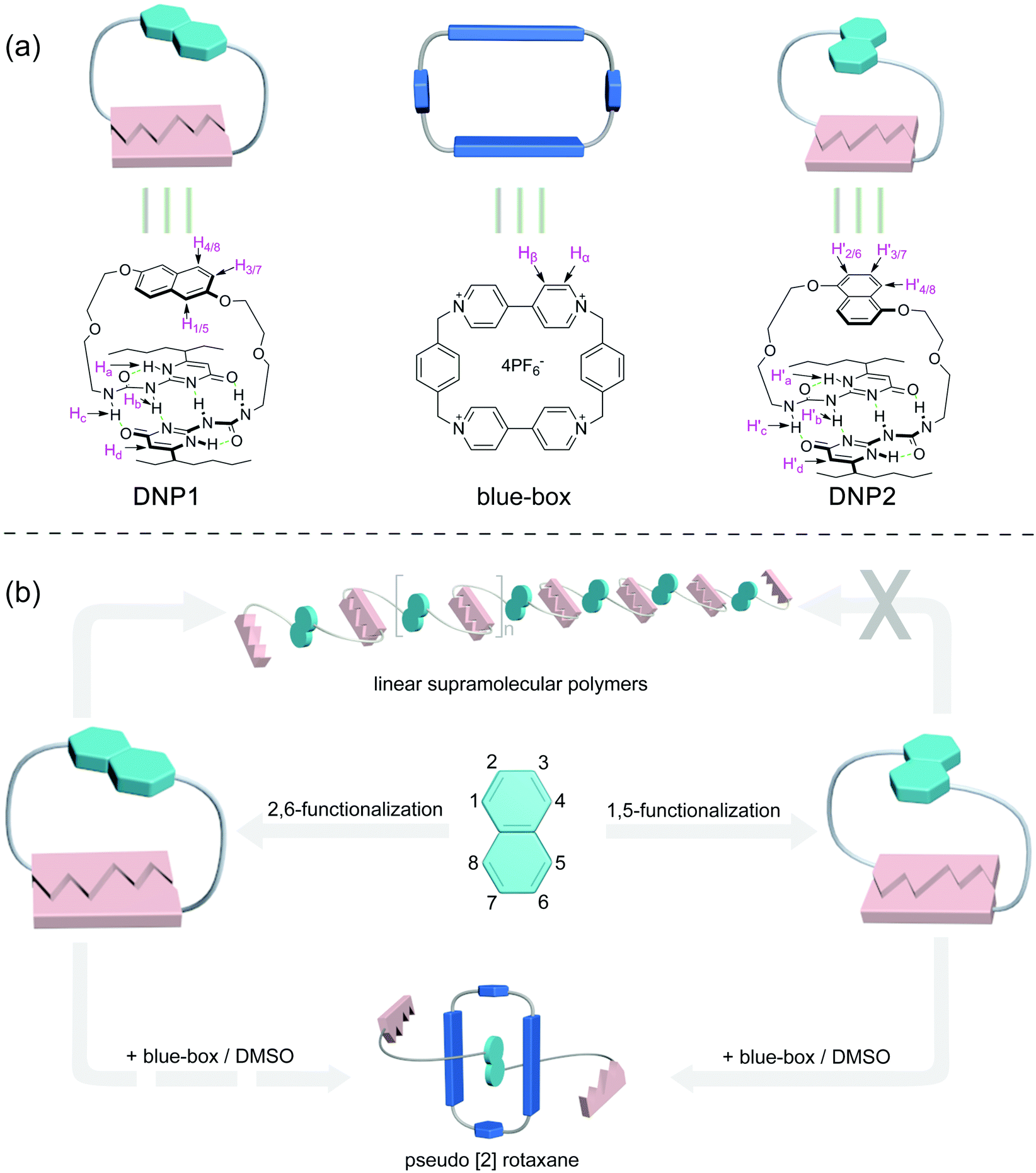 | ||
| Fig. 1 (a) Chemical structures and cartoon representations of DNP1, DNP2, and the blue-box, and (b) schematic illustration of the supramolecular polymerizability of DNP1 and DNP2, and their potential host–guest behaviors with the blue-box. | ||
Results and discussion
The DNP bridged bifunctional UPy compounds DNP1 and DNP2 have been designed (Fig. 1). The synthesis of these UPys is straightforward (see the ESI,† Scheme S3). The starting materials are 1,5-naphthalenediol and 2,6-naphthalenediol, respectively. After a few steps of simple reaction, two DNP group bridged ditopic terminal amine compounds were obtained. Connecting the amine compounds with 1,1′-carbonyldiimidazole activated pyrimidinone (UPy precursor) afforded DNP1 and DNP2. All the compounds were thoroughly characterized by NMR and mass spectrometry.The supramolecular polymerizability of DNP1 and DNP2 was first studied by 1H NMR upon varied monomer concentrations. The chemical shifts of the N–H proton signal in the two monomers are shifted significantly downfield (9.5–13.5 ppm) in CHCl3, indicating the formation of quadruple hydrogen bonding. As shown from the 1H NMR spectra of DNP1 (Fig. 2a), only one set of peaks (marked by red dots) indicates the presence of the cyclic monomer below 350 mM. However, a new set of peaks representing the cyclic dimer appeared upon increasing the concentration (marked by blue squares, see Fig. 6 for further discussion). As the concentration continues to increase, another new set of peaks (marked by green triangles) representing the linear supramolecular polymers occurred. During this process (Fig. 2a, 350–650 mM), the intensity of the polymer peaks increased and their shape became broad gradually. These phenomena suggest that DNP1 follows a typical ring-chain equilibrium supramolecular polymerization mechanism. Subsequently, a CPC value of 347 mM for DNP1 was calculated based on Hb by plotting the cyclic species concentration versus the total monomer concentration (see the ESI,† Fig. S3). Compared with other bifunctional UPys, the CPC of DNP1 is relatively high. This might be due to the π–π stacking interaction between dimerized UPy and DNP groups, which can stabilize the structure of the cyclic monomer and inhibit the ring-opening to some extent.13 It is worth noting that Hb and Hc, both of which are involved in quadruple hydrogen bonding, showed obvious downfield shifts (from 10.82 to 11.00 to 11.70 ppm for Hb, from 10.03 to 10.13 to 10.32 ppm for Hc) upon ring-opening polymerization, indicating the stepwise loss of shielding effect between the dimerized UPy motif and DNP moiety. The peaks of Ha and Hd shifted a little upfield during polymerization, indicating that Ha and Hd were located at the peripheral domain of the DNP group, which is consistent with the structure model of the cyclic monomer.
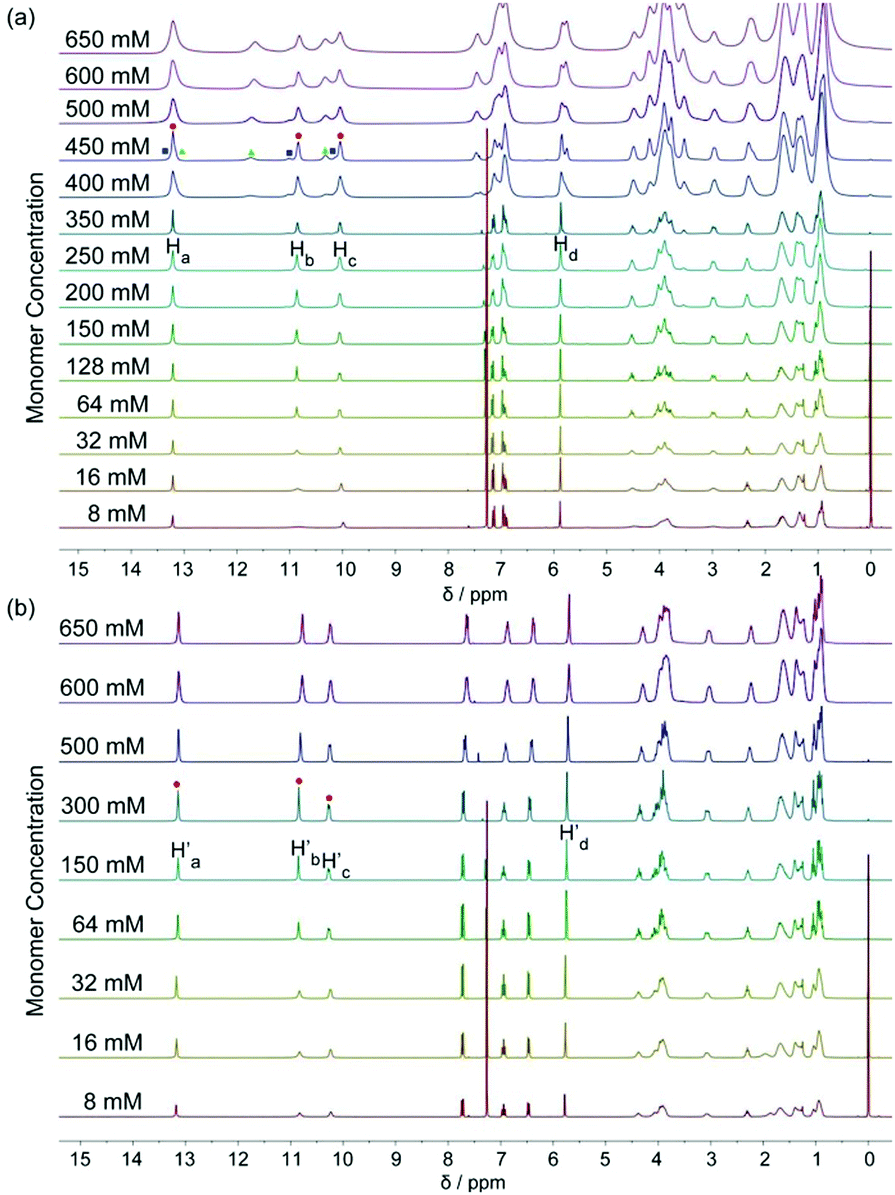 | ||
| Fig. 2 1H NMR spectra (300 MHz, CDCl3, 298 K) of (a) DNP1 and (b) DNP2 at different monomer concentrations. The red dots indicate cyclic monomers and the blue squares stand for cyclic dimers and the green triangles represent polymeric assemblies. | ||
In contrast to DNP1, DNP2 exhibited a completely different self-assembly behavior. As shown in Fig. 2b, only one set of peaks appeared during the whole concentration range (8–650 mM) and no new peaks occurred even in the highest measurable concentration (650 mM). Higher concentration solutions are not suitable for NMR due to the limited solubility of the compound. Notably, the peaks were still sharp even at 650 mM, indicating that no polymeric species were generated. These phenomena suggested that DNP2 existed in the form of a very stable cyclic monomer in solution and no CPC was found, which showed a dramatic difference with DNP1. DNP2 did not show ring-opening supramolecular polymerization behavior mainly due to the stronger π–π interaction between the DNP moiety and the dimerized UPy motif, which greatly enhanced the stability of the monomeric structure.13 Compared to DNP2, the ring-strain of 2,6-substituted DNP1 is much larger than the one of DNP2, which favors the ring-opening of DNP1. It can be understood as follows: (1) the positions of the building blocks DNP and UPy of the two monomers in space are fixed, (2) the diethylene glycol linkers play the role of a “rope” to bundle the DNP and UPy building blocks together, (3) the length of the “rope” is the same, but DNP1 is 2,6-substituted and causes the tension of the rope to increase.
The monomeric form of DNP1 and DNP2 was further evidenced by NOESY NMR. A solution of DNP1 in CDCl3 at 64 mM, which is far from its CPC, was employed for measurement. As expected, strong NOE signals occurred between DNP protons (H1/5, H3/7, and H4/8) and UPy N–H protons (Ha, Hb, and Hc) (Fig. 3a). Additionally, obvious correlations were also observed between the protons from the 1-ethylpentyl group of the UPy and DNP protons (H1/5, H3/7, and H4/8) (see the ESI,† Fig. S1). All these correlations indicate that DNP1 indeed prevails as a cyclic monomer architecture through intramolecular quadruple hydrogen bonding of UPy at the current concentration (64 mM). The NOESY of DNP2 exhibited similar results, which also proved that DNP2 exists in its cyclic monomer form. Obvious NOE correlations were observed between DNP protons (H′2/6, H′3/7, and H′4/8) and UPy N–H protons (H′a, H′b, and H′c) (Fig. 3b) as well as protons from the 1-ethylpentyl chain (see the ESI,† Fig. S2).
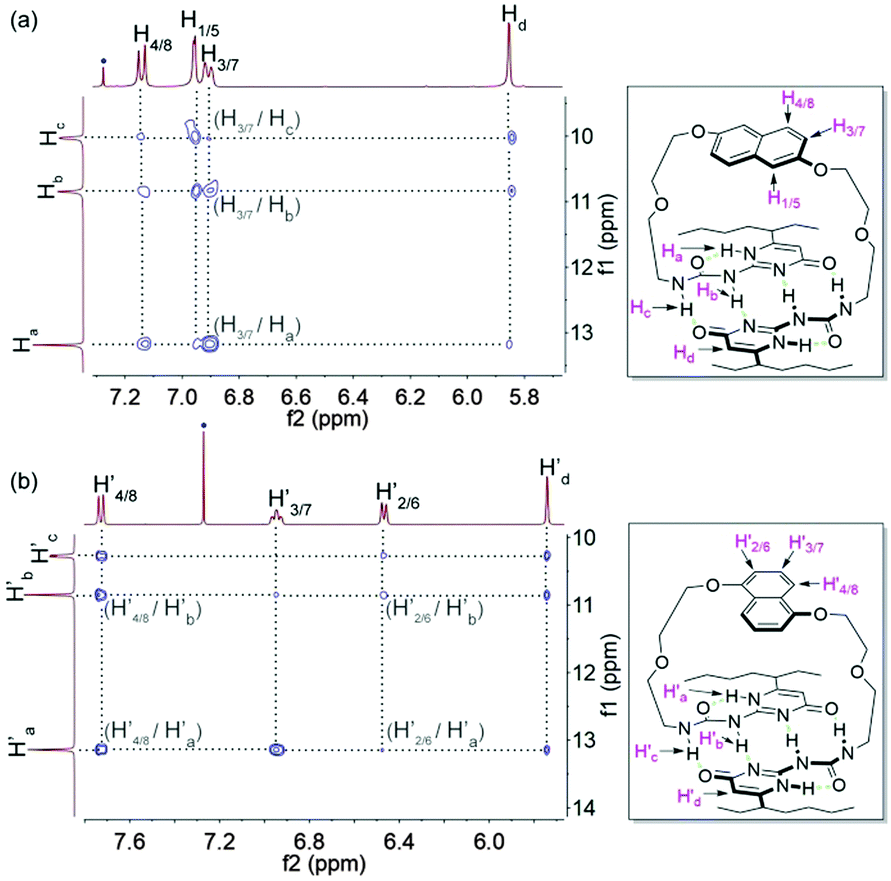 | ||
| Fig. 3 Partial NOESY (400 MHz, CDCl3, 298 K) of (a) DNP1 (64 mM) and (b) DNP2 (64 mM). The blue dots indicate solvent peaks. | ||
The self-assembly behaviors of DNP1 and DNP2 were further tested by temperature-dependent 1H NMR. The concentration of DNP1 was set at 350 mM, which was close to its CPC value. As shown in Fig. S4 (see the ESI†), the peaks belonging to the polymer species are gradually increased and become broad as the temperature increases from 298 K to 333 K, indicating that the cyclic monomer of DNP1 is not stable enough and would undergo ring-opening supramolecular polymerization as the temperature increases. Under the same conditions, DNP2 exhibited totally different behavior. It can be seen from Fig. S5 (see ESI†) that as the temperature increases from 298 K to 333 K, no new peaks appear and the original peaks remain sharp, showing that the cyclic monomer of DNP2 is very stable even at high temperature. These observations again confirmed the drastic differences between DNP1 and DNP2 in their self-assembly processes.
Subsequently, the stability of DNP1 and DNP2 in their cyclic monomer form was studied in CDCl3 with the addition of DMSO-d6 which is a strong hydrogen bond breaking solvent. As χDMSO (percentage of volume) increased to above 0.38, a new set of small peaks representing the open form of DNP2 appeared (Fig. 4). Compared with other UPys that usually start to dissociate at χDMSO < 0.1,51 the cyclic form of DNP2 is much more stable. The simultaneous presence of two sets of peaks indicates that the equilibrium between open form and closed form of monomeric DNP2 is in a slow exchange regime on the time scale of NMR. As the amount of DMSO continued to increase, the proportion of open monomers was also increasing due to more hydrogen bonds being destroyed. The self-assembly behavior of DNP1 in mixed CDCl3/DMSO-d6 solvent showed similar results (see ESI,† Fig. S6). However, DNP1 started to open its ring when χDMSO was about 0.30, which is smaller than DNP2, further suggesting that the cyclic form of DNP1 is less stable than DNP2.
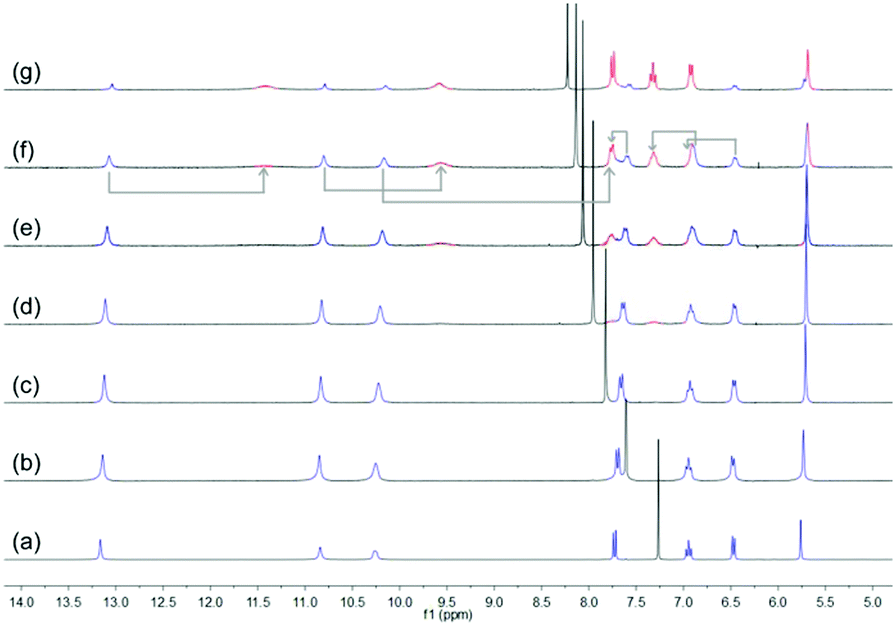 | ||
| Fig. 4 Partial 1H NMR spectra (300 MHz) of DNP2 (16 mM) in mixed CDCl3/DMSO-d6 solvents. From bottom to top: χDMSO = (a) 0, (b) 0.17, (c) 0.29, (d) 0.38, (e) 0.47, (f) 0.58 and (g) 0.71. Blue peaks stand for cyclic monomers and red peaks originate from open monomers. | ||
Diffusion ordered NMR (DOSY) was further performed to study the self-assembly behaviors of DNP1 and DNP2. The size of the self-assemblies was estimated by employing heptakis(2,3,6-tri-O-methyl)-β-cyclodextrin (peralkylated β-CD) as an internal standard. In the DOSY NMR spectrum, the larger the diffusion coefficient, the smaller the molecular weight of the assembly. As shown in Fig. 5a, the DOSY signal of DNP1 below the CPC was regular and the diffusion coefficient (9.33 × 10−10 m2 s−1, 24 mM, MW = 805 Da, 2MW = 1610 Da) displayed a larger value than peralkylated β-CD (6.31 × 10−10 m2 s−1, MW = 1429 Da), indicating that the molecular weights of DNP1 assemblies are smaller than 1429 Da and DNP1 indeed existed as cyclic monomers in solution below the CPC. In stark contrast, the signals for DNP1 in concentrated solution above CPC (350 mM) exhibited a wide distribution, suggesting a broad molecular weight distribution of assemblies (Fig. 5b and see Fig. 6 for details). The DOSY signals of DNP2 both in dilute solution (24 mM, Fig. 5c) and concentrated solution (350 mM, Fig. 5d) displayed a well-defined narrow distribution and their diffusion coefficients were also larger than peralkylated β-CD. These phenomena indicate that DNP2 always maintains a stable cyclic monomer structure, which is consistent with the results of variable concentration 1H NMR and NOESY.
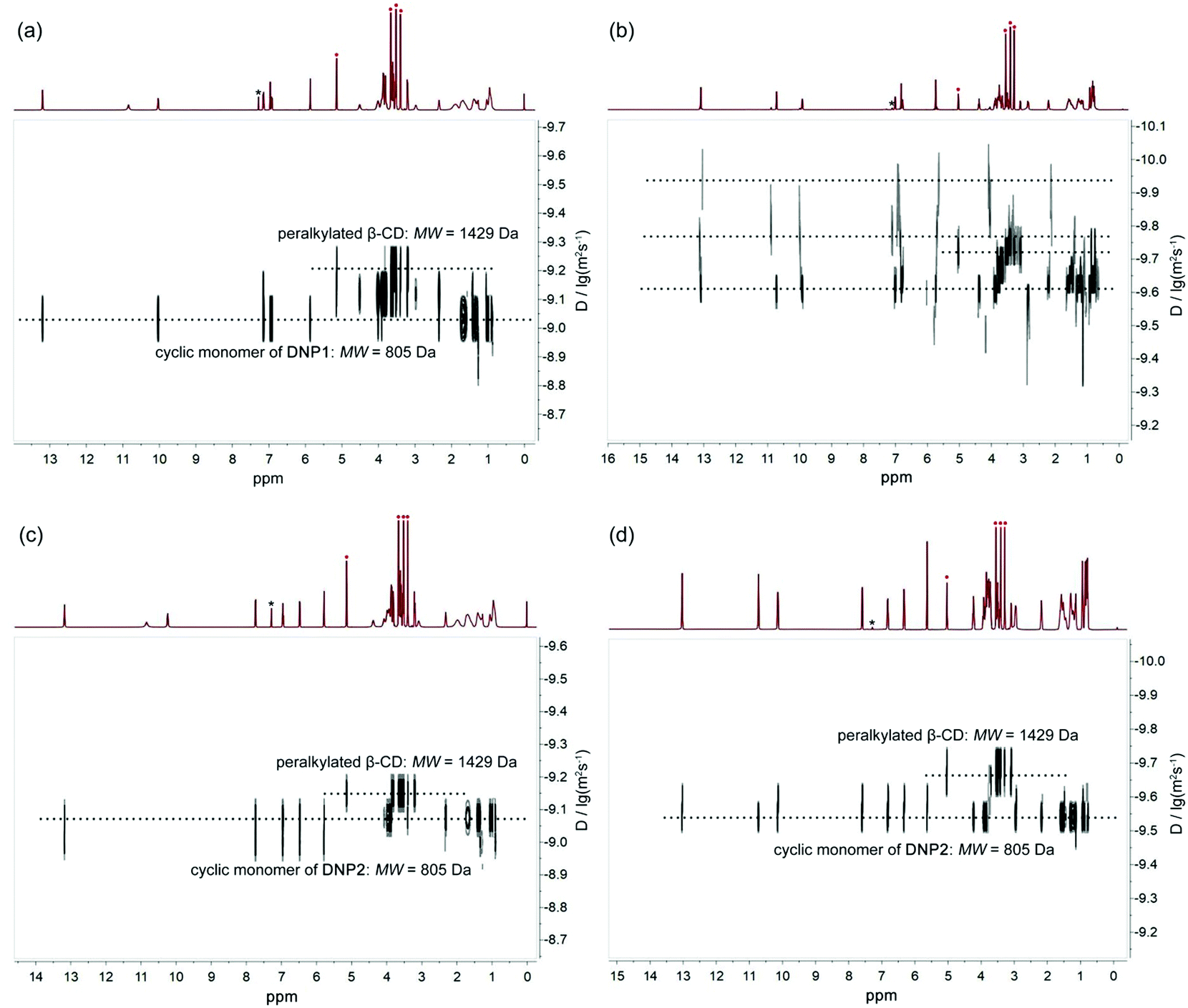 | ||
| Fig. 5 DOSY spectra (600 MHz, CDCl3, 298 K) of (a) DNP1 in 24 mM, (b) DNP1 in 350 mM, (c) DNP2 in 24 mM and (d) DNP2 in 350 mM. Peralkylated β-CD was added to each sample as internal standard. The asterisk symbols indicate solvent peaks and the red dots stand for proton signals from peralkylated β-CD. | ||
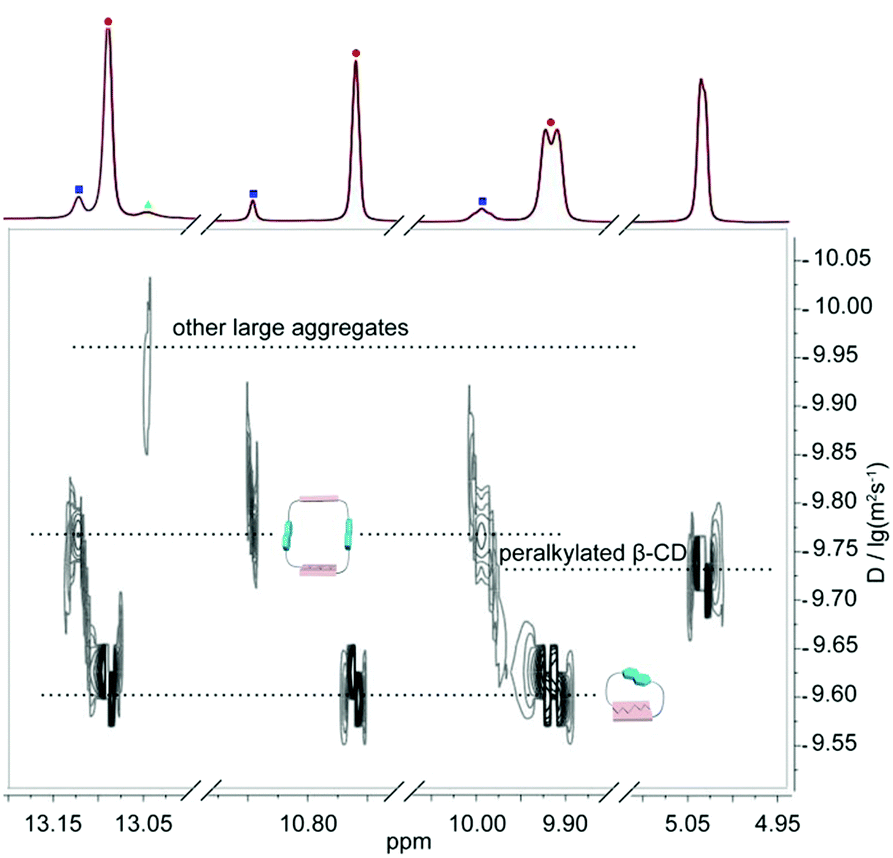 | ||
| Fig. 6 Enlarged DOSY profile (600 MHz, CDCl3, 298 K) of DNP1 in 350 mM with the addition of peralkylated β-CD as internal standard. The red dots indicate cyclic monomers and the blue squares stand for cyclic dimers and the green triangles represent polymeric assemblies. | ||
In order to better understand the self-assembly behavior of DNP1 at relatively high concentration, a partially enlarged view of Fig. 5b was shown in Fig. 6. Proton signals from N—H involved in hydrogen bonding are mainly concerned. A wide distribution was clearly observed, indicating the formation of various assemblies with different size, including monomer (2.45 × 10−10 m2 s−1, MW = 805 Da), dimer (1.58 × 10−10 m2 s−1, MW = 1610 Da), and other larger aggregates (1.12 × 10−10 m2 s−1, MW > 1610 Da). Herein, the signals of the β-CD (1.86 × 10−10 m2 s−1, MW = 1429 Da) fell just in the middle of the signals of the monomer (MW = 805 Da) and the dimer (MW = 1610 Da), which further confirms our hypothesis.
The supramolecular polymerizability of DNP1 and DNP2 was further tested by viscosity measurements in CHCl3. Double logarithmic plots of specific viscosity versus monomer concentration are shown in Fig. 7. For DNP1, a slope of 1.01 was found in the initial stage upon concentration increasing, which is characteristic of cyclic monomers with constant size. As the concentration increased, a turning point occurred at 345 mM (CPC), which is close to the CPC determined by 1H NMR measurements (347 mM). It is noteworthy that an extremely high slope of viscosity (k′DNP1 = 13.85) was obtained, indicating the formation of entangled supramolecular polymers with high molecular weight in concentrated solution. To the best of our knowledge, this slope value is the highest one for bifunctional UPy compounds.13,52 The inset in Fig. 7a directly showed a highly viscous solution of DNP1 (650 mM) in CHCl3. Due to the relatively high CPC, the extra monomers will undergo rapid ring-opening polymerization to form large aggregates once the concentration reached the CPC. In contrast, DNP2 showed a yellowish solution with good fluidity, even in very concentrated solutions (650 mM), implying that supramolecular polymerization did not occur. The viscosity profile of DNP2 further explained this special phenomenon. As shown in Fig. 7b, the line displayed no turning point and the slope was 1.05 over the entire measurable range of concentration (0–300 mM), suggesting that a highly stable cyclic monomer always existed in solution, which was in line with concentration-dependent 1H NMR. Attempts to test at higher concentrations (>300 mM) had failed because DNP2 readily crystallized in the capillary of the viscometer. However, this phenomenon gave us another surprising conclusion that DNP2 could easily crystallize out from its concentrated solution, indicating a totally different physicochemical property. Moreover, crystals with larger size were also found in a vial after partial evaporation of solvent from a concentrated solution of DNP2 (Fig. 8b). A microscopic view of the crystals was further recorded by SEM (Fig. 8a).
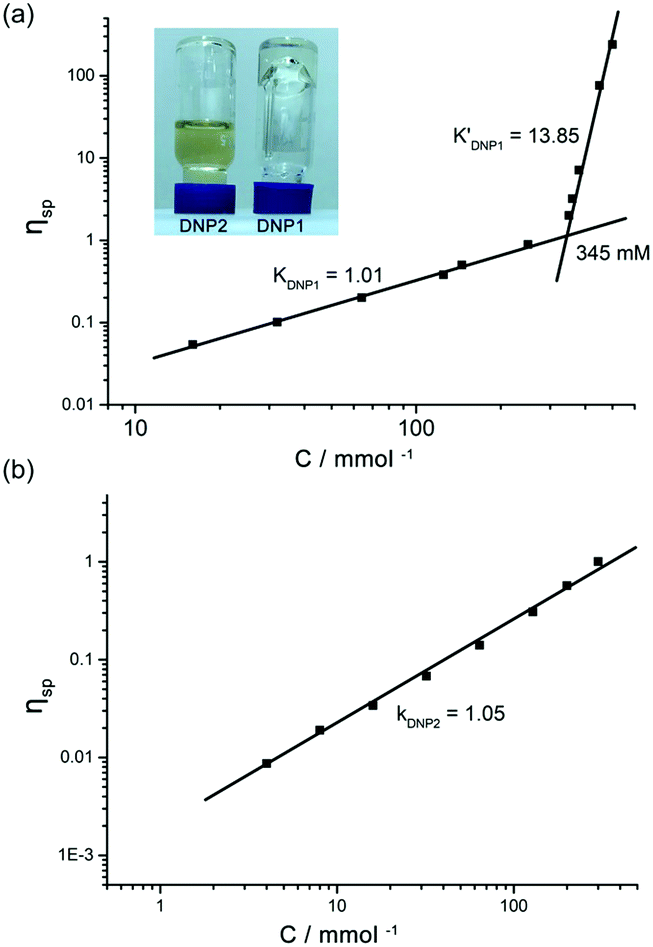 | ||
| Fig. 7 Specific viscosity of chloroform solutions of (a) DNP1 and (b) DNP2versus the monomer concentration (298 K). Values on the curves indicate the slope. Inset: Concentrated solution of DNP1 (650 mM) and DNP2 (650 mM) in CHCl3. | ||
 | ||
| Fig. 8 (a) SEM image of the microscopic crystals of DNP2 and (b) photograph of crystals precipitated from a concentrated solution (650 mM, in CHCl3) of DNP2. | ||
The density functional theory studies of the representative models of mDNP1 (for DNP1) and mDNP2 (for DNP2) reveal that the total energy of mDNP1 is about 12.4 kcal mol−1 higher than that of mDNP2 (Fig. 9). This energy difference can be attributed to the stress of the strained ring or/and the energy difference between the 1,5-di-substituted naphthalene and the 2,6-di-substituted naphthalene. Further calculations of 1,5-dimethoxylnaphthalene and 2,6-dimethoxylnaphthalene indicate that 1,5-disubstituted naphthalene is only 0.6 kcal mol−1 more stable than 2,6-disubstituted naphthalene in energy. Thus, the additional energetic contribution to the destabilization of mDNP1 (as compared to mDNP2) is due to the stress of the strained ring system. The optimized structures of mDNP1 and mDNP2 (Fig. 9 and Table 1) expose that although the H-bond lengths are similar for both compounds, the planes of H-bonding UPy moieties in mDNP1 are highly twisted. The angle of the planar vectors of UPy moieties amounts to 25.7° in mDNP1, while this angle is 8° in mDNP2. The stress of the strained ring system of mDNP1 reduces the H-bonding energy of the UPy moieties by about 11.8 kcal mol−1.
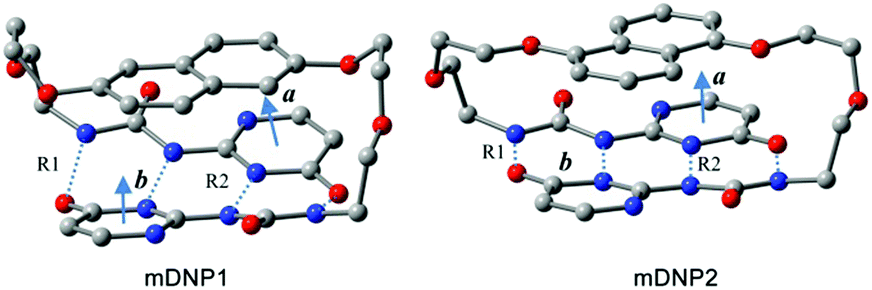 | ||
| Fig. 9 The optimized structures representing models mDNP1 and mDNP2. Color legend: red for O, blue for N, and grey for C. H atoms have been omitted for clarification. a and b represent the planar vectors of UPy moieties. All the optimized structures have C2 symmetry. | ||
Since the DNP moiety is a well-known electron rich group which can complex with electron deficient macrocycles to form mechanically interlocked molecules, such as rotaxanes and catenanes, we envisioned what differences would happen in the host–guest complexation of DNP1 and DNP2 with a blue-box. We first investigated these molecules in mixed CDCl3/CD3CN solvent, hoping that the hydrogen bonds were not destroyed. However, at a low concentration (16 mM) both DNP1 and DNP2 showed no host–guest complexations with the blue-box, even after three days of reflux. This might be due to the high stability of these cyclic structures. Subsequently, we suspected that DNP1 would bind with the blue-box at a high concentration (350 mM), where DNP1 would open its ring owing to ring-opening supramolecular polymerization. However, the poor solubility of DNP1 and the blue-box in mixed CDCl3/CD3CN impeded the investigation. Then, we studied the host–guest behaviors of DNP1 and DNP2 in mixed CDCl3/DMSO-d6 solvent. The results showed that only those open monomers could thread into the blue-box. For DNP2, even at χDMSO = 0.40, no bonded species were observed (see ESI,† Fig. S8) but at χDMSO = 0.45, obvious peaks representing pseudorotaxanes were observed (Fig. 10). Protons from DNP (H-2/6, H-3/7 and H-4/8) and the blue-box (Hα and Hβ) exhibited large upfield shifts (blue peaks in Fig. 10b), indicating that the DNP moiety was encircled by the blue-box. In pure DMSO, the threading efficiency was up to 44% for DNP2 (see ESI,† Fig. S8). In contrast, DNP1 showed less affinity to the blue-box. Even in pure DMSO, only 14% blue-box was complexed with DNP1 (see ESI,† Fig. S7). This should be due to the different association constants of 1,5-DNP and 2,6-DNP with the blue-box.
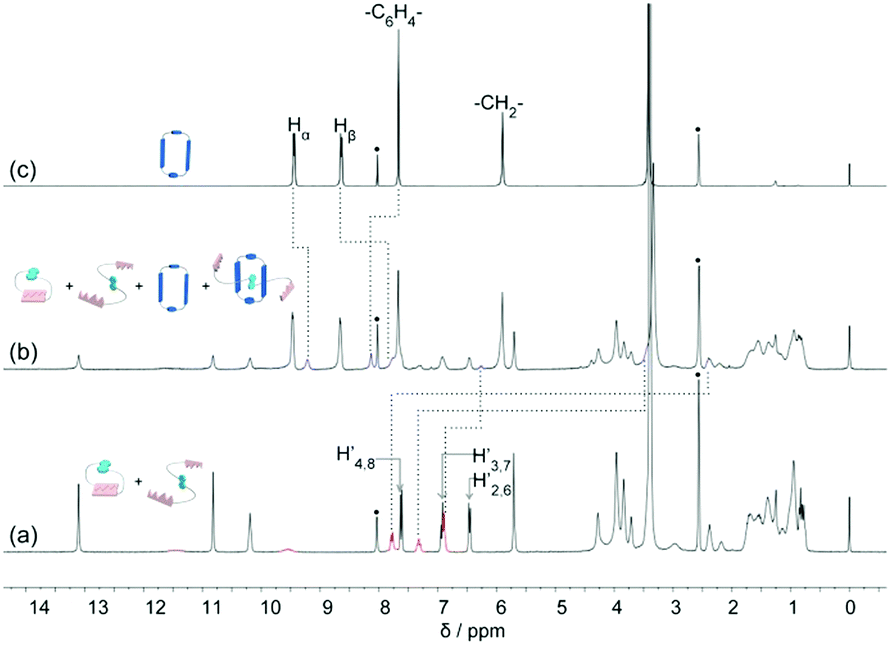 | ||
| Fig. 10 Partial 1H NMR spectra (300 MHz, CDCl3/DMSO-d6, v/v, χDMSO = 0.45) of: (a) DNP2, (b) DNP2 (10 mM) and blue-box (10 mM), and (c) blue-box. The black dots stand for solvent peaks. Red peaks indicate open monomers of DNP2. Blue peaks represent the host–guest complex. | ||
Conclusions
In conclusion, we have demonstrated a dramatic difference in supramolecular polymerizability for two structurally similar building blocks. The 2,6-substituted monomer DNP1 possesses excellent supramolecular polymerizability, leading to the formation of highly viscous supramolecular polymers. The 1,5-substituted monomer DNP2 lacks supramolecular polymerizability, resulting in the production of highly stable cyclic monomers with the capability of precipitation of crystals. Their different self-assembly behaviors have been thoroughly investigated by a series of techniques including concentration-dependent 1H NMR, temperature-dependent 1H NMR, mixed deuterated solvent 1H NMR, NOESY, DOSY, SEM, viscosity measurements and theoretical calculations. Moreover, the host–guest complexation experiments showed that DNP2 had a stronger binding ability with a blue-box than DNP1 to form pseudorotaxanes when the hydrogen bonding was disrupted by addition of the hydrogen bond breaking solvent DMSO. The findings in this work will offer inspiration on supramolecular engineering and dynamic materials design. The construction of quadruple hydrogen bonded polyrotaxanes is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (21702020, 21572026) and the Advanced Catalysis and Green Manufacturing Collaborative Innovation Center (ACGM2016-06-05).Notes and references
- G. Vantomme and E. W. Meijer, Science, 2019, 363, 1396–1397 CrossRef CAS.
- Z. Chen, Y. Liu, W. Wagner, V. Stepanenko, X. Ren, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 5729–5733 CrossRef CAS.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernandez, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS.
- T. Xiao, L. Zhou, X.-Q. Sun, F. Huang, C. Lin and L. Wang, Chin. Chem. Lett., 2019 DOI:10.1016/j.cclet.2019.05.011.
- T. Xiao, W. Zhong, L. Qi, J. Gu, X. Feng, Y. Yin, Z.-Y. Li, X.-Q. Sun, M. Cheng and L. Wang, Polym. Chem., 2019, 10, 3342–3350 RSC.
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- X. Ma, Y. Zhang, Y. Zhang, Y. Liu, Y. Che and J. Zhao, Angew. Chem., Int. Ed., 2016, 55, 9539–9543 CrossRef CAS.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS.
- D.-S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925–1934 CrossRef CAS.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS.
- Q. Zhang, T. Li, A. Duan, S. Dong, W. Zhao and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 8058–8063 CAS.
- S. G. Chen, Y. Yu, X. Zhao, Y. Ma, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2011, 133, 11124–11127 CrossRef CAS PubMed.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- K. C. Bentz and S. M. Cohen, Angew. Chem., Int. Ed., 2018, 57, 14992–15001 CrossRef CAS PubMed.
- L. Xu, X. Shen, Z. Zhou, T. He, J. Zhang, H. Qiu, M. L. Saha, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 16920–16924 CrossRef CAS PubMed.
- Q. Wang, M. Cheng, L. Tian, Q. Fan and J. Jiang, Polym. Chem., 2017, 8, 6058–6063 RSC.
- H. Wang, X. Ji, Z. Li, C. N. Zhu, X. Yang, T. Li, Z. L. Wu and F. Huang, Mater. Chem. Front., 2017, 1, 167–171 RSC.
- L. J. Chen and H. B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- A. T. t. Cate and R. P. Sijbesma, Macromol. Rapid Commun., 2002, 23, 1094–1112 CrossRef.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- X. Zhu, J. X. Wang, L. Y. Niu and Q. Z. Yang, Chem. Mater., 2019, 31, 3573–3581 CrossRef CAS.
- S. J. Rao, K. Nakazono, X. Liang, K. Nakajima and T. Takata, Chem. Commun., 2019, 55, 5231–5234 RSC.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- W.-J. Liang, J.-J. Yu, Q. Zhang, C.-S. Ma, Z.-T. Shi and D.-H. Qu, Polym. Chem., 2018, 9, 4808–4812 RSC.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- A. Goujon, G. Mariani, T. Lang, E. Moulin, M. Rawiso, E. Buhler and N. Giuseppone, J. Am. Chem. Soc., 2017, 139, 4923–4928 CrossRef CAS PubMed.
- T. Xiao, L. Xu, J. Wang, Z.-Y. Li, X.-Q. Sun and L. Wang, Org. Chem. Front., 2019, 6, 936–941 RSC.
- Q. Chen, X. Su, E. Orentas and Q. Shi, Org. Chem. Front., 2019, 6, 611–617 RSC.
- H.-Q. Peng, X. Zheng, T. Han, R. T. K. Kwok, J. W. Y. Lam, X. Huang and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 10150–10156 CrossRef CAS PubMed.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- T. Xiao, X. Feng, S. Ye, Y. Guan, S.-L. Li, Q. Wang, Y. Ji, D. Zhu, X. Hu, C. Lin, Y. Pan and L. Wang, Macromolecules, 2012, 45, 9585–9594 CrossRef CAS.
- D. Guo, R. P. Sijbesma and H. Zuilhof, Org. Lett., 2004, 6, 3667–3670 CrossRef CAS PubMed.
- K. Liu, C. Wang, Z. Li and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 4952–4956 CrossRef CAS PubMed.
- T. Xiao, W. Zhong, L. Zhou, L. Xu, X.-Q. Sun, R. B. P. Elmes, X.-Y. Hu and L. Wang, Chin. Chem. Lett., 2019, 30, 31–36 CrossRef CAS.
- T. Xiao, L. Xu, L. Zhou, X.-Q. Sun, C. Lin and L. Wang, J. Mater. Chem. B, 2019, 7, 1526–1540 RSC.
- T. Xiao and L. Wang, Tetrahedron Lett., 2018, 59, 1172–1182 CrossRef CAS.
- T. F. A. D. Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- S. Dasgupta and J. Wu, Chem. Sci., 2012, 3, 425–432 RSC.
- R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed.
- S.-L. Li, T. Xiao, W. Xia, X. Ding, Y. Yu, J. Jiang and L. Wang, Chem. – Eur. J., 2011, 17, 10716–10723 CrossRef CAS PubMed.
- O. A. Scherman, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 2072–2076 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional NMR spectra, and HR-ESI-MS spectra of individual compounds. See DOI: 10.1039/c9qm00595a |
| This journal is © the Partner Organisations 2019 |