
Palladium-catalyzed regioselective C–H alkynylation of indoles with haloalkynes: access to functionalized 7-alkynylindoles†
Songjia
Fang
b,
Guangbin
Jiang
b,
Meng
Li
b,
Zhenying
Liu
b,
Huanfeng
Jiang
 b and
Wanqing
Wu
*ab
b and
Wanqing
Wu
*ab
aState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: cewuwq@scut.edu.cn; Fax: +86 20-87112906
bKey Laboratory of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China
First published on 23rd October 2019
Abstract
A palladium-catalyzed uniquely regioselective C–H alkynylation of indoles has been described. In this protocol, simple and readily available haloalkynes are employed as efficient alkynylating reagents, affording a series of functionalized 7-alkynylindoles in moderate to good yields. Moreover, further transformations of 7-alkynylated products were performed, which demonstrated the potential application of this method in organic synthesis.
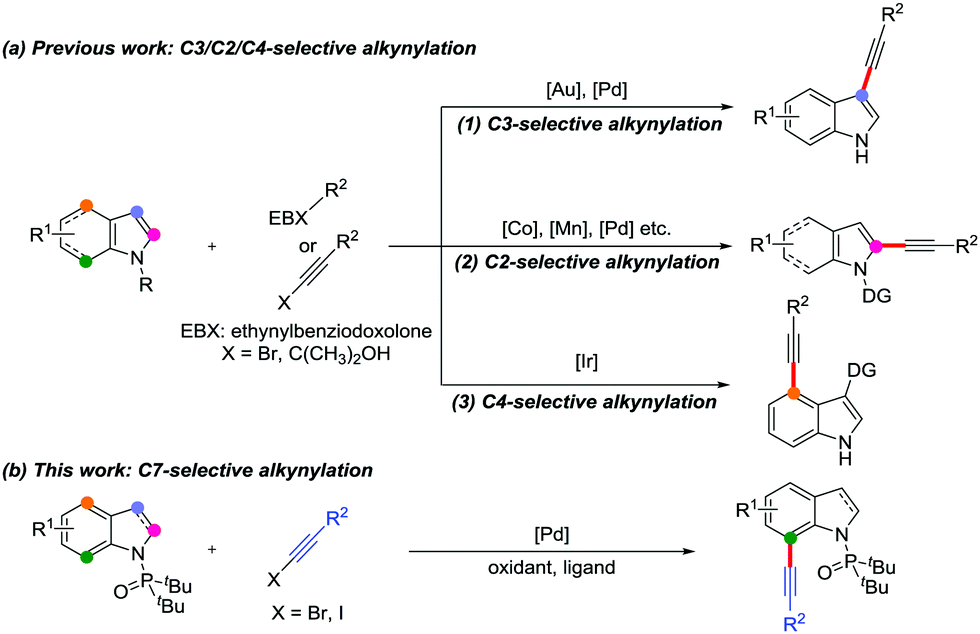
Indole skeletons are core motifs in medicinal chemistry, organic synthesis and materials science due to their individual biological activity.1 Consequently, the development of synthetic methods for functionalized indole derivatives has become one of the research hotspots.2,3 Among which, most of the studies focus on the synthesis of high-value alkynylated indoles due to the significant chemical properties of the alkyne moiety.3 In recent years, regioselective direct C–H alkynylation has been regarded as an efficient and powerful approach to construct alkynylindoles.4–6 Generally, for indole skeletons, there are multiple C–H bonds including pyrrole rings (C2–C3 positions) and benzenoid rings (C4–C7 positions) that can be alkynylated. However, the direct C–H alkynylation of indoles is mainly at the C3 position5 owing to the abundant electron effects and C2 position6 near the nitrogen atom due to the intrinsic reactivity7 of the indole ring (Scheme 1a, eqn (1) and (2)). Moreover, utilizing a carbonyl directing group (DG) at the C3 position results in the C4-selective alkynylation (Scheme 1a, eqn (3)).8 Despite considerable progress made with indole alkynylation, the selective C7 alkynylation of indoles is rarely reported owing to the following issues: (i) the C–H bond at this electron-deficient position is difficult to activate due to the inherently poor reactivity; (ii) the high selectivity is interfered by the reactivity of C2 and C3 postions.9 Recently, Miura et al. developed an iridium-catalyzed and sulfur-directed C4/C7–H alkynylation between indoles and ethynylbenziodoxole.10 In view of the importance of alkynes, novel and convenient methods for the selective installation of alkynyl groups at the C7 position of indoles is still desirable and challenging.
 | ||
| Scheme 1 Regioselective C–H alkynylations of indoles. | ||
Over the past few years, haloalkynes as valuable building blocks, featuring synthetic convenience and high practicability, have exhibited versatile reactivities in organic chemistry.11 In particular, in cross-coupling reactions, haloalkynes can be used as simple and effective alkynylating reagents to obtain the desired alkynyl products.12 In recent years, we have investigated a series of cross-coupling reactions involving haloalkynes, including palladium-catalyzed bromoalkynylation of norbornenes,13 directed alkynylation of biaryl compounds14 and C2-selective alkynylation of indoles.15 Based on our continuous interest in haloalkyne chemistry, herein, we disclose a novel palladium-catalyzed C7-selective alkynylation of indoles with di-tert-butylphosphinoyl as an effective directing group16 and simple haloalkynes as alkynylating reagents (Scheme 1b). It is noteworthy that this protocol shows specific regioselectivity to form 7-alkynylated indoles. In addition, the availability of starting materials and the derivatization of alkynylated products show the practicability of this method.
Initially, the directed C7-alkynylation of indoles was tested using di-tert-butyl(1H-indol-1-yl)phosphine oxide (1a) and (bromoethynyl)triisopropylsilane (2a) as the coupling models (Table 1). Delightfully, in the presence of Pd(OAc)2 (10 mol%) as the catalyst, Ag2CO3 (2 equiv.) and Cu(OTf)2 (1 equiv.) as additives, the desired product 3a was obtained in 43% yield at 120 °C (Table 1, entry 1). However, employing AgF instead of Ag2CO3 reduced the yield of 3a to 15% and changing Cu(OTf)2 to CuO inhibited the formation of 3a (Table 1, entries 2 and 3). Next, the exploration of different catalysts showed that Pd(0) catalysts suppressed the formation of C3-alkynylated product and Pd2(dba)3 could maintain the yield of 3a at 43% (Table 1, entries 4 and 5). To promote this reaction, a series of N-ligands were studied and L5 was proved to be the most suitable ligand for the alkynylation, which might be caused by the optimum balance between the electronic and steric properties (Table 1, entries 6–10). The transformation was further improved by the screening of other reaction parameters, such as the ratio of additives, substrate amounts and dosage of toluene, giving the desired product 3a in 81% isolated yield (Table 1, entry 11). Besides, control experiments showed that the co-existence of Ag2CO3 and Cu(OTf)2 was critical for this alkynylation (Table 1, entries 12 and 13) and no reaction occurred without palladium catalyst (Table 1, entry 14) (see the ESI† for details).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additives | Ligand | Yieldb (%) |
| a Conditions: unless otherwise noted, all reactions were performed with 1a (0.1 mmol), 2a (2 equiv.), catalyst (10 mol%), ligand (20 mol%), Ag salt (2 equiv.), and Cu salt (1 equiv.) in toluene (1.0 mL), under air at 90 °C for 12 h. b Monitored by NMR using CH2Br2 as the internal standard. c 2a (1.8 equiv.), toluene (1.5 mL). d Ag2CO3 (1.8 equiv.). e Cu(OTf)2 (1.5 equiv.). f Isolated yield. | ||||
| 1 | Pd(OAc)2 | Ag2CO3/Cu(OTf)2 | — | 43 |
| 2 | Pd(OAc)2 | AgF/Cu(OTf)2 | — | 15 |
| 3 | Pd(OAc)2 | Ag2CO3/CuO | — | n.d. |
| 4 | Pd2(dba)3 | Ag2CO3/Cu(OTf)2 | — | 43 |
| 5 | Pd(PPh3)4 | Ag2CO3/Cu(OTf)2 | — | 34 |
| 6 | Pd2(dba)3 | Ag2CO3/Cu(OTf)2 | L1 | 38 |
| 7 | Pd2(dba)3 | Ag2CO3/Cu(OTf)2 | L2 | Trace |
| 8 | Pd2(dba)3 | Ag2CO3/Cu(OTf)2 | L3 | 50 |
| 9 | Pd2(dba)3 | Ag2CO3/Cu(OTf)2 | L4 | 49 |
| 10 | Pd2(dba)3 | Ag2CO3/Cu(OTf)2 | L5 | 51 |
| 11 , , | Pd 2 (dba) 3 | Ag 2 CO 3 /Cu(OTf) 2 | L5 | 83 (81 ) |
| 12c,d | Pd2(dba)3 | Ag2CO3 | L5 | n.d. |
| 13c,e | Pd2(dba)3 | Cu(OTf)2 | L5 | n.d. |
| 14c | — | Ag2CO3/Cu(OTf)2 | L5 | n.d. |
|
||||
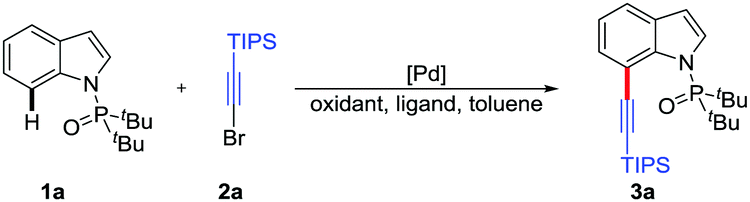
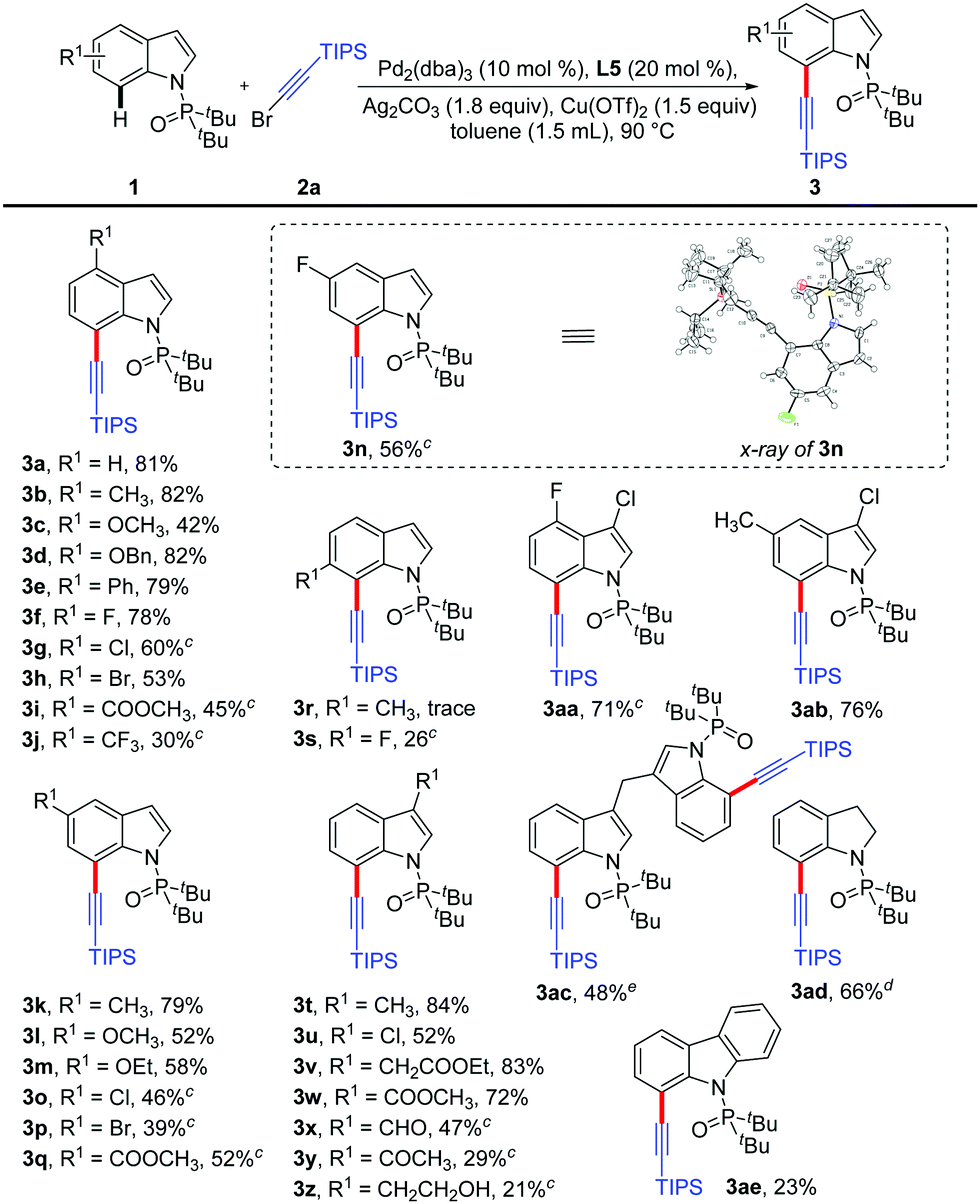
With the optimized reaction conditions in hand, a systemic investigation of the substrate scope was implemented (Table 2). Gratifyingly, various substitution patterns of the indole backbone were applicable in this reaction and the desired C7 alkynylated products could be obtained in moderate to excellent yields. It was found that the indole derivatives 1 with either electron-donating (–CH3, –OCH3, –OBn and –OCH2CH3) or electron-withdrawing (–Ph, –F, –Cl, –Br, –COOCH3 and –CF3) substituents at the C4 and C5 positions were successfully transformed to the corresponding C7-alkynylated products 3b–3q in 30–82% yields. The molecular structure could be verified by X-ray crystallography of 3n (CCDC 1915232†). However, the substitution at the C6 position did not show good tolerance (3r–3s). Moreover, the substrates 1 bearing various substituents (–CH3, –Cl, –CH2COOEt, –COOCH3, and –CHO) at the C3 position were smoothly alkynylated to provide the products 3t–3x. The desired products 3y and 3z were obtained in low yields without C2-alkynylated products detected. Additionally, the 3-Cl-4-F and 3-Cl-5-CH3 disubstituted indole substrates also showed favorable reactivity and the corresponding products 3aa and 3ab could be obtained in 71% and 76% yields, respectively. Then, the alkynylation between the 3,3′-diindolylmethane derivative and two molecules of bromoalkyne afforded 3ac in 48% yield. When the indoline substrate was subjected to this alkynylation protocol, 3ad was formed quickly in 66% yield within 2 h. It should be noted that a sterically demanding carbazole substrate could also be transformed to the monoalkynylation product 3ae, albeit in a low yield.
| a Conditions: unless otherwise noted, all reactions were performed with 1 (0.1 mmol), 2a (0.18 mmol), Pd2(dba)3 (10 mol%), Ag2CO3 (1.8 equiv.), Cu(OTf)2 (1.5 equiv.), and L5 (20 mol%) in toluene (1.5 mL) under air at 90 °C for 12 h. b Isolated yield. c L4 (20 mol%). d 2 h. e 2a (0.36 mmol), Pd2(dba)3 (15 mol%), L5 (30 mol%), Ag2CO3 (3.6 equiv.), Cu(OTf)2 (3.0 equiv.), and toluene (2.0 mL). |
|---|
|
After evaluating the scope of indole derivatives 1, we further investigated the effects of various haloalkynes 2 (Table 3). Ethynyltriisopropylsilanes with different halogen atoms were first examined. Pleasingly, the desired product 3a could be obtained in 44% yield when using (iodoethynyl)triisopropylsilane as the alkynylating reagent (Table 3, entry 1). However, no reaction occurred with (chloroethynyl)triisopropylsilane as the substrate (Table 3, entry 2). Moreover, the effects of substituents at silane were also examined. (Bromoethynyl)(tert-butyl)dimethylsilane was compatible with this catalytic system and the corresponding product 3af could be obtained in 67% yield, while replacing the isopropyl group to triethyl or trimethyl just showed poor reactivity (3ag–3ah), which might be caused by the coordination between low sterically hindered bromoalkynes and palladium catalyst via π bonding.17 Unfortunately, ethyl 3-bromopropiolate and (bromoethynyl)benzene were found to be not suitable for the C7 alkynylation.
|
|
|||
|---|---|---|---|
| Entry | Haloalkyne 2 | Product 3 | Yield of 3b (%) |
| a Conditions: unless otherwise noted, all reactions were performed with 1a (0.1 mmol), 2 (0.18 mmol), Pd2(dba)3 (10 mol %), Ag2CO3 (1.8 equiv.), Cu(OTf)2 (1.5 equiv.), and L5 (20 mol %) in toluene (1.5 mL), under air at 90 °C for 12 h. b Isolated yield. | |||
| 1 | X = I, R2 = TIPS | 3a | 44 |
| 2 | X = Cl, R2 = TIPS | 3a | n.d. |
| 3 | X = Br, R2 = TBDMS | 3af | 67 |
| 4 | X = Br, R2 = TES | 3ag | Trace |
| 5 | X = Br, R2 = TMS | 3ah | n.d. |
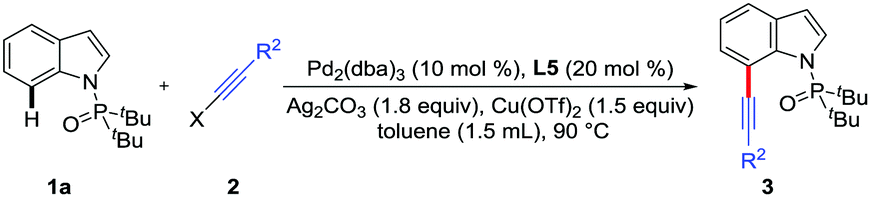
Furthermore, the potential applications of C7-alkynlated products as useful synthetic blocks are illustrated (Scheme 2). With appropriate reaction temperature and time, both the triisopropylsilyl group and the directing group could be easily removed upon treatment with TBAF to deliver the desilylation product 4a or 7-ethynyl-1H-indole 5a. The Sonogashira coupling reaction of 4a offered the phenylacetylene product 6a in 67% yield. Additionally, in the presence of CuI, 5a could react with BnN3 to give triazole indole 7a in 65% yield via a Click reaction, which might be used for medicinal chemistry and materials science.18
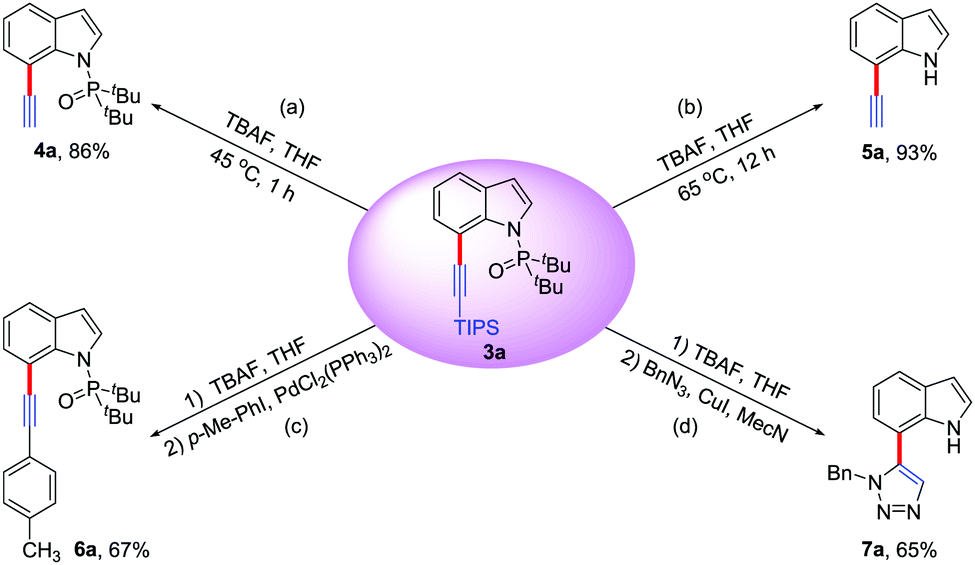 | ||
| Scheme 2 Derivatizations of alkynylation product 3a (conditions: see the ESI† for details). | ||
Several control experiments were then carried out to shed light on the reaction mechanism (Scheme 3). When this C7 alkynylation was respectively carried out in the presence of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxyl) or BHT (2,6-di-tert-butyl-p-cresol), the yields of alkynylation product 3a dramatically dropped. However, 3a could be obtained in 73% yield when 1,1-diphenylethylene was added in this reaction under the optimized conditions (Scheme 3a), indicating that the oxidation system was important and specific for this transformation, and the radical pathway should not be involved in this process. Then, an intermolecular KIE of kH/kD = 3.5 suggested that the rate-determining step plausibly was the cleavage of the C7–H bond of substrate 1a in this alkynylation reaction (Scheme 3b). In addition, the competition experiment indicated that electron-rich indole substrates reacted preferentially (Scheme 3c).
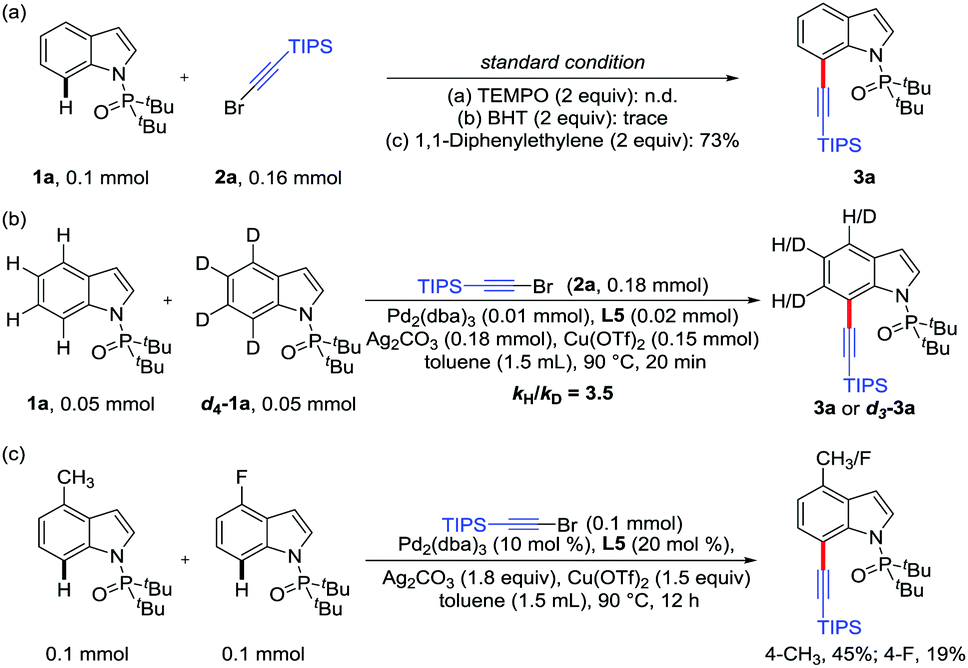 | ||
| Scheme 3 Mechanistic studies. | ||
On the basis of the experimental results and related studies,14,19 a plausible catalytic cycle is proposed for this C7-selective alkynylation (Scheme 4). First, the Pd(II) species is formed by the oxidation of Pd2(dba)3 in the presence of Ag2CO3 and Cu(OTf)2. Then the intermediate Int-1 is generated by complexation between the Pd(II) species and 1a. Subsequently, as the rate-determining step, the palladacycle Int-2 is obtained by the intramolecular selective C–H activation of Int-1. After 2a is activated by Ag(I) and Cu(II), the Int-2 can further undergo oxidative addition to form the Pd(IV) complex Int-3. Finally, the AgBr precipitate will promote the reductive elimination of Int-3, which leads to the formation of the alkynylation product, along with the regeneration of the Pd(II) catalyst to complete this catalytic cycle.
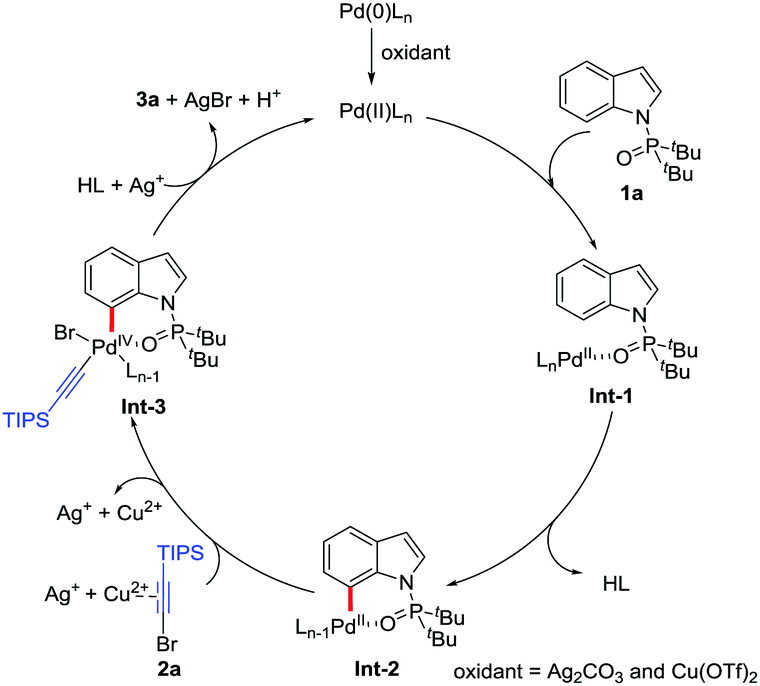 | ||
| Scheme 4 Proposed mechanism. | ||
In conclusion, we have developed a Pd-catalyzed di-tert-butylphosphinoyl directed C7-selective activation/alkynylation between indoles and haloalkynes, affording a series of highly functionalized 7-alkynylindole derivatives in good yields. The remarkable regioselectivity and good substrate compatibility have been highlighted in this reaction. In addition, the easy availability of starting materials and the functionalization of the newly formed alkynylated products show the synthetic practicality of this protocol. Further investigations of this method in pharmacochemistry are currently underway in our laboratory.
The authors thank the National Key Research and Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (21672072 and 21472051), and the Guangdong Natural Science Foundation (2018B030308007) for financial support.
Conflicts of interest
We have a patent (CN Pat., 109867694A, 2019) relevant to this work.Notes and references
- (a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (b) M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421 CrossRef CAS PubMed; (c) T. Zhang, Z. Qiao, Y. Wang, N. Zhong, L. Liu, D. Wang and Y.-J. Chen, Chem. Commun., 2013, 49, 1636 RSC; (d) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS PubMed; (e) M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604 CrossRef CAS PubMed; (f) M. A. Corsello, J. Kim and N. K. Garg, Chem. Sci., 2017, 8, 5836 RSC; (g) A. Palmieri and M. Petrini, Nat. Prod. Rep., 2019, 36, 490 RSC.
- (a) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403 CrossRef CAS; (b) Ł. W. Ciszewski, J. Durka and D. Gryko, Org. Lett., 2019, 21, 7028 CrossRef PubMed; (c) I. A. Utepova, M. A. Trestsova, O. N. Chupakhin, V. N. Charushin and A. A. Rempel, Green Chem., 2015, 17, 4401 RSC; (d) S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2017, 53, 6544 RSC; (e) H.-R. Wu, L. Cheng, D.-L. Kong, H.-Y. Huang, C.-L. Gu, L. Liu, D. Wang and C.-J. Li, Org. Lett., 2016, 18, 1382 CrossRef CAS PubMed.
- (a) Q.-J. Liu, W.-G. Yan, L. Wang, X. P. Zhang and Y. Tang, Org. Lett., 2015, 17, 4014 CrossRef CAS PubMed; (b) J. Luo, Y.-B. Fang, X.-Y. Mao, M. Yang, Y.-L. Zhao, Y.-L. Liu, G.-S. Chen and Y.-F. Ji, Adv. Synth. Catal., 2019, 361, 1408 CrossRef CAS; (c) P. P. Sharp, M. G. Banwell, J. Renner, K. Lohmann and A. C. Willis, Org. Lett., 2013, 15, 2616 CrossRef CAS PubMed; (d) A. J. Pearson and Y. Zhou, J. Org. Chem., 2009, 74, 4242 CrossRef CAS PubMed; (e) N. Kumarswamyreddy, K. Lokesh and V. Kesavan, Tetrahedron Lett., 2018, 59, 4344 CrossRef CAS; (f) O. Gherbovet, F. La Spisa, S. Thoret, M. C. García Alvarez, H. Levaique, J. Bignon and F. Roussi, Bioorg. Med. Chem. Lett., 2015, 25, 1771 CrossRef CAS PubMed.
- (a) E. Dickson, B. R. Copp and D. Barker, Tetrahedron Lett., 2013, 54, 5239 CrossRef CAS; (b) E. Brachet and P. Belmont, J. Org. Chem., 2015, 80, 7519 CrossRef PubMed; (c) C. Chen, P. Liu, J. Tang, G. Deng and X. Zeng, Org. Lett., 2017, 19, 2474 CrossRef CAS PubMed; (d) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 2722 CrossRef CAS PubMed; (e) E. Tan, A. I. Konovalov, G. A. Fernandez, R. Dorel and A. M. Echavarren, Org. Lett., 2017, 19, 5561 CrossRef CAS PubMed; (f) G. L. Tolnai, S. Ganss, J. P. Brand and J. Waser, Org. Lett., 2013, 15, 112 CrossRef CAS PubMed; (g) L. Yang, L. Zhao and C. Li, Chem. Commun., 2010, 46, 4184 RSC.
- (a) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (b) G. N. Hermann, M. T. Unruh, S.-H. Jung, M. Krings and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 10723 CrossRef CAS PubMed; (c) Y. Gu and X.-M. Wang, Tetrahedron Lett., 2009, 50, 763 CrossRef CAS.
- (a) Z.-Z. Zhang, B. Liu, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094 CrossRef CAS PubMed; (b) Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172 CrossRef CAS PubMed; (c) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780 CrossRef CAS PubMed; (d) T. Li, Z. Wang, W.-B. Qin and T.-B. Wen, ChemCatChem, 2016, 8, 2146 CrossRef CAS; (e) S. M. Khake, V. Soni, R. G. Gonnade and B. Punji, Chem. – Eur. J., 2017, 23, 2907 CrossRef CAS PubMed; (f) N. Sauermann, M. J. González and L. Ackermann, Org. Lett., 2015, 17, 5316 CrossRef CAS PubMed.
- (a) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS PubMed; (b) S. G. Modha and M. F. Greaney, J. Am. Chem. Soc., 2015, 137, 1416 CrossRef CAS PubMed; (c) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742 RSC; (d) T. Li, Z. Wang, M. Zhang, H.-J. Zhang and T.-B. Wen, Chem. Commun., 2015, 51, 6777 RSC.
- X. Li, G. Wu, X. Liu, Z. Zhu, Y. Huo and H. Jiang, J. Org. Chem., 2017, 82, 13003 CrossRef CAS PubMed.
- (a) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618 CrossRef CAS; (b) C. G. Hartung, A. Fecher, B. Chapell and V. Snieckus, Org. Lett., 2003, 5, 1899 CrossRef CAS PubMed; (c) L. Xu, C. Zhang, Y. He, L. Tan and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 321 CrossRef CAS PubMed; (d) X.-F. Yang, X.-H. Hu, C. Feng and T.-P. Loh, Chem. Commun., 2015, 51, 2532 RSC; (e) Y. Wu, Y. Yang, B. Zhou and Y. Li, J. Org. Chem., 2015, 80, 1946 CrossRef CAS PubMed; (f) N. Jin, C. Pan, H. Zhang, P. Xu, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2015, 357, 1149 CrossRef CAS.
- C. N. Kona, Y. Nishii and M. Miura, Angew. Chem., Int. Ed., 2019, 58, 9856 CrossRef CAS PubMed.
- (a) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC; (b) W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483 CrossRef CAS PubMed; (c) H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374 CrossRef CAS PubMed; (d) G. Jiang, S. Fang, W. Hu, J. Li, C. Zhu, W. Wu and H. Jiang, Adv. Synth. Catal., 2018, 360, 2297 CrossRef CAS; (e) Y.-B. Bai, Z. Luo, Y. Wang, J.-M. Gao and L. Zhang, J. Am. Chem. Soc., 2018, 140, 5860 CrossRef CAS PubMed; (f) Y. Zhao, J. Jin and P. W. H. Chan, Adv. Synth. Catal., 2019, 361, 1313 CrossRef CAS; (g) H. Jiang, C. Zhu and W. Wu, Haloalkyne Chemistry, Springer Berlin Heidelberg, 2016, pp. 1–77 CrossRef.
- (a) G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661 CrossRef CAS PubMed; (b) Y. Zhao, Ji. Jin and P. W. H. Chan, Adv. Synth. Catal., 2019, 361, 1313 CrossRef CAS; (c) Y.-H. Xu, Q.-C. Zhang, T. He, F.-F. Meng and T.-P. Loh, Adv. Synth. Catal., 2014, 356, 1539 CrossRef CAS; (d) F. Besselièvre and S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553 CrossRef PubMed; (e) L. D. Caspers and B. J. Nachtsheim, Chem. – Asian J., 2018, 13, 1231 CrossRef CAS PubMed; (f) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC.
- Y. Li, X. Liu, H. Jiang, B. Liu, Z. Chen and P. Zhou, Angew. Chem., Int. Ed., 2011, 50, 6341 CrossRef CAS PubMed.
- G. Jiang, W. Hu, J. Li, C. Zhu, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 1746 RSC.
- W. Wu, S. Fang, G. Jiang, M. Li and H. Jiang, Org. Chem. Front., 2019, 6, 2200 RSC.
- Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387 CrossRef CAS PubMed.
- (a) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC; (c) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
- (a) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed; (b) P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092 CrossRef CAS PubMed; (c) S.-W. Tao, J.-Y. Zhou, R.-Q. Liu and Y.-M. Zhu, J. Org. Chem., 2019, 84, 8121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1915232. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07263b |
| This journal is © The Royal Society of Chemistry 2019 |