
Hydrogen-bonding regulated assembly of molecular and macromolecular amphiphiles
Amrita
Sikder
and
Suhrit
Ghosh
 *
*
School of Applied and Interdisciplinary Sciences, Indian Association for the Cultivation of Science, 2A and 2B Raja S. C. Mullick Road, Kolkata, 700032, India. E-mail: psusg2@iacs.res.in
First published on 20th September 2019
Abstract
Amphiphilic small molecules and polymers produce diverse nanostructures in an aqueous medium. Emerging reports suggest that there is a great opportunity to enrich the structure and function of amphiphilic aggregates by suitable molecular engineering in the building block to enable directional non-covalent interaction based on specific molecular recognition to circumvent the immiscibility driven aggregation governed by the critical packing parameter. This review discusses representative recent examples on self-assembly of small molecule and polymeric amphiphiles by H-bonding interaction in an aqueous medium. The specific impact of H-bonding on regulating morphology, stability, dynamics, thermodynamics, surface functional group display, stimuli-responsiveness and precision of amphiphilic nanostructures has been highlighted and correlated with their potential application as supramolecular biomaterials.
 Amrita Sikder | Amrita Sikder obtained Master of Science in 2003 from the Indian Institute of Technology Madras, India. Subsequently she joined the group of Professor Suhrit Ghosh at the Indian Association for the Cultivation of Science, Kolkata for PhD. She has worked on directional supramolecular assembly of π-amphiphiles and multivalent interaction with suitable biological targets and obtained a PhD in 2019. At present, she is a senior research fellow at the Indian Association for the Cultivation of Science, Kolkata. Her research interests include self-assembly of amphiphilic systems, supramolecular chemistry and biological applications. |
 Suhrit Ghosh | Suhrit Ghosh was born in 1976 in India. After completion of undergraduate education in Kolkata, India, he was admitted to the integrated PhD program (Chemical Science) at the Indian Institute of Science, Bangalore in 1997. He received an MS degree (Chemistry) in 2000 and continued for a PhD until 2005 under the supervision of Professor S. Ramakrishnan. Then he moved to the group of Professor S. Thayumanavan at the University of Massachusetts, Amherst, USA, for postdoctoral studies (2005–2007). Subsequently he worked as an Alexander von Humboldt postdoctoral fellow (2007–2008) with Professor Frank Würthner at the University of Würzburg, Germany. In 2008 he joined the Indian Association for the Cultivation of Science (IACS), Kolkata, India, as an Assistant Professor where he currently holds the position of a Professor and Chair of the School of Applied and Interdisciplinary Sciences. Research interest of his group includes supramolecular polymerization of donor–acceptor π-systems, H-bonding driven assembly of amphiphilic π-systems/macromolecules and biologically relevant stimuli responsive aggregation of amphiphilic polymers (polydisulfides, polyurethanes). |
Introduction
Self-assembly is ubiquitous in living organisms and allows construction of cell membranes, protein folding, creation of the DNA double helix and other elegant structures. Inspired by their structures and complex function, a wide range of synthetic building blocks have been explored1 for creating programmable nanostructures with the aim to address pressing issues in biology and materials science. Amongst them, amphiphiles are probably the most versatile building blocks for the creation of aqueous nanostructures. A large number of small molecule and polymeric amphiphiles have been reported2,3 to show spontaneous aggregation in water producing a range of nanostructures including spherical micelles, nanoparticles, vesicles/polymersomes, worm like structures, 2D-sheets, nanotubes and others which exhibit a promising future in biological applications4–7 including drug delivery, gene delivery, tissue engineering, antimicrobial activity and others. Although structural diversity of amphiphiles has been explored to a great extent, in most of the examples, the specific nature of the nanostructure relies on the critical packing parameter (p)8 which depends on the ratio of hydrophobic and hydrophilic segments. A particular value of p can be translated via simple geometrical relations into a specific shape of the aggregate (Scheme 1). Generally, spherical micelles, cylindrical micelles and vesicles are formed when p ≤ 1/3, 1/3 < p ≤ 1/2 and 1/2 <p ≤ 1, respectively.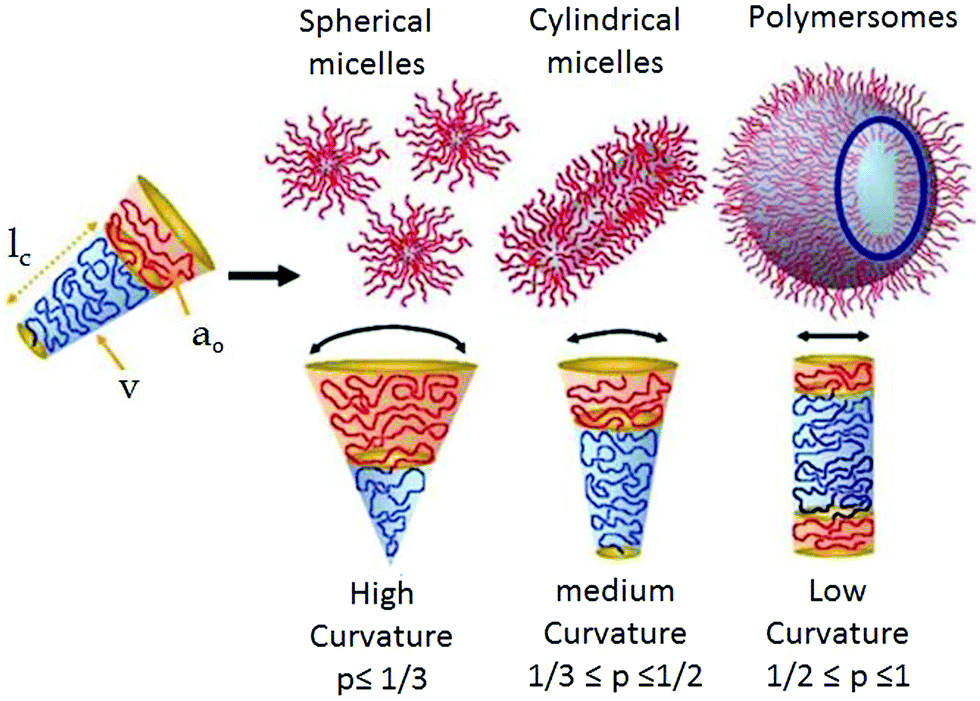 | ||
| Scheme 1 Schematic showing the critical packing parameter dependent expected morphology of amphiphilic aggregates. Adapted and modified with permission from ref. 8c. Copyright 2009. Wiley VCH. | ||
Such packing parameter based morphology control is unpredictable for new molecular/macromolecular scaffolds and does not endow molecular scale precision in designing a building block for a specific nanostructure. In the recent past, it has been demonstrated that in a special class of amphiphilic block copolymers in which the water insoluble block consists of a crystalline polymer, it is possible to control the self-assembly primarily by crystallization9 of the core which may be considered a significant step forward compared to immiscibility driven aggregation of amorphous systems. Such crystallization driven supramolecular polymerization has been explored to create an elegant nanostructure with a predictable size and shape. In a different research field, significant effort has been devoted to employing various directional supramolecular interactions such a donor–acceptor charge-transfer interaction,10 π-stacking,11 metal–ligand coordination12 and others13 to engineer the structure of amphiphilic building blocks which strongly influence the morphology, stability, stimuli-responsiveness and thermodynamic properties of the resulting nanostructures in water. Amongst these supramolecular interactions, hydrogen-bonding is particularly attractive as it provides a strong motivation for specific molecular interaction between the amphiphilic building blocks resulting in precise nanostructures with control over size, shape and surface functional group displays, which is highly important for multivalent interaction with biological targets.14 Typical hydrogen-bond energies vary between 5 and 65 kcal mol−1 which provides a rather large window to fine-tune the properties of the nanostructures constructed by H-bonds.15 The association constants of complementary H-bonds among various units have been reported to be in the window of 103–108 M−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 which depends on multiple factors including chemical nature of the H-bonding donor and acceptor groups, solvent polarity and the number and sequence of H-bonding groups in a particular molecule. Stability of H-bonds is usually higher in non-polar solvent while in a polar medium (including water) it is less pronounced. However, in biological systems, stable H-bonds are formed by shielding the H-bonding sites from the competing water molecules within the hydrophobic pockets. For example, the DNA double helix is constructed by the complementary H-bonding between the specific base pairs (pairing enthalpies of 21.0 kcal mol−1 for guanine–cytosine and 13.0 kcal mol−1 for adenine–thymine),17 which remain located inside the double helix by exposing the hydrophilic sugar-phosphate backbone on the surface. Likewise H-bonding between the amide groups occurs in the hydrophobic domain of the secondary structure of a protein. These examples have inspired chemists to design new synthetic amphiphilic building blocks containing H-bonding units in specific locations for precision self-assembly in an aqueous medium by directional molecular interaction rather than immiscibility driven aggregation. We have described such recent examples in this review with specific emphasis on the impact of H-bonding on the structure formation of abiotic amphiphilic small molecules and polymers in water. Related topics such as peptide-amphiphiles,18 or hydrogels19 also involve a significant role of H-bonding. However, these are distinct and extensive topics which have been reviewed elsewhere18,19 and therefore not included in this review.
16 which depends on multiple factors including chemical nature of the H-bonding donor and acceptor groups, solvent polarity and the number and sequence of H-bonding groups in a particular molecule. Stability of H-bonds is usually higher in non-polar solvent while in a polar medium (including water) it is less pronounced. However, in biological systems, stable H-bonds are formed by shielding the H-bonding sites from the competing water molecules within the hydrophobic pockets. For example, the DNA double helix is constructed by the complementary H-bonding between the specific base pairs (pairing enthalpies of 21.0 kcal mol−1 for guanine–cytosine and 13.0 kcal mol−1 for adenine–thymine),17 which remain located inside the double helix by exposing the hydrophilic sugar-phosphate backbone on the surface. Likewise H-bonding between the amide groups occurs in the hydrophobic domain of the secondary structure of a protein. These examples have inspired chemists to design new synthetic amphiphilic building blocks containing H-bonding units in specific locations for precision self-assembly in an aqueous medium by directional molecular interaction rather than immiscibility driven aggregation. We have described such recent examples in this review with specific emphasis on the impact of H-bonding on the structure formation of abiotic amphiphilic small molecules and polymers in water. Related topics such as peptide-amphiphiles,18 or hydrogels19 also involve a significant role of H-bonding. However, these are distinct and extensive topics which have been reviewed elsewhere18,19 and therefore not included in this review.
Probing H-bonds in an aqueous medium: a small shift of electron density from the proton acceptor to the donor during H-bonding helps to probe it by spectroscopy techniques. Vibrational spectroscopy is a key technique to determine H-bonds. The formation of H-bonds causes a large red-shift (>100 cm−1) of the X–H stretching vibration due to a lengthening of the X–H bond. In addition, the intensity of the new band is significantly increased and broadened where the magnitude of the red-shift correlates linearly with the H-bond strength.201H NMR spectroscopy is another useful method to detect H-bonding. The sharpness of the peak and relative downfield position of the –NH protons can give an indication of H-bonds involving amide, urea or urethane functionalities.21 The magnitude of the chemical shift is indicative of the strength of the H-bond. Meijer and co-workers investigated dimerization constants of a series of oligo(ethylene oxide) substituted 2-ureido-pyrimidones, differing in the length of ethylene oxide and aliphatic chain length, by probing the H-bonding by concentration dependent NMR and IR spectroscopy.22 They also demonstrated probing H-bonding in water soluble 1,3,5-tricarboxamide (BTA) derivatives23 by solvent dependent IR spectroscopy in D2O and correlated the results obtained from previous findings of theoretical studies. Sánchez and co-workers reported thorough investigation on H-bonding by FT-IR spectroscopy and concentration/solvent dependent NMR spectroscopy in the context of supramolecular polymerisation of a C3-symmetric OPE based trisamide.24,25
H-Bonding mediated assembly of small molecule amphiphiles
One of the early examples of H-bonding mediated supra-amphiphilic assembly in an aqueous medium has been demonstrated by Kunitake and co-workers.26,27 A melamine derivative containing two pendant hydrocarbon chains and an ammonium-salt derivatized cyanuric acid (1 and 2, respectively, in Fig. 1) were co-assembled in water by complementary H-bonding between melamine–cyanuric acid to generate a supramolecular two-tail surfactant which produced a bilayer membrane.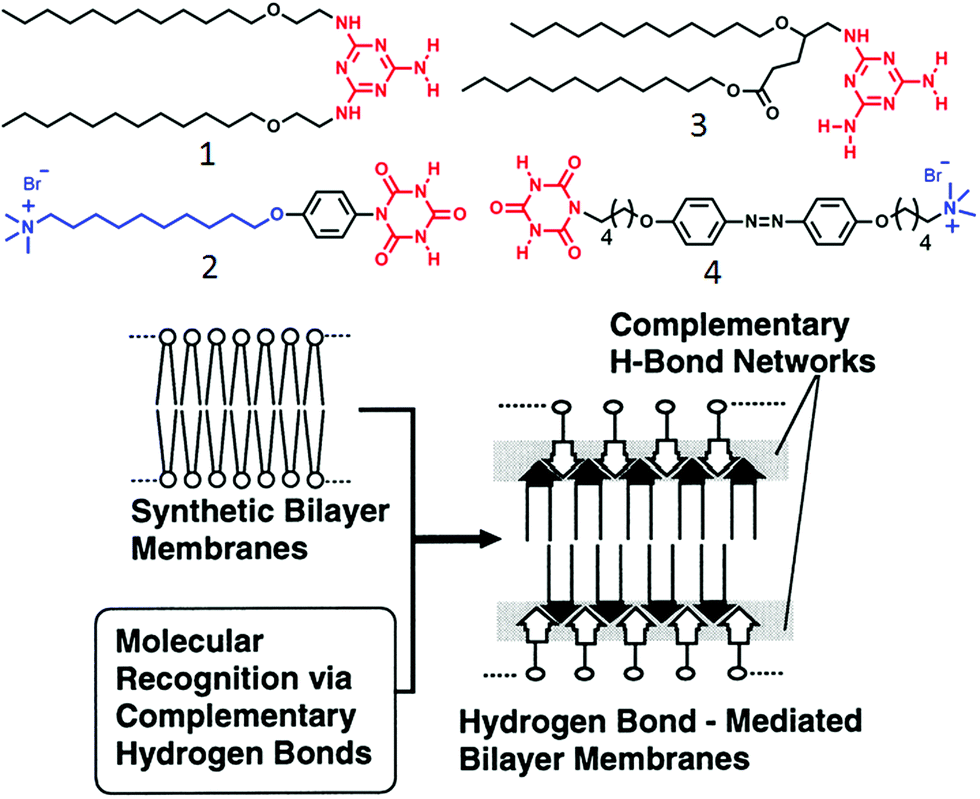 | ||
| Fig. 1 (top) Structure of amphiphiles containing complementary H-bonding functionalities. (bottom) Schematic illustration of supramolecular membrane formation via complementary H-bonding. Adapted and modified with permission from ref. 27. Copyright 1998, American Chemical Society. | ||
In this molecular design, the presence of alkyl chains promoted the pre-organization of the amphiphiles and the phenyl unit introduced a hydrophobic pocket which further facilitated extended intermolecular H-bonding network in a competitive aqueous environment to make a thermodynamically stable bilayer nanostructure as illustrated in Fig. 1. The self-assembled aqueous dispersion showed a reversible phase transition from highly ordered to a liquid crystalline state at 53 °C and dissociated on further increase in the temperature.21 Subsequently, they extended the study28 with melamine–cyanuric acid derivatives (3, 4 in Fig. 1) wherein a photo-responsive azobenzene moiety was incorporated in the cyanuric acid derived amphiphile. Self-assembly of compound 4 alone generated a globular nanostructure whereas a 1:1 mixture of 3 and 4 was found to form a supra-amphiphile which further assembled to produce photo-responsive helical nano-fibers. When the azobenzene moiety was in a trans-form, a stable network was produced but its isomerization to the cis-form destroyed the self-assembly motif due to ineffective packing of the molecules.
In a different approach single stranded DNA/analogues have been used as effective templates for constructing supramolecular amphiphiles by virtue of complementary H-bonding. Shimizu and coworker reported29 the formation of double helical nanofibers by H-bonding between thymidine-appended bolaamphiphiles with adenylic acid oligomers of varying chain length (Fig. 2). Assembly of compound 5 with short oligomers exhibited intertwined fibrillar morphology whereas with the longer chain oligomers, discrete helical nanofibers were formed with helical pitch of approximately 20 nm. Detailed structural analysis demonstrated that two strands of oligomer 6 were involved in complementary H-bonding with bolaamphiphile 5 through the adenine–thiamine complementary base pairs which further wound around the vertically stacked bolaamphiphile along the long axis to produce helical nanofibers. An alkyl chain of bolaamphiphile provided a hydrophobic pocket for effective base pairing and the hydrophilic deoxyribose and phosphodiester moieties were exposed to water (Fig. 2). Subsequently, they studied30 similar supramolecular assembly of H-bonded complexes between various bolaamphiphiles consisting of different nucleobases (guanylic acid-, adenylic acid, thymidylic acid, and cytidylic acid) and complementary 20-meric oligonucleotides in aqueous solution. Diverse morphologies could be produced including vesicles, 2D nano-sheets or fibers depending on the appended base to the bolaamphiphile and its complementary oligonucleotide base pairs.
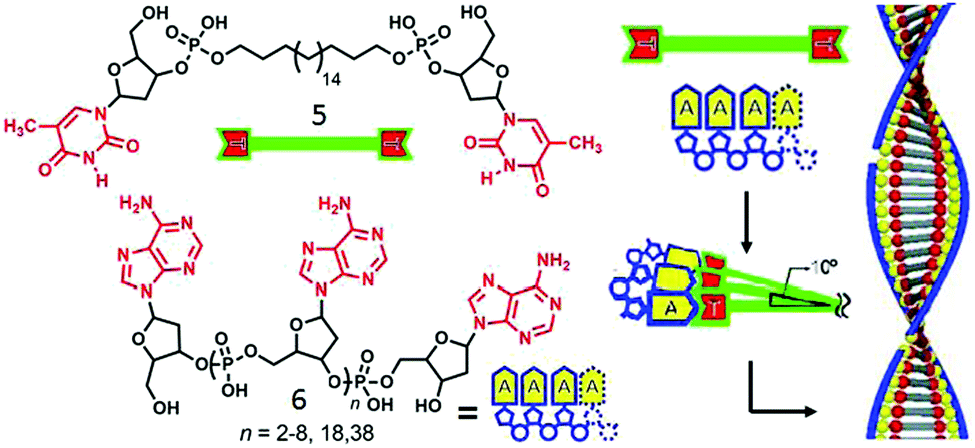 | ||
| Fig. 2 (left) Structure of thymidine-appended bolaamphiphile and adenylic acid oligomers. (right) Proposed mechanism of templated assembly. Adapted and modified with permission from ref. 23. Copyright 2003, Wiley-VCH. | ||
Such templating approach based on H-bonding has been further exploited by Schenning and coworkers31,32 using nucleic acid appended bolaamphiphiles containing a π-conjugated oligo(p-phenylenevinylene) (OPV) core and a complementary oligo-nucleotide (Fig. 3). Oligo-oxyethylene derivatized diamino-triazine containing OPV (7, Fig. 3) could be anchored to the oligo-thymine template (8) to produce a two-component assembled structure, stabilized by complementary H-bonding as well as π–π interaction between the OVP units (Fig. 3).
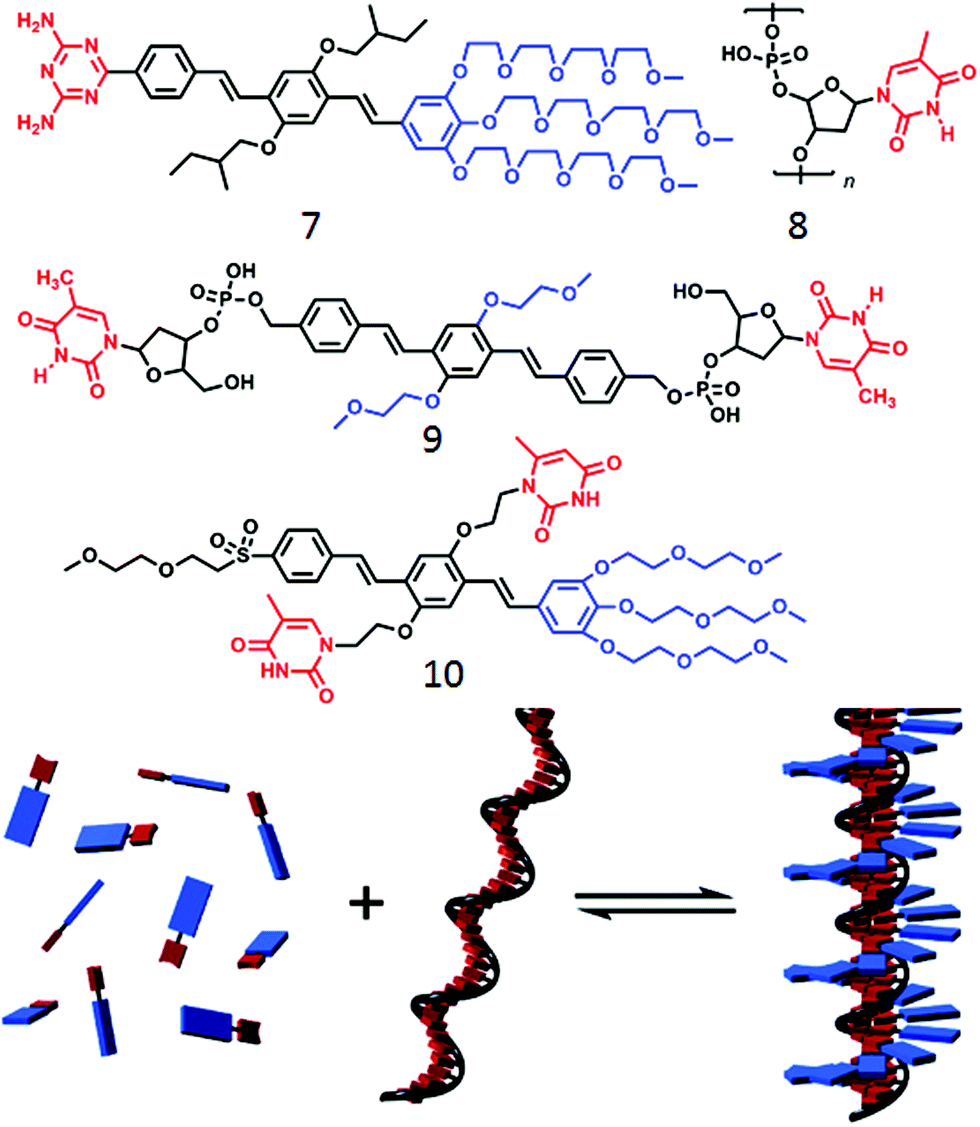 | ||
| Fig. 3 (top) Molecular structure of various OPV unit as reported in ref. 25–27. (bottom) Schematic representation of ssDNA templated assembly between compounds 8 and 9. Adapted with permission from ref. 31. Copyright 2007, American Chemical Society. | ||
In a slightly different design,32 the ssDNA-templated assembly was explored using thymine appended OVP (9) wherein the thymine moiety was made to interact with the oligo-deoxyadenylic acid by a complementary H-bond (A–T base pair) to produce helical fibers. In a related work,33 Lo and co-workers demonstrated co-assembly of a thymine-substituted donor–acceptor π-conjugated amphiphile (10) with oligoadenine templates producing stable tubular assemblies through various noncovalent interactions including π–π stacking, dipole–dipole interactions, and adenine–thymine (A–T) H-bonding. While the forgone discussion showed examples on two-component supramolecular assembly of amphiphiles by complementary H-bonding, Ferniri and coworkers reported34 an interesting example of a cationic amphiphile (11, Fig. 4) with a hydrophobic bi-cyclic unit that contains a Watson–Crick donor–donor–acceptor (DDA) array of guanine as well as acceptor–acceptor–donor (AAD) array of cytosine in the same molecule. It self-assembled via complementary H-bonding in water to form a six-membered macrocycle (rosette structure), which hierarchically organized to form nanotubes. Compound 12 (Fig. 4) with two such bi-cyclic units produced thermodynamically more stable rosette nanotubes via the formation of an energetically favorable syn-stacked parallel dimer.35
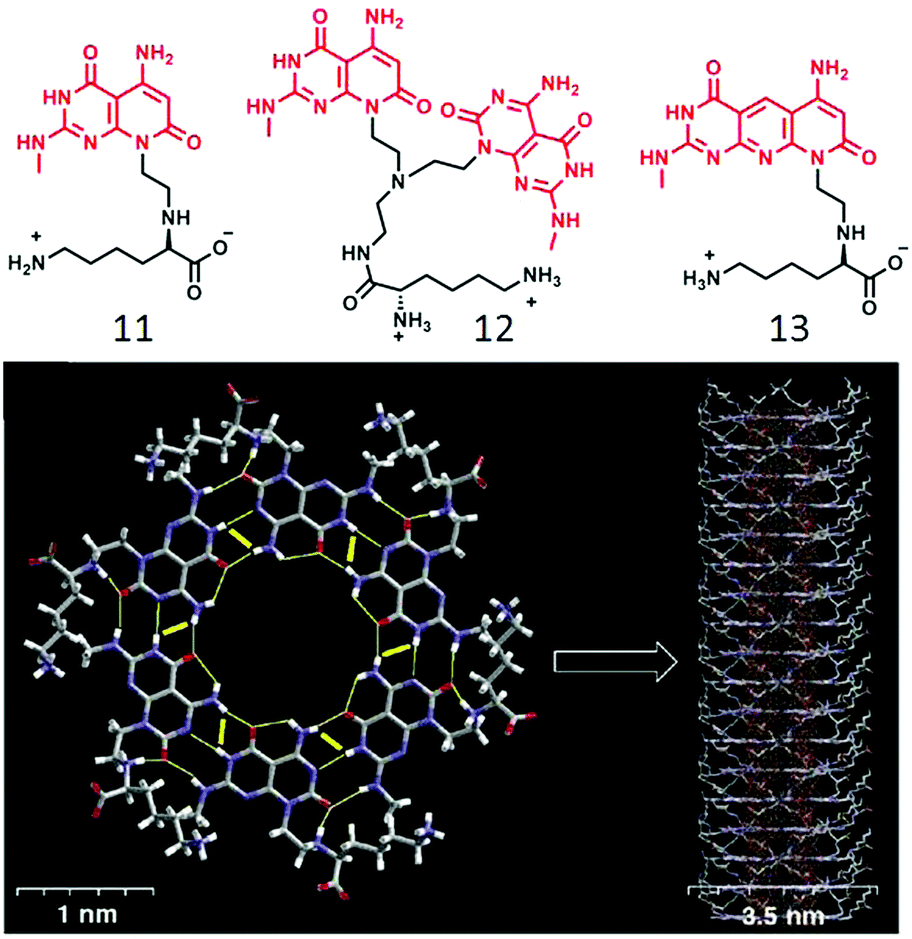 | ||
| Fig. 4 (top) Molecular structure of compounds 12, 13 and 14. (bottom) Hierarchical self-assembly of compound 12 to form rosette nanotubes. Adapted with permission from ref. 34. Copyright 2001, American Chemical Society. | ||
Enhanced stability of the resulting nanotube originated from stronger H-bonding network due to the presence of two bases as well as better π–π interaction. They also have shown,36 that the diameter of the nanotubes could be altered by fine-tuning the molecular structure of the amphiphilic building block. Compound 13 (Fig. 4) has similar H-bonding array like compound 11, but separated by an internally fused pyridine ring, which resulted in the formation of nanotubes with a relatively wider inner diameter than that formed by 11.
We have studied the impact of H-bonding on the supramolecular assembly of π-amphiphiles in which naphthalene-diimide (NDI) serves as the hydrophobic chromophore.37,38 NDI-bolaamphiphile 14 (Fig. 5) consists of a central hydrophobic NDI chromophore which is attached to two hydrophilic wedges by a hydrazide group. It showed spontaneous vesicle-like assembly in water by synergistic H-bonding among the hydrazide groups and extended π-stacking between the NDI chromophores. NDI is well-known to form a charge-transfer (CT) complex with a pyrene donor.10 To investigate if that has any effect on the self-assembly process, 14 was co-assembled with pyrene (1:1) in an aqueous medium which resulted in deep green color solution indicating NDI–pyrene CT-complex formation which resulted in transformation of initially formed spherical nanostructures to elongated fibrils after aging and hydrogelation. To examine the actual role of H-bonding in the self-assembly process, compound 15 (Fig. 5) was studied as a control molecule as it lacks the hydrazide groups.38
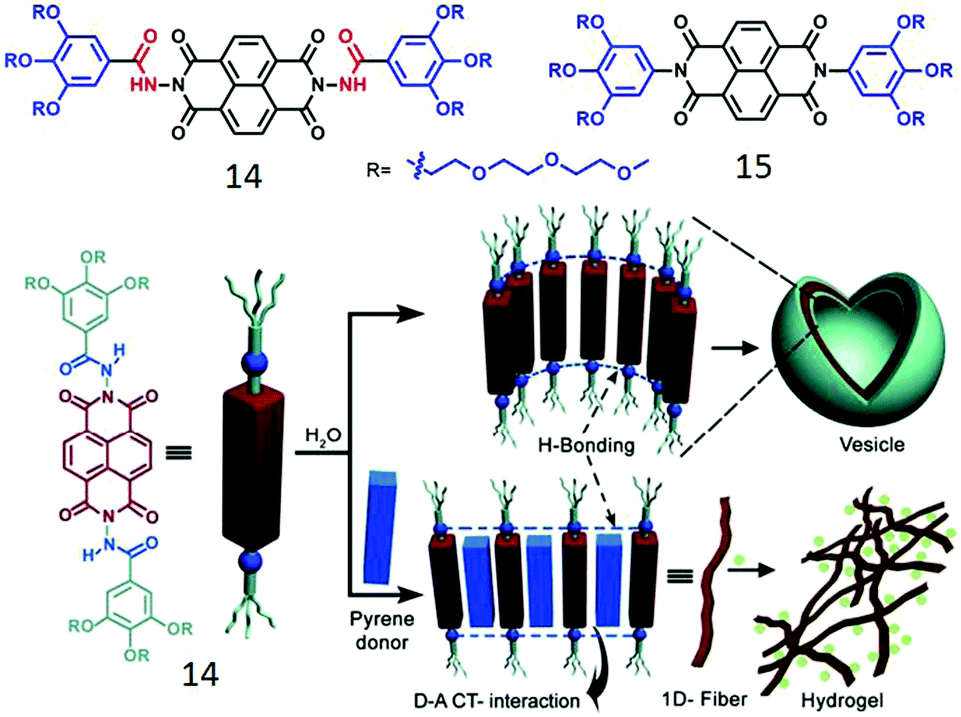 | ||
| Fig. 5 (top) Structure of NDI-derived bolaamphiphiles. (bottom) Schematic showing H-bonded assembly of 14 and its donor intercalation driven morphology transition. Adapted with permission from ref. 37. Copyright 2012, Wiley-VCH. | ||
Although 15 showed similar vesicle like morphology, its critical aggregation concentration was significantly lower than that of 14 due to increased hydrophobicity in the absence of the two hydrazide groups. Interestingly, 14 exhibited a lower critical solution temperature at 65 °C while 15 did not show any such behavior and neither was it able to intercalate a pyrene donor although having the same acceptor NDI chromophore. These results suggest that H-bonding among the hydrazide groups loosens the π-stacking a bit and enables intercalation of a donor chromophore.
Inspired by these results we extended the study to unsymmetric π-amphiphiles, containing a single H-bonding group. It was anticipated that such molecular design might be useful to control the lateral orientation of the amphiphiles. Two such systems (16 and 17, Fig. 6a), differing only by a single functionality (amide or ester), were compared to probe the specific role of H-bonding on self-assembly.39 Compound 16 containing the amide group formed a hydrogel and cryo-TEM images (Fig. 6b) revealed a long fibrillar structure confirming formation of a one dimensional supramolecular polymer while 17 did not show any gelation and formed a spherical micellar structure. To further enhance the complexity, co-assembly of 16 was studied with a donor (pyrene)-containing amphiphile 18 (Fig. 6a) in which the pyrene chromophore was attached with a dicarboxylic acid containing hydrophilic head group with an amide group. The relative spacer lengths between the NDI/pyrene chromophore and the amide groups were adjusted in such a way that it could facilitate synergistic H-bonding and CT-interaction (Fig. 6c).40 Indeed 16 + 18 (1:1) produced a deep red color indicating CT interaction and in contrast to the fibrillar structure exhibited by 16, the mixture showed polymersome-like spherical assembly while 18 alone produced relatively small micellar aggregates. Zeta potential measurements showed highly negative surface charge indicating display of the carboxylate ions at the outer surface which was useful to inhibit the activity of an enzyme Chymotrypsin which has positive charges near its active site. Interestingly the control molecule 17 (without H-bonding unit) with the same pyrene derivative did not form an effective CT-complex and the surface charge of the nanostructure was found to be negligible, indicating the predominant role of H-bonding in controlling the lateral orientation of the NDI and pyrene chromophores. The H-bonded co-assembled structure could be destroyed under a reducing environment as it reduced the NDI chromophore and thus diminished the alternating stacking propensity with electron rich pyrene.
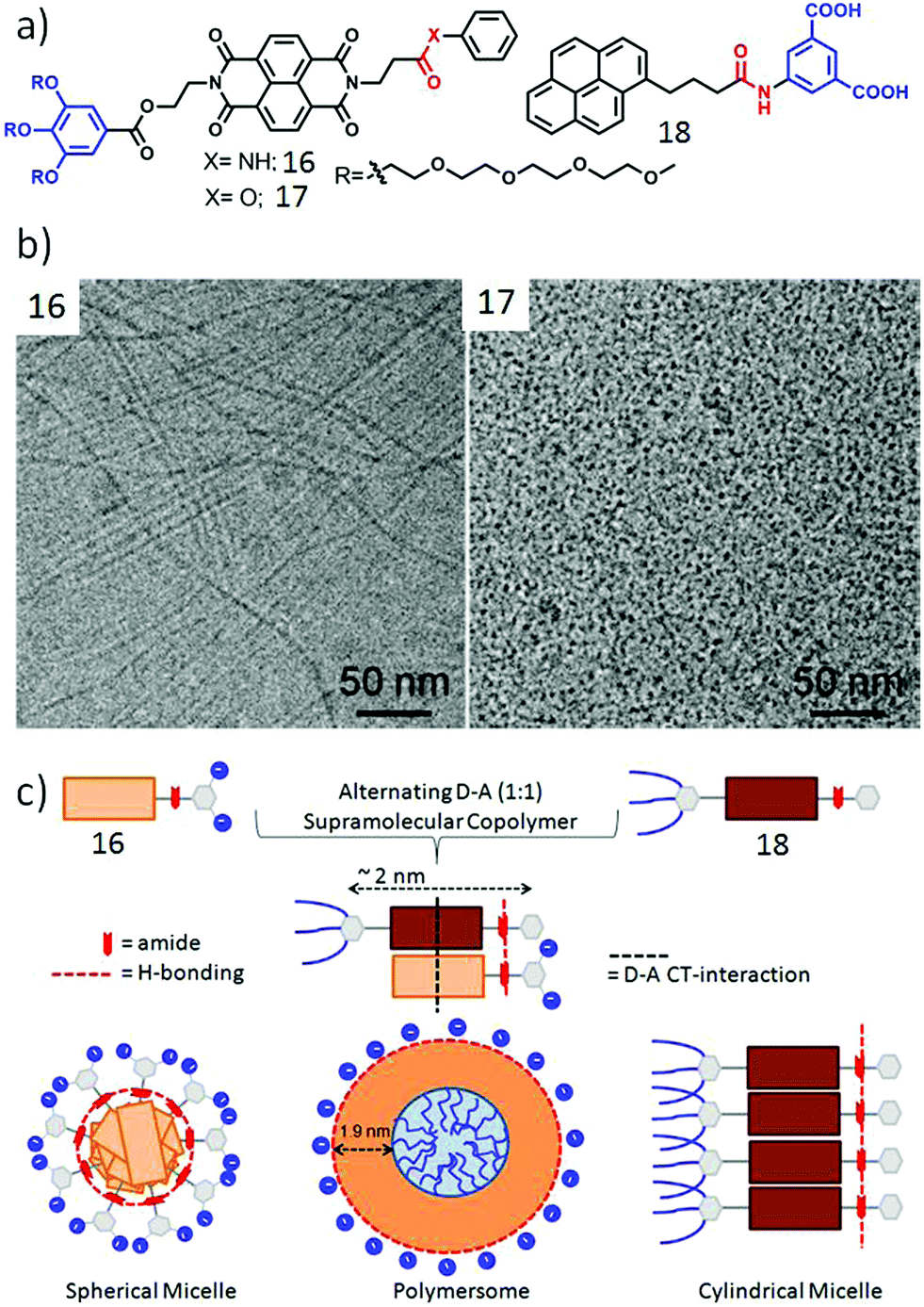 | ||
| Fig. 6 (a) Structure of NDI derivative 16, 17 and pyrene derivative 18. (b) Cryo-TEM images of aqueous assemblies of compounds 16 and 17. Adapted with permission from ref. 39. Copyright 2017, American Chemical Society. (c) Schematic presentation of H-bonding driven homo-assembly and co-assembly. Adapted with permission from ref. 40. Copyright 2018, Wiley-VCH. | ||
We further extended the unsymmetric design to bola-shape NDI-derived amphiphiles (19, 20, Fig. 7) containing a non-ionic hydrophilic wedge and an anionic head group.41 Both of these compounds showed unilamellar polymersome like assembly with Dh in the range of 120–160 nm and identical physical properties except for the zeta potential which suggested a highly negative surface charge for 19 while almost neutral surface for the assembled structure of 20. This was also reflected in their ability to inhibit the enzymatic activity of Chymotrypsin; while 19 was highly effective, 20 merely inhibited it. Such fully contrasting surface charge could only be rationalized by H-bonding driven head-to-head orientation of both the compounds and further control on the direction of the curvature of the membrane ensuring that the H-bonded chain remains at the inner-wall of the polymersome membrane. Subsequent studies42 with different aromatic anionic head groups demonstrated the possibilities of precisely tuning the size and surface charge density of such polymersome-like structures by systematic variation of the ionic head groups. Control over surface functional group display has been attempted before and appeared to be a challenging task.43
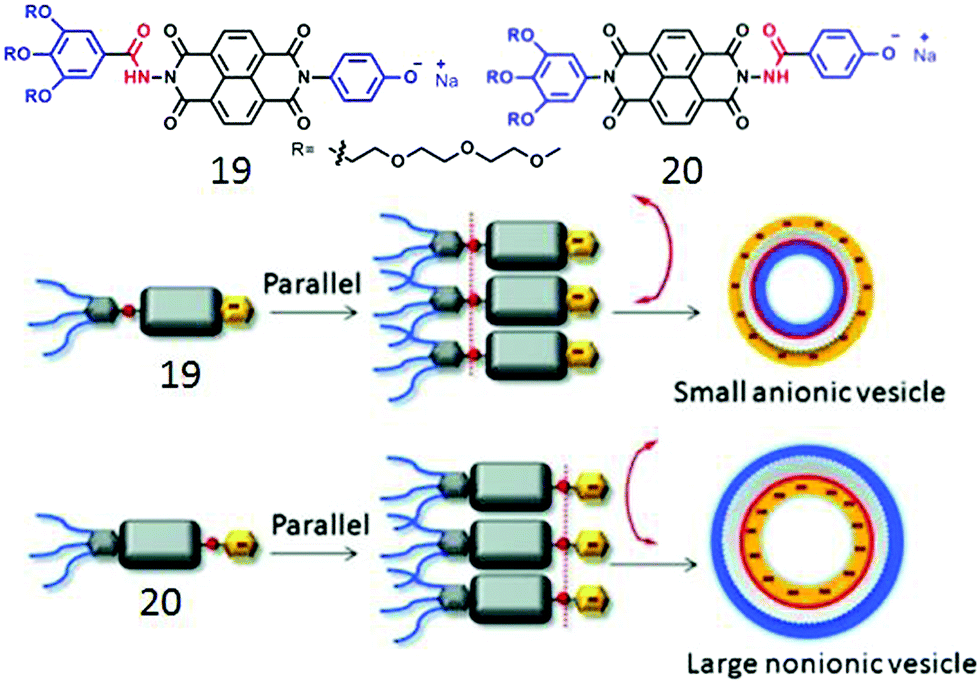 | ||
| Fig. 7 (top) Structure of bolaamphiphile 19 and 20. (bottom) Proposed model of H-bonding driven directional self-assembly. Adapted with permission from ref. 41. Copyright 2015, Wiley-VCH. | ||
The present design based on H-bonding appeared promising to address this long standing issue and thus to examine its general applicability we examined similar NDI-based amphiphiles with different head group structures (Fig. 8). For example, 21 showed similar polymersome-like structures in acidic water with cationic surface charge confirming the display of the pyridinium groups on the outer surface. In a curiosity driven experiment its assembly was compared in water and tetrachloroethylene (TCE).
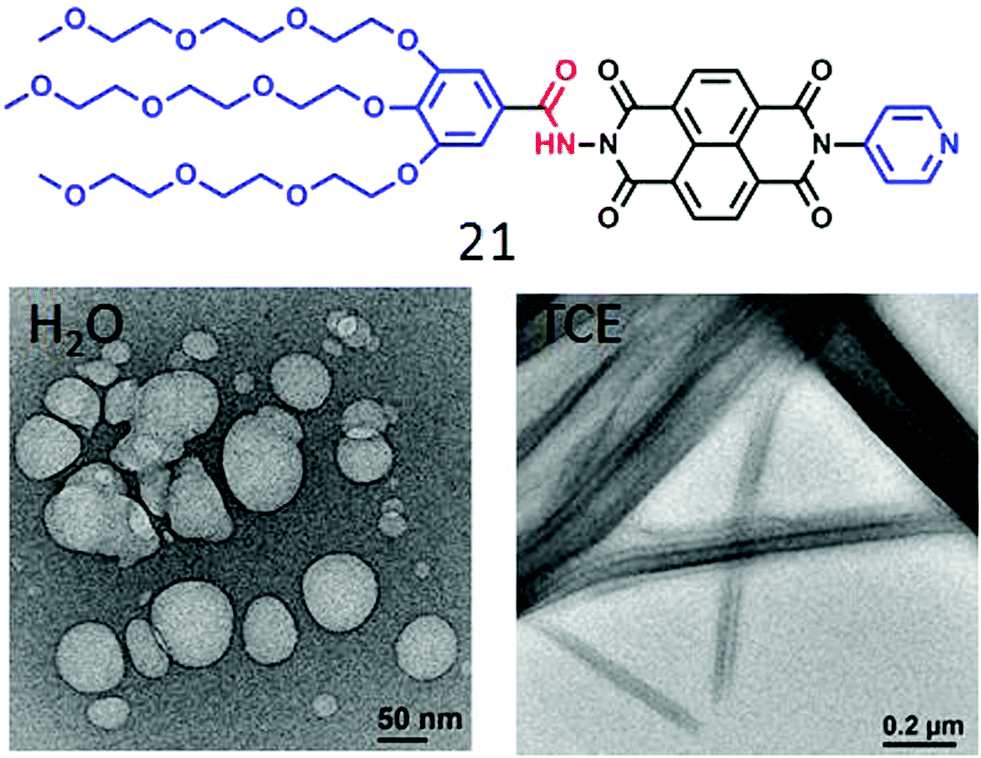 | ||
| Fig. 8 Structure of a pyridine head group containing NDI-bolaamphiphile 21 and its nanostructure morphology in H2O and TCE. Adapted with permission from ref. 44. Copyright 2018, Royal Society of Chemistry. | ||
Unlike the polymersome-like structure in water, it formed well-defined nanotubes (Fig. 8) in TCE with highly crystalline packing of the molecules in the wall of the tubes and this particular architecture was proved to be relatively better for providing percolated charge transport pathways compared to polymersome-like assembly.44 We have also investigated a series of glucose appended NDI-bolaamphiphiles (22, 22a, 23 and 24 in Fig. 9)45 with specific objectives to understand the effect of different H-bonding functionalities (hydrazide or amide) on morphology, thermodynamics and multivalent binding with a lectin protein ConA.
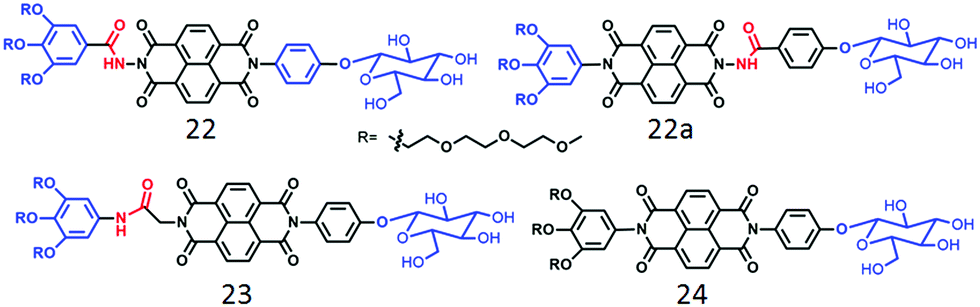 | ||
| Fig. 9 Structure of carbohydrate-appended unsymmetric NDI-bolaamphiphile as reported in ref. 45. | ||
First of all, both compounds 22 and 22a, containing the hydrazide group, showed polymersome-like assembly as before and the full contrast in the surface functional group display was evident by their ability to interact with ConA. While compound 22 showed strong binding affinity, 22a did not interact at all, indicating full control over functional group display and multi-valent binding by H-bonding. In contrast to 22, compound 23 with a relatively flexible amide group lead to the formation of 1D nanostructures with significantly stronger binding propensity with ConA which was attributed to the more flexible and adaptive cylindrical structure than the polymersome morphology. On the other hand, compound 24, lacking any H-bonding group, produced nanoparticles by random lateral orientation of the building block and exhibited negligible binding with ConA.
Bhosale and coworkers have reported46 an interesting flower-shape supramolecular nanostructure formed by co-assembly of phosphonic acid appended NDI (25, Fig. 10) with complementary H-bonding moiety melamine. Systematic study revealed that π–π interaction along with van der Waals interaction constructed the core of the nanoflowers whereas, the phosphonic acid groups formed strong H-bonds with melamine and favored the directional growth in a 2D fashion. Finally the steric hindrance among the large phosphonate groups induced a twist and folding of the sheets causing 3D fractal growth to generate such elegant nanostructures.
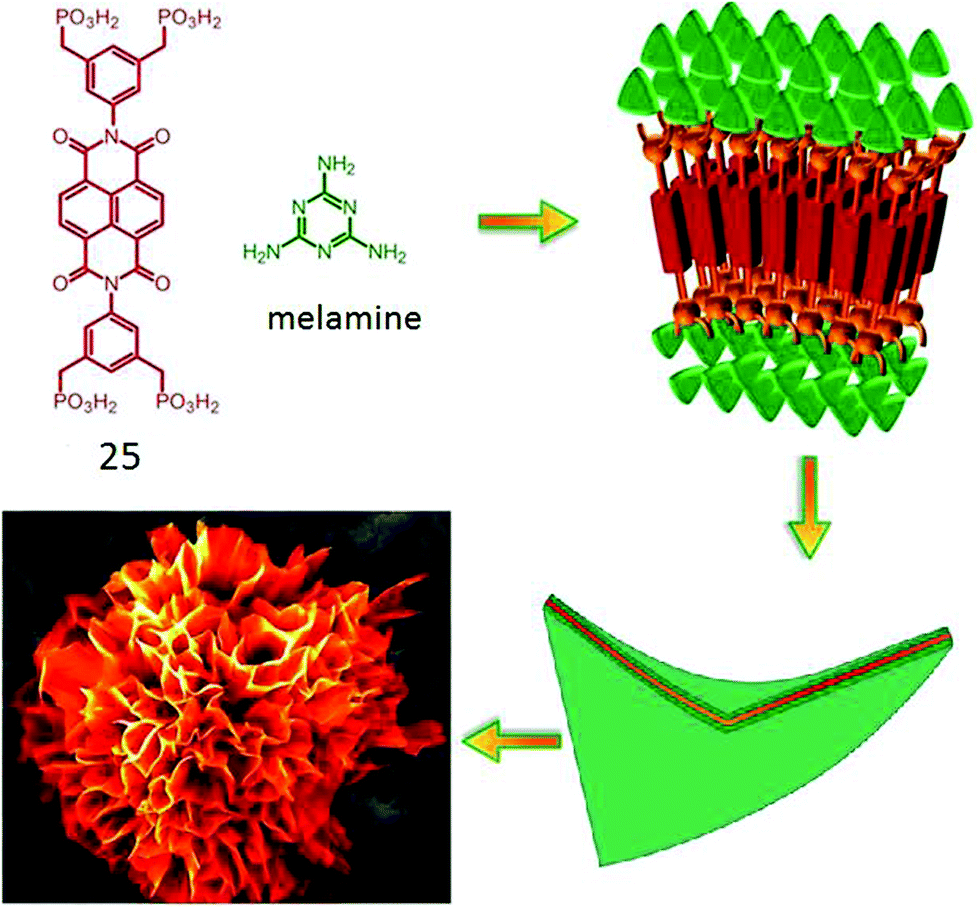 | ||
| Fig. 10 Structure of phosphonoic acid appended NDI-amphiphile and its proposed self-assembly pathway to produce nano-flower morphology. Adapted with permission from ref. 46. Copyright 2015, Nature Publishing Group. | ||
For another well-known dye in the rylene family, perylene-bisimide (PBI), although it has been studied extensively in the context of supramolecular assembly in organic solvents,47 H-bonding driven assembly of PBI derivatives in water is relatively less explored. Würthner and co-workers48 have synthesized an oligo-oxyethylene-aryl jacketed PBI derivative (compound 26, Fig. 11) bearing free NH groups at the imide positions, which produced a hydrogel due to formation of an entangled fibrillar network via self-assembly. Interestingly, upon heating, the red hydrogel reversibly transformed to blue hydrogel accompanied by enhanced fluorescence caused by chromophoric arrangement from co-facial packing to entropically driven H-bonding assisted slipped stacking arrangement (J type aggregation).
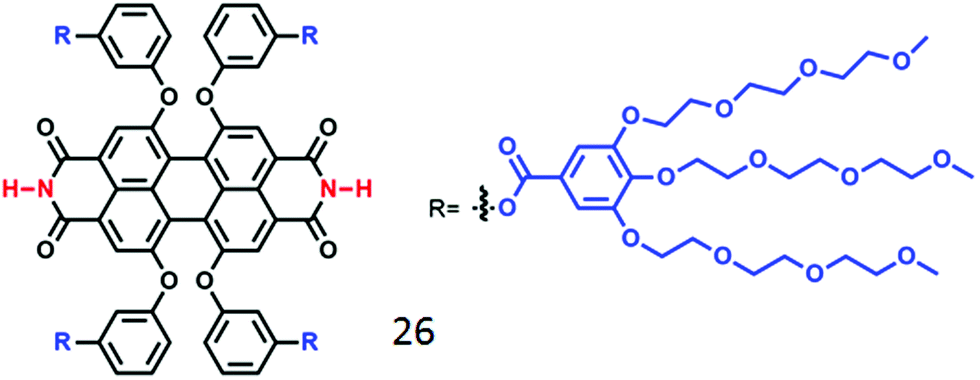 | ||
| Fig. 11 Structure of the water-soluble H-bonding containing PBI derivative as reported in ref. 43. | ||
Meijer, Palmans and co-workers have extensively studied supramolecular polymerization and chirality issues with benzene 1,3,5-tricarboxamide (BTA) derivatives in organic solvents49 and amphiphilic BTA-derivatives in water (Fig. 12).50 In the compound 27, the BTA-core was attached to amphiphilic chains composed of hydrocarbon spacer (to create hydrophobic pocket) followed by oligo-oxyethylene chains (to improve water solubility). Compound 28 contains a stereogenic center to probe the self-assembly by CD spectroscopy. Both 27 and 28 were found to form micrometer long fibers by three-fold intermolecular H-bonding along with π-staking. Control molecule 29, with methylated amide groups, did not show such 1D supramolecular polymerization indicating a specific role of H-bonding in supramolecular assembly.
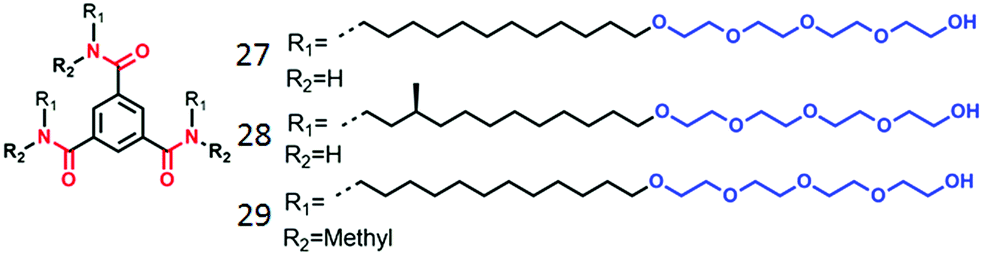 | ||
| Fig. 12 Structure of amphiphilic BTA-derivatives as reported in ref. 45. | ||
Systematic studies with different peripheral chains confirmed an important role of the hydrophobic/hydrophilic balance in the occurrence of H-bonded assembly. It was found that the undecyl spacer is the minimum length required for this kind of BTA-amphiphile to self-assemble in an aqueous medium.23 Furthermore, the monomer exchange of the dye-labeled BTA molecules in the self-assembled fibers was investigated by Förster resonance energy transfer (FRET) and more elegantly visualized using super-resolution stochastic optical reconstruction microscopy (STROM) and stochastic simulation by studying the co-assembly of 27–27a and 27–27b (Fig. 13).51 Studies revealed that the dye labelled monomers exchange (half-life of one hour) from fibre to fibre by a release-incorporation mechanism producing mixed polymers with homogeneous distribution of dye molecules within each fiber.
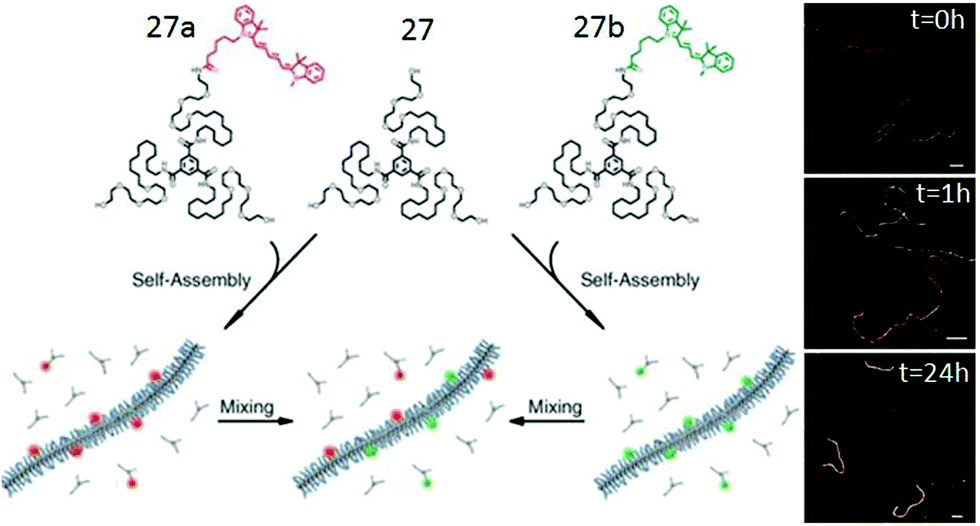 | ||
| Fig. 13 Schematic overview of the mixing experiment and time resolved reconstruction microscopy (STORM). Adapted with permission from ref. 51. Copyright 2014. The American Association for the Advancement of Science. | ||
In a recent study,52 this group has unraveled the nanoscale organization and the structural dynamics of a supramolecular polymer of BTA-amphiphiles by hydrogen/deuterium exchange (HDX) mass spectrometry. For this study, BTA polymers (Fig. 14) prepared in H2O were diluted 100 times in D2O for hydrogen–deuterium exchange to occur, where a maximum of six hydrogen atoms are able to exchange. The three OH groups exchanged immediately as they are exposed to surrounding D2O and on the other hand it took one hour to many days for the amide exchange due to reduced D2O accessibility inside the hydrophobic pocket.
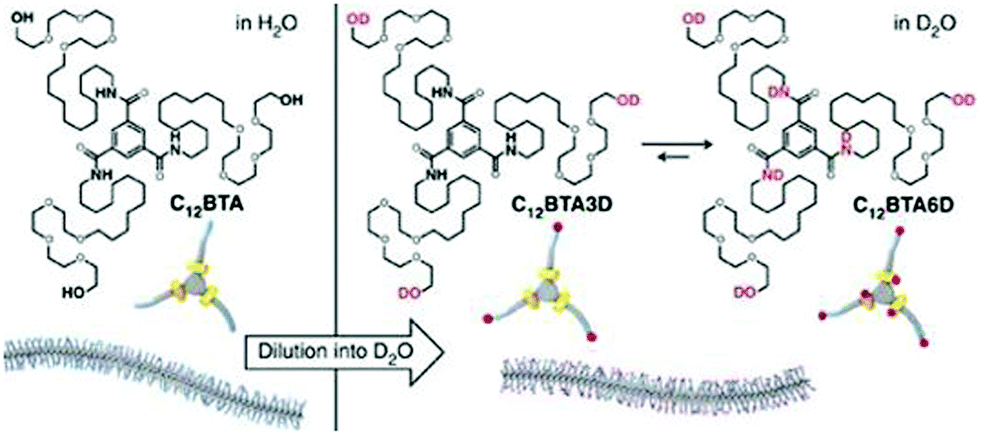 | ||
| Fig. 14 Schematic representation of the supramolecular polymer formed in water and H/D exchange process in D2O. Adapted with permission from ref. 52. Copyright 2017, Nature Publishing Group. | ||
Zhang and co-workers demonstrated53 aqueous self-assembly of diketopyrrolopyrrole (DPP)-chromophore based bolaamphiphile 30 (Fig. 13) which produced supramolecular nanofibers by H-bonding interaction. Very recently, Yamaguchi and co-workers demonstrated controlled supramolecular polymerization of amide-functionalized DPP-derived amphiphile 31 (Fig. 15).54 Kinetically trapped monomeric species were obtained via rapid cooling of the aqueous solution of 31. Addition of seed initiated the supramolecular assembly with structural precision in the same system (Fig. 15).
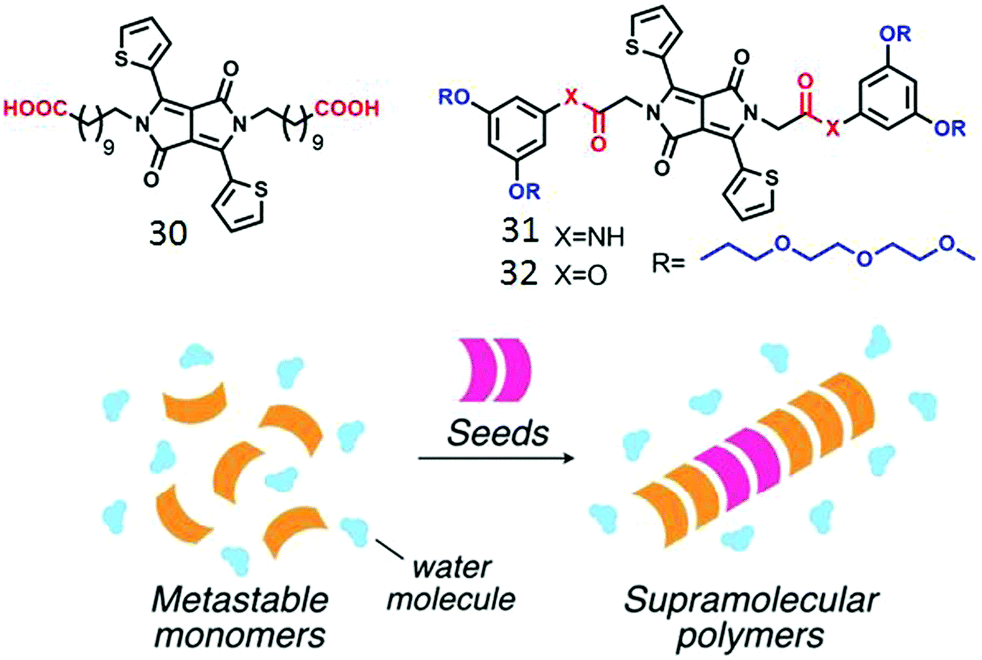 | ||
| Fig. 15 (top) Structure of the various amphiphilic DPPE derivatives. (bottom) Schematic illustration of seeded polymerization. Adapted with permission from ref. 53. Copyright 2017, Wiley-VCH. | ||
However no assembly was observed for the control molecule 32 without the amide units which further justified the importance of H-bonding in supramolecular polymer formation. While H-bonding driven seeded supramolecular polymerization in an organic medium has been demonstrated with a few different types of building blocks,55 examples in aqueous medium are limited so far.
Zhang and co-workers have demonstrated56 interfacial polymerization of two miscible supramolecular H-bonding motifs. They have synthesized bifunctional supra-monomer (33, Fig. 16) containing ureidopyrimidinone units with isocyanate end groups and a series of diamine containing monomers (34). Quadruple H-bonding between the two ureidopyrimidinone groups along with isocyanate–amine reaction lead to the formation of a dynamic supramolecular polymer with tailor made structure and functions (Fig. 16).
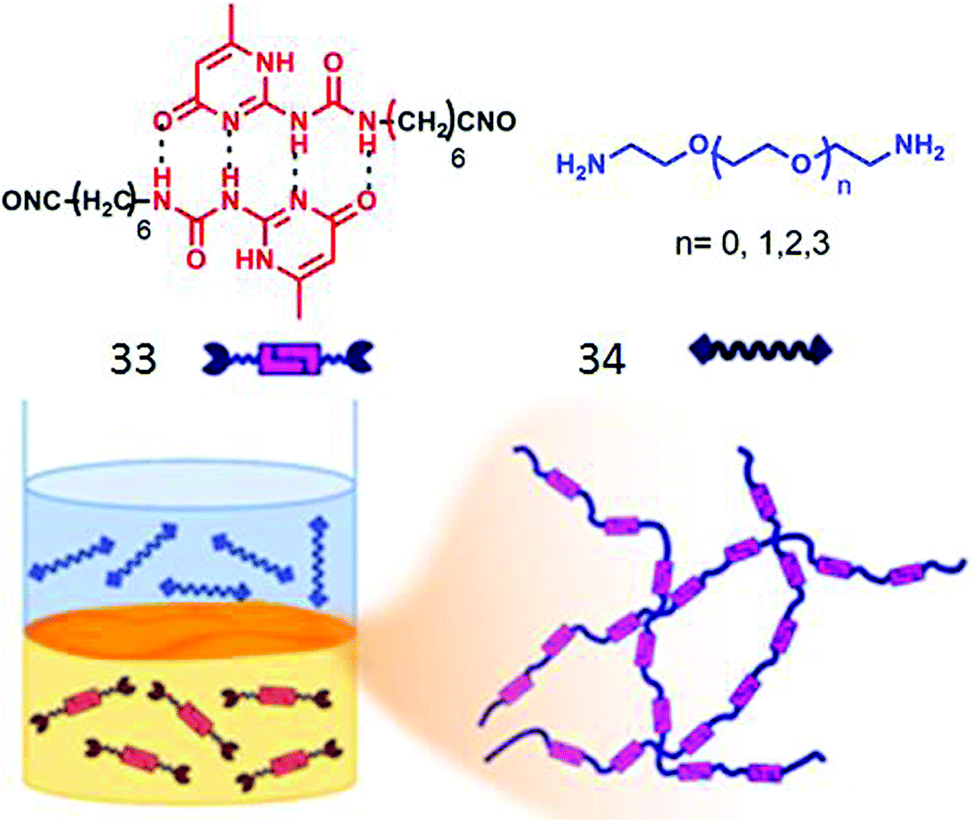 | ||
| Fig. 16 (top) Structure of the supramolecular monomer. (bottom) Schematic presentation of interfacial polymerization. Adapted with permission from ref. 56. Copyright 2018, American Chemical Society. | ||
In a subsequent report,57 they demonstrated “supramolecular emulsion interfacial polymerization” by polymerizing a supramonomer bearing two thiol groups connected to quadruple H-bonded ureidopyrimidinone groups with water soluble cationic monomers connected to maleimide end groups. At the oil–water interface, thiol–maleimide reaction of the two monomers generated supramolecular polymeric nanospheres. The size of the aggregates could be further tuned by changing the concentration of the two monomers.
Farnández and co-workers reported58 self-assembly of amphiphilic BODIPY-derived bolaamphiphile by H-bonding, π-stacking and hydrophobic interaction producing well-defined spherical micelles. It was postulated that initially formed small micelles produced larger nanostructures with further inter-particle H-bonding interaction.
H-Bonding mediated macromolecular assembly
In the past three decades aggregation of amphiphilic polymers3 with different connectivity (block copolymer, homopolymers, hyperbranched polymer, star polymer, bottle brush polymer, alternating copolymer and others) has been extensively studied. They generally exhibit much lower critical aggregation concentration, slower exchange dynamics and suitable aggregate size for bio-medical applications.4–7 However, the primary motivation for aggregation depends on the critical packing parameter similar to small molecule surfactants which limits the scope for predictable molecular design for a particular morphology or efficient functional group display for a multivalent binding with biological targets.In the domain of the immiscibility driven aggregation in conventional amphiphilic polymers, it has been envisioned that specific molecular recognition may offer new possibilities for realizing unchartered structure and function. Van Hest and coworkers59 have studied aqueous aggregation of two amphiphilic block copolymers (Fig. 17), namely poly(ethylene glycol)(PEG)-b-poly (adenine) and (PEG)-b-poly (thymine) (P1 and P2, respectively in Fig. 17) individually and in a mixture. Both produced micellar structures in water driven by hydrophobic collapse. Upon mixing the two polymers, micelles with different sizes were observed and the critical aggregation constant (CAC) increased significantly.
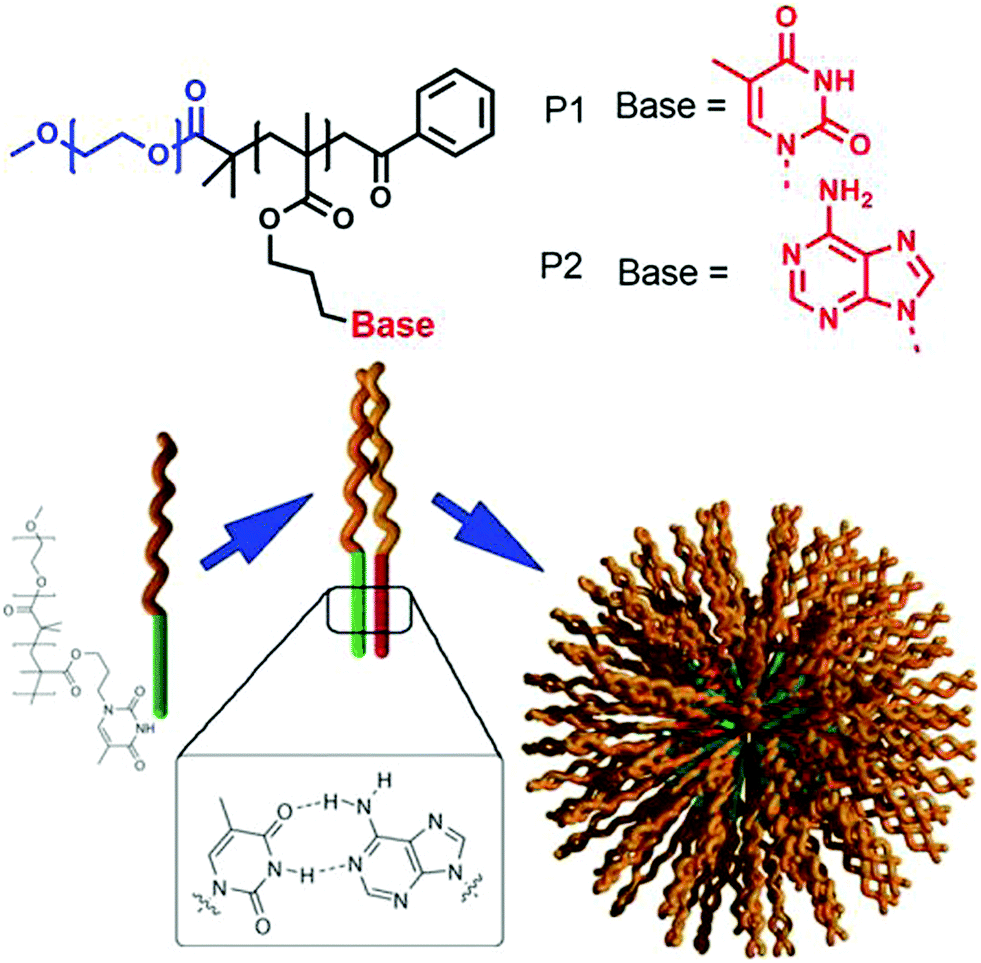 | ||
| Fig. 17 Structure of adenine and thymine appended amphiphilic block-copolymers and schematic representation of H-bonding driven co-assembly. Adapted with permission from ref. 59. Copyright 2006, Wiley-VCH. | ||
Systematic studies revealed that the complementary adenine (A)–thymine (T) H-bonding played a crucial role in co-assembly of the polymers and increased the solubility of the block-copolymers resulting in a shift of the CAC to a higher value.
O’Reilly and coworkers also studied nucleobase60a containing amphiphilic block-copolymers containing hydrophilic poly(oligo(ethylene glycol) methyl ether methacrylate) blocks and adenine and/or thymine containing hydrophobic blocks (P3–P5 in Fig. 18). These polymers formed spherical micelles or bi-continuous micelles by individual self-assembly or co-assembly in water, depending on the co-solvent used in the sample preparation by the solvent exchange method. Individual adenine or thymine assembly resulted into poor stability of the micelles whereas H-bonding interaction of the complementary base pair in co-assembly played the key role in forming well organized and stable nanostructures.
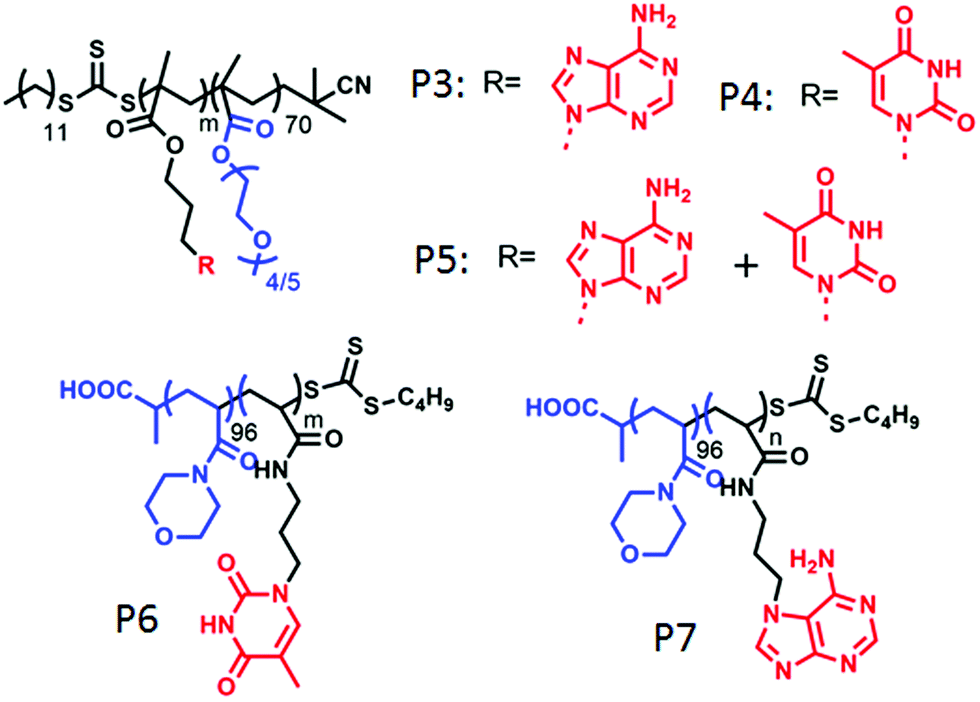 | ||
| Fig. 18 Structure of various nucleobase appended amphiphilic polymers reported by O’Reilly and co-workers. | ||
In a subsequent report,60b they have demonstrated morphological transformation of self-assembled nucleobase-containing amphiphilic polymers by a second polymer containing the complementary nucleobase. The amphiphilic polymer P6 (Fig. 18) showed spherical micellar aggregates in water with a nucleic acid core and hydrophilic (poly(4-acryloylmorpholine)-b-poly(3-(thymin-1-yl)propyl acrylamide) (PNAM-b-PTAm) corona. Addition of another diblock copolymer containing the complementary nucleobase (poly(4-acryloylmorpholine)-b-poly(3-(adenine-9-yl)propyl acrylamide) (PNAM-b-PAAm)) (P7 in Fig. 18) caused a morphological change in these micelles.
For micelles with moderate hydrophobic TAm blocks, with insertion of a complementary base containing polymer, a decrease in the micellar size was observed. Scattering study together with SLS confirmed a decrease in aggregation number (Nagg) with addition of complementary polymer, which was triggered by low interfacial tension of the hydrophobic chain that finally induced the micellar reorganization by reducing corona-chain repulsion and core-chain stretching. On the other hand, micelles with a long hydrophobic TAm block having a higher energy barrier of exchange, transformed to cylindrical micelles in order to reduce the total free energy of the system.
In a recent work,61 they have utilized a “grafting to” methodology to form mixed corona micelles by exploiting multiple hydrogen bonding between thymine (T) containing preformed micelles and adenine (A) conjugated polymers. To a pre-formed micelle of poly(4-acryloylmorpholine) polymer (P8, Fig. 19) with thymine units at the core, addition of complementary diblock copolymers P9 containing thermo-responsive poly(N-isopropylacrylamide) (PNIPAM) produced mixed corona micelles with higher hydrodynamic radius (Fig. 19). The size of the micelles was successfully tuned by changing the length of the complementary di-block polymer. Moreover, the thermo-responsive behavior of the PNIPAM block was used to reversibly display or conceal a desire functional group at the surface of the micelles (Fig. 19).
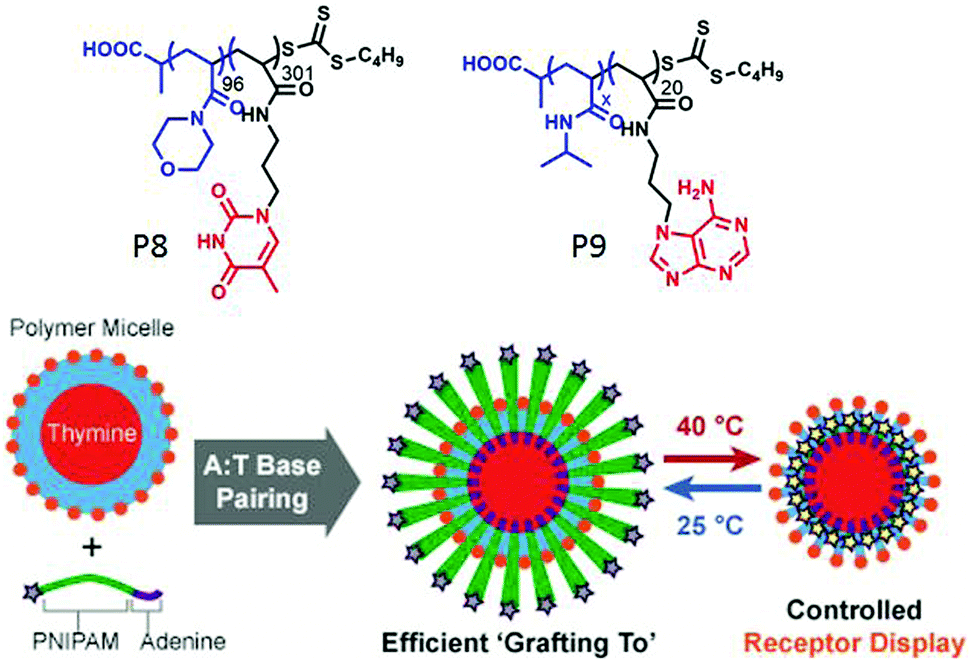 | ||
| Fig. 19 Structure of polymer P8 and P9 and schematic representation of the grafting to approach to form mixed corona micelles with thermo-responsive properties. Adapted with permission from ref. 61. Copyright 2017, Wiley-VCH. | ||
Zhu and co-workers62 have demonstrated a different strategy for supramolecular engineering in amphiphilic block copolymers. They prepared acid responsive supramolecular block co-polymer micelles, in which the adenine-terminated hydrophobic poly-(caprolactone) and uracil-terminated hydrophilic poly(ethylene glycol) were glued by multiple complementary H-bonding (Fig. 20). Disruption of the H-bonding in an acidic medium lead to the disassembly of the micellar aggregates, which was exploited for pH-responsive drug delivery applications. P12 (Fig. 21) represents one such system in which two hydrophilic polyethylene-oxide chains are attached with a hydrophobic chain containing two urea groups in a specific location. The P13 structure is similar to P12 except for the absence of the urea groups. Experimental results revealed formation of cylindrical micelles for both the polymers in water but with different dimensions of the nanoscopic structure. Complementary H-bonding between the bis-urea moieties resulted in significantly higher viscosity for the aqueous solution of P12. Furthermore, a pyrene containing polymer P14 was synthesized to probe the possibility of its inclusion into the amphiphilic polymer aggregates by H-bonding.
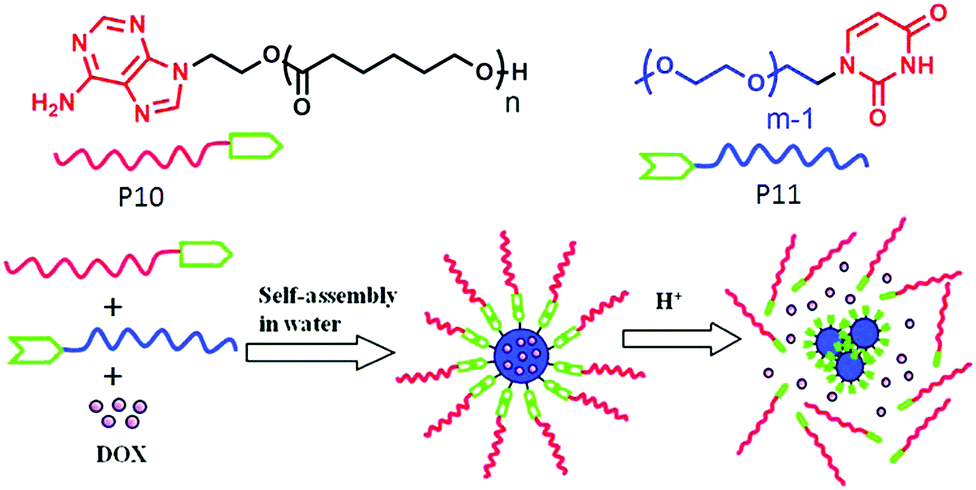 | ||
| Fig. 20 Structure of the adenine terminated polymer P10 and uracil terminated polymer P11 and schematic representation of their acid responsive supramolecular co-polymer micelle formation. Adapted with permission from ref. 62. Copyright 2011, American Chemical Society. | ||
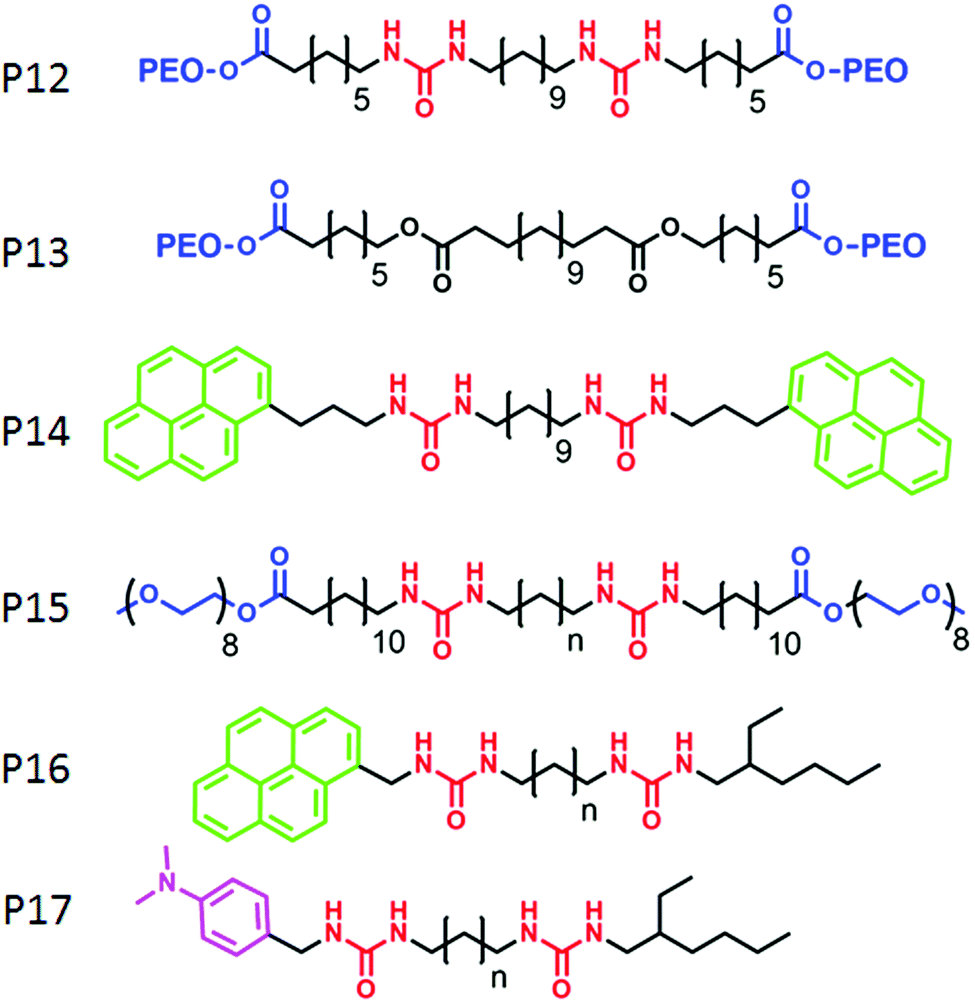 | ||
| Fig. 21 Structure of amphiphilic polymers reported by Sijbesma and co-workers. | ||
Sijbesma and co-workers have elucidated specific molecular recognition via H-bonding in amphiphilic triblock co-polymers.63 They have shown that the presence of H-bonding between urea moieties constrained the packing of the hydrophobic unit and the incorporation of P14 into H-bonded assembly of P12 resulted into unfolded conformation which kept the pyrene moieties apart. As a result, although the excimer band for pyrene was observed in the co-mixture of P14 with P13, no excimer band was found in the presence of P12.
They have further investigated64 a self-sorting phenomenon among these polymers (P15) by systematic variation of the spacer length between the urea groups. P16 and P17 were mixed with P15 so that the mixed assembly and self-sorting could be probed by the presence and the absence of exciplex formation between the pyrene and di-methyl-aniline chromophore. By detailed fluorescence experiments it was concluded that for matching spacer length, mixed assembly was formed but self-sorting was observed with high efficiency for the non-matching spacer.
Meijer and co-workers reported65 a new class of supramolecular materials which can form a transient network via multiple hydrogen bonding between the polymer chains. The polymer (P18 in Fig. 22) contains multiple urea groups in the backbone, separated by an alkyl chain and end functionalized with a ureidopyrimidinone (UPy) group. Macroscopic properties of the supramolecular network could be modulated by varying the alkyl spacer length. In a subsequent report66 they have studied the multicomponent assembly of P18 with mono UPy functionalized polymer (P19) to regulate the structural and mechanical properties, which is highly relevant in biochemical and biological applications.
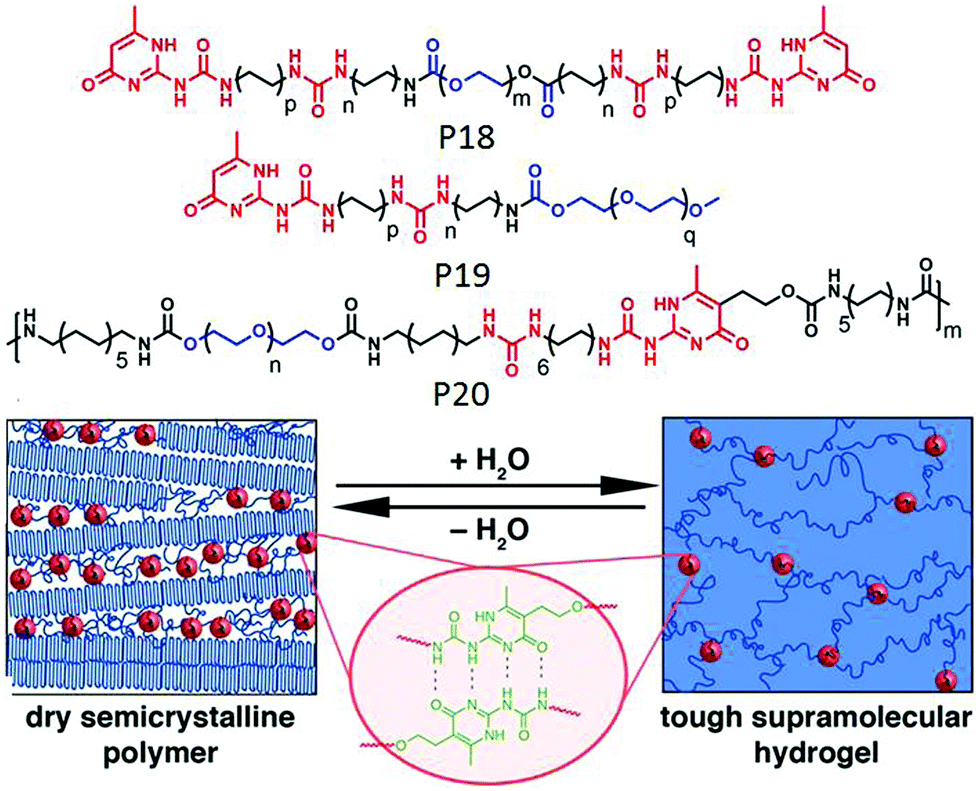 | ||
| Fig. 22 Structure of P18, P19 and P20 and schematic representation of cross-linked hydrogel and phase separated domain in a dry polymer. Adapted with permission from ref. 67. Copyright 2014, American Chemical Society. | ||
In a slightly different design, polymer P20 was synthesized with H-bonding UPy groups in the backbone of the polymer and it was found to form a stable supramolecular hydrogel by employing the complementary UPy–UPy H-bonding as a physical crosslinker and produced phase separated domains in a xerogel (Fig. 22).67
We have reported68 H-bonding driven assembly of amphiphilic polyurethane P21 (Fig. 23) which contains a segmented hydrophobic backbone and appended carboxylic acid groups which acts as the hydrophilic head group under basic pH. In basic water, it adopted a pleated structure by intra-chain H-bonding among the urethane groups which further assembled to a polymersome like nanostructure with negative surface charge.
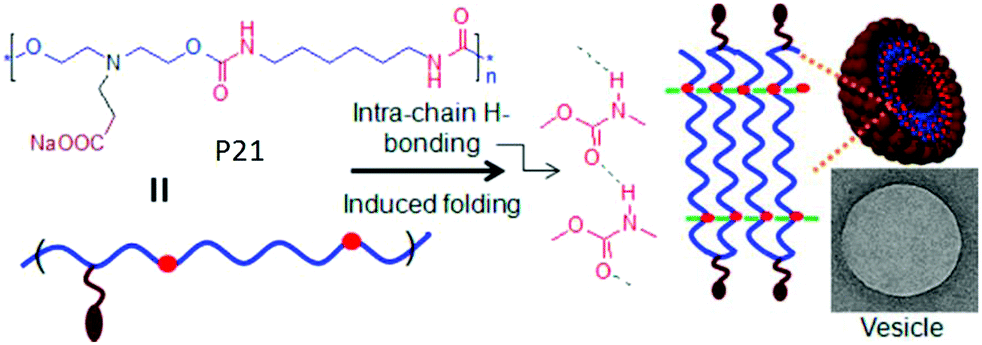 | ||
| Fig. 23 Schematics of intrachain H-bonding induced polymersome assembly of amphiphilic polyurethane. Adapted with permission from ref. 68. Copyright 2011, American Chemical Society. | ||
Kieltyka and co-workers explored69 the combined effect of aromatic gain and intermolecular H-bonding in supramolecular assembly of squaramide attached amphiphilic polymer P22 (Fig. 24). Squaramides are known to produce partial aromatic character and are able to form intermolecular H-bonding between the amide groups arranged in a head to tail fashion. The cryo-TEM image of P22 exhibited a stiff fibrillar nanostructure in aqueous solution, whereas the IR study revealed intermolecular H-bonding. Another control polymer with a urea analogue was prepared to establish the added advantage of aromaticity in the formation of robust supramolecular architecture with enhanced thermodynamic stability.
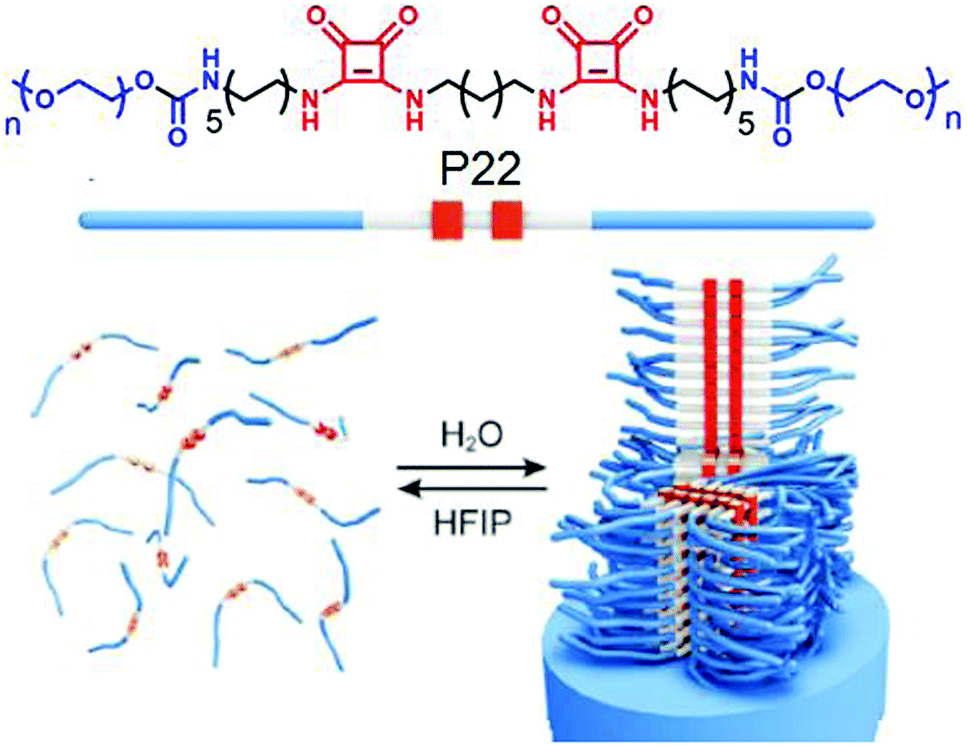 | ||
| Fig. 24 (top) Structure of the squaramide-based polymer P22. (bottom) Self-assembly of P22 into fibrillar structures. Adapted with permission from ref. 69. Copyright 2015, Wiley-VCH. | ||
Meijer and coworkers have reported70 the formation of dendrimers based on non-covalent synthetic procedures via complementary H-bonding in water by using urea-adamantyl-functionalized poly-(propylene imine) dendrimers (generations 1–5), combined with ureidoacetic acid guest molecules (Fig. 25).
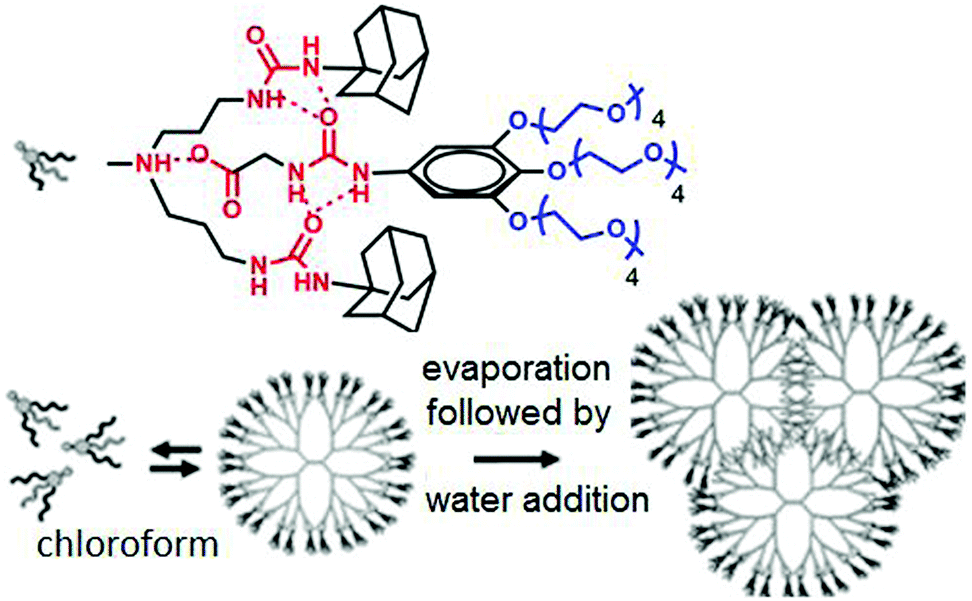 | ||
| Fig. 25 Structure of supra-dendron and schematic of supramolecular dendrimer assembly in water. Adapted with permission from ref. 70. Copyright 2007, American Chemical Society. | ||
A supramolecular dendrimer was generated via multiple H-bonding among the two units in CHCl3. After evaporation of CHCl3, followed by dissolution of this in water, the preformed dendrimer structure hierarchically assembled to form a small dendrimer based spherical assembly.
We have explored the opportunity of regulating self-assembly events of hydrophilic polymers by attaching it with a hydrophobic supramolecular structure directing unit (SSDU).71P23 (Fig. 26) shows an example of such a polymer in which the SSDU is consisting of an NDI chromophore and H-bonding functional groups. Spectroscopy and microscopic study revealed the formation of a stable polymersome structure in water via J-aggregation of the SSDU unit, driven by H-bonding among the hydrazide groups, located in the hydrophobic pocket of this largely hydrophilic polymer.
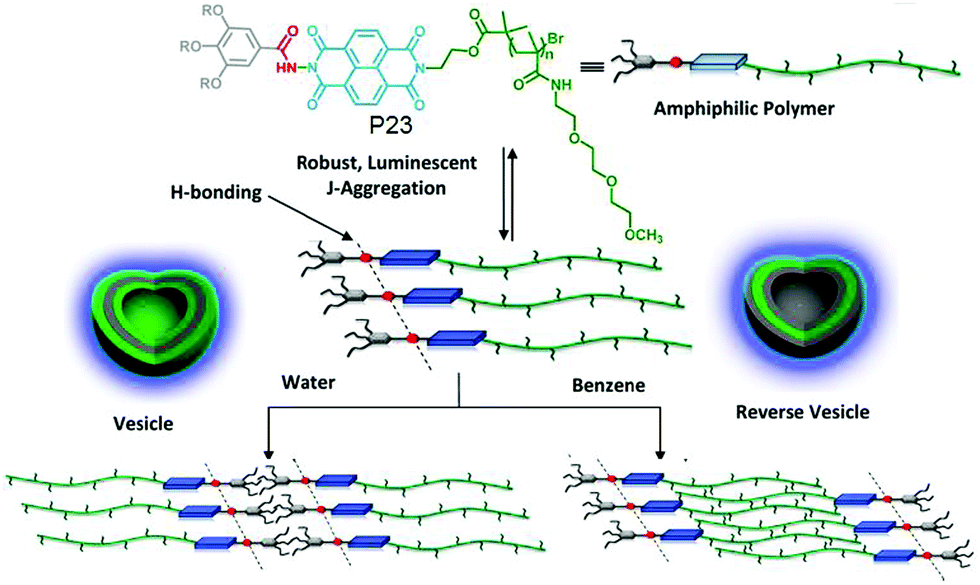 | ||
| Fig. 26 Structure and solvent dependent self-assembly of P23. Adapted with permission from ref. 71. Copyright 2013, American Chemical Society. | ||
It also showed similar polymersome like assembly in benzene with identical photophysical properties which indirectly suggested that the assembly is not governed by the critical packing parameter because in that case in water and oil, the nature of the assembly is unlikely to be same as the solvophobic and solvophilic segments are exactly opposite in these two solvents of contrasting polarity. Rather specific molecular recognition between the SSDU was responsible for highly stable self-assembly of the polymer in both solvents. To further expand this concept, aqueous self-assembly of P24 and P25 (Fig. 27) was compared.72 These two polymers differ only in the nature of the H-bonding groups which is hydrazide and amide, respectively. TEM images revealed stable polymersome assembly for P24, similar to P23. But P25 showed spherical micelle like morphology at the beginning which after aging produced cylindrical micelles which was further supported by DLS, SANS, viscosity and other studies.
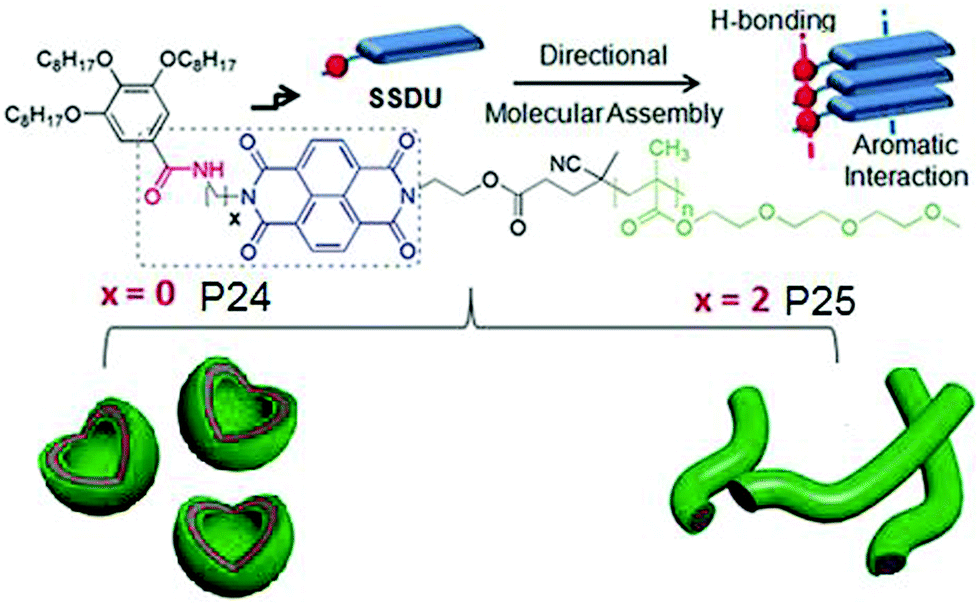 | ||
| Fig. 27 Structure of P24 and P25 and a schematic showing directional molecular interaction-driven distinct self-assembly. Adapted with permission from ref. 72. Copyright 2017, Wiley-VCH. | ||
These results unambiguously established the vital role of H-bonding in regulating the self-assembly process. In each case, a series of polymers were studied with varying degree of polymerization for the hydrophilic block which showed no impact on the morphology revealing that H-bonding is the key and the self-assembly does not depend on the critical packing parameters. FRET studies between fluorescently labeled derivatives of P24 and P25 indicated self-sorting among these two polymers in the aqueous medium.
In a subsequent study we examined the general applicability of this strategy with SSDU-attached water soluble polymers of diverse nature which indicated that the H-bonding regulated morphology rule generally holds true for all the polymers.73 A detailed thermodynamic analysis of self-assembly by ITC studies revealed that the highly stable self-assembly of these unconventional amphiphilic polymers with merely 2–4 wt % hydrophobic content is driven by the entropy factor, originating from the escape of water molecules during the self-assembly process (Fig. 28). Inspired by these results, we have further extended the SSDU-regulated supramolecular assembly of protein (BSA).74P26 (Fig. 29) represents the SSDU–protein conjugate which differs with the previous examples in the sense that the synthetic water soluble polymer has been replaced by a protein and also the peripheral hydrophobic chains of the SSDU are replaced by a hydrophilic oligo-oxyethylene wedge. It produced well defined and homogeneous nanoparticles in water with the surface decorated by the protein with no adverse effect on the conformation of the protein or its enzyme-like activity. In fact DSC and temperature dependent CD studies confirmed in the assembled state that the thermal stability of the protein was enhanced as no signature of denaturation was visible even at a temperature which is significantly higher than the denaturation temperature of the native BSA.
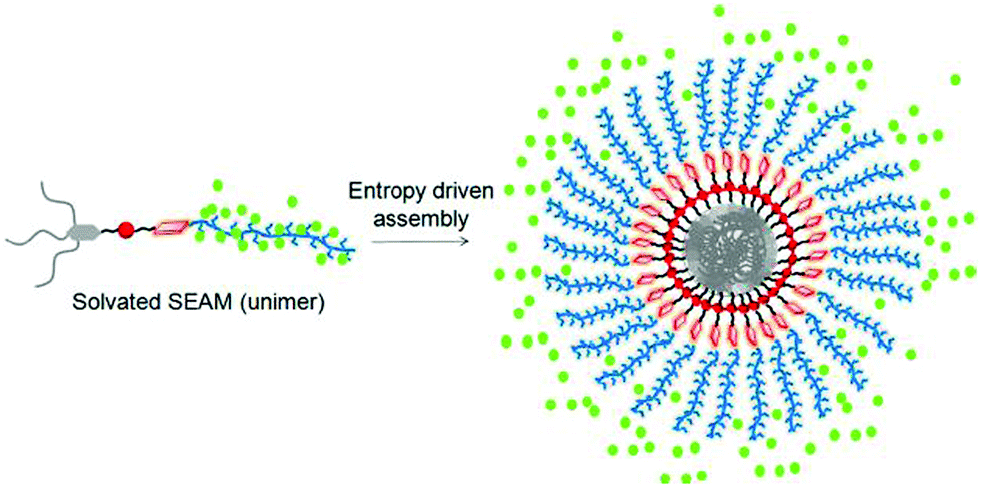 | ||
| Fig. 28 Schematic showing origin of entropy gain in SSDU-appended amphiphilic polymers by release of the water molecules (represented by green dot). Adapted with permission from ref. 73. Copyright 2018, American Chemical Society. | ||
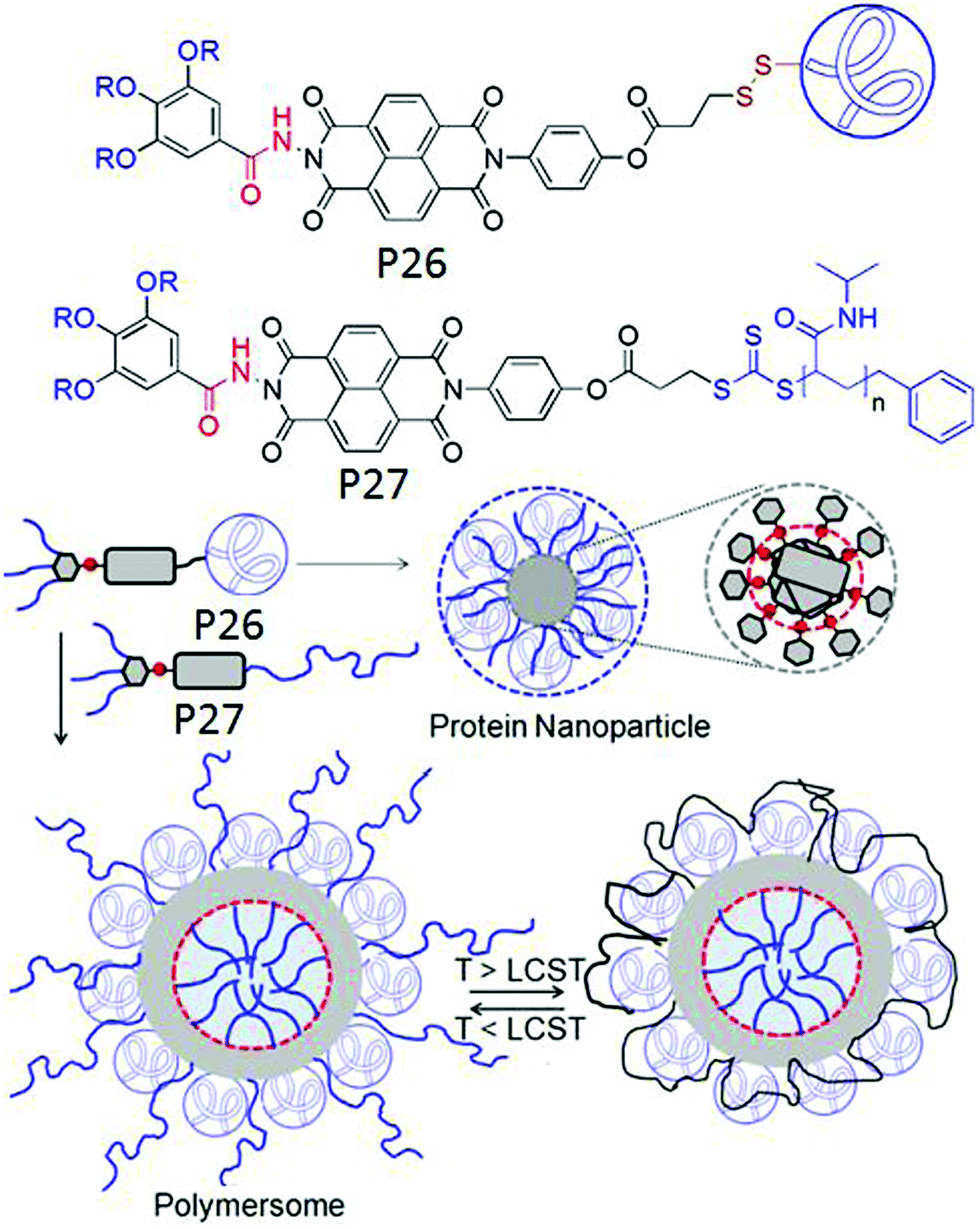 | ||
| Fig. 29 (top) Structure of P26 and P27. (bottom) Schematic representation of the supramolecular assembly of P26 and P26 + P27 (1:1). Adapted with permission from ref. 74. Copyright 2019, Wiley-VCH. | ||
Furthermore it was demonstrated that P27 in which the protein was replaced by a thermo-responsible water soluble PNIPAM, could form co-assembly with P26 resulting in significantly larger polymersome like assembly in which the outer surface was decorated by BSA and PNIPAM (Fig. 29). Resultantly at T > LCST of PNIPAM, the protein active site was partially blocked and its enzyme like activity was reduced significantly.
Very recently, Reiger and co-workers have demonstrated75 H-bonding driven supramolecular assembly of chain-end functionalized hydrophilic polymers (Fig. 30) which in a broad sense is similar to what has been demonstrated by us.
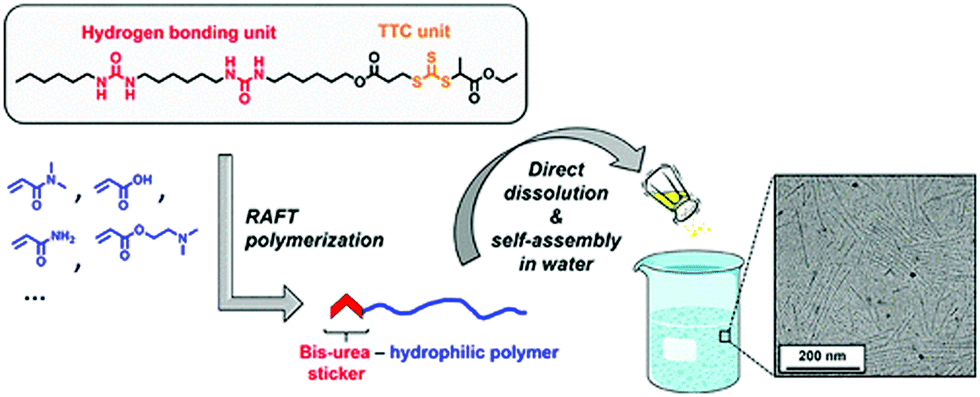 | ||
| Fig. 30 Schematic presentation showing formation of cylindrical micelles from bis-urea containing SSDU. Adapted with permission from ref. 75. Copyright 2018, American Chemical Society. | ||
They have designed a functional chain transfer agent that contains a segmented hydrocarbon tail containing two urea groups. These polymers produced cylindrical micellar structures in water. In contrast, polymers synthesized from a similar RAFT agent without the bis-urea moiety resulted in spherical micelles, confirming the crucial role of the H-bonding in self-assembly.
Summary
In this review article we have presented various examples of H-bonding regulated self-assembly of amphiphilic molecules and macromolecules. In contrast, common perception, H-bonding indeed can be effectively utilized in water as long as it is located in a hydrophobic domain of the building block. In one class of systems, H-bonding has helped to construct the amphiphile by gluing the hydrophobic and hydrophilic segments while in another class of systems it has been used to drive the self-assembly of an amphiphilic building block. Many examples have demonstrated the specific role of H-bonding in morphology regulation beyond the classical theory based on critical packing parameter. Based on these results, H-bonding appears to be a very attractive and fruitful arsenal in the designer's tool box to design predictable supramolecular nanostructures with high stability, precision in macroscopic structure and desired functional group display, which enhances the potential of these materials to be useful as supramolecular biomaterials.76Conflicts of interest
There are no conflicts to declare.Acknowledgements
AS thanks IACS for a research fellowship. SG thanks DST, Government of India for funding through the SwarnaJayanti Fellowship (DST/SJF/CSA-01/2-14-15).Notes and references
-
(a) T. Aida, E. W. Meijer and S. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed
; (b) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed
-
K. Holmberg, B. Jonsson, B. Kronberg and B. Lindman, Surfactants and Polymers in Aqueous Solution, John Wiley & Sons, Ltd, New York, 2nd edn, 2003 Search PubMed
-
(a) Y. Y. Maia and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
-
(a) M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed
- H. Sun, Y. Hong, Y. Xi, Y. Zou, J. Gao and J. Du, Biomacromolecules, 2018, 19, 1701–1720 CrossRef CAS PubMed
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed
- A. V. Ambade, E. N. Savariar and S. Thayumanavan, Mol. Pharmaceutics, 2005, 2, 4264–4272 Search PubMed
-
(a) J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans., 1976, 72, 1525–1568 RSC
-
(a) C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785–792 CrossRef CAS PubMed
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed
-
(a) H.-J. Kim, T. Kim and M. Lee, Acc. Chem. Res., 2011, 44, 72–82 CrossRef CAS PubMed
-
(a) J.-F. Gohy, B. G. G. Lohmeijer and U. S. Schubert, Macromolecules, 2002, 35, 4560–4563 CrossRef CAS
-
(a) C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed
-
(a) A. Barnard and D. K. Smith, Angew. Chem., Int. Ed., 2012, 52, 6572–6581 CrossRef PubMed
-
(a)
S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University Press, New York, 1997 Search PubMed
-
(a) R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC
- E. Stofer, C. Chipot and R. Lavery, J. Am. Chem. Soc., 1999, 121, 9503–9508 CrossRef CAS
-
(a) M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed
-
(a) E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC
-
S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University Press, New York, 1997 Search PubMed
-
P. Schuster, G. Zundel and C. Sandorfy, The Hydrogen Bond: Recent Developments in Theory and Experiments, North-Holland Pub. Co., New York, 1976 Search PubMed
- T. F. A. de Greef, M. M. L. Nieuwenhuizen, R. P. Sijbesma and E. W. Meijer, J. Org. Chem., 2010, 75, 598–610 CrossRef CAS PubMed
- C. M. A. Leenders, M. B. Baker, I. A. B. Pijpers, R. P. M. Lafleur, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, Soft Matter, 2016, 12, 2887–2893 RSC
- F. García, J. Buendía and L. Sánchez, J. Org. Chem., 2011, 76, 6271–6276 CrossRef PubMed
- J. Buendía and L. Sánchez, Org. Lett., 2013, 15, 5746–5749 CrossRef PubMed
- N. Kimizuka, T. Kawasaki and T. Kunitake, J. Am. Chem. Soc., 1993, 115, 4387–4388 CrossRef CAS
- N. Kimizuka, T. Kawasaki, K. Hirata and T. Kunitake, J. Am. Chem. Soc., 1998, 120, 4094–4104 CrossRef CAS
- T. Kawasaki, M. Tokuhiro, N. Kimizuka and T. Kunitake, J. Am. Chem. Soc., 2001, 123, 6792–6800 CrossRef CAS PubMed
- M. Yoshida and T. Shimizu, Angew. Chem., Int. Ed., 2003, 42, 1009–1012 CrossRef PubMed
- T. Iizawa, H. Minamikawa, M. Ohnishi-Kameyama, T. Shimizu and R. Iwaura, Small, 2010, 6, 1131–1139 CrossRef PubMed
- P. G. A. Janssen, J. Vandenbergh, J. L. J. van Dongen, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2007, 129, 6078–6079 CrossRef CAS PubMed
- R. Iwaura, F. J. M. Hoeben, M. Masuda, A. P. H. J. Schenning, E. W. Meijer and T. Shimizu, J. Am. Chem. Soc., 2006, 128, 13298–13304 CrossRef CAS PubMed
- W. Yang, Y. Chen, M. S. Wong and P. K. Lo, Biomacromolecules, 2012, 13, 3370–3376 CrossRef CAS PubMed
- P. Mathivanan, K. L. Vidale, D. M. Sherman, K. Hallenga, K. V. Wood, J. G. Stowell and H. Fenniri, J. Am. Chem. Soc., 2001, 123, 3854–3855 CrossRef PubMed
- J. G. Moralez, J. Raez, T. Yamazaki, R. K. Motkuri, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2005, 127, 8307–8309 CrossRef CAS PubMed
- G. Borzsonyi, R. L. Beingessner, T. Yamazaki, J.-Y. Cho, A. J. Myles, M. Malac, R. Egerton, M. Kawasaki, K. Ishizuka, A. Kovalenkoand and H. Fenniri, J. Am. Chem. Soc., 2010, 132, 15136–15139 CrossRef CAS PubMed
- M. R. Molla and S. Ghosh, Chem. – Eur. J., 2012, 18, 9860–9869 CrossRef CAS PubMed
- P. Rajdev, M. R. Molla and S. Ghosh, Langmuir, 2014, 30, 1969–1976 CrossRef CAS PubMed
- P. Rajdev, S. Chakraborty, M. Schmutz, P. Mesini and S. Ghosh, Langmuir, 2017, 33, 4789–4795 CrossRef CAS PubMed
- S. Chakraborty, D. Ray, V. K. Aswal and S. Ghosh, Chem. – Eur. J., 2018, 24, 16379–16387 CrossRef CAS PubMed
- A. Sikder, A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2015, 54, 6755–6760 CrossRef CAS PubMed
- A. Sikder, D. Ray, V. K. Aswal and S. Ghosh, Langmuir, 2018, 34, 868–875 CrossRef CAS PubMed
- D. Itsch and J. H. Fuhrhop, Acc. Chem. Res., 1986, 19, 130–137 CrossRef
- A. Sikder, J. Sarkar, T. Sakurai, S. Seki and S. Ghosh, Nanoscale, 2018, 10, 3272–3280 RSC
- A. Sikder, D. S. Ray, V. K. Aswal and S. Ghosh, Angew. Chem., Int. Ed., 2019, 58, 1606–1611 CrossRef CAS
- R. S. Bhosale, M. Kobaisi, S. V. Bhosale, S. Bhargava and S. V. Bhosale, Sci. Rep., 2015, 5(1-11), 14609 CrossRef CAS PubMed
- C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat, D. Schmidt and F. Würthner, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed
- V. Grande, B. Soberats, S. Herbst, V. Stepanenkoaand and F. Wurthner, Chem. Sci., 2018, 9, 6904–6911 RSC
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC
- C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC
- L. Albertazzi, D. van der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. van der Hofstad and E. W. Meijer, Science, 2014, 344, 491–495 CrossRef CAS PubMed
- X. Lou, R. P. M. Lafleur, C. M. A. Leenders, S. M. C. Schoenmakers, N. M. Matsumoto, M. B. Baker, J. L. J. van Dongen, A. R. A. Palmans and E. W. Meijer, Nat. Commun., 2017, 15, 8–15420 Search PubMed
- B. Song, H. Wei, Z. Wang, X. Zhang, M. Smet and W. Dehaen, Adv. Mater., 2007, 19, 416–420 CrossRef CAS
- S. Ogi, N. Fukaya, Arifin, B. B. Skjelstad, Y. Hijikata and S. Yamaguchi, Chem. – Eur. J., 2019, 25, 7303–7307 CrossRef CAS PubMed
-
(a) F. Würthner, Nat. Chem., 2014, 6, 171 CrossRef PubMed
- B. Qin, S. Zhang, Z. Huang, J.-F. Xu and X. Zhang, Macromolecules, 2018, 51, 1620–1625 CrossRef CAS
- S. Zhang, B. Qin, Z. Huang, J.-F. Xu and X. Zhang, ACS Macro Lett., 2019, 8, 177–182 CrossRef CAS
- A. Rödle, M. Lambov, C. Mück-Lichtenfeld, V. Stepanenko and G. Fernández, Polymer, 2017, 128, 317–324 CrossRef
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS
-
(a) Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O’Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC
- Z. Hua, R. Keogh, Z. Li, T. R. Wilks, G. Chen and R. K. O’Reilly, Macromolecules, 2017, 50, 3662–3670 CrossRef CAS PubMed
- D. Wang, Y. Su, C. Jin, B. Zhu, Y. Pang, L. Zhu, J. Liu, C. Tu, D. Yan and X. Zhu, Biomacromolecules, 2011, 12, 1370–1379 CrossRef CAS PubMed
- N. Chebotareva, P. H. H. Bomans, P. M. Frederik, N. A. J. M. Sommerdijka and R. P. Sijbesma, Chem. Commun., 2005, 4967–4969 RSC
- A. Pal, S. Karthikeyan and R. P. Sijbesma, J. Am. Chem. Soc., 2010, 132, 7842–7843 CrossRef CAS PubMed
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed
- R. E. Kieltyka, A. C. H. Pape, L. Albertazzi, Y. Nakano, M. M. C. Bastings, I. K. Voets, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS PubMed
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankersand and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed
- T. Mondal, K. Dan, J. Deb, S. S. Jana and S. Ghosh, Langmuir, 2013, 29, 6746–6753 CrossRef CAS PubMed
- V. S. Talens, P. Englebienne, T. T. Trinh, W. E. M. Noteborn, I. K. Voetsand and R. E. Kieltyka, Angew. Chem., Int. Ed., 2015, 54, 10502–10506 CrossRef PubMed
- T. M. Hermans, M. A. C. Broeren, N. Gomopoulos, A. F. Smeijers, B. Mezari, E. N. M. Van Leeuwen, M. R. J. Vos, P. C. M. M. Magusin, P. A. J. Hilbers, M. H. P. Van Genderen, N. A. J. M. Sommerdijk, G. Fytas and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 15631–15638 CrossRef CAS PubMed
- A. Das and S. Ghosh, Macromolecules, 2013, 46, 3939–3949 CrossRef CAS
- P. Pramanik, D. Ray, V. K. Aswal and S. Ghosh, Angew. Chem., Int. Ed., 2017, 56, 3516–3520 CrossRef CAS PubMed
- P. Dey, P. Rajdev, P. Pramanik and S. Ghosh, Macromolecules, 2018, 51, 5182–5190 CrossRef CAS
- A. Sikder, D. Ray, V. K. Aswal and S. Ghosh, Chem. – Eur. J., 2019, 25, 10464–10471 CrossRef CAS PubMed
- G. Mellot, J.-M. Guigner, J. Jestin, L. Bouteiller, F. Stoffelbach and J. Rieger, Macromolecules, 2018, 51, 10214–10222 CrossRef CAS
-
(a) M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2019 |