
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production of renewable long-chained cycloalkanes from biomass-derived furfurals and cyclic ketones†
Qiying Liu *a,
Caihong Zhangab,
Ning Shiab,
Xinghua Zhang*a,
Chenguang Wanga and
Longlong Maa
*a,
Caihong Zhangab,
Ning Shiab,
Xinghua Zhang*a,
Chenguang Wanga and
Longlong Maa
aCAS Key Laboratory of Renewable Energy, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou, 510650, P. R. China. E-mail: liuqy@ms.giec.ac.cn; zhangxh@ms.giec.ac.cn; Fax: +86-20-87057737; Tel: +86-20-87057751 Tel: +86-20-87057790
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 12th April 2018
Abstract
Developing renewable long-chain cycloalkanes from lignocellulosic biomass is of significance because it offers huge resource storage, wide applications in aviation/diesel fuels and mitigation of CO2 emissions. In this paper, cycloalkanes with carbon chain lengths of 13–18 were produced from biomass-derived furfural species (furfural and 5-hydroxymethylfurfural) and cyclic ketones (cyclopentanone and cyclohexanone) via aldol condensation, followed by hydrogenation to saturate the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds, and hydrodeoxygenation to remove oxygen atoms. The aldol condensation of the furfural species with cyclic ketones was catalyzed by NaOH and the target condensation intermediates were obtained in yields of more than 90% at room temperature (30 °C) with a short reaction time (40 min). By using amorphous zirconium phosphate combined with Pd/C as the catalyst, liquid cycloalkanes were produced at the optimal conditions with a yield of 76%. When the combined solid catalyst was reused, the target products reduced after the second run but the initial yield could be largely recovered by recalcination of the spent zirconium phosphate. Considering that cyclopentanone and cyclohexanone can be easily produced from furfural (originating from hemicellulose) and phenol (originating from lignin), respectively, this condensation has the potential to achieve the integrated conversion of biomass-derived cellulose, hemicellulose and lignin to jet fuel and/or diesel additives.
C and CO bonds, and hydrodeoxygenation to remove oxygen atoms. The aldol condensation of the furfural species with cyclic ketones was catalyzed by NaOH and the target condensation intermediates were obtained in yields of more than 90% at room temperature (30 °C) with a short reaction time (40 min). By using amorphous zirconium phosphate combined with Pd/C as the catalyst, liquid cycloalkanes were produced at the optimal conditions with a yield of 76%. When the combined solid catalyst was reused, the target products reduced after the second run but the initial yield could be largely recovered by recalcination of the spent zirconium phosphate. Considering that cyclopentanone and cyclohexanone can be easily produced from furfural (originating from hemicellulose) and phenol (originating from lignin), respectively, this condensation has the potential to achieve the integrated conversion of biomass-derived cellulose, hemicellulose and lignin to jet fuel and/or diesel additives.
1. Introduction
Lignocellulosic biomass is a renewable resource with huge storage, wide distribution and fast carbon cycles during biomass growth and utilization. Biomass-derived fuels and chemicals are regarded as an alternative to fossil energy, and their development has obvious significance for environmental protection and reduced dependence on fossil energy. Therefore, converting lignocellulosic biomass into fuels has attracted worldwide attention during recent decades.1–4Cellulose, hemicellulose and lignin are the three main components of a typical lignocellulosic biomass, however, the monosaccharide units (typically, glucose and xylose) and derivatives (5-hydroxymethylfurfural (HMF) and furfural (FF)) are limited to carbon chains of length 6, while the phenolic units (typically, phenol-, guaiacol- and syringyl-based primary structures) are limited to carbon chains of length 9. This results in the fuels from direct hydrodeoxygenation of these platforms having short carbon chains.5,6 To synthesize long-chained alkanes with carbon numbers of more than 10, for jet fuel and diesel use, condensations between suitable biomass-derived platforms with special groups are necessary to extend the carbon chains.6–9 Typically, the production of long-chained alkanes involves three steps: depolymerization of the lignocellulosic biomass into oxygenated platforms (FF, HMF, levulinic acid, γ-valerolactone and 2-methylfuran et al.); fuel precursor formation by condensation; and hydrodeoxygenation of the fuel precursors.9,10 Herein, the second step of platform condensation is the key to obtain structurally diversified fuel precursors; the desired platforms and condensation methods must be chosen in order to produce versatile hydrocarbon fuels by hydrodeoxygenation.11 Aldol condensations of FF/HMF and/or their derivatives with carbonyl groups like acetone,12–16 hydroxyacetone,17 butaldehyde,18 2-butanone,19 methyl isobutyl ketone,20 levulinic acid11,21,22 and angelica lactone23 have commonly been conducted to obtain the C9–C16 fuel precursors, but these carbonyl compounds originate from fossil resources or are produced from biomass by multi-step reactions, leading to low efficiency and high cost for the entire synthesis. As an alternative, hydroxyalkylation-alkylation of FF and 2-methylfuran obtained by hydrodeoxygenation of FF was reported to produce C15 fuel precursors with a branched carbon framework.24–27 Despite the fact that the precursor could be converted into branched long-chain hydrocarbons, the main drawback was that only hemicellulose (the least abundant component in a typical lignocellulosic biomass) could be utilized. Interestingly, alternative routes including direct C–C bond formation by condensation of monosaccharides/furfurals,28 α-alkylation of ketones with 1-butanol,29 self-coupling by two HMF molecules30 and oligomerization of angelica lactone31,32 were used to produce C10–C15 precursors with high yields, and hydrodeoxygenation to the relative long-chained hydrocarbons was then conducted over supported metal catalysts. Instead of the common furfurals, these routes extended the platform molecules for target hydrocarbons by using diversified structures.
Compared to branched and normal hydrocarbons with long chains, cyclic counterparts show particular advantages in view of their low freezing points and high energy densities, which are preferential for drop-in jet and/or diesel fuels.33 Carbonyl-containing HMF and FF are the two main platforms which can be easily produced from biomass through a hydrolysis–dehydration process. On the other hand, cyclopentanone (CP) and cyclohexanone (CH) are two important cyclic ketones that have special carbons with α-Hs. In particular, CP could be selectively produced by hydrogenation-rearrangement of hemicellulose-derived FF,34,35 and CH could be prepared with very high yield from lignin-derived phenol.36,37 Thus, production of cyclic hydrocarbons with long chains from these furfural species and cyclic ketones are attractive for the integrated utilization of biomass.38–40
Here, we developed a new route for the production of cycloalkanes with carbon chain lengths of 13–18 from FF/HMF and CP/CH (Scheme 1). Aldol condensations of these furfural species with cyclic ketones (CP and CH) were first conducted over a base catalyst to produce the oxygenated precursors with cyclic structures. Then these precursors were hydrogenated to thermally stable intermediates by saturating the CC and CO bonds over a metal catalyst. Finally they were transformed to the long-chained cycloalkanes by using a catalyst of metal combined with solid acid. The reaction parameters of each step were screened and the pathway for long-chained cycloalkane formation is discussed.
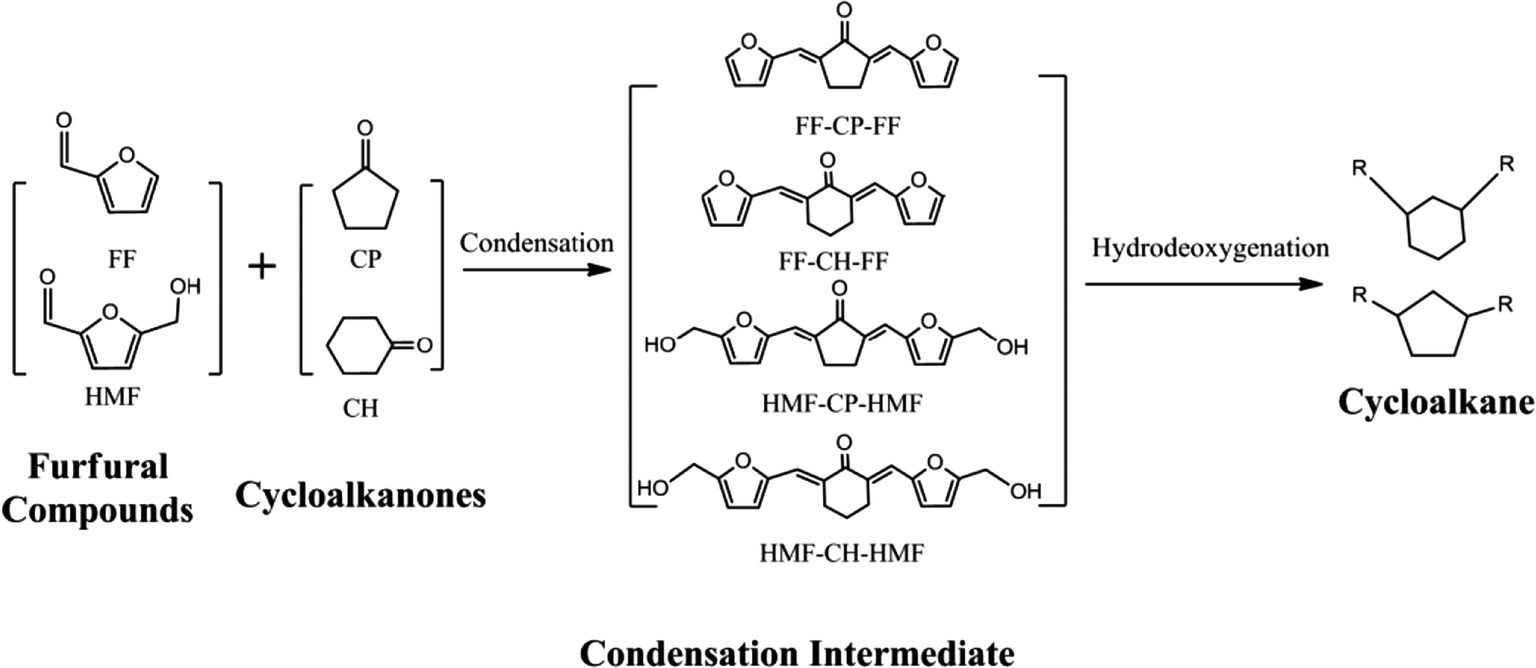 | ||
| Scheme 1 Routes for long-chained cycloalkane production from furfural species and cyclic ketones. | ||
2. Experimental section
2.1. Materials
FF (99%), HMF (98%), CP (99%), CH (99%), ZrOCl2·8H2O, Pt/C (5 wt%), Pd/C (5 wt%) and Ru/C (5 wt%) were purchased from Shanghai Crystal Pure Reagent Co., Ltd., China. Analytical grade NaOH, KOH, Ba(OH)2, LiOH, K2CO3, Na2CO3, Mg(OH)2, H3PO4 and NH4H2PO4 were purchased from Tianjin Fuchen Chemical Reagent Factory, China. Nb2O5·H2O was purchased from the Conghua Tantalum Niobium Factory (Guangzhou, China). All these chemicals and reagents were used without further purification.2.2. Catalyst preparation
2.3. Catalyst characterization
X-ray powder diffraction (XRD) patterns of catalysts were obtained using an X-ray diffractometer (X'Pert Pro MPD, Philip) with Cu Kα radiation (λ = 0.154 nm) operated at 40 kV and 100 mA.The Brunauer–Emmett–Teller (BET) specific surface areas were obtained from N2 isothermal adsorption–desorption measurements at 77 K using a QUADRASORB SI-MP-10/PoreMaster analyzer equipped with QuadraWin software. The mesoporous volumes and pore size distributions were calculated by the desorption branch using the Barrett–Joyner–Halenda (BJH) method.
Ammonia-temperature programmed desorption (NH3-TPD) was conducted in a U-tube quartz reactor (inner diameter 0.6 cm) using an ASIQACIV200-2 automatic physical/chemical adsorption analyzer (Quantachrome, US). Catalyst (0.15 g) was loaded into the U-tube quartz reactor and heated to 400 °C for 0.5 h (heating ramp of 10 °C min−1) under 30 mL min−1 of He flow. The system was cooled to 50 °C, NH3 was adsorbed onto the catalyst for 0.5 h until the saturation state was obtained and then the catalyst was flushed with a He flow of 30 mL min−1 to remove the physically adsorbed NH3. The adsorption and removal processes were monitored by the TCD signals. Then the system was heated to 825 °C with a heating ramp of 10 °C min−1 under the same He flow. The online MS (AMETEK DYCOR LC-D200, US) was used to determine the effluent NH3 desorbed from the catalyst. The quantitative analysis of acidic sites was performed using a calibration loop of 250 μL in which 5% NH3-95% Ar mixed gas was used for the single point calibration.
Transmission electron microscopy (TEM) images were gained on a Gatan Ultra scan camera using a JEOL JEM-2100F instrument operated at 200 kV. The samples were ultrasonically dispersed in ethanol, and drops of each suspension were placed on carbon-coated copper grids and then dried in air.
Scanning electron microscopy (SEM) images were recorded using an S-4800 instrument operated at 2 kV. The samples were placed on a conductive carbon tape adhered to an aluminium sample holder.
Fourier transform infrared (FT-IR) spectra were recorded on a Bruker TENSOR27 spectrometer with a resolution of 4 cm−1. The samples were pelleted with KBr before measurement.
The Pd dispersion in Pd/C was tested by H2-chemisorption using a Quantachrome-ASIQACIV200-2 automated gas sorption analyzer. The catalyst (0.2 g) was treated at 300 °C with a heating ramp of 5 °C min−1 under a hydrogen atmosphere (30 mL min−1). To clean the catalyst surface, the catalyst was further heated to 350 °C for 0.5 h under nitrogen (30 mL min−1), and then cooled to 50 °C for the H2-chemisorption test.
2.4. Aldol condensation of furfural species with cyclic ketones
Aldol condensation of furfural species and cyclic ketones was conducted in a 250 mL conical flask under atmospheric pressure. Using FF and CP as example, 2 mmol FF and 1 mmol CP were introduced into the flask, followed by the addition of 15 mL H2O and a certain amount of basic catalyst. The condensation was conducted at 30 °C for 40–120 min in a water bath with vigorous stirring (800 rpm). After the reaction, the solid product was filtrated, fully washed with a 5% aqueous ethanol solution and dried in a freeze dryer for 48 h. A yellowish solid was finally obtained and was labeled as FF-CP-FF. The condensations of FF and CH, HMF and CP and HMF and CH were carried out by the procedure mentioned above, and the solid intermediates were labeled as FF-CH-FF, HMF-CP-HMF and HMF-CH-HMF, respectively.The structures of the aldol condensation intermediates were identified using 13C NMR analysis (Bruker BioSpin GmbH) using CDCl3 or DMSO as the solvent.
The yield of each condensation intermediate was calculated by the following equation:
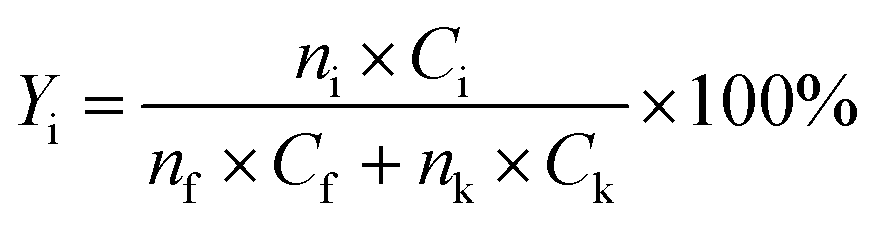 | (1) |
2.5. Hydrodeoxygenation of condensation intermediates to cycloalkanes
The hydrodeoxygenation of the condensation intermediates was carried out in a 50 mL stainless steel reactor with a mechanical stirrer. The condensation intermediate (1 mmol), 15 mL H2O and a certain amount of catalyst (solid acid and metal) were added to the reactor. After purging five times with H2, the reaction system was pressured to 4 MPa with H2 (room temperature). Then the reactor was heated to the target temperature and maintained at that temperature for the set time under vigorous stirring (800 rpm). After the reaction, the reactor was cooled to room temperature, and 15 mL ethyl acetate was used to extract the organic products three times. The organic layer was analyzed by gas chromatography (GC, GC-2014C, Shimadzu, Japan) and gas chromatography-mass spectrometry (GC-MS, Agilent 7890, US). For calculating the conversion percentage, the solid residue was collected by filtration, further rinsed with deionized water, collected by filtration again and dried in a freeze dryer for 48 h.For reuse, the combined catalyst (ZrP and Pd/C) was recovered by centrifugation from the reaction mixture, thoroughly rinsed with deionized water and directly used for the next cycle. Considering that the components of the combined catalyst were hard to separate from each other, to ascertain which catalyst led to deactivation, comparative experiments were conducted by hydrothermal treatment of ZrP and Pd/C at 300 °C, followed by either recalcination at 400 °C in static air (for ZrP) or 300 °C in 30 mL min−1 of H2 flow (for Pd/C); heating ramps of 10 °C min−1 were used for both. For monitoring the saturation of CC and CO bonds by hydrogenation in the solid intermediates, the sole Pd/C catalyst was used at relatively low temperatures according to the hydrodeoxygenation process. The structures of these hydrogenation intermediates were identified using 13C NMR analysis (Bruker BioSpin GmbH) using CDCl3 or DMSO as the solvent.
The quantitative analysis of the extracted liquid products was determined by GC (SHIMADZU 2014C) with a flame ionization detector (FID) and a HP-INNOWAX column (30 m × 0.25 mm × 0.25 μm). The oven temperature was programmed initially at 60 °C for 2 min, and then ramped up to 260 °C at a heating rate of 10 °C min−1 and held at this temperature for another 10 min. The injector was kept at 280 °C and used in splitting mode (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with helium as the carrier gas. Tridecane was used as the internal standard. The qualitative analysis of liquid products was carried out by GC-MS (Agilent 7890, US) used with the same capillary column and analytical conditions to those of the GC analysis. The gas products (CH4, C2H6 and C3H8, etc.) were collected with a sampling bag and analyzed by a GC 9800 chromatography tester equipped with a FID and a packed column (Porapak-Q column, 3 m × 3 mm). The quantification of gas products was conducted using a typical external standard.
:1) with helium as the carrier gas. Tridecane was used as the internal standard. The qualitative analysis of liquid products was carried out by GC-MS (Agilent 7890, US) used with the same capillary column and analytical conditions to those of the GC analysis. The gas products (CH4, C2H6 and C3H8, etc.) were collected with a sampling bag and analyzed by a GC 9800 chromatography tester equipped with a FID and a packed column (Porapak-Q column, 3 m × 3 mm). The quantification of gas products was conducted using a typical external standard.
The conversion of intermediates and yield of liquid product was calculated by the following equations:
| Conversion (%) = mintermediate residue/mintermediate loaded × 100% | (2) |
| mintermediate residue = msolid residue − mcatalyst | (3) |
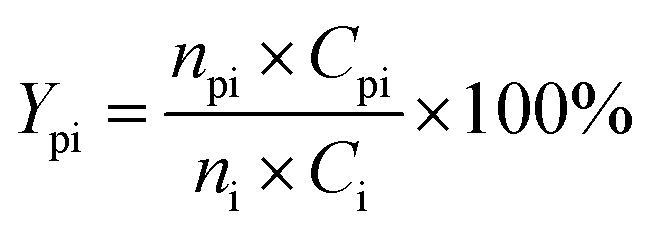 | (4) |
The turnover frequency (TOF) of the Pd/C based catalysts was determined by using the following equation:
| TOF (s−1) = converted FF-CP-FF in mmol/((exposed Pd atom) × (acid in mmol) × second) | (5) |
3. Results and discussion
3.1. Catalyst characterization
Fig. 1 shows the XRD patterns of freshly prepared NbP, ZrP and Pd/C catalysts. The obvious diffractions at 2θ = 19.8°, 28.0°, 40.2°, 44.9°, 57.8° and 65.1° indicate the crystalline nature of NbP, consistent with the previous report.41 On the other hand, the very weak and broadened diffractions observed in the range of 15–70° for ZrP, show that ZrP presents an amorphous nature. For Pd/C, peaks at 2θ = 40.1°, 43.9° and 68.3° were obtained but the intensities were weak, implying that the Pd particles are tiny and highly dispersed on the activated carbon surface.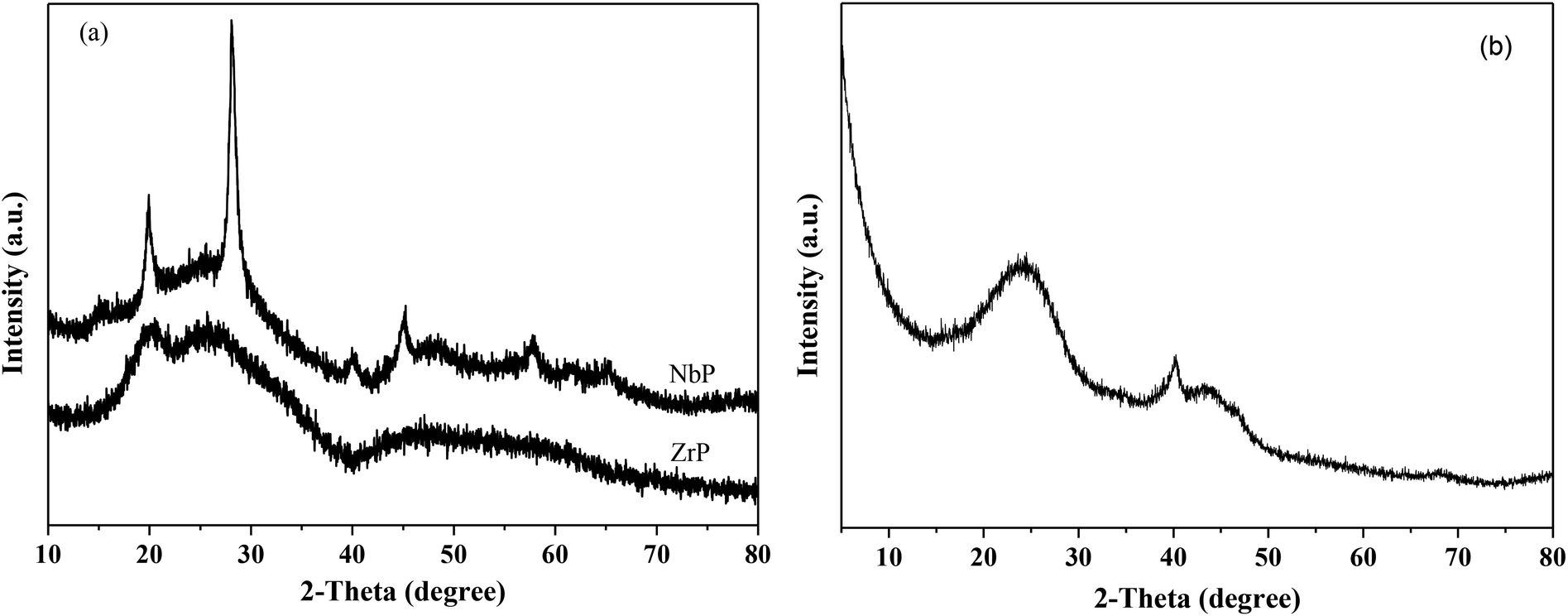 | ||
| Fig. 1 XRD patterns of fresh ZrP and NbP (a), and fresh Pd/C (b). | ||
The SEM images of NbP and ZrP, the TEM images of Pd/C and ZrP, and the N2 adsorption–desorption curve of ZrP are shown in Fig. 2. Based on the SEM analysis, the particle size of the NbP was in the range of 10–20 nm, and that of ZrP was about 20 nm. The particle size of ZrP was consistent with the results obtained from TEM measurement. For Pd/C, the Pd particles were observed and their size was in the range of 2–3 nm, which is consistent with the result of XRD. As shown in Fig. 2e and Table 1, the surface area, pore capacity and diameter of ZrP were 112 m2 g−1, 0.25 mL and 6.8 nm, respectively, which are larger than those of NbP aside from the pore diameter. Both NbP and ZrP showed similar acidity, and both weak acidic sites (observed from NH3 desorption at temperatures below 350 °C) and strong acidic sites (observed from NH3 desorption at temperatures higher than 450 °C) were simultaneously presented on the two catalysts' surfaces (Fig. 3). As compared with NbP (0.58 mmol g−1), ZrP presented a much higher acid density of 1.55 mmol g−1 (Table 1). The Pd/C showed a large surface area of more than 1000 m2 g−1 with significant numbers of mesopores. Meanwhile, the Pd dispersion was as high as 0.49 according to the H2-chemisorption analysis. After hydrothermal treatment and recalcination, the surface area and acidic sites of the used ZrP were reduced, but more than 80% of the original values were still retained while the pore volume and diameter changed a little. The surface area of the used Pd/C was 90% of that of the fresh catalyst. The Pd dispersion decreased to 0.36, although high dispersion still remained during the hydrothermal treatment.
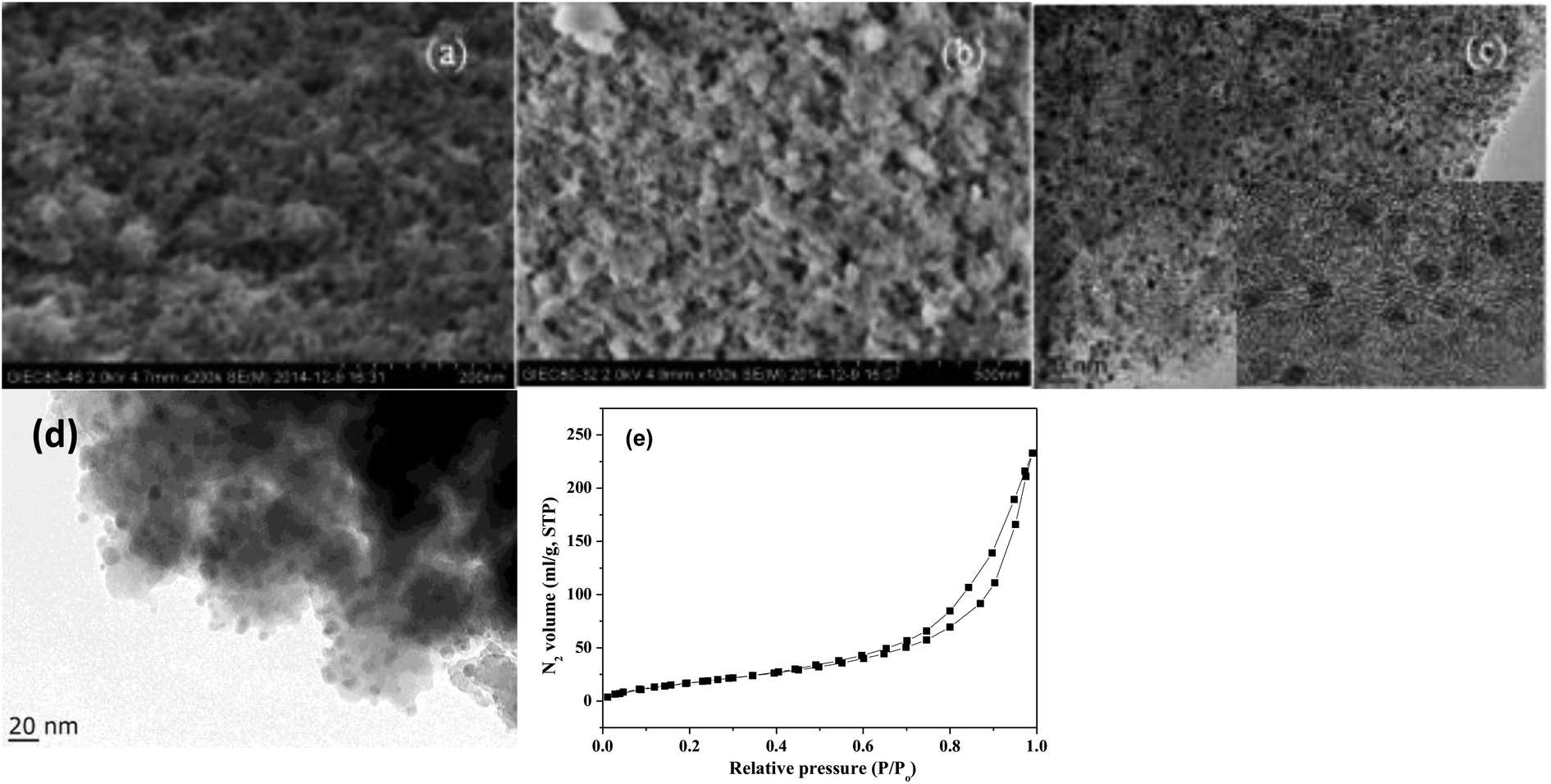 | ||
| Fig. 2 SEM images of fresh NbP (a) and ZrP (b). TEM images of Pd/C (c) and ZrP (d). N2 adsorption–desorption curve of ZrP (e). The inset in (c) shows the HRTEM image of Pd/C. | ||
| Entry | Catalyst | Surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | Average pore diameterb (nm) | Acidic densityc (mmol g−1) | Dd |
|---|---|---|---|---|---|---|
| a The surface areas were estimated by the BET method using N2 at −196 °C.b The pore volume and average pore diameter were determined by using the desorption branch and BJH method.c The acidic densities were calculated by NH3-TPD measurements and a calibration loop of 250 μL.d The dispersion of Pd was determined by H2-chemisorption.e The spent ZrP was treated at 300 °C for 3 h, dried and recalcinated at 400 °C for 4 h. The spent Pd/C was treated at 300 °C for 3 h, dried and recalcinated at 300 °C for 3 h under H2 flow. | ||||||
| 1 | ZrP | 112.7 | 0.25 | 6.8 | 1.55 | — |
| 2 | Spent ZrPe | 93.9 | 0.27 | 5.6 | 1.31 | — |
| 3 | NbP | 78.1 | 0.20 | 7.9 | 0.58 | — |
| 4 | Pd/C | 1025.9 | 0.28 | 3.8 | — | 0.49 |
| 5 | Spent Pd/Ce | 932.5 | 0.25 | 4.3 | — | 0.36 |
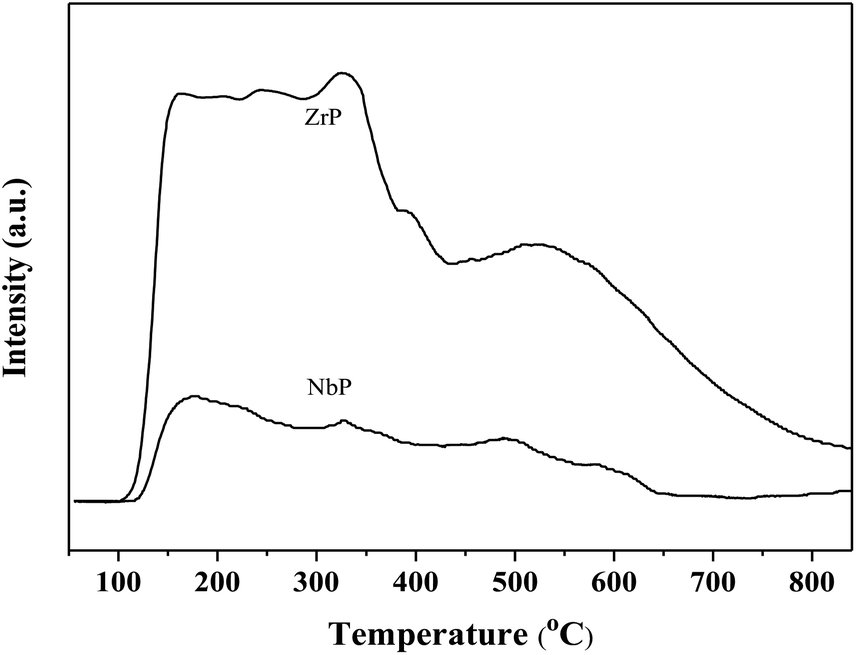 | ||
| Fig. 3 NH3-TPD profiles of fresh ZrP and NbP. | ||
Fig. 4 presents the FT-IR spectra of fresh, hydrothermally treated, and regenerated ZrP. The three samples presented the stretching vibrations of P–OH (the Bronsted acidic centers, 2354 cm−1), and tetrahedral P–O (1082 cm−1), and the bending vibration of P–O–P (752 cm−1), similar to the previous reports.42–45 After hydrothermal treatment, the peak intensity corresponding to P–O–P significantly reduced while that corresponding to P–OH increased, indicating that ZrP hydrolysis occurred during the treatment process. After recalcination, the P–O–P was regenerated at the expense of P–OH, based on the corresponding intensity changes. This means that the hydrolyzed P–OH could be re-condensed to recover the original structure. It was noted that the peak associated with the Zr–O–P framework was largely unchanged for the three samples, showing the hydrothermal stability of the prepared ZrP.
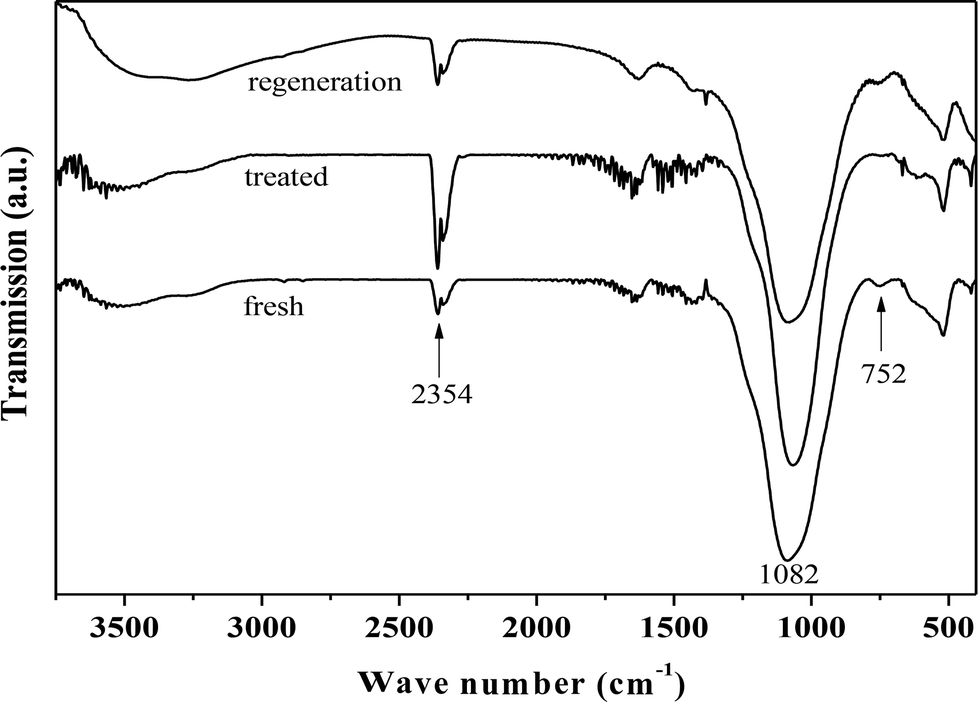 | ||
| Fig. 4 FT-IR spectra of fresh, hydrothermally treated, and regenerated ZrP. | ||
3.2. Condensation of furfural species with cyclic ketones
Aldol condensations of biomass-derived aldehydes (FF and HMF) with cyclic ketones (CP and CH) with mole ratios of 2:1 were used to synthesize C10–C18 precursors for downstream long-chained cycloalkane production. The condensation mechanism using FF and CP as an example is shown in Scheme S1.† The production of FF-CP-FF originates from consecutive C–C bond formation via a classical carbanion mechanism followed by dehydration over a base catalyst.
We began our study by choosing the aldol condensation of FF and CP as the model reaction to investigate. Various alkali catalysts (LiOH, NaOH, KOH, Ba(OH)2, Mg(OH)2, Na2CO3, Na2CO3) were used to catalyze this reaction at 30 °C (Table 2). The aldol condensation of FF with CP could not take place without any catalyst. However, when only a 5% molar equivalent of NaOH (relative to CP) was added into the mixture of FF and CP (at a FF:CP mole ratio of 2:1), the reaction took place quickly, producing a 68% yield of solid product in 40 min. After increasing the NaOH dosage to 10%, the conversion of FF and CP reached 100% (based on GC measurements; data not shown), and was accompanied by a high FF-CP-FF yield of 94%. Further increasing the NaOH to 20% slightly increased the yield of isolated condensation product FF-CP-FF to 96%. Other strong bases such as LiOH, KOH and Ba(OH)2 also showed excellent performance resulting in target product yields of 90% and above. However, no condensation product was observed when using the weak alkalis Mg(OH)2, Na2CO3 and K2CO3 as the catalysts even after 2 h, showing that such condensation preferentially takes place over a strong base. Because the strong base NaOH showed an excellent performance in the aldol condensation of FF with CP, we further employed NaOH for aldol condensations of FF with CH, and HMF with CP/CH. No obvious difference was observed when CH was used to replace CP as the starting material, which is in accordance with results from a previous study.46 Condensation products (FF-CP-FF and FF-CH-FF) were obtained in yields of over 90% from the condensation of FF with both CH and CP. When HMF was used as the starting material instead of FF, isolated condensation products (HMF-CP-HMF and HMF-CH-HMF) were obtained in over 95% yield. The condensation intermediates were yellowish for FF-CP-FF, FF-CH-FF and HMF-CH-HMF, and red for HMF-CP-HMF, as seen in the photographs of the isolated solid intermediates (Fig. S1†). The molecular structures of these intermediates were determined by 13C NMR measurement, which confirmed the synthesis of the desired products by aldol condensation without any impurities (Fig. S2–S4 and S6(a)†).
| Entry | Furfural species | Cyclic ketones | Catalyst dosageb (mol%) | Time (min) | Product | Isolated yieldc (C-mol%) |
|---|---|---|---|---|---|---|
| a All the reactions were conducted at 30 °C and the mole ratio of furfural compounds to cyclic ketone was 2:1.b Relative to cyclic ketone.c The isolated yield was calculated for the target product based on the cyclic ketone. |
||||||
| 1 | FF | CP | — | 40 | FF-CP-FF | 0 |
| 2 | FF | CP | NaOH, 5 | 40 | FF-CP-FF | 68 |
| 3 | FF | CP | NaOH, 10 | 40 | FF-CP-FF | 94 |
| 4 | FF | CP | NaOH, 20 | 40 | FF-CP-FF | 96 |
| 5 | FF | CP | KOH, 10 | 40 | FF-CP-FF | 93 |
| 6 | FF | CP | LiOH, 10 | 40 | FF-CP-FF | 87 |
| 7 | FF | CP | Ba(OH)2, 10 | 40 | FF-CP-FF | 92 |
| 8 | FF | CP | Mg(OH)2, 10 | 120 | FF-CP-FF | 0 |
| 9 | FF | CP | Na2CO3, 10 | 120 | FF-CP-FF | 0 |
| 10 | FF | CP | K2CO3, 10 | 120 | FF-CP-FF | 0 |
| 11 | FF | CH | NaOH, 10 | 40 | FF-CH-FF | 92 |
| 12 | HMF | CP | NaOH, 10 | 40 | HMF-CP-HMF | 96 |
| 13 | HMF | CH | NaOH, 10 | 40 | HMF-CH-HMF | 98 |
3.3. Long-chain cycloalkanes by hydrodeoxygenation of condensation intermediates
After obtaining the intermediates from the condensation of furfural species with cyclic ketones, we explored the hydrodeoxygenation of the condensation intermediates to cycloalkanes in an autoclave equipped with a mechanical stirrer.Solvent is necessary for the hydrodeoxygenation process because the condensation products are all solid. The common oxy-organic solvents (methanol, ethanol and tetrahydrofuran) are not suitable because they could be digested under the hydrodeoxygenation conditions. Water is comparatively stable during intermediate hydrodeoxygenation, and is suitable as a green and economical solvent. Supported metal combined with solid acid is typically used as an effective catalyst for this process. For example, Huang et al. reported that carbon-supported noble metals (Pd/C and Pt/C) combined with solid acids (NbOPO4 and TaOPO4) are efficient in dehydration/hydrogenation of fuel precursors into liquid alkanes with water as the reaction medium.47
Using water as the solvent, we employed a one-pot hydrodeoxygenation of the FF-CP-FF intermediate into long-chained cycloalkanes. A low intermediate conversion of less than 10% as well as negligible cycloalkane production (with a summed yield below 5%) were obtained when using Pd/C, Pt/C or Ru/C alone as catalysts (Fig. 5a), suggesting that oxygen removal is a main barrier for alkane formation in the current reaction conditions. The addition of solid acid significantly increased the conversion and the cycloalkane yield. We calculated the average TOF of Pd/C combined with ZrP and NbP catalysts from the Pd dispersion, respective acidic sites and FF-CP-FF conversion at 3 h. The values of 0.47 s−1 and 0.87 s−1 were obtained for Pd/C plus ZrP, and Pd/C plus NbP, respectively; NbP has a much lower value for acidic density. Fixing Pd/C as the hydrogenation catalyst, 38.7% and 70.0% of cycloalkanes were obtained with the addition of NbP and ZrP, respectively, evidencing their obvious promotion effect in this process. The yield of cycloalkanes obtained with ZrP was much higher than that with NbP, because the superior acidic properties of ZrP could enhance dehydration and produce more cycloalkanes. As shown in Fig. S7,† the cycloalkanes with 13–15 carbon atoms were the main products apart from some minor oxygenated compounds at retention times of more than 15 min. Because the carbon chain of the FF-CP-FF intermediate contains 15 carbon atoms, the formation of C13 and C14 was ascribed to hydrocracking during the hydrodeoxygenation process. Among these products, the C13 and C14 cycloalkanes presented as the major products. 54% of C13–C15 cycloalkanes were produced when using Pt/C combined with ZrP as the catalyst. This indicates that Pd/C and ZrP are preferable for this hydrodeoxygenation. Additionally, gas products including C1–C5 light alkanes were observed; the existence of solid acid promotes the formation of such products.
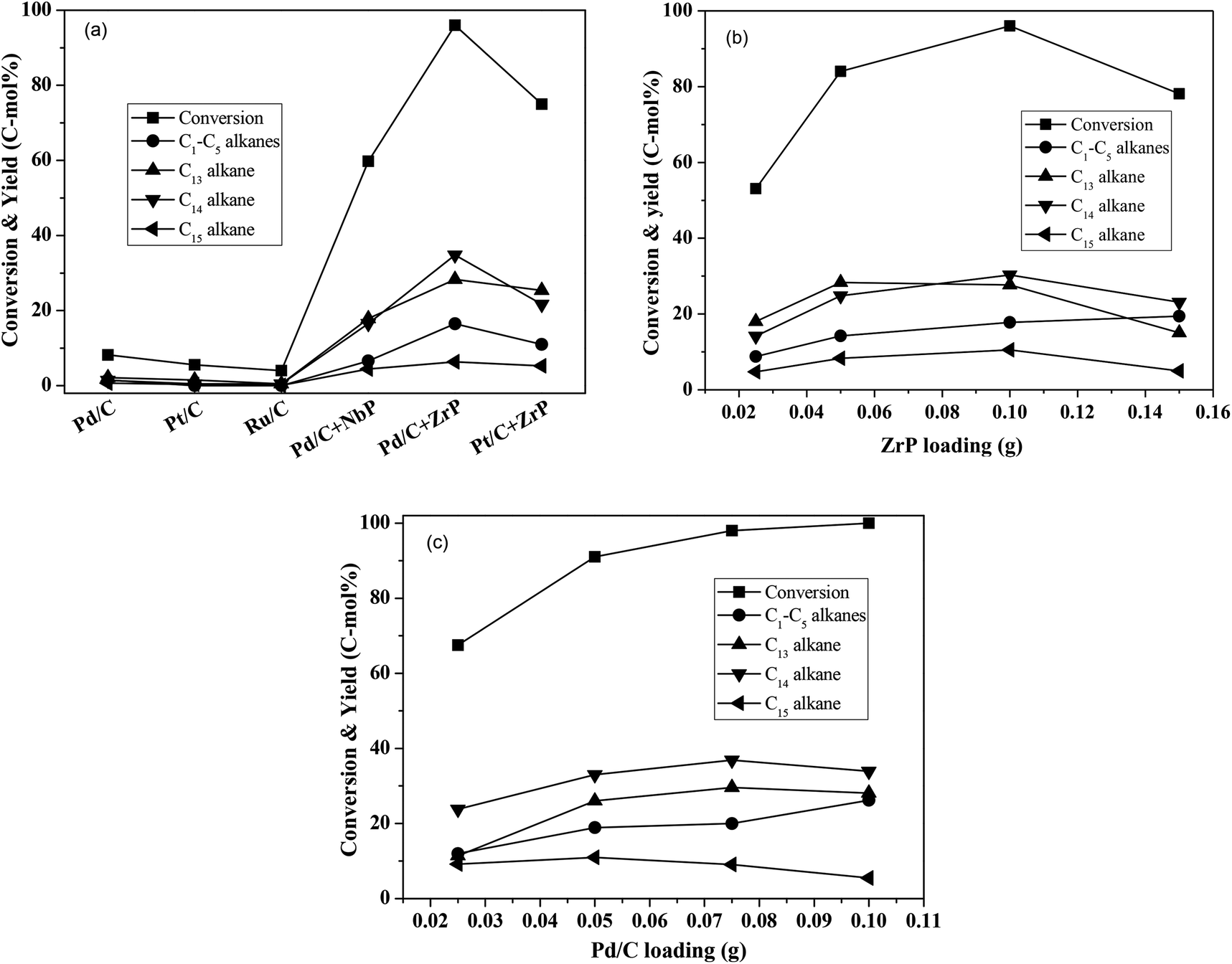 | ||
| Fig. 5 Hydrodeoxygenation of the FF-CP-FF intermediate to hydrocarbons over different solid acids and metals supported on activated carbon catalysts (a), with different ZrP loadings (b), and with different Pd/C loadings (c). Reaction conditions: 1.0 mmol FF-CP-FF, 15 mL H2O, 0.05 g metal supported on carbon, 0.10 g solid acid, 4 MPa H2 pressure, 300 °C, 3 h. | ||
ZrP and Pd/C loadings were further investigated and the results are shown in Fig. 5b–c. With increasing ZrP from 0.025 g to 0.1 g (fixing the Pd/C dosage at 0.05 g), the intermediate conversion and cycloalkane yield significantly increased from 53% to 96% and 47–76%, respectively, suggesting that the acid catalyst ZrP plays the critical role in oxygen removal. However, further increasing ZrP to 0.15 g led to a significant decrease in the cycloalkane yield, to 43%, as well as reduced conversion, which is perhaps caused by the intermediate coking and/or the produced cycloalkane hydrocracking to short-chain alkanes. In addition to the target products, the light alkanes (mostly CH4, C2H6, C3H8 and trace butane, pentane) increased with increasing ZrP and reached the highest proportion of 19% at 0.15 g ZrP. When the ZrP amount was fixed at 0.1 g, the cycloalkane yield increased with increasing amounts of Pd/C and the highest yield of 74% was obtained at 0.075 g Pd/C. When Pd/C was further increased to 0.1 g, the target products decreased but to a lesser degree than occurred with excess ZrP. This result shows that hydrocracking more likely occurred over solid acid. Based on the conversion efficiency and detected products by GC analysis, more than 90% of the carbon balance was obtained under the optimized conditions, showing that coking was suppressed during the process. At the same time, a decreased carbon balance of about 80% was observed in the case of excess ZrP or at a lower ratio of Pd/C to ZrP indicating that coking takes places with excessive solid acid. It is noted that the highest yield of 76% of cycloalkanes is comparable to that obtained in previous reports.18–20
By using the combined catalyst (0.05 g Pd/C and 0.1 g ZrP), we studied the influences of reaction temperature and time on cycloalkane production from the FF-CP-FF intermediate (Fig. 6). At a low temperature of 260 °C, a total cycloalkane yield of only 13% was obtained, but this greatly increased to 36% and 70% when increasing the temperature to 280 °C and 300 °C, respectively. However, further increasing the reaction temperature to 320 °C, decreased the cycloalkane yield to 49%. Additionally, the proportion of light alkanes increased with increasing temperature, reaching the highest value of 29% at 320 °C due to the enhanced hydrocracking at higher temperatures. Among these light alkanes, CH4 was selectively produced (>85% of the gaseous products) no matter what temperature was used (Fig. 7). The impact of reaction time was also investigated at 300 °C. At the short time of 1 h, the yield of C13–C15 cycloalkanes was 34%. The cycloalkane yield increased to 56% and 70% when prolonging the reaction time to 2 h and 3 h, respectively. However, further increasing the time led to a decreased cycloalkane yield. Similar to the effect of temperature, the lengthened time induced hydrocracking during the hydrodeoxygenation process and produced more light alkanes, resulting in reduced target products.
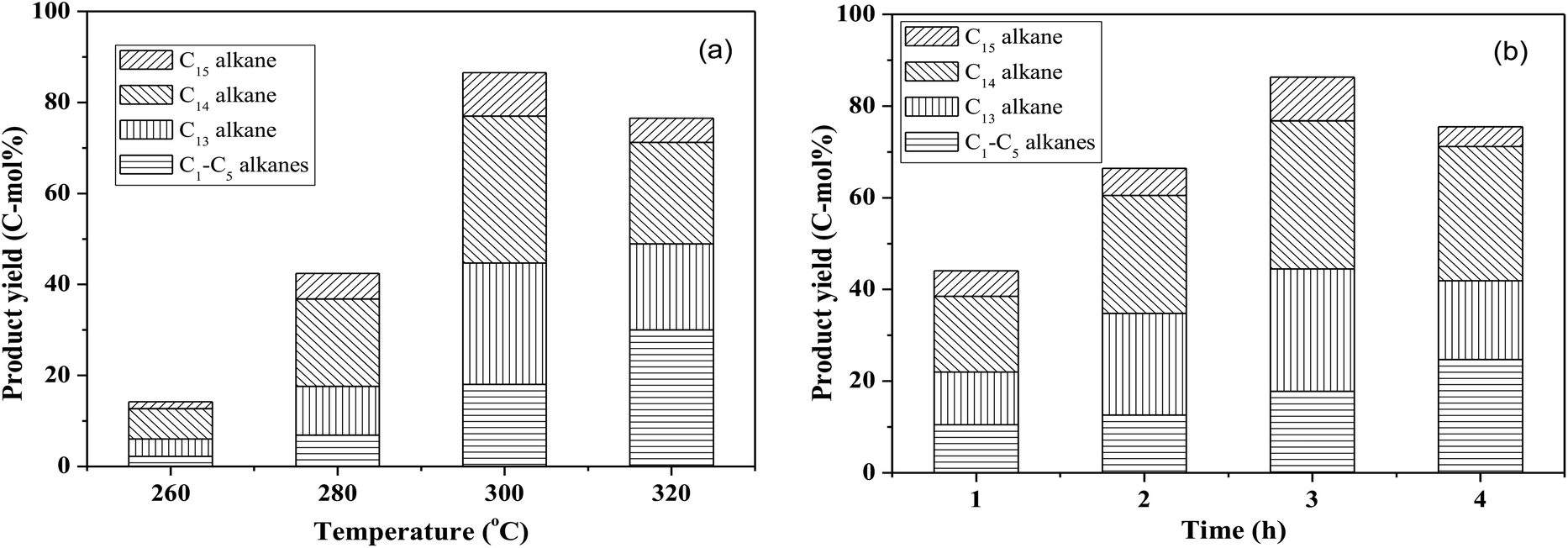 | ||
| Fig. 6 The influence of reaction temperature (a) and time (b) on the hydrodeoxygenation of the FF-CP-FF intermediate over ZrP and Pd/C. Reaction conditions: 1.0 mmol FF-CP-FF, 15 mL H2O, 0.05 g Pd/C, 0.10 g ZrP, 4 MPa H2 pressure. | ||
 | ||
| Fig. 7 The influence of reaction temperature on gas product distribution in FF-CP-FF hydrodeoxygenation. The reaction conditions are as stated for Fig. 6(a). | ||
Hydrodeoxygenation of FF-CP-FF to produce cycloalkanes involves saturation of CO/CC bonds followed by oxygen removal. To ascertain this process, FF-CP-FF was hydrogenated into water-soluble oxygenated chemicals at the low temperature of 150 °C when using the single Pd/C catalyst (Fig. S5†) and the structures of the products were analyzed by the 13C NMR technique (Fig. S6†). The peaks at around 200 ppm, corresponding to CO bonds, had not disappeared, suggesting that the CO bonds in the FF-CP-FF were not efficiently hydrogenated into C–O bonds. The peaks in the range of 100 ppm to 170 ppm, which are associated with CC bonds, were greatly decreased, indicating the hydrogenation of CC bonds to C–C bonds. For the products obtained at the higher temperature of 250 °C, the peaks higher than 100 ppm all disappeared, confirming the saturation of these CC and CO bonds into C–O and C–C bonds, respectively. The quantified analysis shown in Table 3 further demonstrated the sequential CC and CO bond hydrogenation. At the reaction temperature of 300 °C, most of the oxygen was removed by cracking the C–O bonds, with only 3.5% of C–O bonds remaining in the final products. Introduction of solid acid promotes the dehydration of hydrogenation-derived C–OH, to produce the target products with high yields.
| CO (%) |
CC (%) |
C–O (%) | C–C (%) | |
|---|---|---|---|---|
| FF-CP-FF | 6.8 | 80.1 | 0 | 13.1 |
| Hydrogenation at 150 °C | 1.9 | 0.8 | 26.3 | 72.2 |
| Hydrogenation at 250 °C | 0 | 0 | 17.2 | 82.6 |
| Hydrogenation at 300 °C | 0 | 0 | 3.5 | 96.6 |
Fig. 8 shows the catalytic stability and regeneration performance of Pd/C combined with ZrP. The cyclic alkanes decreased from the initial 70% to a stable yield of 57% while simultaneously the light alkanes reduced from the initial 19% to below 10% during cycling. This decrease is due to a reduction in the acidic sites caused by P leaching in this hydrothermal hydrodeoxygenation according to our previous report.48 Considering that mixed Pd/C and ZrP are hardly separable from each other, we simulated the catalyst regeneration by hydrothermal treatment of the individual Pd/C and ZrP followed by recalcination according to the optimum reaction conditions. Interestingly, no matter which catalyst was regenerated, the activity of the combined catalyst could be recovered to the original level, demonstrating the excellent regeneration performance. After regeneration, the ZrP showed a similar amorphous nature to the fresh catalyst (Fig. 9). The weak and broad diffractions of the regenerated Pd/C show that Pd particles were still highly dispersed on the carbon surface although their intensities were slightly higher than those in the fresh catalyst. The good retention of the physicochemical properties for both regenerated catalysts is the main reason for their excellent performance.
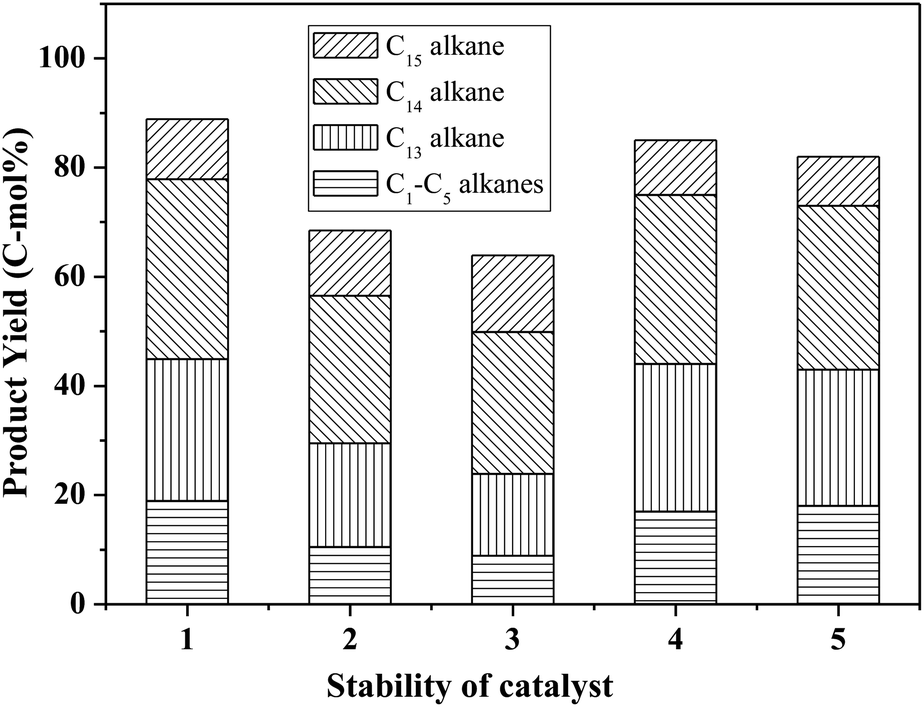 | ||
| Fig. 8 Catalytic stability of ZrP and Pd/C in the hydrodeoxygenation of FF-CP-FF to cyclic alkanes. Reaction conditions: 1.0 mmol FF-CP-FF, 15 mL H2O, 0.05 g Pd/C, 0.10 g ZrP, 4 MPa H2 pressure, 300 °C, 3 h. Bars 1–3 show successive cycles, bar 4 represents the regenerated ZrP combined with fresh Pd/C and bar 5 represents the regenerated Pd/C combined with fresh ZrP. | ||
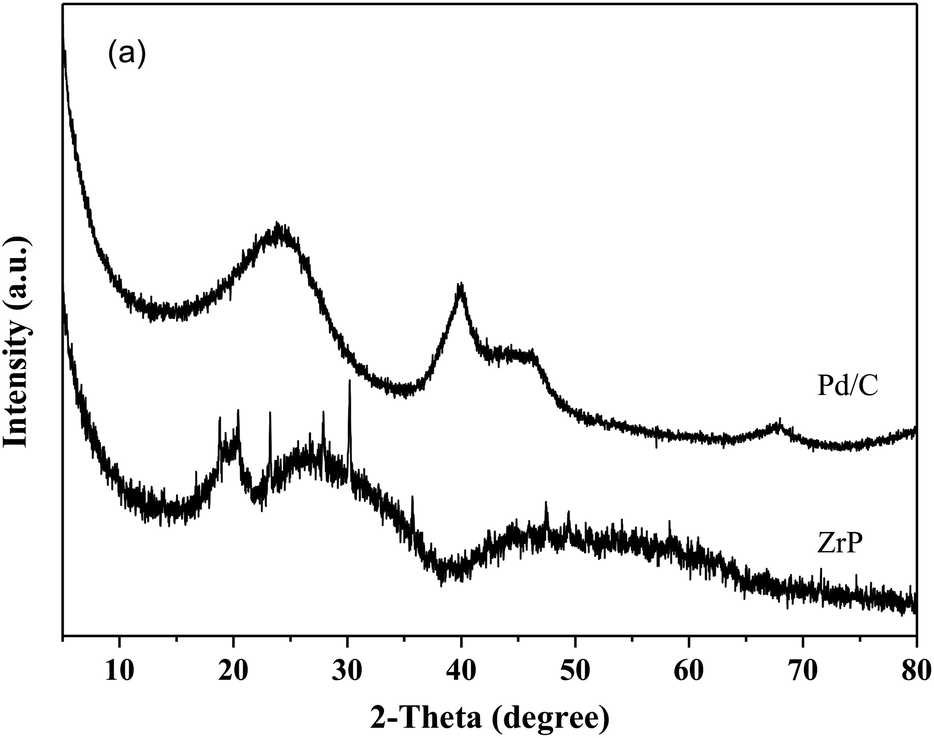 | ||
| Fig. 9 XRD patterns of hydrothermally treated ZrP and Pd/C followed by recalcination. | ||
Hydrodeoxygenation of other aldol condensation intermediates (FF-CH-FF, HMF-CP-HMF, HMF-CH-HMF) were also investigated under the optimized reaction conditions by using Pd/C combined with ZrP as the catalyst. Cycloalkanes with carbon chain lengths of between 13 and 18 were obtained. In the cases of FF-CH-FF and HMF-CP-HMF, 55% and 72% yields, respectively, of cycloalkanes were obtained. When using HMF-CH-HMF as the feedstock, a 43% yield of cycloalkanes was obtained. This indicates that the developed catalyst of Pd/C combined with ZrP is generally efficient for the production of jet fuel and/or diesel hydrocarbons by hydrodeoxygenation of these condensation intermediates (Fig. 10).
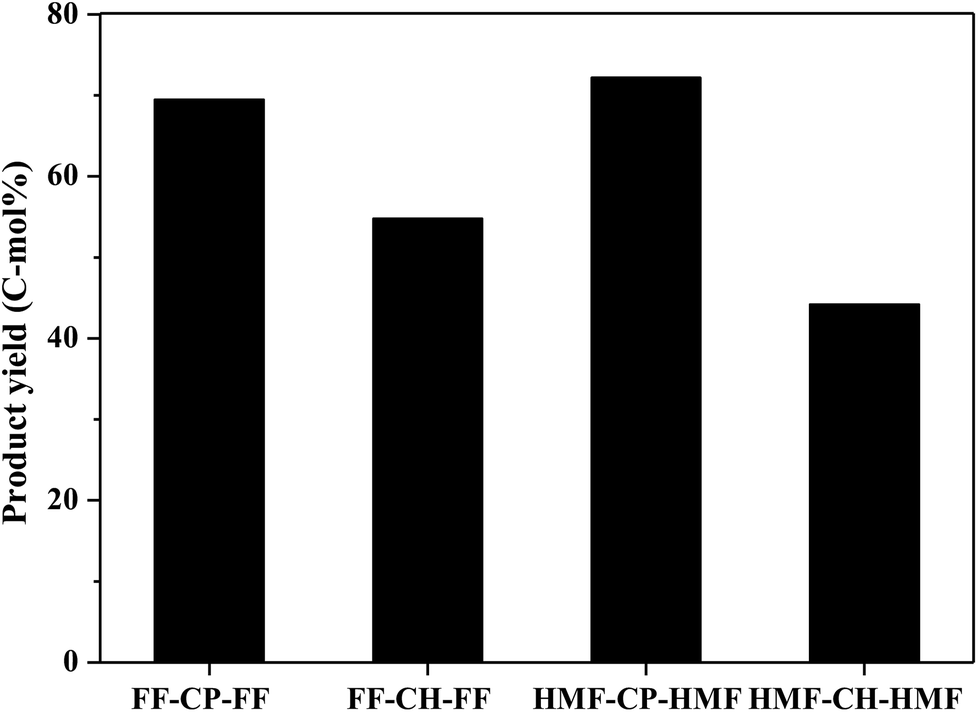 | ||
| Fig. 10 C13–C18 cycloalkane production from various condensation intermediates. Reaction conditions: 1.0 mmol condensation intermediates, 15 mL H2O, 0.05 g Pd/C, 0.1 g ZrP, 4 MPa H2 pressure, 300 °C, 3 h. | ||
4. Conclusions
Long-chained cycloalkanes with carbon chain lengths of 13–18 were obtained from furfural species (FF and HMF) and cyclic ketones (CP and CH), all of which could be obtained from the renewable lignocellulosic biomass. The aldol condensation of the furfural species and cyclic ketones was conducted at 30 °C with a strong basic catalyst, to obtain over 90% yields of the desired condensation intermediates. The long-chained cycloalkanes with carbon chain lengths of 13–18 were produced via hydrodeoxygenation of the condensation intermediates at temperatures of 280–320 °C and times of 1–4 h by using a catalyst of Pd/C combined with the solid acid ZrP. Under the optimized conditions, the highest yield of 76% of target products could be obtained. The combined catalyst showed reduced yields of cycloalkanes after the second and third cycles, but its performance could be largely recovered to its original level by hydrothermal treatment followed by recalcination of Pd/C and ZrP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of China (51576199, 51536009 and 51376185) and the Natural Science Foundation of Guangdong Province (2017A030308010).References
- R. A. Sheldon, Green Chem., 2014, 16, 950 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516 RSC.
- N. Shi, Q. Y. Liu, Q. Zhang, T. J. Wang and L. L. Ma, Green Chem., 2013, 15, 1967 RSC.
- Y. H. Liao, Q. Y. Liu, T. J. Wang, J. X. Long, L. L. Ma and Q. Zhang, Green Chem., 2014, 16, 3305 RSC.
- Z. R. Zhang, J. L. Song and B. X. Han, Chem. Rev., 2017, 117, 6834 CrossRef CAS PubMed.
- F. Cheng and C. E. Brewer, Renewable Sustainable Energy Rev., 2017, 72, 673 CrossRef CAS.
- A. Bohre, S. Dutta, B. Saha and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2015, 3, 1263 CrossRef CAS.
- P. Y. Bi, J. C. Wang, Y. J. Zhang, P. W. Jiang, X. P. Wu, J. X. Liu, H. Xue, T. J. Wang and Q. X. Li, Bioresour. Technol., 2015, 183, 10 CrossRef CAS PubMed.
- J. P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150 CrossRef CAS PubMed.
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Y. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500 CAS.
- C. R. Li, D. Q. Ding, Q. N. Xia, X. H. Liu and Y. Q. Wang, ChemSusChem, 2016, 9, 1712 CrossRef CAS PubMed.
- Y. P. Li, X. M. Huang, Q. Zhang, L. G. Chen, X. H. Zhang, T. J. Wang and L. L. Ma, Appl. Energy, 2015, 160, 990 CrossRef CAS.
- A. Bohre, B. Saha and M. M. Abu-Omar, ChemSusChem, 2015, 8, 4022 CrossRef CAS PubMed.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446 CrossRef CAS PubMed.
- L. Faba, E. Díaz and S. Ordóñez, ChemSusChem, 2014, 7, 2816 CrossRef CAS PubMed.
- M. X. Su, W. Z. Li, T. W. Zhang, H. S. Xin, S. Li, W. Fan and L. L. Ma, Catal. Sci. Technol., 2017, 7, 3555 CAS.
- G. Y. Li, N. Li, S. S. Li, A. Q. Wang, Y. Cong, X. D. Wang and T. Zhang, Chem. Commun., 2013, 49, 5727 RSC.
- G. Y. Li, N. Li, J. F. Yang, A. Q. Wang, X. D. Wang, Y. Cong and T. Zhang, Bioresour. Technol., 2013, 134, 66 CrossRef CAS PubMed.
- M. H. Chávez-Sifontes, M. E. Domine and S. Valencia, Catal. Today, 2015, 257, 305 CrossRef.
- J. F. Yang, N. Li, G. Y. Li, W. T. Wang, A. Q. Wang, X. D. Wang, Y. Cong and T. Zhang, ChemSusChem, 2013, 6, 1149 CrossRef CAS PubMed.
- C. L. Cai, Q. Y. Liu, J. Tan, T. J. Wang, Q. Zhang and L. L. Ma, Korean Chem. Eng. Res., 2016, 54, 519 CrossRef CAS.
- J. Tan, Q. Y. Liu, C. L. Cai, S. B. Qiu, T. J. Wang, Q. Zhang, L. L. Ma and G. Y. Chen, RSC Adv., 2015, 5, 58784 RSC.
- J. L. Xu, N. Li, X. F. Yang, G. Y. Li, A. Q. Wang, Y. Cong, X. D. Wang and T. Zhang, ACS Catal., 2017, 7, 5880 CrossRef.
- A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chem., Int. Ed., 2011, 50, 2375 CrossRef CAS PubMed.
- A. Corma, O. de la Torre and M. Renz, ChemSusChem, 2011, 4, 1574 CrossRef CAS PubMed.
- G. Y. Li, N. Li, Z. Q. Wang, C. Z. Li, A. Q. Wang, X. D. Wang, Y. Cong and T. Zhang, ChemSusChem, 2012, 5, 1958 CrossRef CAS PubMed.
- M. Balakrishnan, E. R. Sacia and A. T. Bell, ChemSusChem, 2014, 7, 1078 CrossRef CAS PubMed.
- A. K. Misra and G. Agnihotri, Carbohydr. Res., 2004, 339, 1381 CrossRef CAS PubMed.
- G. Q. Xu, Q. Li, J. G. Feng, Q. Liu, Z. J. Zhang, X. C. Wang, X. Y. Zhang and X. D. Mu, ChemSusChem, 2014, 7, 105 CrossRef CAS PubMed.
- D. J. Liu and Y. X. E. Chen, ACS Catal., 2014, 4, 1302 CrossRef CAS.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., Int. Ed., 2014, 53, 1854 CrossRef CAS PubMed.
- J. Y. Xin, S. J. Zhang, D. X. Yan, O. Ayodele, X. M. Lu and J. J. Wang, Green Chem., 2014, 16, 3589 RSC.
- G. Y. Li, N. Li, X. K. Wang, X. R. Sheng, S. S. Li, A. Q. Wang, Y. Cong, X. D. Wang and T. Zhang, Energy Fuels, 2014, 28, 5112 CrossRef CAS.
- M. Hronec and K. Fulajtarova, Catal. Commun., 2012, 24, 100 CrossRef CAS.
- M. Hronec, K. Fulajtarova and T. Liptaj, Appl. Catal., A, 2012, 437, 104 CrossRef.
- H. Z. Liu, T. Jiang, B. X. Han, S. G. Liang and Y. X. Zhou, Science, 2009, 326, 1250 CrossRef CAS PubMed.
- G. Y. Xu, J. H. Guo, Y. Zhang, Y. Fu, J. Z. Chen, L. L. Ma and Q. X. Guo, ChemCatChem, 2015, 7, 2485 CrossRef CAS.
- M. Hronec, K. Fulajtarova, T. Liptaj, M. Stolcova, N. Pronayova and T. Sotak, Biomass Bioenergy, 2014, 63, 291 CrossRef CAS.
- J. F. Yang, N. Li, G. Y. Li, W. T. Wang, A. Q. Wang, X. D. Wang, Y. Cong and T. Zhang, Chem. Commun., 2014, 50, 2572 RSC.
- T. V. Bui, T. Sooknoi and D. E. Resasco, ChemSusChem, 2017, 10, 1631 CrossRef CAS PubMed.
- J. C. G. Da Silva, S. Folgueras-Dominguez and A. C. B. Dos Santos, J. Mater. Sci. Lett., 1999, 18, 197 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, B. Roy, P. Pal, H. C. Bajaj and A. B. Panda, Appl. Catal., B, 2011, 103, 378 CrossRef CAS.
- H. N. Kim, S. W. Keller, T. E. Mallouk, J. Schmitt and G. Decher, Chem. Mater., 1997, 9, 1414 CrossRef CAS.
- H. M. Gan, X. G. Zhao, B. N. Song, L. Guo, R. Zhang, C. Chen, J. Z. Chen, W. W. Zhu and Z. S. Hou, Chin. J. Catal., 2014, 35, 1148 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, B. Roy, A. Tarafdar, H. C. Bajaj and A. B. Panda, Appl. Catal., A, 2010, 385, 22 CrossRef CAS.
- J. T. Li, G. F. Chen, J. X. Wang and T. S. Li, Synth. Commun., 1999, 29, 965 CrossRef CAS.
- Y. B. Huang, Z. Yang, J. J. Dai, Q. X. Guo and Y. Fu, RSC Adv., 2012, 2, 11211 RSC.
- Q. Y. Liu, T. Zhang, Y. H. Liao, C. L. Cai, J. Tan, T. J. Wang, S. B. Qiu, M. H. He and L. L. Ma, ACS Sustainable Chem. Eng., 2017, 5, 5940 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01723a |
| This journal is © The Royal Society of Chemistry 2018 |