
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing stereoretention for the synthesis of E-macrocycles with ruthenium-based olefin metathesis catalysts†
Tonia S.
Ahmed
,
T. Patrick
Montgomery
and
Robert H.
Grubbs
 *
*
Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: rhg@caltech.edu
First published on 14th March 2018
Abstract
The synthesis of E-macrocycles is achieved using stereoretentive, Ru-based olefin metathesis catalysts supported by dithiolate ligands. Kinetic studies elucidate marked differences in activity among the catalysts tested, with catalyst 4 providing meaningful yields of products in much shorter reaction times than stereoretentive catalysts 2 and 3. Macrocycles were generated with excellent selectivity (>99% E) and in moderate to high yields (47–80% yield) from diene starting materials bearing two E-configured olefins. A variety of rings were constructed, ranging from 12- to 18-membered macrocycles, including the antibiotic recifeiolide.
Introduction
Ring-closing metathesis (RCM) has gained widespread use in organic synthesis for the production of macrocyclic frameworks.1 This transition metal-catalyzed reaction is commonly used in the synthesis of many biologically active and olfactory compounds.2 The stereochemistry of the olefin often governs the properties of these cyclic molecules. Consequently, the stereochemical purity of olefin mixtures is important. The separation of E- and Z-isomers can be challenging, and thus methods for stereoselectively generating macrocycles are desirable.The synthesis of Z-macrocycles has been reported using an array of Ru-,3 W-, and Mo-based4 olefin metathesis catalysts. The steric environment of each of these catalysts is tuned such that the syn metallacyclobutane pathway is favored over the anti metallacyclobutane pathway (Fig. 1). Cycloreversion of this syn intermediate gives the macrocycle with Z-configuration.
 | ||
| Fig. 1 Key metallacyclobutane intermediates for making Z-macrocycles using Mo-, W-, and Ru-based olefin metathesis catalysts. | ||
In 2013, Hoveyda and co-workers reported Ru-based catalysts bearing dithiolate ligands that were able to perform highly Z-selective cross metathesis from Z-olefin starting materials.5 In 2015, we demonstrated that these catalysts were further capable of cross metathesis between two E-olefins or between an E-olefin and a terminal olefin to generate E-products with high selectivity (>98% E).6 This was the first reported example of highly E-selective cross metathesis through kinetic control.
The stereoretention exhibited by these complexes is proposed to arise from the α substituents of the metallacyclobutane pointing away from the large N-aryl groups of the N-heterocyclic carbene (NHC) ligand (Fig. 2a). Depending on the stereochemistry of the starting olefin, the β substituent can either point down or up into the open space between the two N-aryl groups and in front of the imidazol-2-ylidene ring. Given that the olefin starting material stereochemistry is Z, the β substituent in the favored intermediate will point down, and the product formed after cycloreversion will be Z. Conversely, if the starting olefin has E stereochemistry, the β substituent will point up, and the product from the favored intermediate will be E. Soon after this report, Schrock and Hoveyda described Mo-based catalysts capable of a similar transformation, by which E-selectivity in cross metathesis is achieved from E-alkenyl halide starting materials (Fig. 2b).7
 | ||
| Fig. 2 Models for selectivity in cross metathesis using stereoretentive olefin metathesis catalysts for (a) Ru catalysts and (b) Mo catalysts. | ||
E-selective cross metathesis using stereoretentive Ru-based catalysts was often marred by low yields in reactions involving functionalized substrates or terminal olefins. Although the low yields observed in reactions with terminal olefins were attributed to decomposition of Ru methylidenes,6 studies performed in our group showed that a large contributing factor to low activity with functionalized substrates is slow catalyst initiation in reactions of these catalysts with E-olefins.8 A series of fast-initiating catalysts 1–4 was reported to significantly improve activity of stereoretentive catalysts in highly E-selective reactions (Fig. 3).
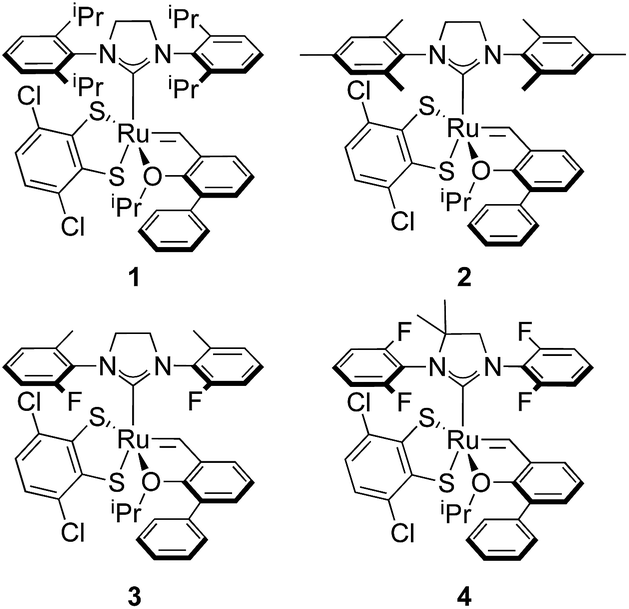 | ||
| Fig. 3 Series of fast-initiating, Ru-based catalysts used in achieving efficient synthesis of E-products in cross metathesis. | ||
The first example of the stereoselective synthesis of macrocycles using stereoretention was reported using Mo-based catalysts to generate E-macrocycles (91–98% E) from diene substrates containing an E-alkenyl-B(pinacolato) moiety and a terminal olefin.9 We recently reported a highly efficient synthesis of Z-macrocycles (95–99% Z) using stereoretention with Ru-based catalysts and substrates bearing a Z-olefin and a terminal olefin.10 Hoveyda and co-workers then provided further examples of Z-macrocyclizations using other stereoretentive Ru catalysts and one example of E-macrocycle synthesis using 3.11 Additional methods of obtaining E-macrocycles include Z-selective ethenolysis of stereochemical mixtures of macrocycles3 and alkyne metathesis followed by E-selective semihydrogenation catalyzed by Cp*Ru(COD)Cl/AgOTf.12
Results and discussion
We anticipated that E-macrocycles could be generated from diene starting materials containing an E-olefin using stereoretentive Ru catalysts. The proposed favored metallacyclobutane intermediate avoids steric clashing of the α substituent with the N-aryl group as proposed in the disfavored intermediate (Fig. 4). Catalysts 2–4 were chosen for studying RCM with these substrates as they had previously exhibited remarkable activity and selectivity in cross metathesis of E-olefins.8 Based on the aforementioned proposed model for stereoretention, we expected that reducing the ortho substituent size of the N-aryl groups would allow for better accommodation of the β substituent in E-selective RCM. Therefore the reactivity of 4 with E-olefins would be greater than 3, which would furthermore be greater than that of 2.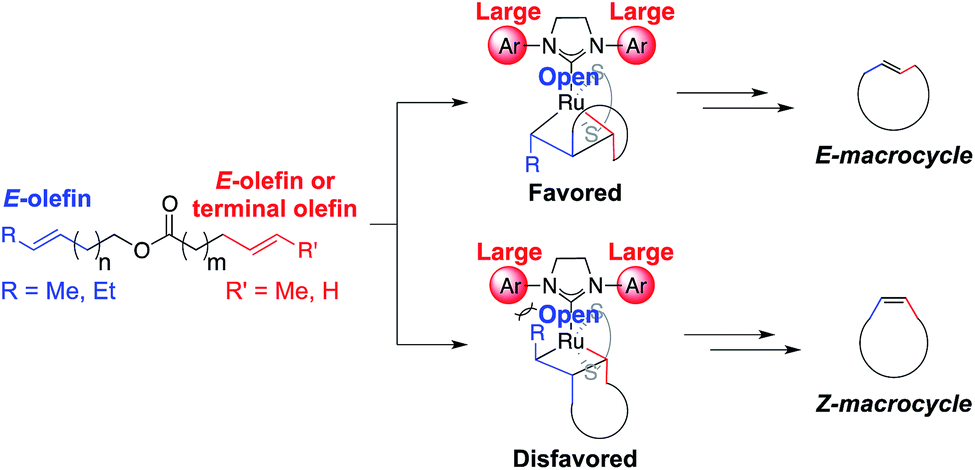 | ||
| Fig. 4 Proposed favored and disfavored intermediates in the formation of macrocycles from dienes containing an E-olefin. | ||
Using the approach we established in the synthesis of Z-macrocycles, we attempted to make E-macrocycle 6 from diene starting material 5 bearing an E-olefin and a terminal olefin (Scheme 1). Using a standard 7.5 mol% catalyst loading typical of macrocyclization reactions,13 catalysts 2, 3, and 4 reached a maximum of 4%, 30%, and 39% conversion, respectively, to the desired product with high E-selectivity (>99% E) at 35 °C. This low conversion is presumably a result of the instability and decomposition of unstable Ru methylidenes formed in this reaction.14 Previous studies have shown that reaction of these catalysts with E-olefins is considerably slower than with Z-olefins.8 Therefore it is proposed that Ru methylidenes persist longer in solution in reactions with E-olefins and are accordingly more prone to decomposition in reactions with E-olefins than in those with Z-olefins.
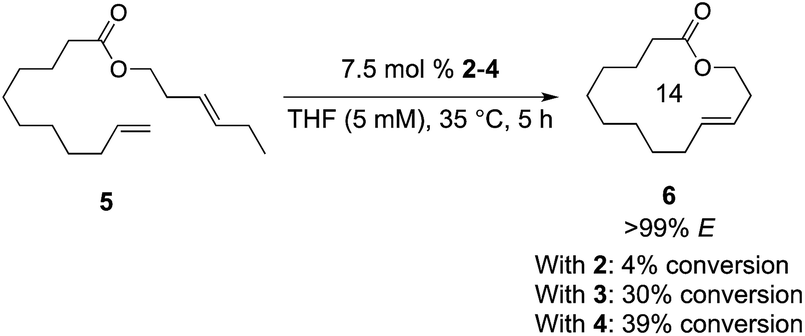 | ||
| Scheme 1 Synthesis of E-macrocycle 6 from 5 using 2–4. Conversion determined by 1H NMR. Selectivity determined using gas chromatography. | ||
To avoid the formation of Ru methylidenes, diene substrates containing two E-olefins were synthesized. Using substrate 7, the formation of 12-membered macrocycle 8 was monitored using catalysts 2–4 at 35 °C (Fig. 5a). Catalyst 4 displayed remarkable activity in this reaction. After 30 minutes, the reaction reached 30% conversion to 8 with 4, while 3 provided just 5% conversion and 2 reached 2% conversion. After just 1 h, 4 achieves 57% conversion. To reach the same conversion, 3 requires 9 h, while 2 never attains this level of conversion. A maximum of 70% conversion to 8 is achieved using 4, while 3 reaches a maximum of 62% conversion and 2 gives 51% conversion. With each of these catalysts, high E-selectivity (>99% E) was maintained throughout the course of the reaction.
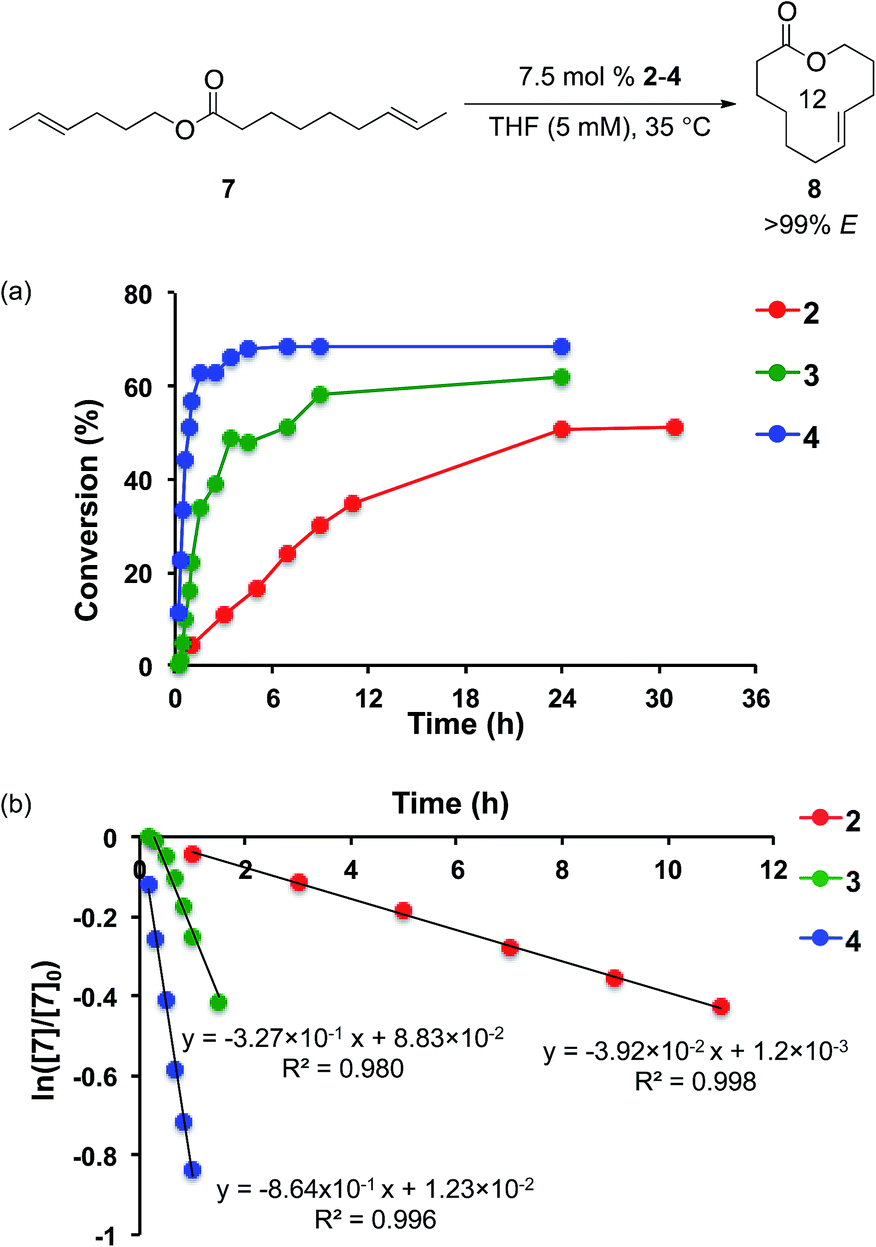 | ||
| Fig. 5 (a) Plot of conversion vs. time for RCM of 7 to 8 using catalysts 2–4. (b) Plot of ln([7]/[7]0) vs. time for this reaction. Conversion determined by 1H NMR. Selectivity determined using gas chromatography. | ||
Assuming first-order kinetics with respect to diene 7, rate constants were measured for catalysts 2, 3, and 4 under these reaction conditions and were determined to be 3.92 × 10−2 s−1, 3.27 × 10−1 s−1, and 8.64 × 10−1 s−1, respectively (Fig. 5b). The relative rate constants hence have values of krel4 = kobs4/kobs2 = 22.0 and krel3 = kobs3/kobs2 = 8.46. Given the previously observed low reactivity of E-olefins with these catalysts,6,8 these large disparities in activity highlight the difference in the ability of each of these catalysts to provide significant yields of products.
An array of diene substrates bearing two E-olefins were synthesized from internal alkyne starting materials which could be reduced by Li/NH3 or LiAlH4 (Scheme 2). Subsequent Jones oxidation was used to generate the carboxylate moiety of the desired ester product. EDC coupling of this carboxylic acid with the corresponding alcohol gives the substrate in a scalable synthesis.
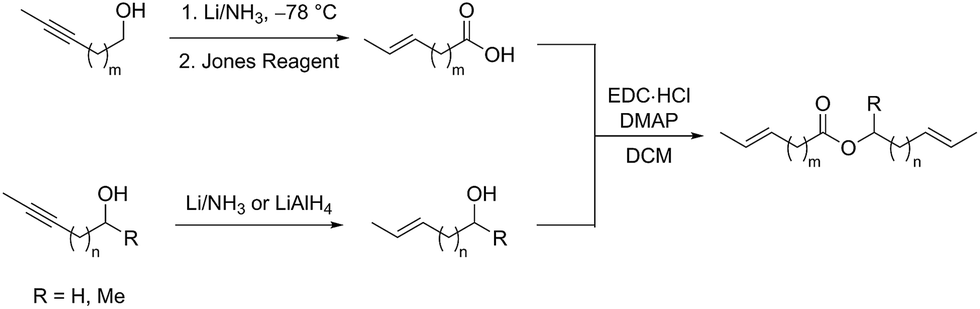 | ||
| Scheme 2 Synthesis of diene substrates bearing two E-olefins. | ||
Using these substrates, a variety of macrocyclic lactones were synthesized, ranging in size from 12- to 18-membered rings (Table 1). Each of these macrocycles was generated with the concomitant loss of gaseous trans-2-butene, which could be easily removed from the reaction mixture. Using catalyst 2, products were obtained in moderate to good yields (47–66%) and with high E-selectivity (>99% E) in 24 h at 35 °C. Much shorter reaction times could be achieved using 4, which provided these macrocycle products in 5 h in good to high yields (60%–80%) while high E-selectivity was maintained (>99% E). Using 4, the antibiotic recifeiolide 9 was synthesized in 80% yield with >99% E-selectivity in 8 h. Longer reaction times were likely required for this reaction due to steric encumbrance of the methyl group in the starting material.
| a Yields shown are isolated yields. Stereoselectivity determined by gas chromatography. |
|---|
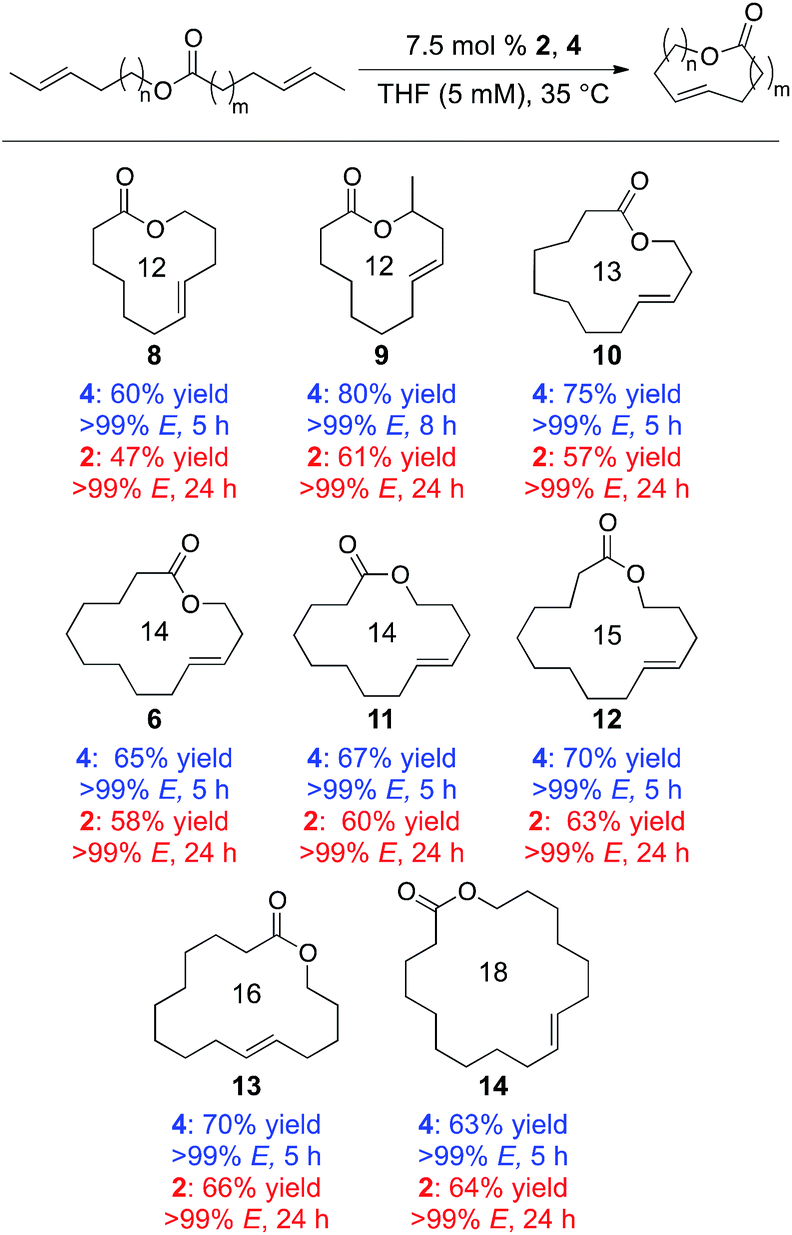
|
Conclusions
We have demonstrated that stereoretentive Ru catalysts supported by dithiolate ligands can be used in the synthesis of E-macrocycles with exceptional selectivity (>99% E) from diene starting materials bearing two E-olefins. Catalyst 4 delivers meaningful yields of macrocyclic products in appreciably shorter reaction times than other stereoretentive Ru catalysts 2 and 3 as evidenced by kinetic studies. Using this method, 12- to 18-membered macrocycles including recifeiolide were synthesized in moderate to high yield (47–80% yield).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Office of Naval Research (N00014-14-1-0650), NIH (GM031332), and NSF (CHE-1502616). T. S. A. recognizes NSF for support through a Graduate Research Fellowship. T. P. M. would like to acknowledge the Arnold and Mabel Beckman Foundation for funding. The authors would like to thank W. J. Wolf, C. Xu, C. W. Lee, J. Li, and N. F. Nathel for helpful suggestions. N. Torian and M. Shahgholi are thanked for their help with high-resolution mass spectrometry. Materia, Inc. is acknowledged for generous donations of Ru catalyst precursors.Notes and references
-
Handbook of Metathesis, ed. Grubbs, R. H., Wenzel, A. G., O'Leary, D. J. and Khosravi, E., Wiley-VCH, Weinheim, 2015 Search PubMed
.
-
(a) A. Michrowska, P. Wawrzyniak and K. Grela, Eur. J. Org. Chem., 2004, 2053 CrossRef CAS
- V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94 CrossRef CAS PubMed
- M. Yu, C. Wang, A. F. Kyle, P. Jakubec, D. J. Dixon, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 479, 88 CrossRef CAS PubMed
- M. J. Koh, R. K. M. Khan, S. Torker, M. Yu, M. S. Mikus and A. H. Hoveyda, Nature, 2015, 517, 181 CrossRef CAS PubMed
-
(a) A. M. Johns, T. S. Ahmed, B. W. Jackson, R. H. Grubbs and R. L. Pederson, Org. Lett., 2016, 18, 772 CrossRef CAS PubMed
- T. T. Nguyen, M. J. Koh, X. Shen, F. Romiti, R. R. Shrock and A. H. Hoveyda, Science, 2016, 352, 569 CrossRef CAS PubMed
- T. S. Ahmed and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 1532 CrossRef CAS PubMed
- X. Shen, T. T. Nguyen, M. J. Koh, D. Xu, A. W. H. Speed, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 541, 380 CrossRef CAS PubMed
- T. S. Ahmed and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 11213 CrossRef CAS PubMed
- C. Xu, X. Shen and A. H. Hoveyda, J. Am. Chem. Soc., 2017, 139, 10919 CrossRef CAS PubMed
- K. Radkowski, B. Sundararaju and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 355 CrossRef CAS PubMed
- High catalyst loadings are often used in macrocyclization reactions because of the dilute conditions necessary for preventing oligomerization. For examples see ref. 3, 4, and 9.
- This result was in agreement with previous studies showing that cross metathesis between E-olefins and terminal olefins using these catalysts is challenging (ref. 6 and 8).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00435h |
| This journal is © The Royal Society of Chemistry 2018 |