
Photoredox catalysts based on earth-abundant metal complexes
Bryony M.
Hockin†
ab,
Chenfei
Li†
 a,
Neil
Robertson
*b and
Eli
Zysman-Colman
*a
a,
Neil
Robertson
*b and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK
bEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, Scotland, UK. E-mail: neil.robertson@ed.ac.uk
First published on 17th January 2019
Abstract
Over the last decade, visible light photoredox catalysis has exploded into the consciousness of the synthetic chemist. The principal photocatalysts used are based on rare and toxic ruthenium(II) and iridium(III) complexes. This critical review focusses on Earth-abundant metal complexes as potential replacement photocatalysts and summarizes the use of photoactive Cu(I), Zn(II), Ni(0), V(V), Zr(IV), W(0), W(VI), Mo(0), Cr(III), Co(III) and Fe(II) complexes in photoredox reactions. The optoelectronic properties of these complexes and relevant structurally related analogs, not yet used for photoredox catalysis, are discussed in combination with the reaction scope reported for each photocatalyst. Prospects for the future of photocatalyst design are considered.
 Bryony May Hockin | Bryony May Hockin graduated with an MChem degree with honours from the University of Durham in the UK in 2016, where she studied physical organic chemistry. She moved further north to St Andrews to undertake a Ph.D. at the CRITICAT Centre for Doctoral Training, co-supervised by Eli Zysman-Colman at St Andrews and Neil Robertson at the University of Edinburgh. Her research interests are centred on the development of new copper(I) complexes for use as photoredox catalysts in small molecule reactions. |
 Chenfei Li | Chenfei obtained his bachelor's degree at Nanjing University of Posts and Telecommunications where he studied organic semiconductors. His M.Sc. project at Durham University dealt with the synthesis and photophysical properties of novel iridium complexes. His Ph.D. project now at St Andrews is about the chemistry and photophysics of Earth abundant metal complexes and photoredox catalysis using noble metal free photocatalysts. |
 Neil Robertson | Neil Robertson is Chair of Molecular Materials in the School of Chemistry, University of Edinburgh. His interests are in the synthesis, characterisation and application of new molecular and nano-materials for optoelectronic applications including solar photovoltaics, solar photocatalysis, conducting, luminescent and magnetic materials and devices. He studied at the University of Edinburgh and worked in the Freie Universität Berlin, University of Wales Bangor and Imperial College London before returning to Edinburgh. |
 Eli Zysman-Colman | Eli Zysman-Colman obtained his Ph.D. from McGill University. He then completed two postdoctoral fellowships, one with Jay Siegel at the University of Zurich and the other with Stefan Bernhard at Princeton University. He started his career in Canada in 2007. In 2013, he moved to the University of St Andrews where he is Reader in Optoelectronic Materials and Fellow of the Royal Society of Chemistry. His research program focuses on the rational design of: (I) luminophores for displays and lighting based on organic light-emitting diodes and light-emitting electrochemical cells; (II) light harvesting dyes for solar cells; and (III) photoredox catalysts. |
Introduction
Photoredox catalysis, first mentioned by Michael Grätzel et al. in 1982, implies a catalysis system that can accelerate redox reactions by electron transfer between a photocatalyst and an organic substrate, driven by light.1 These photocatalysts (PC) can take the form of organometallic or coordination complexes,2,3 organic dye molecules,4 inorganic semiconductors,5–7 or hybrid systems that are combinations of the three. The wide use of d-block metal complexes as PC implies that the process of their visible light excitation relies both on the nature of the metal and the identity of the ligands. An attractive feature of this class of PCs is that the photoredox properties of these complexes can be judiciously tuned through a combination of metal and ligand choice, thereby rendering these photocatalysts highly adaptable.Since 2007, there has been a dramatically increasing number of reports of visible light photoredox-catalyzed reactions (Fig. 1). Iridium(III) and ruthenium(II) complexes remain the two most popular families of PCs, in part due to the maturity of the field of the study of these photoactive complexes that underpins these photocatalytic studies. Despite their capacity to catalyse a wide number of distinct transformations, the intrinsic cost of the noble metal PCs and their toxicity profiles represent barriers to adoption of this technology in industry. Thus, the design of viable alternative PCs based on Earth-abundant elements is presently a very active area of research.
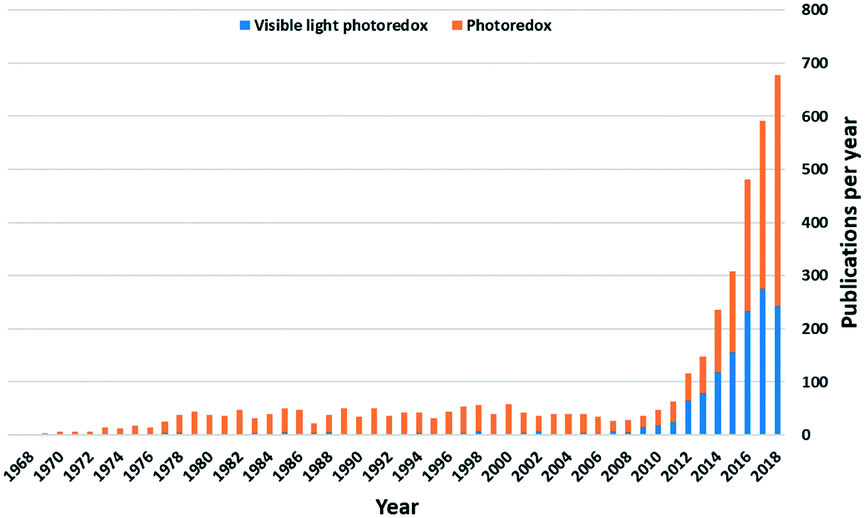 | ||
| Fig. 1 Number of photoredox publications per year. Blue part using keyword: “visible light photoredox”, while the orange part represents the rest of publications using keyword: “photoredox” Scifinder search conducted: 28/10/2018. | ||
Popular PCs such as fac-Ir(ppy)3 and [Ru(bpy)3]2+ possess the following features: (1) absorption in the visible region, where most organic substrates are transparent; (2) long-lived photoexcited states that are sufficiently persistent to diffuse to and react with the organic substrate; common iridium(III) and ruthenium(II) PCs possess microsecond lifetimes as a function of the spin-forbidden nature of both the intersystem crossing (ISC) and emission processes (τPL of 2000 ns for Ir(ppy)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 and of 1100 ns for [Ru(bpy)3]2+)9 but organic photocatalysts such as Eosin-Y (τPL of 2 ns) have lifetimes in the nanosecond regime;10 (3) suitably highly oxidizing or reducing species in either the ground or the excited state compared to the organic substrate; (4) sufficiently reversible electrochemical behaviour and photostability such that there is no degradation of the photoactivated, oxidized or reduced species.
8 and of 1100 ns for [Ru(bpy)3]2+)9 but organic photocatalysts such as Eosin-Y (τPL of 2 ns) have lifetimes in the nanosecond regime;10 (3) suitably highly oxidizing or reducing species in either the ground or the excited state compared to the organic substrate; (4) sufficiently reversible electrochemical behaviour and photostability such that there is no degradation of the photoactivated, oxidized or reduced species.
Visible light photoredox catalysis reactions can roughly be classified according to one of three reaction mechanisms (Fig. 2). Firstly, the most common is photoinduced electron transfer (PeT), in which an electron is transferred between substrate and photocatalyst. Secondly, there is atom (or group) transfer, in which an atom or a group is transferred from substrate to the excited photocatalyst to form a radical (ATRA). Finally, an inner sphere reaction can occur on a photoexcited metal center.11
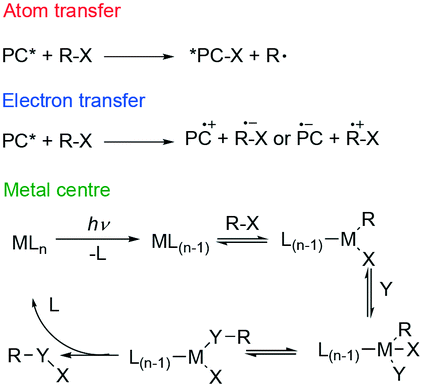 | ||
| Fig. 2 Different possible mechanisms of photoredox catalysis. | ||
It is useful at this point to introduce the principle of photo-induced redox chemistry using a case study of [Ir(ppy)2(bpy)]+ (Fig. 3);12 there are a few examples of photoinduced energy transfer (PET) catalysed reactions, but the vast majority of reports implicate PeT processes.13 In the ground state, [Ir(ppy)2(bpy)]+ is chemically stable with an oxidation potential Eox1/2 = +1.27 V versus a saturated calomel electrode (SCE, SCE is used as the reference throughout this review), and a reduction potential Ered1/2 = −1.38 V vs. SCE in MeCN.14 The ground state oxidation and reduction potentials (Eox and Ered) are frequently solvent-sensitive and affect the ground state reducing and oxidizing power of the photocatalyst, and also determine the energy required for regeneration of the PC from its oxidized/reduced species. According to the equations 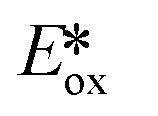 = Eox − E0,0,
= Eox − E0,0, 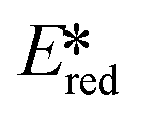 = Ered + E0,0 (a simplified version of the equation developed by Rehm and Weller
= Ered + E0,0 (a simplified version of the equation developed by Rehm and Weller 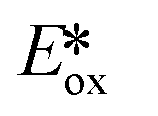 = Eox − E0,0 + wr,
= Eox − E0,0 + wr, 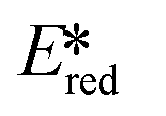 = Ered + E0,0 + wr),15,16 for photocatalysts with similar E0,0, those with a more negative
= Ered + E0,0 + wr),15,16 for photocatalysts with similar E0,0, those with a more negative 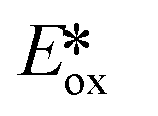 are stronger photoreductants while those with a more positive
are stronger photoreductants while those with a more positive 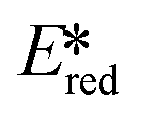 are stronger photooxidants (
are stronger photooxidants (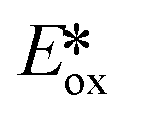 = the excited state oxidation potential;
= the excited state oxidation potential; 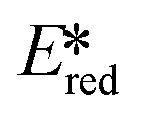 = the excited state reduction potential; E0,0 = the energy between the zeroth vibrational states of the ground and excited states). For example, 9-mesityl-10-methylacridinium (Mes-Acr+) has a
= the excited state reduction potential; E0,0 = the energy between the zeroth vibrational states of the ground and excited states). For example, 9-mesityl-10-methylacridinium (Mes-Acr+) has a 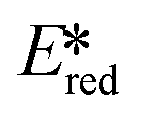 of 2.06 V, which makes it a particularly strong photooxidant.17 However, the Ered of −0.57 V makes the catalyst regeneration more difficult.18
of 2.06 V, which makes it a particularly strong photooxidant.17 However, the Ered of −0.57 V makes the catalyst regeneration more difficult.18
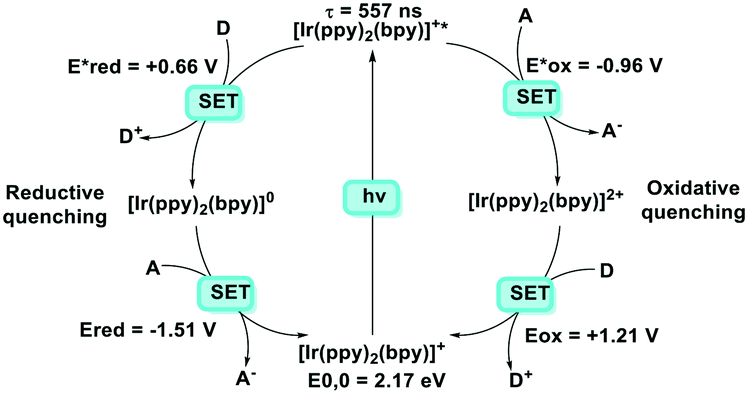 | ||
| Fig. 3 Oxidative and reductive quenching cycles of [Ir(ppy)2(bpy)]+. | ||
Upon absorption of a photon into the mixed charge transfer band (λabs = 420 nm, E0,0 = 2.17 eV) an electron in one of the photocatalyst's dπppy orbitals is excited into a ligand-centered 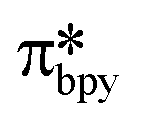 orbital, generating a species that initially exists in the singlet excited state and can be described as [IrIV(ppy)2(bpy˙−)]+*. Given the presence of the heavy metal in the complex, intersystem crossing is fast, on the order of tens to hundreds of fs,9 generating the corresponding triplet mixed charge transfer state. This excited state can then relax radiatively back to the ground state in the form of phosphorescence (λPL = 602 nm). The excited state energy plays an important role in photoredox catalysis and refers to the energy gap between zeroth vibrational states of the electronic ground and excited states (E0,0). Accordingly, a larger excited state energy will increase the power of photocatalysts, regardless of whether it is a photoreductant or photooxidant. However, a higher excited state energy implicates a higher required energy to photoexcite the PC. The highest energy visible light that is usually used for photoredox catalytic reactions is violet light (395 nm, 3.14 eV). Therefore, PCs with very high E0,0 will possess stronger photooxidizing and reducing power but with the disadvantage of loss of chemoselectivity. In the case of [Ir(ppy)2(bpy)]+,
orbital, generating a species that initially exists in the singlet excited state and can be described as [IrIV(ppy)2(bpy˙−)]+*. Given the presence of the heavy metal in the complex, intersystem crossing is fast, on the order of tens to hundreds of fs,9 generating the corresponding triplet mixed charge transfer state. This excited state can then relax radiatively back to the ground state in the form of phosphorescence (λPL = 602 nm). The excited state energy plays an important role in photoredox catalysis and refers to the energy gap between zeroth vibrational states of the electronic ground and excited states (E0,0). Accordingly, a larger excited state energy will increase the power of photocatalysts, regardless of whether it is a photoreductant or photooxidant. However, a higher excited state energy implicates a higher required energy to photoexcite the PC. The highest energy visible light that is usually used for photoredox catalytic reactions is violet light (395 nm, 3.14 eV). Therefore, PCs with very high E0,0 will possess stronger photooxidizing and reducing power but with the disadvantage of loss of chemoselectivity. In the case of [Ir(ppy)2(bpy)]+, 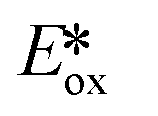 = −0.96 V and
= −0.96 V and 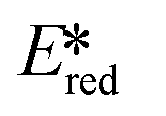 = +0.66 V and define the thermodynamic driving forces for PeT. Whether a PC acts as a photoreductant (via an oxidative quenching mechanism) or a photooxidant (via a reductive quenching mechanism) depends on the relative thermodynamics of the PC and the targeted species that will be attacked. The complex [Ir(ppy)2(bpy)]+ is thus viewed as a medium strength photooxidant and photoreductant and there are examples of reactions implicating both oxidative and reductive quenching mechanisms. The excited state redox properties of this complex compare favourably to the well-studied [Ru(bpy)3]2+ where
= +0.66 V and define the thermodynamic driving forces for PeT. Whether a PC acts as a photoreductant (via an oxidative quenching mechanism) or a photooxidant (via a reductive quenching mechanism) depends on the relative thermodynamics of the PC and the targeted species that will be attacked. The complex [Ir(ppy)2(bpy)]+ is thus viewed as a medium strength photooxidant and photoreductant and there are examples of reactions implicating both oxidative and reductive quenching mechanisms. The excited state redox properties of this complex compare favourably to the well-studied [Ru(bpy)3]2+ where 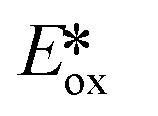 = −0.72 V and
= −0.72 V and 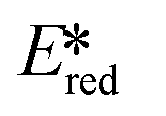 = +0.73 V.
= +0.73 V.
Reports of Earth-abundant PCs has increased significantly in recent years and include complexes based on vanadium(V), chromium(III), iron(II), cobalt(III), copper(I), zinc(II), zirconium(IV), and tungsten(VI). Of the most Earth-abundant transition-metal elements (Fig. 4), only PCs based on manganese and molybdenum have not as of yet been reported; however, there are examples of photoluminescent manganese(II)19,20 and molybdenum(0)21 complexes as well as examples using a manganese complex as a metallocatalyst while the intermediate is photoactive.22 Previously, Wenger et al. reviewed photoredox catalytic reactions with PCs based on zinc(II), iron(II), chromium(III), copper(I), zirconium(IV), molybdenum(0) and uranium(VI) complexes,23 and more recently, Wenger reviewed the photophysics of photoactive Earth-abundant complexes.24 Herein, we review photoredox catalytic reactions with PCs based on Earth-abundant metal complexes. Reactions and PCs are categorized by PeT mechanism: either via oxidative quenching or reductive quenching. Additionally, we review the optoelectronic properties of some other related photoactive Earth-abundant metal complexes in the literature that have the potential to be used as photocatalysts.
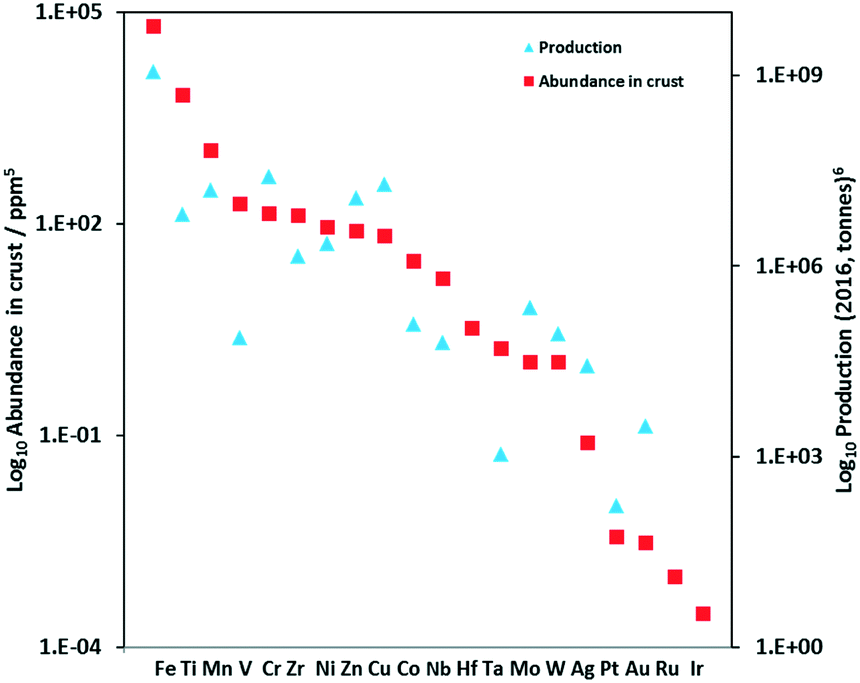 | ||
| Fig. 4 Abundance of selected elements for use in the formation of PCs. | ||
Photoredox catalysts that proceed by an oxidative quenching pathway
The non-emissive triplet metal-centered state (3MC) state and the emissive 3MLCT of first row transition metal complexes are typically both closer in energy to each other and less energetic than those of second and third row metal complexes (Fig. 5). In many cases, the 3MC state in fact lies lower in energy than the 3MLCT state, rendering these complexes both non-emissive and with poor photostability. To illustrate this point, many luminescent platinum(II) and palladium(II) complexes have been reported,25,26 while there exist far fewer examples of room temperature luminescent nickel(II) complexes.18 Similarly, there are a plethora of emissive ruthenium(II) complexes while the longest-lived excited state for an iron(II) complex is only 528 ps.27 To mitigate easily accessible 3MC states, metals with a d10 electronic configuration that perforce do not have MC states can be used.28Table 1 summarizes the optoelectronic properties of PCs and related complexes that proceed via an oxidative quenching mechanism. The two largest families of complexes in Table 1 contain low oxidation state copper(I) and zinc(II) metals possessing a d10 electronic configuration; there are also some examples of nickel(0) complexes. However, due to the lack of metal-centred redox processes for Zn(II) and the limited number of stable Ni(0) complexes with redox-active ligands, copper(I) complexes remain the largest subset of Earth-abundant PCs. Further, the ample resources of copper in the Earth's crust at an abundance of 68 ppm29 coupled with its industrial use dictates the commodity pricing of $6238 USD per tonne (LME November data).30 | ||
| Fig. 5 Schematic representation of the relative position of the potential energy curves for the ground state (GS) and the lowest electronic excited states of Fe(II), Ru(II), and Os(II) polypyridine complexes, to illustrate the change of the MLCT–MC energy gap and ordering. Reproduced from ref. 28. | ||
| Compound | λ abs/nm | λ PL/nm | τ PL/μs | E (0,0)/eVa | E ox/Vb |
|
Ref. |
|---|---|---|---|---|---|---|---|
a
E
(0,0) usually provided in the literature. In the cases where this value is not explicitly reported it has been approximated by the energy of the onset absorption band extracted by Digitize function of Origin 9.
b
E
ox are referenced vs. SCE.
c
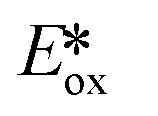 = Eox − E(0,0)
d In degassed CHCl3.
e In degassed DCM.
f In degassed THF.
g In degassed MeCN.
h In THF at 77 K.
i In a PVK:PBD doped film.
j In degassed PhMe.
k In a PMMA doped film. = Eox − E(0,0)
d In degassed CHCl3.
e In degassed DCM.
f In degassed THF.
g In degassed MeCN.
h In THF at 77 K.
i In a PVK:PBD doped film.
j In degassed PhMe.
k In a PMMA doped film.
|
|||||||
| Zn(NMe2-phpo)2 | 372d | 482d | 3.01 | 0.54 | −2.47 | 47 | |
| [Cu(dppp)2]BF4 | 347e | 556e | 0.24e | 3.57 | 1.13 | −2.44 | 55 |
| Cu(XyBnta)(dppb) | 500f | 632f | 0.06f | 2.49 | 0.09 | −2.4 | 35 |
| [Cu(POP)2]BF4 | 332f | 3.74 | 1.40 | −2.34 | 56 | ||
| Cu(Xypta)(dppb) | 520f | 631f | 2 | 2.39 | 0.09 | −2.3 | 35 |
| Ni(dicdmqp)2 | 424f | 513h | 1.1h | 2.53 | 0.24 | −2.29 | 48 |
| Ni(dicmbpt)2 | 422f | 558h | 1.2h | 2.53 | 0.26 | −2.27 | 48 |
| Zn(OMe-phpo)2 | 369d | 440d | 3.2 | 0.94 | −2.26 | 47 | |
| Zn(phpo)2 | 363d | 428d | 3.33 | 1.07 | −2.26 | 47 | |
| Zn4(Q)6(Ac)2 | 372i | 503i | 2.86 | 0.61 | −2.25 | 57 | |
| [Cu2(Mebpy)2(tdppc)]BF4 | 410g | 3.03 | 0.80 | −2.23 | 58 | ||
| Zn(dF-phpo)2 | 357d | 420d | 3.37 | 1.14 | −2.23 | 47 | |
| Zn2(MeQ)2(Ac)2(Me)2 | 361i | 534i | 2.78 | 0.58 | −2.2 | 57 | |
| [Cu(dppb)2]BF4 | 374f | 3.32 | 1.13 | −2.19 | 56 | ||
| Zn(Cl-phpo)2 | 362d | 423d | 3.27 | 1.09 | −2.18 | 47 | |
| Zn(Cz-pimp)2 | 409f | 496f | 3 | 0.84 | −2.16 | 59 | |
| Zn(CN-phpo)2 | 326d | 416d | 3.32 | 1.18 | −2.14 | 47 | |
| [Zn(MeQ)2]Br | 365i | 497i | 2.94 | 0.84 | −2.1 | 57 | |
| Cu(OMe-ppta)(dppb) | 520f | 633f | 0.07f | 2.39 | 0.35 | −2.04 | 35 |
| [Cu(bpy)(SIPr)]PF6 | 370d | 3.35 | 1.33 | −2.02 | 39 | ||
| [Cu(dppp)(POP)]BF4 | 363e | 494e | 2.44e | 3.42 | 1.44 | −1.98 | 55 |
| Cu(Me-ppta)(dppb) | 520f | 633f | 0.35f | 2.39 | 0.41 | −1.98 | 35 |
| [Zn(MeQ)2]Br2 | 353i | 543i | 2.79 | 0.81 | −1.98 | 57 | |
| W(CNdipp)6 | 575j | 0.12j | 2.28 | −0.16 | −1.98 | 50 | |
| [Cu(bath)(ThioPOP)]PF6 | 386f | 545f | 16.3f | 3.21 | 1.28 | −1.93 | 60 |
| Cu(ppta)(dppb) | 520f | 632f | 0.57f | 2.39 | 0.46 | −1.93 | 35 |
| Cu(Cl-ppta)(dppb) | 500f | 631f | 0.75f | 2.48 | 0.56 | −1.92 | 35 |
| [Cu(dmbpy)(IPr)]PF6 | 381d | 3.26 | 1.37 | −1.89 | 39 | ||
| [Cu(bath)(Xant)]PF6 | 389f | 569f | 6.4f | 3.19 | 1.31 | −1.88 | 60 |
| [Cu(dpp)(binc)]BF4 | 17k | 2.57 | 0.69 | −1.88 | 61 | ||
| Zn(diPy)2 | 489e | 508e | 0.002j | 2.62 | 0.74 | −1.88 | 62 |
| [Cu(bpy)(IPr)]PF6 | 382d | 3.25 | 1.40 | −1.85 | 39 | ||
| Zn(Cz-pimn)2 | 430f | 503f | 2.73 | 0.92 | −1.81 | 59 | |
| [Cu(dBdPP)(Xant)]PF6 | 387f | 546f | 54.1f | 3.2 | 1.40 | −1.8 | 60 |
| W(CNdippPhOMe3)6 | 612j | 1.83j | 2.15 | −0.09 | −1.78 | 50 | |
| [Cu2(dmphen)2(tdapc)]BF4 | 420g | 2.95 | 1.18 | −1.77 | 58 | ||
| W(CNdippPhPh)6 | 629j | 1.73j | 2.08 | −0.11 | −1.73 | 50 | |
| [Cu(pytz)(POP)]BF4 | 2.71 | 1.00 | −1.71 | 63 | |||
| [Cu(phen)(IPr)]PF6 | 384d | 3.23 | 1.54 | −1.69 | 39 | ||
| Zn(Cz-bip)2 | 346e | 422e | 3.24 | 1.62 | −1.62 | 64 | |
| Zn(OMe2Ph-diPy)2 | 621e | 639e | 0.005j | 1.93 | 0.37 | −1.56 | 62 |
| Zn(DPA-pimp)2 | 422f | 518f | 2.67 | 1.12 | −1.56 | 59 | |
| [Cu(bpy)(Xant)]BF4 | 480k | 2.58 | 1.04 | −1.54 | 65 | ||
| [Cu(4-Mebpy)(POP)]BF4 | 480k | 2.58 | 1.05 | −1.53 | 65 | ||
| [Cu(bpy)(POP)]BF4 | 480k | 2.58 | 1.10 | −1.48 | 65 | ||
| Mo(CNAr3NC)3 | 419f | 609f | 0.22f | 2.2 | 0.16 | −1.48 | 21 |
| [Cu(4-Mebpy)(Xant)]BF4 | 480k | 2.58 | 1.12 | −1.46 | 65 | ||
| Zn(DPA-pimn)2 | 441f | 544f | 2.53 | 1.07 | −1.46 | 59 | |
| [Cu(6-Mebpy)(Xant)]BF4 | 470k | 2.64 | 1.19 | −1.45 | 65 | ||
| [Cu(dmphen)Xant]BF4 | 378g | 545g | 2.64 | 1.20 | −1.44 | 66 | |
| Zn(OMe2St-diPy)2 | 656e | 673e | 1.82 | 0.38 | −1.44 | 62 | |
| [Cu(6-Mebpy)(POP)]BF4 | 470k | 2.64 | 1.21 | −1.43 | 65 | ||
| [Cu(dap)2]Cl | 0.27 | 2.05 | 0.62 | −1.43 | 46 | ||
| [Cu(pytz)(dppe)]BF4 | 2.67 | 1.25 | −1.42 | 63 | |||
| [Cu(4-Mebpy)(PPh3)2]BF4 | 480k | 2.58 | 1.19 | −1.39 | 65 | ||
| [Cu(bpy)(PPh3)2]BF4 | 480k | 2.58 | 1.20 | −1.38 | 65 | ||
| [Cu(odmdpya)(IPr)]PF6 | 379d | 3.27 | 1.93 | −1.34 | 39 | ||
| Zn(TPP) | 588e | 597, 647e | 2.16 | 0.82 | −1.34 | 67 | |
| [Cu(mdmdpya)(IPr)]PF6 | 375d | 3.31 | 1.99 | −1.32 | 39 | ||
| Cr(CNtBuAr3NC)3 | 468f | 630f | 0.002f | 2.05 | 0.18 | −1.31 | 54 |
| [Cu(pytz)(dppm)]BF4 | 2.76 | 1.49 | −1.27 | 63 | |||
| [Cu(dppb)(POP)]BF4 | 544e | 0.002e | 2.38 | 1.11 | −1.27 | 68 | |
| [Cu(dppe)(POP)]BF4 | 546e | 0.23e | 2.38 | 1.13 | −1.25 | 68 | |
| [Cu(pdmdpya)(IPr)]PF6 | 389d | 3.19 | 1.94 | −1.25 | 39 | ||
| Zn(Ph-diPy)2 | 639e | 655e | 0.004e | 1.93 | 0.72 | −1.21 | 62 |
| Zn(Tol-ethynyl-Ph-diPy)2 | 658e | 673e | 0.004j | 1.85 | 0.65 | −1.2 | 62 |
| [Cu(dmphen)(POP)]BF4 | 383e | 565e | 14.3e | 2.60 | 1.40 | −1.20 | 69 |
| WO2(dhfcdbzbmp) | 427e | 598e | 62.0e | 2.64 | 1.45 | −1.19 | 70 |
| WO2(cdbzbmp) | 400e | 595e | 14.6e | 2.70 | 1.53 | −1.17 | 70 |
| [Cu(bath)(POP)]PF6 | 0.81 | 2.23 | 1.21 | −1.02 | 71 | ||
| Zn(Pc) | 1.77 | 0.78 | −0.99 | 72 | |||
| TiO(Pc) | 704e | 1.7 | 0.96 | −0.74 | 72 | ||
| Zn(Et-bip)2 | 344e | 422e | 3.24 | 2.52 | −0.72 | 64 | |
| [Cu(dpya)(IPr)]PF6 | 473d | 2.62 | 1.92 | −0.7 | 39 | ||
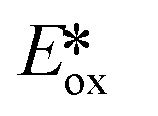
The nature of the emission in copper(I) complexes is complex and is usually a combination of phosphorescence and thermally activated delayed fluorescence (TADF).31,32 This is a result of the smaller spin–orbit coupling than 2nd or 3rd row transition metals and an MLCT excited state, a consequence of which is that the exchange integral of the frontier molecular orbitals is small, leading to a minuscule singlet-triplet energy gap (ΔEST) and therefore a facile thermally activated reverse intersystem crossing (rISC). The small ΔEST may also be advantageous for driving photocatalytic processes, since less of the excitation energy is lost through intersystem crossing compared with similar 2nd and 3rd row transition metals. The majority of copper(I) complexes used as PCs are either homoleptic bis(diimine) complexes of the form [Cu(N^N)2]+ with 2,9-dimethyl-1,10-phenanthroline (dmphen) as a prototypical N^N ligand; or heteroleptic tri- and tetracoordinate complexes composed of a combination of diimine, mono or bisphosphine, P^P, hybrid P^N, and N-heterocyclic carbene (NHC) ligands. Heteroleptic [Cu(N^N)(P^P)]+ complexes (Fig. 6) show promising and tunable photophysics,33 which has made this class popular as emitters in electroluminescent devices.34 Cu(XyBnta)(dppb) has the most negative excited state oxidation potential ( = −2.40 V) amongst heteroleptic copper(I) complexes. However, the very low ground state oxidation potential indicates that the reduction of the oxidized species of this copper(I) complex would be difficult, which would thus potentially limit the substrate scope; this complex has not been used as a PC. There are, on the other hand, several reports of the use of [Cu(dmphen)(Xant)]+ as a PC and photosensitizer in polymerisations,35 in oxidative-cyclisations of derivatives of triarylamines to make N-substituted carbazoles and stilbenoid to make helicenes,36,37 and oxygen sensing.38
= −2.40 V) amongst heteroleptic copper(I) complexes. However, the very low ground state oxidation potential indicates that the reduction of the oxidized species of this copper(I) complex would be difficult, which would thus potentially limit the substrate scope; this complex has not been used as a PC. There are, on the other hand, several reports of the use of [Cu(dmphen)(Xant)]+ as a PC and photosensitizer in polymerisations,35 in oxidative-cyclisations of derivatives of triarylamines to make N-substituted carbazoles and stilbenoid to make helicenes,36,37 and oxygen sensing.38
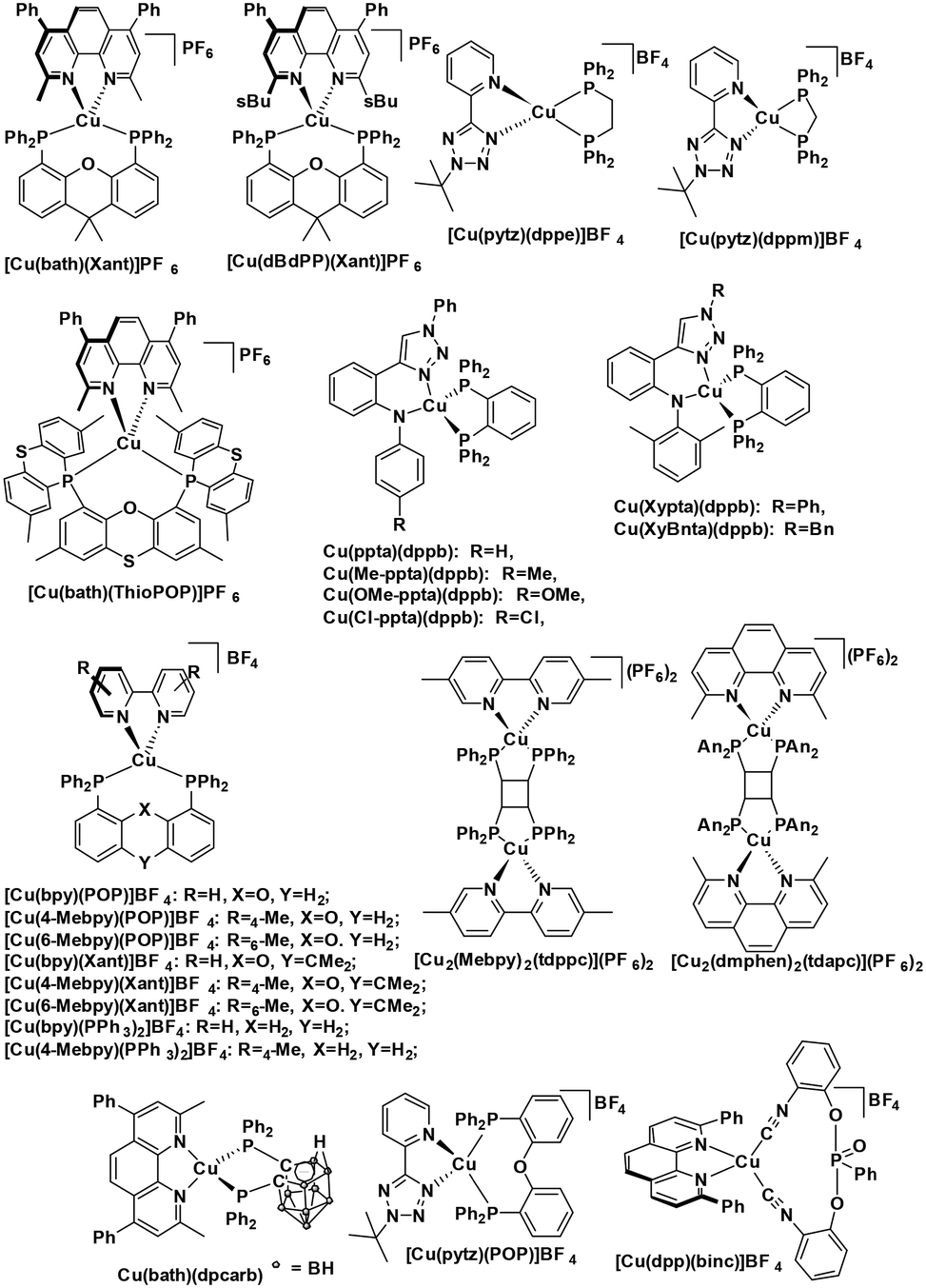 | ||
| Fig. 6 [Cu(N^N)(P^P)]+ and [Cu(N^N−)(P^P)] type complexes. | ||
Heteroleptic NHC copper(I) complexes (Fig. 8) also show the potential to be strong photoreductants, [Cu(bpy)(SIPr)]PF6 shows the most negative excited state oxidation potential amongst copper(I) NHC complexes of −2.02 V,39 while [Cu(mdmdpya)(IPr)]PF6 shows the highest ground state oxidation potential of 1.99 V, which indicates that the oxidized photocatalyst could work as a very strong oxidant.39
Other studies of similar heteroleptic NHC copper(I) complexes bearing N^N ligands do not investigate the electrochemical properties in such detail as this aforementioned study, however these related complexes are likely to be strong oxidants.40–45 Substitution of the N^N ligand using different bridging groups or substituents on the pyridyl moieties can tune the emission colour and intensity and also affect the electrochemistry of the complexes.
Homoleptic copper(I) complexes have also been intensively studied with [Cu(N^N)2]+ type of complexes (Fig. 7) such as [Cu(dap)2]Cl usually possessing a low excited state energy (E(0,0) = 2.05 eV)46 and are poorly emissive in solution. By contrast, [Cu(P^P)2]+ type of complexes usually have a much larger bandgap and show weak absorption in the visible region.
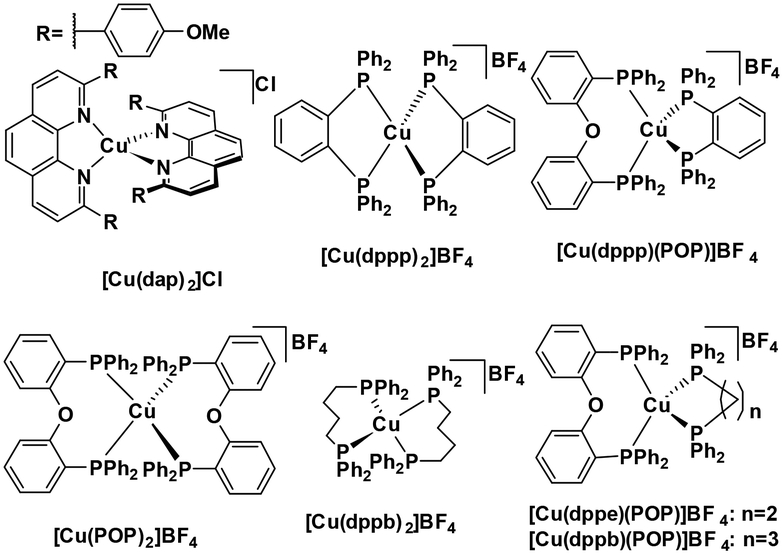 | ||
| Fig. 7 [Cu(N^N)2]+ and [Cu(P^P)2]+ complexes. | ||
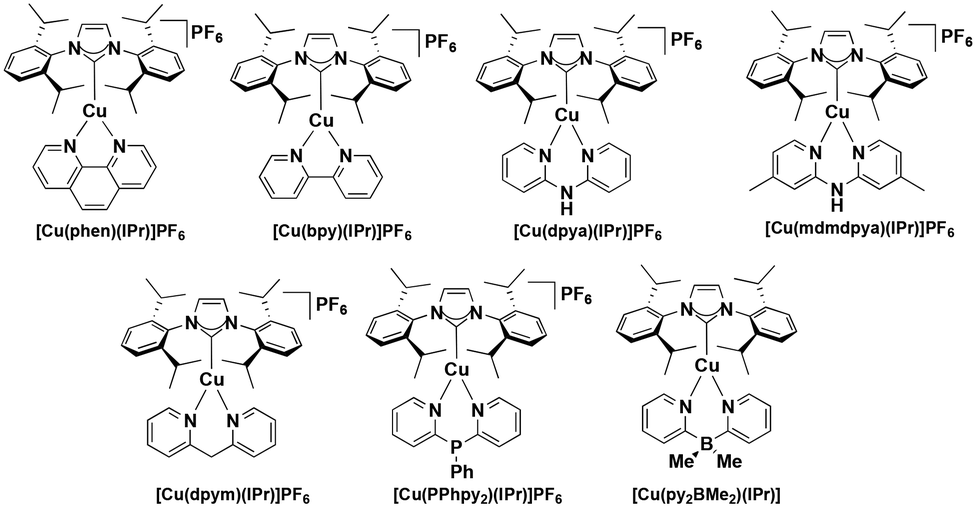 | ||
| Fig. 8 Copper(I) NHC complexes. | ||
As with Cu(I) complexes, the zinc(II) ion also has a d10 electronic configuration, and hence there is no accessible MC excited state. The high redox potential of zinc(II) leads to excited states that are localized on the ligands, which are ligand-centered (LC) or intra-ligand charge transfer (ILCT) in nature. In these complexes, the metal essentially only acts as a Lewis acid, stabilizing the ligand-centered orbitals, and is otherwise electronically benign. Zinc(II) complexes (Fig. 9) are usually recognized as fluorescent materials with τPL in the nanosecond regime.
 | ||
| Fig. 9 Luminescent zinc(II) complexes. | ||
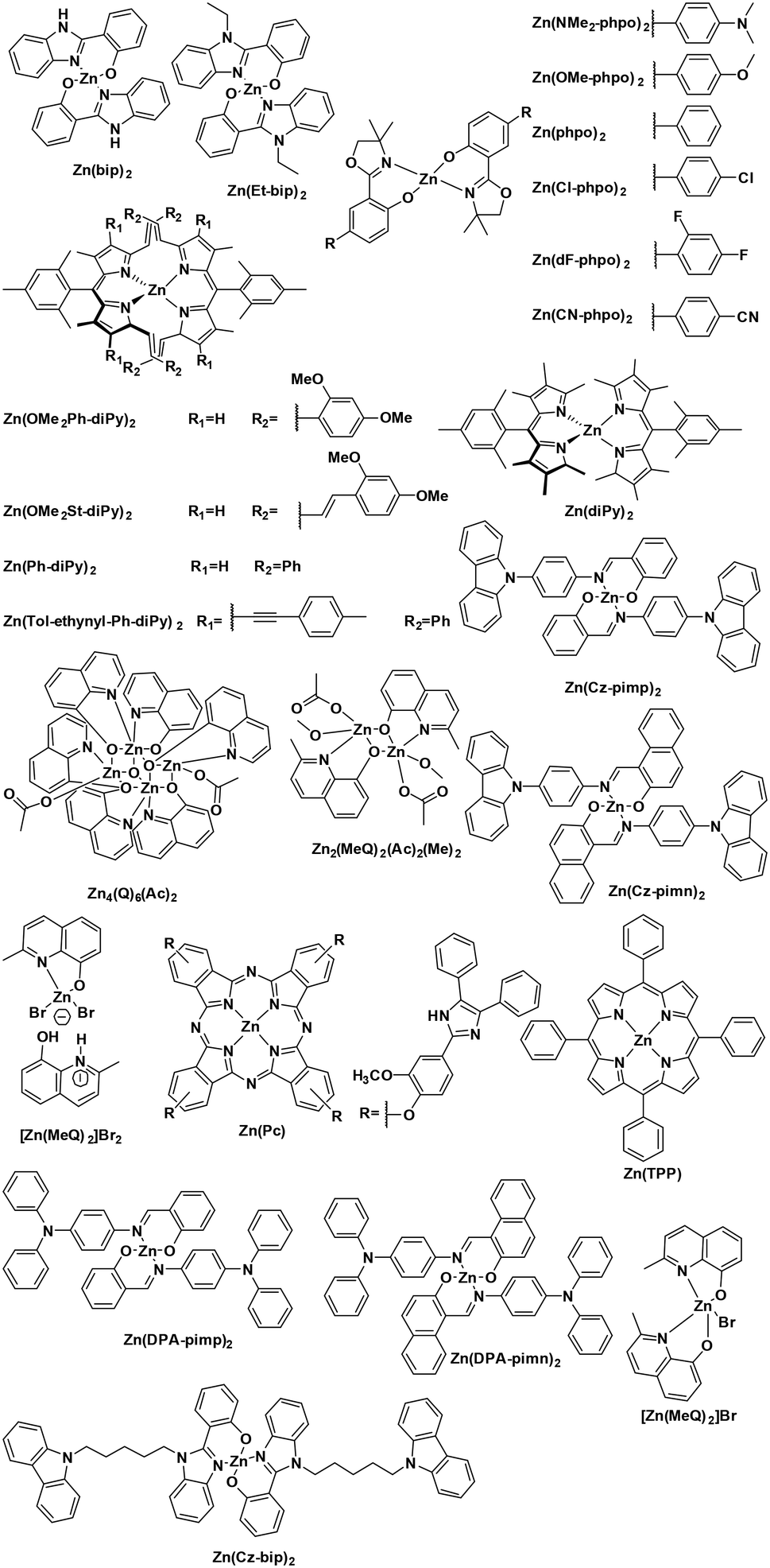
Despite their fluorescent nature, Zn(II) complexes remain attractive as photocatalysts. In particular the PC Zn(TPP) has an 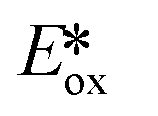 significantly more negative than that of the widely used [Ir(dF(CF3)ppy)2(dtbubpy)]PF6 (for Zn(TPP):
significantly more negative than that of the widely used [Ir(dF(CF3)ppy)2(dtbubpy)]PF6 (for Zn(TPP): 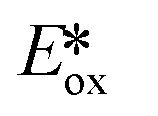 = −1.34 V; for [Ir(dF(CF3)ppy)2(dtbubpy)]PF6:
= −1.34 V; for [Ir(dF(CF3)ppy)2(dtbubpy)]PF6: 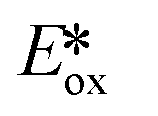 = −0.89 V) and hence is a stronger reducing agent. A great number of zinc complexes exist with similar structures and similar, or even more negative,
= −0.89 V) and hence is a stronger reducing agent. A great number of zinc complexes exist with similar structures and similar, or even more negative, 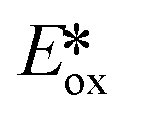 values (Table 1) and these could potentially also be applied as PCs. Zn(NMe2-phpo)2, first reported by Sang et al.,47 has the most negative
values (Table 1) and these could potentially also be applied as PCs. Zn(NMe2-phpo)2, first reported by Sang et al.,47 has the most negative 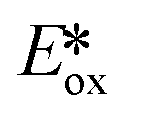 (−2.47 V) of the Zn complexes surveyed. Due to the combination of an electron-rich ligand and low oxidation state metal centre, this
(−2.47 V) of the Zn complexes surveyed. Due to the combination of an electron-rich ligand and low oxidation state metal centre, this 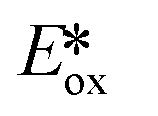 value is more negative than that of most common photoreductants based on noble metal complexes that utilise a triplet excited state (e.g., fac-Ir(ppy)3
value is more negative than that of most common photoreductants based on noble metal complexes that utilise a triplet excited state (e.g., fac-Ir(ppy)3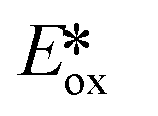 = −1.73 V). The Eox of Zn(NMe2-phpo)2 is 0.54 V, which implies that the regeneration of this PC would be difficult.
= −1.73 V). The Eox of Zn(NMe2-phpo)2 is 0.54 V, which implies that the regeneration of this PC would be difficult.
Nickel(0) also has a d10 electronic configuration; however, Ni(0) complexes have not attracted nearly as much attention as those based on copper(I) or zinc(II). Recently, Wenger et al. reported the first two examples of luminescent nickel(0) complexes, although they only emit at low temperature. They both have fairly low oxidation potentials at about 0.25 V while their  are −2.29 and −2.27, respectively. Considering their reported photophysics and electrochemistry data, these nickel(0) complexes may be good photoreductants (Fig. 10).48
are −2.29 and −2.27, respectively. Considering their reported photophysics and electrochemistry data, these nickel(0) complexes may be good photoreductants (Fig. 10).48
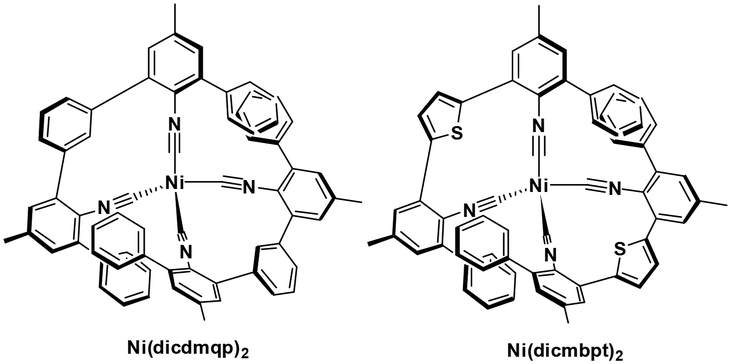 | ||
| Fig. 10 Nickel isocyanide complexes. | ||
Luminescent isocyanide-containing group 6 d6 complexes have been studied for more than 40 years, yet it is only recently that these complexes have been assessed as possible PCs and photosensitizers.49 Among chromium, molybdenum and tungsten complexes those based on W(0) showed the longest τPL of 1.83 μs for W(CNdippPhOMe3)6, which is similar to the lifetimes of many photocatalysts based on iridium(III) (e.g. fac-Ir(ppy)3τPL = 1.9 μs (ref. 8)). Both W(0)50,51 and Mo(0)21,52 complexes show strong phosphorescence in THF solution, with ΦPL of up to 44% and 2.3%, respectively. Though these complexes are strong reductants, they are air-sensitive, which is an undesirable trait for wide use as PCs.53 Chromium(0) complexes possess much shorter excited state lifetimes, illustrated by the τPL for Cr(CNtBuAr3NC)3 of only 2.2 ns. This value is, however, orders longer than that of iron(II) complexes (e.g. [Fe(btz)3]2+τPL = 0.5 ns).54 These complexes all possess very negative excited state oxidation potentials ( : −1.31 V to −1.98 V), which indicates that they are potentially powerful photoreductants (Fig. 11).
: −1.31 V to −1.98 V), which indicates that they are potentially powerful photoreductants (Fig. 11).
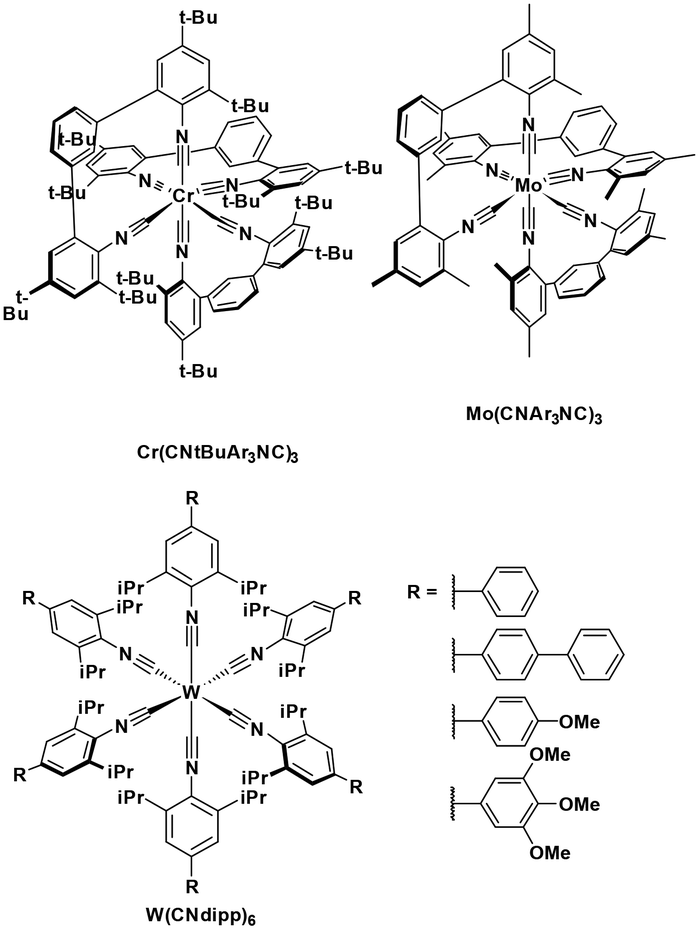 | ||
| Fig. 11 Chromium, molybdenum, tungsten isocyanide complexes. | ||
Photoredox catalytic reactions by oxidative quenching pathway
Recently there has been increasing interest in luminescent copper(I) complexes, which have mainly been used as emitters in electroluminescent devices. Copper(I) complexes represent the largest subset of Earth-abundant PCs, which is a function of both the favourable photophysical properties of copper(I) complexes and of the abundance of copper in the Earth's crust. It is best to begin the analysis of Cu(I) PCs by examining early studies in photoinduced electron transfer (PeT) by copper(I) complexes before moving on to discuss their use in photoredox catalysis.Photoinduced electron transfer between copper(I) complexes and derivatives of viologen, which act as electron acceptors, were first studied by Sakaki et al. in 1994.73 Both the cationic [Cu(dmphen)(PPh3)2]+ and the anionic [Cu(dmphen)(PPh2(m-C6H4SO3))2]− complexes were used as photosensitizers for the reduction of dicationic methyl viologen, MV2+, and neutral propylviologen disulfonate, PVS0. The authors found that the quantum yield of photoreduction of [Cu(dmphen)(PPh2(m-C6H4SO3))2]− was about 10 times higher than that of [Cu(dmphen)(PPh3)2]+. They concluded that the efficiency of the PeT was found to be governed mainly by the electrostatic interaction between the photosensitizer and the viologen rather than the thermodynamic driving force linked to the optoelectronic properties of the copper complexes.73 In 1996, Sakaki et al. studied the relationship between the quantum yield of photoreduction of viologen and the electron-donating ability of the phosphine ligands.74 They found that although the rate of encounter-complex formation is not sensitive to the nature of phosphine ligands, the more electron-donating phosphine ligands led to higher ΦPL and longer τPL, which resulted in a smaller deactivation rate constant – the sum of radiative and non-radiative decay rate constants – and a higher photoreduction quantum yield. These two early studies demonstrated how heteroleptic copper(I) complexes can act as photoreductants.
One of the applications of copper complexes as photocatalysts is in the water splitting reaction towards the generation of H2 as a solar fuel. Beller et al.75 have shown homogeneous photocatalytic hydrogen evolution using heteroleptic copper complexes, with turnover numbers (TON) varying from 224 to 1330 (moles of H+ reduced per mole of copper). Of the complexes tested [Cu(bath)(Xant)]PF6 gave the highest TON. This complex is a bright yellow emitter with a long-lived excited state in degassed THF (λPL = 546 nm, τPL = 54.1 μs, ΦPL = 75%).
The first example of photoredox catalysis using a copper(I) complex was reported by Sauvage et al. in 1987.46 They reported a photoactivated C–C coupling of benzylic bromides to form bibenzyl derivatives using [Cu(dap)2]Cl as the photocatalyst (Fig. 12). In the example of bis(p-nitro)bibenzyl, the authors report an isolated yield of 47%, demonstrating near quantitative conversion of the radical intermediate to the product. The TON of the copper catalyst was 30 and the starting complex was photostable (95% recovery) according to the solution analysis. In the case of the reaction in the presence of oxygen, the p-nitrobenzyl radical was converted to p-nitrobenzaldehyde nearly quantitatively (yield = 95%).
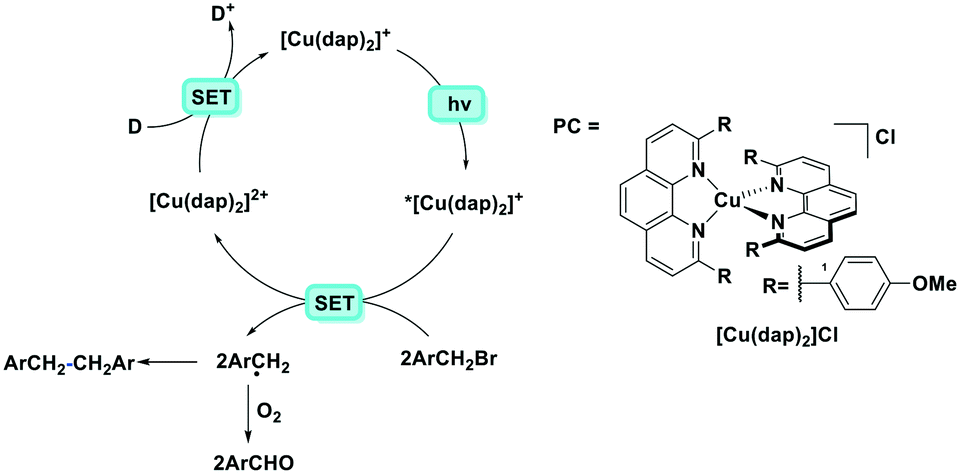 | ||
| Fig. 12 Photoredox catalysed reaction using a copper(I) complex reported by Sauvage et al. D represents an electron donor such as alkyl amines.46 | ||
More recently, the same complex, [Cu(dap)2]Cl, was employed by Reiser et al. towards the photoinduced atom-transfer radical addition (ATRA) between alkenes and alkyl bromides as well as between α-haloketones and allyl stannanes. Under the ATRA conditions high chemoselectivity was obtained (Fig. 13).68 According to the proposed mechanism, the highly reducing [Cu(dap)2]Cl* (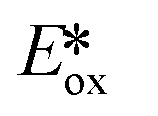 = −1.43 V) enabled the reduction of various organohalides including tetrabromomethane (Ered = −0.48 V), 2-bromo-1-phenylethan-1-one (Ered = −0.49 V) and nonafluoro-4-iodobutane (Ered = −1.41 V) to their radical species, which can then participate in radical addition reactions. The ATRA allylation of α-halocarbonyl compounds proceeded in good to excellent yield (63–89%). Classically, allylations of organohalides by allyltributyltin have proceeded through radical initiation using azobisisobutyronitrile (AIBN) at elevated temperature or using Et3B at ambient temperature.76–79 The work by Reiser et al. demonstrates that these sorts of reactions can proceed under much milder conditions and in good yields.
= −1.43 V) enabled the reduction of various organohalides including tetrabromomethane (Ered = −0.48 V), 2-bromo-1-phenylethan-1-one (Ered = −0.49 V) and nonafluoro-4-iodobutane (Ered = −1.41 V) to their radical species, which can then participate in radical addition reactions. The ATRA allylation of α-halocarbonyl compounds proceeded in good to excellent yield (63–89%). Classically, allylations of organohalides by allyltributyltin have proceeded through radical initiation using azobisisobutyronitrile (AIBN) at elevated temperature or using Et3B at ambient temperature.76–79 The work by Reiser et al. demonstrates that these sorts of reactions can proceed under much milder conditions and in good yields.
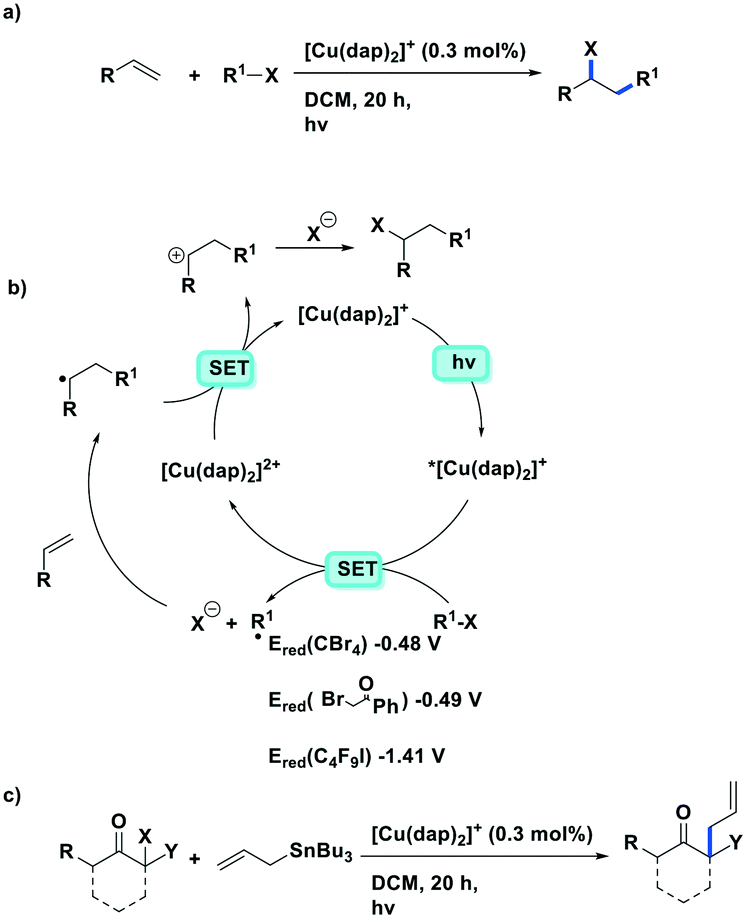 | ||
| Fig. 13 a) ATRA reactions of halides and olefins; b) proposed mechanism by Reiser et al.; c) visible-light-induced [Cu(dap)2]Cl catalysed allylation of α-halocarbonyl compounds. | ||
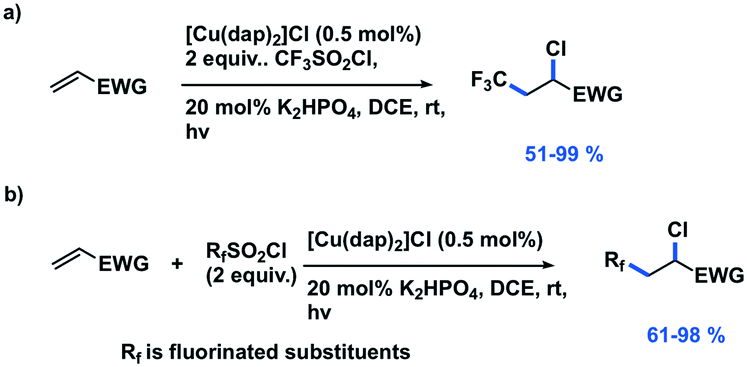
Reiser et al. expanded the scope of reactions with [Cu(dap)2]Cl as the PC in the context of the trifluoromethylation of alkenes (TfCl Ered = −0.18 V) using triflyl chloride (Fig. 14).80 Unlike most of the other photoredox trifluoromethylation conditions, this reaction was proposed to proceed through an inner-sphere PeT mechanism where the sulfonyl chloride coordinates to the metal center. The striking difference of [Cu(dap)2]Cl to Ru(II), Ir(III), or Eosin Y PCs points toward the essential role of copper in the overall process. This reaction proceeded in moderate to excellent yield of 42–87% and with good chemoselectivity (mostly affording the 1-trifluoromethyl-2-sulfonyl chloride product) over a large substrate scope. A similar conversion was obtained using [Ru(bpy)3]Cl2 as the PC; however, the chemoselectivity is different (mostly affording the 1-trifluoromethyl-2-chloride product). The quantum yield of this process was determined to be 12%, likely ruling out an efficient free radical chain mechanism. [Cu(dap)2]Cl was identified as a unique catalyst for this transformation, which in contrast to other PCs suppresses SO2 extrusion, thus providing access to α-trifluoromethylethylsulfonyl chloride skeletons. More recently, Hu et al. reported the synthesis of 1-trifluoromethyl-2-chloride products using a similar protocol with a series of heteroleptic [Cu(N^N)(Xant)]+ derivatives (Fig. 14c).81
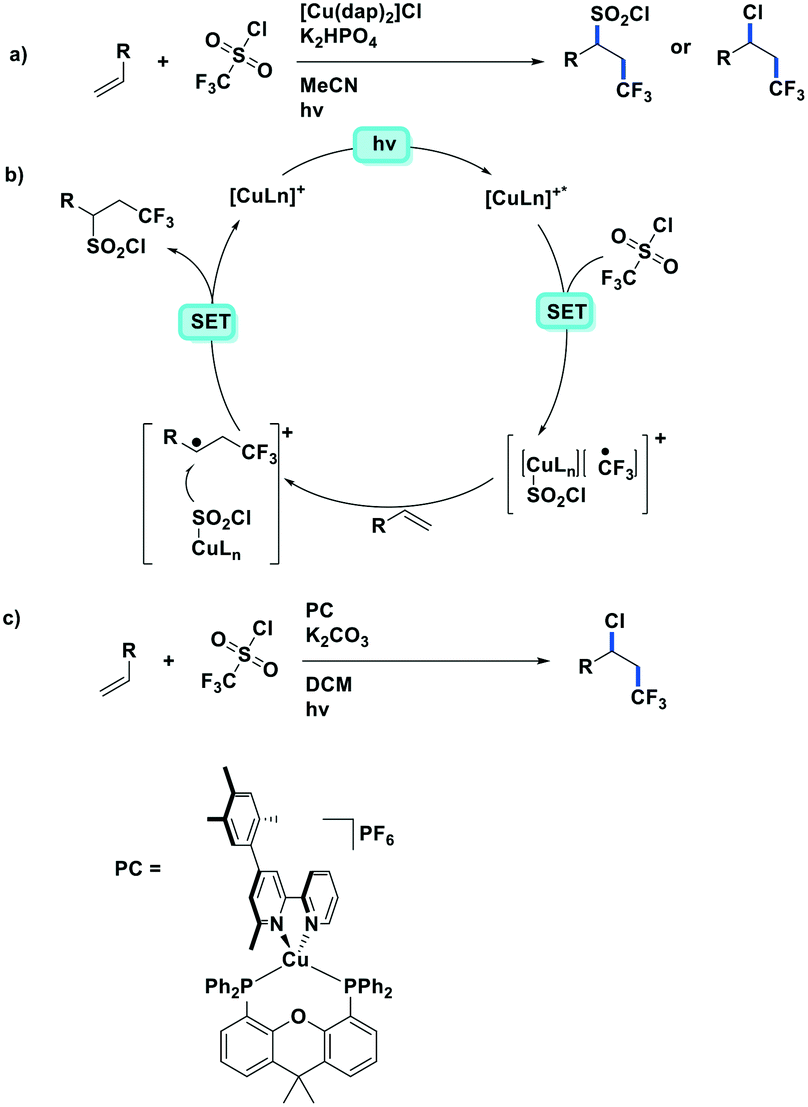 | ||
| Fig. 14 Visible-light-induced [Cu(dap)2]Cl catalysed trifluoromethylations of alkene. | ||
Reiser et al. expanded their PC methodology further towards a two-step synthesis of β-hydroxysulfones from sulfonyl chlorides and alkenes (Fig. 15a).82 The first step exploited their previously developed [Cu(dap)2]+-photocatalyzed ATRA reaction between triflyl chloride and alkenes, while the second step implicated a second photoredox catalytic ATRA reaction, this time using an Ir PC, with the sulfonyl chloride intermediate and substituted styrene substrates. In this second step, they screened a number of PCs, with fac-Ir(ppy)3 (95%) and [Ru(bpy)3]2+ (90%) performing better than [Cu(dap)2]Cl (61%); no further insight was provided to rationalize the difference in yields amongst the three PCs. The tandem PC protocol tolerated a wide range of sulfonyl chlorides and afforded the β-hydroxysulfones in good to excellent yields (49–84% for the first step, and 84–92% for the second step). For the second step, the yields for the scope of different sulfonyl chlorides reacting with prop-1-en-2-ylbenzene varied between 78–97%, while the alkenes scope has the limitation of only working with aryl-substituted alkenes (yield: 66–86%).
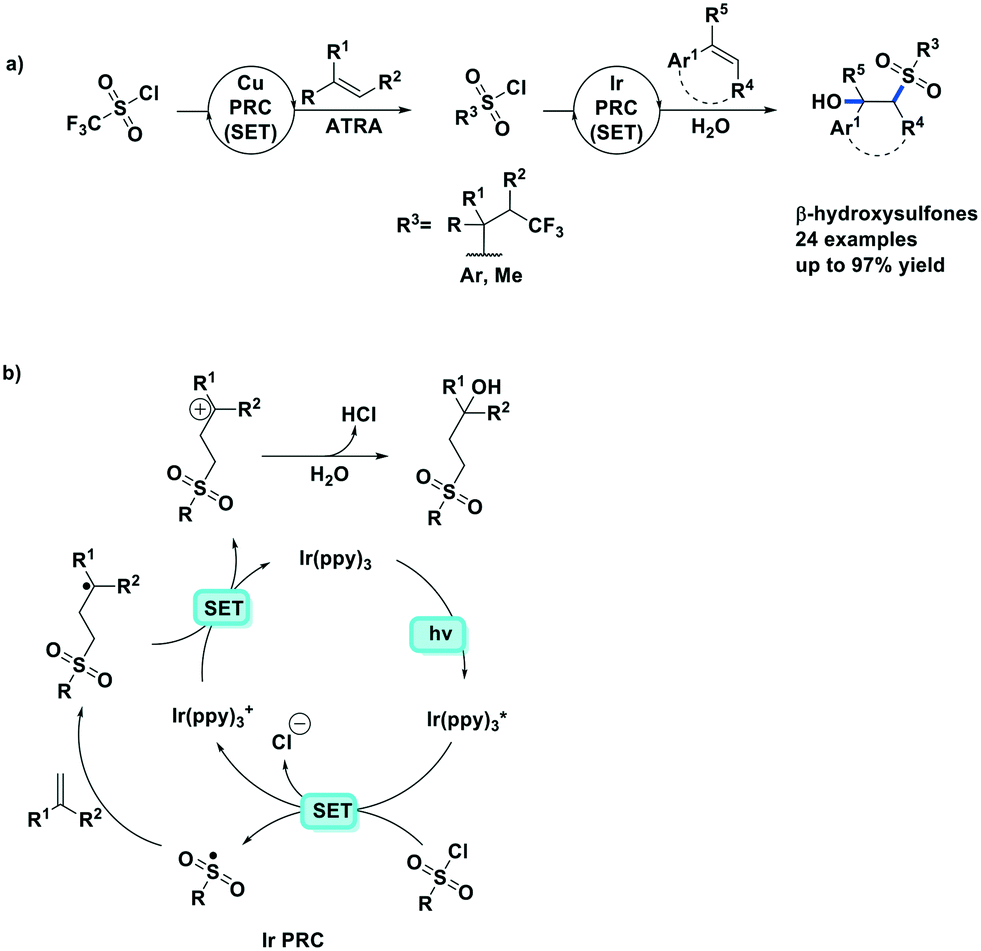 | ||
| Fig. 15 Synthesis of β-hydroxysulfones from sulfonyl chlorides and alkenes,82 Cu PC (SET) see Fig. 14b. | ||
In another communication by the same group,83 the same ATRA process was employed using benzyl halides as the coupling partner, a la Sauvage et al., with alkenes (Fig. 16a). They demonstrated the successful coupling of a large scope of styrenyl derivatives with electron-deficient benzyl halides in generally good yields (35–95%). Reiser et al. extend their photocatalytic ATRA reaction methodology to the coupling of benzyl halides with silyl enol ethers (Fig. 16b). Reiser et al. also showed how the products formed from these photoredox catalysed reactions can be used further towards the synthesis of 2-substituted tetrahydroquinoline compounds.
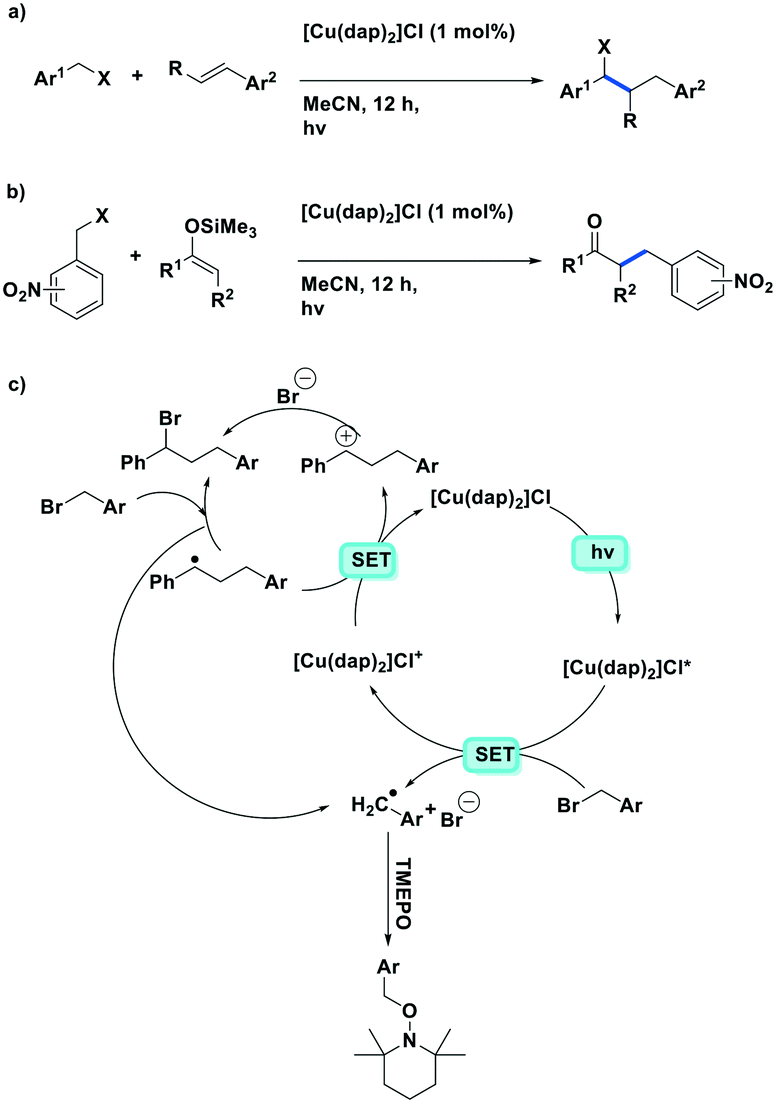 | ||
| Fig. 16 ATRA of benzyl halides to aryl olefins. ATRA reaction of electron-deficient benzyl halides with silyl enol ethers. | ||
The same group developed five new copper(I) complexes employing strongly π-accepting diisonitrile ligands. The complex [Cu(dpp)(binc)]BF4 exhibits reversible redox behaviour at Eox = 0.69 V for the CuI/II couple, a correspondingly highly negative 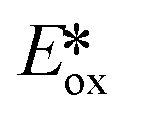 of −1.88 V and a long τPL of 17 μs, thereby making it a much stronger photoreductant than [Cu(dap)2]Cl (
of −1.88 V and a long τPL of 17 μs, thereby making it a much stronger photoreductant than [Cu(dap)2]Cl (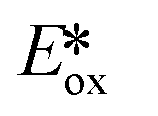 = −1.43 V, τPL = 560 ns for [Cu(dap)2]Cl).61 Using [Cu(dpp)(binc)]BF4, ten examples of coupling between allylic amines and bromomalonates were presented with yields ranging widely from 29–88% (Fig. 17). A notable disadvantage of this reaction is that it will not work in the presence of cyano and methylsulfonyl groups at the para-position of the benzylbromide substrate.61
= −1.43 V, τPL = 560 ns for [Cu(dap)2]Cl).61 Using [Cu(dpp)(binc)]BF4, ten examples of coupling between allylic amines and bromomalonates were presented with yields ranging widely from 29–88% (Fig. 17). A notable disadvantage of this reaction is that it will not work in the presence of cyano and methylsulfonyl groups at the para-position of the benzylbromide substrate.61
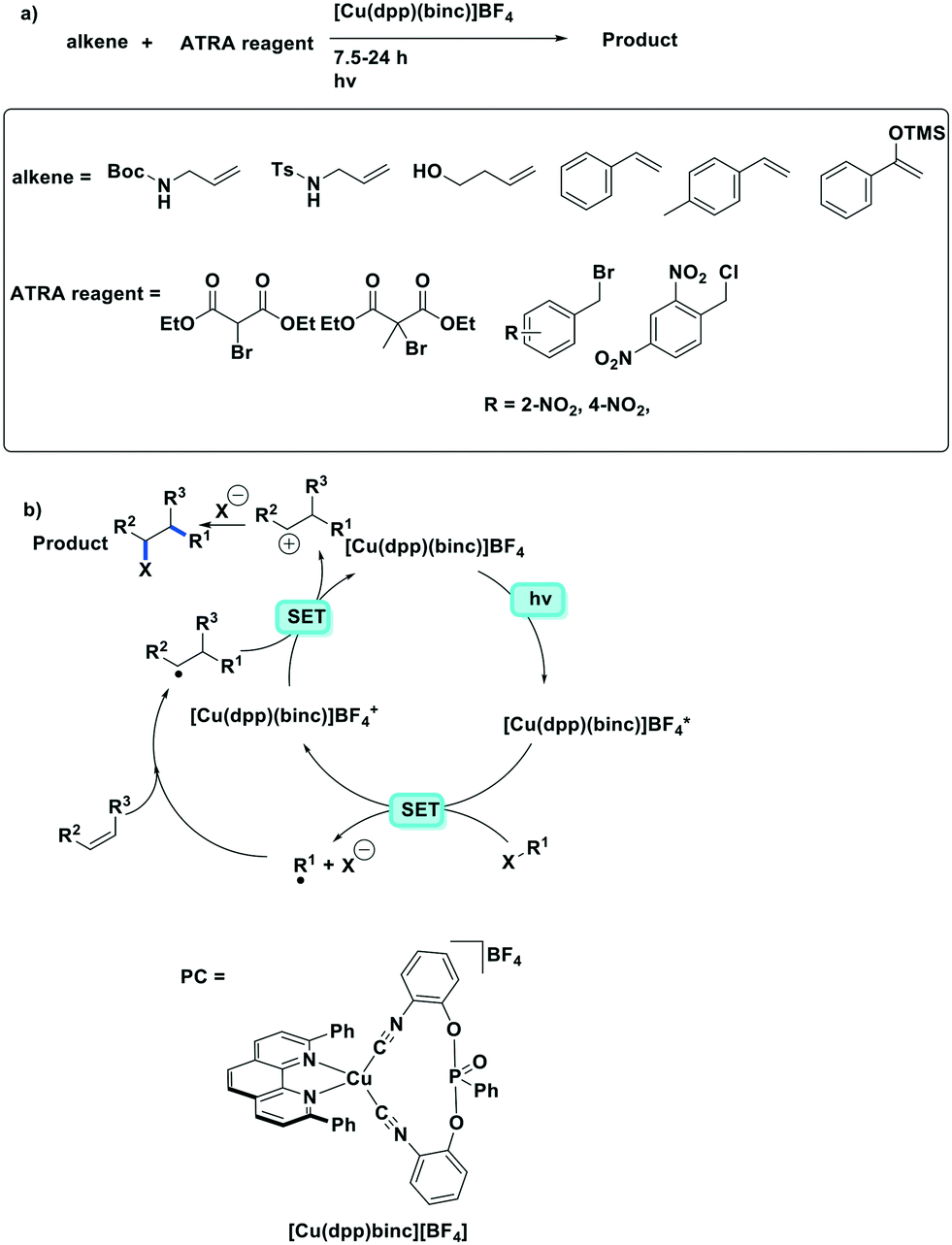 | ||
| Fig. 17 Visible-light-induced [Cu(dpp)(binc)]Cl catalytic ATRA reaction. | ||
Dolbier et al. reported ATRA reactions of fluoroalkylsulfonyl chlorides with electron-deficient alkenes,84 expanding on the previous work of Reiser. Moderate yields of 51–59% were obtained when TfCl was used with a wide range of alkenes. When combined with heating to 100 °C other fluoroalkylsulfonyl chlorides can also be activated. Under these modified conditions, twenty different alkenes and three different fluoroalkylsulfonyl chlorides could be coupled together in good to excellent yields of 61–98% (Fig. 18). The use of [Cu(dap)2]Cl as the PC resulted in significantly improved yields compared to other PCs such as [Ru(phen)3]Cl2, [Ru(bpy)3]Cl2, fac-Ir(ppy)3, as well as thermally activated reactions with AIBN and dilauroyl peroxide (DLP). The authors proposed that a combination of the use of the stronger photoreductant and improved Cl˙ transfer properties of [Cu(dap)2]+ resulted in the higher yields.
 | ||
| Fig. 18 Photoredox atom transfer radical addition reactions of fluoroalkylsulfonyl chlorides with electron-deficient alkenes.84 | ||
Dolbier et al. expanded their synthetic methodology towards the aminodifluoromethylation of unactivated alkenes (Fig. 19a).85 This reaction also used [Cu(dap)2]Cl as the photoreductant to generate difluoromethyl radicals from difluoromethylsulfonyl chloride before subsequent reaction with N-substituted-2,2-disubstituted-pent-4-en-1-amine substrates. Silver carbonate was used as a base and to suppress the chloride addition byproduct (Fig. 19b). Moderate to excellent yields (48–95%) were obtained amongst the 19 substrates. However, the reaction does not work well in the presence of the more electron-deficient N-substituents such as BOC-protected amines. Similar to their previous studies, [Cu(dap)2]+ was found to work better as the PC than fac-Ir(ppy)3, which gave chlorodifluoromethylated byproducts.
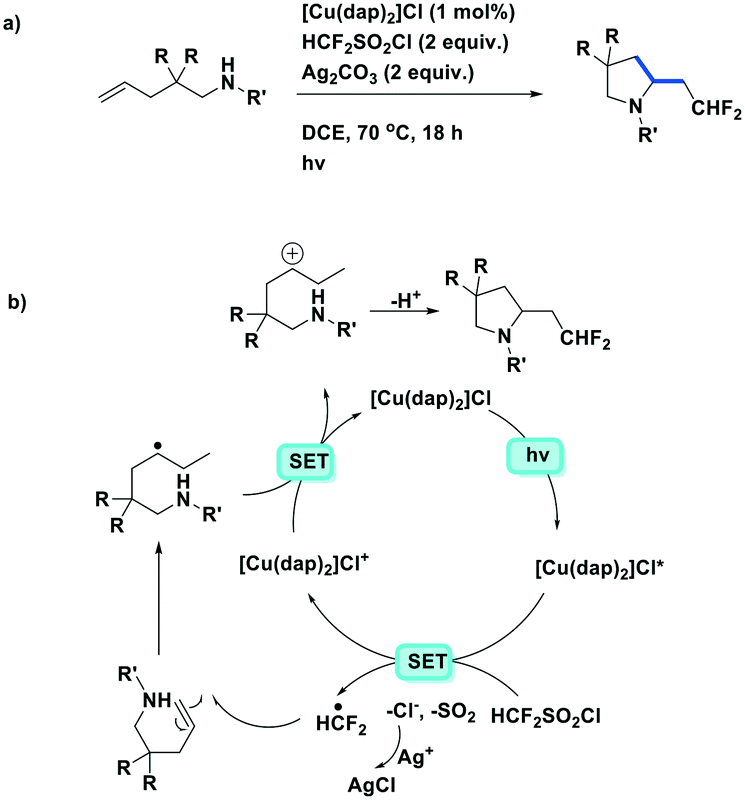 | ||
| Fig. 19 Photoredox-catalyzed intramolecular aminodifluoromethylation of unactivated alkenes.85 | ||
Expanding on the work of Dolbier as well as other research in photoredox trifluoromethylations, Reiser et al. published the synthesis of trifluoromethylated sultones from alkenols (Fig. 20).86 Similar to their previous work,80 the Cu PC actively participates in the catalysis through weak SO2Cl− coordination. By contrast, both [Ru(bpy)3]Cl2 and fac-Ir(ppy)3 showed very small conversion to the desired product. Good yields were obtained throughout their substrate scope of alkenols (50–90%) except when the resulting product forms 4-membered and 7-membered rings. The reaction could be generalized to other sulfonyl chlorides such as undecafluorohexane-1-sulfonyl chloride; however, both pentafluorobenzenesulfonyl chloride are trichloromethanesulfonyl chloride were incompatible reactants for this transformation.
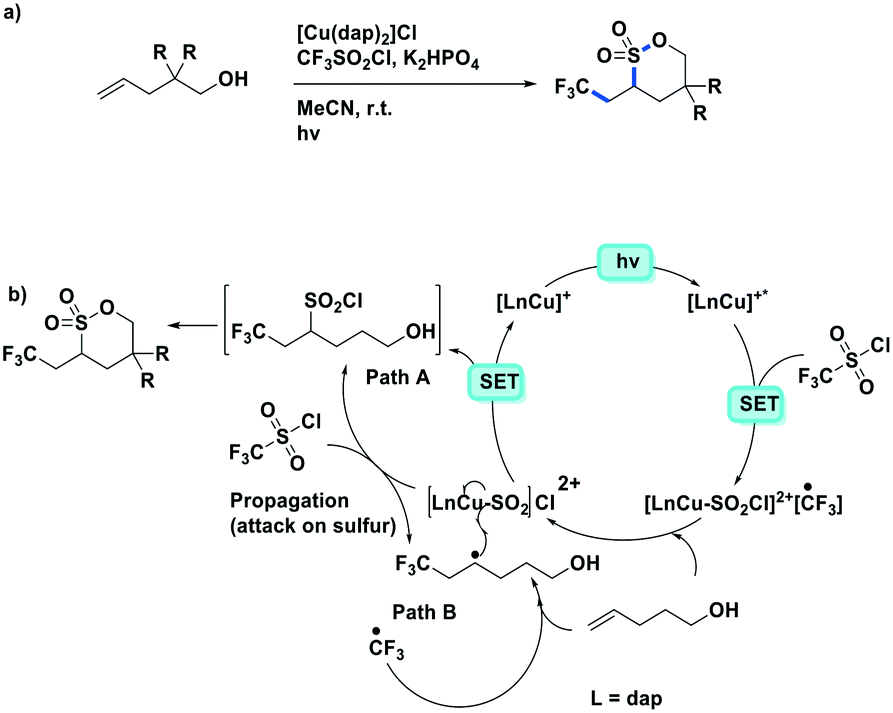 | ||
| Fig. 20 Synthesis of trifluoromethylated sultones from alkenols using a copper photoredox catalyst.86 | ||
Ollivier et al. reported the photocatalytic reduction of diaryliodonium ions with [Cu(dap)2]+ and the reaction of the corresponding aryl radicals with allyl sulfones to afford functionalized allylated products (Fig. 21).87 The same group also investigated the related PC [Cu(dpp)2]+ (dpp = 2,9-diphenyl-1,10-phenanthroline). Both Cu PCs were comparable in terms of the yield of the desired product and these performed similarly well to [Ru(bpy)2]Cl2 (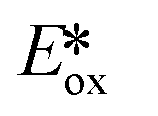 = −0.81 V) and fac-Ir(ppy)3 (
= −0.81 V) and fac-Ir(ppy)3 (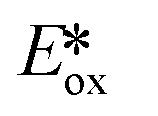 = −1.73 V). Five different allyl tosylates and eight different iodonium salts were used in the substrate scope study with yields ranging from 43% to 82%. Fig. 21b shows the authors' proposed mechanism for the reaction, with evidence presented supporting an oxidative quenching cycle.
= −1.73 V). Five different allyl tosylates and eight different iodonium salts were used in the substrate scope study with yields ranging from 43% to 82%. Fig. 21b shows the authors' proposed mechanism for the reaction, with evidence presented supporting an oxidative quenching cycle.
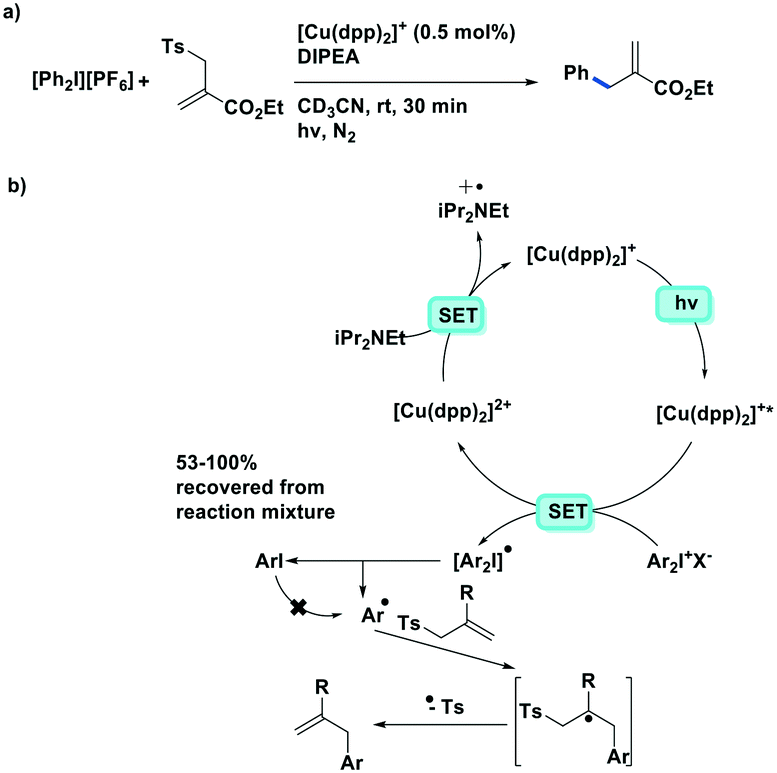 | ||
| Fig. 21 Reductive arylation of allyl-tosylates by Ollivier et al.; the photoreduction of aryl iodides was discussed by Sauvage et al.87 | ||
Xiao et al. studied the photopolymerization of both acrylates and epoxides using heteroleptic complexes [Cu(dmphen)(Xant)]BF4, [Cu(dmphen)(POP)]BF4 and [Cu(Czbp)(POP)]BF4.35 This polymerization is initiated by a cation generated from the photoredox catalytic reaction between an iodonium salt and a Cu(I) PC while N-vinyl-9H-carbazole (NVK) can be used to accelerate the reaction. For the polymerization of trimethylolpropane triacrylate (TMPTA), the highest conversion rates of 63% and 56% (upon photoirradiation at 405 nm and 455 nm, respectively), were obtained with [Cu(dmphen)(POP)]BF4 as the PC, which mediated the polymerization of Iod2 (Fig. 22) and NVK. Similar to TMPTA, the highest conversion (∼56%) for the polymerisation of (3,4-epoxycyclohexane)methyl-3,4-epoxy cyclohexylcarboxylate (EPOX) was achieved with the [Cu(dmphen)(POP)][BF4]/Iod2/NVK system (Fig. 22). In terms of the photoredox initiation, both iodonium salts have very small negative reduction potentials (Ered = −0.20 V), which indicates that the photoreduction is thermodynamically favoured. In this case, [Cu(dmphen)(POP)]BF4 obtained the highest conversion because of its correspondingly large extinction coefficient of the MLCT band.
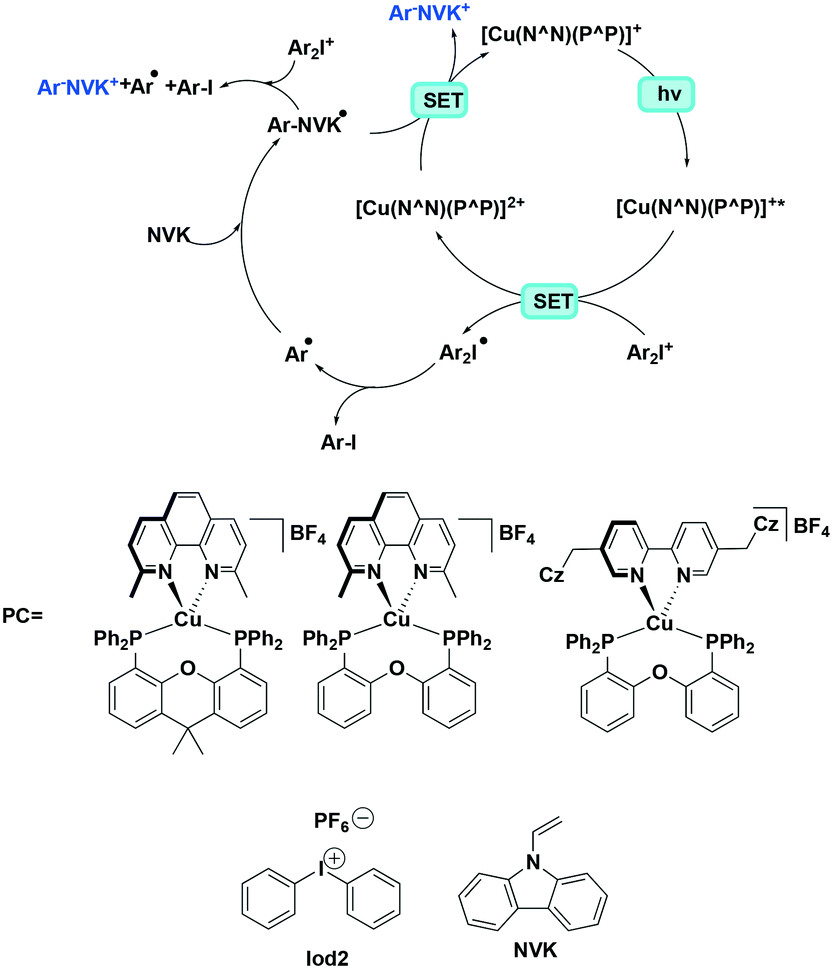 | ||
| Fig. 22 The mechanism of cation formation. | ||
Collins et al. reported several examples of oxidative photocyclization reactions of Z-dinaphthylethenes to form helicenes using heteroleptic copper(I) PCs.37,88 Their first report concerned the formation of [5]helicene in the presence of a mixture of iodine and propylene oxide as the co-oxidants (Fig. 23a). Collins et al. found that the use of in situ-generated Cu(I) photocatalysts showed comparable or even higher yields of the desired product. It was found that among the PCs tested, the use of [Cu(dmphen)(Xant)]BF4 showed the best yield (yield: 57%) while <10% conversion was observed with each of [Ru(bpy)3](PF6)2 or [Ir(ppy)2(dtbubpy)]PF6. The authors also demonstrated a scale-up synthesis of [5]helicene (yield: 42%) as well as one conducted using continuous flow chemistry (yield: 40%) (Fig. 23b). Although the yields of both were reduced compared to the small-scale reaction, the advantages of the flow reactor setup were manifest in the much shorter reaction times (10 h vs. 120 h). The authors expanded their substrate scope towards the photocyclization of stilbenes (Fig. 23c); however, only the trimethoxy derivative could be converted to the phenanthrene product, albeit in low yield (23%) while the parent stilbene was unreactive.
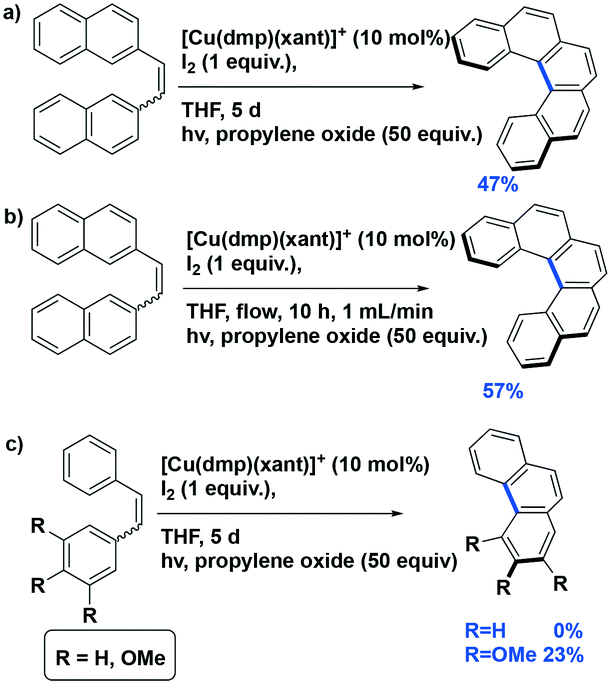 | ||
| Fig. 23 a) Photocyclization of [5]helicene; b) photocyclization of [5]helicene with flow reactor; c) photocyclization of stilbenes. | ||
The Collins group subsequently showed that these photocyclization reactions can be applied towards the synthesis of carbazoles from triarylamines, both in batch and in flow.37 Among the photocatalysts tested, the use of [Cu(dmphen) (Xant)]BF4 resulted in the highest yield of 85% (reaction time of 14 days), which was higher than that [Ru(bpy)3](PF6)2 where the yield was only 27%. By using a flow reactor, the reaction time was not only expectedly reduced to 20 h but after the optimization of the reaction conditions, the substrate scope was expanded to include diarylamines. A range of eight triphenylamine and eight diarylamine substrates were converted to the corresponding carbazoles in 50–95% yield. The photocyclization of unsymmetrical substrates was also investigated. Good chemoselectivity was found when 4-methyl-N,N-diphenylaniline and 4-methoxy-N,N-diphenylaniline were used as substrates, with N-para-substituted-phenylcarbazoles obtained in a ratio of 7:1 for 4-methyl-N,N-diphenylaniline and 9:1 for 4-methoxy-N,N-diphenylaniline. In the major isomers, the electron-rich N-tolyl and N-anisyl rings were not incorporated in the carbazole core but were instead positioned exo to the carbazole skeleton. The proposed reaction mechanism is shown in Fig. 24a. Similar to the helicene formation mechanism, an oxidative quenching pathway with the concomitant reduction of I2 was proposed. Subsequent oxidation of the di- or triarylamine via SET would afford the nitrogen-centered radical cation with concomitant reduction of the PC to its ground state electronic configuration. The preference for this radical cation species to be delocalized preferentially to one of the aromatic rings is responsible for the observed chemoselectivity for the different constitutional isomers.
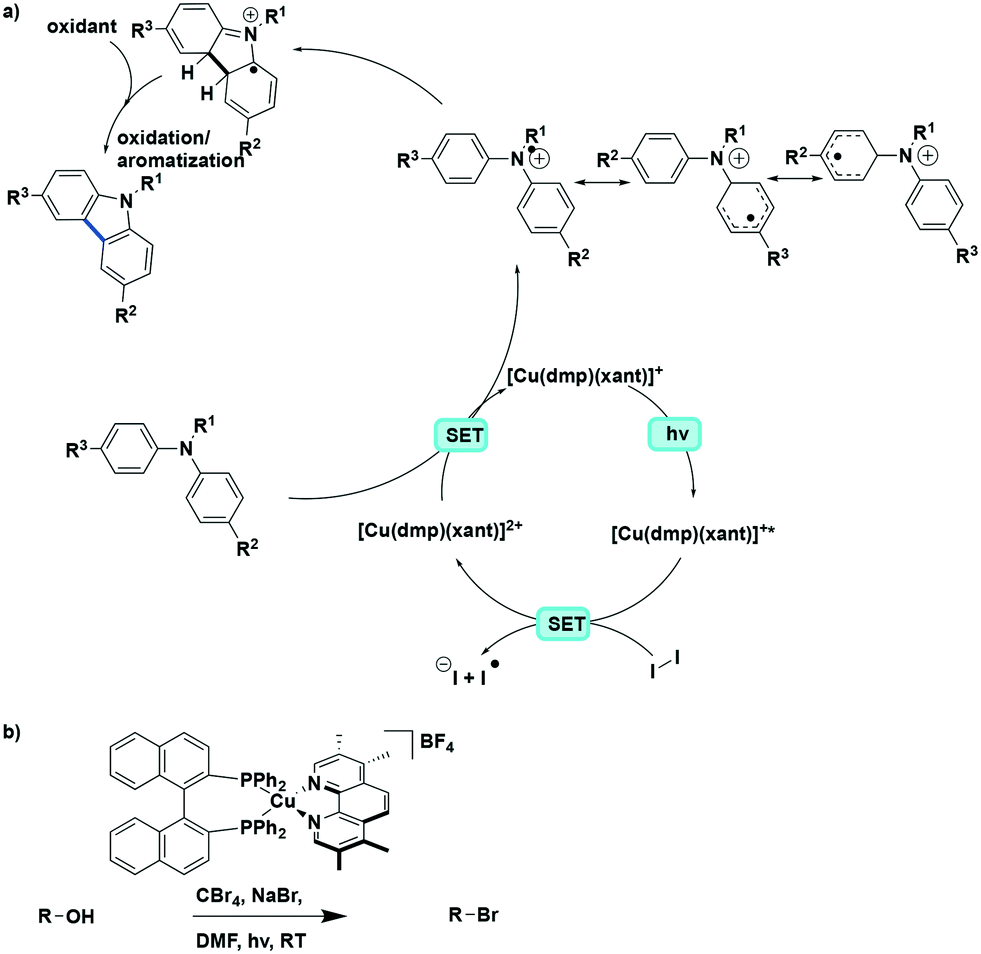 | ||
| Fig. 24 a) Proposed mechanism for the photoredox cyclization of arylamines to carbazoles, b) photoredox Appel reaction with same continuous flow setup. | ||
More recently, the same group reported an example of a photoredox Appel reaction under continuous flow conditions. The Collins group screened the reactivity of a wide range of different heteroleptic copper(I) complexes generated through a combination of 10 different N^N ligands and 4 different P^P ligands. With the selected photocatalyst [Cu(tmp)(BINAP)]+, the yield of alkyl halides from alkyl alcohol reached up to 99% (Fig. 24b).89
Greaney et al. reported a three-component azidation of styrene and analogous aryl-substituted olefins90 using the hypervalent iodine reagent 1-azido-1-λ3-benzo[d][1,2]iodaoxol-3(1H)-one as an N3 delivery vehicle and [Cu(dap)2]Cl as the PC (Fig. 25). Nineteen different olefins were evaluated using these photochemical azidation conditions with yields ranging from 34–89%. The reaction conditions tolerated the use of both bulky and mildly electron-withdrawing substituents. In the absence of light, the reaction mechanism changed and di(azide) products were formed in moderate to excellent yields of 49–97%. The mechanistic details remain to be established.
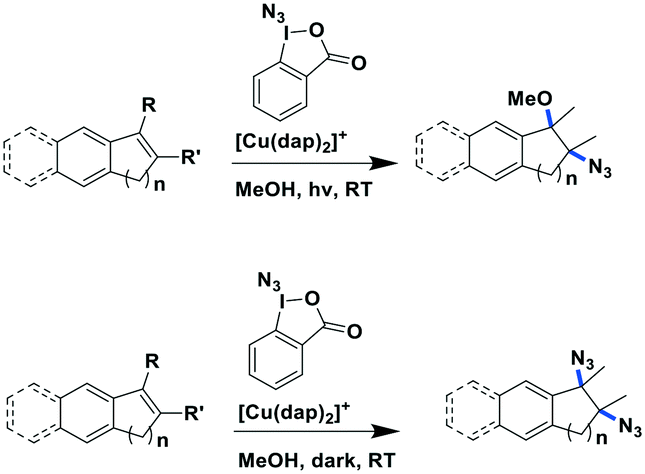 | ||
| Fig. 25 Three-component azidation of styrene-type double bonds.90 | ||
The same group extended their methodology towards the photoredox catalytic benzylic azidation.91 Herein, they reported the efficient synthesis of 20 primary azides (yields: 22–85%), five secondary and tertiary azides (yields: 66–82%) and five heterocylic and thioanisyl azides (yields: 50–93%). Their proposed radical chain reaction mechanism is shown in Fig. 26.
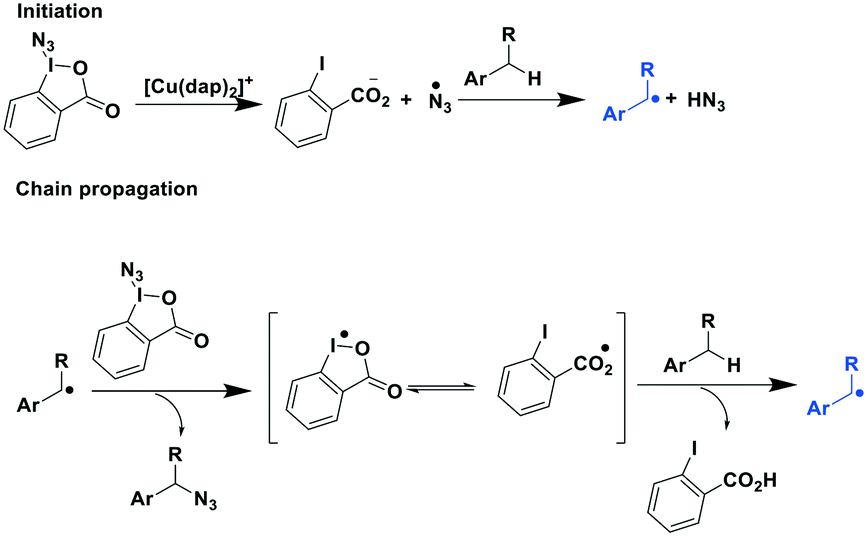 | ||
| Fig. 26 Proposed mechanism of photoredox catalytic benzylic azidation.91 | ||
Bissember et al. reported a copper(I)-photocatalyzed α-amino C–H bond functionalization using [Cu(dap)2]Cl as the photocatalyst.92 They found that due to the small 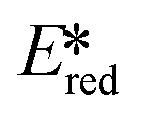 value of [Cu(dap)2]Cl, the PC would be unlikely to proceed via a reductive quenching pathway directly with the substrate. Bissember et al. therefore designed a synthetic strategy of firstly using [Cu(dap)2]Cl to photoreduce oxygen and then use the oxidized species, [Cu(dap)2]2+, to oxidize a N,N-dimethylaniline substrate to the radical cation (Fig. 27). Another facet of this work is the use of trifluoroacetic acid as a Brønsted acid co-catalyst to activate 1-phenyl-1H-pyrrole-2,5-dione. Moderate to good yields were obtained over most of the aniline substrates (yields: 42–80%). However, reaction of 1-benzyl-1H-pyrrole-2,5-dione and N,N-dimethylaniline gave a yield of only 35% while the use of 1-(p-substituted phenyl)pyrrolidine also led to low yields (yields: 21–36%). It is unclear what is the reason for these low yields observed for these three substrates.
value of [Cu(dap)2]Cl, the PC would be unlikely to proceed via a reductive quenching pathway directly with the substrate. Bissember et al. therefore designed a synthetic strategy of firstly using [Cu(dap)2]Cl to photoreduce oxygen and then use the oxidized species, [Cu(dap)2]2+, to oxidize a N,N-dimethylaniline substrate to the radical cation (Fig. 27). Another facet of this work is the use of trifluoroacetic acid as a Brønsted acid co-catalyst to activate 1-phenyl-1H-pyrrole-2,5-dione. Moderate to good yields were obtained over most of the aniline substrates (yields: 42–80%). However, reaction of 1-benzyl-1H-pyrrole-2,5-dione and N,N-dimethylaniline gave a yield of only 35% while the use of 1-(p-substituted phenyl)pyrrolidine also led to low yields (yields: 21–36%). It is unclear what is the reason for these low yields observed for these three substrates.
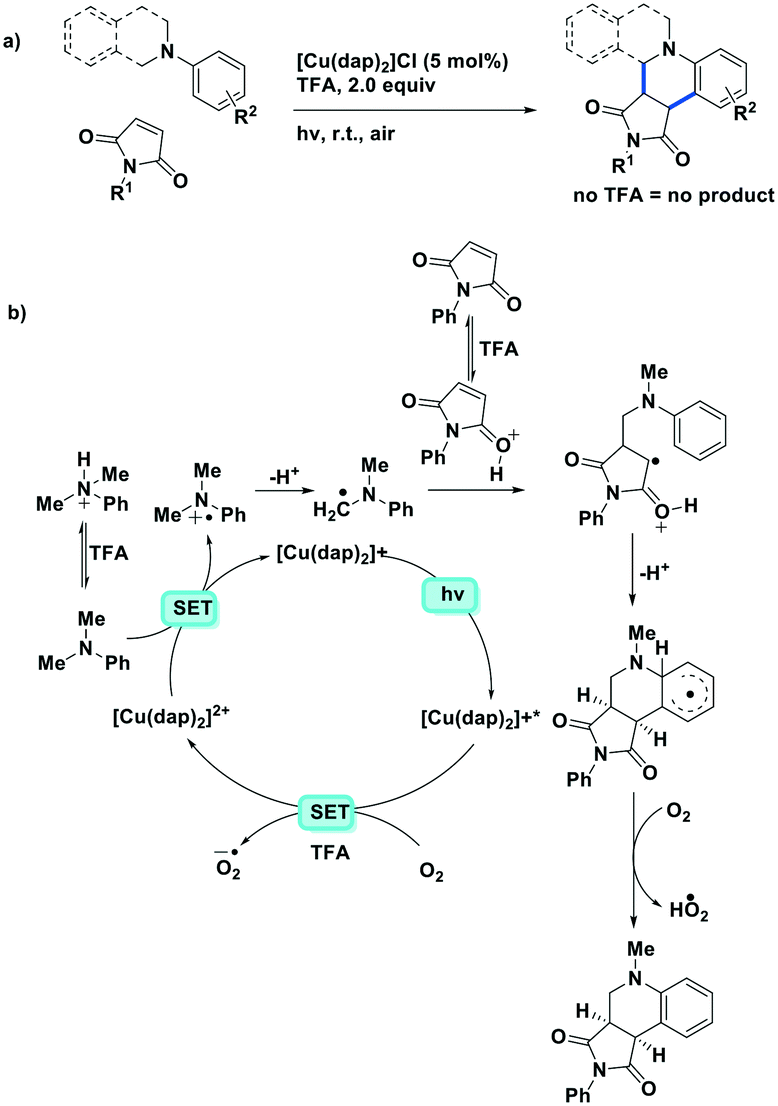 | ||
| Fig. 27 Brønsted acid co-catalysis in copper(I)-photocatalyzed α-amino C–H bond functionalization.92 | ||
Apart from several examples of Zn(II) complexes used in artificial photosynthesis,93 often in tandem with other metals such as manganese, few reports of Zn(II) PCs exist. One notable example employing the well-known zinc porphyrin complex Zn(TPP) for polymerisation reactions was recently reported by Boyer et al.67 In a study investigating a variety of metal porphyrins, the authors showed that Zn(TPP) selectively activates photoinduced electron transfer-reversible addition-fragmentation chain transfer (PeT-RAFT) polymerization of trithiocarbonate compounds for the polymerisation of styrene, (meth)acrylates and (meth)acrylamides using a broad range of excitation wavelengths (435–655 nm). Zn(TPP), unlike Ir-based PCs, is capable of chemoselective activation of trithiocarbonates over other thiocarbonyl compounds such as dithiobenzoate, dithiocarbamate and xanthate. This is based largely on favourable zinc–sulfur soft–soft interactions that are also important in biological systems. An examination of the mechanism of the reaction (Fig. 28) reveals that Zn(TPP) acts as a PC, regulating living radical polymerization under air. The polymerization rates can be tuned as a function of excitation energy while the polymerization can be arrested and then restarted by turning the light off/on.
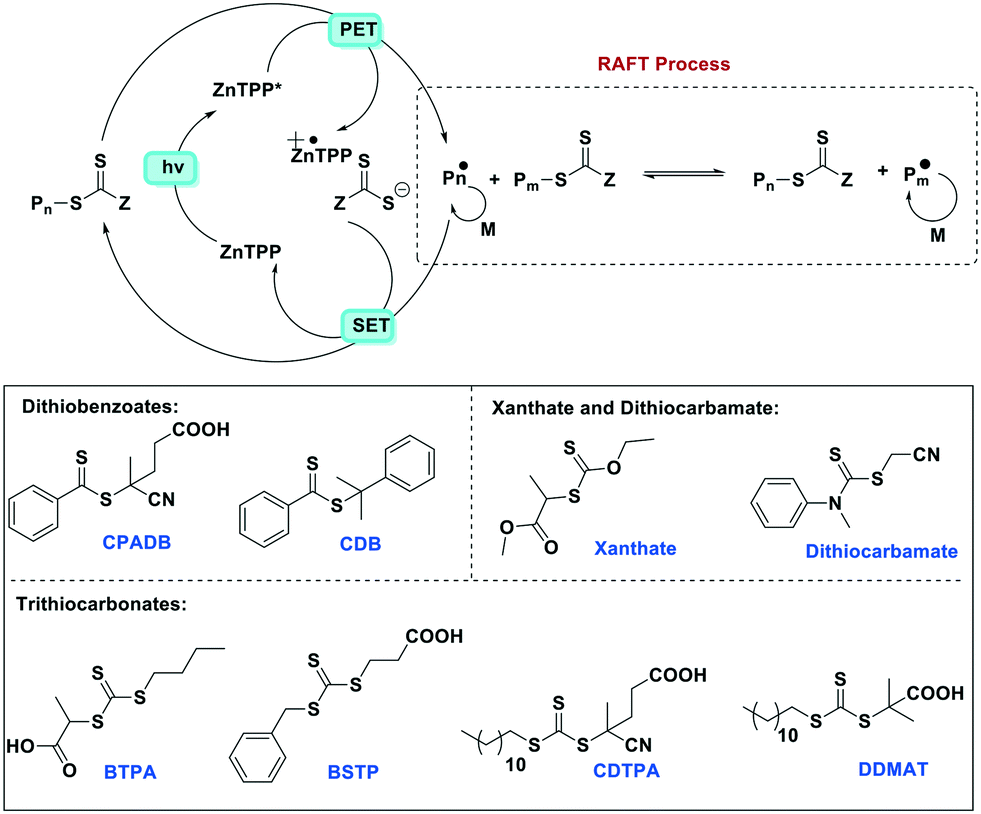 | ||
| Fig. 28 Proposed mechanism for PeT-RAFT polymerization mediated by Zn(TPP) and different thiocarbonylthio compounds (M = monomer). | ||
Presently, photoredox catalysts that proceed via an oxidative quenching pathway are dominated by two well-studied copper complexes in [Cu(dap)2]Cl or [Cu(dmphen)(Xant)]+ with only a handful of reactions using zinc(II) photocatalysts; however, according to our survey, abundant MLCT nickel(0), zinc(II) and other copper(I) complexes are featured with more negative 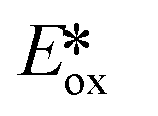 , longer τPL, which would suggest that they are of great potential as photoredox catalysts.
, longer τPL, which would suggest that they are of great potential as photoredox catalysts.
Photoredox catalysts that proceed by a reductive quenching pathway
There are many photoredox reactions that proceed by a reductive quenching pathway.94 Currently, one of the most widely used photooxidants is the sky-blue emitting iridium complex [Ir(dF(CF3)ppy)2(dtbubpy)]+ (λPL = 470 nm, Ered = −1.37 V,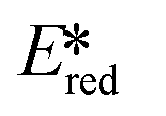 = 1.21 V).95 Unlike Table 1 which summarized PCs that proceed via an oxidative quenching pathway where the PCs are mostly complexes with a MLCT excited state, many of the metal complexes in this section possess a LMCT excited state. The design paradigm to possess low-lying accessible LMCT excited states requires a complex with high valent metal ions and strongly σ-donating ligands. These types of complexes have been studied for many years though there are only a few examples of emitters and PCs based on LMCT luminescent metal complexes. After early studies of Re(II) complexes,96,97 some photoluminescent tantalum98,99 and niobium98 oxalate and imido complexes were reported, along with metallocene complexes based on tantalum.99
= 1.21 V).95 Unlike Table 1 which summarized PCs that proceed via an oxidative quenching pathway where the PCs are mostly complexes with a MLCT excited state, many of the metal complexes in this section possess a LMCT excited state. The design paradigm to possess low-lying accessible LMCT excited states requires a complex with high valent metal ions and strongly σ-donating ligands. These types of complexes have been studied for many years though there are only a few examples of emitters and PCs based on LMCT luminescent metal complexes. After early studies of Re(II) complexes,96,97 some photoluminescent tantalum98,99 and niobium98 oxalate and imido complexes were reported, along with metallocene complexes based on tantalum.99
Although these elements (rhenium, scandium, niobium, tantalum) are not as abundant as copper or iron, they still qualify as Earth-abundant. Indeed, over the last two decades, metallocene complexes of scandium, titanium, zirconium, tungsten and hafnium were all shown to have low-lying LMCT excited states.100–102 Although these metallocenes showed favourable photophysics consisting of an emissive LMCT state with τPL ranging from hundreds of nanoseconds to microseconds, they have not yet been explored as PCs. It is at present difficult to assess their potential given the absence of electrochemical data reported in the literature (Fig. 29 and Table 2).
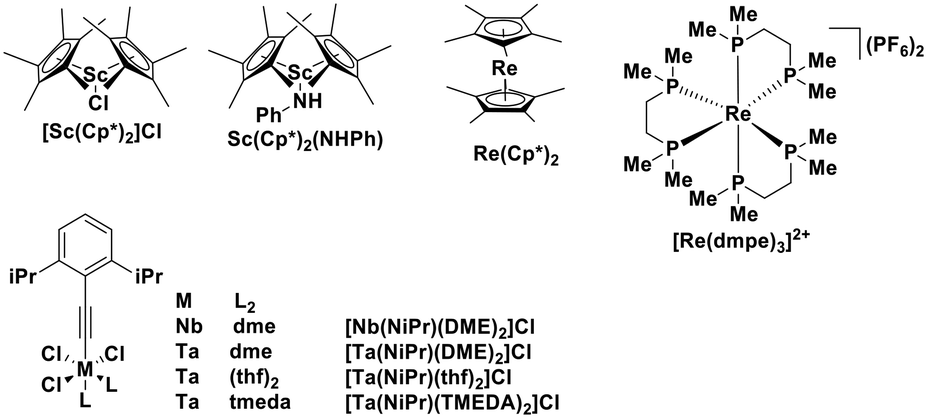 | ||
| Fig. 29 Luminescent rhenium, scandium, niobium, tantalum complexes. | ||
| Compound | λ abs/nm | λ PL/nm | τ PL/μs | Ref. |
|---|---|---|---|---|
|
a In isooctane/methylcyclohexane 1:1 (v/v).
b In PhMe.
c In PhH.
|
||||
| [Sc(Cp*)2]Cl | 390a | 521a | 2.0a | 100 |
| Sc(Cp*)2(NHPh) | 391a | 600a | 2.0a | 100 |
| [Nb(NiPr)(DME)2]Cl3 | 323b | 483c | 0.046c | 98 |
| [Ta(NiPr)(DME)2]Cl3 | 280b | 404c | 0.135c | 98 |
| [Ta(NiPr)(thf)2]Cl3 | 280b | 429c | 0.074c | 98 |
| [Ta(NiPr)(TMEDA)2]Cl3 | 297b | 429c | 0.071c | 98 |
More recently, with the development of photoredox catalytic reactions, LMCT complexes have gained increasing attention. Low-spin iron(III) complexes with NHC ligands have also been shown to have an albeit short-lived LMCT excited state (τPL of 100 ps), observable room temperature photoluminescence and the highest 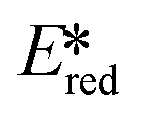 amongst the complexes surveyed in this review (
amongst the complexes surveyed in this review (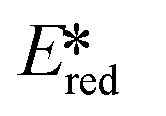 = 1.96 V).103 Thus far, only cobalt(III), vanadium(V) and zirconium(IV) complexes have been employed as PCs amongst all LMCT complexes with Earth-abundant metal ions: the cobalt(III) complexes were used in photoredox trifluoromethylation, the vanadium(V) complexes in photocatalytic C–C cleavage reactions104–106 and the zirconium(IV) complexes in photoredox dehalogenation, reduction of electron-deficient olefin and reductive coupling of benzyl bromide to bibenzyl reaction.107 In particular, thanks to the combination of high-valent vanadium(V) ion, an electron-rich Schiff base ligand with electron-deficient substituents (F, NO2), vanadium(V) complexes have been found to have very small Ered and very high
= 1.96 V).103 Thus far, only cobalt(III), vanadium(V) and zirconium(IV) complexes have been employed as PCs amongst all LMCT complexes with Earth-abundant metal ions: the cobalt(III) complexes were used in photoredox trifluoromethylation, the vanadium(V) complexes in photocatalytic C–C cleavage reactions104–106 and the zirconium(IV) complexes in photoredox dehalogenation, reduction of electron-deficient olefin and reductive coupling of benzyl bromide to bibenzyl reaction.107 In particular, thanks to the combination of high-valent vanadium(V) ion, an electron-rich Schiff base ligand with electron-deficient substituents (F, NO2), vanadium(V) complexes have been found to have very small Ered and very high 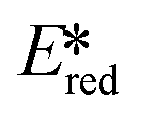 values, making them potent photooxidants.106
values, making them potent photooxidants.106
Milsmann et al. reported titanium(IV) and zirconium(IV) complexes carrying two 2,6-bis(pyrrolyl)pyridine ligands.107 Zr(MePDP)2 shows an intense red emission centered at 594 nm while Ti(MePDP)2 shows an absorption band at 800 nm without any detectable emission. In terms of Zr(MePDP)2, its Ered is −1.60 V, which is similar to that of [Cu(dmphen)(Xant)]+ (Ered = −1.73 V); however, the smaller excited state energy of Zr(MePDP)2 (E(0,0) = 2.09 eV) limits its 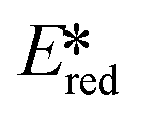 to 0.49 V compared to that of [Cu(dmphen)(Xant)]+ at 0.91 V (Fig. 31).
to 0.49 V compared to that of [Cu(dmphen)(Xant)]+ at 0.91 V (Fig. 31).
Though TiO2 is widely used in heterogeneous photoredox catalytic reactions,108 research into photoactive titanium complexes and their downstream homogeneous photoredox catalysis is presently absent. In 2013, Kantekin et al. reported a titanium(IV) complex bearing a tetradentate phthalocyanine ligand TiO(Pc) which is red-emissive (Fig. 32).72
It should be noted that although these LMCT complexes show a great potential as PCs, there remain some weakness in terms of their optoelectronic properties. For instance, iron(III) and vanadium(V) complexes have only very short τPL (100 ps for the iron(III) NHC complex, and 420 ps for the vanadium(V) complex, VOLF, see Fig. 30104), while the E0,0 of iron (III) and zirconium(IV) complexes are small (2.14 eV for [Fe(btz)3]3+ and 2.09 eV for Zr(MePDP)2). Thus, further efforts are required to develop higher excited state energy LMCT complexes with improved lifetimes if this class of compounds is to find use in photoredox catalysis.
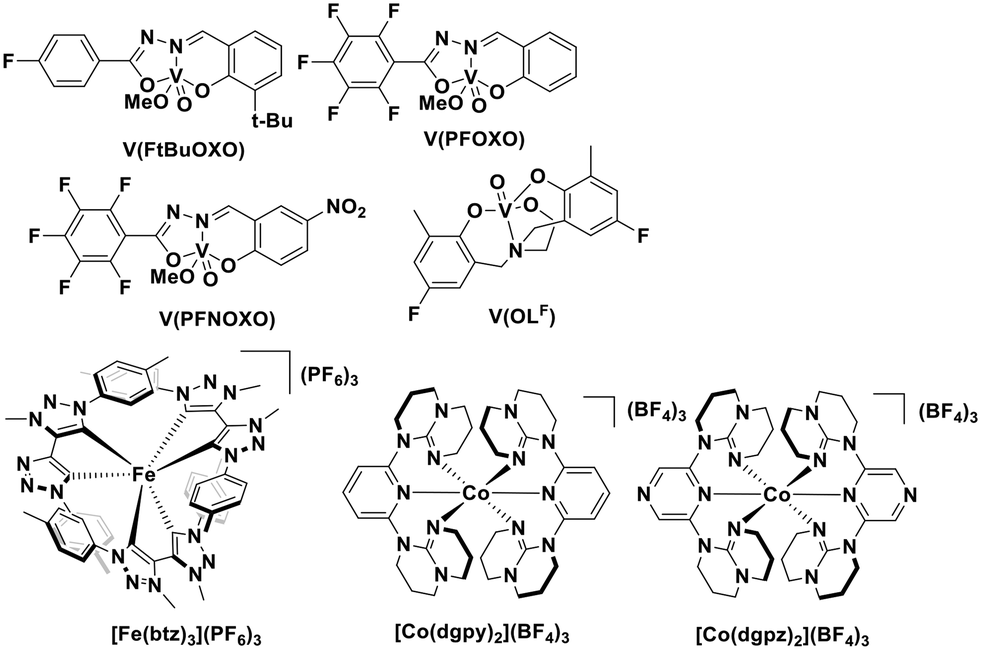 | ||
| Fig. 30 Vanadium(V), iron(III) and cobalt(III) complexes. | ||
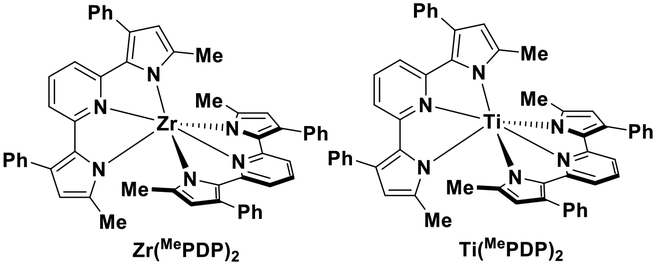 | ||
| Fig. 31 Titanium(IV) and zirconium(IV) complexes.107 | ||
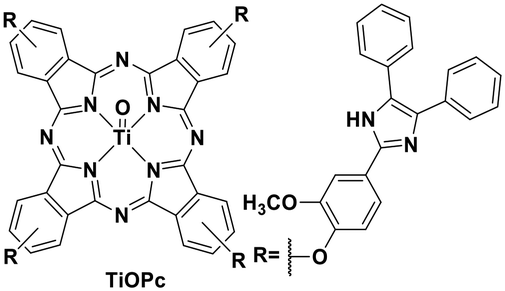 | ||
| Fig. 32 Photoluminescent titanium(IV) complex. | ||
Using complexes with the 3rd row element tungsten is a potential solution towards these issues. Three new families of tungsten(III) compounds (Fig. 33) were reported by Che et al. and their effectiveness as PCs investigated.70 Both classes of complex absorb strongly in the UV with λabs values between 240–290 nm. These complexes also exhibit absorption in the visible region. In CH2Cl2, the lowest energy absorption band for WO2(dhfcdbzbmp) resides at 427 nm while for WO2(dpb)2 an intense ILCT absorption band is observed for both complexes at 396 nm. These complexes display weak photoluminescence at room temperature, with ΦPL ranging from 0.1–2.8% in dichloromethane, though up to 22% in a 5 wt% doped mCP thin film. Complexes WO2(dhfcdbzbmp) and WO2(cdbzbmp) exhibit orange to red emission while WO2(dbp)2 derivatives give what the authors refer to as “dual fluorescence–phosphorescence” leading to a range of different colours. The dual emission was identified based on time-resolved photoluminescence spectra where τPL at 490–520 nm was <0.1 μs while that at 585–616 nm was significantly longer, ranging from 10–76 μs (Table 3). While these properties are interesting, more important for photocatalysis are the excited state reduction potentials. For WO2(dhfcdbzbmp) and WO2(dbp)2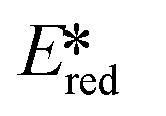 are 0.59 and 0.40 V, respectively, which are sufficiently high for the complexes to be used as photooxidants.
are 0.59 and 0.40 V, respectively, which are sufficiently high for the complexes to be used as photooxidants.
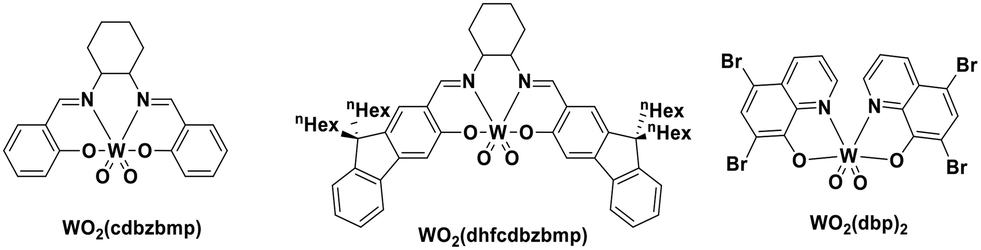 | ||
| Fig. 33 Tungsten(VI) complexes. | ||
| Compound | λ abs/nm | λ PL/nm | τ PL/μs | E (0,0)/eVa | E red/Vb |
|
Ref. |
|---|---|---|---|---|---|---|---|
a
E
0,0 usually provided in the literature. In the cases where this value is not explicitly reported it has been approximated by the energy of the onset absorption band extracted by Digitize function of Origin 9.
b
E
red are referenced vs. SCE.
c
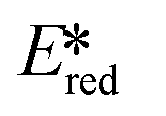 = Ered + E0,0.
d In degassed MeCN.
e In degassed DCM.
f In degassed THF.
g In degassed CHCl3.
h In a PMMA doped film.
i In degassed H2O. = Ered + E0,0.
d In degassed MeCN.
e In degassed DCM.
f In degassed THF.
g In degassed CHCl3.
h In a PMMA doped film.
i In degassed H2O.
|
|||||||
| V(PFNOXO) | 373d | 507d | 2.86 | 0.82 | 3.68 | 105 | |
| [Co(dgpy)2](BF4)3 | 311d | 440d | 5.07 × 10−3d | 3.30 | −0.58 | 2.72 | 113 |
| V(PFOXO) | 389d | 507d | 2.69 | 0.52 | 3.21 | 105 | |
| V(FtBuOXO) | 398d | 507d | 2.64 | 0.29 | 2.93 | 105 | |
| [Co(dgpz)2](BF4)3 | 340d | 412d | 5.30 × 10−3d | 2.99 | −0.39 | 2.60 | 113 |
| [Fe(btz)3]3+ | 558d | 633d | 1.00 × 10−4d | 2.14 | −0.18 | 1.96 | 103 |
| WO2(dbp)2 | 375e | 490, 585e | 75.9e | 3.02 | −1.19 | 1.83 | 70 |
| [Cu(bath)(ThioPOP)]PF6 | 386f | 545f | 16.3f | 3.21 | −1.65 | 1.56 | 60 |
| Zn(bip)2 | 364e | 416e | 3.22 | −1.33 | 1.55 | 64 | |
| [Cu(bpy)(SIPr)]PF6 | 370g | 3.35 | −1.83 | 1.52 | 39 | ||
| [Cu(bath)(Xant)]PF6 | 389f | 569f | 6.4f | 3.19 | −1.67 | 1.52 | 60 |
| [Cu(dBdPP)(Xant)]PF6 | 38f | 546f | 54.1f | 3.2 | −1.74 | 1.47 | 60 |
| [Cu(4-Mebpy)(POP)]BF4 | 480h | 2.58 | −1.16 | 1.42 | 65 | ||
| [Cr(bpm)3]3+ | 728d | 50d | 1.7 | −0.28 | 1.42 | 115 | |
| [Cu(4-Mebpy)(PPh3)2]BF4 | 480h | 2.58 | −1.18 | 1.4 | 65 | ||
| [Cu(phen)(IPr)]PF6 | 384g | 3.23 | −1.84 | 1.39 | 39 | ||
| [Cu(bpy)(IPr)]PF6 | 382g | 3.25 | −1.87 | 1.38 | 39 | ||
| [Cu(6-Mebpy)(Xant)]BF4 | 470h | 2.64 | −1.26 | 1.38 | 65 | ||
| [Cu(bpy)(PPh3)2]BF4 | 480h | 2.58 | −1.2 | 1.38 | 65 | ||
| [Cu(odmdpya)(IPr)]PF6 | 379g | 3.27 | −1.9 | 1.37 | 39 | ||
| [Cu(mdmdpya)(IPr)]PF6 | 375g | 3.31 | −1.95 | 1.36 | 39 | ||
| [Cu(dmbpy)(IPr)]PF6 | 381g | 3.26 | −1.93 | 1.33 | 39 | ||
| [Cu(dppp)2]BF4 | 347e | 556e | 0.24e | 3.57 | −2.26 | 1.32 | 55 |
| Cu(bath)(dpcarb) | 449e | 602e | 1.4e | 2.4 | −1.09 | 1.31 | 116 |
| [Cu(bpy)(Xant)]BF4 | 480h | 2.58 | −1.28 | 1.3 | 65 | ||
| WO2(dhfcdbzbmp) | 427e | 598e | 62.0e | 2.64 | −1.39 | 1.25 | 70 |
| [Cu2(dmphen)2(tdapc)]BF4 | 420d | 2.95 | −1.7 | 1.25 | 58 | ||
| [Cu(4-Mebpy)(Xant)]BF4 | 480h | 2.58 | −1.33 | 1.25 | 65 | ||
| [Cu(bpy)(POP)]BF4 | 480h | 2.58 | −1.35 | 1.23 | 65 | ||
| TiO(Pc) | 704e | 1.7 | −0.47 | 1.23 | 72 | ||
| [Cr(bpy)3]3+ | 728d | 187d | 1.7 | −0.47 | 1.23 | 115 | |
| [Cr(phen)3]3+ | 728d | 199d | 1.7 | −0.48 | 1.22 | 115 | |
| [Cr(dip)3]3+ | 732d | 317d | 1.69 | −0.47 | 1.22 | 115 | |
| [Cr(dmphen)3]3+ | 730d | 259d | 1.7 | −0.5 | 1.2 | 115 | |
| [Cr(dmb)3]3+ | 730d | 91d | 1.7 | −0.5 | 1.2 | 115 | |
| WO2(cdbzbmp) | 400e | 595e | 14.6e | 2.70 | −1.51 | 1.19 | 70 |
| [Cr(mphen)3]3+ | 729d | 208d | 1.7 | −0.51 | 1.19 | 115 | |
| [Cu(pdmdpya)(IPr)]PF6 | 389g | 3.19 | −2.01 | 1.18 | 39 | ||
| [Cr(aphen)3]3+ | 730d | 0.21d | 1.7 | −0.53 | 1.17 | 115 | |
| [Cr(tmp)3]3+ | 731d | 187d | 1.7 | −0.54 | 1.16 | 115 | |
| [Cu(dppp)(POP)]BF4 | 363e | 494e | 2.44e | 3.42 | −2.29 | 1.13 | 55 |
| Zn(dF-phpo)2 | 357g | 420g | 3.37 | −2.26 | 1.11 | 47 | |
| Zn(CN-phpo)2 | 326g | 416g | 3.32 | −2.21 | 1.11 | 47 | |
| [Cu(6-Mebpy)(POP)]BF4 | 470h | 2.64 | −1.55 | 1.09 | 65 | ||
| [Cu(dppb)2]BF4 | 374f | 3.32 | −2.26 | 1.06 | 56 | ||
| Zn(phpo)2 | 363g | 428g | 3.33 | −2.28 | 1.05 | 47 | |
| Zn(Cl-phpo)2 | 362g | 423g | 3.27 | −2.24 | 1.03 | 47 | |
| Zn(TPP) | 588e | 597, 647e | 2.16 | −1.2 | 0.96 | 67 | |
| Zn(Pc) | 1.77 | −0.82 | 0.95 | 72 | |||
| [Cu(dmphen)(Xant)]BF4 | 378d | 545d | 2.64 | −1.73 | 0.91 | 66 | |
| [Cu(pytz)(dppm)]BF4 | 2.76 | −1.86 | 0.9 | 63 | |||
| Zn(OMe-phpo)2 | 369g | 440g | 3.2 | −2.31 | 0.89 | 47 | |
| [Cr(ddpd)3]3+ | 435i | 778i | 0.90i | 1.6 | −0.73 | 0.87 | 117 |
| Zn(diPy)2 | 489e | 508e | 0.002g | 2.62 | −2.35 | 0.73 | 62 |
| [Cu(pytz)(POP)]BF4 | 2.71 | −1.99 | 0.72 | 63 | |||
| Zn(Ph-diPy)2 | 639e | 655e | 0.004e | 1.93 | −1.67 | 0.72 | 62 |
| [Cu(pytz)(dppe)]BF4 | 2.67 | −2.02 | 0.65 | 63 | |||
| Zn(Tol-ethynyl-Ph-diPy)2 | 658e | 673e | 0.004d | 1.85 | −1.66 | 0.65 | 62 |
| [Cu(bath)(POP)]PF6 | 0.81d | 2.27 | −1.64 | 0.63 | 71 | ||
| [Cu(dpya)(IPr)]PF6 | 473g | 2.62 | −2 | 0.62 | 39 | ||
| [Cu2(Mebpy)2(tdppc)][BF4] | 410d | 3.03 | −2.42 | 0.61 | 58 | ||
| Zn(Et-bip)2 | 344e | 422e | 3.24 | −2.34 | 0.56 | 64 | |
| Zn(NMe2-phpo)2 | 372g | 482g | 3.01 | −2.5 | 0.51 | 47 | |
| Zr(MePDP)2 | 528f | 594f | 2.09 | −1.6 | 0.49 | 107 | |
| Zn(OMe2St-diPy)2 | 656e | 673e | 1.82 | −1.9 | 0.38 | 62 | |
| Zn(OMe2Ph-diPy)2 | 621e | 639e | 0.005g | 1.93 | −2.02 | 0.37 | 62 |
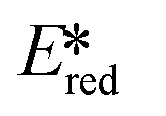
Chromium(III) complexes (Fig. 34) with π-accepting ligands have been recognized as photoluminescent materials for many years.109,110 Its 2E excited state endows chromium(III) complexes with a particularly long-lived excited state compared to most of LMCT complexes. Recently, Piguet et al. reported several heteroleptic chromium(III) complexes that exhibited red emission with millisecond-long τPL at room temperature.89 Recently, chromium(III) complexes have also been investigated as PCs in Diels–Alder-type reactions.23 In most cases, the ligand sphere about the Cr ion is composed of polypyridyl ligands capable of forming an octahedral complex. The CrIII/II redox couple in these complexes is anodically shifted (Ered ranging from −0.28 to −0.73 V) compared to [Ru(bpy)3]2+ (Ered = −1.33 V). Although the ground state reduction potentials make these chromium(III) polypyridyl complexes reasonably powerful photooxidants, the excited state energies of these complexes are low. Thus, it is considerably more difficult to oxidize the reduced chromium(II) species and this is a potential drawback for photoredox catalytic reactions. There are also examples of chromium(III) complexes that have been used as photosensitizers for energy transfer to an energy acceptor, rather than as an electron acceptor in a PeT reaction.111,112
 | ||
| Fig. 34 Chromium(III) complexes. | ||
We recently reported the first examples of room temperature luminescent Co(III) complexes.113 These complexes exhibit intense ligand-to-metal and ligand-to-ligand charge transfer absorption bands (λabs ∼ 360–400 nm) and low-negative quasi-reversible reduction waves (Ered1/2 = −0.58 V and −0.39 V for [Co(dgpy)2](BF4)3 and [Co(dgpz)2](BF4)3, respectively, Fig. 30). The blue emission of these complexes at room temperature is due to the larger bite angles and strong σ-donation of the six-membered chelate ligands, the combined effect of which helps to separate the emissive 3LMCT and the non-emissive 3MC states. These complexes were found to be very powerful photooxidants (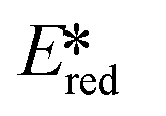 = 2.72 V and 2.60 V for [Co(dgpy)2](BF4)3 and [Co(dgpz)2](BF4)3, respectively). These values are significantly higher than those of photocatalysts such as [Ir(dF(CF3)ppy)2(dtbubpy)]+ (
= 2.72 V and 2.60 V for [Co(dgpy)2](BF4)3 and [Co(dgpz)2](BF4)3, respectively). These values are significantly higher than those of photocatalysts such as [Ir(dF(CF3)ppy)2(dtbubpy)]+ (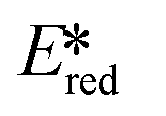 = 0.89 V)114 and [Ru(bpy)3]2+ (
= 0.89 V)114 and [Ru(bpy)3]2+ (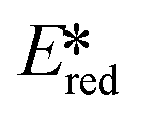 = 0.77 V).9
= 0.77 V).9
A handful of MLCT complexes such as copper(I) and zinc(II) complexes also show high excited state reduction potentials and are potential candidate photooxidants. For an instance, [Cu(bath)(ThioPOP)]PF6 has an excited state reduction potential of 1.56 V, which is higher than that of [Ir(dF(CF3)ppy)2(dtbubpy)]+ (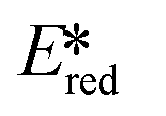 = 1.21 V). Comparing this complex with its xantphos analog ([Cu(bath)(Xant)]PF6Ered = −1.67 V,
= 1.21 V). Comparing this complex with its xantphos analog ([Cu(bath)(Xant)]PF6Ered = −1.67 V, 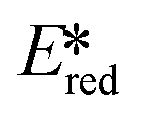 = 1.52 V), [Cu(bath)(ThioPOP)]PF6 shows a less negative ground state reduction potential and a more positive excited state reduction potential as a function of the bulkier ThioPOP ligand. Cu(I) NHC complexes, attributed to their high excited state energies, are also amongst highly oxidizing photooxidants (
= 1.52 V), [Cu(bath)(ThioPOP)]PF6 shows a less negative ground state reduction potential and a more positive excited state reduction potential as a function of the bulkier ThioPOP ligand. Cu(I) NHC complexes, attributed to their high excited state energies, are also amongst highly oxidizing photooxidants (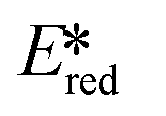 ≈ 1.4 V).
≈ 1.4 V).
Zinc(II) complexes, such as Zn(bip)2 (Ered = −1.33 V, 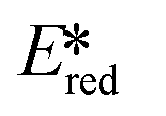 = 1.55 V), also show potential as strong photooxidants.
= 1.55 V), also show potential as strong photooxidants.
Photoredox catalytic reactions that proceed by a reductive quenching pathway
Photoredox catalytic reactions proceeding via a reductive quenching pathway represent a large and diverse family of reactions where the reductive quenching is usually used to generate a radical cation in the reaction118 or use the reduced photocatalyst as a strong ground state reductant.119Recently, Lloret-Fillol et al. reported a dual cobalt/copper photocatalytic reduction of aldehydes and aromatic ketones in water (Fig. 35).119 The proposed mechanism contains two separate photoredox catalytic cycles reducing both cobalt(II) and cobalt(III) species in the cobalt catalytic cycle. The photocatalytic cycle contains a reductive quenching by a sacrificial electron donor, triethylamine or triisopropylethylamine, and the reduced photocatalyst then acts as a SET reductant of the Co(II) co-catalyst, generating the active Co(I) species. Moderate to excellent yields were obtained among the 30 aldehyde and ketone substrates (yields: 40–99%), except for 2-methylbenzaldehyde (32%) and 2,2-dimethyl-1-phenylpropan-1-one (3%) where the substrate is too sterically encumbered to efficiently be reduced.
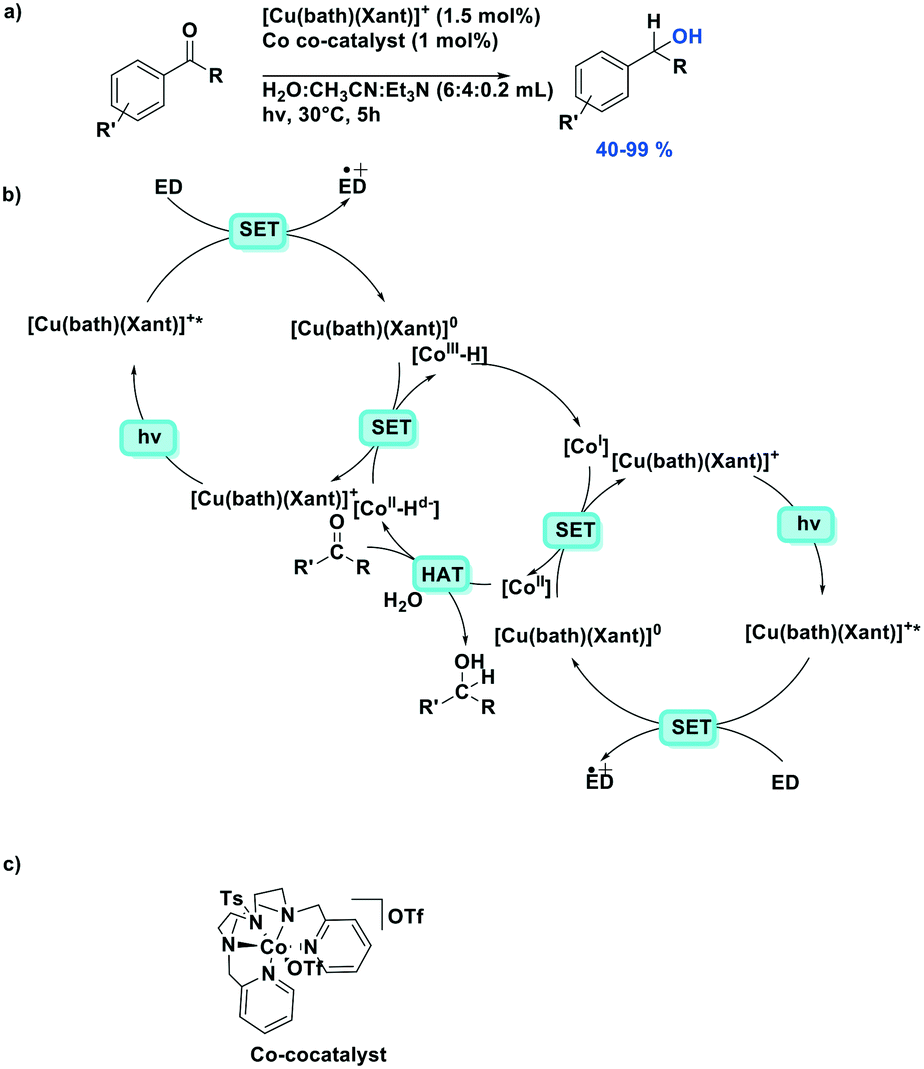 | ||
| Fig. 35 Dual cobalt–copper light-driven catalytic reduction of aldehydes and aromatic ketones in aqueous media.119 | ||
Evano et al. reported the copper(I) catalyzed photoredox C–C bond formation from aryl halides and aromatic or heteroaromatic coupling partners via dehalogenation and subsequent aryl radical formation.71 This is a rare example of using a copper(I) PC that proceeds via a reductive quenching pathway. The PC possesses too small an 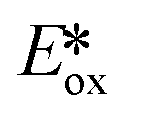 value (−1.02 V) to photoreduce the aryl halides. However, the PC is sufficiently strong to first photooxidize ethyldiisopropylamine, and the thus-formed Cu(0) species can then reduce the aryl halides, concomitantly completing the photocatalytic cycle. Evano et al. screened the reaction with different copper(I) PCs. There was no conversion with homoleptic copper(I) complexes including those of the form [Cu(N^N)2]+ and [Cu(P^P)2]+. However, amongst heteroleptic copper(I) complexes, [Cu(bath)(POP)]+ showed the highest conversion to the dehalogenated product. When it comes to the coupling reactions, aryl iodides are well tolerated substrates (yields: 60–99%). Most aryl bromides could be coupled (yields: 55–95%) though the acetylamide substrate showed no conversion and the bromoanisole showed only 10% conversion to the dehalogenated product. In the case of aryl chlorides, only a 15% yield was obtained for methyl 4-chlorobenzoate while no conversion was observed for chloroanisole. Based on their photodehalogenation study, Evano et al. also reported the C–C formation using the intercepted aryl radicals with four different pyrroles and three different arenes, with yields ranging from 47–74% (Fig. 36).
value (−1.02 V) to photoreduce the aryl halides. However, the PC is sufficiently strong to first photooxidize ethyldiisopropylamine, and the thus-formed Cu(0) species can then reduce the aryl halides, concomitantly completing the photocatalytic cycle. Evano et al. screened the reaction with different copper(I) PCs. There was no conversion with homoleptic copper(I) complexes including those of the form [Cu(N^N)2]+ and [Cu(P^P)2]+. However, amongst heteroleptic copper(I) complexes, [Cu(bath)(POP)]+ showed the highest conversion to the dehalogenated product. When it comes to the coupling reactions, aryl iodides are well tolerated substrates (yields: 60–99%). Most aryl bromides could be coupled (yields: 55–95%) though the acetylamide substrate showed no conversion and the bromoanisole showed only 10% conversion to the dehalogenated product. In the case of aryl chlorides, only a 15% yield was obtained for methyl 4-chlorobenzoate while no conversion was observed for chloroanisole. Based on their photodehalogenation study, Evano et al. also reported the C–C formation using the intercepted aryl radicals with four different pyrroles and three different arenes, with yields ranging from 47–74% (Fig. 36).
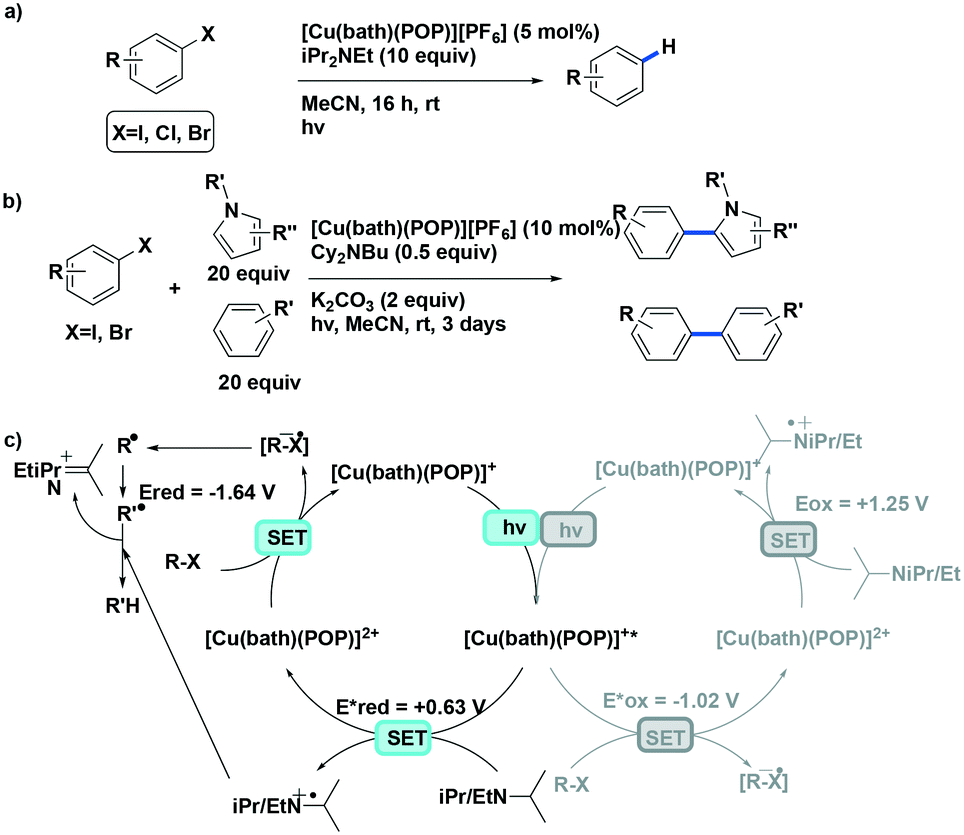 | ||
| Fig. 36 Photoredox transformations of organic halides with copper(I) photocatalyst.71 | ||
Cross-dehydrogenative coupling (CDC) reactions have been widely studied and are frequently promoted with photocatalysts based on iridium (e.g., [Ir(ppy)2(dtbubpy)]+) and ruthenium (e.g., [Ru(bpy)3]2+) complexes. Che et al. reported four zwitterionic carborane-containing heteroleptic copper(I) complexes and their use as photocatalysts in the aza-Henry type CDC reaction between tetrahydroisoquinolines and nitroalkanes.116 According to their catalyst screening, the highest yield of 87% for the CDC reaction of 2-phenyl-1,2,3,4-tetrahydroisoquinoline and nitromethane was achieved with Cu(bath)(dpcarb), showing 100% conversion; the isolated yield was slightly lower despite quantitative conversion when Cu(dmphen)(dpcarb) was used as the PC (Fig. 37a). Using Cu(bath)(dpcarb), fifteen different tetrahydroisoquinolines were shown to react with nitroalkanes in moderate to very good yield (yields: 48–87%). The coupling partner was also changed from a nitroalkane to an aromatic indole, with similarly high yields reported (yields: 70–78%). Based on EPR spectroscopy measurements, the proposed mechanism (Fig. 37b) proceeds via trapping of a peroxo intermediate subsequent to the single electron transfer (SET) from the substrate to the PC and the resulting formation of the radical cation.
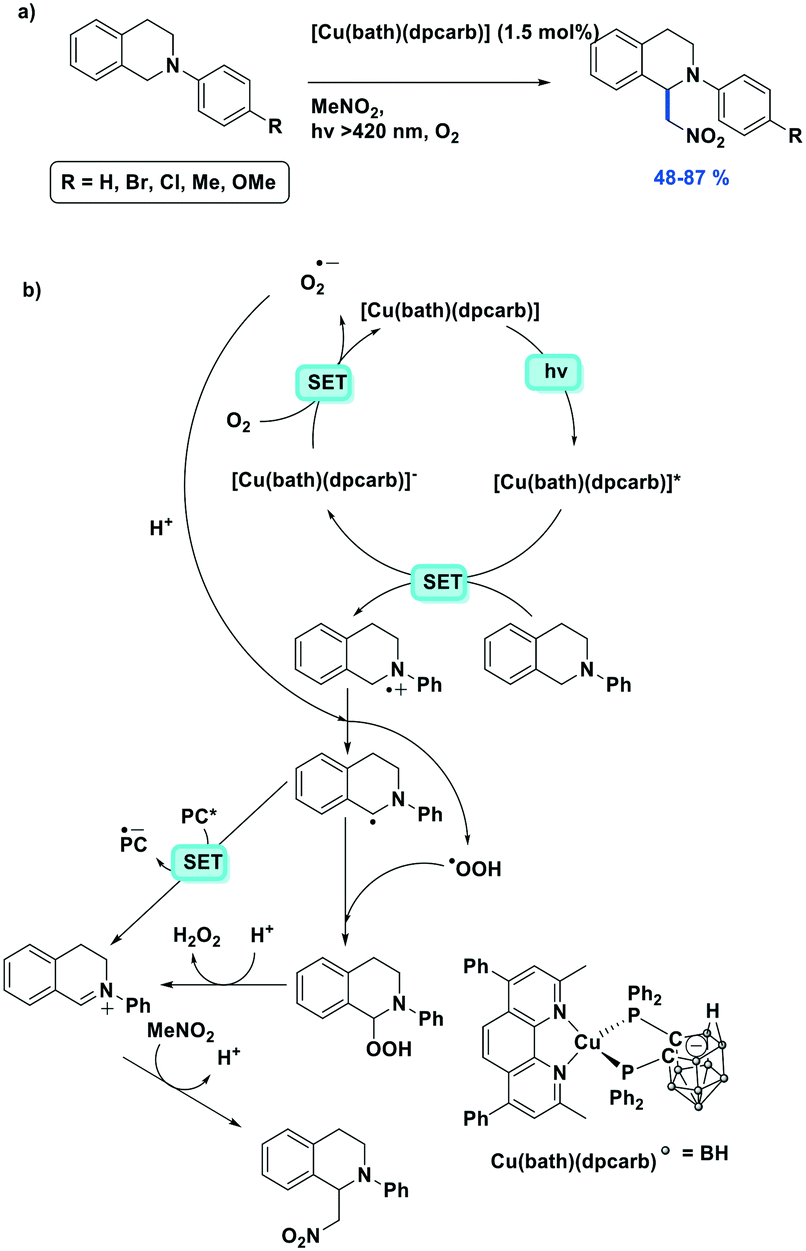 | ||
| Fig. 37 a) Visible-light induced CDC reaction; b) the proposed mechanism. | ||
While W(0) and Mo(0) complexes are potential powerful photoreductants, high oxidation state analogs can act as strong photooxidants. Air-stable W(VI) complexes with Schiff-base ligands,70 WO2(dhfcdbzbmp) and WO2(cdbzbmp) were both used as photocatalysts (λexc > 370 nm) for the cyanation of N-aryltetrahydroisoquinolines, affording the desired compounds in very good yields [77–88% with WO2(dhfcdbzbmp); 78–95% with WO2(R-dbp)2]. The mechanism of this reaction is thought to proceed via reaction with singlet oxygen, itself sensitized by the triplet-excited tungsten(VI) complex.120 Previously, the fluorinated porphyrin complex PdF20TPP had been used as the PC for this transformation.121 The yields employing the tungsten PC were comparable to those using PdF20TPP (yields: 47–86%) but required significantly higher catalyst loading (2 mol% for the W(VI) PC compared to 0.05 mol% of PdF20TPP) and varied depending on the nature of the substrate. The PC WO2(R-dbp)2 which bears phenyl substituents on the dpp backbone has also been shown to catalyze the aerobic oxidative hydroxylation of aryl boronic acids to phenol derivatives in moderate to very good yields (57–82%)45 (Fig. 38). Yields using the tungsten PC were slightly lower than when [Ru(bpy)3]Cl2 was employed as the PC (69–96%) under otherwise similar reaction conditions.122 This first study shows a promising future for W(VI) complexes in photocatalysis as direct replacements for ruthenium PCs.
 | ||
| Fig. 38 a) Oxidative cyanation of tertiary amines photocatalyzed by WO2(dhfcdbzbmp) and WO2(dbp)2 (where R = C6H5 instead of Br); b) oxidative hydroxylation of aryl boronic acids photocatalyzed by WO2(R-dbp)2 where R = C6H5. | ||
The complex Zr(MePDP)2 shows a strong absorption at 520 nm and an emission centered at 594 nm when excited at 520 nm.107 The corresponding titanium complex Ti(MePDP)2 is not photoluminescent. Photoexcitation of Zr(MePDP)2 at 520 nm promoted several photoredox reactions such as dehalogenation, reduction of diethyl maleate to diethyl succinate and reductive coupling of benzyl bromide to bibenzyl (Fig. 39). During cyclic voltammetry studies, both the Zr and Ti complexes showed an irreversible ligand-based oxidation wave with a peak potential around 0.94 V vs. SCE For Zr(MePDP)2, reversible reduction waves at −1.72 and −2.19 V along with a quasi-reversible wave at −2.78 V were observed. The initial wave at −1.72 V is predominantly metal-centered. The second and third reduction waves are primarily ligand-centered, indicating MLCT excited states. The 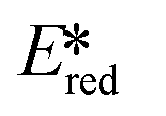 at −0.026 V is very small. Therefore, the easily oxidizable sacrificial reductant MeBIH (Eox = −0.116 V vs. SCE in MeCN), was chosen as the quencher for
at −0.026 V is very small. Therefore, the easily oxidizable sacrificial reductant MeBIH (Eox = −0.116 V vs. SCE in MeCN), was chosen as the quencher for 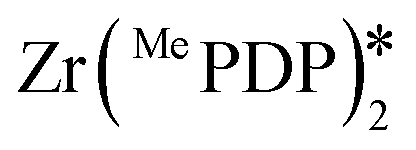 (Fig. 39).123
(Fig. 39).123
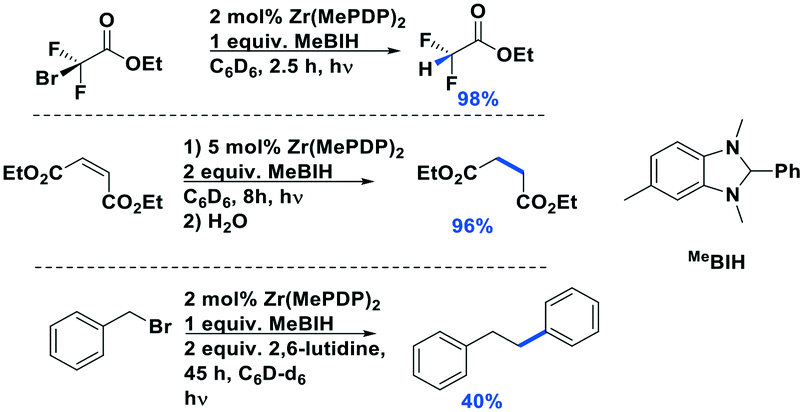 | ||
| Fig. 39 Reactions tested using Zr(MePDP)2 as photocatalyst. | ||
The same group recently reported two new luminescent zirconium complexes, Zr(HCNN)2 and Zr(MeCNN)2, the latter of which has been applied as a PC for the reductive homocoupling of benzyl bromide to bibenzyl.124 Using MeOBIH as a sacrificial reductant and triethylamine as the base, the coupling product was obtained in 40% yield (Fig. 40). The authors suggest that the steric hindrance afforded by the methyl groups on the ligand allows for an additional reversible electrochemical/chemical process in Zr(MeCNN)2 and this in turn allows for photoinduced outersphere electron transfer, which catalyses the reaction. The complex Zr(HCNN)2 exhibits C–C reductive elimination upon photoexcitation, leading to rapid decomposition when tested as a photoredox catalyst.
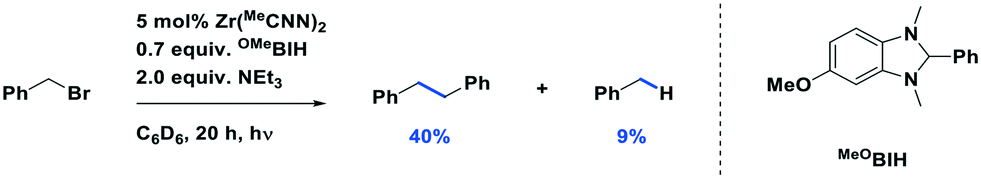 | ||
| Fig. 40 Photoredox catalytic homocoupling of benzyl bromide with Zr(MeCNN)2 as the photocatalyst. | ||
Shores, Ferreira et al. first demonstrated the utility of Cr(III) polypyridyl complexes as PCs, which were employed in the photocatalyzed formal [4 + 2] dimerization of 1,3-cyclohexadiene.125 The Cr(III) complexes (Fig. 41) feature 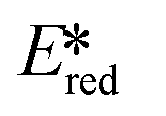 ranging from +1.40 to +1.84 V, which are comparable if not higher than that of the widely used [Ir(dF(CF3)ppy)2(dtbubpy)]+ (
ranging from +1.40 to +1.84 V, which are comparable if not higher than that of the widely used [Ir(dF(CF3)ppy)2(dtbubpy)]+ (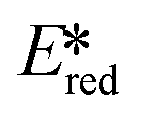 = +1.21 V)95 and [Ru(bpz)3]2+ (
= +1.21 V)95 and [Ru(bpz)3]2+ (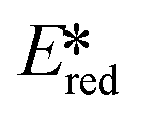 = +1.45 V) PCs. The use of the PC [Cr(dmcbpy)3](BF4)3 gave a 55% yield after 24 h of irradiation using a 23W CFL. This can be compared to the use of the strongly oxidizing [Ru(bpz)3](PF6)2, which gave a 21% yield under identical conditions. The mechanism of the reaction, along with the role of atmospheric oxygen, was not immediately clear. Notably, under the initial set of conditions near UV light (419, 350, or 300 nm) was used as the excitation source, which represents a disadvantage of the protocol.
= +1.45 V) PCs. The use of the PC [Cr(dmcbpy)3](BF4)3 gave a 55% yield after 24 h of irradiation using a 23W CFL. This can be compared to the use of the strongly oxidizing [Ru(bpz)3](PF6)2, which gave a 21% yield under identical conditions. The mechanism of the reaction, along with the role of atmospheric oxygen, was not immediately clear. Notably, under the initial set of conditions near UV light (419, 350, or 300 nm) was used as the excitation source, which represents a disadvantage of the protocol.
 | ||
| Fig. 41 Formal [4 + 2] dimerization of cyclohexadiene. | ||
Shores et al. subsequently determined that oxygen acts as an electron shuttle following photoexcitation of the Cr(III) complex.112 It was theorized that 3O2 quenches the excited state of the catalyst, forming singlet oxygen, and this protects the catalyst from decomposition (Fig. 42). The correspondingly formed superoxide was hypothesized to reduce the Diels–Alder cycloadduct radical cation to the final product, hence reforming triplet oxygen. The substrates from this study are more restricted than those investigated by Yoon et al., who demonstrated the formal Diels–Alder reaction between electronically mismatched substrates using [Ru(bpz)3]2+ in excellent yields (42–98%).126
 | ||
| Fig. 42 Proposed roles of oxygen in cycloaddition pathways, where the green boxes denote the initial form of the Cr-containing catalyst. Reprinted with permission from R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappé and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451–5464. Copyright (2016) American Chemical Society. | ||
There remains some debate as to the role of O2 in the Cr-photocatalyzed Diels–Alder reaction. Using similar Cr(III) complexes as Shores et al., Dang et al. suggested dioxygen mediation proceeding via an inner-sphere mechanism.127 The PC catalyzes a formal Diels–Alder reaction between two electron-rich substrates. Prior work on the photocatalyzed Diels–Alder reaction using Ru(II) photoredox catalysts126,128 afforded both inter- and intramolecular products. The authors carried out extensive DFT studies to calculate the energy barriers and compared them with experimental results. Hence, their mechanistic study relies entirely on a theoretical model. The proposed mechanism using the [Cr(Ph2phen)3]3+ complex (Fig. 43) implicates a dual role for oxygen after the catalyst is excited from its ground state (4ΔCrL33+) to its doublet excited state (2ΔCrL33+) upon visible light irradiation. The excited doublet state can either be quenched by dioxygen via an inner-sphere energy transfer process to regenerate the 4ΔCrL33+ (red arrows) or can be consumed via a competitive SET with the dienophile to give 3ΔCrL33+, which then initiates a radical-mediated [4 + 2] cycloaddition processes (blue arrows). Dioxygen is proposed to stabilize the 3ΔCrL33+ to form the quintet 5Δ[CrL3–O2] complex, which can then easily donate an electron to the cycloadduct cation to give 4Δ[CrL3–O2]. Spin inversion can then occur, resulting in the regeneration of the ground state chromium complex and triplet dioxygen. This mechanism is much more complex than the one proposed by Shores et al., but it does perhaps explain the moderate yields (yields: 55%) due to non-productive PC quenching pathways.
 | ||
| Fig. 43 Proposed mechanism of photoredox catalysis using [Cr(Ph2phen)3]3+. | ||
The complex [Cr(bpy)3](OTf)3 has also been used to photocatalyze aza-Diels–Alder type cycloaddition reactions (λexc = 455 nm).129 This reaction is remarkable in that conventional PCs such as [Ru(bpy)3]Cl2 and [Ir(dF(CF3)ppy)2(bpy)]PF6 were not effective. The authors note that there is a dark background reaction where small amounts of product (yield: 35%) are generated. Using a range of different substrates, good to excellent yields (yields: 26–97%) with high cis:trans selectivity are obtained, where the cis isomer is favoured in all cases. Little mechanistic detail is provided in this study, but a SET from the alkenes to the excited Cr(III) complex is proposed, which is analogous to previous studies of photocatalytic aza-Diels–Alder type reactions using 2,4,6-triphenylpyrylium tetrafluoroborate or similar organic PCs.130–133
As well as Cr(III) PCs employing π-accepting ligands similar to those used in common Ru(II)- and Ir(III)-containing PCs, Cr(III) complexes using electron-rich ligands have also been investigated, particularly by the group of Heinze.134 The photophysics of Cr(III) complexes is non-trivial and there are a diverse range of potential mechanisms concerning the initial photoactivation step of the reaction, which is governed by fast intersystem crossing (ISC) and reverse ISC (rISC) between the two lowest lying excited states: the 2E, which is redox inert, and the 4T2, which is a strong photooxidant. The PC [Cr(ddpd)2]3+ was used for the photocyanation of a variety of amine substrates using trimethylsilylcyanide (TMSCN) as the cyanide source, dioxygen from the air as the terminal oxidant and a compact fluorescent lamp as the visible light source134 (Fig. 44). These initial results are promising, particularly as the PC was found to be highly photostable and reusable.
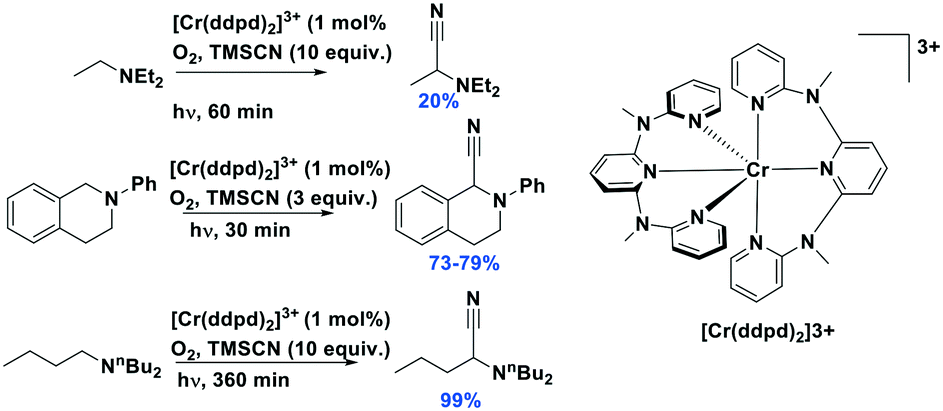 | ||
| Fig. 44 α-Aminonitrile formation using [Cr(ddpd)2]3+, as the PC. | ||
Development of vanadium(V) complexes as PCs has intensified after an initial study of a d0 V(V) oxido LMCT complex V(F2OXO) gave favourable results (see Table 3) suggesting its potential as a photoredox catalyst.104 Beginning with TD-DFT studies, followed by transient absorption and X-ray absorption spectroscopy to trace the intermediate states in photochemical reactions, the authors found that the excited state dynamics of these vanadium(V) complexes were very promising. The complex is highly absorptive in the visible region between 400–500 nm and has a very strong excited state reduction potential of −1.43 V vs. SCE, which makes it a promising PC for oxidative coupling reactions.
Further studies into the use of vanadium(V) oxido complexes as PCs demonstrated their ability to selectively cleave C–C bonds under ambient conditions.106 Soo et al. chose a representative lignin model substrate, which gave aldehyde products upon photodegradation that are very useful building blocks in organic synthesis. The current conditions for chemical degradation require a multistep reaction sequence that is significantly less selective than the photocatalytic protocol described in this work. Indeed, the selective bond cleavage of lignin is a hot topic in chemistry given the sustainable nature of the starting material,135,136 but so far only one other photocatalytic example exists where [Ir(ppy)2(dtbubpy)]PF6 was used by Stephenson et al. as the PC in a tandem TEMPO-oxidation and photocatalytic C–O bond cleavage under visible light irradiation.137 Although this reaction is high-yielding (up to 95% in some cases) and preserves various functional groups on the model lignin compound, it is also notable in its use of sacrificial amine-formate reductants, which are themselves not sustainable reagents.
Initially, Soo et al. screened the reaction at temperatures between 25–80 °C, demonstrating that the catalyst works under thermal as well as photochemical initiation. A set of mild photocatalytic conditions were then developed for the selective C–C bond cleavage on a range of substrates (Fig. 45). This Earth-abundant complex is supported by a redox non-innocent salicylaldimine ligand, the nature of which was confirmed by extensive mechanistic studies. The yields of the photodegradation of model substrates were moderate to good, and while not as high as those reported by Stephenson et al. are nevertheless promising, particularly considering the sustainable nature of the PC. It should be noted that the excitation source differed in both studies; the vanadium PC was excited using a solar simulator, a much more intense light source with a higher incident flux than the blue LEDs used for the iridium PC.
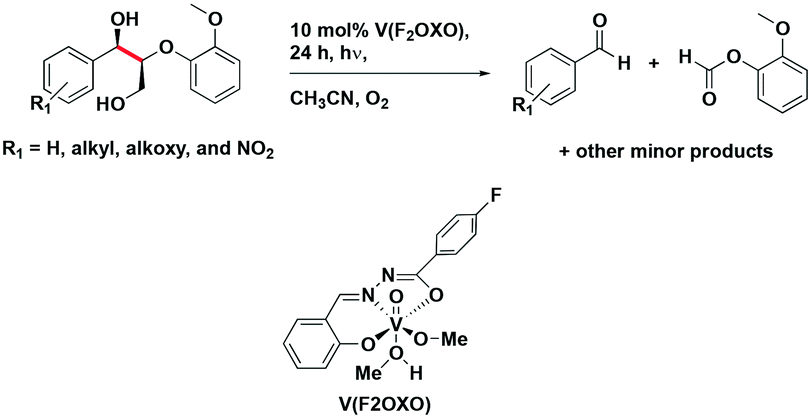 | ||
| Fig. 45 The new procedure involves mild, photocatalytic C–C cleavage with earth abundant vanadium oxo catalysts. | ||
The related oxido complexes V(PFOXO) and V(PFNOXO), bearing the highly electron-withdrawing pentafluorophenyl group, and in the latter complex an additional nitrated phenyl group, possessed significantly more positive (VV/VIV) reduction potentials [Eox of V(PFOXO) = 0.5 V; Eox of V(PFNOXO) = 0.82 V] compared to V(F2OXO) (Eox = 0.29 V), and very high excited state reduction potential (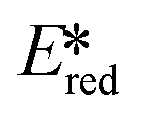 = 3.68 V for V(PFNOXO)).105 The more positive excited state reduction potentials of these PCs translate into their becoming more powerful oxidizing agents. This leads directly to increased catalyst stability, increased reactivity towards C–C bond cleavage and an increased diversity in substrate reactivity.
= 3.68 V for V(PFNOXO)).105 The more positive excited state reduction potentials of these PCs translate into their becoming more powerful oxidizing agents. This leads directly to increased catalyst stability, increased reactivity towards C–C bond cleavage and an increased diversity in substrate reactivity.
We recently reported the first examples of deep blue luminescent Co(III) complexes using strongly σ-donating 6-membered guanidine-based chelating ligands.113 These complexes are some of the most powerful photooxidants reported to date and act as visible light photocatalysts in the regioselective mono(trifluoromethylation) of conjugated arenes (Fig. 30 and 46). The proposed mechanism of the photocatalysis proceeds via a reductive quenching pathway. Although their ΦPL values are low (0.7% and 0.4%, respectively, for [Co(dgpy)2](BF4)3 and [Co(dgpz)2](BF4)3) their τPL are within the nanosecond regime, which are far longer than other 3d LMCT photoactive complexes. Previous work on the monotrifluoromethylation of PAHs involved very harsh conditions and poor yields and chemoselectivity. The Co(III)-based photocatalytic reaction takes place at room temperature and pressure under nitrogen and uses TfCl instead of CF3I. The yields were moderate to good regardless of which PC was used. The proposed reaction mechanism is an unusual one as it involves two photons.
 | ||
| Fig. 46 Reaction conditions, catalyst structure and proposed mechanism for photocatalysis using a Co(III) complex. | ||
Non-emissive iron(II) complexes as photosensitizers/photocatalysts
The case of iron(II) complexes as photocatalysts is more complex. Since most of these complexes are non-emissive, it is difficult to determine if the mechanism of photocatalysis is oxidative quenching or reductive quenching in nature. Hence, they are presented separately from the previous two sections.A unique case utilizing an Fe(II) photosensitizer combined with an organocatalytic system is described by Cozzi et al.138 The PC [Fe(bpy)3]Br2 (2.5 mol%) in concert with an organocatalyst (20 mol%) previously reported by MacMillan139 were highly effective in promoting the enantioselective alkylation of aldehydes with various different α-bromo carbonyl compounds.138 This reaction was highly tolerant of various functional groups on both the aldehydes and the α-bromo carbonyls, and gave very high e.e. values and yields that are comparable to those when using [Ru(bpy)3]2+ as the photosensitizer. Using this iron catalyst, the reaction gave moderate to very good yields (40–83%) while use of [Ru(bpy)3]2+ gave slightly more elevated yields (63–92%).140 The preparation of enantioenriched lactones via alkylation of aldehydes also gave very high yields and good e.e. values (64–80%). Employing these conditions, the synthesis of (−)-isodehydroxypodophyllotoxin was also reported. The authors propose a radical chain mechanism for this reaction, based on extensive EPR and radical clock experiments (Fig. 47).
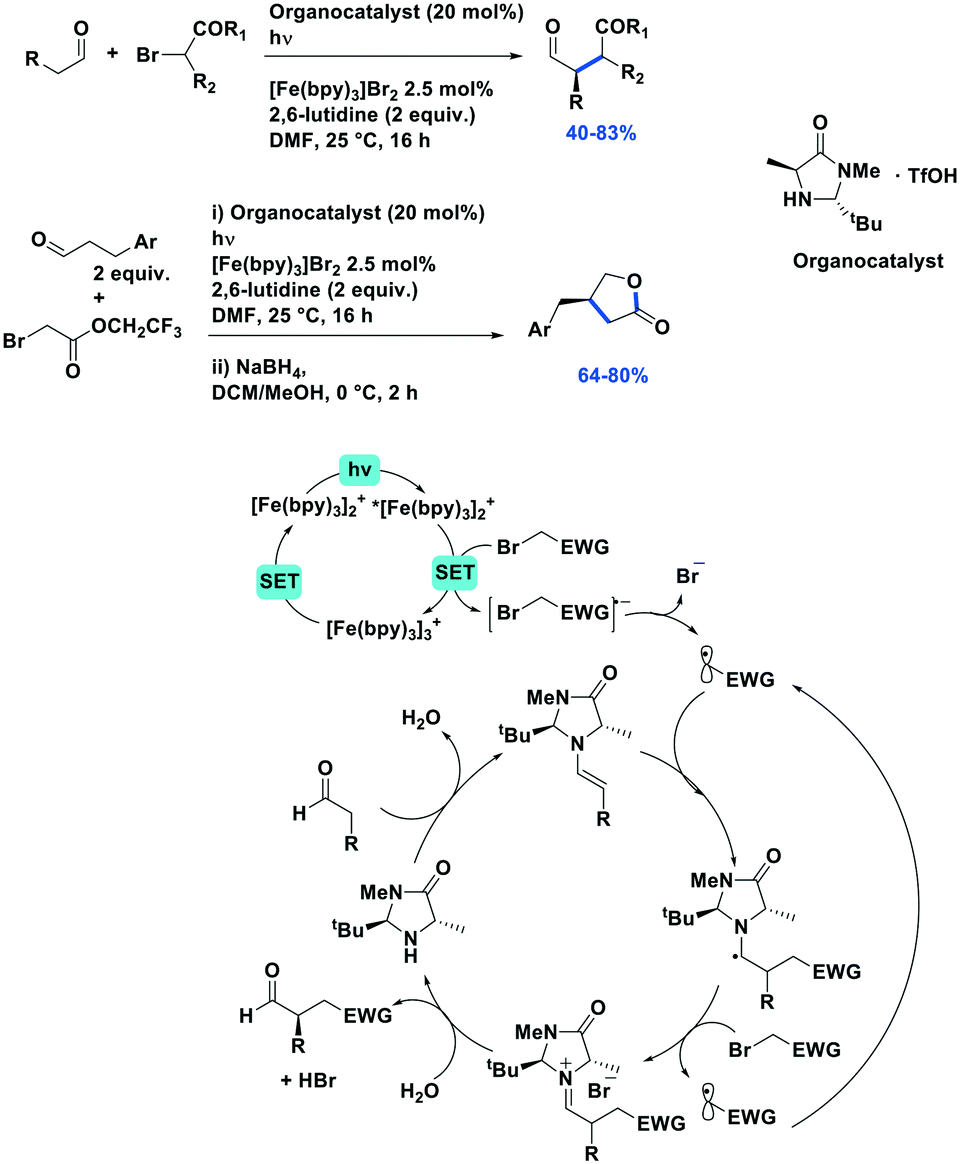 | ||
| Fig. 47 a) Scope of the stereoselective alkylation promoted by [Fe(bpy)3]Br2; b) preparation of enantioenriched lactones via alkylation of aldehydes. | ||
A recent paper by Collins et al.36 demonstrates the use of the PC [Fe(phen)3](NTf2)2 which catalyses carbazole formation under continuous flow conditions, using visible light and molecular oxygen as the oxidant (Fig. 48). Previous work in the same group employed56 [Cu(dmphen)(Xant)]+, which gave a maximum yield of 85%. The use of the [Fe(phen)3](NTf2)2 PC gave an excellent yield of 90% at gram scale whereas yields using [Cu(neo)(Xant)]BF4 varied from moderate to excellent (55–95%). It could be inferred that the iron PC is more attractive, particularly at larger scale as it reliably gives higher yields rather than the wide range given by the copper PC. The related PC [Fe(bpy)3](NTf2)2 only afforded a 37% yield of carbazole.
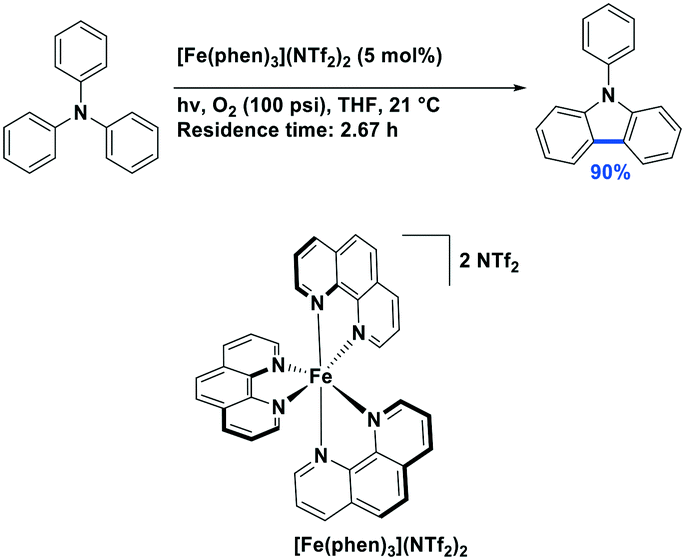 | ||
| Fig. 48 Carbazole formation via photocatalysis using a novel Fe based catalyst. | ||
Mechanistically, it is unlikely for the excited [Fe(N^N)3]2+ complexes to participate in SET from a MLCT state, as this state is too short-lived (ca. τPL = 650 ps).53 Rather a 5T2 excited state is formed.141,142 The authors ruled out the likelihood of Fe(III)-based impurities assisting the photocatalysis. Instead they suggested that the reaction proceeds via superoxide formation on the basis that the presence of oxygen at higher pressures in flow appears to improve the yield of carbazole formation.
Zhang et al., reported a series of Fe(II) PCs developed to initiate different polymerization processes.143,144 Initial studies demonstrated that the complexes Fe(C_a) and Fe(C_f) (Fig. 49) could generate radical species and gave a final conversion (Cf) of above 60% for the cationic polymerization of epoxides. Reaction conditions consisted of low concentrations of the iron(II) PC (0.2 wt%) in the presence of several additives that act as sacrificial oxidants: 2 wt% of an iodonium salt was always present, and in addition there was either 3 wt% of an N-vinylcarbazole (NVK) or a 2,4,6-tris(trichloromethyl)-1,3,5-triazine (R-Cl). The authors varied the combination of PC, iodonium salt, NVK or R-Cl along with the excitation wavelength (either 405 nm or 520 nm) to determine the optimal reaction conditions. LED light excitation either in the near-UV (385 nm) or visible region (405 nm) photoinitiated the polymerization. These PCs were successful in activating both cationic polymerisation (CP) and free-radical-promoted cationic polymerisation (FRPCP) of epoxides, as well as free radical polymerisation of acrylates.
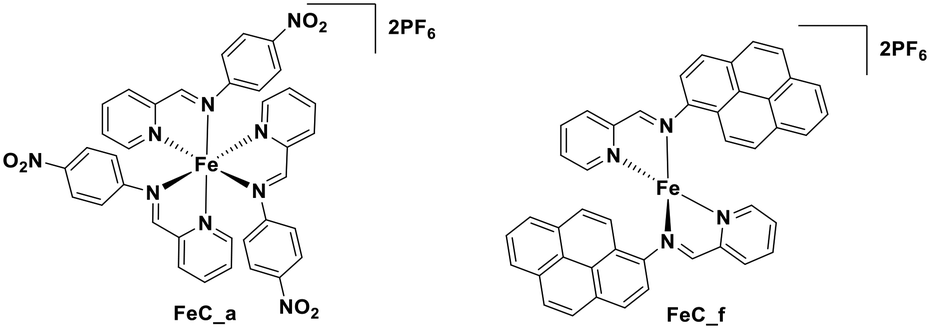 | ||
| Fig. 49 Chemical structures of iron complexes FeC_x which are active photoinitiators. | ||
Another polymerization reaction is the ATRP of methacrylate monomers.145 In this case, FeBr3, which has a very broad optical absorption, (Fig. 50) was used as the PC under low catalyst loading and mild conditions in the presence of oxygen to afford low polydispersity (Đ < 1.20) co-polymers. There was also good retention of polymer chain end functionality as revealed by in situ studies of block copolymer formation, i.e. the terminal bromine and ester groups were retained even after polymer formation.
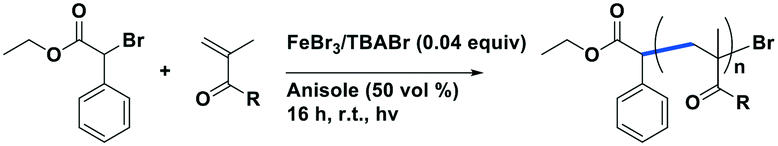 | ||
| Fig. 50 ATRP catalysed by FeBr3. | ||
Overview
The ground state and excited state redox potentials for 76 photoreductants and 64 photooxidants are summarized in Fig. 51. Candidate PCs based on different metal ions are represented by differently coloured symbols. Dashed lines indicate threshold energies of 2.0 eV, 2.5 eV and 3.0 eV for the E(0,0), which correspond roughly to red, green and blue emitter. The red dots on the figure represent commonly used PCs based on iridium(III) or ruthenium(II). In particular, given their popularity, Ir(ppy)3, [Ru(bpy)3]2+ and [Ir(dF(CF3)ppy)2(dtbubpy)]+ are explicitly identified.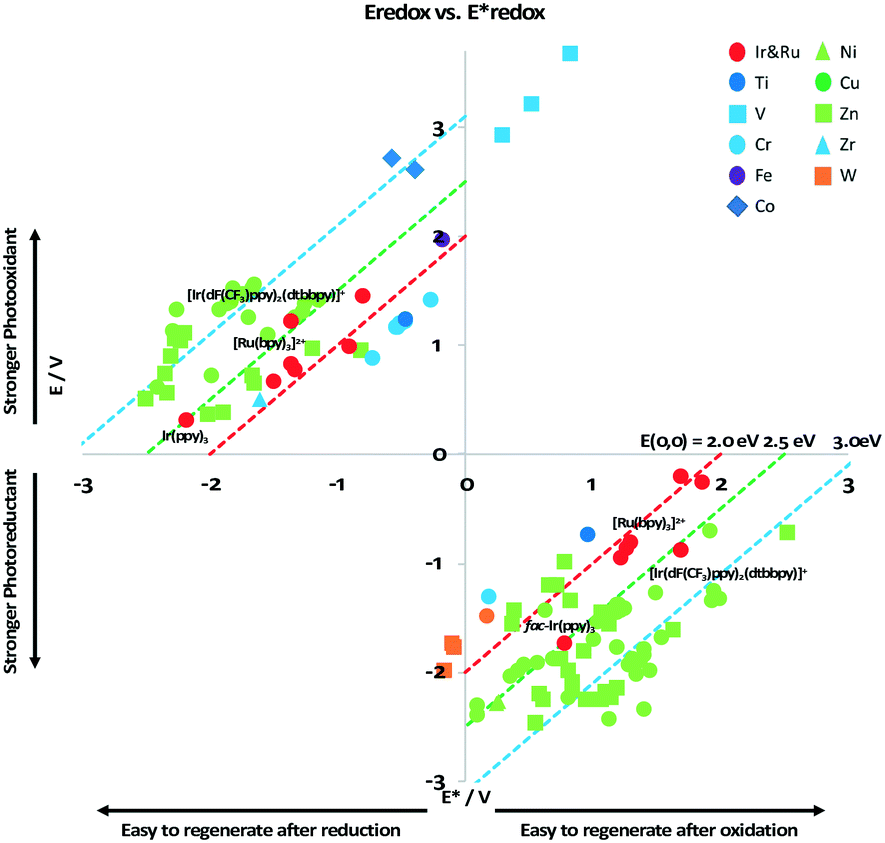 | ||
| Fig. 51 A summary of the photophysics and electrochemistry of the complexes discussed in this review. | ||
For [Ir(ppy)2(bpy)]+ and [Ru(bpy)3]2+ the low oxidation state and electron-rich nature of the metal coupled with the strong π*-accepting nature of the ligand(s) contributed to an excited state with strong metal-to-ligand charge transfer (MLCT) character. The green-emitting complex fac-Ir(ppy)3 (λPL = 494 nm, Eox = +0.77 V, 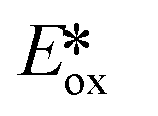 = −1.73 V) is thus one of the most powerful photoreductants. From an analysis of Fig. 51, it becomes evident that there are several candidate Cu(I) and Zn(II) complexes with similar E0,0 that are comparable or even stronger photoreductants. More recently, yellow-to-green photoluminescent MLCT Mn(II) complexes were reported by Zhao et al.146 Although the electrochemistry of these complexes has not been reported and they have not been investigated as photocatalysts, these complexes along with Cu(I) and Zn(II) complexes could potentially replace fac-Ir(ppy)3 in existing reactions and could even be used to activate substrates that fac-Ir(ppy)3 could not.
= −1.73 V) is thus one of the most powerful photoreductants. From an analysis of Fig. 51, it becomes evident that there are several candidate Cu(I) and Zn(II) complexes with similar E0,0 that are comparable or even stronger photoreductants. More recently, yellow-to-green photoluminescent MLCT Mn(II) complexes were reported by Zhao et al.146 Although the electrochemistry of these complexes has not been reported and they have not been investigated as photocatalysts, these complexes along with Cu(I) and Zn(II) complexes could potentially replace fac-Ir(ppy)3 in existing reactions and could even be used to activate substrates that fac-Ir(ppy)3 could not.
However, when the oxidation state of the metal is high and the complex contains strongly electron-donating ligands, the nature of the excited state typically adopts a ligand-to-metal charge transfer (LMCT) character. For instance, the sky-blue-emitting [Ir(dF(CF3)ppy)2(dtbubpy)]+ (λPL = 470 nm, Ered = −1.37 V, 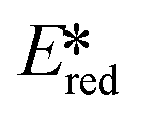 = 1.21 V) is a strong photooxidant used in a wide range of reductive quenching photoredox reactions. Here too, there exist potential Earth-abundant replacement Cu(I), Zn(II), V(V) and Co(III) complexes (Fig. 51). In terms of its excited state reduction potential, an oxo-titanium phthalocyanine complex and several LMCT chromium(III) complexes show similar or even higher values, and a much smaller optical gap (E(0,0) < 2 eV) that provides an avenue for chemoselectivity in photocatalytic reactions; however, the oxidation of their corresponding reduced species is more difficult, which may be an issue for closure of the photocatalytic cycle. Nonetheless, further study is warranted based on the appraisal of the photophysics of these complexes.
= 1.21 V) is a strong photooxidant used in a wide range of reductive quenching photoredox reactions. Here too, there exist potential Earth-abundant replacement Cu(I), Zn(II), V(V) and Co(III) complexes (Fig. 51). In terms of its excited state reduction potential, an oxo-titanium phthalocyanine complex and several LMCT chromium(III) complexes show similar or even higher values, and a much smaller optical gap (E(0,0) < 2 eV) that provides an avenue for chemoselectivity in photocatalytic reactions; however, the oxidation of their corresponding reduced species is more difficult, which may be an issue for closure of the photocatalytic cycle. Nonetheless, further study is warranted based on the appraisal of the photophysics of these complexes.
Many of these photoreductants have an 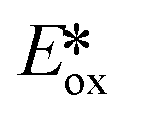 ≤ −2 V and there are currently no photooxidants based on Earth-abundant metal complexes that have an
≤ −2 V and there are currently no photooxidants based on Earth-abundant metal complexes that have an 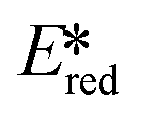 ≥ 2 V, save three vanadium(V) complexes reported by Soo et al.106 This is mainly because most of the photoluminescent Earth-abundant metal complexes are based on Cu(I) and Zn(II), whose excited states are difficult to reduce. The successes of vanadium(V) complexes demonstrate the potential of green- or blue-emitting complexes based on high valent metal ions and a LMCT excited state. However, regeneration of the reduced species of these vanadium(V) complexes are quite difficult (Ered ≥ 0). Thus, LMCT complexes with finely tailored photophysics and redox properties are still required.
≥ 2 V, save three vanadium(V) complexes reported by Soo et al.106 This is mainly because most of the photoluminescent Earth-abundant metal complexes are based on Cu(I) and Zn(II), whose excited states are difficult to reduce. The successes of vanadium(V) complexes demonstrate the potential of green- or blue-emitting complexes based on high valent metal ions and a LMCT excited state. However, regeneration of the reduced species of these vanadium(V) complexes are quite difficult (Ered ≥ 0). Thus, LMCT complexes with finely tailored photophysics and redox properties are still required.
Moreover, unlike robust cyclometalated iridium(III) complexes, poor chemical and photostability has been observed for many Earth abundant metal complexes due to two main reasons. Firstly, although the electron-rich low valent metal ions [e.g. nickel(0), copper(I)] usually lead to a very negative 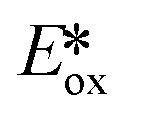 value, these complexes are sometimes air-sensitive. Secondly, unlike cyclometalated complexes, ligand disassociation can be problematic and this has proven challenging in related areas such as the application of Cu(I) sensitizers in dye-sensitized solar cells.147
value, these complexes are sometimes air-sensitive. Secondly, unlike cyclometalated complexes, ligand disassociation can be problematic and this has proven challenging in related areas such as the application of Cu(I) sensitizers in dye-sensitized solar cells.147
Conclusions
As a function of this survey, several factors have become evident in terms of photocatalyst design and choice. Firstly, more often than not, photocatalysts are chosen by brute PC screening or serendipity, rather than systematically considering their optoelectronic properties and how they relate to the reaction of interest.12 This potentially leads to sub-optimal choice in PC and is an evident area for improvement. Secondly, oxidative photoredox reactions are better explored than reductive photoredox reactions. More generally, most photocatalytic reactions are based on a PeT mechanism. A broadened reaction scope is also warranted, with particular emphasis on both photoinduced energy transfer and atom transfer reactions.148 Finally, for this technology to become more widely adopted in the commercial sector it must be scalable, which requires solutions unique to photochemistry that represent a continuing challenge.149 Both fine and commodity chemicals have the potential to be synthesized in a much greener fashion using photoredox catalysis coupled with continuous flow methods, conditions that are compatible with industrial scale applications.150,151From this review it is evident that there exist many photoactive. Earth-abundant metal complexes, only a small fraction of which have been actively developed as PCs. Some of these PCs have favourable optoelectronic properties compared to the commonly used Ir(III) and Ru(II) PCs and so could readily act as replacements. The dearth of reactions studied is a present impediment to wider adoption. However, an appreciation of the thermodynamic criteria underpinning the choice of PCs for a particular reaction makes Earth-abundant photoactive complexes particularly attractive.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council and CRITICAT Centre for Doctoral Training for financial support [Ph.D. studentship to B. H.; Grant code: EP/L016419/1]. C. L. thanks the Prof. & Mrs Purdie Bequests Scholarship and AstraZeneca for his PhD Studentship.Notes and references
- J. Kiwi, K. Kalyanasundaram and M. Grätzel, in Solar Energy Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 1982, pp. 37–125 Search PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- X. Wu, C. Meng, X. Yuan, X. Jia, X. Qian and J. Ye, Chem. Commun., 2015, 51, 11864–11867 RSC.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
- W. J. Finkenzeller and H. Yersin, Chem. Phys. Lett., 2003, 377, 299–305 CrossRef CAS.
- N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Science, 1997, 275, 54–57 CrossRef CAS PubMed.
- A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS.
- Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514–11526 CrossRef CAS PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC.
- M. Bortoluzzi, J. Castro, F. Enrichi, A. Vomiero, M. Busato and W. Huang, Inorg. Chem. Commun., 2018, 92, 145–150 CrossRef CAS.
- M. Bortoluzzi, J. Castro, E. Trave, D. Dallan and S. Favaretto, Inorg. Chem. Commun., 2018, 90, 105–107 CrossRef CAS.
- L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 11247–11250 CrossRef PubMed.
- Y. F. Liang, R. Steinbock, L. Yang and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10625–10629 CrossRef CAS PubMed.
- C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed.
- J. A. Gareth Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef CAS.
- P. Chábera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Persson and K. Wärnmark, J. Phys. Chem. Lett., 2018, 9, 459–463 CrossRef PubMed.
- A. Barbieri, G. Accorsi and N. Armaroli, Chem. Commun., 2008, 2185–2193 RSC.
- M. Winter, WebElements Periodic Table»Abundance in Earth's crust»periodicity, https://www.webelements.com/periodicity/abundance_crust/, (accessed 22 November 2017) Search PubMed.
- LME, London Metal Exchange, https://www.lme.com/Metals/Non-ferrous/Copper#tabIndex=0, (accessed 6 November 2018) Search PubMed.
- F. Dumur, Org. Electron., 2015, 21, 27–39 CrossRef CAS.
- R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- F. Dumur, Org. Electron., 2015, 21, 27–39 CrossRef CAS.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- P. Xiao, F. Dumur, J. Zhang, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2014, 47, 3837–3844 CrossRef CAS.
- S. Parisien-Collette, A. C. Hernandez-Perez and S. K. Collins, Org. Lett., 2016, 18, 4994–4997 CrossRef CAS PubMed.
- A. C. Hernandez-Perez and S. K. Collins, Angew. Chem., Int. Ed., 2013, 52, 12696–12700 CrossRef CAS PubMed.
- C. S. Smith, C. W. Branham, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2010, 132, 14079–14085 CrossRef CAS PubMed.
- R. Marion, F. Sguerra, F. Di Meo, E. Sauvageot, J. F. Lohier, R. Daniellou, J. L. Renaud, M. Linares, M. Hamel and S. Gaillard, Inorg. Chem., 2014, 53, 9181–9191 CrossRef CAS PubMed.
- J. Nitsch, F. Lacemon, A. Lorbach, A. Eichhorn, F. Cisnetti and A. Steffen, Chem. Commun., 2016, 52, 2932–2935 RSC.
- V. A. Krylova, P. I. Djurovich, M. T. Whited and M. E. Thompson, Chem. Commun., 2010, 46, 6696–6698 RSC.
- M. D. Weber, E. Fresta, M. Elie, M. E. Miehlich, J. L. Renaud, K. Meyer, S. Gaillard and R. D. Costa, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef.
- V. A. Krylova, P. I. Djurovich, B. L. Conley, R. Haiges, M. T. Whited, T. J. Williams and M. E. Thompson, Chem. Commun., 2014, 50, 7176–7179 RSC.
- M. Elie, J. L. Renaud and S. Gaillard, Polyhedron, 2018, 140, 158–168 CrossRef CAS.
- M. Elie, F. Sguerra, F. Di Meo, M. D. Weber, R. Marion, A. Grimault, J. F. Lohier, A. Stallivieri, A. Brosseau, R. B. Pansu, J. L. Renaud, M. Linares, M. Hamel, R. D. Costa and S. Gaillard, ACS Appl. Mater. Interfaces, 2016, 8, 14678–14691 CrossRef CAS PubMed.
- J. M. Kern and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1987, 546–548 RSC.
- H. J. Son, W. S. Han, J. Y. Chun, B. K. Kang, S. N. Kwon, J. Ko, J. H. Su, C. Lee, J. K. Sung and O. K. Sang, Inorg. Chem., 2008, 47, 5666–5676 CrossRef CAS PubMed.
- L. A. Büldt, C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2017, 23, 8541 CrossRef.
- L. A. Büldt and O. S. Wenger, Angew. Chem., Int. Ed., 2017, 56, 5676–5682 CrossRef PubMed.
- W. Sattler, L. M. Henling, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2015, 137, 1198–1205 CrossRef CAS PubMed.
- W. Sattler, M. E. Ener, J. D. Blakemore, A. A. Rachford, P. J. Labeaume, J. W. Thackeray, J. F. Cameron, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 10614–10617 CrossRef CAS PubMed.
- L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., 2016, 128, 11413–11417 CrossRef.
- K. R. Mann, M. Cimolino, G. L. Geoffroy, G. S. Hammond, A. A. Orio, G. Albertin and H. B. Gray, Inorg. Chim. Acta, 1976, 16, 97–101 CrossRef CAS.
- L. A. Büldt, X. Guo, R. Vogel, A. Prescimone and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 985–992 CrossRef PubMed.
- O. Moudam, A. Kaeser, B. Delavaux-Nicot, C. Duhayon, M. Holler, G. Accorsi, N. Armaroli, I. Séguy, J. Navarro, P. Destruel and J. F. Nierengarten, Chem. Commun., 2007, 3077–3079 RSC.
- A. Kaeser, O. Moudam, G. Accorsi, I. Séguy, J. Navarro, A. Belbakra, C. Duhayon, N. Armaroli, B. Delavaux-Nicot and J. F. Nierengarten, Eur. J. Inorg. Chem., 2014, 2014, 1345–1355 CrossRef CAS.
- M. Janghouri, E. Mohajerani, M. M. Amini and E. Najafi, J. Lumin., 2014, 154, 465–474 CrossRef CAS.
- C. Bizzarri, C. Strabler, J. Prock, B. Trettenbrein, M. Ruggenthaler, C. H. Yang, F. Polo, A. Iordache, P. Brüggeller and L. De Cola, Inorg. Chem., 2014, 53, 10944–10951 CrossRef CAS PubMed.
- Y. Z. Xie, G. G. Shan, P. Li, Z. Y. Zhou and Z. M. Su, Dyes Pigm., 2013, 96, 467–474 CrossRef CAS.
- E. Mejía, S. P. Luo, M. Karnahl, A. Friedrich, S. Tschierlei, A. E. Surkus, H. Junge, S. Gladiali, S. Lochbrunner and M. Beller, Chem. – Eur. J., 2013, 19, 15972–15978 CrossRef PubMed.
- M. Knorn, T. Rawner, R. Czerwieniec and O. Reiser, ACS Catal., 2015, 5, 5186–5193 CrossRef CAS.
- R. Sakamoto, T. Iwashima, J. F. Kögel, S. Kusaka, M. Tsuchiya, Y. Kitagawa and H. Nishihara, J. Am. Chem. Soc., 2016, 138, 5666–5677 CrossRef CAS PubMed.
- C. Femoni, S. Muzzioli, A. Palazzi, S. Stagni, S. Zacchini, F. Monti, G. Accorsi, M. Bolognesi, N. Armaroli, M. Massi, G. Valenti and M. Marcaccio, Dalton Trans., 2013, 42, 997–1010 RSC.
- H. Xu, Z. F. Xu, Z. Y. Yue, P. F. Yan, B. Wang, L. W. Jia, G. M. Li, W. Bin Sun and J. W. Zhang, J. Phys. Chem. C, 2008, 112, 15517–15525 CrossRef CAS.
- I. Andrés-Tomé, J. Fyson, F. Baiao Dias, A. P. Monkman, G. Iacobellis and P. Coppo, Dalton Trans., 2012, 41, 8669–8674 RSC.
- Y. Zhang, M. Heberle, M. Wächtler, M. Karnahl and B. Dietzek, RSC Adv., 2016, 6, 105801–105805 RSC.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 7336–7340 CrossRef CAS PubMed.
- D. G. Cuttell, S. M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6–7 CrossRef CAS PubMed.
- K. T. Yeung, W. P. To, C. Sun, G. Cheng, C. Ma, G. S. M. Tong, C. Yang and C. M. Che, Angew. Chem., Int. Ed., 2017, 56, 133–137 CrossRef CAS PubMed.
- B. Michelet, C. Deldaele, S. Kajouj, C. Moucheron and G. Evano, Org. Lett., 2017, 19, 3576–3579 CrossRef CAS PubMed.
- H. T. Akçay, R. Bayrak, Ü. Demirbaş, A. Koca, H. Kantekin and I. Değirmencioğlu, Dyes Pigm., 2013, 96, 483–494 CrossRef.
- S. Sakaki, H. Mizutani, Y. Ichi Kase, T. Arai and T. Hamada, Inorg. Chim. Acta, 1994, 225, 261–267 CrossRef CAS.
- S. Sakaki, H. Mizutani, Y. I. Kase, K. J. Inokuchi, T. Arai and T. Hamada, J. Chem. Soc., Dalton Trans., 1996, 1909–1914 RSC.
- A. J. J. Lennox, S. Fischer, M. Jurrat, S. P. Luo, N. Rockstroh, H. Junge, R. Ludwig and M. Beller, Chem. – Eur. J., 2016, 22, 1233–1238 CrossRef CAS PubMed.
- N. A. Porter, I. J. Rosenstein, R. A. Breyer, J. D. Bruhnke, W. X. Wu and A. T. McPhail, J. Am. Chem. Soc., 1992, 114, 7664–7676 CrossRef CAS.
- D. P. Curran, W. Shen, J. Zhang and T. A. Heffner, J. Am. Chem. Soc., 1990, 112, 6738–6740 CrossRef CAS.
- G. E. Keck and J. B. Yates, J. Am. Chem. Soc., 1982, 104, 5829–5831 CrossRef CAS.
- M. Kosugi, H. Arai, A. Yoshino and T. Migita, Chem. Lett., 1978, 7, 795–796 CrossRef.
- D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999–7002 CrossRef CAS PubMed.
- M. Alkan-Zambada and X. Hu, Organometallics, 2018, 37, 3928–3935 CrossRef CAS.
- S. K. Pagire, S. Paria and O. Reiser, Org. Lett., 2016, 18, 2106–2109 CrossRef CAS PubMed.
- S. Paria, M. Pirtsch, V. Kais and O. Reiser, Synthesis, 2013, 45, 2689–2698 CrossRef CAS.
- X. J. Tang and W. R. Dolbier, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed.
- Z. Zhang, X. Tang, C. S. Thomoson and W. R. Dolbier, Org. Lett., 2015, 17, 3528–3531 CrossRef CAS PubMed.
- T. Rawner, M. Knorn, E. Lutsker, A. Hossain and O. Reiser, J. Org. Chem., 2016, 81, 7139–7147 CrossRef CAS PubMed.
- A. Baralle, L. Fensterbank, J. P. Goddard and C. Ollivier, Chem. – Eur. J., 2013, 19, 10809–10813 CrossRef CAS PubMed.
- A. C. Hernandez-Perez, A. Vlassova and S. K. Collins, Org. Lett., 2012, 14, 2988–2991 CrossRef CAS PubMed.
- J.-R. Jiménez, B. Doistau, C. Besnard and C. Piguet, Chem. Commun., 2018, 54, 13228–13231 RSC.
- G. Fumagalli, P. T. G. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11481–11484 CrossRef CAS PubMed.
- P. T. G. Rabet, G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2016, 18, 1646–1649 CrossRef CAS PubMed.
- T. P. Nicholls, G. E. Constable, J. C. Robertson, M. G. Gardiner and A. C. Bissember, ACS Catal., 2016, 6, 451–457 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- J. Lantrip, M. Griffin and A. Aly, World Water Congr. 2005 Impacts Glob. Clim. Chang. - Proc. 2005 World Water Environ. Resour. Congr., 2005, vol. 17, p. 436 Search PubMed.
- Y. Foo Lee and R. K. Jon, J. Am. Chem. Soc., 1994, 116, 3599–3600 CrossRef.
- J. A. Bandy, F. G. N. Cloke, G. Cooper, J. P. Day, R. B. Girling, R. G. Graham, J. C. Green, R. Grinter and R. N. Perutz, J. Am. Chem. Soc., 1988, 110, 5039–5050 CrossRef CAS.
- K. S. Heinselman and M. D. Hopkins, J. Am. Chem. Soc., 1995, 117, 12340–12341 CrossRef CAS.
- S. Paulson, B. P. Sullivan and J. V. Caspar, J. Am. Chem. Soc., 1992, 114, 6905–6906 CrossRef CAS.
- B. W. Pfennig, M. E. Thompson and A. B. Bocarsly, Organometallics, 1993, 12, 649–655 CrossRef CAS.
- B. W. Pfennig, M. E. Thompson and A. B. Bocarsly, J. Am. Chem. Soc., 1989, 111, 8947–8948 CrossRef CAS.
- G. V. Loukova and V. A. Smirnov, Chem. Phys. Lett., 2000, 329, 437–442 CrossRef CAS.
- P. Chábera, Y. Liu, O. Prakash, E. Thyrhaug, A. El Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. B. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695–699 CrossRef PubMed.
- S. N. Choing, A. J. Francis, G. Clendenning, M. S. Schuurman, R. D. Sommer, I. Tamblyn, W. W. Weare and T. Cuk, J. Phys. Chem. C, 2015, 119, 17029–17038 CrossRef CAS.
- S. Gazi, M. Dokić, A. M. P. Moeljadi, R. Ganguly, H. Hirao and H. Sen Soo, ACS Catal., 2017, 7, 4682–4691 CrossRef CAS.
- S. Gazi, W. K. Hung Ng, R. Ganguly, A. M. Putra Moeljadi, H. Hirao and H. Sen Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- Y. Zhang, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2016, 138, 13115–13118 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- A. D. Kirk and G. B. Porter, J. Phys. Chem., 1980, 84, 887–891 CrossRef CAS.
- N. Serpone, M. A. Jamieson, M. S. Henry, M. Z. Hoffman, F. Bolletta and M. Maestri, J. Am. Chem. Soc., 1979, 101, 2907–2916 CrossRef CAS.
- L. A. Büldt and O. S. Wenger, Chem. Sci., 2017, 8, 7359–7367 RSC.
- R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappé and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451–5464 CrossRef CAS PubMed.
- A. K. Pal, C. Li, G. S. Hanan and E. Zysman-Colman, Angew. Chem., Int. Ed., 2018, 57, 8027–8031 CrossRef CAS PubMed.
- J. Lantrip, M. Griffin and A. Aly, World Water Congr. 2005 Impacts Glob. Clim. Chang. - Proc. 2005 World Water Environ. Resour. Congr., 2005, vol. 17, p. 436 Search PubMed.
- M. Isaacs, A. G. Sykes and S. Ronco, Inorg. Chim. Acta, 2006, 359, 3847–3854 CrossRef CAS.
- B. Wang, D. P. Shelar, X. Z. Han, T. T. Li, X. Guan, W. Lu, K. Liu, Y. Chen, W. F. Fu and C. M. Che, Chem. – Eur. J., 2015, 21, 1184–1190 CrossRef CAS PubMed.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS.
- S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222–226 CrossRef CAS.
- A. Call, C. Casadevall, F. Acuña-Parés, A. Casitas and J. Lloret-Fillol, Chem. Sci., 2017, 8, 4739–4749 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- W. P. To, Y. Liu, T. C. Lau and C. M. Che, Chem. – Eur. J., 2013, 19, 5654–5664 CrossRef CAS PubMed.
- Y. Q. Zou, J. R. Chen, X. P. Liu, L. Q. Lu, R. L. Davis, K. A. Jørgensen and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- X. Q. Zhu, M. T. Zhang, A. Yu, C. H. Wang and J. P. Cheng, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed.
- Y. Zhang, J. L. Petersen and C. Milsmann, Organometallics, 2018, 37(23), 4488–4499 CrossRef CAS.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- Y. Yang, Q. Liu, L. Zhang, H. Yu and Z. Dang, Organometallics, 2017, 36, 687–698 CrossRef CAS.
- S. Lin, C. E. Padilla, M. A. Ischay and T. P. Yoon, Tetrahedron Lett., 2012, 53, 3073–3076 CrossRef CAS PubMed.
- N. Arai and T. Ohkuma, J. Org. Chem., 2017, 82, 7628–7636 CrossRef CAS PubMed.
- X. Jia, H. Lin, C. Huo, W. Zhang, J. Lü, L. Yang, G. Zhao and Z. L. Liu, Synlett, 2003, 2003, 1707–1709 Search PubMed.
- X. Jia, B. Han, W. Zhang, X. Jin, L. Yang and Z. L. Liu, Synthesis, 2006, 2006, 2831–2836 CrossRef.
- W. Zhang, Y. Guo, Z. Liu, X. Jin, L. Yang and Z. L. Liu, Tetrahedron, 2005, 61, 1325–1333 CrossRef CAS.
- W. Zhang, X. Jia, L. Yang and Z. L. Liu, Tetrahedron Lett., 2002, 43, 9433–9436 CrossRef CAS.
- S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- X. Solans, L. Ruiz-Ramirez, R. Moreno-Esparza, M. Labrador and A. Escuer, J. Solid State Chem., 1994, 109, 315–320 CrossRef CAS.
- J. K. McCusker, K. N. Walda, R. C. Dunn, J. D. Simon, D. Magde and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 298–307 CrossRef CAS.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevee, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 42–49 CrossRef CAS.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 2227–2233 CrossRef CAS.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Macromolecules, 2017, 50, 7967–7977 CrossRef CAS.
- Y. Qin, P. Tao, L. Gao, P. She, S. Liu, X. Li, F. Li, H. Wang, Q. Zhao, Y. Miao and W. Huang, Adv. Opt. Mater., 2018, 1801160 Search PubMed.
- Y. Liu, S. C. Yiu, C. L. Ho and W. Y. Wong, Coord. Chem. Rev., 2018, 375, 514–557 CrossRef CAS.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC.
- F. R. Bou-Hamdan and P. H. Seeberger, Chem. Sci., 2012, 3, 1612–1616 RSC.
- R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 4140–4143 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the review. |
| This journal is © The Royal Society of Chemistry 2019 |