
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective bromination of sp3 C–H bonds†
Shyam
Sathyamoorthi
 a,
Shibdas
Banerjee
b,
J.
Du Bois
*a,
Noah Z.
Burns
*a and
Richard N.
Zare
*a
a,
Shibdas
Banerjee
b,
J.
Du Bois
*a,
Noah Z.
Burns
*a and
Richard N.
Zare
*a
aStanford University, Department of Chemistry, 333 Campus Drive, Stanford, CA 94305-4401, USA. E-mail: zare@stanford.edu; jdubois@stanford.edu; nburns@stanford.edu
bIndian Institute of Science Education and Research Tirupati, Karakambadi Road, Tirupati-517507, India
First published on 20th November 2017
Abstract
A method for converting sp3 C–H to C–Br bonds using an N-methyl sulfamate directing group is described. The reaction employs Rh2(oct)4 and a mixture of NaBr and NaOCl and is performed in aqueous solution open to air. For all sulfamates examined, oxidation occurs with high selectivity at the γ-carbon, affording a uniquely predictable method for C–H bond halogenation. Results from a series of mechanistic experiments suggest that substrate oxidation likely proceeds by a radical chain process. Initial formation of an N-halogenated sulfamate followed by Rh-mediated homolysis generates an N-centered radical, which serves as the active oxidant.
Introduction
Oxidation of a specific sp3 C–H bond in a complex molecule remains an outstanding challenge in reaction methods development.1–3 While several protocols for the selective conversion of sp3 C–H centers to C–N and C–O bonds are now available,4–16 fewer methods for the synthesis of C–halogen bonds17–36 have been described despite the fact that molecules bearing halogen functional groups are prevalent in nature. In addition, as alkyl halides are versatile precursors for a variety of synthetic transformations, including cross-coupling, substitution, elimination, and the installation of boron-, silicon-, nitrogen-, and oxygen-based groups, methods for accessing these types of materials have value in synthesis.37–41 Here, we describe a reaction protocol for the site-selective bromination of sp3 C–H bonds using an N-methyl sulfamate directing group. This auxiliary is facile to install on 1° and 2° alcohol derivatives and can be removed through nucleophilic displacement. Mechanistic studies suggest that the reaction proceeds through an N-centered radical, reminiscent of the Hoffman–Löffler–Freytag amine synthesis.42–55 With all sulfamates tested, oxidation occurs preferentially at the γ-carbon, offering a predictable and precise method for oxidative C–H bond halogenation under mild reaction conditions.Results and discussion
Initial optimization of conditions for alkylbromide formation was performed with isopentyl methylsulfamate 1, a simple, unfunctionalized substrate with a single tertiary C–H bond. A variety of transition metal salts were tested in conjunction with 3 equivalents each of NaBr and NaOCl, a reagent combination known to generate hypobromite in situ and used previously for the oxidative cyclization of sulfamate esters.56 While little to no reaction ensued in the presence of catalytic Mn3+, Co2+, Cu2+, and Ni2+ (Table 1, entries 1–4), switching to Rh2(oct)4 (Table 1, entry 5) afforded a marked increase in conversion to brominated product 2. The choice of dirhodium complex had a clear influence on reaction performance, as catalysts bearing hydrophobic ligands such as octanoate or triphenylacetate (Table 1, entries 5–7) out-performed others tested (Table 1, entries 8, 9), likely owing to the greater solubility of these complexes in dichloromethane. In the complete absence of transition metal, conversion to product still occurred (Table 1, entry 10) in a process that appears to be promoted by ambient light (Table 1, entry 11).A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 biphasic mixture of CH2Cl2/saturated aqueous Na2HPO4 was found to be the optimal solvent combination (Table 1, entry 5). In neat CH2Cl2, conversion to product was significantly reduced, and in unbuffered water with CH2Cl2 a mixture of both brominated and chlorinated products was obtained (Table 1, entries 12 and 13). Using organic co-solvents other than CH2Cl2 was similarly deleterious to reaction performance (Table 1, entries 14 and 15).
:1 biphasic mixture of CH2Cl2/saturated aqueous Na2HPO4 was found to be the optimal solvent combination (Table 1, entry 5). In neat CH2Cl2, conversion to product was significantly reduced, and in unbuffered water with CH2Cl2 a mixture of both brominated and chlorinated products was obtained (Table 1, entries 12 and 13). Using organic co-solvents other than CH2Cl2 was similarly deleterious to reaction performance (Table 1, entries 14 and 15).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solventa | [2]/[1]b |
| a All reactions were performed in a biphasic solvent mixture with the indicated solvent and an equivalent volume of saturated aqueous Na2HPO4 unless otherwise noted. b Product ratio determined by 1H NMR integration, see ESI for details. c Reaction flask wrapped in foil. d Reaction performed with no added co-solvent. e Reaction conducted with an equivalent volume of deionized H2O. f A small amount of the corresponding chloride product is also formed. RSM = recovered starting material. | |||
| 1 | (R,R)-Mn-Jacobsen (5%) | CH2Cl2 | RSM |
| 2 | Co(OAc)2·4H2O (5%) | CH2Cl2 | 1/8 |
| 3 | CuBr2 (5%) | CH2Cl2 | 1/8 |
| 4 | Ni(OAc)2·4H2O (5%) | CH2Cl2 | RSM |
| 5 | Rh 2 (oct) 4 (5%) | CH 2 Cl 2 | 4/1 |
| 6 | Rh2(O2CtBu)4 (5%) | CH2Cl2 | 2/1 |
| 7 | Rh 2 (O 2 CCPh 3 ) 4 (5%) | CH 2 Cl 2 | 4/1 |
| 8 | Rh2(OAc)4 (5%) | CH2Cl2 | 1/5 |
| 9 | Na4Rh2(CO3)4 (5%) | CH2Cl2 | 1/7 |
| 10 | None | CH2Cl2 | 1/4 |
| 11 | Nonec | CH2Cl2 | RSM |
| 12 | Rh2(oct)4 (5%) | CH2Cl2d | 1/3 |
| 13 | Rh2(oct)4 (5%) | CH2Cl2e | 1/2f |
| 14 | Rh2(oct)4 (5%) | iPrOAc | 1/2 |
| 15 | Rh2(oct)4 (5%) | Benzene | 1/2 |
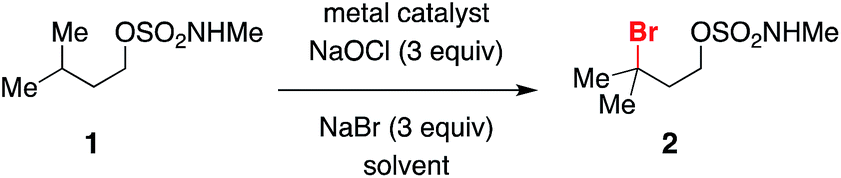
A range of structurally diverse N-methyl sulfamates has been prepared by condensation of the corresponding 1° and 2° alcohols with ClSO2NHMe and subjected to the optimized halogenation protocol (Table 2). Oxidation of both tertiary and benzylic C–H bonds is possible in moderate to good yields, even with Rh2(oct)4 loadings as low as 0.1 mol% (entry 2). Different functional groups, including alkyl and benzyl esters, epoxides, trichloroethylsulfamate-protected aziridines, and silylated alcohols, are compatible with these conditions (Table 2, entries 3–6). For all substrates examined, C–H bond oxidation occurs nearly exclusively at the γ-carbon. This finding compares favorably with other directed C–H halogenation methods, which afford mixtures of constitutional isomers.15,20 The directed nature of this process is further highlighted in entries 8, 9, and 10. Each of these substrates furnishes the product of γ-C–H bond bromination despite possessing an activated benzylic C–H center. Positional selectivity is also noted in entries 11 and 12. Experiments with the latter substrate show that oxidation of an optically active 3° C–H bond gives racemic alkylbromide, a result consistent with the formation of a carbon-centered radical intermediate (vide infra). In addition, we have found that the absence of NaBr leads to generation of the corresponding chlorinated product (entry 12), albeit in reduced yield. All told, this new protocol offers an efficient, predictable, operationally simple method for C–H bond functionalization.
|
a Isolated product yield unless otherwise indicated.
b Reaction performed with 0.1 mol% Rh2(oct)4.
c Yield estimated by 1H NMR integration using an internal standard.
d Product isolated as a 1:1 mixture of diastereomers.
e Product isolated as a mixture of diastereomers, ratio undetermined.
f Product yield estimated by 1H NMR integration using an internal standard. Chromatography on SiO2 facilitates bromide elimination, see Fig. S1 for details.
g Product isolated as a racemic mixture, see Fig. S2 for details.
h Yield of corresponding chloride product obtained from a reaction performed without NaBr.
|
|||
|---|---|---|---|
|
Displacement of the N-methyl alkoxysulfonyl auxiliary can be achieved in a single-flask, two-step protocol that involves initial N-carbamoylation with Boc2O followed by an SN2 reaction (Scheme 1). The N-acylated sulfamate undergoes smooth reaction with nucleophiles such as N3− and I− to give the corresponding alkylazide and alkyliodide products, respectively. This method for excising the sulfamate directing group should add to the overall utility of the C–H halogenation process.
 | ||
| Scheme 1 Representative sulfamate displacement reactions. | ||
Previous work exploring the use of NaBr/NaOCl for the synthesis of [1,2,3]-oxathiazinane-2,2-dioxide heterocycles56 suggested the formation of an N-halogenated species as a first step in the reaction pathway. In accord with this hypothesis, we have demonstrated that the N,N-dimethyl sulfamate 3 is not a competent substrate for oxidation. Additionally, we have prepared an N-brominated sulfamate 5 and have shown that this compound will react with 5 mol% Rh2(oct)4 to form alkylbromide 6 in 40% yield (Scheme 2 and Fig. S3†). Although the efficiency of this process is reduced from that of the catalytic protocol (entry 1, Table 2), these findings establish the N-brominated species as a chemically competent intermediate on the reaction pathway.
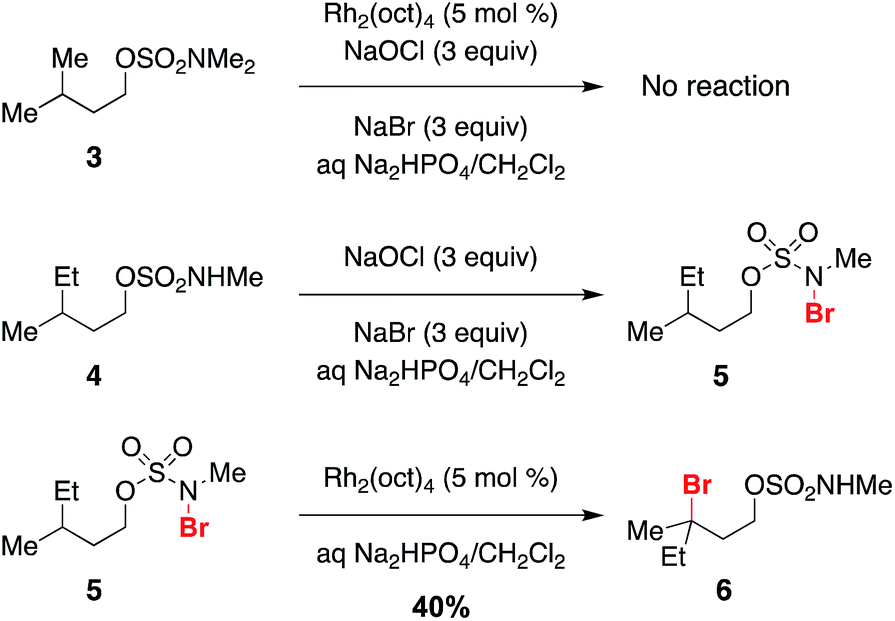 | ||
| Scheme 2 N-Bromo sulfamate 5 is a chemically competent intermediate. | ||
The ability to access N-brominated sulfamate 5 has enabled a series of experiments to determine the role of Rh2(oct)4 in the oxidation reaction. UV/Visible spectroscopic monitoring of the reaction of 5 with Rh2(oct)4 in CH2Cl2 reveals a distinct change in the absorption spectrum, evidenced by the disappearance of the feature at λmax = 418 nm, shifting of the λmax at 655 to 595 nm, and the appearance of a new λmax at 985 nm (Fig. 1a). The final absorption spectrum is indicative of a mixed-valent Rh2+/Rh3+ tetracarboxylate dimer,57,58 consistent with a mechanism involving one-electron reduction of the N–Br bond to generate an N-centered radical. Support for this conclusion has been obtained through electrospray ionization mass spectrometric (ESI-MS) analysis, which confirms the presence of both the Rh2+/Rh3+ complex and free Br− (Fig. 1b–c) resulting from this reaction.
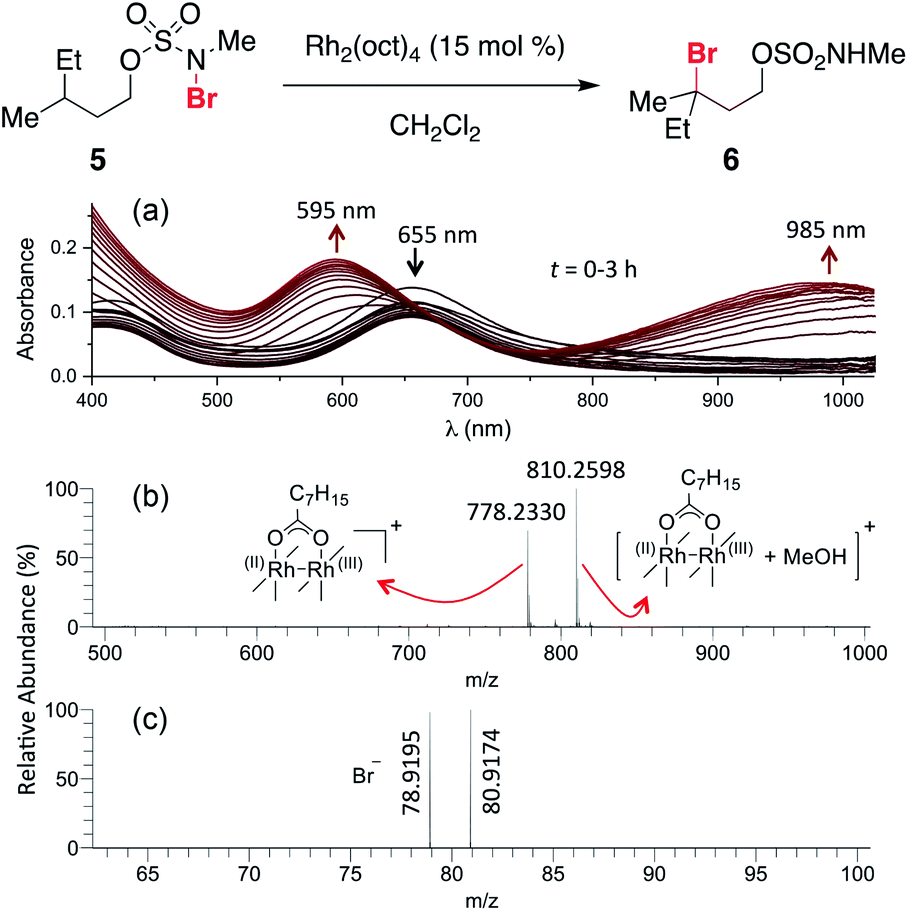 | ||
| Fig. 1 (a) Continuous UV/vis spectrophotometric monitoring of the reaction of 5 shows an absorption spectrum characteristic of the conversion of a dinuclear Rh2+/Rh2+ complex (λmax = 418 nm and 655 nm) to a Rh2+/Rh3+ complex (λmax = 595 nm and 985 nm). High-resolution ESI-MS detected ion signals of (b) Rh2+/Rh3+ complex in positive ion mode, and (c) Br− in negative ion mode; see ESI† for experimental details. | ||
In a reaction mixture containing 5 and Rh2(oct)4, the red color ascribed to the mixed-valent dirhodium species persists for several hours. Under standard catalytic reaction conditions, however, the deep green color of intact Rh2(oct)4 bleaches to pale yellow within 30 min following NaOCl addition. A UV/vis spectrum of the reaction mixture at this time point shows a featureless spectrum, consistent with decomposition of the rhodium dimer (Fig. S4a†). Interestingly, at 30 min, product conversion is only ∼30%, with starting material accounting for the remainder of the mass balance (Fig. S4b†). After the full reaction time (15 h), the isolated product yield is 61%. Thus, the reaction appears to proceed beyond the lifetime of Rh2(oct)4, suggesting its role as an initiator rather than as a catalyst for oxidative halogenation (Scheme 3). Accordingly, these data have led us to favor a mechanism for C–H bromination through a chain transfer process involving N- and C-centered radical intermediates, as depicted in Scheme 3. We cannot, however, discount the possibility that the intermediate carbon radical could also react with [Rh2(oct)4Br] to give the brominated product.
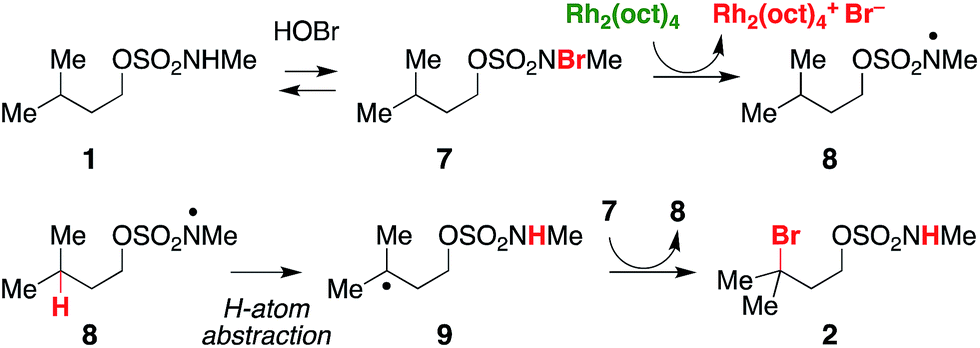 | ||
| Scheme 3 A proposed radical-chain transfer process for C–H bromination. | ||
To test for a radical chain mechanism, a 1:1 mixture of brominated sulfamate 5 and chlorinated sulfamate 10 was stirred with catalytic Rh2(oct)4. ESI-MS analysis of the reaction mixture at 2 h revealed brominated products 2 and 6 and chlorinated products 11 and 12 (Fig. 2 and S5†). Such a product distribution lends strong support to our mechanistic scheme, as only an intermolecular collision between intermediates derived from 5 and 10 could lead to cross-halogenated products 2 and 12.
 | ||
| Fig. 2 High-resolution ESI-MS analysis shows that cross-halogenated products form in a competition experiment between bromosulfamate 5 and chlorosulfamate 10. Experimental m/z values (in the spectrum) agree well with the theoretical m/z values (underneath the chemical structures). | ||
As a final piece of mechanistic insight, a kinetic isotope effect (KIE) of [PH]/[PD] = 3 (Scheme 4 and Fig. S6†) has been measured in a competition experiment between protio- and deutero-sulfamate substrates, 13 and 14. This result suggests that N-centered radical formation is not a committed, irreversible step. Reactions of 15 with a second N-halogenated sulfamate (or HOBr) to form 16 or with solvent to regenerate 13 are possible pathways that apparently compete with intramolecular γ-C–H abstraction, thus giving rise to a non-unitary KIE value in the competition experiment.59
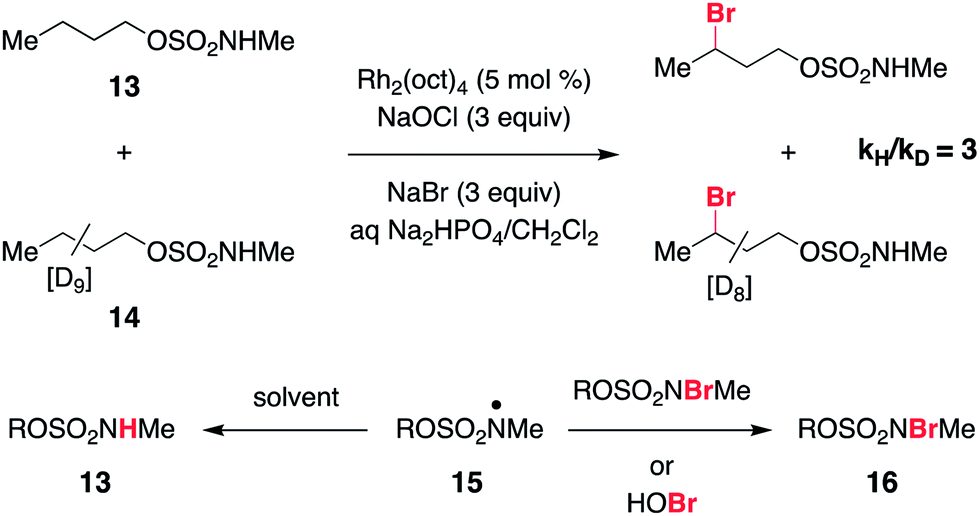 | ||
| Scheme 4 KIE study suggests reversibility of N-centered radical formation. | ||
Given a radical chain mechanism for C–H halogenation, it is possible that metal complexes other than Rh2(oct)4 could serve as initiators. We have found that treatment of 5 with a combination of 15 mol% CuBr2 and 1,10-phenanthroline forms the tertiary bromide product 6 in 31% yield (Scheme 5 and Fig. S7†).60 While the efficiency of this reaction is lower than that with Rh2(oct)4 (Scheme 2), formation of 6 suggests that, at least in principle, new reaction manifolds utilizing first-row transition metals can be optimized for the oxidative halogenation of sp3 C–H bonds.
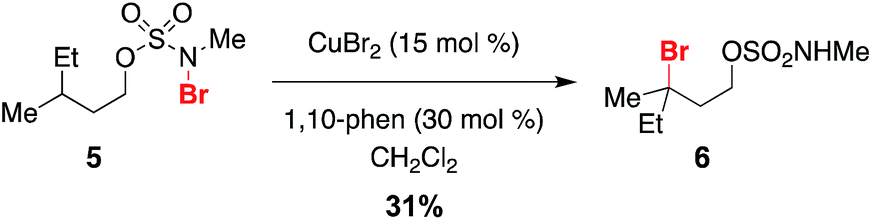 | ||
| Scheme 5 CuBr2/phenanthroline as an alternate metal complex for reaction initiation. | ||
Conclusions
A method for site-selective bromination of sp3 C–H bonds using N-methyl sulfamate substrates is presented. Following halogenation, the sulfamate directing group can be displaced with nucleophiles to generate value-added alkylbromide products. The scope and predictability of this oxidation reaction distinguish these findings. UV/visible spectroscopy, ESI-MS analysis, and substrate probe experiments implicate a radical chain mechanism for C–H halogenation, initiated by Rh2(oct)4. Further exploration of sulfamate directing groups in C–H functionalization catalysis is warranted and should lead to high-precision methods for modifying sp3 carbon centers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Will Keown for assisting with UV/visible measurements. This work has been supported by the NSF under the CCI Center for Selective C–H Functionalization (CHE-1205646) and by the Air Force Office of Scientific Research through Basic Research Initiative grant (AFOSR FA9550-16-1-0113).Notes and references
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- J. B. C. Mack, J. D. Gipson, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 9503–9506 CrossRef CAS PubMed.
- M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed.
- A. Company, J. Lloret, L. Gómez and M. Costas, in Alkane C–H Activation by Single-Site Metal Catalysis, ed. P. J. Pérez, Springer Netherlands, Dordrecht, 2012, pp. 143–228, DOI:10.1007/978-90-481-3698-8_5.
- J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed.
- S. Sathyamoorthi and J. Du Bois, Org. Lett., 2016, 18, 6308–6311 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- R. T. Gephart and T. H. Warren, Organometallics, 2012, 31, 7728–7752 CrossRef CAS.
- E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974–9978 CrossRef CAS PubMed.
- E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204–10207 CrossRef CAS PubMed.
- B. J. Stokes and T. G. Driver, Eur. J. Org. Chem., 2011, 4071–4088 CrossRef CAS.
- G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed.
- L. Gómez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720–5723 CrossRef PubMed.
- C. Kim, K. Chen, J. Kim and L. Que, J. Am. Chem. Soc., 1997, 119, 5964–5965 CrossRef CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed.
- C. L. Hill, J. A. Smegal and T. J. Henly, J. Org. Chem., 1983, 48, 3277–3281 CrossRef CAS.
- X. Yang, Y. Sun, T. Sun and Y. Rao, Chem. Commun., 2016, 52, 6423–6426 RSC.
- R. K. Rit, M. R. Yadav, K. Ghosh, M. Shankar and A. K. Sahoo, Org. Lett., 2014, 16, 5258–5261 CrossRef CAS PubMed.
- R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724–5727 CrossRef CAS PubMed.
- A. E. Shilov and A. A. Shteinman, Coord. Chem. Rev., 1977, 24, 97–143 CrossRef CAS.
- R. K. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 696–702 CrossRef CAS PubMed.
- G. I. Nikishin, E. I. Troyansky and M. I. Lazareva, Tetrahedron Lett., 1985, 26, 3743–3744 CrossRef CAS.
- J. Ozawa and M. Kanai, Org. Lett., 2017, 19, 1430–1433 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS PubMed.
- R. Kundu and Z. T. Ball, Org. Lett., 2010, 12, 2460–2463 CrossRef CAS PubMed.
- K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247–7249 CrossRef CAS PubMed.
- T. Liu, M. C. Myers and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306–309 CrossRef CAS PubMed.
- Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef CAS PubMed.
- L. R. Reddy, B. V. S. Reddy and E. J. Corey, Org. Lett., 2006, 8, 2819–2821 CrossRef CAS PubMed.
- (a) G. W. Gribble, in Naturally Occurring Organohalogen Compounds – A Comprehensive Update, ed. G. W. Gribble, Springer Vienna, Vienna, 2010, pp. 9–348 Search PubMed; (b) B. Gál, C. Bucher and N. Z. Burns, Mar. Drugs, 2016, 14, 206 CrossRef PubMed.
- K. Chen and P. S. Baran, Nature, 2009, 459, 824–828 CrossRef CAS PubMed.
- J. Richers, M. Heilmann, M. Drees and K. Tiefenbacher, Org. Lett., 2016, 18, 6472–6475 CrossRef CAS PubMed.
- L. Han, J.-B. Xia, L. You and C. Chen, Tetrahedron, 2017, 73, 3696–3701 CrossRef CAS PubMed.
- H.-Y. Xiong, D. Cahard, X. Pannecoucke and T. Besset, Eur. J. Org. Chem., 2016, 3625–3630 CrossRef CAS.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483–11498 CrossRef CAS.
- A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693–10697 CrossRef CAS PubMed.
- S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624–627 CrossRef CAS PubMed.
- X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed.
- C. K. Chu, Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 6404–6407 CrossRef CAS PubMed.
- D. M. Peacock, C. B. Roos and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 647–652 CrossRef CAS PubMed.
- M. E. Wolff, Chem. Rev., 1963, 63, 55–64 CrossRef CAS.
- C. Betancor, J. I. Concepcion, R. Hernandez, J. A. Salazar and E. Suarez, J. Org. Chem., 1983, 48, 4430–4432 CrossRef CAS.
- C. Q. O'Broin, P. Fernández, C. Martínez and K. Muñiz, Org. Lett., 2016, 18, 436–439 CrossRef PubMed.
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- D. H. R. Barton, A. L. J. Beckwith and A. Goosen, J. Chem. Soc., 1965, 181–190 RSC.
- A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1879, 12, 984–990 CrossRef.
- K. Löffler and C. Freytag, Ber. Dtsch. Chem. Ges., 1909, 42, 3427–3431 CrossRef.
- G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095–7129 CrossRef CAS.
- C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef PubMed.
- P. Becker, T. Duhamel, C. J. Stein, M. Reiher and K. Muñiz, Angew. Chem., Int. Ed., 2017, 56, 8004–8008 CrossRef CAS PubMed.
- R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994–8997 CrossRef CAS PubMed.
- D. Šakić and H. Zipse, Adv. Synth. Catal., 2016, 358, 3983–3991 CrossRef.
- M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201710322.
- D. N. Zalatan and J. Du Bois, Synlett, 2009, 143–146 CAS.
- C. R. Wilson and H. Taube, Inorg. Chem., 1975, 14, 2276–2279 CrossRef CAS.
- D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 7558–7559 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- The combination of CuBr and 1,10-phenanthroline also promotes conversion of 5 to 6 (Fig. S8†).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04611a |
| This journal is © The Royal Society of Chemistry 2018 |